

ABSTRACT
IL13Rα2 is a cell surface tumor antigen that is overexpressed in multiple tumor types. Here, we studied biodistribution and targeting potential of an anti-IL13Rα2 antibody (Ab) and anti-tumor activity of anti-IL13Rα2-antibody-drug conjugate (ADC). The anti-IL13Rα2 Ab was labeled with fluorophore AF680 or radioisotope 89Zr for in vivo tracking using fluorescence molecular tomography (FMT) or positron emission tomography (PET) imaging, respectively. Both imaging modalities showed that the tumor was the major uptake site for anti-IL13Rα2-Ab, with peak uptake of 5–8% ID and 10% ID/g as quantified from FMT and PET, respectively. Pharmacological in vivo competition with excess of unlabeled anti-IL13Rα2-Ab significantly reduced the tumor uptake, indicative of antigen-specific tumor accumulation. Further, FMT imaging demonstrated similar biodistribution and pharmacokinetic profiles of an auristatin-conjugated anti-IL13Rα2-ADC as compared to the parental Ab. Finally, the anti-IL13Rα2-ADC exhibited a dose-dependent anti-tumor effect on A375 xenografts, with 90% complete responders at a dose of 3 mg/kg. Taken together, both FMT and PET showed a favorable biodistribution profile for anti-IL13Rα2-Ab/ADC, along with antigen-specific tumor targeting and excellent therapeutic efficacy in the A375 xenograft model. This work shows the great potential of this anti-IL13Rα2-ADC as a targeted anti-cancer agent.
KEYWORDS: Antibody drug conjugate, cancer, efficacy, fluorescence molecular tomography, IL13rα2, imaging, pharmacokinetics, positron emission tomography, tumor targeting
Introduction
While surgery, chemotherapy, and radiation therapy have been used to treat cancers for decades or even centuries, monoclonal antibody (mAb)-based therapeutics are a relatively new, yet still rapidly growing, category.1,2 The discovery of tumor-associated antigens was first made in the 1960s, when cancer cells were observed to over-express or preferentially express secreted or cell surface-bound targets that were not as commonly found as in normal tissues.1,3 Since the late 1990s, several types of antibody-based cancer therapeutics have been established that exploit various functions of the antibody: agonist or antagonist antibodies that function through modulating the target itself; antibodies that elicit or modulate immune reactions against the target; and antibody-drug conjugates (ADCs) that deliver highly cytotoxic drugs (payloads) to the target-expressing tumor cells. Compared to conventional chemotherapies, payloads delivered by ADCs are conceptualized to have reduced systemic exposure and toxicity, resulting in a broader therapeutic index.4–6 Here, we report a mAb against a tumor antigen, IL13Rα2. We examined the pharmacologic properties of the antibody, as well as its therapeutic potentials in the cancer-targeted ADC setting.
There are two types of receptors (R) for human interleukin 13 (IL13). First, the “shared” IL4/IL13 receptor, which consists of an IL4Rα chain and an IL13Rα1 chain (also known as IL13R alpha’).7 This heterodimeric receptor is responsible for signal transduction and effector function of IL13.8 IL13 also binds to a second type of receptor, an IL4 independent, “restrictive”, monomeric receptor, IL13Rα2, which has a short cytoplasmic tail (17 amino acids), and therefore is incapable of signaling via the canonical JAK/STAT pathway, but has an extraordinarily high binding affinity toward IL13 compared to the shared receptor.7,9 Because of these features, IL13Rα2 was once considered a decoy receptor that can sequester and inhibit the signaling of IL13.10,11 Recent studies have demonstrated that IL13Rα2 stimulates a signaling cascade that is separate from the STAT6 pathway. Depending on the cell context, the expression of IL13Rα2 can be increased by tumor necrosis factor (TNF) alone or in synergy with IL17, IL13 or IL4.11,12
In addition to IL13, chitinase 3-like 1 has been identified as another binding ligand for IL13Rα2.13,14 Upon ligand binding, IL13Rα2 is able to elicit activator protein 1 (AP-1) activation and subsequent transforming growth factor (TGF)β induction, as well as MAPK, Akt/PKB, and Wnt/β-catenin signaling and to promote cancer metastasis.12,13,15 In oncology, over-expression of IL13Rα2 was found in the majority of glioblastoma multiforme patients. The expression pattern, assessed in tumor sections from patients, was reported to be abundant and relatively homogeneous.16 The over-expression of IL13Rα2 has also been linked to rapid growth, metastasis, and/or poor prognosis in breast, lung, gastric, pancreatic, and ovarian cancers,15,17–20 as well as increased tumorigenicity in melanoma models.21 Further, several groups have validated that IL13Rα2 undergoes rapid internalization following ligand, peptide, or antibody interactions, which suggests that this target could be amenable to antibody/ADC targeting.22,23 Taken together, these data supported the exploitation of IL13Rα2 as a cancer antigen in the construction of an ADC, to promote tumor targeted delivery.24–26
We previously demonstrated the strong expression of IL13Rα2 on A375 tumors compared to U87MG or H460 tumor cell lines.27 To determine the utility of our anti-IL13Rα2 antibody (Ab) in the ADC setting, we first examined the pharmacokinetics (PK) and biodistribution characteristics of this antibody (without the drug load) in nude mice bearing IL13Rα2-positive A375 xenograft tumors. Further, IL13Rα2-specific tumor targeting was confirmed by in vivo blocking experiments. In both sets of experiments, the antibody was labeled using two methods, near-infrared fluorophore AF680 and radioisotope 89Zr chelated via deferoxamine (DFO), to enable fluorescence molecular tomography (FMT) and positron emission tomography (PET) imaging, respectively. Both imaging modalities can generate three-dimensional longitudinal images, and PET imaging is readily translatable to the clinic. Finally, we asked whether the anti-IL13Rα2 ADC with auristatin as the cytotoxic payload (PF-06473811) exhibited similar pharmacological properties, and more importantly, could convey therapeutic efficacy in xenograft tumor models.
Results
Conjugation did not affect antigen binding for anti-IL13Rα2-Ab-AF680 or anti-IL13Rα2-Ab-DFO
The effect of fluorophore or DFO conjugation on antigen binding was accessed by cell binding assay using flow cytometry. Note that fluorophore and DFO conjugations were done separately, and thus the antibody in the research work was not double labeled. Both AF680- and DFO-conjugated anti-IL13Rα2-Ab showed only a marginal peak shift in the cell binding curves (Figure 1(a,b)), and Figure S1A). The mean channel intensities for samples incubated with the fluorophore- or DFO-conjugated anti-IL13Rα2-Ab were about 60–70% of the samples incubated with the unconjugated Ab (data not shown). These results were consistent with previous reports27 and confirmed that neither AF680 nor DFO conjugation changed the antigen binding properties of the anti-IL13Rα2 Ab.
Figure 1.
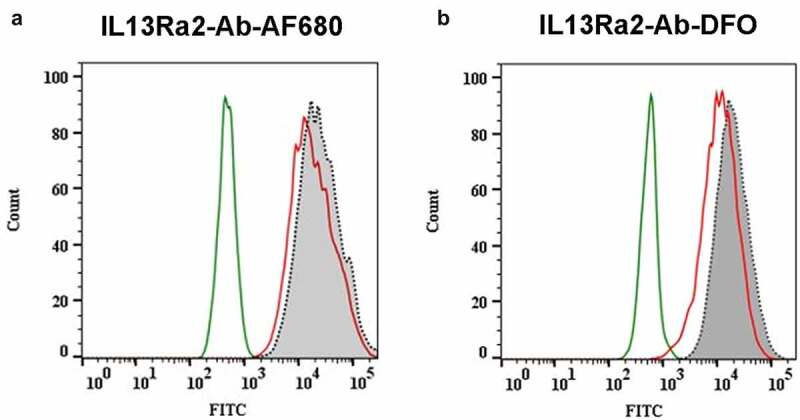
Effect of AF680 and DFO conjugation on antigen binding of the anti-IL13Rα2 Ab by flow cytometry. IL13Rα2-expressing A375 cells were incubated with 8.8-mAb (isotype control), anti-IL13Rα2-Ab, anti-IL13Rα2-Ab-AF680, or anti-IL13Rα2-Ab-DFO, followed by detection with a FITC-labeled secondary Ab. (a) AF680 conjugation to anti-IL13Rα2-Ab had no impact on A375 tumor cell binding. (b) DFO conjugation to anti-IL13Rα2-Ab had no impact on A375 tumor cell binding. Histogram labeling: Green: isotype control 8.8-mAb; Gray: anti-IL13Rα2-Ab; Red: anti-IL13Rα2-Ab-AF680 or anti-IL13Rα2-Ab-DFO
FMT-based biodistribution and PK of anti-IL13Rα2-Ab-AF680
Anti-IL13Rα2-Ab-AF680 was injected at 1, 3, and 10 mg/kg dose levels in the xenograft-bearing mice, followed by longitudinal FMT imaging. Representative time-course images of the mice from all three dose groups are shown in Figure 2(a,b). FMT imaging showed high fluorescence throughout the torso at 5 min post-injection (Figure 2a). Timepoints afterward showed a gradual reduction in torso and heart fluorescence signal. Interestingly, the rate of decrease in fluorescence over time was lower as dose increased. For example, the fluorescence in the low-dose group (1 mg/kg) was very negligible at 48 h post-injection, whereas significant fluorescence was observed in the high-dose group (10 mg/kg) at 240 h post-injection (Figure 2a). A three-dimensional (3D) region of interest (ROI) around the tumor demonstrated that the uptake peaked between 6–48 h for all the dose groups, with gradual reduction thereafter (Figure 2b). A dose-dependent higher signal was observed in the tumors at later timepoints. The quantification of FMT signal for heart, a surrogate for blood profile, showed the highest %ID at all timepoints for the 10 mg/kg dose group, and the lowest %ID for the 1 mg/kg dose group, suggesting that anti-IL13Rα2-Ab-AF680 was being eliminated faster at lower dose levels as compared to higher doses (Figure 2c). The heart concentrations also showed a biphasic decline at all dose levels, suggesting a rapid distribution phase followed by steady elimination phase from blood (Figure 2c). At 240 h post-injection, anti-IL13Rα2-Ab-AF680 was only detectable in blood in the 10 mg/kg dose group while the other two groups were below level of detection. The peak tumor uptake (%ID) was observed to be similar at all three dose levels, which ranged from 5– 8%ID (Figure 2d) and between 6- and 48-h post-injection. However, tumor uptake curves showed that the rate of elimination from tumors was higher at lower doses, as compared to the high-dose group (Figure 2d), a phenomenon consistent with heart clearance.
