

Abstract
Iron–sulfur (Fe-S) clusters (ISCs) are ubiquitous cofactors essential to numerous fundamental cellular processes. Assembly of ISCs and their insertion into apoproteins involves the function of complex cellular machineries that operate in parallel in the mitochondrial and cytosolic/nuclear compartments of mammalian cells. The spectrum of diseases caused by inherited defects in genes that encode the Fe-S assembly proteins has recently expanded to include multiple rare human diseases, which manifest distinctive combinations and severities of global and tissue-specific impairments. In this review, we provide an overview of our understanding of ISC biogenesis in mammalian cells, discuss recent work that has shed light on the molecular interactions that govern ISC assembly, and focus on human diseases caused by failures of the biogenesis pathway.
Biological Roles of Iron–Sulfur Proteins and Relevance to Human Diseases
ISCs are among the oldest cofactors known in biology and have been proposed to have had a critical role in the emergence of life on Earth more than 3 billion years ago, when they were incorporated into early metabolic pathways by primitive organisms [1]. Their versatile chemical properties have fostered their pervasive use in almost all organisms to execute an impressive number of reactions involved in fundamental cellular processes, such as respiration, photosynthesis, metabolism, and nitrogen fixation. They have also been shown to have roles in DNA replication and repair, tRNA modifications, and the cell cycle. Given the involvement of Fe-S enzymes in several fundamental cellular processes (see specific examples detailed later), it is not surprising that defects in the components of the ISC biogenesis pathway cause an increasingly recognized number of rare human diseases (Table 1).
Table 1.
Proteins Involved in ISC Biogenesis and Diseases Caused by Mutations that Inactivate their Function
| Gene symbol | S. cerevisiae ortholog | Chromosomal localization (exon count) | Function | Disorder | Refs |
|---|---|---|---|---|---|
| NFS1 | Nfs1 | 20q11.22 (14) | Cysteine desulfurase; provides sulfur for ISC biogenesis | Infantile respiratory complexes defects (p.Arg72Gln) | [106] |
| ISD11, also known as LYRM4 | Isd11 | 6p25.1 (13) | Stabilizes NFS1 | Combined oxidative phosphorylation deficiency 19 (COXPD19) p.Arg68Leu | [107,108] |
| COXPD19 (p.Tyr31Cys) | |||||
| NDUFAB1 | Acp | 16p12.2 (6) | Binds LYRM4 in ISC core complex | None reported | [31–34] |
| FXN | Yfh1 | 9q21.11 (5) | Transiently binds to NFS1/LYRM4/ACP/ISCU complex at interface between NFS1 and ISCU and enhances rate of persulfide transfer from NFS1 to ISCU | Friedreich ataxia (FRDA) (GAA expansion in intron 1 of FXN) | [109,110] |
| ISCU | Isu1; Isu2 | 12q23.3 (8) | Main scaffold for de novo ISC assembly | Hereditary myopathy with exercise intolerance, Swedish type, characterized by loss of aconitase and succinate dehydrogenase (IVS5 + 382 G>C; deep intronic splicing) | [96,98,111] |
| In two cases, compound heterozygosity for intronic variant and missense mutation ISCUG50E | |||||
| In one case, a single dominant mutation in ISCU (p.Gly96Val) was reported | |||||
| HSC20 | Jac1 | 22q12.1 (7) | Cochaperone involved in cluster transfer | Nonsyndromic microcytic to normocytic sideroblastic anemia [a promoter variant (c.−134C>A) predicted to disrupt a conserved transcription factor binding site, and a frameshift (c.259dup, p.T87fs*27)] | [112] |
| HSPA9 | Ssq1 | 5q31.2 (17) | Chaperone that works with HSC20 to facilitate cluster transfer | Nonsyndromic microcytic to normocytic sideroblastic anemia (several mutations reported) | [113] |
| NFU1 | Nfu1 | 2p13.3 (10) | Secondary carrier | Multiple Mitochondria Dysfunctions Syndrome 1 (MMDS1) (c.545GNA predicted to result in R182Q substitution. substitution is also adjacent to splice site leading to excision of exon 6; p.Gly208Cys) | [65,66,114] |
| MMDS1 and pulmonary hypertension (p.Gly208Cys) | |||||
| GLRX5 | Grx5 | 14q32.13 (2) | ISC carrier | Nonsyndromic sideroblastic anemia (homozygous c.294A>G mutation that interferes with intron 1 splicing and drastically reduces GLRX5 mRNA) | [94,97,115] |
| Nonsyndromic sideroblastic anemia due to missense mutations (p.K101Q; p.L148S) | |||||
| Spasticity, childhood-onset, with hyperglycinemia (p.K51del) | |||||
| FDX1; FDX1L also known as FDX2 | Yah1 | 11q22.3 (4); 19p13.2 (5) | Provides reducing equivalents for initial cluster assembly | Mitochondrial myopathy, episodic, with or without optic atrophy and leukoencephalopathy (c.1A>T predicted to disrupt ATG translation initiation site; p.P144L) | [116,117] |
| FDXR | Arh1 | 17q25.1 (14) | Ferredoxin reductase that serves as first electron transfer protein that donates reducing equivalents to FDX1/2 | Auditory neuropathy and optic atrophy (p.Arg306Cys) | [118] |
| Auditory neuropathy and optic atrophy (p.Arg306Cys/p.Gln419*) | |||||
| Auditory neuropathy and optic atrophy (p.Arg242W/Arg327Ser) | |||||
| Auditory neuropathy and optic atrophy (p. Leu215Val/p.Glu477Lys) | |||||
| ISCA1 | Isa1 | 9q21.33 (4) | Serves as secondary carrier | MMDS5 (p.Glu87Lys) | [75] |
| ISCA2 | Isa2 | 14q24.3 (4) | Serves as secondary carrier of the cluster | MMDS4 (p.Gly77Ser; c.295delT, predicted to result in frameshift and premature termination (Phe99LeufsTer18), and c.334A-G transition, resulting in Ser112Gly) | [119] |
| MMDS4 (p.Leu52Phe; p.Arg105Gly) | |||||
| IBA57 | Iba57 | 1q42.13 (4) | Late-acting ISC biogenesis component | MMDS3 (p.Q314P; homozygous c.678A-G transition resulting in splicing defect) | [73,120–123] |
| MMDS3 (p.R146W) | |||||
| MMDS3 (p.W196G) | |||||
| MMDS3 (p.Pro229Leu) | |||||
| MMDS3 (p.Pro236Ser) | |||||
| MMDS3 (p.Tyr96His) | |||||
| MMDS3 (p.Thr106Ala) | |||||
| MMDS3 (p.Arg223*) | |||||
| MMDS3 (p.Tyr108Ser) | |||||
| MMDS3 (p.Gln314*) | |||||
| MMDS3 (p.Cys50*) | |||||
| BOLA1 | Bola1 | 1q21.2 (2) | Involved in biogenesis of subsets of mitochondrial Fe-S proteins; probably acts together with GLRX5 | None reported | |
| BOLA2 | Bola2 | 16p11.2 (2) | In complex with PCBP1, delivers iron for cytosolic ISC biogenesis | Haploinsufficiency causes anemia, microcytosis, low serum iron, or low blood hemoglobin | [124] |
| BOLA3 | Bola3 | 2p13.1 (4) | Involved in biogenesis of subsets of mitochondrial Fe-S proteins | MMDS2 (c.123dupA in exon 2 of BOLA3, resulting in frameshift and premature termination affecting both isoforms; p.Arg46*) | [65,115] |
| PCBP1 | Not present | 2p13.3 (1) | In complex with BOLA2, delivers iron for cytosolic ISC biogenesis | None reported; embryonically lethal (early gestation) | [125,126] |
| NUBP1 | Nbp35 | 16p13.13 (11) | Member of NUBP/MRP subfamily of ATP-binding proteins. Yeast ortholog has been reported to function as a scaffold for cytosolic ISC assembly | None reported; yeast ortholog Nbp35 is an essential gene | [67,127] |
| NUBP2 | Cfd1 | 16p13.3 (10) | In yeast, encoded protein functions in heterotetrameric complex with Nbp35 as scaffolds for cytosolic ISC assembly | None reported; yeast ortholog Cfd1 is an essential gene | [67,128] |
| CIAO1 | Cia1 | 2q11.2 (7) | Component of CIA machinery | None reported; yeast ortholog Cia1 is an essential gene | [19,20,129] |
| CIAO2B (FAM96B) | Cia2b | 16q22.1 | DUF59 family protein that functions as component of CIA machinery | None reported | [19,20] |
| (CIAO4) MMS19 | Mms19 | 10q24.1 (33) | Component of CIA machinery | None reported; embryonically lethal (before implantation stage) | [19,20] |
| CIAO3 (NARFL) | Nar1 | 16p13.3 (12) | Hydrogenase-like protein; yeast ortholog Nar1 has been reported to be involved in cytosolic ISC assembly | None reported; embryonically lethal (embryonic day 10.5) | [19,20,130] |
| CIAPIN1 (Anamorsin) | Dre2 | 16q21 (9) | In yeast, Dre2 forms complex with diflavin reductase Tah18, thus constituting an electron-transfer chain for assembly of [4Fe-4S] clusters. CIAPIN in mammalian cells may perform a similar function working with NDOR (human ortholog of Tah18) | None reported; embryonically lethal (late gestation) | [131–133] |
| NDOR | Tah18 | 9q34.3 (14) | In yeast, Tah18/Dre2 complex functions as electron transfer chain for cytosolic ISC assembly | None reported | [133] |
| GLRX3 | Grx3 | 10q26.3 | In complex with BOLA2, can transfer [2Fe-2S] clusters to recipients | None reported; embryonically lethal (late gestation) | [134–136] |
ISCs comprise iron and inorganic sulfide that are typically, but not exclusively, ligated to cysteinyl sulfur in proteins [2] (Box 1). The most common types are the rhombic [2Fe-2S] clusters, which are present in enzymes such as mammalian ferrochelatase, mitochondrial respiratory complexes I and II, ferredoxins (FDXs), and Rieske proteins, and the tetranuclear [4Fe-4S] clusters, which can readily accept or donate single electrons [2–8].
Box 1. Why Are Iron-Sulfur Cofactors Ubiquitous and Indispensable in Metabolism?
