

ABSTRACT
Hypoxia develops in germinal centers (GCs) induced by model antigens; however, it is unknown whether tumor-reactive GCs are also hypoxic. We identified GC hypoxia in lymph nodes (LNs) draining murine mammary tumors and lethally irradiated tumor cells, and found that hypoxia is associated with the levels of antibody-secreting B cells. Hypoxic culture conditions impaired the proliferation of activated B cells, and inhibited class-switching to IgG1 and IgA immunoglobulin isotypes in vitro. To assess the role of the hypoxic response in tumor-reactive GCs in vivo, we deleted von Hippel-Lindau factor (VHL) in class-switched B cells and found decreased GC B cells in tumor-draining LNs, reduced class-switched and tumor-specific antibodies in the circulation, and modified phenotypes of tumor-infiltrating T cells and macrophages. We also detected the hypoxia marker carbonic anhydrase IX in the GCs of LNs from breast cancer patients, providing evidence that GC hypoxia develops in humans. We conclude that GC hypoxia develops in TDLNs, and that the hypoxic response negatively regulates tumor-induced humoral immune responses in preclinical models.
KEYWORDS: B cells, germinal centers, hypoxia, breast cancer, lymph nodes
Introduction
Despite their central role in the adaptive immune system, the impact of B cells on anti-tumor immunity remains controversial, with studies showing that B cells can have both pro and anti-tumor immune functions.1 Recent work has shown that accumulation of B cells and tertiary lymphoid structures in tumors is associated with improved responses to immune checkpoint blockade (ICB) in cancer patients, suggesting that B cells help promote an effective antitumor response in this context.2–4 It is currently unknown how the activation of B cells in specialized secondary lymphoid organs (SLOs) such as the lymph nodes (LNs) affects tumor-targeted immunity, and the microenvironmental conditions governing optimal B cell selection in tumor-draining LNs (TDLNs) are incompletely understood.
B cells expressing surface immunoglobulin with high affinity for an antigen are selected in the germinal center (GC) reactions of LNs and spleens. Recent studies in C57BL/6 mice have shown that GCs responding to model foreign antigens, such as sheep red blood cells (SRBCs) and chicken ovalbumin, develop regions of low oxygen (hypoxia) within the GC light zone.5,6 However, the effects of hypoxia on GC B cell function are not fully understood, with one paper reporting that hypoxia negatively regulates class-switch recombination and affinity maturation in the GC,5 and a subsequent study reporting that hypoxia potentiated B cell class-switching and antigen-specific responses.6 It is currently unknown whether GC hypoxia can develop in response to an infection or other active pathology such as cancer, and how hypoxic exposure affects the ability of GC B cells to manage an infection or malignancy. In addition, there is currently no evidence that hypoxia can develop in human GCs.
In this study, we assess whether GC hypoxia can develop in response to tumors using preclinical mouse models and determine how hypoxia affects B cell phenotype ex vivo. We also develop a Cre-Lox mouse to model the hypoxia response system in class-switched B cells and assess the consequent effect on GC reactions, mammary tumor growth, and tumor-infiltrating immune cells. We then show evidence of hypoxia in GCs from LNs resected from triple-negative breast cancer patients, validating our mouse models and supporting further work to study the influence of GC hypoxia in cancer patients.
Results
Hypoxia can develop in the GCs of tumor-draining LNs and associates with antibody-secreting cell differentiation in vivo
To determine whether hypoxia could develop among B cells responding to tumors, we injected BALB/c mice bearing orthotopic 4T07 mammary tumors with pimonidazole (Hypoxyprobe), a 2-nitroimidazole that is reduced and bound in cells under low oxygen conditions.7 We focused our analysis on the B cells of tumor-draining lymph nodes (TDLNs) since sentinel LNs are routinely sampled from breast cancer patients, and lymph is more concentrated for tumor-secreted factors relative to plasma.8 All inguinal and axillary TDLNs contained an expanded population of B cells (Figure 1(a)) and GL7+ GC B cells (Figure 1(b)) relative to naïve LNs. In approximately 50–60% of 4T07-bearing mice, a portion of B cells in TDLNs were also positive for Hypoxyprobe, while B cell hypoxia was not detected in the LNs of naïve, tumor-free mice (Figure 1(c-d)). The development of B cell hypoxia only occurred in the inguinal and axillary LNs ipsilateral to 4T07 tumors, as B cells in the corresponding contralateral LNs were not expanded nor hypoxic (Figure 1(e-f)). We assessed whether B cell hypoxia also developed in response to 4T1 mammary tumors, a cell line that is more metastatic and less immunogenic than the related 4T07 cell line.9,10 In line with the results from 4T07-bearing mice, LNs draining 4T1 mammary tumors also displayed an expansion of B cells and GC B cells relative to LNs from naïve mice (Supplemental Figure 1(a-b)), and Hypoxyprobe-binding among a portion of B cells (Figure 1(g)).
Figure 1.
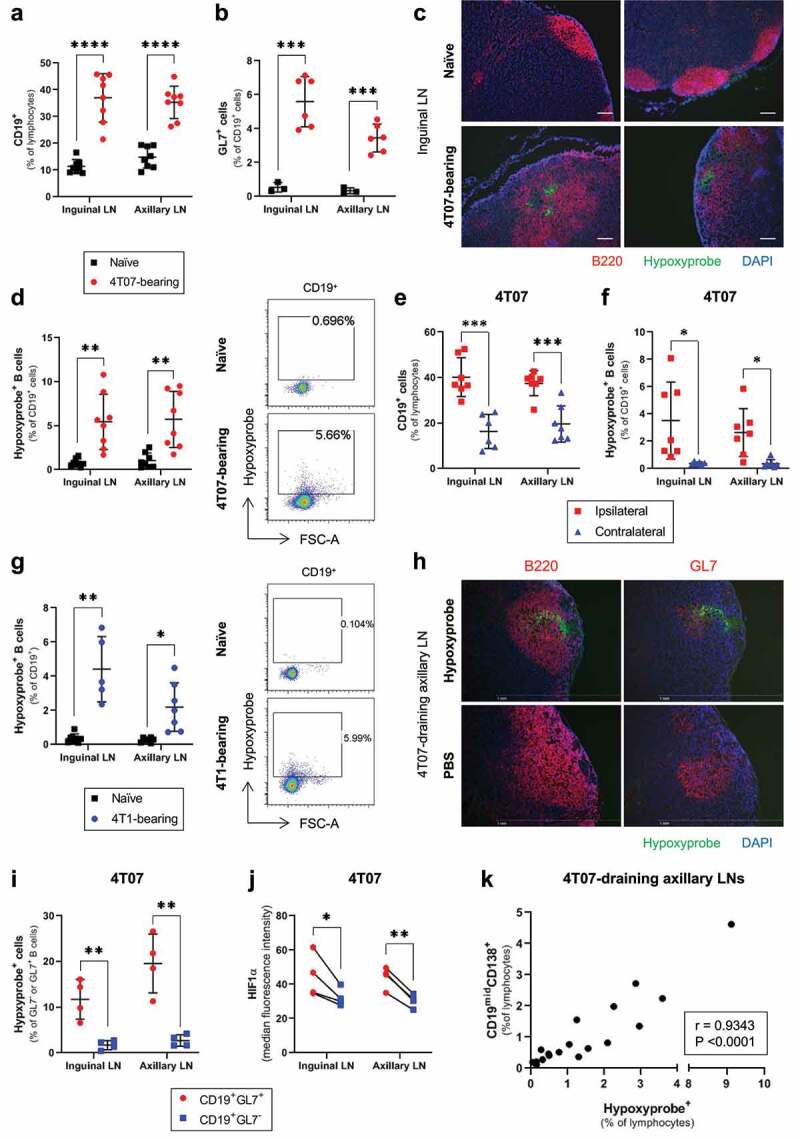
Hypoxia develops in the GCs of LNs draining murine mammary tumors and associates with the levels of antibody-secreting cells
Flow cytometry data evaluating the frequency of (a) CD19+ B cells (% of lymphocytes; n = 8/group), and (b) GL7+ GC B cells (% of CD19+; n = 3–6/group), in the inguinal and axillary LNs of naïve and 4T07 tumor-bearing mice. (c) Immunofluorescence microscopy on sections of frozen inguinal LNs from naïve and 4T07-bearing mice stained for B cells (B220), the exogenous hypoxia marker pimonidazole (Hypoxyprobe), and nuclear counterstain (DAPI). Scale bar = 100 μm. (d) Frequency of Hypoxyprobe+ B cells (% of CD19+) in inguinal and axillary LNs from naïve and 4T07 tumor-bearing mice (n = 8/group). (e) Frequency of CD19+ B cells (% of lymphocytes) and (f) Hypoxyprobe+ B cells (% of CD19+) in ipsilateral and contralateral inguinal and axillary LNs from 4T07-bearing mice (n = 6–7/group). (g) Frequency of Hypoxyprobe+ B cells (% of CD19+) in inguinal and axillary LNs from naïve and 4T1 tumor-bearing mice (n = 5–8/group). (h) Immunofluorescence microscopy on sections of frozen axillary LNs from 4T07-bearing mice, injected with Hypoxyprobe or vehicle control (PBS), and stained for B cells (B220), GCs (GL7), hypoxia (Hypoxyprobe), and nuclear counterstain (DAPI). Scale bar = 1 mm. (i) Frequency of Hypoxyprobe+ cells in the GC (CD19+GL7+) and non-GC (CD19+GL7−) B cells of LNs draining 4T07 tumors (n = 4/group). (J) Median fluorescence intensity of HIF1ɑ among GC (CD19+GL7+) and non-GC (CD19+GL7−) B cells of LNs draining 4T07 tumors (n = 4/group). (k) Correlation plot between the frequency of Hypoxyprobe+ cells and ASCs in axillary LNs draining 4T07 tumors (n = 18 mice; mice were 21 weeks old). Two-tailed Student’s t-test (with Welch’s correction applied when groups had unequal variances) was used to assess statistical significance in (A), (B), (D), (E), (F) and (G), paired t-test was used in (I), and (J). Pearson correlation coefficient (r) is shown in (K). Data are mean ± SD.
