

Abstract
The widespread emergence of antibiotic resistance in pathogens necessitates the development of antibacterial agents inhibiting underexplored targets in bacterial metabolism. One such target is MraY (phospho-MurNAc-pentapeptide translocase), an essential integral membrane enzyme that catalyzes the first committed step of peptidoglycan biosynthesis. MraY has long been considered a promising candidate for antibiotic development in part because it is the target of five classes of naturally occurring nucleoside inhibitors with potent in vivo and in vitro antibacterial activity. Although these inhibitors each have a nucleoside moiety, they vary dramatically in their core structures, and they have different activity properties. Until recently, the structural basis of MraY inhibition was poorly understood. Several recent structures of MraY and its human paralog, GPT (GlcNAc-1-P-transferase), have provided insights into MraY inhibition that are consistent with known inhibitor activity data and can inform rational drug design for this important antibiotic target.
Keywords: Bacterial cell wall synthesis, antibiotics, membrane enzyme, nucleoside inhibitors, MraY
Introduction
The widespread incidence of drug-resistant infections worldwide has raised concerns that the global community will enter a “pre-antibiotic” era in which life-threatening communicable diseases are untreatable with the current arsenal of antimicrobial agents. The predominant technical strategy to address this global health crisis has been to modify existing antibiotics, leading to modest extension of their use; however, the Holy Grail of antibiotic development is to identify entirely new classes of compounds with novel mechanisms of action that circumvent targets already implicated in resistance mechanisms. The central difficulty of this approach is that only a handful of bacterial metabolic processes are vulnerable to chemotherapeutic intervention [1]. One of these is the formation of the bacterial cell wall, of which peptidoglycan is the major constituent. Peptidoglycan biosynthesis consists of three stages based on where they physically take place in the cell: the cytoplasmic, the membrane-associated, and the extracellular/periplasmic steps. While enzymes in the cytoplasmic and extracellular stages of peptidoglycan biosynthesis have been extensively targeted, with the penicillin-binding proteins being the most widely studied and exploited for antibiotic development, enzymes in the membrane-associated step have been underexplored, primarily due to technical difficulties in obtaining stable preparations of these proteins in sufficient quantities for drug design studies.
Phospho-MurNAc-pentapeptide translocase (MraY) is an integral membrane enzyme essential for bacterial survival, which catalyzes the first membrane-associated and committed step of peptidoglycan formation. For several decades, MraY has been considered a promising target in peptidoglycan biosynthesis for antibiotic development because it is inhibited by five classes of naturally-occurring nucleoside inhibitors isolated from Streptomyces species with promising activity against pathogenic bacteria: the muraymycins[2, 3], tunicamycins, mureidomycins[4–6], capuramycins[7–12], and liposidomycins/caprazamycins [13–15]. MraY transfers phospho-MurNAc-pentapeptide from the hydrophilic substrate uridine diphosphate-MurNAc-pentapeptide (UM5A), to the lipid carrier undecaprenyl phosphate (C55-P) in the presence of a Mg2+ cofactor; the resulting product is Lipid I, a key intermediate in peptidoglycan biosynthesis (Figure 1a). Despite the common uridine substructure nucleoside MraY inhibitors share, they each exhibit differing mechanisms of action, inhibitor kinetics, antibacterial activity, and structure-activity-relationship (SAR) profiles. Muraymycin [16] and tunicamycin [17, 18] are competitive for the hydrophilic substrate, UM5A; liposidomycin [17] and tunicamycin [18] are competitive for the lipid carrier substrate, C55-P; capuramycin is mixed type and noncompetitive for UM5A and C55-P, respectively [18] and mureidomycin is competitive for both UM5A and C55-P [17]. Liposidomycin and mureidomycin are reversible slow binding inhibitors of MraY [17, 19] while capuramycin does not exhibit time-dependent inhibition of MraY [18]. Some nucleoside natural product inhibitors of MraY are broadly active against various pathogens, such as the muraymycins and liposidomycins/caprazamycins, which demonstrate potent antibacterial activity against a variety of Gram-negative and Gram-positive bacteria, mycobacteria, and various drug-resistant strains, including MRSA and VRE [16, 20–25]. Mureidomycin and its analogs appear to have a narrower spectrum of activity, primarily against Pseudomonas species [5, 6], while the capuramycins are particularly effective against mycobacteria [9, 10, 26–29]. Recently SQ641, a capuramycin analogue was shown to be effective in a murine model of Clostridium difficile infection[11]. The tunicamycins inhibit not only MraY, but also its eukaryotic homolog GPT (GlcNAc-1-P-transferase; the human DPAGT1 gene or ALG7 in yeast), which catalyzes the committed step of N-linked glycosylation, leading to the Unfolded Protein Response [30–34]. As a result, tunicamycin is cytotoxic to human cells, while muraymycin [16], liposidomycin [35], mureidomycin [36], and capuramycin [18] are non-toxic and are selective for bacterial MraY.
Figure 1.
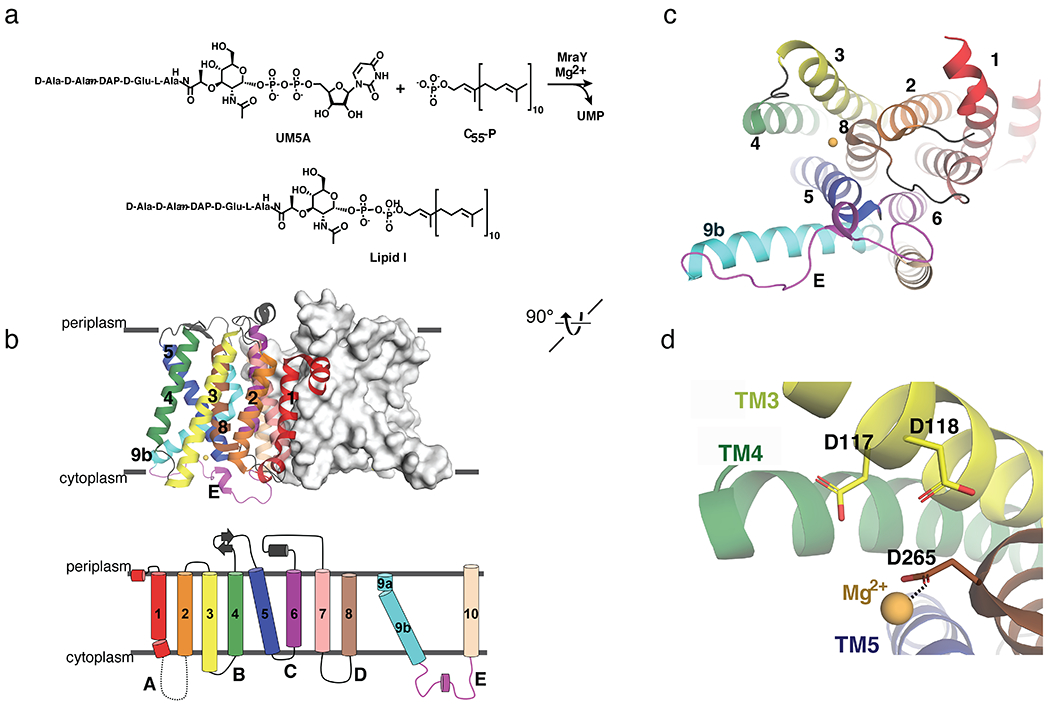
Structural architecture and biochemical function of MraY. (a) Scheme of chemical reaction catalyzed by MraY. (b) Top, structure of MraYAA, viewed from the membrane. One protomer is shown as surface representation and the other as cartoon representation (PDB ID: 4J72). The TMs and loops are colored distinctly, corresponding to the topology diagram below. (c) Cytoplasmic view of MraYAA, rotated 90° relative to the orientation in (b). One protomer is shown for simplicity. TMs and Loops are labelled throughout. The color coding is the same as in (b). The magnesium ion is shown in yellow-orange. (d) Zoomed-in view of the magnesium binding site in MraYAA with the orientation the same as in (c).