Cells are able to capture energy by storing the reducing power of electrons released during metabolism of organic substrates in forms of intracellular energy currency, such as reduced molecules (e.g., the pyridine nucleotide cofactors NADH and NADPH) and ATP. Electrons are relayed along positions of increasing electron avidity in the respiratory chain complexes, which require 12 ISCs for function, by coupling proton extrusion into the intermembrane space to ATP synthesis when protons return to the matrix through the ATP synthase of the inner membrane by moving down a favorable concentration gradient [39]. Only a few cofactors have the ability to transiently bind single electrons. These include ISCs, heme (comprising an iron ion coordinated to a porphyrin ring and to one or two axial ligands), and flavin and pyridine mono- and di-nucleotides.
In this short list, ISCs are by far the most flexible cofactors in facilitating a variety of chemical activities. Examples of ISCs include the common rhombic [2Fe-2S] and cubane [4Fe-4S] clusters (Figure IA,B) [2]. The tetranuclear or cubane clusters shown in Figure IB have the capability to delocalize electrons between iron sites, conferring the ability to accommodate the addition of a single electron (as depicted by the tan cloud-like frames in Figure IB, which illustrate that the iron-associated electrons are highly delocalized between neighboring iron atoms). Because of their flexibility, ISCs can function as modules that reassemble into more complex structures [2]. Two rhombic [2Fe-2S] clusters can form a cubane cluster through reductive coupling [93]. The complexity of Fe-S cofactors can be greater in the P-cluster of nitrogenase (Figure IC) or in the iron-molybdenum cofactor of nitrogenase (Figure ID), where an interstitial carbon atom occupies the core of the cofactor. Other important functions facilitated by Fe-S cofactors include binding of substrates to one of the irons of the cubane cluster to perform dehydration reactions, enabling radical reactions, and functioning as iron and oxygen sensors [2].
Figure I. Examples of Iron-Sulfur Clusters (ISCs) Found in Proteins.
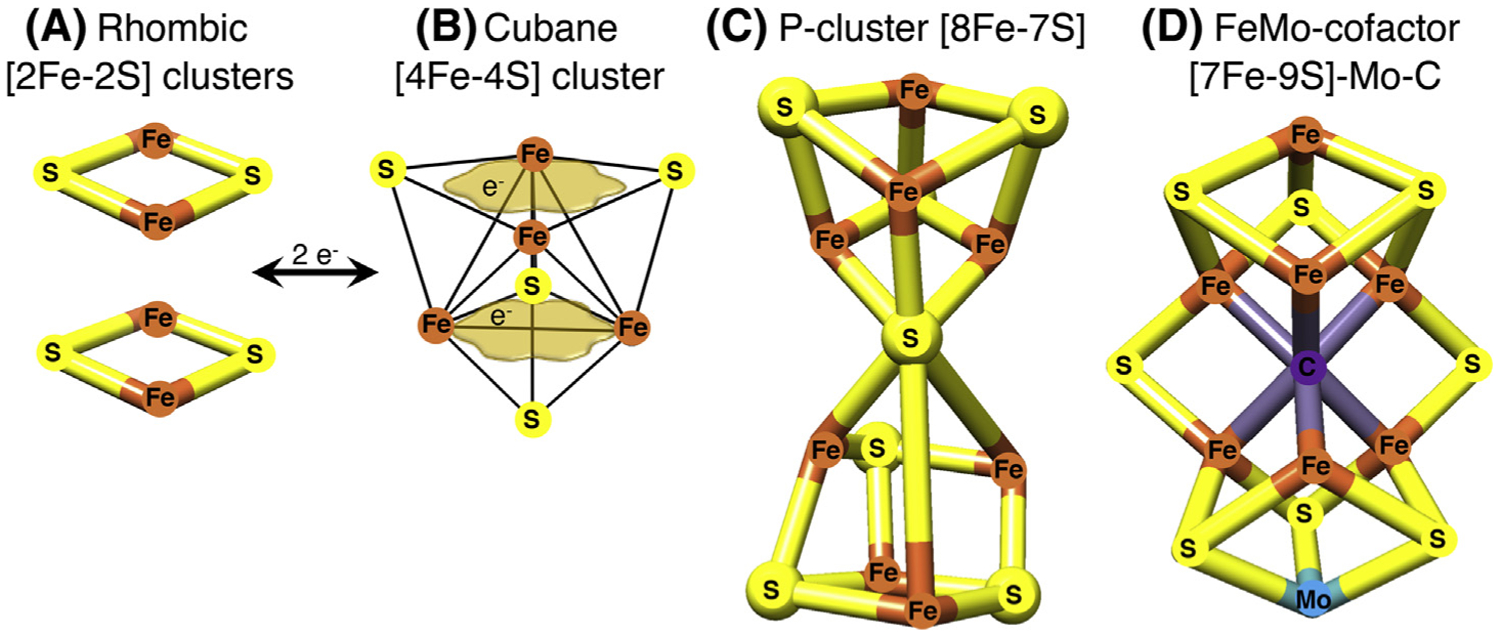
(A) The common rhombic [2Fe-2S] clusters are initially assembled from inorganic iron and sulfur upon the main scaffold protein ISCU and they can be utilized to generate more complex Fe-S cofactors. (B) The tetranuclear or cubane clusters are formed by reductive coupling of two [2Fe-2S] clusters, in a process that requires two electrons (2e−). [4Fe-4S] clusters have the capability to delocalize electrons between iron sites (as depicted by the tan cloud-like frames, which illustrate that the iron-associated electrons are highly delocalized between neighboring iron atoms). (C) The configurations of Fe-S cofactors can be more complex in the P-cluster of nitrogenase or in the iron-molybdenum cofactor of nitrogenase depicted in (D), where an interstitial carbon atom occupies the core of the cofactor.
In this review, we discuss the elaborate process that leads to de novo ISC assembly (see Glossary) on the main scaffold protein, ISCU, with discussion of recent insights into the role of frataxin (FXN) that have emerged from crystal structures of the initial core biogenesis complex. We then explore how ISCs are delivered to recipient proteins in both the mitochondrial matrix and cytosolic compartments of mammalian cells, and outline the molecular pathways that assemble [4Fe-4S] clusters in mitochondria through the involvement of the secondary carriers, all of which are essential for human physiology, because mutations in any of these proteins cause human diseases (Table 1).
Specific Examples of Dysfunctional Fe-S Proteins in Human Diseases
ISCs commonly mediate redox reactions in mitochondrial respiration, photosynthesis, and nitrogen fixation, where they act as electron relay chains that are ensheathed within an otherwise impervious protein matrix. A redox switch in the oxidation state of [4Fe-4S] clusters was also recently found to modulate the DNA-binding affinity of DNA-processing enzymes, including glycosylases, helicases, and primases [5]. Additionally, Fe-S enzymes have crucial roles in translation, as in the case of the ATP-binding cassette sub-family E member 1 (ABCE1), also known as RNase L inhibitor (Rli) in Saccharomyces cerevisiae [9]. Given the essential requirement of ISC enzymes in all aspects of DNA metabolism, mutations in the [4Fe-4S] DNA-helicases ERCC2 or FANCJ, involved in nucleotide excision repair of damaged DNA, were found to cause genome instability and a high predisposition to cancer [10,11].
Besides their involvement in redox reactions, Fe-S enzymes are known to have essential roles in several metabolic pathways, with one of the most notable examples being mitochondrial aconitase of the tricarboxylic acid cycle (TCA) cycle, which ligates a solvent-exposed [4Fe-4S] cluster that directly participates in substrate binding and conversion of citrate to isocitrate through acid-base catalysis [3]. Cytosolic aconitase is a bifunctional enzyme (iron regulatory protein 1, IRP1/cytosolic aconitase, ACO1) with a crucial role in the regulation of cytosolic iron homeostasis [12]. Several bacterial dehydratases share with aconitase the ability to bind and activate substrates at the uniquely open iron site of their [4Fe-4S] clusters, in which the other three iron atoms are ligated to cysteines of the peptide backbone; these include the isopropylmalate isomerase involved in leucine biosynthesis, the Escherichia coli fumarase A that converts fumarate to malate, the dihydroxyacid dehydratase that is involved in the biosynthesis of branched chain amino acids, and the L-serine dehydratase that catalyzes the irreversible deamination of serine to pyruvate [2].
Additionally, ISCs are essential cofactors of the radical S-adenosylmethionine (SAM) superfamily of enzymes, eight of which have so far been reported to occur in pathways that directly affect human health and disease (reviewed in [13]), including molybdenum and lipoic acid cofactor biosynthesis [14,15]. They are also involved in the generation of specific tRNA modifications that affect codon–anticodon recognition and translation fidelity [16–18]. Furthermore, Fe-S enzymes are responsible for DNA replication and the maintenance and/or repair of the nuclear genome [19,20], as well as for cell division, given that characterization of the chromosome-associated kinesin KIF4A recently revealed that it was a [4Fe-4S] cluster protein [21].
As a final example, inactivating mutations in CISD2 (also known as NAF1), initially annotated as a zinc-finger protein [22] and later characterized as a member of the NEET family of proteins that coordinates a labile [2Fe-2S] cluster [23], were recently reported to cause Wolfram syndrome 2 [24]. This is an autosomal recessive neurodegenerative disorder characterized by diabetes mellitus, sensorineural hearing loss, optic atrophy, and neuropathy and it represents an additional example of the essential role of Fe-S proteins in human physiology.
ISC Biogenesis on the Main Scaffold Protein ISCU in Mammalian Cells
ISC biogenesis is an evolutionarily highly conserved process. The first insights into the proteins that contributed to the pathway were obtained through analysis of a bacterial operon needed for Fe-S biogenesis in nitrogen-fixing bacteria [25]. Most protein functions defined in bacterial ISC biogenesis have been conserved in mammalian cells.
De novo ISC assembly is a complex, multiprotein-mediated process that involves an initial critical step catalyzed by a pyridoxal-phosphate (PLP)-dependent homodimeric transaminase, the cysteine desulfurase NFS1, which converts cysteine into alanine, thereby donating sulfur to generate a persulfide intermediate on a conserved cysteine residue (Cys381 of human NFS1) of a mobile S-transfer loop of the enzyme [26] (Figure 1). The persulfide sulfur is then transferred to a conserved cysteine of the main scaffold protein ISCU (Cys138 of human mitochondrial ISCU) through a conformational change that is enhanced by FXN [27,28]. NFS1 is stabilized by the binding of the accessory protein ISD11, also known as LYRM4 [29,30]. The acyl-carrier protein ACP (NDUFAB1 in human) was also recently found to be part of the initial ISC core biosynthetic complex and to bind ISD11 [31]. Combined structural and biochemical analyses showed that a critical aspect of the ISD11–ACP interaction involved the insertion of the entire acyl-chain and the 4′-phosphopantotheine (4′-PP) group of ACP into a hydrophobic barrel created by the triple-helical structure of ISD11 (Figure 2A), through a proposed mechanism known as chain-flipping [32–35]. Mitochondrial ACP is a bacterial-type acyl-carrier protein involved in mitochondrial fatty acid biosynthesis (mFAS), a pathway that provides octanoic acid, the precursor of lipoic acid, through elongation of nascent acyl-chains bound to the 4′-PP cofactor attached to Ser112 of human ACP [15,36].