To assess whether hypoxia was developing in the GC reactions of TDLNs, we stained sections of frozen LNs from 4T07-bearing mice for Hypoxyprobe and found that hypoxic regions could overlap with both GL7+ GC B cells and non-GC B cells (Figure 1(h)). Analysis by flow cytometry showed that a greater proportion of the GL7+ GC B cell population was positive for Hypoxyprobe (Figure 1(i)) and displayed a higher median fluorescence intensity (MFI) for the oxygen-labile hypoxia-inducible factor-1ɑ (HIF1ɑ) (Figure 1(j)) than non-GC B cells. We also verified increased Hypoxyprobe staining among B cells positive for additional GC markers (IgD−CD38−/dimCD95+GL7+Bcl6+; Supplemental Figure 1(c)). The development of GC hypoxia in response to model foreign antigens was previously reported in C57BL/6 mice, with no experiments performed in the BALB/c background.5,6 Given the skew toward Th1 immunity in C57BL/6 mice and toward Th2 responses in BALB/c mice,11 we assessed whether B cell hypoxia was also observed in C57BL/6 LNs responding to syngeneic E0771 mammary tumors. Concomitant with only a modest GC response (Supplemental Figure 1(d)), B cell hypoxia was not induced in LNs draining E0771 mammary tumors (Supplemental Figure 1(e-f)), suggesting that the proclivity to develop B cell hypoxia in TDLNs may be affected by the genetic background of different mouse strains. Taken together, the data show that GC hypoxia can develop in the TDLNs of BALB/c mice bearing 4T07 and 4T1 mammary tumors.
Given the variable Hypoxyprobe-binding in TDLNs (Figure 1(d)), we assessed whether the level of hypoxia was associated with a particular B cell subtype. Across 4T07-bearing mice (n = 18), the levels of hypoxia in TDLNs were not associated with the overall frequency of GC B cells (Supplemental Figure 2(a)), demonstrating that GCs could form in TDLNs without developing hypoxia. Tumor burden did appear to correlate with the levels of hypoxia in TDLNs (r = 0.5792; Supplemental Figure 2(b)), although the correlation was attenuated as tumors reached approximately 0.8 g. Across the replicates, the frequency of CD19midCD138+ antibody-secreting cells (ASCs) was more strongly correlated with TDLN hypoxia (r = 0.9343; Figure 1(k)), than primary tumor weight (r = 0.5420; Supplemental Figure 2(c)), suggesting that TDLN hypoxia associates with the development of ASCs. To assess whether other markers of hypoxia also associated with ASC levels, we quantified the expression of HIF1α and hypoxia-inducible factor-ɑ (HIF2α) in the B cells of LNs draining 4T07 mammary tumors. The frequency of HIF2αhi B cells correlated more strongly with Hypoxyprobe staining than the frequency of HIF1αhi B cells (Supplemental Figure S2(d-e)), and we observed a significant correlation between HIF2αhi B cells and the levels of ASCs in TDLNs (Supplemental figure S2f). Together, our data show that a hypoxic environment in TDLNs associates with the local development of ASCs during immune responses to tumors.
Exposure to lethally irradiated tumor cells is sufficient to induce GC hypoxia
While we established that B cell hypoxia could develop in TDLNs, it was unclear whether this phenotype arose due to an immune response against the tumor cells. To address this question, we assessed whether an immune response to dying tumor cells was sufficient to induce GC hypoxia in the absence of tumor formation. We used a lethal dose (20 Gy) of ionizing radiation to kill tumor cells in vitro prior to injection into mice, as radiation is known to induce an immunogenic form of cell death.12 We empirically determined that injection of mice with three doses of lethally irradiated 4T07 cells into the mammary fat pad (Figure 2(a)) induced B cell expansion and GC formation in both inguinal and axillary LNs (Figure 2(b,c)). Using fluorescence microscopy, we observed Hypoxyprobe binding localized within the boundaries of GC reactions of LNs responding to irradiated 4T07 cells (Figure 2(d)). Analysis by flow cytometry confirmed the presence of Hypoxyprobe positive B cells in inguinal and axillary LNs (Figure 2(e)), where hypoxia primarily affected GC B cells (Figure 2(f)). We also observed GC formation (Figure 2(g)) and B cell hypoxia (Figure 2(h,i) in LNs responding to lethally irradiated 4T1 cells, although the frequency of Hypoxyprobe positive B cells was lower than the levels induced by irradiated 4T07 cells. We found that injection of lethally irradiated E0771 mammary tumor cells into C57BL/6 mammary fat pads induced a weaker GC response (Supplemental Figure 3a) and lower levels of B cell hypoxia (Supplemental Figure 3b-c) than 4T07 or 4T1 cells in BALB/c mice, consistent with our observations in E0771 tumor-bearing mice. These results show that GC development in response to lethally irradiated tumor cells is sufficient to induce hypoxia among GC B cells, particularly in BALB/c mice.
Figure 2.
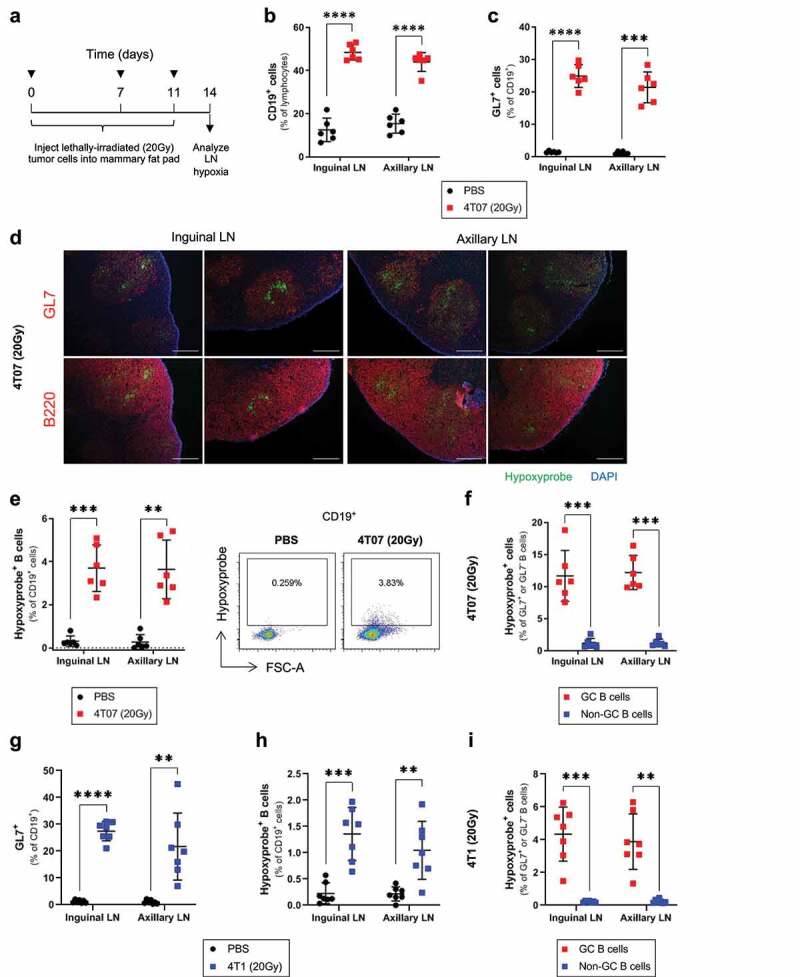
Injection of lethally irradiated tumor cells is sufficient to induce GC hypoxia
(a) Injection schedule used to expose mice to tumor cells irradiated in vitro (20 Gy). (b) Flow cytometry data evaluating the frequency of B cells (CD19+, % of lymphocytes) and (c) GC (GL7+) B cells (% of CD19+) in inguinal and axillary LNs of mice injected with lethally irradiated 4T07 tumor cells, or PBS controls; n = 6/group. (d) Immunofluorescence microscopy on sections of frozen inguinal and axillary LNs of mice injected with lethally irradiated 4T07 cells, and stained for GCs (GL7), B cells (B220), hypoxia (Hypoxyprobe), and nuclear counterstain (DAPI). Scale bar = 100 μm. (e) Frequency of Hypoxyprobe+ B cells (% of CD19+) in inguinal and axillary LNs draining lethally irradiated 4T07 tumor cells, or PBS controls (n = 6/group). (f) Frequency of Hypoxyprobe+ cells in the GC (CD19+GL7+) and non-GC (CD19+GL7−) B cells of LNs draining lethally irradiated 4T07 tumors (n = 6/group). (g) Frequency of GC (GL7+) B cells (% of CD19+) and (h) Hypoxyprobe+ B cells (% of CD19+) in inguinal and axillary LNs of mice injected with lethally irradiated 4T1 tumor cells, or PBS controls; n = 7/group. (i) Frequency of Hypoxyprobe+ cells in the GC (CD19+GL7+) and non-GC (CD19+GL7−) B cells of LNs draining lethally irradiated 4T1 tumors (n = 7/group). Two-tailed Student’s t-test was used to assess statistical significance between groups in (B), (C), (E), (G), and (H) with Welch’s correction applied to groups with unequal variances. Paired t-test was used in (F) and (I). Data are mean ± SD.
