

Abstract
Two Italian patients with the initial clinical diagnosis of Rothmund-Thomson syndrome were negative for RECQL4 mutations but showed in peripheral blood cells a spontaneous chromosomal instability significantly higher than controls. Revisiting after time their clinical phenotype, the suggestive matching with the autosomal dominant syndrome Poikiloderma, Hereditary Fibrosing with Tendon Contracture, Myopathy and Pulmonary fibrosis (POIKTMP) was confirmed by identification of the c.1879A>G (p.Arg627Gly) alteration in FAM111B. We compare the overall clinical signs of our patients with those of reported carriers of the same mutation and present the up-to-date mutational repertoire of FAM111B and the related phenotypic spectrum. Our snapshot highlights the age-dependent clinical expressivity of POIKTMP and the need to follow-up patients to monitor the multi-tissue impairment caused by FAM111B alterations. We link our chromosomal instability data to the role of FAM111B in cancer predisposition, pointed out by its implication in DNA-repair pathways and the outcome of pancreatic cancer in 2 out of 17 adult POIKTMP patients. The chromosomal instability herein highlighted well connects POIKTMP to cancer-predisposing syndromes, such as Rothmund-Thomson which represents the first hereditary poikiloderma entering in differential diagnosis with POIKTMP.
Keywords: POIKTMP, FAM111B pathogenic variants, chromosomal instability, cancer predisposition, pancreatic carcinoma
Blood samples of two unrelated patients with a phenotype suggestive of a genodermatosis reminiscent of Rothmund-Thomson syndrome (RTS, MIM #268400) were sent to our laboratory in 2002 to perform RECQL4 (MIM *603780) molecular analysis, after obtainment of informed consents. Briefly, the two Italian patients, a male (patient A) and a female (patient B) both representing the only child born to non-consanguineous parents, presented typical RTS clinical signs, such as poikiloderma, sparse and rare hair, eyelashes and eyebrows, short stature, and photosensitivity, combined with peculiar features, mainly oedema, muscular weakness and liver dysfunction with vanishing bile duct syndrome since infancy, rarely or never reported in RTS. Their clinical conditions deteriorated with time and the liver involvement in patient A caused ascites and sepsis that led him to premature death at age 17 years. Patient B also had a liver impairment: she suffered from idiopathic inflammatory cholangiopathy with ductopenia, as attested by hepatic biopsy, but had a much less impaired liver function; unfortunately, she prematurely died following a trauma that occurred when she was 15 years old. Sanger sequencing of the entire RECQL4 gene (NG_016430.1), 21 exons and 20 introns, except for IVS12 minisatellite (Colombo et al., 2018), was carried out but did not detect any pathogenic alteration.
In parallel to RECQL4 gene scan, spontaneous and induced chromosomal instability, a cellular hallmark of RTS syndrome (Larizza et al., 2010) and other autosomal recessive syndromes driven by defects in RECQ helicase genes (such as Werner Syndrome, MIM #277700) or entering in differential diagnosis with RTS (such as Fanconi Anemia, MIM Phenotypic Series-PS227650) was monitored on heparinized peripheral blood lymphocytes from the two probands and six healthy controls, including both probands’ parents. A minimum of 50 metaphase spreads prepared according to standard procedures and QFQ-banded were analysed per sample. As shown in Table 1, a significantly higher number of chromatid and chromosome gaps and breaks per metaphase was highlighted in both probands (patient A: 0.25 (13/52), patient B: 0.22 (11/51)) as compared to controls (0.11 (34/318)) (p<.05 at the chi-square test). The chromosomal lesions, widespread in the genome, involved different chromosomal locations, rarely coincident with fragile sites, as apparent in the set of eight different Q-banded chromosomes (Figure 1). Conversely, the search for chromosomal fragility after aphidicolin treatment (0.4 M for 24 hr) did not evidence a rate of chromosome lesions significantly higher in the probands than in parallel control lymphocytes cultures as the ratio of aberrations/metaphases was 3.21 in patient A, 4.96 in patient B, and 4.52 in controls.