Figure 1. Iron-Sulfur Cluster (ISC) Biogenesis in Mammalian Mitochondria: An Overview of the Main Steps.
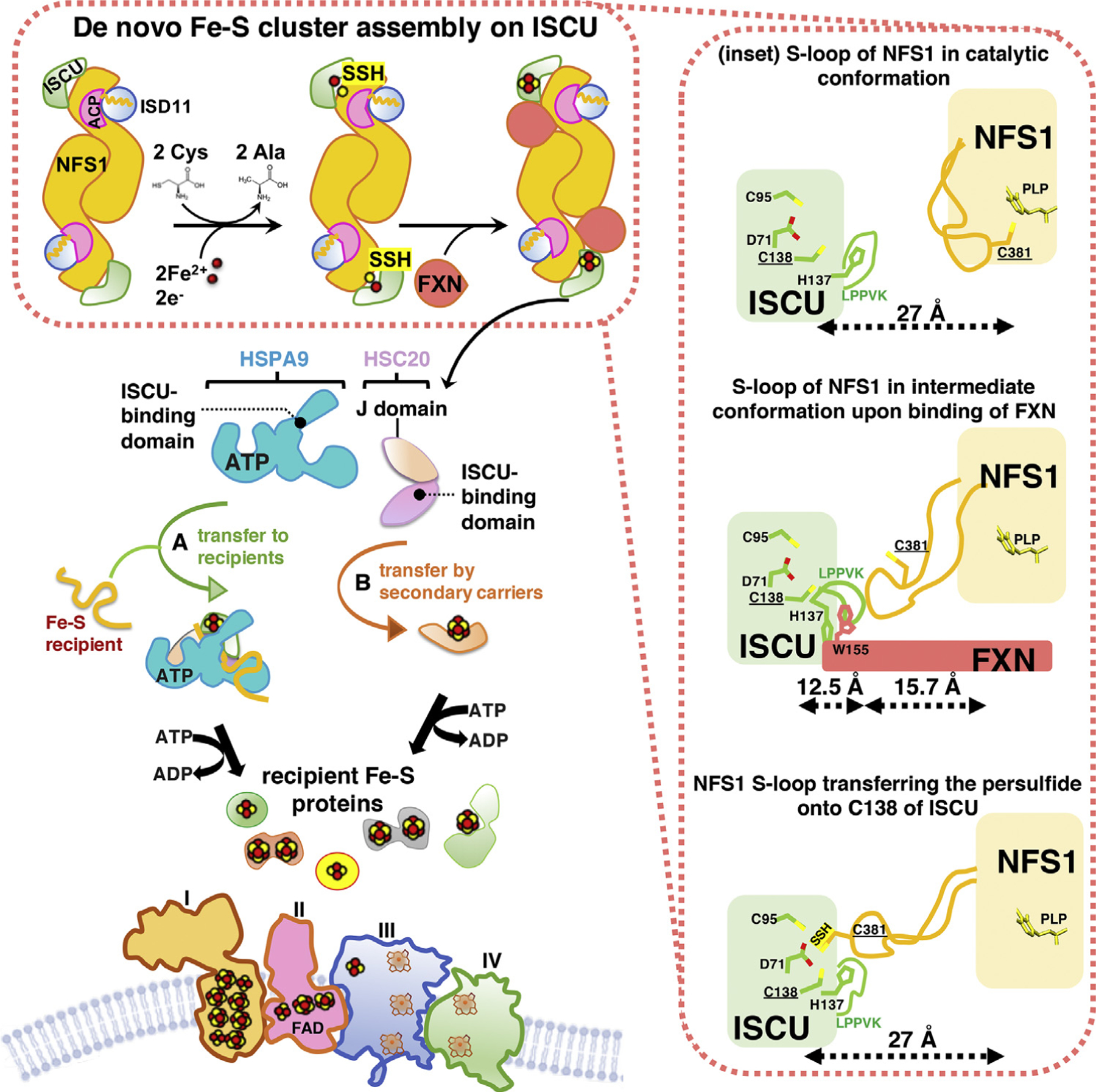
(Top left) Nascent ISCs are assembled de novo on the main scaffold protein ISCU. A cysteine desulfurase, NFS1, forms a dimer to which monomers of the primary scaffold ISCU bind at either end. ISD11 (also known as LYRM4) and acyl carrier protein (ACP) with its bound acyl chain are structural components of the core complex in eukaryotes. NFS1, aided by its cofactor pyridoxal phosphate (not shown), provides inorganic sulfur, removed from cysteine, to the nascent cluster. Transient binding of frataxin (FXN) in a pocket-like region between NFS1 and ISCU promotes sulfur transfer from NFS1 to ISCU. The cluster assembles upon ISCU when iron is provided together with the reducing equivalents needed to generate the final electronic configuration of the cluster. (Bottom left) A chaperone-cochaperone complex binds to the LPPVK motif of ISCU and either facilitates direct transfer of ISCs to recipient proteins (pathway A) or mediates transfer to secondary carriers (e.g., NFU1, GLRX5, ISCA1, ISCA2, and BOLA3; pathway B), which then donate ISCs to specific recipients (e.g., lipoic acid synthase and subunits of the respiratory chain complexes). (Right inset) Three different conformations of the mobile S-transfer catalytic loop of NFS1 during de novo ISC assembly upon ISCU in its trajectory from the catalytic conformation, in which Cys381 of NFS1 is close to pyridoxal-phosphate (PLP) and the substrate cysteine, to the intermediate conformation, recently structurally characterized [34] produced by FXN-binding, to the final ISC assembly conformation, in which the catalytic Cys381 of NFS1 donates sulfur to Cys138 of ISCU [32].
Figure 2. Comparison of the Three Recently Solved Crystal Structures of the Human Iron-Sulfur Cluster (ISC) Core Complex.
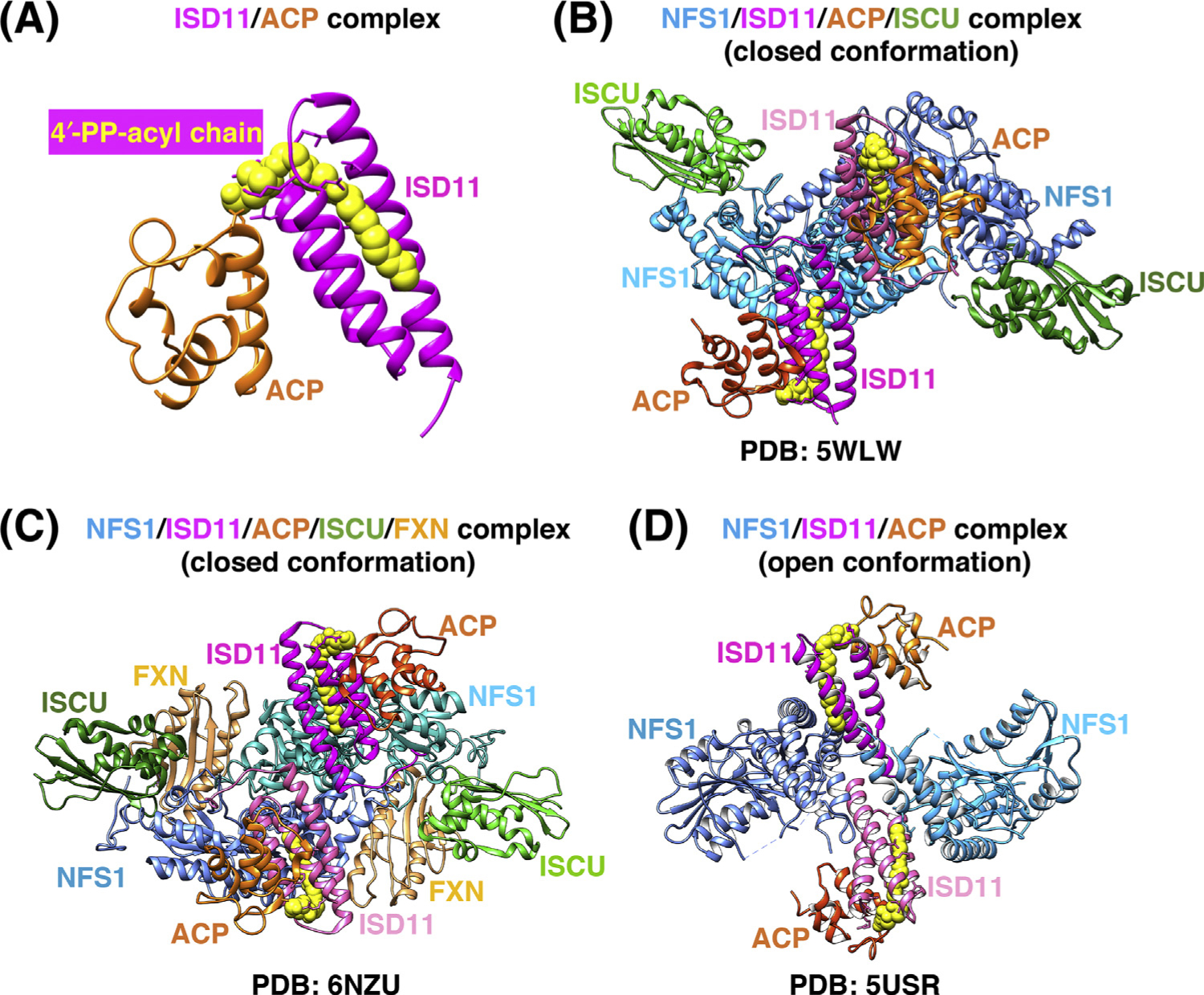
(A) Structure of the ISD11/acyl carrier protein (ACP) complex [Protein Data Bank (PDB) ID: 5USR]. The acyl chain (yellow) is covalently attached to the 4′-phosphopantotheine (4′-PP) group of ACP (orange) and fits within the three-helical structure of ISD11 (magenta). (B) Structure of the homodimeric (NFS1/ISD11/ACP/ISCU-Zn2+)2 complex [32] (PDB ID: 5WLW). NFS1 protomers (shades of blue), ISD11 (magenta), and ACP (orange) form a homohexameric core, with ISCU (green) bound at each end of the complex. (C) Structure of the homodimeric (NFS1/ISD11/ACP/ISCU-Zn2+-FXN)2 complex [34] (PDB ID: 6NZU). Frataxin (FXN) (tan) binds in a pocket-like region between ISCU and NFS1 protomers (colored as in B). (D) Structure of the (NFS1/ISD11/ACP)2 homodimeric complex in the ‘open’ conformation [33] (PDB ID: 5USR), in which the two NFS1 protomers make minimal contacts with each other, and ISD11 mediates the interactions between the NFS1 subunits.