Since the discovery of these nucleoside natural product inhibitors several decades ago, major efforts within academia and industry have been directed toward synthesizing analogs and carrying out structure-activity relationship (SAR) studies without a clear understanding of the protein-inhibitor interactions that govern the inhibition of MraY. This knowledge gap has hampered the development of nucleoside MraY inhibitors into antibiotics that can be used to treat pathogenic bacterial infections.
Recent crystal structures of MraY bound to representative members of each inhibitor class, muraymycin [37], tunicamycin [38], lipodisomycin/caprazamycin, mureidomycin, and capuramycin [39], reveal critical cryptic binding sites in the shallow, surface exposed binding site of MraY that can be exploited for rational inhibitor design. In addition, crystal structures of human MraY paralog GPT (GlcNAc-1-P-transferase) bound to tunicamycin [40, 41], have allowed for structural comparisons between the two orthologs that reveal the key to selectively targeting MraY for antibiotic development. Here we summarize these recent works, which have advanced our structural and functional understanding of MraY and GPT inhibition and can be leveraged for antibiotic development.
Structural architecture and inhibitor dependent conformational changes of MraY
The apoenzyme structure of MraY [42] constituted a major conceptual and technical advance in the field of MraY-targeted antibiotic design. Previous efforts to study MraY had been hampered by technical difficulties in generating and handling large quantities of stable MraY. This challenge was overcome by identifying a stable ortholog of the enzyme from Aquifex aeolicus (MraYAA) that is codon-optimized for recombinant expression in E. coli [42]. The apoenzyme structure provides fundamental information about the overall architecture of the enzyme, its active site, and the location of the Mg2+ cofactor. Supported by cross-linking studies, the structure of MraYAA demonstrated this enzyme exists as a homodimer in the membrane and that each protomer contains 10 transmembrane helices (TM 1-10) and five cytoplasmic loops (Loop A-E) (Figure 1b). On the cytoplasmic face of MraY there is a large hydrophilic cleft, which is formed by TMs 5, 8, and 9b and Loops C, D, and E, which is highly conserved among Gram-positive and Gram-negative bacteria (Figure 1c). The conserved hydrophilic cleft consists of with 34 invariant amino acid residues, including the catalytically-critical DDD motif [43] (D117, D118, and D265 in MraYAA). D265 was found to coordinate the Mg2+ cofactor via anomalous scattering experiments [42] (Figure 1d). Although crystal structures of MraY in complex with its natural substrates are not yet available, it has been proposed that this cleft is the binding site of the hydrophilic substrate, UM5A. The Mg2+ cofactor coordinated by D265 may interact with the diphosphate of UM5A, thereby orienting the substrate for catalysis. Furthermore, TM9b and LoopE, which are part of this hydrophilic region, have been predicted to play a role in the recognition of sugar moieties, such as the MurNAc in UM5A [44].
The first reported structure of MraY in complex with a nucleoside inhibitor was MraYAA bound to muraymycin D2, a member of the muraymycin class [37]. Comparison of the apoenzyme and muraymycin D2-bound structures of MraYAA demonstrate dramatic conformational changes occur in the hydrophilic cleft of the enzyme upon nucleoside inhibitor binding [37] . Large movements in Loops A, C, D, and E, as well as TM9b are observed. TM5 helix partially unwinds at the C-terminus and a helical segment is introduced in Loop E [37] (Figure 2a–b). A similar inhibitor-bound conformation in the hydrophobic cleft was observed in later structures of MraY from Clostridium boltea (MraYCB) bound to tunicamycin [38] (Figure 2c), as well as MraYAA bound to carbacaprazamycin (a member of the liposidomycin/caprazamycin class), capuramycin, and 3’-hydroxymureidomycin A [39] (Figures 2d–f). Overall, the inhibitor-bound structural conformations of MraY are more similar to one another than they are to the unliganded structure; however, each complex exhibits variation in the degree of TM9b bending and in the conformation of Loop E (Figure 2g). Major differences among the inhibitor-bound structures are observed in the binding sites accessed by each of the five MraY inhibitors. These sites are cryptic in that they are not exposed in or appreciable from the apoenzyme structure. We describe each of these cryptic druggable sites, both unique and overlapping among the inhibitor-bound MraY structures, in the following sections.
Figure 2.
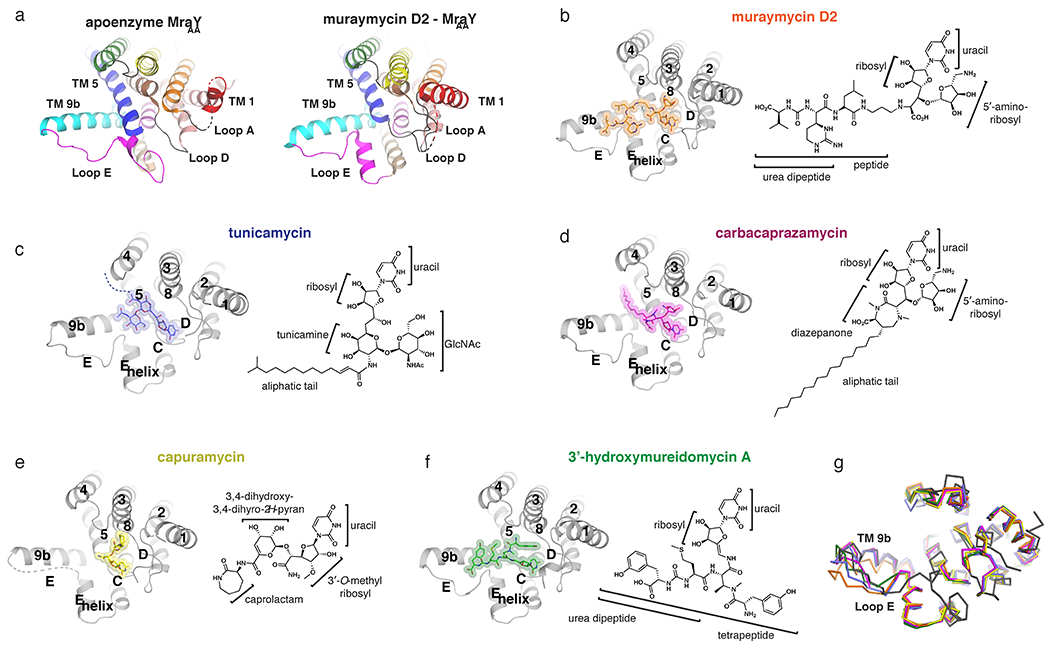
Inhibitor-bound structures of MraY. (a) Comparison of the apoenzyme (PDB ID: 4J72) and muraymycin D2-bound (PDB ID: 5CKR) structures of MraYAA, viewed from the cytoplasm. Color coding of the TMs and Loops is the same as in Figure 1. In the muraymycin D2-bound structure, the inhibitor is removed for simplicity. (b-f) Structures of MraY bound to each inhibitor, viewed from the cytoplasm, with TMs and Loops labelled throughout: muraymycin D2- MraYAA (orange), tunicamycin- MraYCB (PDB ID: 5JNQ; blue), carbacaprazamycin- MraYAA (PDB ID: 6OYH; magenta), capuramycin- MraYAA (PDB ID: 6OYZ; yellow), and 3′-hydroxymureidomycin A- MraYAA (PDB ID: 6OZ6; green). Next to each inhibitor-bound MraY structure is the corresponding inhibitor chemical structure, with the moieties and substructures labelled. (g) Structural superposition of apoenzyme MraYAA (dark grey), muraymycin D2-MraYAA (orange), tunicamycin- MraYCB (blue), carbacaprazamycin- MraYAA (magenta), capuramycin- MraYAA (yellow), and 3’-hydroxymureidomycin A- MraYAA (green).