Table 1. Spontaneous chromosomal instability in peripheral blood cells from two unrelated patients with a Rothmund-Thomson-like phenotype.
| Patients | Controls | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Observed aberrations | Patient A | Patient B | Controls total | Parents of patient A | Parents of patient B | Control 1 | Control 2 | ||
| Mother | Father | Mother | Father | ||||||
| Chromatid Gaps | 7 | 4 | 19 | 1 | 2 | 5 | 3 | 5 | 3 |
| Chromatid Breaks | 1 | 0 | 5 | 1 | 0 | 3 | 0 | 0 | 1 |
| Chromosome Gaps | 1 | 4 | 6 | 1 | 0 | 2 | 0 | 2 | 1 |
| Chromosome Breaks | 4 | 3 | 4 | 1 | 1 | 1 | 1 | 0 | 0 |
| Total aberrations | 13 | 11 | 34 | 4 | 3 | 11 | 4 | 7 | 5 |
| No. scored metaphases | 52 | 51 | 318 | 50 | 54 | 53 | 56 | 54 | 51 |
| Aberrations/ metaphase | 0.25* | 0.22* | 0.11* | 0.08 | 0.06 | 0.21 | 0.07 | 0.13 | 0.10 |
* Significant at p < .05 at the chi-square test
Figure 1 -. Spontaneous breaks and gaps in two unrelated Italian patients with a clinical presentation reminiscent of Rothmund-Thomson syndrome. Selected representative images document the spontaneous breaks and gaps observed in the QFQ-banded chromosome spreads from blood samples of the two patients herein reported.
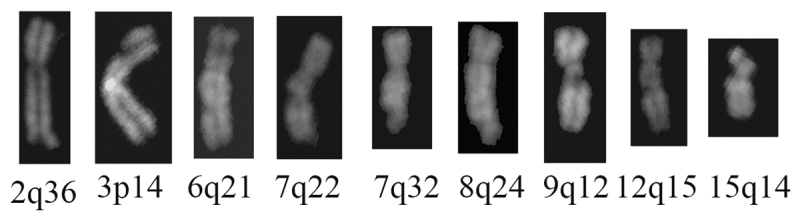
As the molecular identity of the syndromic condition of the two patients was unknown at that time this data could neither be associated with a specific rare syndrome nor corroborated by testing similarly affected patients.
Many years later, the phenotype of our probands was revisited in the context of the literature. We noted a reasonable match between the phenotype of our cases and that of patients with the ultra-rare autosomal dominant syndrome Poikiloderma, Hereditary Fibrosing, with Tendon contracture, Myopathy and Pulmonary fibrosis (POIKTMP, MIM #615704) (Khumalo et al., 2006; Mercier et al., 2013). Hence, targeted Sanger sequencing of the POIKTMP-causative gene FAM111B (MIM #615584), encoding a trypsin-like cysteine/serine peptidase (Mercier et al., 2013), highlighted on both our patients the monoallelic c.1879A>G alteration (NG_034129.1) (Figure 2A). This pathogenic variant is not present in the DNA of patient B’ mother, the only parent we could test. Moreover, we detected this alteration in the probands’ lymphoblastoid cell lines RNA (data not shown) confirming the stability of the mutated FAM111B transcript r.1879A>G (p.Arg627Gly).
Figure 2 -. The mutational repertoire of FAM111B. A) Electropherograms showing the c.1879A>G FAM111B missense variant (framed by the red rectangle) detected in the two unrelated Italian patients. B) Picture summarizing the FAM111B mutational repertoire. The dark blue bar indicates the coding regions of the gene while the non-coding regions are in light blue. The pink bar indicates the protein: the punctate boxes correspond to the protease domain, spanning from amino acid residues 471 to 664 and interrupted by a linker region (dotted bar). Each change is described at DNA (top) and protein level (below). Numbers within the circles specify the number of patients carrying the variant. Pathogenic variants associated with POIKTMP, POH, and colon cancer are written in black, blue and red, respectively.

The identified missense pathogenic variant affects a highly conserved amino acid residue placed in the loop that splits into two parts the functional enzymatic domain of FAM111B (Mercier et al., 2013) (Figure 2B). Of note, six additional FAM111B mutations map in this linker region (41 amino acids in length) that can be considered the major mutation cluster; a minor cluster of three pathogenic variants maps upstream the catalytic domain (Figure 2B).
Including our cases, the c.1879A>G (p.Arg627Gly) mutation has been reported so far in five patients from four families (Figure 2B and Table 2): our two from Italy, namely patient A and patient B (alias F3-II1 reported by Mercier et al., 2013, 2015), a third one from France (patient C, F1-II2, Mercier et al., 2013, 2015), and two from Algeria (patient D, F2-II4, Mercier et al., 2013, 2015, 2019, and his son patient E, F2-III1, Mercier et al., 2015). Table 2 summarizes the clinical signs of the reported POIKTMP cases sharing the hot spot c.1879A>G (p.Arg627Gly) pathogenic variant. A detailed clinical report of patient B marking the evolution of the disease can be found in Supplementary File 1.