Hypoxia inhibits B cell proliferation and reduces class-switching in vitro
Previous work has shown that hypoxia can inhibit IgG2c class-switching in activated C57BL/6 B cells, but does not affect class-switching to IgA.5 To resolve whether hypoxia affects class-switching to IgG and IgA isotypes in BALB/c B cells, we activated B cells with stimuli that promote IgG1 or IgA class-switching under normoxic (21% O2) or hypoxic (1% O2) conditions. Hypoxia reduced the frequency of both IgG1+ and IgA+ B cells after a 4-day stimulation period (Figure 3(a,b)). We noted that the class-switched isotype appeared in B cells that underwent more rounds of cell division based on CFSE staining (flow plots in Figure 3(a,b)), and hypoxia reduced B cell proliferation under both IgG1 and IgA-inducing stimuli (Figure 3(c,d)). When class switching was analyzed on a per cell division basis, we found that hypoxia reduced the frequency of IgG1+ B cells at later cell divisions (Figure 3(e)), while the number of IgA+ B cells in each cell cycle generation was not significantly affected by hypoxia (Figure 3(f)). Taken together, our data indicate that hypoxic conditions can inhibit the proliferation of activated B cells and decrease immunoglobulin class-switching.
Figure 3.
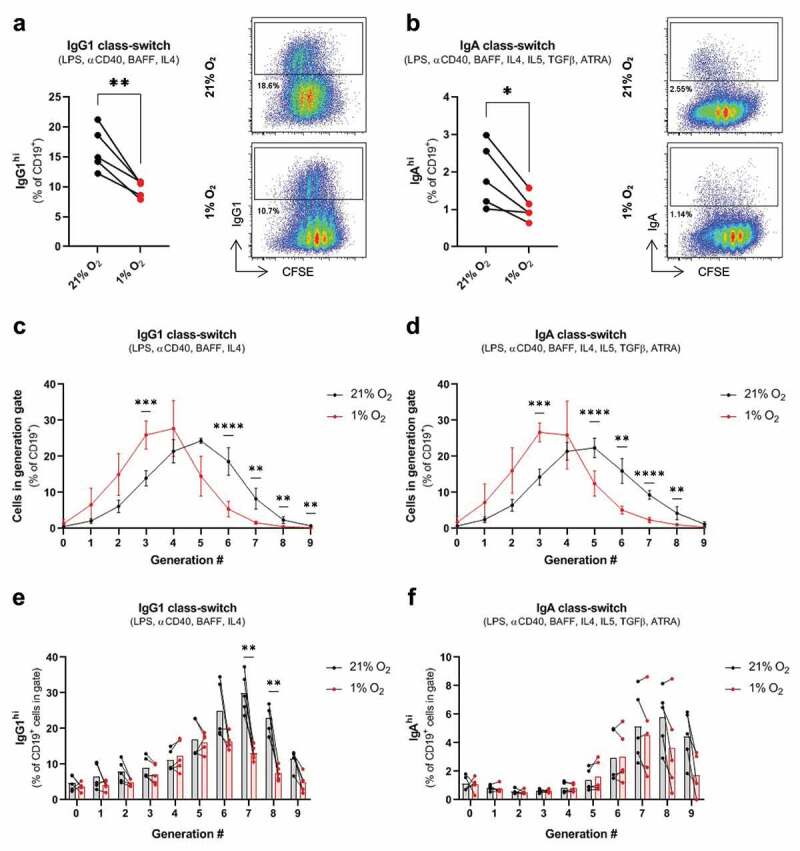
Hypoxia inhibits B cell proliferation and class-switching in vitro.
(a) Frequency of IgG1hi cells (% of CD19+) amongst mouse BALB/c B cells cultured with IgG1 class-switch inducing stimuli (LPS, ɑCD40, BAFF, IL4) for 4 days under normoxic (21% O2) or hypoxic conditions (1% O2). (b) Frequency of IgAhi cells (% of CD19+) amongst B cells cultured with IgA class-switch inducing stimuli (LPS, ɑCD40, BAFF, IL4, IL5, TGFβ, ATRA) for 4 days under normoxic or hypoxic conditions. (c) Frequency of B cells in CFSE-defined cell-division gates (% of total CD19+ cells) cultured with IgG1 or (d) IgA class-switch inducing stimuli under normoxic or hypoxic conditions, with statistical significance assessed between normoxic and hypoxic conditions at each cell division. (e) Frequency of IgG1hi or (f) IgAhi B cells on a per cell-division basis following stimulation under normoxic or hypoxic conditions. Two-tailed paired t-test was used to assess statistical significance in (A) and (B), while a two-tailed paired t-test followed by the Holm-Bonferroni correction to control for the family-wise error rate was used to assess statistical significance between normoxic and hypoxic cells within a cell-division gate in (C), (D), (E), and (F). n = 5 paired samples for all panels. Data are mean ± SD.
The hypoxia response system negatively regulates tumor-induced GC reactions
To better delineate the functional role of hypoxia in tumor-reactive GCs in vivo, we developed a BALB/c mouse model where the hypoxia response system is induced in class-switched B cells. Von Hippel-Lindau factor (VHL) promotes the oxygen-dependent degradation of the alpha subunit of hypoxia-inducible factor-1 (HIF1), and VHL deletion results in constitutive stabilization of HIF1ɑ and HIF1 activation despite the presence of molecular oxygen.13 Mice with loxP sites flanking the promoter and first exon of VHL were crossed with mice expressing Cre recombinase from the IgG1 (Cγ1) locus, resulting in VHL deletion and HIF1ɑ stabilization when activated B cells initiate antibody-class switching (Figure 4(a)).
Figure 4.
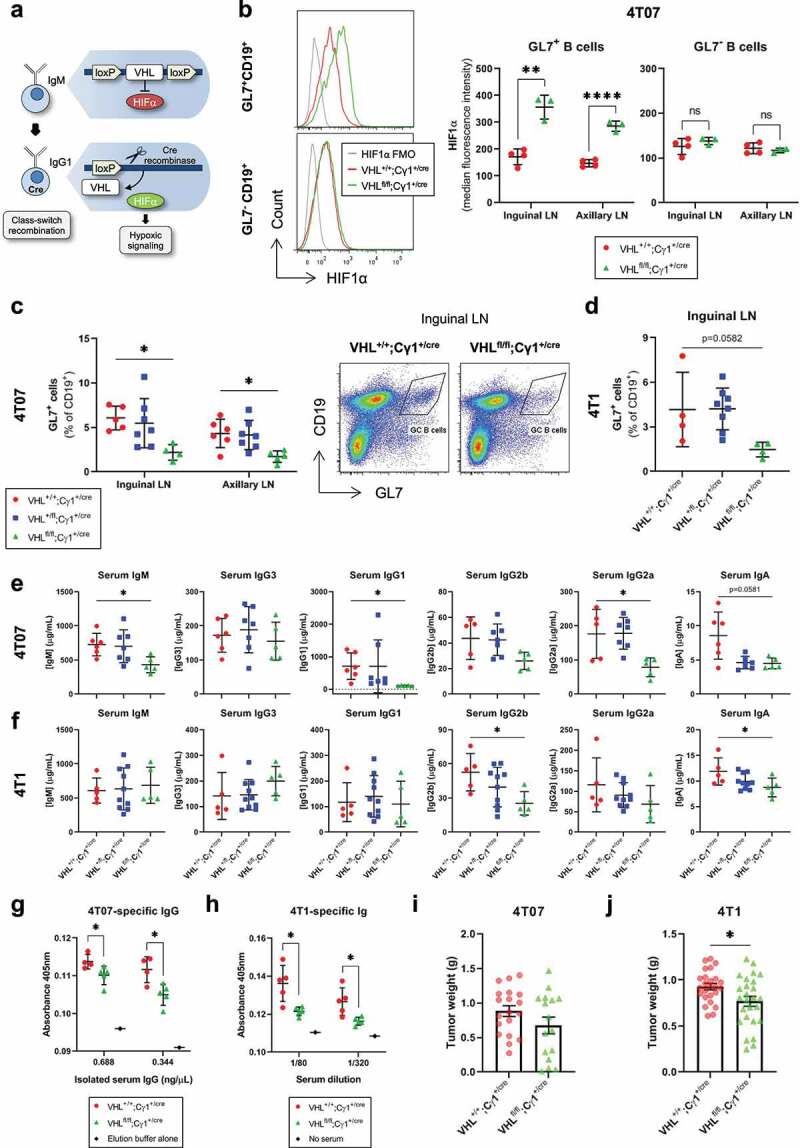
pVHL deletion in class-switched B cells inhibits tumor-induced humoral immune responses
(a) Schematic of VHL 2-lox;Cγ1-cre mice used to model the effects of the hypoxia response system on GC B cells through deletion of the promoter and exon 1 of the VHL gene in class-switched, IgG1+ B cells. (b) HIF1ɑ median fluorescence intensity in GC (GL7+CD19+) and non-GC B cells (GL7−CD19+) of 4T07 TDLNs from VHL+/+;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice (n = 3–4/group). (c) Flow cytometry data evaluating the frequency of GL7+ GC B cells (% of CD19+) in TDLNs from VHL+/+;Cγ1+/cre, VHL+/fl;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice bearing 4T07 tumors or (d) 4T1 mammary tumors (n = 4–8/group). The represented GL7+ gate was applied to the CD19+ population to evaluate the frequency of GC B cells. (e) ELISA-based quantification of different immunoglobulin subclasses in the serum of VHL+/+;Cγ1+/cre, VHL+/fl;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice bearing 4T07 or (f) 4T1 tumors (n = 5–10/group). (g) In-cell ELISA-based quantification of 4T07-binding IgG or (h) 4T1-binding immunoglobulin in the serum of VHL+/+;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice bearing 4T07 or 4T1 tumors (n = 4–5/group). (i) Weights of 4T07 tumors (n = 16–19/group) and (j) 4T1 tumors (n = 25–26/group) from VHL+/+;Cγ1+/cre or VHLfl/fl;Cγ1+/cre mice 3 weeks post-tumor implant. Two-tailed Student’s t-test was used to assess statistical significance in (B), (G), (H), (I), and (J). One-way ANOVA followed by Dunnett’s multiple comparisons test to controls (VHL+/+;Cγ1+/cre) (or Brown-Forsythe ANOVA, followed by Dunnett’s T3 multiple comparisons test to controls if SDs were significantly different between groups) was used in (C), (D), (E), and (F). Data are mean ± SD in (a)-(H), and mean ± SEM in (I) and (J).