Although shown to interact with the NFS1-stabilizing protein, ISD11, the role of ACP in ISC biogenesis remains unclear. It has been proposed that ACP may link fatty acid metabolism, through its role in mFAS, to ISC biogenesis [31]. Interestingly, an abnormal abundance of lipid droplets was detected in the myocardium of the muscle-specific FXN-knockout mouse model [37], suggesting that one of the many steps of lipid homeostasis is defective in cells with impaired ISC assembly due to loss of FXN. In accordance, it was recently reported that the acute disruption of ISC biogenesis caused a dramatic increase in the intracellular levels of glucose-derived citrate, which was diverted into fatty acid biosynthesis, leading to cytosolic lipid droplet accumulation [38]. Mammalian ACP has also been crystallized as part of mitochondrial complex I, through its binding to the subunits known as LYR proteins NDUFA6 (also known as LYRM6) and NDUFB9 (also known as LYRM3) [7]. Twelve ISCs are required for the assembly of a functional mitochondrial respiratory chain that utilizes the energy molecules (NADH and FADH2) generated from acetyl-CoA in the TCA as primary substrates for oxidative phosphorylation (OXPHOS) [7,39]. Acetyl-CoA is also the precursor for the synthesis of the acyl chain of ACP. Therefore, it has been proposed that acylation of ACP may serve as a functional link between mitochondrial acetyl-CoA abundance and ISC biogenesis [31].
Additionally, the FDX/FDX reductase system assists ISC assembly [40–42] by reducing the persulfide transferred from Cys381 of NFS1 to Cys138 of human ISCU [42]. Combination of sulfane sulfur with Fe2+ on ISCU generates a [2Fe-2S] cluster (Figure 1) that is subsequently transferred to recipient Fe-S apoproteins or to secondary carriers by an ISC biogenesis chaperone/cochaperone system that comprises HSPA9 and HSC20 in mammalian cells [43].
Architecture of the Human Mitochondrial ISC Assembly Machinery
The NFS1/ISD11/ACP/ISCU complex is a symmetric hetero-octamer, comprising two copies of each of the four constituent proteins [32,34] (Figure 2B). NFS1, ISD11, and ACP form a homohexameric core, with ISCU bound to each end of the complex. FXN, which acts as an allosteric regulator of ISC biogenesis by driving efficient sulfur transfer from Cys381 of the mobile S-loop of NFS1 to Cys138 of ISCU (Figure 1), was recently co-crystallized with the NFS1/ISD11/ACP/ISCU complex [34] (Figure 2C). FXN occupied a cavity at the interface between NFS1 and ISCU, and did not make direct physical contact with ISD11, whereas it simultaneously interacted with both NFS1 protomers of the complex [34] (Figure 2C). This is consistent with the structure of the NFS1/ISD11/ACP/ISCU complex [32] (Figure 2B) but not compatible with a previously reported open conformation of the NFS1/ISD11/ACP complex [33] (Figure 2D), in which the two NFS1 protomers made minimal contacts with each other. The unusual arrangement of the NFS1/ISD11/ACP open complex [33] revealed an incomplete substrate-binding pocket, a solvent-exposed PLP cofactor, a disordered mobile S-transfer loop, and an overall open architecture in which the ISD11 protomers mediated the interactions between NFS1 subunits [33] (Figure 2D).
To reconcile the two structures, it is possible that the NFS1/ISD11/ACP complex exists in vivo as a mixture of the open and closed conformations in equilibrium. Given that eukaryotic NFS1 has a turnover rate that is b10% of the prokaryotic cysteine desulfurase, IscS, it is conceivable that FXN stimulates the activity and catalytic turnover rate of IscS [26] by inducing an open-to-closed architectural rearrangement of the complex that repositions the second NFS1 protomer to complete formation of the active site and force a conformational change of the S-transfer loop. By contrast, prokaryotic IscS is proposed to mainly adopt the highly active closed conformation that is not further activated by the bacterial ortholog of FXN, CyaY [19].
Insights into the FXN-Mediated Activation of the Core ISC Complex
In the closed conformation of the NFS1/ISD11/ACP/ISCU/FXN complex captured in a cryo-electron microscopy (cryo-EM) structure [34] (Figure 2C), FXN occupied the interface between NFSI and ISCU and contacted one of the two NFS1 protomers at the catalytic S-loop, supporting the idea that FXN functions as an allosteric modulator of ISC biogenesis [27] (Figures 1 and 2C). FXN also bound to two key regions of ISCU, one of which is the conserved ISCU-Alanine-loop (Ala66-Asp71), containing the Fe-S ligating residues Cys69 and Asp71 [38,44] (Figure 1), and the other is the ISCU-LPPVKLHC138SM140 motif. This motif contains not only the LPPVK consensus sequence recognized by the chaperone HSPA9 for downstream delivery of the cluster to recipient proteins, but also Cys138, which is the proposed sulfur acceptor from NFS1 [27] (Figure 1), and Met140, a residue reported to confer FXN-dependence of ISC biogenesis in S. cerevisiae [45], or FXN independence in prokaryotes [46]. The fact that the LPPVK motif of ISCU, required for the binding of the chaperone, is buried at the FXN interface suggests that FXN has to be displaced before binding of the HSPA9/HSC20 transfer complex can occur.
The labile iron pool of the mitochondrial matrix, which has been calculated to be ~170 μM (15–20% of total mitochondrial iron content), may serve as a source of iron for ISC biogenesis [47]. It remains to be established whether a designated iron chaperone is required to donate iron to ISCU during initial ISC assembly. Therefore, the ISC core complex would cycle between sulfur donation (mediated by FXN) from NFS1 to iron loaded-ISCU, and electron donation (mediated by FDX/FDXR), followed by binding of the chaperone/cochaperone complex (HSPA9/HSC20) that mediates transfer of de novo assembled clusters to recipient proteins. Since only reduced FDX was found to interact with the complex [48], it is possible that, once electron donation is complete, oxidized FDX exits the complex, allowing replacement by another reduced FDX or FXN.
A Specialized Chaperone/Cochaperone System Assists Delivery of ISCs to Recipient Proteins
Transfer of newly assembled ISCs downstream of the main scaffold protein ISCU in mammalian cells relies on the activity of a highly conserved chaperone/cochaperone system analogous to the yeast Ssq1/Jac1 and the bacterial HscA/HscB complexes [49–51]. In mammalian cells, the multifunctional member of the HSP70 family, HSPA9 (also known as mortalin or GRP75), works with the specialized DnaJ-type III protein, HSC20 (also referred to as DNAJC20 or HSCB), to either directly facilitate ISC transfer to recipient proteins (Figure 1, step A) or to secondary carriers, which then target specific recipients [52,53] (Figure 1, step B).
A longstanding question in the field relates to how the biogenesis machinery guides selection of specific subsets of mammalian proteins as Fe-S recipients. Most eukaryotes, including humans, have a single multifunctional mitochondrial chaperone, which participates in diverse cellular functions by virtue of its ability to interact with an array of different J-proteins (also known as HSP40s) (reviewed in [54]). Cochaperones were suggested to have the crucial role of driving the functional specificity of HSP70s into a definite biological process by selectively binding to specific protein targets [54]. Recent studies revealed that HSC20 is the major component of the Fe-S transfer machinery that guides ISC transfer from ISCU to recipients [12,53,55,56]. A stringent yeast two-hybrid (Y2H) screen was used to identify protein targets bound by HSC20, as well as peptide motifs, that might function as molecular signatures that guide the recruitment of the HSC20/HSPA9-transfer complex through direct binding to HSC20 [53]. The tripeptide Leu-Tyr-Arg and/or analogous sequences containing an hydrophobic amino acid in position 1, an aromatic residue (tyrosine or phenylalanine) in position 2, and arginine or lysine in position 3, were found to mediate direct binding of the ISC transfer complex, comprising HSC20, HSPA9, and [2Fe-2S]-ISCU, to recipient Fe-S apoproteins to promote cluster acquisition [53,55,56].
Several late-acting ISC biogenesis factors, including NFU1, BOLA3, IBA57, ISCA1, and ISCA2, have been proposed to function as secondary carriers that acquire [2Fe-2S] clusters from ISCU and assemble [4Fe-4S] cofactors. These are then delivered to specific recipient proteins, such as lipoic acid synthase (LIAS) and respiratory complexes I and II (see later for discussion on interactions, mechanisms, and remaining challenges).
Distinct Pathways Assemble [4Fe-4S] Clusters in Mitochondria for Specific Fe-S Recipient Proteins
The initial model for the biogenesis of [4Fe-4S] clusters in mitochondria, largely based on studies in S. cerevisiae, proposed that three human proteins, ISCA1, ISCA2, and IBA57, function as secondary carriers and form a heterocomplex that enable the assembly of [4Fe-4S] clusters, using as building blocks [2Fe-2S] cofactors assembled upon ISCU and transferred by GLRX5 [57,58]. However, the functional association between ISCA1, ISCA2, and IBA57 has been recently questioned [59–62]. For example, in knockdown experiments in mouse skeletal muscle and in primary cultures of neurons, ISCA1, but not ISCA2 or IBA57, was found to be required for mitochondrial [4Fe-4S] cluster biogenesis [59]. Moreover, biochemical and spectroscopic approaches revealed that, despite sometimes forming a heterodimeric complex, ISCA1 and ISCA2 established distinct interactions with proteins of the late mitochondrial ISC machinery, namely GLRX5, IBA57, and NFU1 [59,60,62]. Compared with the elaborate context of the molecular interactions that take place in vivo in mitochondria, in vitro experiments represent a simplified system in which only a limited number of purified components of the ISC machinery are simultaneously tested.