Figure 2.
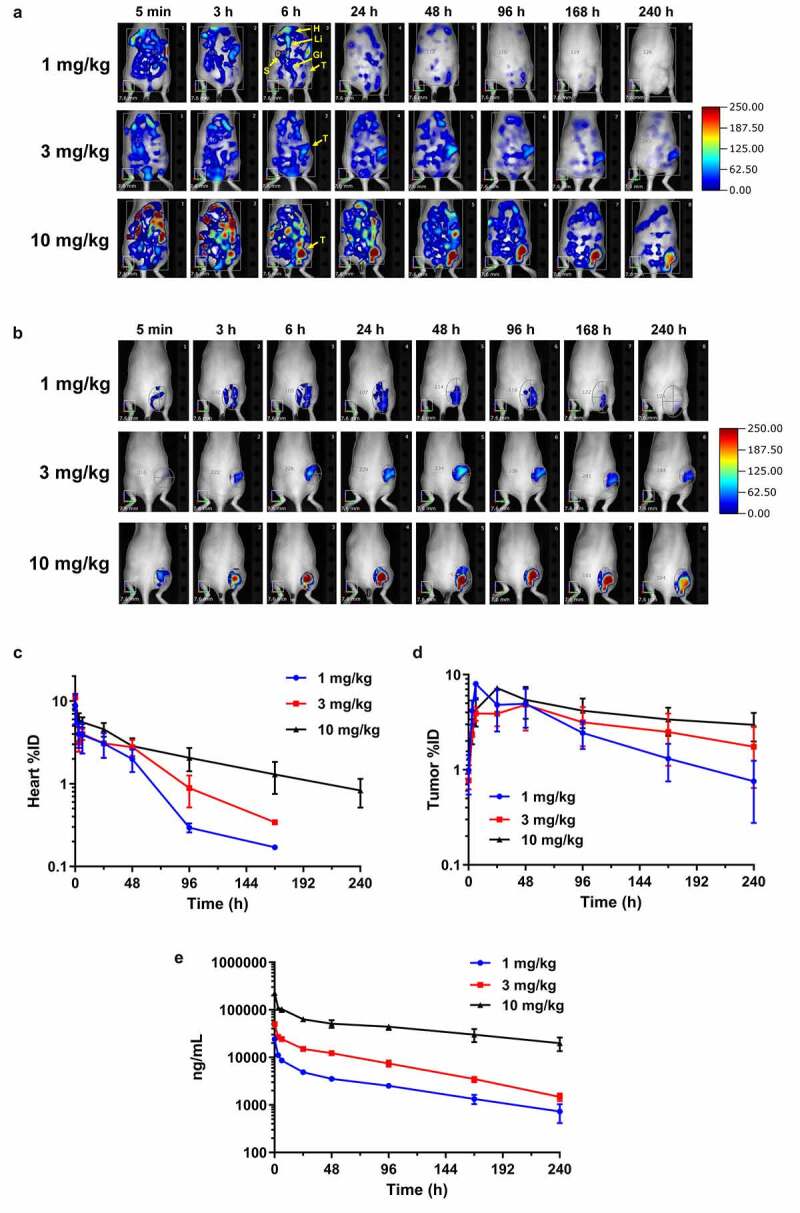
In vivo biodistribution and tumor targeting by FMT imaging. (a) noninvasive longitudinal FMT imaging of whole body at 5 min, 3, 6, 24, 48, 96, 168, and 240 h after injection of anti-IL13Rα2-Ab-AF680 in A375 xenograft-bearing mice at 1, 3, or 10 mg/kg doses. Shown are time course images of one representative mouse per group. T – tumor; H – heart; Li – liver; S – spleen; Gi – gastrointestinal tract. (b) Tumor regions of interest (ROIs) demonstrating targeting profile and increased fluorescence signal with higher doses. (c-d) In vivo ROI quantitation of heart (c) and tumor (d) uptake (%ID) in various dose groups. (e) Plasma pharmacokinetic profile of anti-IL13Rα2-Ab at different doses as evaluated by ligand binding assay. N = 3– 6 mice per group for each time point; data are Mean ± SEM
The plasma concentration of anti-IL13Rα2-Ab-AF680 was assessed by ligand binding assay. Overall concentration profiles showed biphasic curves with a rapid distribution phase that lasted for 24 h, followed by steady elimination from the blood (Figure 2e and Table 1). The half-lives for 1, 3, and 10 mg/kg dose groups ranged from 3–5 days and clearance ranged from 0.7–1.3 mL/h/kg (Table 1). The anti-IL13Rα2 Ab does not cross-react with mouse IL13Rα2, therefore any effect of antigen sink on the PK of this antibody could not be assessed. Of note, as shown in Figure S1B, the AF680-labeled anti-IL13Rα2 Ab showed similar PK profile to the unlabeled anti-IL13Rα2 Ab (parental antibody).
Table 1.
Non-compartmental analysis parameters for the anti-IL13Rα2 Ab at 1, 3, and 10 mg/kg
| Dose (mg/kg) | t½ (h) |
AUC (h.µg/mL) |
Cl (mL/h/kg) |
|
|---|---|---|---|---|
| Anti-IL13Rα2-Ab-AF680 | 1 | 80 | 663 | 1.34 |
| Anti-IL13Rα2-Ab-AF680 | 3 | 63 | 1924 | 1.46 |
| Anti-IL13Rα2-Ab-AF680 | 10 | 125 | 10,462 | 0.71 |
The PK parameters were determined from plasma concentrations of anti-IL13Rα2-Ab-AF680 at different timepoints obtained at three dose levels: 1, 3, and 10 mg/kg. Non-compartmental analysis was performed using software WinNonlin®.
Ex vivo FMT imaging was performed on selected organs after whole-body perfusion at 24, 96, and 240 h for all three dose groups. Representative images in Figure S2 and fluorescence quantitation normalized to tissue weight (Figure S3) showed accumulation of anti-IL13Rα2-Ab-AF680 in different organs. Tumor was the site with the highest uptake at any dose level and at all timepoints. The tumor uptake was similar in the 1- and 3-mg/kg groups and the concentrations remained stable at 24 and 96 h, but declined at 240 h. Interestingly, the high-dose group showed similar and stable tumor uptake at all three timepoints. This could be partly due to saturation of intracellular dye degradation pathways at the high dose level. Tumor uptake ranged between 8– 12%ID/g at doses of 1 and 10 mg/kg. However, tumor uptake was observed to be slightly higher (18– 23%ID/g) at 3 mg/kg, as compared to other dose levels. Among the other organs evaluated, liver showed uptake between 2– 7%ID/g across the three dose levels at 24 h. No significant uptake was observed in other organs in the 1 and 3 mg/kg groups, whereas low uptake was observed in the 10 mg/kg group in spleen, kidneys, and lungs (Figure S3).
Biodistribution of 89Zr-DFO-anti-IL13Rα2-Ab by PET/CT Imaging
Since FMT is limited to preclinical imaging, we performed a biodistribution study of the anti-IL13Rα2 Ab by PET, a clinically translatable imaging modality, after labeling with 89Zr-DFO. In vitro cell binding assessment demonstrated similar binding between DFO-conjugated anti-IL13Rα2-Ab and the unconjugated parental antibody. Additionally, an immuno-reactivity (IR) QC assay with 89Zr-DFO-anti-IL13Rα2-Ab demonstrated 85% IR in the IL13Rα2-expressing A375 cells compared to a background-level 20% IR in the IL13Rα2-negative H460 cells, confirming unaffected antigen binding. To evaluate in vivo biodistribution, 89Zr-DFO-anti-IL13Rα2-Ab (3 mg/kg) was injected into A375 tumor-bearing mice followed by longitudinal PET/CT imaging, which showed diffused distribution of radioactivity throughout the animal at 4 h post-injection, followed by a continued reduction in later timepoints (Figure 3a). The tumor, however, showed a sustained level of activity, with an uptake of 9.23%ID/g at 4 h post-injection, which increased over time and peaked at 96 h at 19.84%ID/g (Figure 3b), a value similar to FMT quantitation (Figure S3B). As observed with FMT and the ligand binding assay, the heart concentrations from PET quantitation also showed a biphasic decline (21.87%ID/g at 4 h versus 7.47%ID/g at 96 h), suggesting rapid distribution into tissues until 24 h, followed by steady elimination from blood (Figure 3b). Additionally, we collected blood and measured the activity ex vivo in a gamma counter as a surrogate for blood PK (Figure 3c). The blood profile recapitulated the biphasic profile of the heart uptake in vivo (Figure 3b), and the blood PK assessed using anti-IL13Rα2-Ab-AF680 (Figure 2e).