The uridine binding pocket is present in each MraY-inhibitor complex.
Upon inhibitor binding, a pocket forms in MraY that binds to the uridine moiety common to all nucleoside natural product inhibitors of this enzyme. The uracil portion of the uridine moiety engages in an extensive hydrogen-bonding network involving amino acid residues in Loop C, including interactions with the backbone of G194, L195 and the side chain of D196 (MraYAA amino acid numbering throughout unless otherwise noted). In addition, the uracil forms a hydrogen bond with residues in Loop D, such as K70 (not resolved in the capuramycin-bound MraYAA structure) and a π-π stacking interaction with F262 (Figure 3a). The uracil moiety makes additional contact with D193 and N255 in some of the ligand-bound structures MraY. Mutagenesis studies of MraYAA demonstrate the importance of this stacking interaction; both the inhibitor activity and binding of muraymycin D2 substantially decrease when F262 is mutated to an alanine, but less so when mutated to another aromatic amino acid residue, such as tyrosine [37]. The key residues that form the uracil binding pocket of MraY (K70, G194, L195, D196, and F262) are likely also involved in binding the uracil moiety of the natural substrate of this enzyme, UM5A. The uridine pocket in MraY is the most well defined and enclosed pocket in the active site of MraY. Even so, it is relatively accommodating to modifications on and spatial positioning of the uracil moiety. Structural overlay of the five inhibitor-bound structures of MraY demonstrates that there is some difference in the positioning of the uracil, with the uracil moiety of mureidomycin and capuramycin occupying positions that diverge from that of the uracil moieties of the other nucleoside inhibitors (Figure 3b). Two naturally occurring analogs of mureidomycin, B and D, contain a dihydrouracil moiety instead of uracil [45, 46] and they exhibit only slightly lower biological activity profiles than the analogous uracil-containing compounds [5, 6]. However, major modifications of the uracil moiety, such as methylation, halogenation, or other bulky functionalization, in the mureidomycins or muraymycins, result in a near complete loss of inhibitor activity [47–49].
Figure 3.
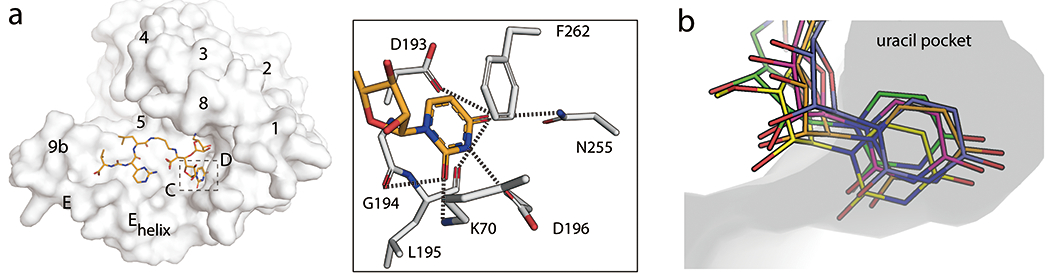
The uridine binding pocket in MraY. (a) Left, Surface representation of MraYAA protomer bound to muraymycin D2 viewed from the cytoplasmic side (PDB ID: 5CKR). The uridine binding pocket is highlighted with dashed lines. TMs and Loops are labelled. Right, interactions between the uridine moiety in muraymycin D2 and MraYAA. (b) Structural overlay of MraY bound to muraymycin D2 (PDB ID: 5CKR; orange), tunicamycin (PDB ID: 5JNQ; blue), carbacaprazamycin (PDB ID: 6OYH; magenta), capuramycin (PDB ID: 6OYZ; yellow), and 3’-hydroxymureidomycin A (PDB ID: 6OZ6; green), focusing on the uridine binding pocket. The ribose moiety is exposed to the cytosol.
The ribosyl group of the uridine moiety (or 3’O-methylated ribosyl group, as is found in capuramycin) assumes a very similar position in each inhibitor-MraY complex. The hydroxyls on the ribosyl moiety in each inhibitor appear to be mostly exposed to the solvent, making no apparent interactions with the enzyme (Figure 3b). As a result, there is some spatial tolerance for modifications at the ribosyl hydroxyl groups, which could introduce favorable biochemical properties into MraY-targeted nucleoside inhibitors. For example, it was shown that capuramycin derivatives with 2’O-alkyl or 2’O-alkoxycarbonyl functional groups of varying chain lengths remained active against MraY and exhibited improved minimum inhibitory concentration (MIC) values against various mycobacterial species [27, 28]. The most optimal capuramycin derivative improved MIC by >120 fold, presumably due to enhanced ability to penetrate the thick and highly hydrophobic cell wall of mycobacteria. Also, removal of 2’ hydroxyl group does not affect the activity of muraymycin significantly [49]. Rather than make specific contacts with the protein, the stereochemistry of the ribosyl moiety appears to direct each inhibitor to additional binding sites on the cytoplasmic face of MraY. This structural observation is supported by SAR studies showing that the stereocenter at the 5’ position of the ribosyl moiety must be (S) for efficient inhibitory activity of MraY within the 5’-aminoribose nucleoside core shared by the muraymycins and liposidomycins [50]. Taken together, we predict that the ribosyl moiety could be replaced by another cyclic moiety provided that it mimics the geometry assumed by the ribosyl group.
Inhibitor interactions at the uridine-adjacent pocket are important for MraY selectivity.
Next to the uridine binding pocket in MraY is a second binding site, called the uridine-adjacent site [39], which comprises of amino acid residues T75, N190, D193, and G264 (Figure 4). The shallow pocket formed by these residues is important for nucleoside natural product binding, as it can accommodate a diverse array of chemical structures. Despite its capacity to recognize a variety of pharmacophores, the residues comprising the uridine-adjacent pocket remain static in each inhibitor-bound structure.
Figure 4.
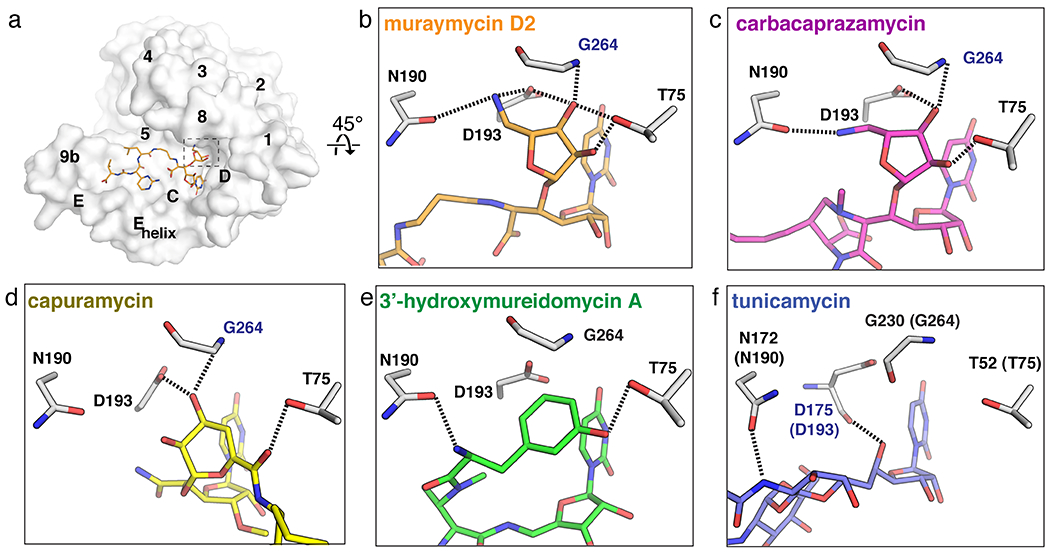
The uridine-adjacent pocket in MraY. (a) Surface representation of the MraYAA protomer bound to muraymycin D2 (cytoplasmic side). The uridine-adjacent binding pocket is hightlighted with dashed lines. (b)-(e) The uridine-adjacent pocket in each inhibitor-bound structure, oriented 45° relative to the structure in (a). Residue numbering is for MraYAA, except for in panel (f), which shows numbering for MraYCB with numbering for MraYAA in brackets. Residues labelled in blue form backbone interactions with the ligand. Muraymycin D2 PDB ID: 5CKR (orange); tunicamycin PDB ID: 5JNQ (blue); carbacaprazamycin PDB ID: 6OYH (magenta); capuramycin PDB ID: 6OYZ (yellow); 3’-hydroxymureidomycin A PDB ID: 6OZ6 (green).