Nonetheless, in vitro assays have helped to shed light on the distinct pathways that assemble [4Fe-4S] clusters in mitochondria and showed that: (i) ISCA1 and ISCA2 form a heterodimeric complex, which is able to assemble [4Fe-4S] clusters through reductive coupling of two [2Fe-2S] clusters donated by GLRX5 in the absence of IBA57 [63,64]; and (ii) ISCA2, but not ISCA1, after receiving a [2Fe-2S] cluster from GLRX5, forms a heterodimeric complex with IBA57 that is stable in highly oxidative environments [61]. These results were consistent with in vivo studies that reported an interaction of ISCA2, but not ISCA1, with IBA57 [59], and with the proposed differential activation of the assembly pathways carried out by ISCA1 or ISCA2 and IBA57 under different physiological conditions [59].
The specific targets of the secondary carriers have not been assigned, although it appears that NFU1 and BOLA3 have essential and nonredundant roles in the biogenesis of the two [4Fe-4S] clusters of lipoic acid synthase [65,66]. Defects in the respiratory complexes I and II, but not in mitochondrial aconitase, have also been reported in association with mutations in NFU1 and BOLA3 [65,66]. However, the interpretation of the biochemical phenotypes associated with mutations in late-acting ISC components is particularly complex because some effects may be indirect. NFU1 was proposed to receive its [4Fe-4S] cluster from the ISCA1-ISCA2 heterocomplex [67], and BOLA3 was thought to mediate cluster transfer from NFU1 to specific recipients [68]. However, the interaction between NFU1 and BOLA3 was not detected in vivo [68], whereas immunoprecipitation experiments in vivo and in vitro studies have confirmed the interaction of BOLA3 with GLRX5 [60,69]. Therefore, it appears that GLRX5, in addition to its role as an early-acting secondary carrier that receives its [2Fe-2S] cluster from ISCU [53,63], has the ability to interact with BOLA3 [60,69]. Two [2Fe-2S]-GLRX5-BOLA3 complexes have been proposed to transfer their clusters to apo-NFU1 to form a [4Fe-4S] cluster on dimeric NFU1 [60]. This pathway may be an alternative to that involving the ISCA1-ISCA2 complex.
Collectively, the late steps of mitochondrial ISC biogenesis constitute a complex and dynamic network system that may rely on distinct pathways that could operate under different physiological conditions or at different levels in different tissues (proliferative, post mitotic, oxidative stress, and hypoxia) to specifically deliver [4Fe-4S] clusters to recipient proteins.
Clinical and biochemical investigations of several human diseases caused by mutations in NFU1, BOLA3, IBA57, ISCA1, or ISCA2 suggest that transfer of ISCs to secondary carriers represents a mechanism for specifically delivering Fe-S cofactors to subsets of recipients [59,60,68,70], which may explain the distinctive phenotypes associated with the different disease gene mutations (Table 1). Multiple mitochondrial dysfunctions syndromes (MMDSs) have recently emerged as a group of mitochondrial disorders that are inherited in an autosomal recessive manner. Six MMDSs have been described thus far (Table 1). These include MMDS1, which is caused by mutations in NFU1 [Online Mendelian Inheritance in Man (OMIM) #605711] [65,66]; MMDS2, caused by BOLA3 gene defects (OMIM #614299) [65,71,72]; MMDS3 due to IBA57 mutations (OMIM #615330) [73]; MMDS4 due to ISCA2 mutations (OMIM #616370) [74]; MMDS5 due to ISCA1 mutations (OMIM #617613) [75]; and MMDS6 due to PMPCB gene defects (OMIM #617954), which encodes the catalytic subunit of the essential mitochondrial-processing protease (MPP). MMDS1–5 manifest with different degrees of defects in the levels of enzymes that depend on [4Fe-4S] clusters for function, including LIAS, which catalyzes the biosynthesis of lipoic acid from octanoic acid and is required for the activity of the lipoylated mitochondrial complexes pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, branched-chain ketoacid dehydrogenase, and the glycine cleavage system, and respiratory complexes I and II [65,66,73].
Longstanding Controversy over the Existence of a Parallel System for ISC Biogenesis in the Cytosolic/Nuclear Compartment of Mammalian Cells
Two contrasting models have emerged to describe cytoplasmic Fe-S biogenesis [67,76] (Figure 3). One model, largely based on studies in yeast, proposed that the assembly of extramitochondrial Fe-S proteins relied on the mitochondrial ISC machinery for the synthesis of a sulfur-containing compound (named X-S) that was exported to the cytoplasm by the ABC transporter Atm1 (ABCB7 in human) and utilized for the assembly of a [4Fe-4S] cluster transiently bound to the heterotetrameric Nbp35/Cfd1 complex (NUBP1/NUBP2 in human; Figure 3A) [67,77]. The NADPH-dependent electron transfer system comprising the diflavin reductase Tah18 (NDOR1 in human) and the Fe-S protein Dre2 (CIAPIN1 in human) was proposed to provide electrons necessary for the assembly of [4Fe-4S] clusters on the NUBP1/NUBP2 heterotetramer, which was considered to function as the primary scaffold complex from which all cytoplasmic and nuclear Fe-S proteins acquired their cofactors [67]; nuclear Fe-S enzymes acquired their clusters from the cytoplasmic Fe-S cluster assembly (CIA) machinery, comprising CIAO1, FAM96B, and MMS19 [19,20] (Figure 3A).
Figure 3. Alternative Proposed Models of Cytoplasmic Iron-Sulfur Cluster (ISC) Biogenesis.
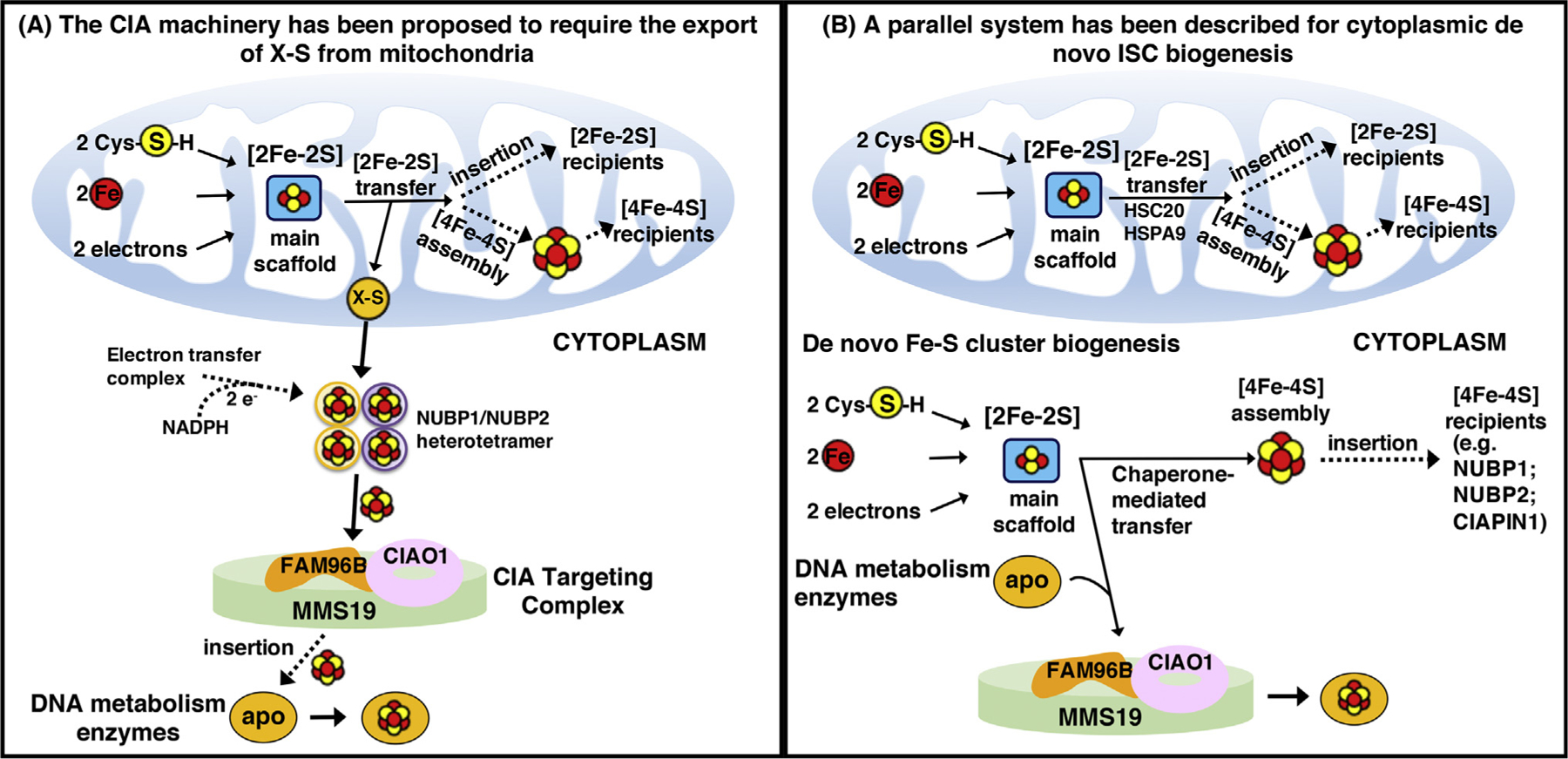
(A) Assembly of cytoplasmic ISCs begins in mitochondria with components of the early ISC machinery synthesizing a sulfur-containing precursor (X-S) that is subsequently exported to the cytosol by the ABC transporter Atm1 (ABCB7 in humans) and utilized by the cytoplasmic ISC assembly machinery for the biosynthesis of [4Fe-4S] clusters upon the main heterotetrameric complex, comprising NUBP1 and NUBP2 [67]. (B) Alternative isoforms of the core early ISC factors are present in the cytosol of mammalian cells, where they initiate de novo assembly of [2Fe-2S] clusters on the main cytosolic scaffold protein ISCU1. A dedicated chaperone/cochaperone system, comprising HSPA9 and HSC20, either facilitates direct ISC transfer to a subset of recipient cytosolic proteins (e.g., CIAPIN1, NUBP1, and NUBP2) or mediates the transfer of ISCs to enzymes involved in DNA metabolism through direct binding of HSC20 to the LYR motif of the CIAO1 component of the cytosolic ISC assembly (CIA)-targeting complex [78].
The second proposed model for cytoplasmic Fe-S biogenesis is based on studies that for more than two decades have demonstrated the existence of a de novo cytosolic ISC pathway in mammalian cells, which can supply the elemental components (i.e., iron and sulfur) and the biogenesis proteins required to build the cluster in the cytosol of mammalian cells [76,78] (Figure 3B). Consistent with this model, the pool of NFS1 that localizes to the cytosol [79] isa functional enzyme that mobilizes sulfur from cysteine [80,81], a notion that questions the need for the export of a sulfur-containing compound out of mitochondria. Moreover, alternative isoforms of the core ISC biogenesis components have been detected in the cytosol of mammalian cells [52,53,79,82–86], suggesting that ISC biogenesis machineries independently operate in parallel to generate nascent ISCs in multiple subcellular compartments of multicellular eukaryotes.