Table 2 -. Summary of the clinical signs observed in patients with the c.1879A>G (p.(Arg627Gly)) FAM111B mutation.
| Patient A | Patient B/F3-II1a,b | Patient C/F1-II2a,b | Patient D/F2-II4a,b,c | Patient E/F2 sonb | |
|---|---|---|---|---|---|
| Origin | Italy | Italy | France | Algeria | Algeria |
| Age at clinical evaluation | 2y | 4y | 7y | 30y | 8m |
| Sex | Male | Female | Male | Male | Male |
| Short stature | + (3° percentile) | + (50° percentile) | - | + | - |
| Skin | |||||
| Poikiloderma | + | + | + | + | + |
| Cellulitis | - | + | + | - | - |
| Dermatitis | + | + | - | - | - |
| Eczematous lesions | - | - | + | + | + |
| Sclerosis of digits | - | - | - | + | - |
| Vitiligo | - | + | - | - | - |
| Sparse scalp hair | + | + | + | + | + |
| Sparse eyelashes | + | + | + | + | + |
| Sparse eyebrows | + | + | + | + | + |
| Heat intolerance | + | + | + | + | n.d. |
| Eyes Cataract | - | + | - | - | - |
| Pancreas | |||||
| Insufficiency | - | - | + | - | n.d. |
| Cancer | - | - | - | + | n.d. |
| Liver | |||||
| Failure | + | - | n.d. | - | - |
| Vanishing bile ducts | + | + | n.d. | - | - |
| Cirrhosis | + | - | n.d. | - | - |
| Cholestasis | + | + | n.d. | - | - |
| Hepatomegaly | - | + | n.d. | - | |
| Lungs | |||||
| Asthma | - | - | + | - | n.d. |
| Restrictive syndrome | - | - | - | + | n.d. |
| Muscle | |||||
| Weakness | + | + | + | + | - |
| Atrophy | - | - | + | + | - |
| Adiposis | - | + | + | + | - |
| Contractures | + | + | + | + | - |
| Lymphedema | + | + | + | - | - |
| Haematological alterations | ↓ IgG1, IgG2, IgG4, ↑ IgE ↑ GoT ↑ GPT ↑ gammaGT | ↓ IgG2, IgG4, ↑ IgG1 ↑ GoT ↑ GPT ↑ gammaGT ↑ LDH ↑ CPK | Eosinophilia | ↑ CPK | n.d. |
| Others | Death at 17y for liver cirrhosis | Scoliosis Nail dysplasia Death at 15y for injury | n.d. | Scoliosis Dysphagia Death at 32y for cancer | n.d. |
n.d.: not documented; a: Mercier et al., 2013; b: Mercier et al., 2015; c: Mercier et al., 2019
Briefly, all patients showed in the first year of life poikiloderma, predominant in the sun-exposed skin areas, and hypotrichosis involving hair, eyelashes, and eyebrows. Then, except patient E who was in the first childhood when the disease is not yet fully expressed, all manifested muscle weakness and limb contractures starting from childhood and/or adolescence. Patient C (F1-II2) suffered from asthma since infancy, while the adult patient D (F2-II4) developed a severe and abnormal lung function (Mercier et al., 2013, 2015). Both the Italian patients showed vanishing bile duct syndrome since infancy and liver impairment. Transaminitis and bile duct loss, not associated with cholestasis, were also detected in a patient with the c.1881C>T mutation affecting the same amino acid residue [p.(Arg627Ser)] (Dokic et al., 2020). Three patients prematurely died: patient A died as above mentioned for decompensated liver cirrhosis at age 17y; patient B had an accidental death at 15y, and patient D, the eldest carrier of c.1879A>G mutation, died at 32y due to intraductal pancreatic adenocarcinoma (Mercier et al., 2019).