The uridine-adjacent pocket in MraY binds the 5’-aminoribose moiety found in carbacaprazamycin and muraymycin D2 (Figure 4b–c). Both SAR and mutagenesis studies suggest that the amino group in this moiety forms a critically important interaction with D193. Near complete abrogation of inhibitor activity is observed if this interaction is disrupted, as seen in enzymatic [37, 51] and direct binding assays [37]. In the capuramycin-bound MraYAA structure, a hydroxyl group in the 3,4-dihydroxy-3,4-dihyro-2H-pyran moiety interacts with the uridine-adjacent pocket by engaging in a hydrogen-bonding interaction with D193 and the backbone amino group of G264 (Figure 4d). Replacing this hydroxyl group with a hydrogen leads to a ten-fold decrease in MraY inhibition by capuramycin [18]. The meta-tyrosine moiety of 3’-hydroxymureidomycin A interacts with N190 and T75 in the uridine-adjacent pocket (Figure 4e). The pacidamycins and napsamycins, which are structurally similar to the mureidomycins, each contain different amino acid residues in place of the meta-tyrosine moiety [52, 53]. It is likely that the uridine-adjacent site accommodates each of the various amino acid residues observed among the mureidomycins, napsamycins, and pacidamycins.
Interestingly, tunicamycin makes limited binding interactions at the uridine-adjacent pocket in MraY and instead forms unique hydrogen bonds not observed among the other nucleoside inhibitors (Figure 4f, Figure 5). For example, the tunicamine sugar moiety of tunicamycin interacts with residues outside the uridine-adjacent site, including K111 (K133 in MraYAA) and a backbone interaction with F173 in MraYCB (L191 in MraYAA) (Figure 5). Tunicamycin is the only known class of promiscuous MraY inhibitors, with activity against GPT, a human paralog of MraY. A uridine-adjacent pocket is not present GPT [40, 41]. The analogous site is partially collapsed in GPT and the sequence of the binding pocket is not conserved; T52 and D175 in MraYCB (T75 and D193 in MraYAA) are E56 and A188 in human GPT. Inhibitors that can form interactions at the uridine-adjacent site on MraY likely have an improved capacity to selectively target bacterial MraY over GPT. Critically, this pocket is also very druggable by surprisingly diverse chemical moieties, so strategies to target this pocket can be quite amenable to various design and synthesis ideas. The uridine-adjacent pocket is not required for MraY inhibition, as evidenced by muraymycin analogs lacking a 5’-aminoribose moiety that maintain their inhibitory activity [48, 54]. However, targeting the uridine-adjacent site appears to lead to improved inhibitor affinity and selectivity.
Figure 5.
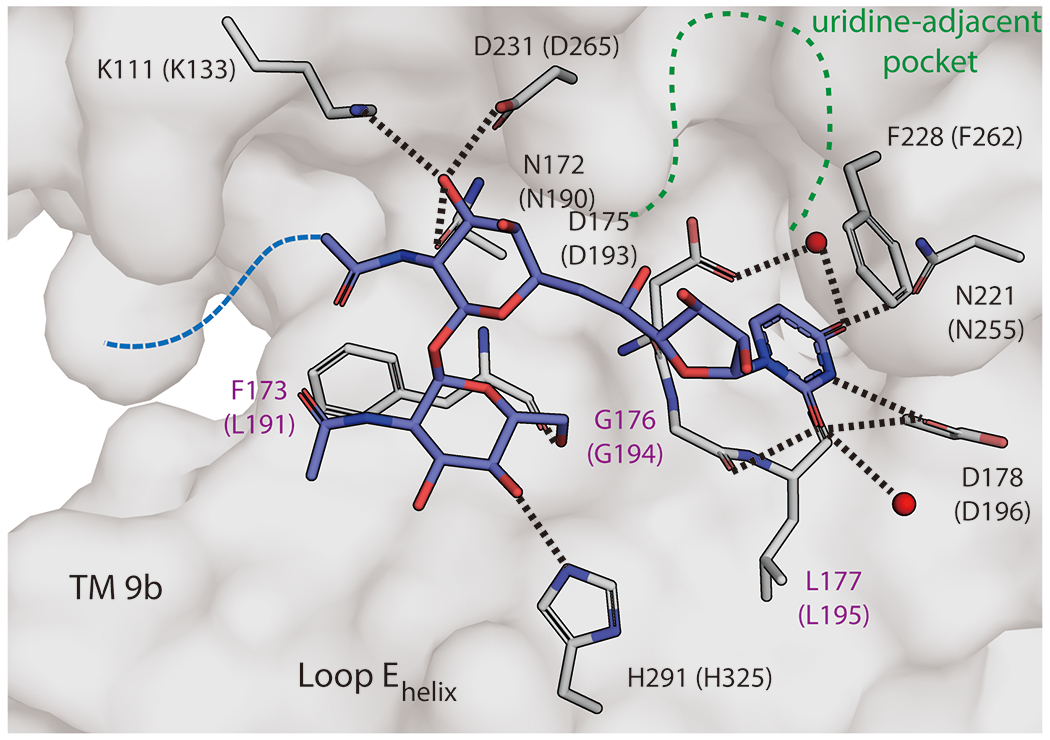
Detailed view of tunicamycin binding in MraY. Zoomed-in view of the tunicamycin binding site in MraYCB (PDB ID: 5JNQ). The residues forming interactions with tunicamycin (blue) are shown in stick representation (numbering for MraYCB, with numbering for MraYAA in brackets). Residues labelled in purple are forming backbone interactions with tunicamyin. Waters are shown as red spheres. The uridine-adjacent site is delineated with dashed green lines. The aliphatic tail of tunicamycin was disordered and is represented by a dashed blue line.
Nucleoside inhibitors make differing interactions with TM9b, Loop E, and the Loop E helix.
The most malleable region of MraY is comprised of TM9b, Loop E and the Loop E helix, which forms a flat, surface-exposed binding site in the inhibitor-bound conformation (Figure 6a). MraY inhibitors make varying contacts with residues Q305, A321, H324, and H325 at this site. Among the five inhibitor classes, muraymycin D2 and 3’-hydroxymureidomycin A form the greatest number of hydrogen bonds in the TM 9b/LoopE pocket (Figure 6b–c). The urea and carboxylate groups common to muraymycin D2 and 3’-hydroxymureidomycin A make contacts with Q305 and A321 in TM9b. These two inhibitors also form an additional hydrogen bond with the Loop E helix via H325, as do carbacaprazamycin and tunicamycin (Figure 6d–e). In muraymycin D2 binding, a water-mediated hydrogen-bonding network forms with H324 and H325, and the L-epi-capreomycidine moiety of the inhibitor packs against H325 as well (Figure 6b).