Specifically, a recent study in mammalian cells revealed the existence of the full complement of initial ISC biosynthetic enzymes for de novo assembly of ISCs in the cytoplasm [78]. A pool of HSC20 that localized to the cytosol was found to bridge components of the initial ISC pathway, including cytosolic ISCU, NFS1, and HSPA9, to the CIA-targeting complex, comprising CIAO1, FAM96B, and MMS19, likely through dimerization of its N-terminal domain [78]. Functional assays confirmed the importance of cytosolic ISCU and HSC20 for the biogenesis of several Fe-S proteins, including POLD1, DPYD, NUBP2, ERCC2, ELP3, and PPAT [78].
Recently, the iron-sensing protein FBXL5, which is the substrate adaptor of a SKP1-CUL1-RBX1 E3-ubiquiting ligase complex that regulates the degradation of IRPs [87], was found to interact with the CIA-targeting complex [88]. Disruption of the interaction between FBXL5 and the CIA-targeting complex, due to either deletion of the CIA-targeting complex-binding domain of FBXL5 or low oxygen tension in cells, severely diminished the ability of FBXL5 to drive polyubiquitination and degradation of IRPs. The interaction of FBXL5 with the CIA-targeting complex demonstrates the existence of crosstalk between ISC biogenesis and iron metabolism, in addition to the already known conversion of IRP1 from cytosolic aconitase into IRE-binding protein under iron-limiting conditions. Further investigations of the physiological significance of the FBXL5-CIA interaction are warranted, particularly in light of the essential link between cellular iron homeostasis and Fe-S biogenesis (Box 2).
Box 2. Defects in ISC Biogenesis Cause Mitochondrial Iron Overload.
Experimental and clinical studies have demonstrated that defects in the early steps of mitochondrial ISC biogenesis lead to mitochondrial iron overload [94–99]. In cell culture models, mitochondrial iron overload occurs in conjunction with cytosolic iron depletion upon knockdown of ISC biogenesis components [53,82,84,100].
In yeast, defective ISC biogenesis increases transcription of the iron-uptake regulon through the activation of the iron-sensing transcriptional factor Aft1 [101]. A [2Fe-2S] cluster, bridged by Grx3/Grx4 complexes, controls the transcriptional activity of Aft1 and causes its dissociation from the target promoters under Fe-replete conditions, preventing excessive iron import [101]. Interestingly, cells that expressed a constitutively active Aft1 accumulated iron mainly in the vacuole, as a protective mechanism that compensated for the increased iron uptake at the plasma membrane [102], whereas cells with defective ISC biogenesis exhibited massive iron import in mitochondria [102], demonstrating that defective ISC biogenesis caused the loss of iron homeostasis regulation.
Unlike yeast, mammalian cellular iron homeostasis is regulated by IRP1 and IRP2, regulatory proteins that are absent in yeast. It remains unexplained how defects in ISC biogenesis lead to mitochondrial iron accumulation. One hypothesis involves the possibility that an Fe-S protein functions as a sensor of mitochondrial iron status, so that decreased biogenesis of this protein in mitochondria could be registered by the cell as a signal of mitochondrial iron deficiency. This may result in the feedback activation of a response that increases iron delivery to mitochondria. In support of this idea, upregulation of the mitochondrial iron importer mitoferrin has been reported in frataxin-deficient mouse hearts [103] and in muscle biopsies from patients with ISCU myopathy [104]. Misregulation of mitochondrial iron homoeostasis and cytosolic iron depletion might engage the cell in a vicious cycle in which increased cellular iron uptake further exacerbates mitochondrial iron overload. Future studies aimed at characterizing the nature of the specific signal that triggers mitochondrial iron overload will provide insights into the pathophysiology of a spectrum of disorders characterized by mitochondrial iron overload, including sideroblastic anemias (reviewed in [105]). Congenital sideroblastic anemias are inherited disorders characterized by mitochondrial dysfunction due to defects in heme biosynthesis, ISC biogenesis, mitochondrial protein synthesis, or in mitochondrial proteins involved in oxidative phosphorylation. It remains unknown why iron accumulates in these dysfunctional mitochondria.
The versatility of ISCs would make them ideal sensors of cellular iron status, since their assembly and trafficking requires integration of iron homeostasis and redox control pathways. Uncovering the molecular mechanisms that coordinate functional ISC biogenesis and maintenance of cellular and mitochondrial iron homeostasis will illuminate the elaborate control mechanisms required to manage iron availability and distribution in the cell.
Concluding Remarks
The fact that an increasing number of newly recognized rare diseases results from compromise of the Fe-S biogenesis pathway offers the prospect that better understanding of the complexity of ISC biogenesis will lead to more effective diagnostics and treatments for a related group of human diseases. However, prior to that, the complexity of these pathways, including identifying the source of iron to build the clusters, characterizing motifs that guide Fe-S cofactors to recipients, and teasing out the details of assembly and delivery of [4Fe-4S] clusters to recipient proteins in the mitochondria and cytoplasm, need to be elucidated (see Outstanding Questions). Analysis of the increased biological complexity from bacteria to eukaryotic species may explain the need for parallel pathways of de novo ISC biogenesis in the different subcellular compartments of mammalian cells.
Outstanding Questions.
What is the source of iron for Fe-S cluster biogenesis? If low-molecular mass iron complexes supply the iron for ISC biogenesis, is a protein chaperone involved in donating iron to the main scaffold ISCU?
What is the role of the acyl carrier protein as a component of the initial Fe-S cluster biogenesis core complex?
Are the two different conformations of the NFS1/ISD11/ACP complexes, ‘open’ and ‘closed’ (Protein Data Bank: 5USR and 5WLW, respectively) both present and functional in vivo?
How are specific Fe-S recipient apoproteins, such as LIAS, targeted by secondary carriers to acquire their Fe-S cofactors?
What is the energy requirement, in terms of ATP molecules hydrolyzed by the chaperone/cochaperone complex, required to facilitate cluster release from ISCU, transfer, and folding of the recipient protein?
Are chaperone/cochaperone complexes other than HSPA9/HSC20 involved in Fe-S cluster delivery to specific subsets of Fe-S recipients?
Are most Fe-S clusters cotranslationally inserted or can some, such as aconitase, acquire the Fe-S cluster after the protein has fully folded?
What are the threshold levels of frataxin that different cell types require to maintain proper function of the ISC biogenesis pathway?
Why does Friedreich ataxia adversely affect a subset of selected neurons and cardiomyocytes far more than other cell types?
What is the signal that triggers iron accumulation in mitochondria upon loss of integrity of the Fe-S biogenesis pathway?
Does mitochondrial iron overload occur in individuals with MMDS1–5?
Are the intersections between ISC biogenesis and lipid synthesis important in regulating respiration and supporting cellular energy supplies?
Integration of in vitro studies, including the reconstitution of the entire ISC biogenesis pathway from the NFS1-mediated release of sulfur to the transfer and insertion of ISCs into apoproteins, and in vivo studies that uncover the requirement of specialized secondary carriers and chaperones for delivery to recipient proteins, will enable insights into how individual ISC biogenesis complexes assist the multiple steps of the elaborate pathway. These insights may guide development of therapeutic interventions to ameliorate human diseases caused by dysfunction of the ISC biogenesis pathway. For example, promising results have been obtained in murine models of the most common ISC-associated human disease, Friedreich’s ataxia, by gene therapy-mediated restoration of FXN expression [89–92].
Highlights.
Iron-sulfur (Fe-S) proteins have critical roles in essential metabolic pathways ranging from DNA and RNA metabolism to mitochondrial function, and regulation of cell growth and division.
Mammalian cells likely contain many more Fe-S proteins than have been identified to date, due to loss of fragile iron–sulfur clusters (ISCs) during purification.
Structural determinations of the architecture of the initial ISC biogenesis complex, comprising NFS1, ISD11, ACP, ISCU, and FXN, have shed light on the interactions that govern de novo ISC assembly.
In mammalian cells, a full complement of initial ISC biogenesis factors localizes to the cytosol, where it initiates de novo ISC assembly.
Further biochemical and clinical investigations of ISC-associated disorders and their phenotypes will elucidate the distinct pathways that assemble and deliver ISCs to the numerous enzymes that require them for function.
Acknowledgments
The authors would like to acknowledge support from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Glossary
- Cysteine desulfurase
an enzyme that catalyzes one of the early steps in Fe-S cluster biogenesis, namely the abstraction of the inorganic sulfur, required to build the cluster, from cysteine. The gene is named NFS1 in humans, Nfs1 in yeast, and IscS in prokaryotes. In humans, the same NFS1 nuclear gene encodes two isoforms that localize either to the cytosol/nucleus or to mitochondria, as a result of initiation of translation from two alternative initiation codons on the mRNA.
- De novo ISC assembly
the pathway that assembles [2Fe-2S] clusters starting from inorganic sulfur and iron through the coordinated action of several multiprotein complexes. [2Fe-2S] clusters provide the building blocks to assemble [4Fe-4S] clusters and clusters with more complex stoichiometries.
- ISC biogenesis chaperone
a member of the HSP70 family that works in concert with a cochaperone to facilitate Fe-S cluster transfer from the scaffold to the recipient protein utilizing ATP to drive the conformational changes required for cluster transfer and ligation by the Fe-S recipient. The chaperone involved in facilitating cluster transfer downstream of the main scaffold ISCU is encoded by the gene named HSPA9 in humans, Ssq1 in yeast, and HscA in prokaryotes.
- ISC biogenesis cochaperone
a member of the Hsp40 family that works in concert with a chaperone and activates the ATPase activity of the cognate chaperone. The cochaperone also specifically recognizes Fe-S clients and the main scaffold ISCU. The cochaperone involved in facilitating cluster transfer downstream of the main scaffold ISCU is encoded by a gene named HSC20 (or HSCB) in humans, Jac1 in yeast, and HscB in prokaryotes.
- Recipient proteins (or Fe-S targets)
proteins that require Fe-S cofactors to be functional and that are recognized by components of the Fe-S biogenesis machinery to acquire their clusters.