We first verified that floxed VHL underwent recombination in the TDLNs of VHL+/fl;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice implanted with 4T07 tumors (Supplemental Figure 4a). HIF1ɑ was stabilized in the GC B cells of VHLfl/fl;Cγ1+/cre mice relative to wildtype controls (VHL+/+;Cγ1+/cre), while there was no difference in HIF1ɑ levels between the non-GC B cells (Figure 4(b)). In response to both 4T07 (Figure 4(c)) and 4T1 (Figure 4(d)) mammary tumors, VHLfl/fl;Cγ1+/cre mice contained reduced GL7+ GC B cells in their TDLNs. We also verified that there was a reduction in B cells labeled with additional GC markers (IgD−CD38−/dimCD95+GL7+Bcl6+) in the TDLNs of VHLfl/fl;Cγ1+/cre mice bearing 4T07 mammary tumors (Supplemental Figure 4b). To assess GC function, we quantified class-switched antibodies and tumor-specific antibodies in the serum of tumor-bearing mice. When implanted with 4T07 tumors, VHLfl/fl;Cγ1+/cre mice displayed significantly lower levels of IgM, IgG1, and IgG2a relative to controls (Figure 4(e)), while IgG2b and IgA were significantly reduced in VHLfl/fl;Cγ1+/cre mice bearing 4T1 mammary tumors (Figure 4(f)). We measured the levels of tumor cell-binding antibodies in the serum of VHLfl/fl;Cγ1+/cre mice using an in-cell ELISA. After purifying and standardizing the levels of whole serum IgG assayed across genotypes (Supplemental Figure 4c-d), we detected lower levels of 4T07-binding IgG in the serum of VHLfl/fl;Cγ1+/cre mice implanted with 4T07 tumors relative to controls (Figure 4(g)). Similarly, VHLfl/fl;Cγ1+/cre mice implanted with 4T1 mammary tumors also contained less 4T1-binding immunoglobulins in the serum than control mice (Figure 4(h)), with no difference observed in whole immunoglobulin levels using a standard ELISA (Supplemental Figure 4e). Interestingly, while GC B cells were reduced in the TDLNs of VHLfl/fl;Cγ1+/cre mice, there was no difference in splenic GC formation between the genotypes (Supplemental Figure 5a-b) and low levels of IgG1 expression in splenic B cells relative to B cells from inguinal TDLNs (Supplemental Figure 5c-d). Thus, the reductions in antibody class-switching and tumor cell-specific antibodies in the serum of VHLfl/fl;Cγ1+/cre mice highlight the significant contribution of TDLNs in mounting humoral immune responses to tumors. Taken together, the data show that VHL deletion in class-switched B cells compromises GC formation and output in tumor-bearing mice, indicating that the hypoxia response system negatively regulates tumor-reactive GCs.
Due to their deficiency in GC development and output, the VHLfl/fl;Cγ1+/cre mouse model can be used to study the impact of humoral B cell responses across different diseases or pathologies. We assessed tumor weights at endpoint (3-week post-tumor cell injection) in VHLfl/fl;Cγ1+/cre mice implanted with 4T07 and 4T1 mammary carcinoma lines. While there was no difference in 4T07 mammary tumor weight between the genotypes (Figure 4(i)), we observed a statistically significant reduction in 4T1 tumor weight in VHLfl/fl;Cγ1+/cre mice relative to controls (Figure 4(j)). These data indicate that pVHL deletion in germinal center B cells reduces GC responses, restricts Ig class switching, reduces tumor-specific antibodies in the circulation, and can reduce primary mammary tumor growth.
pVHL deletion in GC B cells modulates the phenotype of tumor-infiltrating T cells and macrophages in 4T07 mammary tumors
We also assessed whether pVHL deletion in GC B cells affected the levels and phenotypes of tumor-infiltrating immune cells. Compared to VHL+/+;Cγ1+/cre mice, 4T07 tumors in VHLfl/fl;Cγ1+/cre mice had no difference in the levels of tumor-infiltrating CD4+ T cells or F480+CD11b+ macrophages (Supplemental Figure 6a-b) and a trend toward increased tumor-infiltrating CD8+ T cells (Figure 5(a)). Interestingly, we found decreased proportions of activated CD8+ T cells in 4T07 tumors from VHLfl/fl;Cγ1+/cre, including decreased IFNγ (Figure 5(b)), CD44hi (Figure 5(c)), and PD-1 (Figure 5(d)). 4T07 tumors in VHLfl/fl;Cγ1+/cre also contained lower levels of CD44hi and PD-1+ tumor-infiltrating CD4+ T cells (Figure 5(e-f)) and increased proportions of immune suppressive MHC-II+CD206+ M2 macrophages14 (Figure 5(g)). Interestingly, there was no difference in tumor-infiltrating T cells or macrophages in VHLfl/fl;Cγ1+/cre mice bearing 4T1 tumors (Supplemental Figure 6(c-e)), and no difference in IFNγ, CD44, or PD-1 expression in tumor-infiltrating T cells (Supplemental figure 6f-j), or the proportion of M2 macrophages in 4T1 tumors (Supplemental Figure 6k). We did not observe differences in the proliferative fractions or levels of necrosis in 4T07 or 4T1 tumors between VHL+/+;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice (Supplemental Figure 7A-D). Taken together, our data suggest that deletion of pVHL in GC B cells can directly or indirectly impair the activation status of tumor-infiltrating T cells and promote M2 macrophages in immunogenic 4T07 tumors, although the effect appears to be tumor-line dependent.
Figure 5.
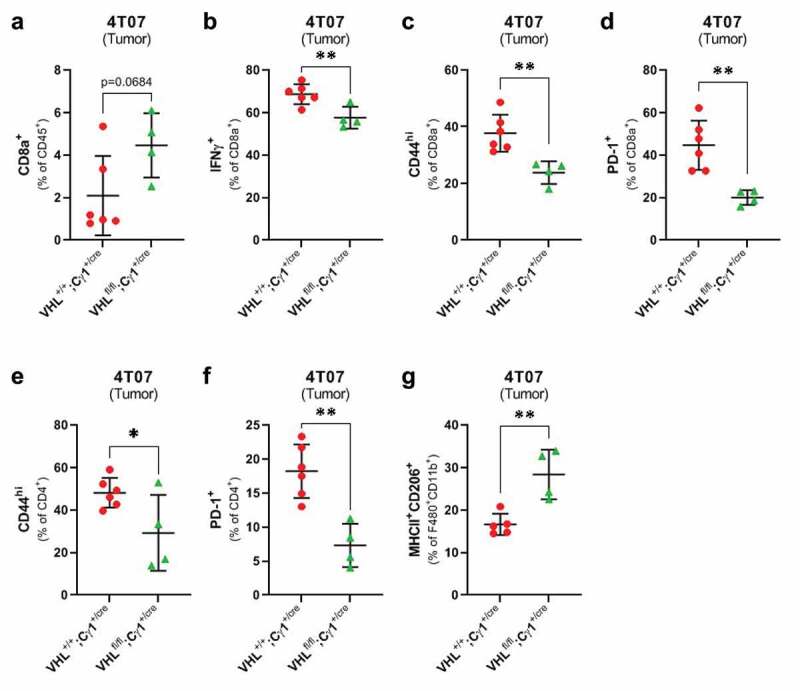
pVHL deletion in class-switched B cells alters the phenotype of T cells and macrophages infiltrating 4T07 mammary tumors
(a) Flow cytometry data quantifying the frequency of CD8+ T cells infiltrating 4T07 tumors in VHL+/+;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice. (b) Frequency of IFNγ+, (c) CD44hi, and (d) PD-1+ tumor-infiltrating CD8+ T cells in 4T07 tumors from VHL+/+;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice. (e) Frequency of CD44hi and (f) PD-1+ tumor-infiltrating CD4+ T cells, and (g) MHCII+ CD206+ F480+ CD11b+ macrophages in VHL+/+;Cγ1+/cre and VHLfl/fl;Cγ1+/cre mice bearing 4T07 tumors. Two-tailed Student’s t-test was used to assess statistical significance in A-G; n = 4–6/group. Data are mean ± SD.
The HIF target gene CA9 is present in GCs of breast cancer patient LNs
To assess whether GC hypoxia develops in a clinical setting, we analyzed LNs from six patients with node-negative or node-positive triple-negative breast cancer (TNBC; Supplemental Table 1). We stained the sections for carbonic anhydrase IX (CA9), a transcriptional target of HIF1 that is commonly used as an endogenous marker of hypoxia.15 To avoid detecting CA9 potentially associated with metastatic tumors, we restricted our analysis to samples of non-involved LNs from each patient. We found that CA9 staining was present in all GCs of TNBC patient lymph nodes (Figure 6), providing the first line of evidence that hypoxia develops within human lymph node germinal centers and validating our observations of hypoxia in TDLNs of pre-clinical BALB/c mammary tumor models.
Figure 6.