Figure 6.
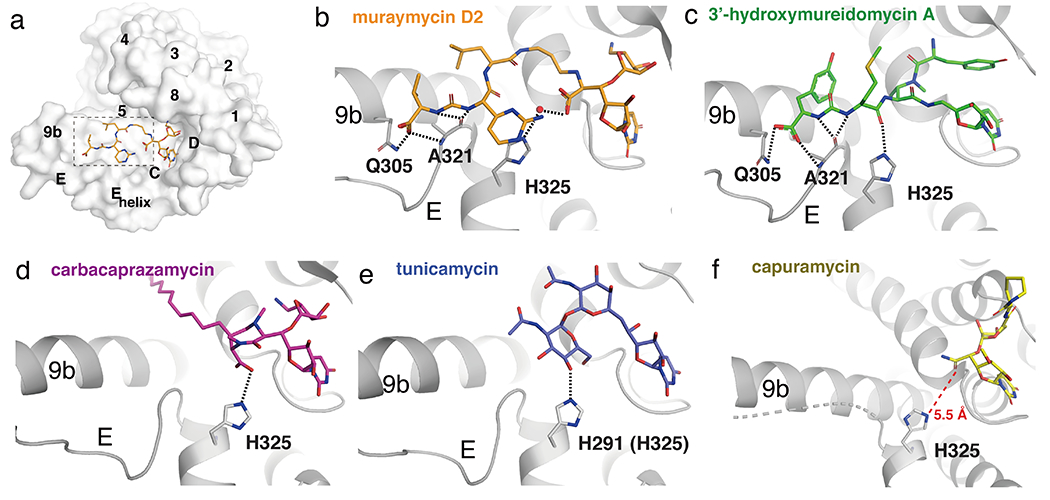
The TM 9b/Loop E binding site in MraY. (a) Surface representation of MraYAA protomer bound to muraymycin D2 (cytoplasmic side). The TM 9b/Loop E binding pocket is highlighted with dashed lines. (b) – (e) Interactions each inhibitor makes in the TM 9b/Loop E pocket, oriented as shown in (a). Residue numbering is for MraYAA, except for in panel (e), which shows numbering for MraYCB with numbering for MraYAA in brackets. Muraymycin D2 PDB ID: 5CKR (orange); 3’-hydroxymureidomycin A PDB ID: 6OZ6 (green); carbacaprazamycin PDB ID: 6OYH (magenta); tunicamycin PDB ID: 5JNQ (blue); capuramycin PDB ID: 6OYZ (yellow).
The core of capuramycin orients away from the Loop E helix and does not form a hydrogen bond with H325, as do the other MraY natural product inhibitors [39]. Loop E in the capuramycin-MraY complex structure is disordered, likely because the inhibitor does not interact with and stabilize it. The carboxamide group of capuramycin is 5.5 Å away from H325 [39] (Figure 6f) and could be modified to pick up a hydrogen bond with this residue, but to our knowledge, SAR has not been explored for this part of the molecule. The carboxamide moiety is an attractive site on capuramycin to functionalize, as extending this inhibitor’s interactions to the Loop E helix and TM9b may improve its potency.
The caprolactam pocket in MraY is uniquely accessed by capuramycin.
Because capuramycin assumes an unusual binding pose away from TM9b and the Loop E helix, it instead occupies a cryptic binding pocket in MraY that is not accessed by other MraY inhibitors. The caprolactam moiety of capuramycin binds to a mostly hydrophobic, surface-exposed pocket on the shallow MraY inhibitor binding site (Figure 7a). The role of the caprolactam moiety of capuramycin has been extensively interrogated via SAR studies. Capuramycin derivatives with a methyl group or alkyl chain in place of the caprolactam are substantially less active than the parent compound [55] and a dramatic loss of inhibitor activity is also observed when the caprolactam is alkylated [26]. In contrast, replacement of the caprolactam with cyclic moieties results in derivatives that are comparably active to the parent compound [26]. The structure of capuramycin bound to MraY is consistent with these SAR trends because the caprolactam pocket is a shallow, hydrophobic groove that is accommodating to small cyclic moieties; long or short alkyl groups would not efficiently fill the pocket (Figure 7b). Part of the caprolactam binding site and some nearby residues are very conserved among MraY orthologs, however, some adjacent residues are not as highly conserved. Therefore, functionalizing the caprolactam or a cyclic moiety in its place to capture interactions with nearby residues outside the caprolactam pocket could be a viable strategy to design capuramycin analogs with species-specific activity.
Figure 7.
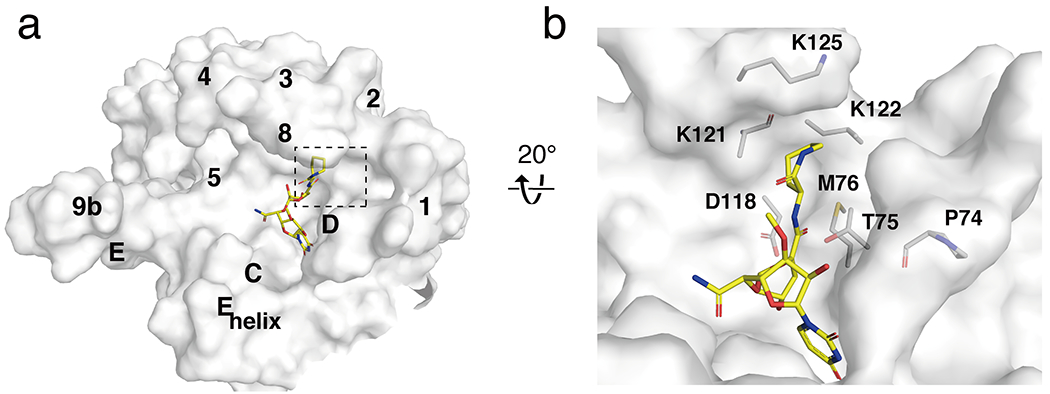
The caprolactam binding pocket in MraY. (a) Surface representation of MraYAA protomer bound to capuramycin (PDB ID: 6OYZ; yellow), viewed from the cytoplasmic side. Visible TMs and Loops are labelled. (b) Detailed view of the capuramycin binding site, with residues around the caprolactam moiety shown in stick representation.
The hydrophobic groove binds some MraY natural product inhibitor classes with aliphatic chains.
Next to the mostly charged nucleoside binding site on the cytoplasmic side of MraY is a long hydrophobic groove, predominantly formed by TMs 5 and 9b, that wraps around from the active site of MraY and opens into the plane of the membrane (Figure 8). This hydrophobic groove is the proposed binding site of C55-P, the membrane-embedded lipid carrier substrate of MraY [42]. The liposidomycins and the tunicamycins contain aliphatic moieties that are thought to compete with C55-P binding [17, 18, 37]. The structure of carbacaprazamycin, a liposidomycin analog, in complex with MraYAA shows that that its alkyl chain does in fact bind to the hydrophobic groove of MraY. The alkyl tail of tunicamycin is disordered in the structure of the tunicamycin-MraYCB complex [38], but the amide group linking the nucleoside core to the aliphatic tail of tunicamycin points toward the hydrophobic groove in MraY (Figure 8).
Figure 8.