The pancreatic dysfunction is considered a relatively frequent clinical findings of POIKTMP: it has been diagnosed in five patients (8, 10, 28, and 58-years old) from a multiplex family co-segregating with the in-frame p.(Lys421del) alteration (Seo et al., 2016). Moreover, a 64-years-old patient with p.(Ser628Arg) mutation died for pancreatic carcinoma pointing to two cases with this rare cancer in the cohort of 17 adult POIKTMP patients who could be followed-up (Goussot et al., 2017; Mercier et al., 2019). Another POIKTMP clinical sign responsible for severe evolution of the disease is pulmonary fibrosis causing respiratory failure in three patients carrying mutations within both the major (p.(Arg627Gly); p.(Thr625Asn)) (Mercier et al., 2013, 2015) or the minor (p.(Gln430Pro)) (Mercier et al., 2015; Sanchis-Borja et al., 2018) mutation clusters (Figure 2B). The position of the mutation per se may not have a great influence on the phenotype as exemplified by the patient carrying the p.(Gln430Pro) mutation deceased for respiratory failure (Sanchis-Borja et al., 2018) and the lack of restrictive pulmonary disease in a 14-years old girl carrying the same variant (Takeichi et al., 2017). Age-dependent clinical expressivity concurs to the phenotype as the multi-tissue/organs impairment caused by FAM111B mutation becomes increasingly manifest with age.
Comprehension of the genotype-phenotype relationship is made further trickier by the identification of the first frameshift FAM111B c.1462delT (p.(Cys488Valfs*21)) variant (Figure 2B), subverting the entire cysteine/serine peptidase domain, in a boy with Progressive Osseous Heteroplasia (POH), who also displayed classic POIKTMP signs such as tendon contractures, muscle weakness and ulcerated erupted skin lesions (Panjawatanan et al., 2019).
Furthermore, FAM111B germline variants have been also detected in two patients with colorectal cancer in the context of a genomic study searching for predisposing variants in genes involved in homologous recombination repair associated with hereditary cancer (Bertelsen et al., 2019). Interestingly the two loss-of-function germline alterations, c.816dup (p.(Ala273Serfs*9)) and c.952G>T (p.Glu318Ter) (Figure 2B), predicting early protein truncation/mRNA degradation, are not associated with a POIKTMP phenotype, raising the possibility that POIKTMP, POH, and non-syndromic cancer predisposition are allelic disorders driven by different types of FAM111B mutation.
The assumption that FAM111B pathogenic variants associated with POIKTMP act by gain-of-function or dominant-negative effect (Mercier et al., 2013) has been recently supported by the discovery that FAM111B mutations exacerbate the catalytic activity repressing DNA replication and transcription and triggering cell death (Hoffmann et al., 2020).
Several data extrapolated from the literature support this hypothesis. An indirect role of FAM111B in the NER and NHEJ repair pathways is inferred by its synthetic lethality with MLN4924, a small molecule of the NEDD8-activating enzyme inhibitor used in clinical trials for solid tumours and haematological malignancies (Blank et al., 2013). Moreover, FAM111B interacts with CAPNS1 (https://www.genecards.org/cgi-bin/carddisp.pl?gene=CAPNS1), a regulator subunit of m-calpain and µ-calpain, the two ubiquitously best-characterized proteases belonging to the calpain family. Interestingly Mercier and collaborators showed a decreased quantity of calpain on immunoblotted protein lysates from the muscle of patient D (F2-II4) (Mercier et al., 2013). A link with DNA-repair pathways may be grasped as the CAPNS1-CAPN1 complex is involved in the stability of the USP1 deubiquitinase that modifies both FANCD2, a central node in different DNA damage response pathways (Lopez-Martinez et al., 2016), and PCNA involved in DNA metabolism (Choe and Moldovan, 2017).
Furthermore, transcriptomic findings of pancreatic cells treated for 48 h with metformin and aspirin had included FAM111B among the 10 top downregulated genes mainly involved in G1/S checkpoint regulation (Yue et al., 2015). Merging observations on a high FAM111B expression during the S phase suggest the protein may play a role in DNA replication (Aviner et al., 2015). Last, FAM111B has been implicated in susceptibility to prostate cancer by a genome-wide association study (Akamatsu et al., 2012) and it has been indicated as an unfavourable prognostic marker in pancreatic and liver cancer (https://www.proteinatlas.org/ENSG00000189057-FAM111B).
Our data evidenced an increased frequency of spontaneous chromosome gaps and breaks in peripheral blood lymphocytes from our two FAM111B-mutated patients as compared to healthy controls. Values of normal controls also appear higher than those reported in other case studies of the literature but they may be explained by physiological issues (including lifestyle) or linked to an overestimate of aberrations due to the evaluation of QFQ-banded chromosomes, instead of Giemsa stained chromosomes, usually preferred to detect gaps and breaks.