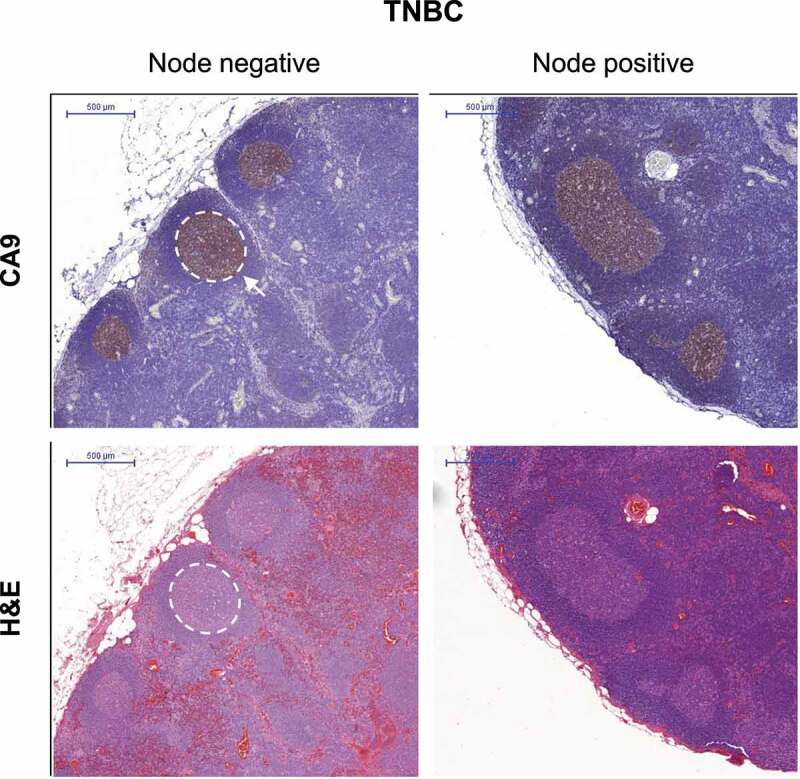
The hypoxia marker CA9 is expressed in breast cancer patient GCs
Representative images of FFPE-sections of non-involved LNs from node-negative or node-positive triple-negative breast cancer patients stained for CA9 (brown) or H&E. White demarcation outlines a GC. Scale bar = 500 μm.
Discussion
Previous reports had shown that hypoxia could develop in the GCs of mice responding to model foreign antigens.5,6 However, it was unknown whether GC hypoxia could also arise in the context of an active pathology, and whether GC hypoxia occurs in humans. We demonstrate that TDLN GCs can develop regions of hypoxia during immune responses to tumors in preclinical mouse models, and that the endogenous hypoxia marker CA9 is present within the GCs of LNs from triple-negative breast cancer patients.
We used two mammary carcinoma models in the BALB/c mouse background to demonstrate that hypoxia could develop in tumor-reactive GCs; however, B cell hypoxia was not detected in C57BL/6 LNs responding to established E0771 mammary tumors, and was only weakly induced in LNs responding to lethally irradiated E0771 cells. We noted a weaker GC response in C57BL/6 LNs relative to BALB/c LNs, which may have restricted their development of B cell hypoxia. Although we only present data from one tumor line in the C57BL/6 host, the known Th1 bias of this mouse background11,16 may generally limit its propensity to develop tumor-reactive GCs and accompanying hypoxia. In contrast, BALB/c immune responses are considered Th2 skewed.11 A comparative study using an autoimmunity model has shown that enhanced secretion of IL4 and IL10 cytokines from BALB/c Tfh cells relative to their C57BL/6 counterparts may augment GC B cell responses.17 Similarly, in response to the same T-dependent antigen, splenic B cells from BALB/c mice were more prone to activation and GC formation than C57BL/6 B cells.18 The BALB/c host genetics may enable the development of a stronger GC reaction in response to tumor cells, allowing the emergence of hypoxia amongst the B cell population. These findings suggest that the BALB/c background may be appropriate for the development of future mouse models attempting to address the impact of GC hypoxia on anti-tumor immunity.
Previous mouse studies investigating the effect of hypoxia on GC responses yielded conflicting results. Cho, et al., reported that hypoxic conditions in vitro impaired immunoglobulin class-switching to IgG2c.5 The authors also used an adoptive transfer model of VHL-deficient B cells to show that the hypoxia response system limited IgG2c class-switching and antigen-specific B cell responses in vivo.5 In contrast, Abbot et al., suggested that hypoxia potentiated IgG1 class-switching at early timepoints and promoted ASC development, and that reversal of tissue hypoxia with supplemental oxygen in vivo reduced antigen-specific IgG1 B cell responses.6 Our results suggest that the hypoxia response system, modeled by the inducible deletion of VHL in class-switched B cells, negatively regulates tumor-induced GC responses. However, we also observed that physiological hypoxia was associated with ASC development. Previous work has shown that the qualitative output of GCs is temporally regulated, where plasma cells are characteristic of late-stage GCs.19 It is possible that hypoxia develops in established, mature GCs, providing environmental cues that promote ASC differentiation, and inducing signaling within GC B cells that modulates a sufficiently developed GC reaction.
Our data show that engagement of the hypoxia response system in class-switched B cells reduces overall GC formation in vivo, rather than only reducing the products of GC reactions as previously reported.5 The increased extent of immunomodulation by the hypoxia response system in our model may arise due to differences in mouse background, the specific B cell population targeted for pVHL deletion, or in the duration of antigenic stimulation. In work by Cho et al., the authors assessed the effect of the hypoxia response system on B cells in vivo by adoptively transferring a mixture of VHLfl/fl B cells and ovalbumin-specific CD4+ T cells into recipient mice, and subsequently measuring antibody responses post-immunization with ovalbumin.5 During a primary immune response, VHL-deficient B cells yielded less IgG2c antibodies of all affinities, while levels of all-affinity IgM, IgG1, and IgA were not affected. Following a secondary immunization, the reduction in IgG2c class switching was more pronounced, and also affected IgM levels,5 suggesting that hypoxia-induced defects in class-switching can accumulate and broaden over the course of several immunizations. While sequential immunization with model antigens yields distinct primary and secondary immune responses,5 the continuous presence of antigen in tumor-bearing mice may lead to a more chronic stimulation of the immune system, which could similarly contribute to broadened defects in class-switching. Indeed, continuous immune stimulation could explain the unexpected reduction of IgM levels in VHLfl/fl;Cγ1+/cre mice bearing 4T07 tumors, as the Cμ locus lies upstream of the IgG1 gene driving Cre-recombinase expression. When B memory cells emerge from a GC, they display a highly antigen-specific immunoglobulin on their surface which allows them to bind and present antigen to T cells. In the continuous presence of antigen, this positive feedback mechanism20 could enable the generation of de novo GC reactions and antibody responses, whose accumulation would be restricted in VHLfl/fl;Cγ1+/cre mice.
In contrast to previous work, we also observed that engagement of the hypoxia response system caused reductions in circulating levels of IgA. Cho et al., showed that activation-induced cytidine deaminase (AID) expression, an enzyme that mediates immunoglobulin class-switching, was only reduced by hypoxia under IgG switch-conditions, and that IgA levels were not affected by the hypoxia response system in vitro or in vivo.5 Our in vitro data partly corroborate the findings by Cho et al., where hypoxia did not reduce IgA class-switching on a per cell-division basis; however, we did observe a reduction in the overall levels of IgA-switched cells under 1% O2 that was concomitant with impaired B cell proliferation. Similarly, a reduction in the overall frequency of GC B cells in the TDLNs of VHLfl/fl;Cγ1+/cre mice may contribute to an impaired IgA response in vivo. Together, our results suggest that the negative effects of the hypoxia response system on B cell proliferation and GC formation may lead to decreases in class-switched immunoglobulins that extend beyond IgG isotypes.
Unlike T cells, the role of B cells in controlling tumor growth is still debated, with data from patient cohorts and preclinical models showing B cells can both promote, and inhibit, effective anti-tumor immunity.1 Over the past few years, it has become increasingly clear that the B cell response is multifaceted, and that the overall effect of B cells on tumor growth partly depends on the types of B cells engaged, the predominant antibody isotype present, and the balance of other immune cell populations in the tumor microenvironment.1 For example, tumor-specific antibodies can enhance anti-tumor immunity by promoting antibody-dependent cellular cytotoxicity (ADCC) when NK cells engage Fc portions of tumor-bound antibodies. Of the different antibody isotypes, IgG1 and IgG3 are most capable of inducing ADCC.21 Elevated levels of IgG1 in particular, are associated with improved survival in cancer patients with high rates of somatic-hypermutation in their tumor-infiltrating B cells,22 and this isotype is thought to promote anti-tumor activity by B cells.1 In contrast, the IgA isotype is frequently associated with immunosuppressive B cells, and high levels of intratumoral IgA1 is associated with poor patient prognosis.1 B cell class-switching to IgA is induced by TGF-β23 and, in the gut, this cytokine mediates a regulatory loop between IgA+ B cells and T regulatory cells that help maintain immune homeostasis.24,25 This regulatory function also helps mediate pro-tumor B cell activity, as TGF-β secretion by B cells is required for their ability to promote the suppressive function of MDSCs.26 Thus, the overall effect of impaired GC responses on tumor growth in preclinical mouse models likely depends on the balance of pro- and anti-tumor functions of B cells induced by individual tumor lines.