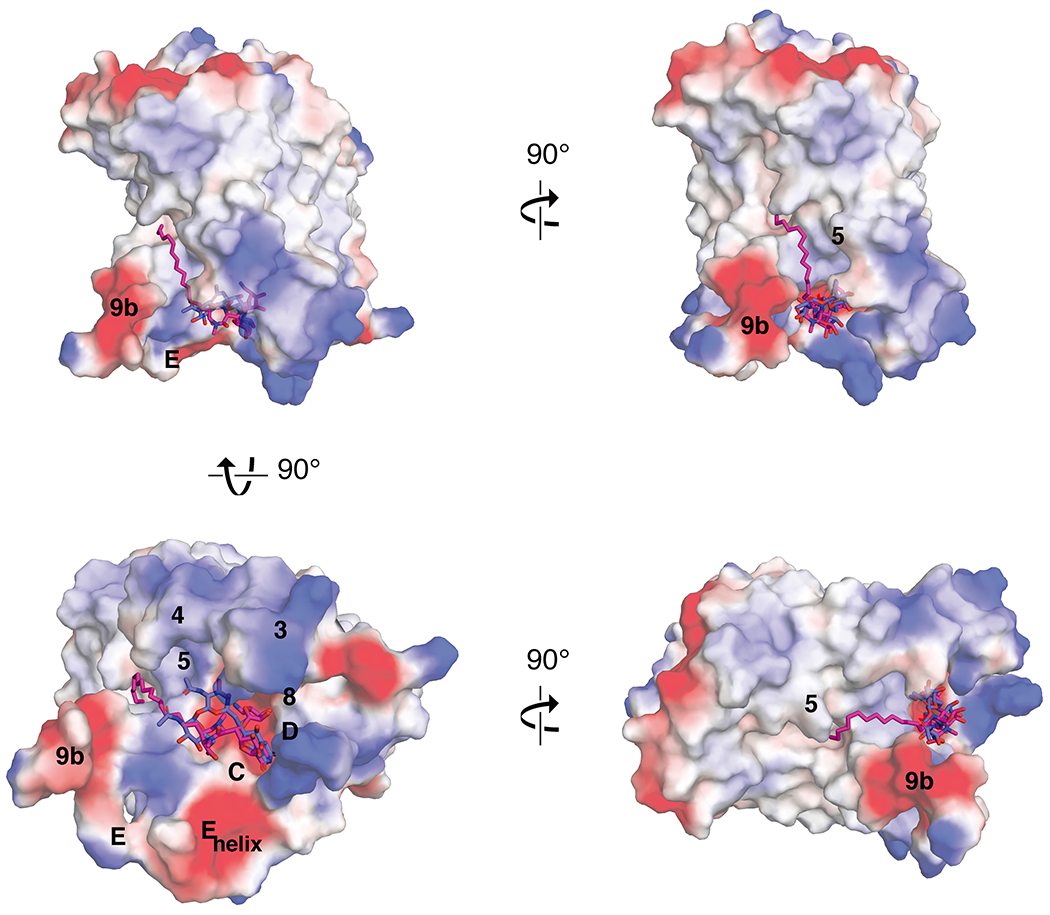
Carbacaprazamycin and tunicamycin bound to MraYAA. Electrostatic surface representation of MraYAA in complex with carbacaprazamycin (PDB ID: 6OYH; magenta sticks) and superimposed with tunicamycin (PDB ID: 5JNQ; blue sticks), seen from the cytoplasm (left, top and bottom) and in the plane of the membrane (right, top and bottom).
The hydrophobic groove recognizes a wide range of liposidomycin analogs with widely varying aliphatic moieties. For example, potent liposidomycins with structural variance in their aliphatic tails have been discovered [56–58]. Liposidomycin Types I and III have a branched lipid tail with ester linkage, while Types II and IV contain a single linear chain; yet, these different classes of liposidomycins exhibit a similar range of inhibitory activity on the enzyme level [35]. Varying the alkyl chain length does not appear to substantially affect activity within a series of structurally-related nucleoside inhibitors; however, deacylation leads to a reduction in potency [59, 60] . Given the broad tolerance for aliphatic chain structures at the hydrophobic groove, this portion of liposidomycin can also be modified to simplify compound synthesis or to engineer favorable properties into the inhibitor. Instead of the more complex aliphatic moieties observed in the liposidomycins and caprazamycins, the derivative carbacaprazamycin has a simplified saturated acyl chain at the 3” position of the diazepanone moiety. This modification makes its chemical synthesis less complicated and improves compound stability, while achieving high in vivo and in vitro potency[22, 25]. The structure of carbacaprazamycin bound to MraY demonstrates its aliphatic tail is well accommodated by the hydrophobic groove (Figure 8).
Importantly, the mere presence of an aliphatic moiety on an MraY natural product does not necessarily direct it to the hydrophobic groove or improve its potency. This is evidenced by several capuramycin analogs with alkyl chains of various lengths in place in caprolactam ring are much less potent than their parent compounds [26]. This is likely because the location of the aliphatic moiety with respect to the nucleoside core is critical in providing the directionality needed to guide the aliphatic moiety into the hydrophobic groove, as is observed in carbacaprazamycin and tunicamycin.
Two classes of MraY-targeted nucleoside inhibitors compete with the Mg2+ cofactor for binding.
Tunicamycin and 3’-hydroxymureidomycin A bind within hydrogen-bonding distance of D265, the Mg2+-coordinating residue in MraY which is a part of the conserved DDD motif (Figure 9). The DDD motif consists of three invariant aspartate residues (D117, D118, and D265 in MraYAA) that are required for MraY activity and likely play a critical role in the catalytic mechanism of the enzyme [43]. In tunicamycin, an interaction is formed between the tunicamine sugar moiety and D265 [38], while in 3’-hydroxymureidomycin A, the amide linkage of the meta-tyrosine interacts with D265 [39]. Independent functional studies show that tunicamycin and mureidomycin compete with Mg2+ cofactor in binding to MraY. The presence of MgCl2 in isothermal titration calorimetry experiments with MraYAA increases the equilibrium binding constant (Kd) of tunicamycin [40] and enzymatic assays performed with MraY from E. coli shows that activity of mureidomycin analogs decreases with increasing concentrations of MgCl2 [61, 62]. These functional data, in combination with the MraYAA-mureidomycin and MraYCB-tunicamycin complex structures, collectively show that targeting the magnesium-coordinating residue is a generalizable strategy for designing MraY inhibitors. This observation is in stark opposition to the previously held notion that Mg2+ is actually required for tunicamycin binding and activity [63–65]. The two other conserved aspartate residues of the DDD motif (D117 and D118 in MraYAA) do not appear to bind any of the five MraY nucleoside inhibitors with known structures (Figure 9). As these residues are also required for catalytic function of MraY, it is possible that targeting these aspartate residues may be a way to increase the potency of MraY-targeted inhibitors.
Figure 9.