Figure 3.
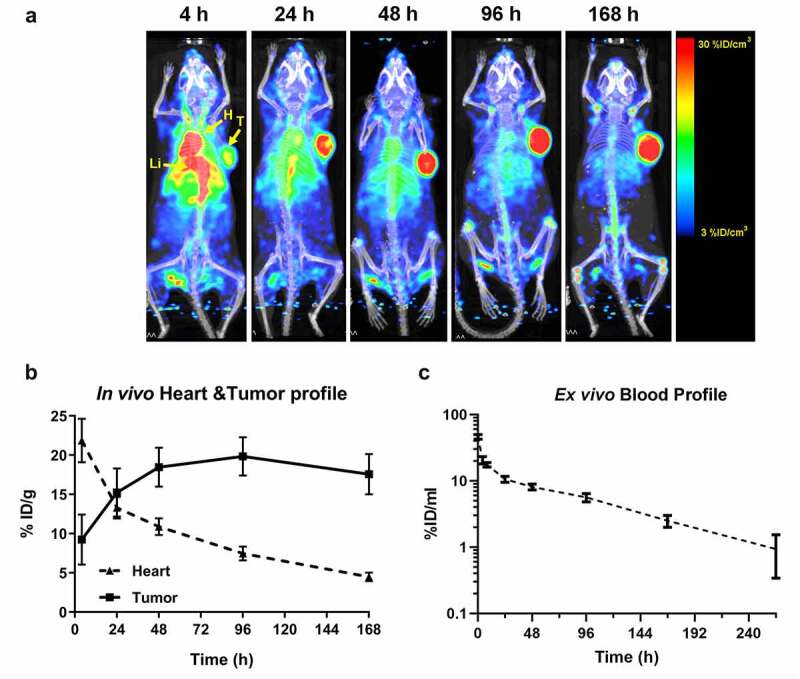
In vivo biodistribution and tumor targeting by PET/CT imaging. (a) In vivo PET/CT imaging was performed in A375 tumor-bearing mice at 4, 24, 48, 96, and 168 h after injection of 89Zr-DFO-anti-IL13Rα2-Ab. Maximum Intensity Projection (MIP) images shown here represent whole-body biodistribution and tumor targeting at different timepoints. T – tumor; H – heart; Li – liver. (b) Quantitative profiling of tumor and heart uptake in vivo showing accumulation in tumor and clearance from heart. Here we use heart as a surrogate for evaluating probe distribution in blood. (c) Ex vivo blood quantitation by gamma counting showing biphasic clearance over time. N = 3– 6 mice per group for each timepoint; data are Mean ± SEM
We quantified the uptake of 89Zr-DFO-anti-IL13Rα2-Ab in tumor and selected tissues (brain, heart, lungs, kidneys, liver, and spleen) ex vivo by gamma counting at 24- and 264 h post-injection (Figure S4). As observed by FMT, tumor was the major tissue for accumulation with 10.05%ID/g at 24 and 8.5%ID/g at 264 h. Liver, spleen, and kidneys were the main sites for nonspecific accumulation and clearance. There was no major difference in the amount of accumulation between 24 and 264 h among these tissues. Although there was signal observed in blood, heart, and lungs at 24 h, the signal drastically decreased by 264 h. No significant signal was observed in the brain at either 24 or 264 h after probe injection. Interestingly, we observed an increased localization of signal to the joints toward the later timepoints. This can be Zr-related artifact, nonspecific to IL13Rα2 expression. Free Zr ions and small metabolites containing Zr are known to distribute to the bone, and, in particular, areas with high bone remodeling activities such as joints.28,29
Tumor-specific targeting of anti-IL13Rα2-Ab-AF680 and 89Zr-DFO-anti-IL13Rα2-Ab
To test tumor-specific uptake and retention of the anti-IL13Rα2 Ab, mice were pre-injected with 10– 30 molar excess of unlabeled competing anti-IL13Rα2-Ab or non-competing isotype control Ab. The baseline group received either phosphate-buffered saline (PBS) (in the FMT study) or no treatment at all (in the PET study). This was followed by the administration of 3 mg/kg anti-IL13Rα2-Ab-AF680 for FMT imaging or 89Zr-DFO-anti-IL13Rα2-Ab for PET imaging. Figure 4a shows representative FMT images for tumor accumulation of anti-IL13Rα2-Ab-AF680 at different timepoints, which was comparable at initial timepoints in all three groups, likely due to significant levels of probe circulating in the blood. However, at 24 h post-injection, the fluorescence signal in the anti-IL13Rα2-Ab competing group was lower compared to the non-competing isotype control group or the PBS group (Figure 4a). Ex vivo FMT quantitation revealed a significant reduction in anti-IL13Rα2-Ab-AF680 uptake in the competing antibody group (10.84%ID/g) compared with PBS (17.99%ID/g) or the non-competing isotype control (17.66%ID/g) (Figure 4b). Corroborating the FMT results, PET imaging also demonstrated that accumulation of 89Zr-DFO-anti-IL13Rα2-Ab could be blocked by competing unlabeled anti-IL13Rα2-Ab, but not the non-competing isotype control Ab (Figure 4c). Ex vivo biodistribution by gamma counting also demonstrated a 34% reduction in tumor uptake in the competition group compared to the non-competing isotype group or the no-pretreatment group (%ID/g: 6.95 versus 10.14 or 10.34, respectively; Figure 4d). Overall, pharmacological competition studies with FMT and PET imaging consistently demonstrated the antigen-specific tumor targeting of anti-IL13Rα2-Ab.
Figure 4.
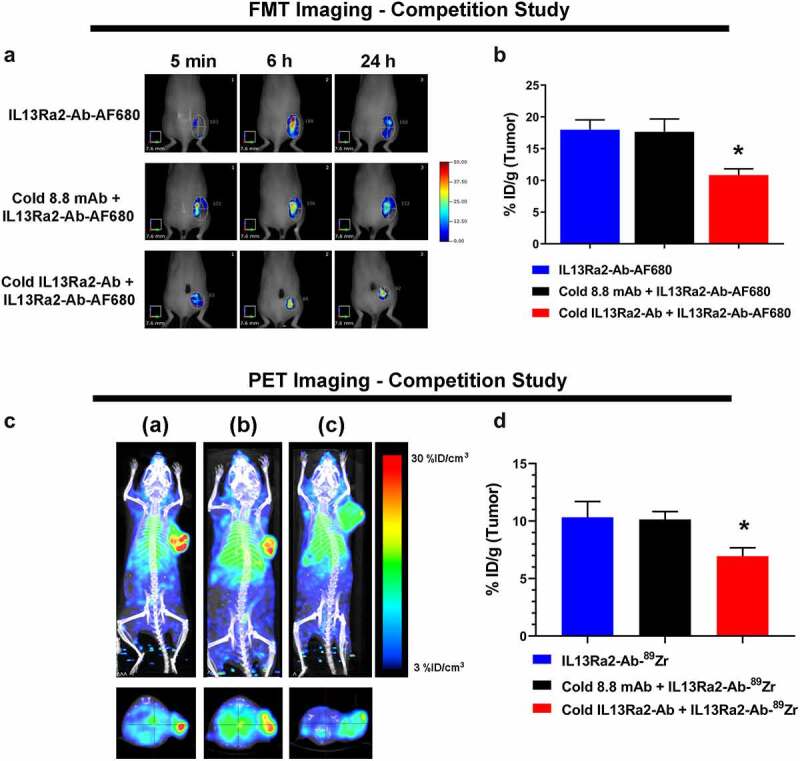
Pharmacological competition showcases antigen specificity of the anti-IL13Rα2 Ab. (a) Representative in vivo FMT images with tumor ROIs at 5 min, 6, and 24 h after injection of anti-IL13Rα2-Ab-AF680 (3 mg/kg) with PBS or pretreatment of 30 mg/kg unlabeled isotype control 8.8-mAb or unlabeled anti-IL13Rα2-Ab in the A375 xenograft model. (b) Ex vivo quantification of tumor signal (%ID/g) at 24 h. (c) Representative in vivo PET/CT images (MIP) at 24 h after injection of 89Zr-DFO-anti-IL13Rα2-Ab (3 mg/kg) without pretreatment (a), or with pretreatment of 30× excess cold isotype control 8.8-mAb (b), or cold anti-IL13Rα2 Ab (c) in the A375 xenograft model. Top panel: coronal view; bottom panel: axial multiplanar reconstruction view. (d) Ex vivo gamma counting quantification of tumor uptake. N = 3– 5 mice per group; *, p-value < 0.05; data are Mean ± SEM
Tumor targeting by anti-IL13Rα2-ADC
After observing the favorable PK profile and antigen-specific uptake of the anti-IL13Rα2 Ab, we next evaluated the biodistribution and tumor uptake of our anti-IL13Rα2 ADC before evaluating the anti-tumor efficacy in vivo. The antibody arm of anti-IL13Rα2-ADC was conjugated to AF680 to enable FMT imaging in mice. Prior to performing in vivo studies, the effects of fluorophore conjugation on the ADC’s antigen binding and cytotoxic activity was evaluated by flow cytometry and cytotoxicity assay, respectively. AF680 conjugation showed no effect on the ADC’s binding to A375 cells (Figure S5). Cytotoxicity assay showed an IC50 of about 0.1 µg/ml (0.7 nM) with anti-IL13Rα2-ADC, as well as anti-IL13Rα2-ADC-AF680, suggesting no change in cytotoxic potential of ADC due to fluorophore conjugation. The anti-IL13Rα2 Ab or anti-IL13Rα2-Ab-AF680 did not have any cytotoxicity (Figure 5a).