In our VHLfl/fl;Cγ1+/cre model, reduced GC formation in tumor-draining lymph nodes was associated with decreased T cell activation within 4T07 tumors, consistent with the emerging hypothesis that B cells may facilitate T cell activation during tumor-induced immune responses.27,28 While it is possible that products of the GC reaction in lymph nodes, such as affinity matured BCRs and secreted antibodies, may modulate T cell activation in the lymph node or at the primary tumor site, it is notable that pVHL deletion in GC B cells only affected tumor-infiltrating T cell phenotypes in 4T07 tumors and not in 4T1 tumors. It is still unknown which conditions enable a T cell-promoting phenotype of B cells during anti-tumor immune responses, but it is plausible that some tumors fail to activate these functions in B cells and it is notable that 4T1 tumors are considered less immunogenic than 4T07 tumors.9,10 Reduced T cell activation and increased M2 macrophages in 4T07 tumors in VHLfl/fl;Cγ1+/cre mice would be hypothesized to enhance tumor growth, although the tendency toward increased overall CD8+ T cell infiltration in these tumors may counteract the effect of reduced T cell activation and lead to no observable difference in 4T07 tumor growth between the two genotypes. The mechanism behind reduced 4T1 tumor growth in VHLfl/fl;Cγ1+/cre mice remains the subject of further investigation in our laboratory. Since recent reports have associated B cell activation with favorable responses to ICB in the clinic,2–4 future work should also assess how impaired GC responses in VHLfl/fl;Cγ1+/cre mice impacts the efficacy of ICB in pre-clinical models.
Upon interpreting our results within the context of previously published literature, we propose the following conceptual model for the role of hypoxia in tumor-reactive GCs. After encountering tumor neoantigens that bind their B cell receptors, B cells in TDLNs form nascent GC reactions. As the GCs grow in size and mature, physiological hypoxia can develop among the GC B cells. The low oxygen conditions may favor the differentiation of antibody-secreting cells (ASCs) from the GC B cells, as preferential output of ASCs is characteristic of later-stage GC reactions.19 The hypoxic conditions also induce the hypoxia response system and HIF activity in GC B cells, which functions to modulate/negatively regulate an established GC reaction. Forcing HIF stabilization by knocking out VHL in GC B cells leads to aberrantly reduced GC formation, decreased circulating class-switched and tumor-specific antibodies, and altered activation of tumor-infiltrating T cells and macrophages. We have also found evidence of hypoxia in GCs of TNBC patients, validating our pre-clinical animal models and supporting future work to investigate how GC hypoxia and GC reactions influence the tumor immune microenvironment and therapeutic outcome in the clinic.
Methods
Animal models of mammary carcinoma and detection of hypoxia.
Female BALB/c and C57BL/6 mice were purchased from Simonsen Laboratories or Jackson Laboratory, and housed under specific pathogen-free conditions in the Animal Resource Center at the BC Cancer Research Center. For the experiments described, mice were between 7 and 10 weeks old unless stated otherwise. 4T1 and 4T07 mammary carcinoma cells, derived from the same spontaneous mammary tumor in a BALB/cfC3H mouse, were a gift from Dr. Fred Miller (Karmanos Cancer Institutes, Detroit, MI). E0771 mammary carcinoma cells syngeneic to the C57BL/6 background were purchased from CH3 Biosystems (Amerherst, NY). Cell lines were maintained in culture with Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS). To establish tumors, the following number of cells resuspended in 50 μL of PBS were implanted orthotopically into the 4th inguinal mammary fat pad (MFP) and allowed to grow for 3 weeks: 4T1 (1×105), 4T07 (1×106), E0771 (5×105). At endpoint, mice were injected intraperitoneally with 100 mg/kg of pimonidazole hydrochloride diluted in PBS, an exogenous hypoxia marker purchased as Hypoxyprobe™-1 (Hypoxyprobe, Inc. Burlington, MA). After at least 90 mins following Hypoxyprobe injections, mice were euthanized and inguinal LNs (which directly drain the MFP), axillary LNs (which receive efferent lymph from inguinal LNs), spleens, and primary tumors were collected and processed as described below.
Immunofluorescence microscopy analysis of LNs.
Inguinal and axillary LNs were collected from mice and submerged in 2% paraformaldehyde/PBS for overnight fixation at 4°C. LNs were subsequently placed in a 30% sucrose/PBS solution overnight, and then embedded in Optimal Cutting Temperature (OCT) compound for freezing at −20°C or −80°C. 10 μm thick cryostat sections were cut from the convex side of each LN and mounted onto Superfrost™ Plus microscope slides (Fisher Scientific). For immunofluorescent staining, sections were fixed in 2% paraformaldehyde/PBS for 15 mins and blocked in a 4% FBS/0.1% Triton X-100/PBS solution for 30 mins. Sections were subsequently incubated in primary antibody (B220 or GL7 (2.5 μg/mL, BioLegend Cat #144601)) for 90 mins at room temperature (RT). Alexa 594 goat anti-rat secondary antibody (20 μg/mL, Invitrogen Cat #A-11007) was applied for 30 mins at RT, followed by incubation with FITC-conjugated anti-pimonidazole mouse antibody (0.5 μg/mL, Hypoxyprobe Cat #HP FITC MAb1) for 90 mins at RT. Sections were stained with nuclear counterstain 4,6-diamidino-2-phenylindole (DAPI) for 5 mins prior to coverslip mounting with VECTASHIELD® Mounting Medium (Vector Laboratories). Stained sections were imaged using a Leica DM2500 microscope.
Flow cytometry analysis of LNs and spleens.
LNs and spleens were mechanically dissociated using the end of a syringe plunger in PBS and resulting cell suspensions were filtered through a 100 μm filter. After counting with a Vi-CELL XR Cell counter (Beckman Coulter), cells were resuspended to 1.0 × 107 cells/mL in PBS and stained for 30 mins with a fixable viability dye (eFluor® 780, eBioscience). Cells were resuspended in Hank’s balanced salt solution with 10 mM HEPES (STEMCELL Technologies), supplemented with 2% FBS and 0.05% NaN3 (HFN). After applying an anti-CD16/32 antibody (eBioscience) to block nonspecific Fc binding, 5 × 105 cells were incubated with primary conjugated antibodies for 15 mins against several surface markers, including CD138 (BV605), CD19 (PE-Cy7), GL7 (PerCP-Cy5.5 or PB), CD38 (APC), CD95 (BV605), IgG1 (BV421) and IgD (BV711). All antibodies against immune cell surface markers were purchased from BioLegend (San Diego, CA). Cells were fixed and permeabilized using the Foxp3/Transcription Factor Buffer Staining Set (eBioscience) and incubated overnight at 4°C in permeabilization buffer and 2% FBS. To detect bound Hypoxyprobe, cells were then stained with FITC-conjugated anti- pimonidazole mouse monoclonal antibody (FITC-MAb1, Hypoxyprobe, Inc. Burlington, MA) at 1 μg/mL in HFN for 90 mins at 4°C. To detect intercellular Bcl6 or HIF, permeabilized cells were also incubated with an antibody against Bcl6 (APC) (358506, BioLegend), HIF1ɑ (APC) (IC1935A, R&D Systems) or HIF2ɑ (AF700) (NB100-122AF700, Novus Biologicals) for 90 mins at 4°C. All samples were analyzed using a BD LSRFortessaTM Cell Analyzer (BD Biosciences). Live, single-cell lymphocytes from each LN or spleen were then analyzed for the cell populations of interest.
Flow cytometry analysis of tumors.
After obtaining tumor weights, tumors were finely minced with a scalpel blade and then agitated in a 37°C incubator for 40 mins with 0.8 mg/mL Collagenase type II (Gibco™, 17101–015) diluted in serum-free RPMI 1640 medium. After addition of 0.5 mg/mL of DNase I (Sigma-Aldrich, DN25) to the tumor digests, the resulting cell suspensions were filtered through a 100 μm filter with RPMI 1640 medium supplemented with 10% FBS, and cells were subsequently stained with fixable viability dye as described above. After resuspension in HFN and applying an anti-CD16/32 antibody, 1.0 × 106 cells were incubated with conjugated antibodies against several surface markers, including CD44 (FITC, BioLegend), CD8a (PE/Dazzle™ 594, BioLegend), CD45 (AF700, BioLegend), CD4 (BV605, BioLegend), PD-1 (eFluor 450, Invitrogen), CD206 (APC, BioLegend), F4/80 (PE, BioLegend), CD11b (PE-Cy7, Invitrogen), and MHC-II (BV510, BioLegend), for 15 mins at 4°C. After fixing and permeabilizing cells as described above, cells were stained for intracellular IFNγ (APC, BioLegend) for 90 mins at 4°C, and analyzed using a BD LSRFortessaTM Cell Analyzer (BD Biosciences). Live, single-cell CD45+ events from each tumor were then analyzed for the population of interest.
Injection of mice with lethally irradiated tumor cells.
Tumor cells were collected from tissue culture dishes by trypsinization, followed by neutralization of trypsin with serum. Cell pellets were washed twice with PBS. Cells were resuspended at a concentration of 4.29 × 107 cells/mL in PBS, and X-irradiated (X-RAD320; Precision X-ray Inc., North Branford, CT) in a 5 mL polystyrene tube with a single 20 Gy dose. After irradiation, cell suspensions were placed into a 96 well, non-tissue culture treated plate (150 μL cell suspension/well) and incubated at 37°C for 2 days. After this incubation period, 70 μL of the cell suspension (~3.0×106 cells) were injected into the MFPs of female BALB/c or C57BL/6 mice. The first injection was considered day 0, and the same procedure was followed for injections on days 7, and 11. 14 days after the first immunization, mice were injected intraperitoneally with 100 mg/kg of hypoxyprobe and euthanized at least 90 mins later. Inguinal and axillary LNs were collected and processed for flow cytometry or microscopy analysis as described above.
In vitro B cell stimulation.