- Scaffold protein
a component of the Fe-S biogenesis pathway upon which a [2Fe-2S] cluster is initially assembled starting from inorganic sulfur and iron. The gene encoding the main scaffold is named ISCU in humans, Isu (1 and 2) in yeast, and IscU in prokaryotes. In humans, the same ISCU nuclear gene encodes two alternative splicing isoforms that localize to either the cytosol or the mitochondrial matrix.
- Secondary carrier
a component of the Fe-S biogenesis pathway that is able to accept Fe-S clusters from a scaffold protein and is able to donate its cluster to recipient proteins.
References
- 1.Martin W et al. (2008) Hydrothermal vents and the origin of life. Nat. Rev. Microbiol 6, 805–814 [DOI] [PubMed] [Google Scholar]
- 2.Beinert H et al. (1997) Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 277, 653–659 [DOI] [PubMed] [Google Scholar]
- 3.Beinert H et al. (1996) Aconitase as iron-sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev 96, 2335–2374 [DOI] [PubMed] [Google Scholar]
- 4.Dailey HA et al. (1994) Human ferrochelatase is an iron-sulfur protein. Biochemistry 33, 403–407 [DOI] [PubMed] [Google Scholar]
- 5.Barton JK et al. (2019) Redox chemistry in the genome: emergence of the [4Fe4S] cofactor in repair and replication. Annu. Rev. Biochem 88, 163–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tse ECM et al. (2017) The oxidation state of [4Fe4S] clusters modulates the DNA-binding affinity of DNA repair proteins. J. Am. Chem. Soc 139, 12784–12792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiedorczuk K et al. (2016) Atomic structure of the entire mammalian mitochondrial complex I. Nature 538, 406–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dos Santos PC and Dean DR (2008) A newly discovered role for iron-sulfur clusters. Proc. Natl. Acad. Sci. U. S. A 105, 11589–11590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mancera-Martinez E et al. (2017) ABCE1: A special factor that orchestrates translation at the crossroad between recycling and initiation. RNA Biol 14, 1279–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Natale V and Raquer H (2017) Xeroderma pigmentosum-Cockayne syndrome complex. Orphanet. J. Rare Dis 12, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Che R et al. (2018) Multifaceted Fanconi anemia signaling. Trends Genet 34, 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rouault TA and Maio N (2017) Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem 292, 12744–12753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Landgraf BJ et al. (2016) Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem 85, 485–514 [DOI] [PubMed] [Google Scholar]
- 14.Hanzelmann P and Schindelin H (2006) Binding of 5’-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc. Natl. Acad. Sci. U. S. A 103, 6829–6834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarthy EL and Booker SJ (2017) Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science 358, 373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei FY et al. (2011) Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Invest 121, 3598–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei FY et al. (2015) Cdk5rap1-mediated 2-methylthio modification of mitochondrial tRNAs governs protein translation and contributes to myopathy in mice and humans. Cell Metab 21, 428–442 [DOI] [PubMed] [Google Scholar]
- 18.Dauden MI et al. (2019) Molecular basis of tRNA recognition by the Elongator complex. Sci. Adv 5, eaaw2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gari K et al. (2012) MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 337, 243–245 [DOI] [PubMed] [Google Scholar]
- 20.Stehling O et al. (2012) MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 337, 195–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben-Shimon L et al. (2018) Fe-S cluster coordination of the chromokinesin KIF4A alters its subcellular localization during mitosis. J. Cell Sci 131, jcs211433 [DOI] [PubMed] [Google Scholar]
- 22.Conlan AR et al. (2009) Crystal structure of Miner1: the redox-active 2Fe-2S protein causative in Wolfram Syndrome 2. J. Mol. Biol 392, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karmi O et al. (2018) The unique fold and lability of the [2Fe-2S] clusters of NEET proteins mediate their key functions in health and disease. J. Biol. Inorg. Chem 23, 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mozzillo E et al. (2014) A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2. BMC Med. Genet 15, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson DC et al. (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem 74, 247–281 [DOI] [PubMed] [Google Scholar]
- 26.Patra S and Barondeau DP (2019) Mechanism of activation of the human cysteine desulfurase complex by frataxin. Proc. Natl. Acad. Sci. U. S. A 116, 19421–19430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bridwell-Rabb J et al. (2014) Human frataxin activates Fe-S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 53, 4904–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parent A et al. (2015) Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun 6, 5686. [DOI] [PubMed] [Google Scholar]
- 29.Adam AC et al. (2006) The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J 25, 174–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiedemann N et al. (2006) Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J 25, 184–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Vranken JG et al. (2016) The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. eLife 5, e17828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boniecki MT et al. (2017) Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun 8, 1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cory SA et al. (2017) Structure of human Fe-S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP-ISD11 interactions. Proc. Natl. Acad. Sci. U. S. A 114, E5325–E5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox NG et al. (2019) Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun 10, 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majmudar JD et al. (2019) 4’-Phosphopantetheine and long acyl chain-dependent interactions are integral to human mitochondrial acyl carrier protein function. Medchemcomm 10, 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masud AJ et al. (2019) Mitochondrial acyl carrier protein (ACP) at the interface of metabolic state sensing and mitochondrial function. Biochim. Biophys. Acta Mol. Cell Res 1866, 118540. [DOI] [PubMed] [Google Scholar]
- 37.Puccio H et al. (2001) Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet 27, 181–186 [DOI] [PubMed] [Google Scholar]
- 38.Crooks DR et al. (2018) Acute loss of iron-sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem 293, 8297–8311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papa S et al. (2012) The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol 942, 3–37 [DOI] [PubMed] [Google Scholar]
- 40.Shi Y et al. (2012) Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim. Biophys. Acta 1823, 484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai K et al. (2017) Human mitochondrial ferredoxin 1 (FDX1) and ferredoxin 2 (FDX2) both bind cysteine desulfurase and donate electrons for iron-sulfur cluster biosynthesis. Biochemistry 56, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gervason S et al. (2019) Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun 10, 3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maio N and Rouault TA (2015) Iron-sulfur cluster biogenesis in mammalian cells: new insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 1853, 1493–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marinoni EN et al. (2012) (IscS-IscU)2 complex structures provide insights into Fe2S2 biogenesis and transfer. Angew. Chem. Int. Ed. Engl 51, 5439–5442 [DOI] [PubMed] [Google Scholar]
- 45.Yoon H et al. (2015) Turning Saccharomyces cerevisiae into a frataxin-independent organism. PLoS Genet 11, e1005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roche B et al. (2015) Turning Escherichia coli into a frataxin-dependent organism. PLoS Genet 11, e1005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindahl PA and Moore MJ (2016) Labile low-molecular-mass metal complexes in mitochondria: trials and tribulations of a burgeoning field. Biochemistry 55, 4140–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai K et al. (2018) Architectural features of human mitochondrial cysteine desulfurase complexes from crosslinking mass spectrometry and small-angle X-ray scattering. Structure 26, 1127–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voisine C et al. (2001) Jac1, a mitochondrial J-type chaperone, is involved in the biogenesis of Fe/S clusters in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A 98, 1483–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dutkiewicz R et al. (2003) Ssq1, a mitochondrial Hsp70 involved in iron-sulfur (Fe/S) center biogenesis. Similarities to and differences from its bacterial counterpart. J. Biol. Chem 278, 29719–29727 [DOI] [PubMed] [Google Scholar]
- 51.Vickery LE and Cupp-Vickery JR (2007) Molecular chaperones HscA/Ssq1 and HscB/Jac1 and their roles in iron-sulfur protein maturation. Crit. Rev. Biochem. Mol. Biol 42, 95–111 [DOI] [PubMed] [Google Scholar]
- 52.Uhrigshardt H et al. (2010) Characterization of the human HSC20, an unusual DnaJ type III protein, involved in iron-sulfur cluster biogenesis. Hum. Mol. Genet 19, 3816–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maio N et al. (2014) Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell Metab 19, 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kampinga HH and Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maio N et al. (2017) A single adaptable cochaperone-scaffold complex delivers nascent iron-sulfur clusters to mammalian respiratory chain complexes I-III. Cell Metab 25, 945–953 [DOI] [PubMed] [Google Scholar]
- 56.Maio N et al. (2016) Disease-causing SDHAF1 mutations impair transfer of Fe-S clusters to SDHB. Cell Metab 23, 292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muhlenhoff U et al. (2011) Specialized function of yeast Isa1 and Isa2 proteins in the maturation of mitochondrial [4Fe-4S] proteins. J. Biol. Chem 286, 41205–41216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sheftel AD et al. (2012) The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fe-4S] protein maturation. Mol. Biol. Cell 23, 1157–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beilschmidt LK et al. (2017) ISCA1 is essential for mitochondrial Fe4S4 biogenesis in vivo. Nat. Commun 8, 15124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nasta V et al. (2019) A pathway for assembling [4Fe-4S](2+) clusters in mitochondrial iron-sulfur protein biogenesis. FEBS J Published online November 14, 2019. 10.1111/febs.15140 [DOI] [PubMed] [Google Scholar]
- 61.Gourdoupis S et al. (2018) IBA57 recruits ISCA2 to form a [2Fe-2S] cluster-mediated complex. J. Am. Chem. Soc 140, 14401–14412 [DOI] [PubMed] [Google Scholar]
- 62.Nasta V et al. (2019) Structural properties of [2Fe-2S] ISCA2-IBA57: a complex of the mitochondrial iron-sulfur cluster assembly machinery. Sci. Rep 9, 18986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brancaccio D et al. (2014) Formation of [4Fe-4S] clusters in the mitochondrial iron-sulfur cluster assembly machinery. J. Am. Chem. Soc 136, 16240–16250 [DOI] [PubMed] [Google Scholar]
- 64.Brancaccio D et al. (2017) [4Fe-4S] cluster assembly in mitochondria and its impairment by copper. J. Am. Chem. Soc 139, 719–730 [DOI] [PubMed] [Google Scholar]
- 65.Cameron JM et al. (2011) Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am. J. Hum. Genet 89, 486–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Navarro-Sastre A et al. (2011) A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am. J. Hum. Genet 89, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Braymer JJ and Lill R (2017) Iron-sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem 292, 12754–12763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melber A et al. (2016) Role of Nfu1 and Bol3 in iron-sulfur cluster transfer to mitochondrial clients. eLife 5, e15991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Floyd BJ et al. (2016) Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell 63, 621–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uzarska MA et al. (2016) Mitochondrial Bol1 and Bol3 function as assembly factors for specific iron–sulfur proteins. eLife 5, e16673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishioka M et al. (2018) An infant case of diffuse cerebrospinal lesions and cardiomyopathy caused by a BOLA3 mutation. Brain and Development 40, 484–488 [DOI] [PubMed] [Google Scholar]