Figure 5.
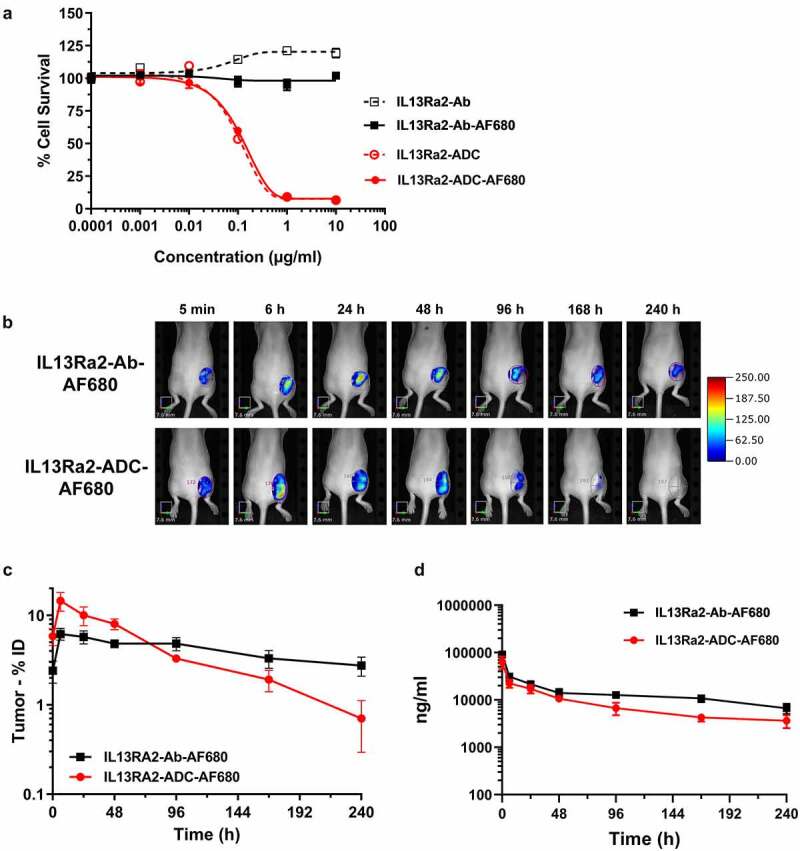
Tumor targeting and PK of the anti-IL13Rα2-ADC. (a) Evaluation of the cytotoxic potential of anti-IL13Rα2-ADC after conjugation with fluorophore AF680 using cell cytotoxicity assay. Anti-IL13Rα2-Ab and IL13Rα2-Ab-AF680 were used as negative controls. (b) Comparison of tumor targeting of anti-IL13Rα2-Ab-AF680 and anti-IL13Rα2-ADC-AF680 in the A375 xenograft model. Mice were administered with anti-IL13Rα2-Ab-AF680 and anti-IL13Rα2-ADC-AF680 at 3 mg/kg via IV injection, and longitudinal FMT imaging was performed. Highlighted are tumor ROIs. (c) Quantitation of anti-IL13Rα2-Ab-AF680 and anti-IL13Rα2-ADC-AF680 tumor uptake from FMT images at different timepoints. (d) Plasma concentrations of anti-IL13Rα2-Ab-AF680 and anti-IL13Rα2-ADC-AF680 were measured by ligand binding assay to compare pharmacokinetic profiles of Ab and the ADC. N = 3– 5 per group; data are Mean ± SEM
To evaluate and compare biodistribution, anti-IL13Rα2-ADC-AF680 and anti-IL13Rα2-Ab-AF680 were intravenously administered to A375 xenograft-bearing mice at a dose of 3 mg/kg and imaged longitudinally by FMT. There was no difference in the whole-body profile of the AF680-labeled ADC and Ab (data not shown). The representative FMT images of tumor ROIs in Figure 5b show accumulation as early as 6 h post-injection for both the Ab and the ADC. Interestingly, the signal in the anti-IL13Rα2-ADC-AF680 group decreased dramatically after 96 h compared to its Ab counterpart. Fluorescence quantification of tumors showed that anti-IL13Rα2-ADC-AF680 signal peaked between 6–24 h post-injection, followed by a steep decline (Figure 5c), whereas anti-IL13Rα2-Ab-AF680 reached the peak tumor uptake between 6–48 h, followed by a gradual decline (Figure 5c). Interestingly, maximum tumor uptake for anti-IL13Rα2-ADC-AF680 was observed to be about 15%ID, as opposed to 6%ID for the anti-IL13Rα2-Ab-AF680 (Figure 5c). This increased accumulation was short-lived and the ADC accumulation in tumor decreased drastically after 48 h. These observations demonstrated the differential tumor uptake kinetics for the Ab and ADC, which is likely attributed to the tumor killing property of the ADC and the differences in tumor volumes between the groups. The ADC-treated tumors were regressing, and their overall retention of the probe decreased. Consistently, ex vivo tumor quantification at 240 h showed accumulation of about 3%ID for anti-IL13Rα2-Ab-AF680, whereas anti-IL13Rα2-ADC-AF680 showed only about 0.4%ID (data not shown). Finally, we compared the plasma PK profile for anti-IL13Rα2-ADC-AF680 and anti-IL13Rα2-Ab-AF680. Although the ADC curve was slightly lower than the Ab curve, no major difference in Ab and ADC profile was observed (Figure 5d).
Anti-tumor efficacy of anti-IL13Rα2-ADC
A375 xenograft tumors were treated with vehicle, isotype control-ADC (3 mg/kg), or anti-IL13Rα2-ADC (three doses: 0.3, 1.0, and 3.0 mg/kg; all doses were administered every 4 days (q4d) × 4 on days 0, 4, 8, and 12) when they reached ~330 mm3. Mice treated with vehicle started to reach maximum tumor size by day 13, while those treated with the isotype control ADC or the low-dose anti-IL13Rα2-ADC started to reach the limit by day 19 (Figure 6a). In sharp contrast, the 1 or 3 mg/kg anti-IL13Rα2-ADC groups showed significant reduction in tumor size and were monitored for longer durations. The % T/C (Tumor/Control; a ratio comparing tumor size of treated groups to the vehicle control) on day 13 for the 1 and 3 mg/kg dose groups was 8% and 3%, respectively, demonstrating significant tumor growth inhibition. Figure 6(b,c,d,e) show the tumor growth curves for individual mice from each group versus the vehicle control to demonstrate the complete regressors (CRs) and long-tern regression. The mice without measurable tumors or with tumors smaller than 2 mm in the largest dimension were considered as complete regressors. The 10 animals receiving isotype control-ADC had 0 CRs, whereas the anti-IL13Rα2-ADC groups showed a dose-dependent effect with 1/10, 3/10 and 9/10 CRs, respectively (Figure 6(b,c,d,e)). Interestingly, anti-IL13Rα2-ADC at 1 mg/kg initially showed tumor regressions in all mice with 6 mice achieving a CR until study day 22, but three of those mice subsequently had tumor growth (Figure 6d). These results demonstrated the ability of anti-IL13Rα2-ADC in inducing long-term regression of melanoma xenograft tumors in an antigen-specific manner, despite a short 12-day course of treatment.
Figure 6.