Spleens were collected from naïve, female BALB/c mice (Jackson Laboratories), and mechanically dissociated using the end of a syringe plunger in PBS. Cell suspensions were filtered through a 100 μm filter and counted using a Vi-CELL XR Cell counter (Beckman Coulter). Splenocytes were resuspended at a concentration of 1.0 × 108 cells/mL in Ca2+Mg2+ free PBS (with 2% FBS and 1 mM EDTA), and B cells were isolated by negative selection using the EasySep™ mouse B cell isolation kit (Stemcell Technologies) according to manufacturer’s instructions. B cells to be analyzed by flow cytometry were resuspended in a 5 μM CFSE solution (CFSE Cell Division Tracker Kit, BioLegend) at a concentration of 1.0 × 107 cells/mL and incubated for 20 mins. 1 × 105 CFSE-labeled or unlabeled B cells were seeded into the wells of a 96-well tissue culture plate in Iscove’s Modified Dulbecco’s Medium with 25 mM HEPES (Stemcell Technologies) supplemented with 10% FBS, 1x Penicillin-Streptomycin (Gibco, 15140122), 1x GlutaMAX™ (Gibco, 35050061), and 55 μM of 2-Mercaptoethanol. To induce IgG1 class-switching, B cells were stimulated with LPS (1 μg/mL, Sigma-Aldrich), LEAF™ purified anti-mouse CD40 antibody (2.5 μg/mL, BioLegend), BAFF (10 ng/mL, BioLegend), and IL4 (10 ng/mL, STEMCELL Technologies). To induce IgA class-switching, cells were stimulated with LPS, anti-mouse CD40 antibody, BAFF, IL4, IL5 (10 ng/mL, STEMCELL Technologies), TGF-β1 (5 ng/mL, BioLegend), and All-Trans Retinoic Acid (10 nM, STEMCELL Technologies). The stimulated B cells were either incubated in normoxic (21% O2) tissue culture incubators or in a humidified hypoxia chamber set to 1% O2. After a 4-day incubation period, B cells were stained with Fixable Viability Dye (eFluor780, eBioScience) for live/dead cell exclusion, followed by staining with anti-CD19 (PE/Cyanine7, BioLegend), anti-IgG1 (BV421, BioLegend), anti-IgA (PE, eBioScience) after CD16/32 block (eBioscience). Stained cells were analyzed by flow cytometry as described above.
Development of VHL 2-lox;Cγ1-cre BALB/c mice.
VHL+/fl mice (C;129S-Vhltm1Jae/J, stock #004081, Jackson Laboratories) and Cγ1+/cre mice (C.129P2(Cg)-Ighg1tm1(cre)Cgn/J, stock #010612, Jackson Laboratories) were purchased from Jackson Laboratories and delivered to the Animal Resource Center at BC Cancer Research Center. Mice were housed under specific pathogen-free conditions, and underwent several rounds of breeding to generate VHL+/+;Cγ1+/cre, VHL+/fl;Cγ1+/cre or VHLfl/fl;Cγ1+/cre littermates. Female mice were used for experiments between 7 and 13 weeks old with either 4T07 or 4T1 orthotopic mammary tumors, and inguinal and axillary LNs, or spleens were harvested and analyzed by flow cytometry as described above.
Validation of VHL gene excision by PCR.
DNA was extracted from archived paraformaldehyde-fixed, frozen scrolls of tumor-draining inguinal LNs using the DNeasy blood and tissue kit (Cat #69506, Qiagen), which included an overnight digest with proteinase K at 56°C, and an RNase A incubation. DNA was quantified using a NanoDropTM spectrophotometer, and 35 ng of DNA was used in the PCR reaction. A three-primer PCR was conducted to detect wild-type VHL (predicted band at 200 bp), floxed VHL (predicted band at 400 bp), and the recombined VHL allele (predicted band at 250 bp) resulting from Cre-mediated recombination, as previously described.29 The following primers were used in the PCR reaction at a concentration of 0.5 μM: forward primer for floxed allele 5ʹ- CCGGAGTAGGATAAGTCAGCTGAG-3′, forward primer for recombined allele 5′- CTGGTACCCACGAAAGTGTC-3′, and the common reverse primer 5′- CTGACTTCCACTGATGCTTGTCACAG-3′. The PCR thermocycling conditions were one cycle of 10 mins at 94°C, 55 cycles of 50 seconds at 95°C, 50 seconds at 58°C, and 60 seconds at 72°C, followed by one cycle of 5 mins at 72°C, and DreamTaq Green PCR Master Mix (Cat #K1081, Thermo Scientific) was included in the PCR reaction. Amplified PCR products were run on a 1.5% agarose gel containing SYBRTM Safe DNA Gel Stain (Cat #S33102, Invitrogen), and bands were visualized using a Molecular Imager® Gel DocTM XR System (Bio-Rad). All VHL 2-lox;Cγ1-cre mice were screened for germline recombination events of the VHL 2-lox allele by three-primer PCR, which included an analysis of DNA extracted from stored tissues and fluids (including frozen LNs, formalin-fixed lungs, and serum).
Quantification of antibody isotypes by ELISA.
Blood was collected via cardiac puncture immediately following euthanasia of tumor-bearing mice. Blood was allowed to coagulate at 4°C overnight, and serum was collected after a 15 min centrifugation at 14000rpm. An ELISA kit for mouse immunoglobulin isotypes (HRP Mouse Clonotyping System Cat #5300-05, SouthernBiotech) was used according to manufacturer’s instructions to evaluate levels of whole Ig, IgM, IgG3, IgG1, IgG2a, IgG2b, and IgA in the serum of tumor-bearing mice. For relative quantitation, a mouse immunoglobulin panel (Mouse Immunoglobulin Panel, Cat #5300-01, SouthernBiotech) was used in the ELISA to create standard curves for each antibody isotype.
Quantification of tumor-specific antibodies with an in-cell ELISA.
96 well tissue-culture plates were seeded with 2.5 × 104 4T1 or 4T07 tumor cells maintained in culture and allowed to adhere to the plate overnight in a 37°C tissue culture incubator. Cells were fixed with 4% paraformaldehyde and stored at 4°C until use. Endogenous cellular peroxidase activity was quenched by incubating fixed cells in a PBS solution with 30% H2O2/5% NaN3/0.05% Tween-20 for 20 mins at RT. Cells were subsequently blocked with 1% BSA/PBS for 2 h at RT. Plates seeded with 4T1 cells were incubated with diluted serum from 4T1-bearing VHL+/+;Cγ1+/cre or VHLfl/fl;Cγ1+/cre mice for 2 h at RT, and after washing, bound antibody was labeled with an HRP-conjugated anti-mouse Ig (HRP Mouse Clonotyping System Cat #5300-05, SouthernBiotech) for 1 h at RT. Absorbance at 405 nm was read following application of the HRP-substrate, ABTS, prepared according to manufacturer’s instructions (HRP Mouse Clonotyping System Cat #5300-05). Prior to the in-cell ELISA with 4T07 cells, serum from 4T07-bearing VHL+/+;Cγ1+/cre or VHLfl/fl;Cγ1+/cre mice underwent IgG purification using NAbTM Protein G Spin Columns, (Cat #89949, ThermoFisher Scientific). Isolated protein was quantified using a DCTM Protein Assay kit (Cat #5000111, Bio-Rad), and standardization of IgG amounts across VHL+/+;Cγ1+/cre and VHLfl/fl;Cγ1+/cre samples was confirmed using an ELISA for mouse immunoglobulin. Standardized IgG was then added to 4T07-coated ELISA plates, and incubated for 2 h at RT, followed by detection with HRP-conjugated anti-mouse Ig as described above.
Quantification of the necrotic and proliferative fraction of tumors.
To evaluate proliferation, 4T1 tumors were harvested and processed for flow cytometry as described above. 1.0 × 106 cells from each tumor sample were incubated with surface antibodies against EpCAM (BV421, BioLegend) and CD45 (AF700, BioLegend) for 15 mins at 4°C. Following fixation and permeabilization, cells were incubated with an antibody against Ki67 (AF488, BioLegend) for 90 mins at 4°C, and analyzed using a BD LSRFortessaTM Cell Analyzer (BD Biosciences). To quantify necrosis, 4T1 tumors were fixed in a 2% paraformaldehyde/PBS solution overnight at 4°C, followed by overnight submersion in a 30% sucrose/PBS solution. Tumors were then embedded in OCT compound for freezing at −80°C. 10 µm-thick cryostat sections were taken 50 µm apart from the core of each tumor, and three step-sections per tumor were stained with DAPI as described above. Stained whole-tumor sections were imaged with a fluorescence microscope (Zeiss Axiomager Z1), and necrotic areas were outlined by hand and analyzed using ImageJ software. The necrotic fraction of each tumor slice was calculated as the necrotic area divided by the total area of the whole tumor section, and the values obtained for the three slices were averaged to yield the necrotic fraction of each tumor sample. 4T07 tumors were fixed in 10% neutral-buffered formalin for proliferation and necrosis assessment. Three step-sections (50 µm apart) were cut from each FFPE-tumor block and stained with an antibody against Ki67 (Spring Bioscience, #M3062; 1/300). MACH 2 rabbit HRP polymer followed by DAB chromogen were used to visualize Ki67 staining, followed by nuclear counterstain hematoxylin. Stained whole-tumor sections were imaged using a digital slide scanner (MOTIC). Ki67 staining and total nuclei were analyzed from a region of interest in each scan using the “color deconvolution” and “analyze particle” function in FIJI/ImageJ. The frequency of Ki67+ nuclei was calculated from each region of interest and averaged across the three step-sections from each tumor sample, and the necrotic fraction of the 4T07 tumors was calculated form the same scans as described above.
Acquisition and immunohistochemical staining of TDLNs from breast cancer patients.