- 72.Yu Q et al. (2019) BOLA (BolA Family Member 3) deficiency controls endothelial metabolism and glycine homeostasis in pulmonary hypertension. Circulation 139, 2238–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ajit Bolar N et al. (2013) Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum. Mol. Genet 22, 2590–2602 [DOI] [PubMed] [Google Scholar]
- 74.Al-Hassnan ZN et al. (2015) ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J. Med. Genet 52, 186–194 [DOI] [PubMed] [Google Scholar]
- 75.Shukla A et al. (2017) Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J. Hum. Genet 62, 723–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rouault TA (2019) The indispensable role of mammalian iron sulfur proteins in function and regulation of multiple diverse metabolic pathways. Biometals 32, 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kispal G et al. (1999) The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J 18, 3981–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim KS et al. (2018) Cytosolic HSC20 integrates de novo iron-sulfur cluster biogenesis with the CIAO1-mediated transfer to recipients. Hum. Mol. Genet 27, 837–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Land T and Rouault TA (1998) Targeting of a human iron-sulfur cluster assembly enzyme, nifs, to different subcellular compartments is regulated through alternative AUG utilization. Mol. Cell 2, 807–815 [DOI] [PubMed] [Google Scholar]
- 80.Marelja Z et al. (2013) The L-cysteine desulfurase NFS1 is localized in the cytosol where it provides the sulfur for molybdenum cofactor biosynthesis in humans. PLoS One 8, e60869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marelja Z et al. (2008) A novel role for human Nfs1 in the cytoplasm: Nfs1 acts as a sulfur donor for MOCS3, a protein involved in molybdenum cofactor biosynthesis. J. Biol. Chem 283, 25178–25185 [DOI] [PubMed] [Google Scholar]
- 82.Shi Y et al. (2009) Human ISD11 is essential for both iron-sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum. Mol. Genet 18, 3014–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li K et al. (2006) Roles of the mammalian cytosolic cysteine desulfurase, ISCS, and scaffold protein, ISCU, in iron-sulfur cluster assembly. J. Biol. Chem 281, 12344–12351 [DOI] [PubMed] [Google Scholar]
- 84.Tong WH and Rouault TA (2006) Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab 3, 199–210 [DOI] [PubMed] [Google Scholar]
- 85.Tong WH et al. (2003) Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proc. Natl. Acad. Sci. U. S. A 100, 9762–9767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thul PJ et al. (2017) A subcellular map of the human proteome. Science 356, eaal3321 [DOI] [PubMed] [Google Scholar]
- 87.Vashisht AA et al. (2009) Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mayank AK et al. (2019) An oxygen-dependent interaction between FBXL5 and the CIA-targeting complex regulates iron homeostasis. Mol. Cell 75, 382–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Belbellaa B et al. (2019) Correction of half the cardiomyocytes fully rescue Friedreich ataxia mitochondrial cardiomyopathy through cell-autonomous mechanisms. Hum. Mol. Genet 28, 1274–1285 [DOI] [PubMed] [Google Scholar]
- 90.Piguet F et al. (2018) Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of Friedreich ataxia. Mol. Ther 26, 1940–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wilson RB (2012) Therapeutic developments in Friedreich ataxia. J. Child Neurol 27, 1212–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perdomini M et al. (2014) Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med 20, 542–547 [DOI] [PubMed] [Google Scholar]
- 93.Chandramouli K et al. (2007) Formation and properties of [4Fe-4S] clusters on the IscU scaffold protein. Biochemistry 46, 6804–6811 [DOI] [PubMed] [Google Scholar]
- 94.Camaschella C et al. (2007) The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 110, 1353–1358 [DOI] [PubMed] [Google Scholar]
- 95.Koeppen AH (2011) Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J. Neurol. Sci 303, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kollberg G et al. (2009) Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain 132, 2170–2179 [DOI] [PubMed] [Google Scholar]
- 97.Liu G et al. (2014) Heterozygous missense mutations in the GLRX5 gene cause sideroblastic anemia in a Chinese patient. Blood 124, 2750–2751 [DOI] [PubMed] [Google Scholar]
- 98.Mochel F et al. (2008) Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet 82, 652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ye H et al. (2010) Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Invest 120, 1749–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li K et al. (2008) Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum. Mol. Genet 17, 2265–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Outten CE and Albetel AN (2013) Iron sensing and regulation in Saccharomyces cerevisiae: ironing out the mechanistic details. Curr. Opin. Microbiol 16, 662–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miao R et al. (2011) Biophysical investigation of the iron in Aft1–1(up) and Gal-YAH1 Saccharomyces cerevisiae. Biochemistry 50, 2660–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huang ML et al. (2009) Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. U. S. A 106, 16381–16386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Crooks DR et al. (2014) Elevated FGF21 secretion, PGC-1alpha and ketogenic enzyme expression are hallmarks of iron-sulfur cluster depletion in human skeletal muscle. Hum. Mol. Genet 23, 24–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ducamp S and Fleming MD (2019) The molecular genetics of sideroblastic anemia. Blood 133, 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Farhan SM et al. (2014) Exome sequencing identifies NFS1 deficiency in a novel Fe-S cluster disease, infantile mitochondrial complex II/III deficiency. Mol. Genet. Genomic Med 2, 73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lim SC et al. (2013) Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum. Mol. Genet 22, 4460–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Coelho MP et al. (2019) Iron-sulfur cluster ISD11 deficiency (LYRM4 gene) presenting as cardiorespiratory arrest and 3-methylglutaconic aciduria. JIMD Rep 49, 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Clay A et al. (2019) New developments in pharmacotherapy for Friedreich ataxia. Expert. Opin. Pharmacother 20, 1855–1867 [DOI] [PubMed] [Google Scholar]
- 110.Delatycki MB and Bidichandani SI (2019) Friedreich ataxia: pathogenesis and implications for therapies. Neurobiol. Dis 132, 104606. [DOI] [PubMed] [Google Scholar]
- 111.Legati A et al. (2017) A novel de novo dominant mutation in ISCU associated with mitochondrial myopathy. J. Med. Genet 54, 815–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Crispin A et al. (2017) Hscb, a mitochondrial iron-sulfur cluster assembly co-chaperone, is a novel candidate gene for congenital sideroblastic anemia. Blood 130, 79 [Google Scholar]
- 113.Schmitz-Abe K et al. (2015) Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9. Blood 126, 2734–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Culley MK et al. (2019) NFU1, iron-sulfur biogenesis, and pulmonary arterial hypertension: a (metabolic) shift in our thinking. Am. J. Respir. Cell Mol. Biol 62, 136–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baker PR 2nd et al. (2014) Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain 137, 366–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Spiegel R et al. (2014) Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur. J. Hum. Genet 22, 902–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gurgel-Giannetti J et al. (2018) A novel complex neurological phenotype due to a homozygous mutation in FDX2. Brain 141, 2289–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Paul A et al. (2017) FDXR mutations cause sensorial neuropathies and expand the spectrum of mitochondrial Fe-S-synthesis diseases. Am. J. Hum. Genet 101, 630–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Alfadhel M (2019) Multiple mitochondrial dysfunctions syndrome 4 due to ISCA2 gene defects: a review. Child Neurol. Open 6 2329048X19847377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Debray FG et al. (2015) Mutation of the iron-sulfur cluster assembly gene IBA57 causes fatal infantile leukodystrophy. J. Inherit. Metab. Dis 38, 1147–1153 [DOI] [PubMed] [Google Scholar]
- 121.Ishiyama A et al. (2017) IBA57 mutations abrogate iron-sulfur cluster assembly leading to cavitating leukoencephalopathy. Neurol. Genet 3, e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu M et al. (2018) Phenotypic spectrum of mutations in IBA57, a candidate gene for cavitating leukoencephalopathy. Clin. Genet 93, 235–241 [DOI] [PubMed] [Google Scholar]
- 123.Torraco A et al. (2017) Novel mutations in IBA57 are associated with leukodystrophy and variable clinical phenotypes. J. Neurol 264, 102–111 [DOI] [PubMed] [Google Scholar]
- 124.Giannuzzi G et al. (2019) The human-specific BOLA2 duplication modifies iron homeostasis and anemia predisposition in chromosome 16p11.2 autism individuals. Am. J. Hum. Genet 105, 947–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Espinoza-Lewis RA et al. (2017) Poly(C)-binding protein 1 (Pcbp1) regulates skeletal muscle differentiation by modulating microRNA processing in myoblasts. J. Biol. Chem 292, 9540–9550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Patel SJ et al. (2019) A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat. Chem. Biol 15, 872–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vitale G et al. (1996) NBP35 encodes an essential and evolutionary conserved protein in Saccharomyces cerevisiae with homology to a superfamily of bacterial ATPases. Gene 178, 97–106 [DOI] [PubMed] [Google Scholar]
- 128.Roy A et al. (2003) A novel eukaryotic factor for cytosolic Fe-S cluster assembly. EMBO J 22, 4826–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Balk J et al. (2005) The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron-sulfur protein assembly. Mol. Cell. Biol 25, 10833–10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Song D and Lee FS (2011) Mouse knock-out of IOP1 protein reveals its essential role in mammalian cytosolic iron-sulfur protein biogenesis. J. Biol. Chem 286, 15797–15805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shibayama H et al. (2004) Identification of a cytokine-induced antiapoptotic molecule anamorsin essential for definitive hematopoiesis. J. Exp. Med 199, 581–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tanimura A et al. (2014) The anti-apoptotic gene Anamorsin is essential for both autonomous and extrinsic regulation of murine fetal liver hematopoiesis. Exp. Hematol 42, 410–422 [DOI] [PubMed] [Google Scholar]
- 133.Camponeschi F et al. (2017) Anamorsin/Ndor1 complex reduces [2Fe-2S]-MitoNEET via a transient protein-protein interaction. J. Am. Chem. Soc 139, 9479–9482 [DOI] [PubMed] [Google Scholar]
- 134.Cha H et al. (2008) PICOT is a critical regulator of cardiac hypertrophy and cardiomyocyte contractility. J. Mol. Cell. Cardiol 45, 796–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Banci L et al. (2015) Elucidating the molecular function of human BOLA2 in GRX3-dependent anamorsin maturation pathway. J. Am. Chem. Soc 137, 16133–16143 [DOI] [PubMed] [Google Scholar]
- 136.Frey AG et al. (2016) A glutaredoxin.BolA complex serves as an iron-sulfur cluster chaperone for the cytosolic cluster assembly machinery. J. Biol. Chem 291, 22344–22356 [DOI] [PMC free article] [PubMed] [Google Scholar]