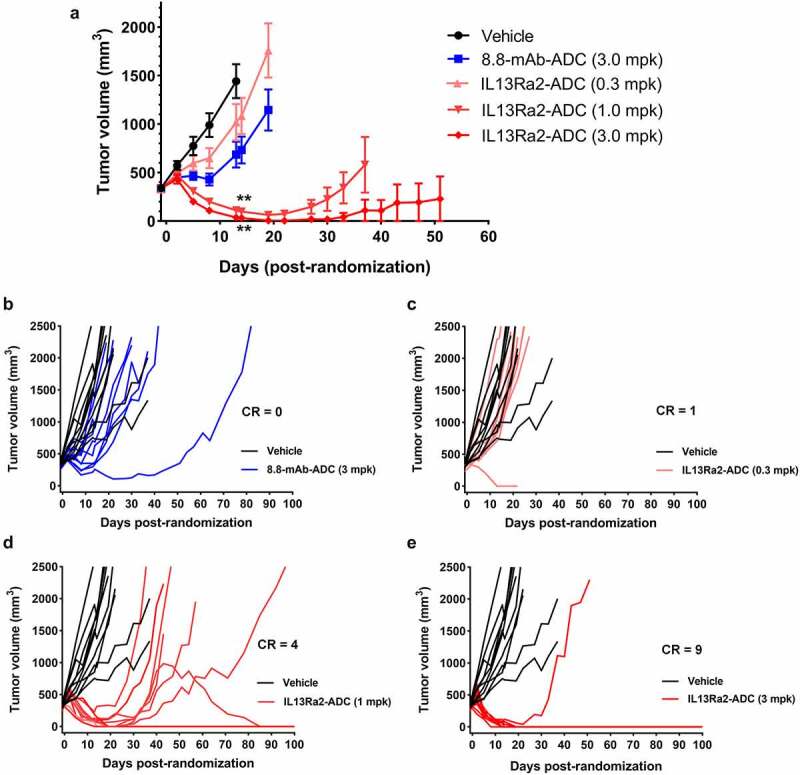
In vivo efficacy of the anti-IL13Rα2 ADC in the A375 xenograft model. A375 tumor-bearing animals were randomized when their tumors reached an average of 330 mm3 in size and treated with vehicle, isotype control 8.8-mAb-ADC (3 mg/kg), or the anti-IL13Rα2 ADC (0.3, 1.0 and 3.0 mg/kg) in a q4d × 4 treatment schedule. (a) Tumor growth inhibition curves demonstrate a dose response in anti-tumor effect with anti-IL13Rα2-ADC. A significant difference in tumor volume was observed in the 1 and 3 mg/kg groups compared to Vehicle on day 14 when the vehicle group animals were euthanized. Tumors treated with 8.8-mAb-ADC (3 mg/kg) and anti-IL13Rα2-ADC (0.3 mg/kg) were not statistically different on day 14, compared to the vehicle group. (b) Spaghetti plot shows no difference in tumor growth curves between mice treated with Vehicle and 8.8-mAb-ADC. (c) Spaghetti plot shows no difference in tumor growth curves between mice treated with Vehicle and anti-IL13Rα2-ADC at 0.3 mg/kg, although one treated mouse showed a complete response (CR). (d) Spaghetti plot showed a significant difference in tumor growth curves between mice treated with anti-IL13Rα2-ADC at 1.0 mg/kg, with 4 CRs, and Vehicle. (e) Spaghetti plot showed a significant difference in tumor growth curves between mice treated with anti-IL13Rα2-ADC at 3.0 mg/kg, with 9 CRs, and Vehicle. **, p-value < 0.001. N = 9– 10 per group
Discussion
We have previously shown that FMT imaging aids understanding of the three pillars of drug development: targeting/distribution, binding to the target, and the expression of pharmacology.30,31 In this study, we again used FMT to demonstrate that the anti-IL13Rα2 Ab was able to target IL13Rα2-expressing tumors, exhibited favorable PK characteristics that are consistent with full-length antibodies, and that anti-IL13Rα2-ADC with an auristatin payload exhibited in vitro and in vivo anti-tumor activity. Since FMT is restricted to use in preclinical imaging, we also studied the biodistribution and tumor targeting of anti-IL13Rα2-Ab by PET imaging, a modality that is readily translatable to the clinic.
Biodistribution studies by noninvasive imaging suggested that anti-IL13Rα2-Ab initially stayed in the blood pool, as reflected by the heart and vena cava uptake at the early timepoints (within the first day); it then gradually distributed to the target site, with the tumor uptake reaching a plateau at around 3–4 days post injection. Remarkably, probe uptake at the tumor site maintained at a high level throughout the study until 7–10 days post-injection, whereas activity in the rest of body was gradually eliminated via the hepatic route. This suggests that anti-IL13Rα2-Ab was being actively retained at the tumor. It is possible that free Zr or Zr-containing metabolites may be contributing to the persistent tumor signals. However, we do not expect this to be the main reason, as signals gradually decreased for the rest of the body.
Ex vivo FMT imaging of organs indicated saturable PK for anti-IL13Rα2-Ab-AF680. When the FMT signal was compared for the three doses for tumor (%ID/g), the 3 mg/kg dose showed the highest uptake at early timepoints, whereas the absolute signal in tumor and whole body increased with dose. This suggested a saturable uptake of anti-IL13Rα2-Ab-AF680 in tumors between a dose of 3 and 10 mg/kg. Although there are a few examples of nuclear imaging techniques used for kinetic modeling,32,33 this study demonstrated the potential of FMT to capture PK characteristics of a biologic. However, the potential impact of antigen sink on plasma PK could not be assessed, as the anti-IL13Rα2 Ab does not cross-react with mouse IL13Rα2.
Pharmacological competition experiments with cold competing and non-competing isotype control antibodies further substantiated the notion that the tumor uptake of anti-IL13Rα2-Ab was at least partially attributed to antigen-specific binding. Large molecules like an antibody must bypass sequential barriers and passively diffuse through the tumor vasculature before encountering the tumor antigen. The residual uptake observed in the competition group is likely due to nonspecific distribution of the antibody as a result of the enhanced permeability and retention at the tumor site.34,35 Of note, no difference was found in the whole-body fluorescence or in non-target organs (e.g., the heart) among the three groups in the FMT competition study. This suggests that the differential tumor uptakes in the PBS, isotype control, and competition antibody groups were not an artifact from differences in total probe availability. Taken together, these findings indicate that anti-IL13Rα2-Ab was able to localize to the IL13Rα2-expressing A375 tumor site in an antigen-specific manner, and this accumulation was persistent over the course of 7–10 days, supporting the desired exposure of anti-IL13Rα2-ADCs. We acknowledge that the experimental system used here consisted of non-human IL13Rα2-expressing NSG mice, human IL13Rα2-expressing xenograft tumors, and an antibody that does not cross-react with murine IL13Rα2. In this contrived model, therefore, we were unable to study any on-target off-site distribution or side effect of our antibody or ADC, respectively.
Both FMT and PET are 3D tomographic modalities that are suitable for assessing the temporal and spatial distribution of large-molecule biologics such as antibodies, ADCs, and nanoparticles, provided that the labeling of the probe is validated. We previously showcased the potential of FMT imaging in such settings.27,36 Meanwhile, although several groups have explored the utility of hybrid or fusion technologies with co-registered optical imaging and nuclear medicine imaging such as PET and SPECT, the literature is relatively scarce in the direct cross-validation of FMT and PET.37–40 Using a nanoparticle imaging probe containing both18F and a far-red fluorophore, VT680, Nahrendorf et al. demonstrated good correlation between FMT and PET in measuring probe concentration and distribution.37 Here, we provided further evidence that the two imaging modalities were well correlated and yielded comparable biodistribution and competitive pharmacology results. It is important to understand some of the merits and limitations of these two imaging modalities in study design and data interpretation, as summarized in Table S1 and detailed below.
Corroborating a previous report,39 in our laboratory the in vivo PET/ex vivo gamma counting workflow appeared to be more sensitive, and it possesses a higher signal-to-noise ratio (SNR) than FMT-mediated in vivo and ex vivo quantifications. The differential activities in the low-uptake organs (kidney, lung, spleen, and brain) were successfully distinguished in the PET experiment, but not in FMT. This could be attributed to the relatively low inherent SNR for AF680 imaging with confounding autofluorescence, which makes it particularly challenging for organs with a smaller mass to be detected above the background.41 Liver, a larger organ, was not as severely affected. Additionally, we found that the terminal (day 10 or 11) tumor uptake was higher in the PET/gamma counting quantification than in FMT. This could be attributed to the differential behaviors of the labeling (89Zr or AF680 fluorophore) of the antibody. 89Zr is a known residualizing radionuclide that is trapped inside the cell following internalization,35 leading to signal that reflects accumulation over time. While AF680 and other cyanine dyes have been shown to have greater cellular residualization rates than other fluorophores (half-life > 24 h), they are still susceptible to structural disruption in the lysosome and the resultant loss of fluorescence.42 Thus, the fluorophore signal detected may only be reflecting a snapshot of the antibody amount in the tissues at that time. An additional technical disparity is that PET, coupled with co-registered CT imaging, was able to generate quantitative data with accurate companion volumetric measurement of the organ or ROI. FMT, on the other hand, was unable to precisely calculate the volume of ROIs due to the lack of an accompanying anatomical imaging module. As a result, the in vivo biodistribution data in the FMT experiment were presented as %ID instead of %ID/g or %ID/cm.3 Other well-recognized advantages of PET imaging include high sensitivity and clinical translatability.40 However, the initial barrier of setting up the PET lab and infrastructure to be compliant with radiation safety and licensing requirements could be arduous, costly, and time consuming. Radioactive isotopes, radiolabeling, and quality control of the probe could also add to the expense for each study. To this end, FMT serves as a less demanding surrogate that still produces high-quality 3D imaging data and is therefore particularly useful in proof-of-concept and probe validation studies. An additional advantage of fluorescent labeling and FMT imaging is that, once labeled, the probe is not subject to radioactive decay. This allows longer shelf-life and greater logistic flexibility.
To evaluate the efficacy of anti-IL13Rα2-ADC, we conjugated the antibody with an auristatin-based cytotoxic agent (payload). Auristatin is an anti-mitotic agent which inhibits tubulin polymerization and arrests the tumor cell proliferation at G2-M phase of cell cycle.43,44 The antibody was conjugated to the payload via a peptide cleavable maleimidocapronic-valinecitruline-p-aminobenzyloxycarbonyl (vc) linker. This cleavable linker is known to release the cytotoxic payload only after internalization and trafficking into the intracellular vesicles which contain cathepsin B and thus reduce any systemic cytotoxicity.45 Indeed, when we evaluated the anti-tumor efficacy in vivo we observed a dose-dependent long-term regression for up to 100 days. We observed some complete regression of tumors in all the doses tested, compared to none with the isotype control-ADC, although the number of complete regressors was also dose-dependent. Although there have been several efforts to therapeutically target IL13Rα2 for cancer,23,46,47 this is the first study showcasing the PK, targeting, and anti-tumor efficacy of an anti-IL13Rα2 ADC.
Taken together, both FMT and PET imaging corroboratively showed a favorable pharmacological profile of anti-IL13Rα2-Ab, which demonstrated persistent and antigen-specific tumor targeting in vivo. Further, anti-IL13Rα2-ADC exhibited desirable PK profile and excellent therapeutic efficacy in the A375 xenograft model. This work shows the great promise of anti-IL13Rα2-ADC as a targeted cancer therapeutic. Further studies are needed to evaluate its safety and therapeutic index before clinical evaluation.