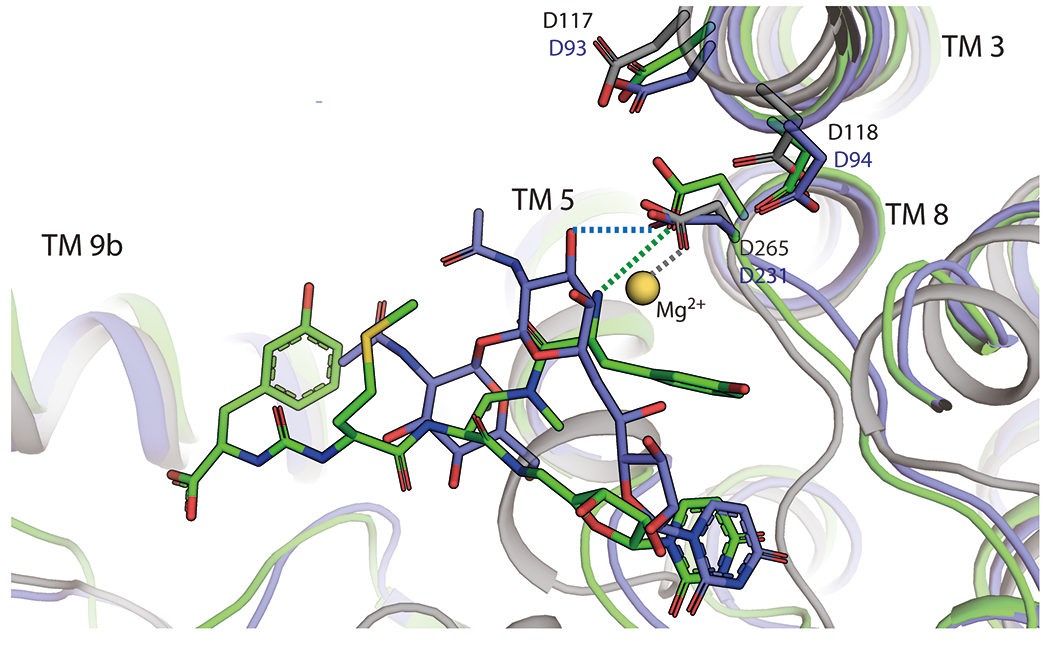
Tunicamycin and 3’-hydroxymureidomycin A compete with the Mg2+ cofactor for binding to MraY. Structural superposition of MraYCB bound to tunicamycin (PDB ID: 5JNQ; blue), MraYAA bound to 3’-hydroxymureidomycin A (PDB ID: 6OZ6; green), and MraYAA bound to Mg2+ (PDB ID: 5JNQ; blue). The magenesium is shown as a yellow-orange sphere. Mg2+ binding is not compatible with Tunicamycin and 3’-hydroxymureidomycin A binding. Hydrogen bonds are represented by dashed lines and are color-coded according to the structures with which they are associated. Amino acid residue numbering is shown in black (MraYAA) and in blue (MraYCB).
Structure-based design of tunicamycin analogs specific for MraY over GPT.
A significant challenge associated with MraY-directed antibiotic development is the potential of engaging the anti-target GPT, a human enzyme that is a key regulator of N-linked protein glycosylation. N-linked glycosylation begins with lipid-linked oligosaccharide (LLO) biosynthesis in the endoplasmic reticulum wherein GPT catalyzes the first and committed step of that pathway. Like MraY, GPT transfers a phospho-sugar moiety from a nucleotide substrate to a lipid carrier in the presence of a Mg2+ cofactor, though each enzyme recognizes different substrates. GPT transfers GlcNAc-1-P from UDP-GlcNAc to the lipid carrier dolichyl phosphate (Dol-P), resulting in the product Dol-PP-GlcNAc (Figure 10a). Both MraY and GPT belong to the polyprenyl-phosphate-N-acetylhexosamine-1-phosphate-transferase (PNPT) superfamily, which also includes the bacterial paralogs WecA and TarO [66]. Recent structures of GPT [40, 41] and MraY [38], each bound to tunicamycin, reveal the structural similarities and differences between these two enzymes, which provide key insights into the rational design of selective MraY inhibitors.
Figure 10.
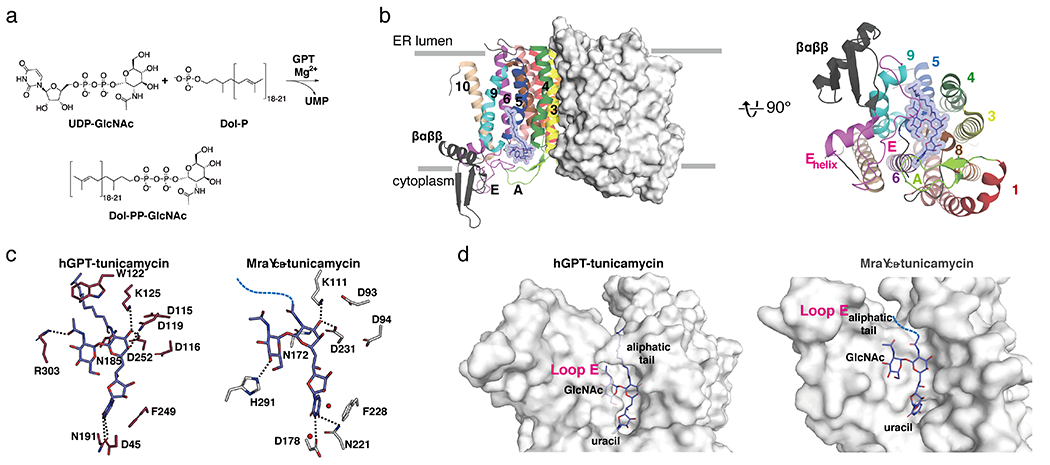
Structural comparison of MraY-tunicamycin and GPT-tunicamycin complexes. (a) Scheme of enzymatic reaction catalyzed by GPT. (b) Left, Structure of hGPT, viewed from the membrane (PDB ID:6BW5). One protomer is shown as surface representation and the other as cartoon representation. The TMs and loops are colored distinctly, corresponding to the coloring used for MraYAA in Figure 1. Right, Cytoplasmic view of hGPT, rotated 90° relative to the orientation in (b). One protomer is shown for simplicity. TMs and Loops are labelled throughout. Tunicamycin is shown in blue. (c) Interactions tunicamycin forms with hGPT (left) and MraYCB (PDB ID: 5JNQ; right). Hydrogen bonds are represented by black dashed lines. The aliphatic tail of tunicamycin is shown as a blue dashed line in the MraYCB structure. (d) Surface representation of hGPT (left) and MraYCB (right), each bound to tunicamycin. Viewed from the cytoplasm. Binding pockets for the uracil, GlcNAc, and aliphatic tail moieties are labelled.
GPT exists as a homodimer and adopts a 10 TM fold, as does MraY, although GPT contains an insertion in Loop E between TMs 9b and 10 a, which is not present in MraY [40, 41]. The tunicamycin binding site in GPT is formed by TMs 4, 5, 6, 8, Loops A and E on the cytoplasmic face of the enzyme (Figure 10b).
The uracil moiety of tunicamycin forms a π- π stacking interaction with F249 in GPT, just as it does with F228 in MraYCB (F262 in MraYAA), and this interaction is also observed in the other nucleoside inhibitor-bound structures of MraY [37, 39]. In both MraY and GPT, the tunicamine sugar forms an interaction with conserved aspartate and lysine residue (D252 and K125 in GPT; D231 and K111 in MraYCB). The amide linker to the aliphatic tail is forming an interaction in GPT with N185, an invariant asparagine. This interaction is likely preserved in the MraY-tunicamycin complex, but the tunicamycin amide was modelled 180° relative to that in the GPT structure, possibly because the aliphatic tail was not resolved in this structure [38] (Figure 10c).
Interactions formed with the GlcNAc moiety of tunicamycin are very different in MraY and GPT[40, 67]. In GPT, an invariant arginine in Loop E (R303) faces inward and interacts directly with the N-acetyl group of the GlcNAc moiety. The corresponding residue in MraY, R282 in MraYCB, is 12 Å away from that site. In addition, the Loop E Helix of MraY is closer to the GlcNAc moiety, providing interactions with histidine residues H290 and H291 in MraYCB, while the Loop E Helix of GPT is oriented at a 30° angle away from the GlcNAc moiety binding site relative to that helix in MraY. These differences could arise from the fact that the natural substrate of GPT contains a GlcNAc moiety. Due to the stark differences in the GlcNAc moiety binding site in MraY and GPT, functionalizing the GlcNAc moiety is a viable strategy for introducing selectivity into the tunicamycin scaffold. Based on this structural insight, we previously designed a derivative of tunicamycin wherein the GlcNAc sugar was modified into a MurNAc moiety. This simple modification of tunicamycin resulted in over a 1000-fold decrease in inhibition of GPT, while the affinity of the tunicamycin-MurNAc analog remained similar to that of the parent compound with MraY [40]. The enhanced selectivity observed is entirely consistent with the structural observation that Loop E packs tightly around the GlcNAc of tunicamycin in GPT and cannot accommodate a bulkier functional group, while the GlcNAc binding site in MraY is more exposed (Figure 10d).