Ethics approval for this study was granted by the British Columbia Cancer Research Ethics Board (UBC BCCA REB, study #H17-01236). Formalin-fixed paraffin embedded sections of non-involved LNs from six triple-negative breast cancer patients were obtained from the Tumor Tissue Repository at the BC Cancer – Deeley Research Center, who obtained patient consent for sample acquisition and anonymized samples. Clinicopathological features of the breast cancer cases used in this study are shown in Supplemental Table 1. All staining was performed at the Molecular and Cellular Immunology Core at the Deeley Research Center (BCCA) in Victoria, Canada, using the Intellipath FLX automated staining platform. Serial sections of LNs were stained with H&E and with an antibody against carbonic anhydrase IX (CA9), using the anti-human CA9 mouse monoclonal antibody (clone M75) at 5 µg/mL (BioScience Slovakia). MACH 2 mouse HRP polymer (BioCare Medical), followed by DAB chromogen was used to detect CA9 binding. After staining, slides were scanned using the Pannoramic MIDI digital slide scanner, and viewed using Pannoramic Viewer software (3DHISTECH Ltd.).
Statistical analysis.
Data included in all graphs are mean ± SD unless otherwise indicated, and each data point represents a distinct biological replicate. Groups were compared with the statistical tests described in each figure legend. * P value ≤0.05; ** P value ≤0.01; *** P value ≤0.001 **** P value ≤0.0001.
Supplementary Material
Acknowledgments
This work was supported by an operating grant from the Canadian Institutes of Health Research (CIHR; PJT-159513). NSF, RAC, RS, and BJW were funded by CIHR Doctoral Research Awards. NSF, BJW, CML, RS, and LRD were scholars in the Strategic Training in Transdisciplinary Radiation Science for the 21st Century (STARS21) program. NSF was funded by the Cordula and Gunter Paetzold Fellowship. KLB was a Michael Smith Foundation for Health Research Biomedical Research Scholar.
Funding Statement
This work was supported by an operating grant from the Canadian Institutes of Health Research (CIHR; PJT-159513). NSF, RAC, RS, and BJW were funded by CIHR Doctoral Research Awards. NSF, BJW, CML, RS, and LRD were scholars in the Strategic Training in Transdisciplinary Radiation Science for the 21st Century (STARS21) program. NSF was funded by the Cordula and Gunter Paetzold Fellowship. KLB was a Michael Smith Foundation for Health Research Biomedical Research Scholar.
Author contributions
N.S.F and K.L.B conceptualized the study. N.S.F. devised the experimental methodology. N.S.F., R.A.C., C. L., R.S., B.J.W., S.E.F., K.N.T., and L.R.D. carried out the investigation. N.S.F. and K.L.B wrote the manuscript. K.L.B. supervised the study.
Competing interests statement
The authors declare no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM.. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol. 2020;20(5):294–14. doi: 10.1038/s41577-019-0257-x. [DOI] [PubMed] [Google Scholar]
- 2.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–565. doi: 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 3.Petitprez F, De Reyniès A, Keung EZ, Chen TWW, Sun C-M, Calderaro J, Jeng Y-M, Hsiao L-P, Lacroix L, Bougoüin A, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 4.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho SH, Raybuck AL, Stengel K, Wei M, Beck TC, Volanakis E, Thomas JW, Hiebert S, Haase VH, Boothby MR, et al. Germinal centre hypoxia and regulation of antibody qualities by a hypoxia response system. Nature. 2016;537(7619):234–238. doi: 10.1038/nature19334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbott RK. germinal center hypoxia potentiates immunoglobulin class switch recombination. J.Immunol. 2016:1601401. doi: 10.4049/jimmunol.1601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raleigh JA, Calkins-Adams D, Rinker LH, Ballenger CA, Weissler MC, Fowler WC, Novotny DB, Varia MA. Hypoxia and Vascular Endothelial Growth Factor Expression in Human Squamous Cell Carcinomas Using Pimonidazole as a Hypoxia Marker. Cancer Res. 1998;58(17):3765.. [PubMed] [Google Scholar]
- 8.Broggi MAS, Maillat L, Clement CC, Bordry N, Corthésy P, Auger A, Matter M, Hamelin R, Potin L, Demurtas D. Tumor-associated factors are enriched in lymphatic exudate compared to plasma in metastatic melanoma patients. J.Exp.Med. 2019;216(5):1091–1107. doi: 10.1084/jem.20181618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res. 2000;2(5):331–334. doi: 10.1186/bcr77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parviz M, Chin CS, Graham LJ, Miller C, Lee C, George K, Bear HD. Successful adoptive immunotherapy with vaccine-sensitized T cells, despite no effect with vaccination alone in a weakly immunogenic tumor model. Cancer Immunology, Immunotherapy. 2003;52(12):739–750. doi: 10.1007/s00262-003-0405-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sellers RS, Clifford CB, Treuting PM, Brayton C. Immunological variation between inbred laboratory mouse strains: points to consider in phenotyping genetically immunomodified mice. Vet.Pathol. 2012;49(1):32–43. doi: 10.1177/0300985811429314. [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 13.Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH. Up-regulation of hypoxia-inducible factors HIF-1α and HIF-2α under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene. 2000;19(48):5435–5443. doi: 10.1038/sj.onc.1203938. [DOI] [PubMed] [Google Scholar]
- 14.Girodet P-O, Nguyen D, Mancini JD, Hundal M, Zhou X, Israel E, Cernadas M. Alternative macrophage activation is increased in asthma. Am J Respir Cell Mol Biol. 2016;55(4):467–475. doi: 10.1165/rcmb.2015-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olive PL, Aquino-Parsons C, MacPhail SH, Liao S-Y, Raleigh JA, Lerman MI, Stanbridge EJ. Carbonic Anhydrase 9 as an Endogenous Marker for Hypoxic Cells in Cervical Cancer. Cancer Res. 2001;61(24):8924.. [PubMed] [Google Scholar]
- 16.Heinzel FP, Sadick MD, Holaday BJ, Coffman RL, Locksley RM. Reciprocal expression of interferon gamma or interleukin 4 during the resolution or progression of murine leishmaniasis. Evidence for expansion of distinct helper T cell subsets. Journal of Experimental Medicine 1989;169(1):59–72. doi: 10.1084/jem.169.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Futatsugi-Yumikura S, Matsushita K, Fukuoka A, Takahashi S, Yamamoto N, Yonehara S, Nakanishi K, Yoshimoto T. Pathogenic Th2-type follicular helper T cells contribute to the development of lupus in Fas-deficient mice. Int.Immunol. 2013;26(4):221–231. doi: 10.1093/intimm/dxt070. [DOI] [PubMed] [Google Scholar]
- 18.Pellegrini A, Guinazu N, Aoki MP, Calero IC, Carrera-Silva EA, Girones N, Fresno M, Gea S. Spleen B cells from BALB/c are more prone to activation than spleen B cells from C57BL/6 mice during a secondary immune response to cruzipain. Int.Immunol. 2007;19(12):1395–1402. doi: 10.1093/intimm/dxm107. [DOI] [PubMed] [Google Scholar]
- 19.Weisel FJ, Zuccarino-Catania G, Chikina M, Shlomchik MJ, Temporal A. Switch in the germinal center determines differential output of memory B and plasma cells. Immunity. 2016;44(1):116–130. doi: 10.1016/j.immuni.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rahman A, Tiwari A, Narula J, Hickling T. Importance of feedback and feedforward loops to adaptive immune response modeling. CPT: Pharmacometrics & Systems Pharmacology. 2018;7:621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irani V, Guy AJ, Andrew D, Beeson JG, Ramsland PA, Richards JS. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol.Immunol. 2015;67(2):171–182. doi: 10.1016/j.molimm.2015.03.255. [DOI] [PubMed] [Google Scholar]
- 22.Hu X, Zhang J, Wang J, Fu J, Li T, Zheng X, Wang B, Gu S, Jiang P, Fan J, et al. Landscape of B cell immunity and related immune evasion in human cancers. Nat.Genet. 2019;51(3):560–567. doi: 10.1038/s41588-018-0339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coffman RL, Lebman DA, Shrader B. Transforming growth factor beta specifically enhances IgA production by lipopolysaccharide-stimulated murine B lymphocytes. J.Exp.Med. 1989;170(3):1039–1044. doi: 10.1084/jem.170.3.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Ray A, Jiang X, Wang J-Y, Basu S, Liu X, Qian T, He R, Dittel BN, Chu Y, et al. T regulatory cells and B cells cooperate to form a regulatory loop that maintains gut homeostasis and suppresses dextran sulfate sodium-induced colitis. Mucosal Immunol. 2015;8(6):1297–1312. doi: 10.1038/mi.2015.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc.Natl.Acad.Sci.U.S.A. 2009;106(46):19256–19261. doi: 10.1073/pnas.0812681106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP, Araki Y, Miyoshi I, Yang L, Trinchieri G, et al. Immunosuppressive and prometastatic functions of myeloid-derived suppressive cells rely upon education from tumor-Associated B cells. Cancer Res. 2015;75(17):3456. doi: 10.1158/0008-5472.CAN-14-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willsmore ZN, Harris RJ, Crescioli S, Hussein K, Kakkassery H, Thapa D, Cheung A, Chauhan J, Bax HJ, Chenoweth A, et al. B cells in patients with melanoma: implications for treatment with checkpoint inhibitor antibodies. Front Immunol. 2021;11:3560. doi: 10.3389/fimmu.2020.622442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, Nelson BH. CD20+Tumor-Infiltrating lymphocytes have an a typical CD27− Memory phenotype and together with CD8+T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. 2012;18(12):3281. doi: 10.1158/1078-0432.CCR-12-0234. [DOI] [PubMed] [Google Scholar]
- 29.Espana-Agusti J, Zou X, Wong K, Fu B, Yang F, Tuveson DA, Adams DJ, Matakidou A. Generation and characterisation of a Pax8-CreERT2 transgenic line and a Slc22a6-CreERT2 Knock-In Line for inducible and specific genetic manipulation of renal tubular epithelial cells. PLoS One. 2016;11(2):e0148055. doi: 10.1371/journal.pone.0148055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.