Materials and methods
Ethics statement
All animal-related procedures in this study were performed in accordance with established guidelines and protocols approved by Pfizer Institutional Animal Care and Use Committee.
Reagents
The anti-IL13Rα2 Ab (IL13Rα2-AB08-v1010-hG1) and anti-IL13Rα2-ADC (IL13Rα2-AB08-v1010-hG1-(C)mcValCitPABC_Aur-06380101; PF-06473811) were generated within Pfizer Inc. The fluorophore, AlexaFluor680® (AF680), was obtained from Life Technologies (Carlsbad, CA). Anti-human FITC-conjugated secondary antibody against human IgG was obtained from Thermo Fisher Scientific Inc. (Waltham, MA; #31,529); BIOT Sheep anti-hIgG pAb and AlexaFluor647® Goat anti-hIgG were obtained from Binding Site (San Diego, CA; #AU003) and Bethyl Labs (Montgomery, TX; #A80-319), respectively.
Cell culture
The A375 human melanoma cell line (ATCC® CRL-1619™) was obtained from the ATCC (Manassas, VA) and was cultured according to ATCC’s instructions. Briefly, A375 cells were cultured in DMEM (Gibco Life Technologies; #2017-01), supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences; #SH30088) and 1% pen/strep (Gibco Life Sciences) in a 37°C incubator with 5% CO2. All experiments were performed before the cell lines reached passage 15.
Antibody and ADC labeling with AF680
The conjugation procedure for the fluorophore has been described earlier.27 Briefly, anti-IL13Rα2-Ab or anti-IL13Rα2-ADC was incubated for 2 h in dark with AF680 in the labeling buffer provided by manufacturer (Perkin Elmer Corporation, Waltham, MA) followed by purification of the conjugates using ZebaTM Spin desalting columns (Thermo Fisher Scientific Inc., Waltham, MA). Degree of labeling (DOL) was determined by HPLC-MS to be 1.7 (Figure S6A; Table S2). Additional measurement by spectrophotometry (NanodropTM 1000, Thermo Fisher Scientific Inc., Waltham, MA) confirmed a DOL lower than 3. Further, the purity of the conjugated products was assessed by size exclusion chromatography (SEC). All species showed excellent purity of >99% (method described in Figure S6B; Table S2).
Antibody-labeling with DFOs and 89Zr
DFO conjugation and radiolabeling were performed by IsoTherapeutics Group, LLC (Angleton, TX). The anti-IL13Rα2 Ab was conjugated with P-SCN-Bn-Deferoxamine (DFO) by incubating 2 mg of Ab and 3 molar excess of DFO (DFO was dissolved in dimethyl sulfoxide) at pH 9.1, 37°C, for 1.5 h. Purification was done on a PD10 Column (6 kDa SEC Column) eluted with 0.1 M HEPES. The conjugation product was estimated to have a 1:1 DFO-to-antibody ratio. 89Zr chelation was performed at pH 6.5– 7.0 under room temperature for 1.5 h. The target-specific activity was 1.3 mCi/mg calibrated to time of injection. Purification was performed using a 30 kDa Amicon® filter and transferred to a final 2 mL glass vial for transportation.
Cell binding flow cytometry
A375 cells were plated in six-well plates and incubated overnight at 37°C. The following day, an isotype control Ab (8.8-mAb), anti-IL13Rα2-Ab, anti-IL13Rα2-Ab-AF680 or anti-IL13Rα2-Ab-DFO were added to the cells at 250 ng/mL (or indicated concentrations). After 2 h incubation at 37°C, the cells were washed and further incubated with FITC-conjugated anti-human secondary Ab for 1 h at 37°C. The cells were washed and analyzed using MACS Quant Analyzer® (Miltenyi Biotec, San Diego, CA). Fluorescence intensity (peak shift) and the mean channel intensities for FITC were determined to compare the cell binding properties of the parental and conjugated Abs.
Cell cytotoxicity assay for anti-IL13Rα2-ADC and fluorophore conjugates
The in vitro efficacy of ADC before and after fluorophore conjugation was measured by cell cytotoxicity assay in A375 cell line using CellTiter-Glo® Luminescent Cell Viability Assay. Approximately 5000 cells/well were seeded overnight in a 96-well plate followed by treatment with varying concentrations (0–10 µg/mL) of unconjugated or fluorophore-conjugated ant-IL13Rα2-Ab or anti-IL13Rα2-ADC. After 96 h of treatment, CellTiter-Glo® reagent was added to each well as per manufacturer’s instructions. Cell viability was measured as luminescence signal after 10 min incubation using a Spectramax® plate reader. The assay was repeated at least twice with triplicates/concentration.
Immuno-reactivity assay for quality control of the radiolabeled antibody
A cell-based QC assay was conducted to verify target binding of anti-IL13Rα2-Ab after DFO conjugation and 89Zr radiolabeling. A trace amount (1 µg) of 89Zr-DFO-anti-IL13Rα2-Ab was incubated with a pair of antigen-positive and -negative cell lines. Following 45–60 min of incubation at room temperature with rotation, the cells were washed twice with PBS and the supernatants from both washes were combined. Radioactivity (in Counts Per Minute, CPM) in the cell pellet versus supernatant portions were then determined by gamma counting. The Immuno-Reactivity (IR) of the probe in each cell line was then calculated as: IR = CPM (Pellet)/[CPM (Pellet) + CPM (Sup)] × 100%. The oversaturating amount of IL13Rα2 expressed on A375 cells was anticipated to sequester the radioactive probe. So, if antigen binding was preserved after radiolabeling, no less than 70% of radioactivity was expected to be captured by the cell pellet. In contrast, when incubating with cells (H460) that do not express IL13Rα2, or if the binding was destroyed, no more than background levels (less than 30%) of radioactivity should be taken up by the cells. In this assay, over 80% of radioactivity was captured by A375 cell pellet and less than 20% of radioactivity was captured by H460 cell pellet, suggesting 89Zr-DFO-anti-IL13Rα2-Ab did not lose binding ability after 89Zr-DFO conjugation.
A375 xenograft development
The biodistribution, tumor targeting of anti-IL13Rα2-Ab and anti-IL13Rα2-ADC were evaluated in the A375 xenograft model, which was established as previously described.27 Briefly, 2 × 106 A375 cells in 50% Cultrex basement membrane extract (R&D Systems, Minneapolis, MN, #3432-005) were injected subcutaneously in the flank of athymic nu/nu mice (Charles River Labs, Wilmington, MA) for FMT imaging or in the upper right shoulder for PET/CT imaging. Tumors were measured at least twice/week with a Vernier caliper (Mitutoyo, Aurora, Illinois) and tumor volume was calculated as width × width × length/2. The mice were enrolled into the study when tumors reached an average size of 200– 500 mm3. All test agents were injected intravenously (IV).
In vivo FMT imaging of anti-IL13Rα2-Ab-AF680 and anti-IL13Rα2-ADC-AF680
FMT imaging and quantification were performed as described before.27,36 Briefly, fluorophore-conjugated Ab or ADC were administered IV at indicated doses. Animals were anesthetized using 1– 3% isoflurane, placed in the FMT cassette and in vivo whole-body imaging was performed using the FMT4000® (Perkin Elmer Corporation, Waltham, MA) at selected timepoints. For ex vivo imaging, the mice were anesthetized with isoflurane and euthanized by whole-body perfusion with saline at 24, 96, and 240 h post-injection and brain, tumor, heart, lungs, kidneys, liver, and spleen were imaged ex vivo. FMT data were analyzed and quantitated using the TrueQuant™ software. Serial blood sampling was performed for each mouse to obtain plasma to quantitate Ab or ADC using ligand binding assay as described.48
Pharmacological competition by FMT imaging
Tumor-bearing mice were randomized in three groups to receive PBS, unlabeled isotype control antibody (30 mg/kg), or unlabeled anti-IL13Rα2-Ab (30 mg/kg). Three hours later, all groups were injected with anti-IL13Rα2-Ab-AF680 at 3 mg/kg. All mice were imaged longitudinally using FMT and the data were quantified to assess target specific uptake in the tumor.
Ligand binding tissue PK assay
The Ab and ADC concentrations in blood samples were quantitated using the Gyrolab™ workstation as described before.27 Briefly, the test compounds in blood samples were captured using biotinylated sheep anti-human IgG (100 µg/mL), followed by detection with goat anti-human IgG AlexaFluor647® (5 µg/mL). The data was analyzed using the Watson v7.4 LIMS system (Thermo Fisher Scientific Inc., Waltham, MA) and the PK parameters were calculated using noncompartmental analysis on WinNonlin® 8.1 (Certara USA, Inc., Princeton, NJ).
In vivo PET/CT imaging and analysis
A375 tumor-bearing mice were enrolled in the PET imaging studies as described before in the FMT imaging methods section. Mice were injected with 3 mg/kg (~ 60 µg, ~ 80 µCi) of 89Zr-DFO-anti-IL13Rα2-Ab. Animals in the competition groups received 30-fold excess of cold isotype control or parental anti-IL13Rα2-Ab 3 h prior to probe injection. Animals were imaged using a G8 PET/CT small-animal scanner (Perkin Elmer/Sofie Biosciences). Static PET/CT images were acquired with 10- or 20-min scan time based on the amount of remaining activity at the timepoint and reconstructed using a vendor-provided 3D ML-EM reconstruction protocol at 1.4 mm resolution. Following the PET scan at each timepoint, a co-registered microCT scan was acquired using these parameters: 50 kVp, 200 µA, and 200 microns on a step-and-shoot (5 degrees/step). PET values were decay- and attenuation-corrected per vendor-supplied algorithm. The VivoQuant™ software (inviCRO LLC., Boston, MA) was used for visualization and quantification of tracer uptake. Circular 3D ROIs were drawn manually on the whole-body microCT images for tumor and heart. PET signal was calculated in the unit of percentage of injected dose per gram (%ID/g) with the assumption that a cm3 equals a gram of tissue.