By linking our data to the above evidence, we speculate that detection of increased chromosome gaps and breaks in peripheral blood lymphocytes from 2 FAM111B-mutated patients could reflect (secondary manifestation) an impairment of one of the DNA replication/DNA-damage repair pathways (primary defects).
Though mechanistic insights on FAM111B role remain to be elucidated and integrated, they are consistent with the increased spontaneous instability noted in FAM111B mutated cells. It could be envisaged that stem/progenitor cells of pancreatic ductal epithelia are especially sensitive to the effect of FAM111B mutations. Further POIKTMP patients and cell types should be analysed to confirm and expand our data. As the instability assay was performed on patients in early childhood, should it be significant in additional POIKTMP patients, it may be used as an adjunct test for diagnosis and oncological surveillance.
The herein highlighted chromosomal instability represents a hallmark that definitely links POIKTMP to cancer-predisposing syndromes driven by disruption of the mechanisms safeguarding genomic stability (Taylor et al., 2019), particularly Rothmund-Thomson type II syndrome where patients cells show centromere mis-division (Larizza et al., 2006) and fragile telomeric ends (Ghosh et al., 2012), and Dyskeratosis Congenita (DK) characterized by short telomeres (Savage, 2018). It is thus not surprising that POIKTMP, RTS-II, and DK display a few overlapping features, such as growth delay and sparse hair, pigmentation changes, and cancer predisposition. POIKTMP and RTS-II patients also share poikiloderma, sparse eyelashes/eyebrows, and the less common sign of sclerosis of the digits, while both POIKTMP and DK patients are prone to pulmonary fibrosis and liver disease. The diagram in Figure S1 illustrates these clinical commonalities, together with the more numerous specific features of POIKTMP, RTS-II, and DK, which should be carefully considered for their differential diagnosis.
Acknowledgments
We thank the patients and their families for their cooperation in the study. We thank Dr. M. Macchiaiolo (Bambino Gesù Hospital, Rome) for personal communication on Patient A. This work was partially supported by Italian Ministry of Health to Istituto Auxologico Italiano (Ricerca Corrente WES-RTS-PN 08C624).
Supplementary material.
The following online material is available for this article:
References
- Akamatsu S, Takata R, Haiman CA, Takahashi A, Inoue T, Kubo M, Furihata M, Kamatani N, Inazawa J, Chen GK, et al. Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nat Genet. 2012;44:426–429. doi: 10.1038/ng.1104. [DOI] [PubMed] [Google Scholar]
- Aviner R, Shenoy A, Elroy-Stein O, Geiger T. Uncovering hidden layers of cell cycle regulation through integrative Multi-omic analysis. PLoS Genet. 2015;11:e1005554. doi: 10.1371/journal.pgen.1005554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertelsen B, Tuxen IV, Yde CW, Gabrielaite M, Torp MH, Kinalis S, Oestrup O, Rohrberg K, Spangaard I, Santoni-Rugiu E, et al. High frequency of pathogenic germline variants within homologous recombination repair in patients with advanced cancer. NPJ Genom Med. 2019;4:13. doi: 10.1038/s41525-019-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, Tayber O, Hather G, Liu R, Narayanan U, et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013;73:225–234. doi: 10.1158/0008-5472.CAN-12-1729. [DOI] [PubMed] [Google Scholar]
- Choe KN, Moldovan GL. Forging ahead through darkness: PCNA, still the principal conductor at the replication fork. Mol Cell. 2017;65:380–392. doi: 10.1016/j.molcel.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo EA, Locatelli A, Cubells Sánchez L, Romeo S, Elcioglu NH, Maystadt I, Esteve Martínez A, Sironi A, Fontana L, Finelli P, et al. Rothmund-Thomson Syndrome: Insights from New Patients on the Genetic Variability Underpinning Clinical Presentation and Cancer Outcome. Int J Mol Sci. 2018;19:1103. doi: 10.3390/ijms19041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokic Y, Albahrani Y, Phung T, Patel K, de Guzman M, Hertel P, Hunt R. Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis: Hepatic disease in a child with a novel pathogenic variant of FAM111B. JAAD Case Rep. 2020;6:1217–1220. doi: 10.1016/j.jdcr.2020.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Rossi ML, Singh DK, Dunn C, Ramamoorthy M, Croteau DL, Liu Y, Bohr VA. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J Biol Chem. 2012;287:196–209. doi: 10.1074/jbc.M111.295063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussot R, Prasad M, Stoetzel C, Lenormand C, Dollfus H, Lipsker D. Expanding phenotype of hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis caused by FAM111B mutations: Report of an additional family raising the question of cancer predisposition and a short review of early-onset poikiloderma. JAAD Case Rep. 2017;3:143–150. doi: 10.1016/j.jdcr.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Pentakota S, Mund A, Haahr P, Coscia F, Gallo M, Mann M, Taylor NM, Mailand N. FAM111 protease activity undermines cellular fitness and is amplified by gain-of-function mutations in human disease. EMBO Rep. 2020:e50662. doi: 10.15252/embr.202050662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khumalo NP, Pillay K, Beighton P, Wainwright H, Walker B, Saxe N, Mayosi BM, Bateman ED. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br J Dermatol. 2006;155:1057–1061. doi: 10.1111/j.1365-2133.2006.07473.x. [DOI] [PubMed] [Google Scholar]
- Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006;232:107–120. doi: 10.1016/j.canlet.2005.07.042. [DOI] [PubMed] [Google Scholar]
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010 doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Martinez D, Liang CC, Cohn MA. Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci. 2016;73:3097–3114. doi: 10.1007/s00018-016-2218-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier S, Küry S, Shaboodien G, Houniet DT, Khumalo NP, Bou-Hanna C, Bodak N, Cormier-Daire V, David A, Faivre L, et al. Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet. 2013;93:1100–1107. doi: 10.1016/j.ajhg.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier S, Küry S, Salort-Campana E, Magot A, Agbim U, Besnard T, Bodak N, Bou-Hanna C, Bréhéret F, Brunelle P, et al. Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet J Rare Dis. 2015;10:135. doi: 10.1186/s13023-015-0352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier S, Küry S, Nahon S, Salort-Campana E, Barbarot S, Bézieau S. FAM111B mutation is associated with pancreatic cancer predisposition. Pancreas. 2019;48:e41-e42. doi: 10.1097/MPA.0000000000001303. [DOI] [PubMed] [Google Scholar]
- Panjawatanan P, Ryabets-Lienhard A, Bakhach M, Pitukcheewanont P. MON-512 a de novo frameshift mutation of FAM111B gene resulting in progressiveosseous heteroplsia in an African American boy: First case report. J Endocr Soc. 2019;3:MON-512 [Google Scholar]
- Sanchis-Borja M, Pastré J, Mercier S, Juvin K, Benattia A, Israël-Biet D. Pulmonary fibrosis associated with hereditary fibrosing poikiloderma caused by FAM111B mutation: A case report. Rev Mal Respir. 2018;35:968–973. doi: 10.1016/j.rmr.2018.09.002. [DOI] [PubMed] [Google Scholar]
- Savage SA. Beginning at the ends: telomeres and human disease. F1000Res. 2018;7:524. doi: 10.12688/f1000research.14068.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo A, Walsh T, Lee MK, Ho PA, Hsu EK, Sidbury R, King MC, Shimamura A. FAM111B mutation is associated with inherited exocrine pancreas dysfunction. Pancreas. 2016;45:858–862. doi: 10.1097/MPA.0000000000000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi T, Nanda A, Yang HS, Hsu CK, Lee JY, Al-Ajmi H, Akiyama M, Simpson MA, McGrath JA. Syndromic inherited poikiloderma due to a de novo mutation in FAM111B. Br J Dermatol. 2017;176:534–536. doi: 10.1111/bjd.14845. [DOI] [PubMed] [Google Scholar]
- Taylor AMR, Rothblum-Oviatt C, Ellis NA, Hickson ID, Meyer S, Crawford TO, Smogorzewska A, Pietrucha B, Weemaes C, Stewart GS. Chromosome instability syndromes. Nat Rev Dis Primers. 2019;5:64. doi: 10.1038/s41572-019-0113-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue W, Wang T, Zachariah E, Lin Y, Yang CS, Xu Q, DiPaola RS, Tan XL. Transcriptomic analysis of pancreatic cancer cells in response to metformin and aspirin: an implication of synergy. Sci Rep. 2015;5:13390. doi: 10.1038/srep13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Internet Resources
- Online Mendelian Inheritance in Man (OMIM) [4 September 2020]. Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/OMIM.
- The Human Protein Atlas [13 July 2020]. The Human Protein Atlas, https://www.proteinatlas.org.
- GeneCard [28 July 2020]. GeneCard, https://www.genecards.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.