Ex vivo gamma counting of tissues and blood
At 24 and 264 h, mice were euthanized and tumor, lung, spleen, brain, heart, kidneys, liver, quadriceps muscle, and blood (via cardiac puncture) were collected for ex vivo quantitation by gamma counter (WIZARD2-2480, Perkin Elmer, Waltham, MA). Radioactivity was determined and %ID/g was calculated after decay correction. Blood (8 µL) was also collected by tail nick at 2 min, 4, 8, 24, 48, 96, 168, and 264 h post probe administration to determine the radioactivity in the blood.
Tumor growth inhibition by anti-IL13Rα2-ADC
Xenografts were developed by implanting 8 × 106 A375 cells in 50% Matrigel (BD Biosciences, Franklin Lakes, NJ) and 50% growth media in 8-week-old female athymic nu/nu mice. Tumors were measured at least twice/week and mice were randomized into study groups when mean tumor volume reached 330 mm3. Mice were dosed IV on a q4d × 4 schedule with either PBS (Thermo Fisher Scientific, Waltham, MA; as vehicle), isotype control-ADC (8-8-hG1-(C)_mcValCitPABC_Aur-06380101; PF-06645272) at 3 mg/kg, or anti-IL13Rα2-ADC (IL13Rα2-AB08-v1010-hG1-(C)_mcValCitPABC_Aur-06380101; PF-06473811) at 0.3, 1, and 3 mg/kg. Tumor sizes were monitored until the tumors reached protocol-defined endpoint or until 100 days post-treatment. The drug-to-antibody ratios (DAR) were determined by HPLC-MS to be 3.2 and 3.7 for the anti-IL13Rα2 and isotype control ADCs, respectively (Figure S6C;Table S2).
Statistical analysis
Statistical analyses were performed using Prism 7.0 (GraphPad software Inc., San Diego, CA, USA). Results were represented as mean ± S.E.M with at least n = 3/group. Tumor and heart uptake data from ROI analysis and biodistribution/gamma counting were tested by One-way ANOVA followed by post-hoc Tukey Test (α = 0.05) and/or Student's t-test.
Supplementary Material
Acknowledgments
We thank the Pfizer - Comparative Medicine, La Jolla Technical Staff for their assistance in the in vivo studies. IsoTherapeutics Group LLC., Angleton, TX for the support with 89Zr-conjugation chemistry.
Funding Statement
This project was funded by Pfizer, Inc.
Authors’ contributions:
Conception and design:
PG; ZJ; ER; PS; AG
Development of methodology:
PG; ZJ; BY; KN; SP; JD; FL; DM; JY; AG
Acquisition of data (provided animals, acquired and provided facilities, etc.):
PG; ZJ; BY; SP; EM; LM; LL; JY; AG
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis):
PG; ZJ; BY; JC; EM; FL; ML; JY; ER; AG
Writing, review, and/or revision of the manuscript:
PG; ZJ; BY; JC; ER; ML; PS; AG
Disclosure statement
All authors are or were full time or part-time employees of Pfizer, Inc.
Abbreviations:
| Abbreviations | Meaning |
|---|---|
| 3D | 3-dimensional |
| Ab | antibody |
| ADC | antibody-drug conjugate |
| AP-1 | activator protein 1 |
| AUC | area under the curve |
| Cl | clearance |
| CPM | counts per minute |
| CR | complete regressor |
| CT | computed tomography |
| DAR | drug-to-antibody ratio |
| DFO | deferoxamine |
| DOL | degree of labeling |
| FMT | fluorescence molecular tomography |
| HC | heavy chain |
| HPLC-MS | high performance liquid chromatography–mass spectrometry |
| IC50 | half-maximal inhibitory concentration |
| ID | injected dose |
| ID/g | injected dose per gram |
| IL13 | interleukin 13 |
| IR | immuno-reactivity |
| IV | intravenous, intravenously |
| JAK | janus kinases |
| LC | light chain |
| mAb | monoclonal Ab |
| MAPK | mitogen-activated protein kinase |
| MIP | Maximum Intensity Projection |
| PBS | phosphate-buffered saline |
| PET | positron emission tomography |
| PK | pharmacokinetics |
| PKB | protein kinase B |
| q4d | every 4 days |
| QC | quality control |
| R | receptor |
| ROI | region of interest |
| SEC | size exclusion chromatography |
| SEM | standard error of the mean |
| SNR | signal-to-noise ratio |
| SPECT | single photon emission computed tomography |
| STAT | signal transducer and activator of transcription proteins |
| T/C | Tumor/control |
| t1/2 | half-life |
| TGF | transforming growth factor |
| TNF | tumor necrosis factor |
| Zr | zirconium |
Supplementary materials
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Scott AM, Wolchok JD, Old LJ.. Antibody therapy of cancer. Nat Rev Cancer. 2012. Mar 22;12(4):278–13. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 2.Hajdu SI, Vadmal M. A note from history: landmarks in history of cancer, part 6. Cancer. 2013. Dec 1;119(23):4058–82. [DOI] [PubMed] [Google Scholar]
- 3.Wold ED, Smider VV, Felding BH. Antibody therapeutics in oncology. Immunotherapy (Los Angeles). 2016. Mar;2(1). doi: 10.4172/2471-9552.1000108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. 2019. Aug 31;394(10200):793–804. doi: 10.1016/S0140-6736(19)31774-X. [DOI] [PubMed] [Google Scholar]
- 5.Birrer MJ, Moore KN, Betella I, Bates RC. Antibody-drug conjugate-based therapeutics: state of the science. J Natl Cancer Inst. 2019. Jun 1;111(6):538–49. doi: 10.1093/jnci/djz035. [DOI] [PubMed] [Google Scholar]
- 6.Anagnostis P, Efstathiadou ZA, Polyzos SA, Adamidou F, Slavakis A, Sapranidis M, Litsas ID, Katergari S, Selalmatzidou D, Kita M. Acromegaly: presentation, morbidity and treatment outcomes at a single centre. Int J Clin Pract. 2011. Aug;65(8):896–902. doi: 10.1111/j.1742-1241.2011.02682.x. [DOI] [PubMed] [Google Scholar]
- 7.Roy B, Bhattacharjee A, Xu B, Ford D, Maizel AL, Cathcart MK. IL-13 signal transduction in human monocytes: phosphorylation of receptor components, association with Jaks, and phosphorylation/activation of stats. J Leukoc Biol. 2002. Sep;72(3):580–89. [PubMed] [Google Scholar]
- 8.Debinski W, Gibo DM. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor-associated cancer/testis antigen. Mol Med. 2000. May;6(5):440–49. doi: 10.1007/BF03401786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupardus PJ, Birnbaum ME, Garcia KC. Molecular basis for shared cytokine recognition revealed in the structure of an unusually high affinity complex between IL-13 and IL-13Ralpha2. Structure. 2010. Mar 10;18(3):332–42. doi: 10.1016/j.str.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabata Y, Khurana Hershey GK. IL-13 receptor isoforms: breaking through the complexity. Curr Allergy Asthma Rep. 2007. Sep;7(5):338–45. doi: 10.1007/s11882-007-0051-x. [DOI] [PubMed] [Google Scholar]
- 11.McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine. 2015. Sep;75(1):38–50. doi: 10.1016/j.cyto.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006. Jan;12(1):99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 13.He CH, Lee CG, Dela Cruz CS, Lee CM, Zhou Y, Ahangari F, Ma B, Herzog EL, Rosenberg SA, Li Y et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor alpha2. Cell Rep. 2013. Aug 29;4(4):830–41. doi: 10.1016/j.celrep.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee CM, He CH, Nour AM, Zhou Y, Ma B, Park JW, Kim KH, Dela Cruz C, Sharma L, Nasr ML, et al. IL-13Ralpha2 uses TMEM219 in chitinase 3-like-1-induced signalling and effector responses. Nat Commun. 2016. Sep 15;7:12752. doi: 10.1038/ncomms12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujisawa T, Joshi B, Nakajima A, Puri RK. A novel role of interleukin-13 receptor alpha2 in pancreatic cancer invasion and metastasis. Cancer Res. 2009. Nov 15;69(22):8678–85. doi: 10.1158/0008-5472.CAN-09-2100. [DOI] [PubMed] [Google Scholar]
- 16.Debinski W, Gibo DM, Slagle B, Powers SK, Gillespie GY. Receptor for interleukin 13 is abundantly and specifically over-expressed in patients with glioblastoma multiforme. Int J Oncol. 1999. Sep;15(3):481–86. [DOI] [PubMed] [Google Scholar]
- 17.Papageorgis P, Ozturk S, Lambert AW, Neophytou CM, Tzatsos A, Wong CK, Thiagalingam S, Constantinou AI. Targeting IL13Ralpha2 activates STAT6-TP63 pathway to suppress breast cancer lung metastasis. Breast Cancer Res. 2015. Jul 25;17:98. doi: 10.1186/s13058-015-0607-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin C, Liu H, Zhang H, He H, Li H, Shen Z, Qin J, Qin X, Xu J, Sun Y. Interleukin-13 receptor alpha2 is associated with poor prognosis in patients with gastric cancer after gastrectomy. Oncotarget. 2016. Aug 2;7(31):49281–88. doi: 10.18632/oncotarget.10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie M, Wu XJ, Zhang JJ, He CS. IL-13 receptor alpha2 is a negative prognostic factor in human lung cancer and stimulates lung cancer growth in mice. Oncotarget. 2015. Oct 20;6(32):32902–13. doi: 10.18632/oncotarget.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujisawa T, Joshi BH, Puri RK. IL-13 regulates cancer invasion and metastasis through IL-13Ralpha2 via ERK/AP-1 pathway in mouse model of human ovarian cancer. Int J Cancer. 2012. Jul 15;131(2):344–56. doi: 10.1002/ijc.26366. [DOI] [PubMed] [Google Scholar]
- 21.Okamoto H, Yoshimatsu Y, Tomizawa T, Kunita A, Takayama R, Morikawa T, Komura D, Takahashi K, Oshima T, Sato M et al. Interleukin-13 receptor alpha2 is a novel marker and potential therapeutic target for human melanoma. Sci Rep. 2019. Feb 4;9(1):1281. doi: 10.1038/s41598-019-39018-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Debinski W, Dickinson P, Rossmeisl JH, Robertson J, Gibo DM, Lesniak M. New agents for targeting of IL-13RA2 expressed in primary human and canine brain tumors. PLoS One. 2013;8(10):e77719. doi: 10.1371/journal.pone.0077719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sattiraju A, Solingapuram Sai KK, Xuan A, Pandya DN, Almaguel FG, Wadas TJ, Herpai DM, Debinski W, Mintz A. IL13RA2 targeted alpha particle therapy against glioblastomas. Oncotarget. 2017. Jun 27;8(26):42997–3007. doi: 10.18632/oncotarget.17792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, Zhou J, Sahin A, Carter BS, Brem H et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res. 2012. Nov 1;18(21):5949–60. doi: 10.1158/1078-0432.CCR-12-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014. Oct;16(10):1304–12. doi: 10.1093/neuonc/nou045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki A, Leland P, Joshi BH, Puri RK. Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine. 2015. Sep;75(1):79–88. doi: 10.1016/j.cyto.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 27.Gupta P, Wentland JA, Leal M, Ma D, Roach R, Esparza A, King L, Spilker ME, Bagi C, Winkelmann CT, et al. Assessment of near-infrared fluorophores to study the biodistribution and tumor targeting of an IL13 receptor alpha2 antibody by fluorescence molecular tomography. Oncotarget. 2017. Aug 22;8(34):57231–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abou DS, Ku T, Smith-Jones PM. In vivo biodistribution and accumulation of 89Zr in mice. Nucl Med Biol. 2011. Jul;38(5):675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berg E, Gill H, Marik J, Ogasawara A, Williams S, van Dongen G, Vugts D, Cherry SR, Tarantal AF, Pet T-B. Highly stable chelators together enable meaningful (89) Zr-AntibodyPET studies up to 30 days after injection. J Nucl Med. 2020. Mar;61(3):453–60. doi: 10.2967/jnumed.119.230961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta VR, Root A, Fisher T, Norberg R, David J, Clark T, Cohen J, May C, Giddabasappa A. Molecular imaging reveals biodistribution of P-cadherin LP-DART bispecific and trafficking of adoptively transferred T cells in mouse xenograft model. Oncotarget. 2020. Apr 14;11(15):1344–57. doi: 10.18632/oncotarget.27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD, Street SD. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today. 2012. May;17(9–10):419–24. doi: 10.1016/j.drudis.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 32.Daldrup H, Shames DM, Wendland M, Okuhata Y, Link TM, Rosenau W, Lu Y, Brasch RC. Correlation of dynamic contrast-enhanced MR imaging with histologic tumor grade: comparison of macromolecular and small-molecular contrast media. AJR Am J Roentgenol. 1998. Oct;171(4):941–49. doi: 10.2214/ajr.171.4.9762973. [DOI] [PubMed] [Google Scholar]
- 33.Watabe H, Ikoma Y, Kimura Y, Naganawa M, Shidahara M. PET kinetic analysis–compartmental model. Ann Nucl Med. 2006. Nov;20(9):583–88. doi: 10.1007/BF02984655. [DOI] [PubMed] [Google Scholar]
- 34.Heneweer C, Holland JP, Divilov V, Carlin S, Lewis JS. Magnitude of enhanced permeability and retention effect in tumors with different phenotypes: 89Zr-albumin as a model system. J Nucl Med. 2011. Apr;52(4):625–33. doi: 10.2967/jnumed.110.083998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heskamp S, Raave R, Boerman O, Rijpkema M, Goncalves V, Denat F. (89)Zr-Immuno-Positron emission tomography in oncology: state-of-the-art (89)Zr radiochemistry. Bioconjug Chem. 2017. Sep 20;28(9):2211–23. doi: 10.1021/acs.bioconjchem.7b00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giddabasappa A, Gupta VR, Norberg R, Gupta P, Spilker ME, Wentland J, Rago B, Eswaraka J, Leal M, Biodistribution SP. Targeting of Anti-5T4 antibody-drug conjugate using fluorescence molecular tomography. Mol Cancer Ther. 2016. Oct;15(10):2530–40. doi: 10.1158/1535-7163.MCT-15-1012. [DOI] [PubMed] [Google Scholar]
- 37.Nahrendorf M, Keliher E, Marinelli B, Waterman P, Feruglio PF, Fexon L, Pivovarov M, Swirski FK, Pittet MJ, Vinegoni C et al. Hybrid PET-optical imaging using targeted probes. Proc Natl Acad Sci U S A. 2010. Apr 27;107(17):7910–15. doi: 10.1073/pnas.0915163107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solomon M, Nothdruft RE, Akers W, Edwards WB, Liang K, Xu B, Suddlow GP, Deghani H, Tai YC, Eggebrecht AT, et al. Multimodal fluorescence-mediated tomography and SPECT/CT for small-animal imaging. J Nucl Med. 2013. Apr;54(4):639–46. doi: 10.2967/jnumed.112.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ocak M, Gillman AG, Bresee J, Zhang L, Vlad AM, Muller C, Schibli R, Edwards WB, Anderson CJ, Gach HM. Folate receptor-targeted multimodality imaging of ovarian cancer in a novel syngeneic mouse model. Mol Pharm. 2015. Feb 2;12(2):542–53. doi: 10.1021/mp500628g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marti-Bonmati L, Sopena R, Bartumeus P, Sopena P. Multimodality imaging techniques. Contrast Media Mol Imaging. 2010. Jul-Aug;5(4):180–89. doi: 10.1002/cmmi.393. [DOI] [PubMed] [Google Scholar]
- 41.Cilliers C, Nessler I, Christodolu N, Thurber GM. Tracking antibody distribution with near-infrared fluorescent dyes: impact of dye structure and degree of labeling on plasma clearance. Mol Pharm. 2017. May 1;14(5):1623–33. doi: 10.1021/acs.molpharmaceut.6b01091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cilliers C, Liao J, Atangcho L, Thurber GM. Residualization rates of near-infrared dyes for the rational design of molecular imaging agents. Mol Imaging Biol. 2015. Dec;17(6):757–62. doi: 10.1007/s11307-015-0851-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Damelin M, Bankovich A, Bernstein J, Lucas J, Chen L, Williams S, Park A, Aguilar J, Ernstoff E, Charati M et al. A PTK7-targeted antibody-drug conjugate reduces tumor-initiating cells and induces sustained tumor regressions. Sci Transl Med. 2017. Jan 11;9(372):eaag2611. doi: 10.1126/scitranslmed.aag2611. [DOI] [PubMed] [Google Scholar]
- 44.Sapra P, Shor B. Monoclonal antibody-based therapies in cancer: advances and challenges. Pharmacol Ther. 2013. Jun;138(3):452–69. doi: 10.1016/j.pharmthera.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 45.Rosen LS, Wesolowski R, Baffa R, Liao KH, Hua SY, Gibson BL, Pirie-Shepherd S, Tolcher AW. A phase I, dose-escalation study of PF-06650808, an anti-Notch3 antibody-drug conjugate, in patients with breast cancer and other advanced solid tumors. Invest New Drugs. 2020. Feb;38(1):120–30. doi: 10.1007/s10637-019-00754-y. [DOI] [PubMed] [Google Scholar]
- 46.Saka M, Amano T, Kajiwara K, Yoshikawa K, Ideguchi M, Nomura S, Fujisawa H, Kato S, Fujii M, Ueno K, et al. Vaccine therapy with dendritic cells transfected with Il13ra2 mRNA for glioma in mice. J Neurosurg. 2010. Aug;113(2):270–79. doi: 10.3171/2009.9.JNS09708. [DOI] [PubMed] [Google Scholar]
- 47.Bartolome RA, Jaen M, Casal JI. An IL13Ralpha2 peptide exhibits therapeutic activity against metastatic colorectal cancer. Br J Cancer. 2018. Oct;119(8):940–49. doi: 10.1038/s41416-018-0259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joyce AP, Wang M, Lawrence-Henderson R, Filliettaz C, Leung SS, Xu X, O’Hara DM. One mouse, one pharmacokinetic profile: quantitative whole blood serial sampling for biotherapeutics. Pharm Res. 2014. Jul;31(7):1823–33. doi: 10.1007/s11095-013-1286-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.