Mg2+ is required for both GPT- and MraY-mediated enzymatic activity. Both Mg2+ - coordinating acidic residues in MraY (D265 in MraYAA) [42] and GPT (D252 and N185 in hGPT)[41] are involved in tunicamycin binding, however their respective contributions to tunicamycin binding differ [40, 67]. We showed that while increase in the concentration of MgCl2 substantially lowers the affinity of tunicamycin for MraYAA, its effect on hGPT is not comparable [40]. Consistent with this observation, D265A in MraYAA substantially lowers the affinity of tunicamycin while D252A in hGPT has more mild effect on tunicamycin binding.
MraY and GPT utilize distinct lipid carrier substrates and the architecture of their respective hydrophobic pockets is different. The lipid carrier for MraY is C55-P, while the analogous substrate for GPT is Dol-P; the two molecules differ with respect to the saturation in the α-isoprene unit and to their chain length. Given an equivalent number of carbons in each substrate, MraY shows strong preference for C55-P over C55-Dol-P, while GPT demonstrates high selectivity for its substrate, C55-Dol-P [40]. The clear recognition each enzyme exhibits for its natural lipid carrier is understandable in light of the structures of GPT and MraY each bound to tunicamycin. The aliphatic tail of tunicamycin is capped by a conserved tryptophan in GPT; by contrast in MraY, the aliphatic tail binding site is a hydrophobic groove, exposed to the membrane (Figure 10d). The more defined lipid tunnel in GPT likely constrains Dol-P, since it has a higher degree of rotational freedom than C55-P due to the saturated bond in the α isoprene unit. The unsaturation at that site in C55-P rigidifies it and restricts its movement in the more shallow, exposed MraY hydrophobic groove.
Overall, a major structural difference between the tunicamycin binding site in MraY and GPT is its solvent accessibility, including at the hydrophobic and GlcNAc binding sites (Figure 10d). This observation has been leveraged to design MraY-selective tunicamycin analogs without off-target effects. By exploiting the difference in the GlcNAc binding site between MraY and tunicamycin, we showed that the tunicamycin-MurNAc analog is highly selective for MraY, demonstrating that MraY-selective inhibitors can be developed using this principle [40]. In parallel, Davis and coworkers designed a series of “lipid-altered” tunicamycin derivatives [41], which engage MraY and have potent antibiotic activity, all while avoiding the cytotoxicity in eukaryotic cells resulting from GPT inhibition. This series of lipid-altered tunicamycins, similar to the tunicamycin-MurNAc analog, are likely accommodated by the surface-exposed binding site on MraY, but not the enclosed, more defined pocket observed in GPT.
Analyzing the druggable hot spots in MraY reveals the principles of MraY inhibition.
Critical learnings for MraY-targeted drug design can be gleaned from trends observed in the available structures of this enzyme in complex with its inhibitors. In order to summarize the similarities and differences in the hot spots accessed by each of the five different classes of nucleoside natural products, we previously created a “barcode” tool of MraY inhibition [39], reproduced here (Figure 11). In addition to the uridine binding pocket, which is similar in all five inhibitor-bound MraY structures, we have identified six druggable hot spots, named HS1-6, which respectively represent the uridine-adjacent, TM9b/Loop E, caprolactam, hydrophobic, Mg2+ cofactor, and tunicamycin binding pockets. Each inhibitor forms interactions with at least two HSs plus the uridine pocket. For example, capuramycin binds HS1, the uridine-adjacent pocket, and HS3, the caprolactam pocket, in addition to the uridine pocket. Carbacaprazamycin makes extensive interactions with HS1 and HS4, the hydrophobic pocket, and forms one hydrogen bond in HS2, the TM9b/Loop E site. Both muraymycin D2 and 3’-hydroxymureidomycin A recognize HS1 and HS2, but 3’-hydroxymureidomycin A forms two fewer hydrogen bonds at these sites and instead makes an additional interaction in HS5, the Mg2+ cofactor binding site (Figure 11). Interestingly, tunicamycin exhibits a very different HS recognition pattern than do the four other MraY inhibitors. Tunicamycin makes limited contacts in HS1 and HS2, probably binds HS4, interacts with HS5, and also forms hydrogen bonds in HS6, the tunicamycin site, which is not recognized by other nucleoside inhibitors. The tunicamine sugar and GlcNAc moieties of tunicamycin facilitate the unique binding pattern exhibited by this promiscuous inhibitor; these two pharmacophores are also recognized by the human enzyme GPT. The unique binding mode of tunicamycin is likely related to its off-target activity on MraY paralog GPT.
Figure 11.
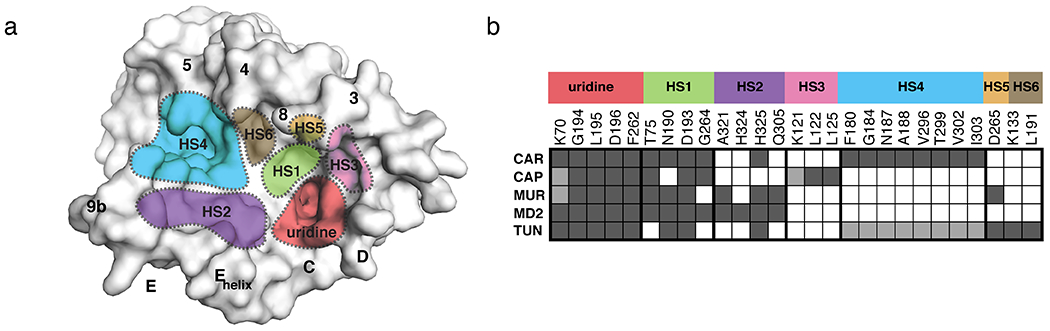
Hot spots of MraY inhibition. (a) Surface representation of MraYAA (muraymycin D2-bound, inhibitor removed for clarification). The inhibitor binding site hot spots (HSs) are color-coded and labeled: uridine (red), uridine-adjacent (HS1; lime green), TM9b/LoopE (HS2; purple), caprolactam (HS3; pink), hydrophobic (HS4; cyan), Mg2+ (HS5; gold), and tunicamycin (HS6; brown). Visible TMs and Loops are labelled. (b) A barcode tool summarizing the interactions each nucleoside inhibitor makes with HS1–6 in MraY. The residues shown corresponding to each HS label (MraYAA numbering) comprise that site in MraY. Each row corresponds to a different compound: carbacaprazamycin (CAR), capuramycin (CAP), 3′-hydroxymureidomycin A (MUR), muraymycin D2 (MD2), and tunicamycin (TUN). A dark gray square indicates an interaction between the corresponding inhibitor and residue is formed and a white square means that no contact is made. Squares colored light gray represent that either the amino acid residue side chain or the inhibitor substructure is not resolved in the crystal structure, but likely makes the binding interaction.
Discussion.
The barcode summarizing the cryptic inhibitor binding sites on MraY (Figure 11b) clearly reveals a structure-based principle of MraY inhibition: each MraY inhibitor binds at least two HSs in addition to the uridine pocket. This structural observation may be critical for the design of potent MraY-targeted antibiotics with favorable physiochemical properties. A modular design strategy may be fruitful, wherein inhibitors are developed to target different combinations of cryptic hotspots on the cytoplasmic surface of MraY. This approach can also shed light on the basis of the biochemical and biological activity differences observed among the nucleoside MraY inhibitors. For example, perhaps targeting a certain combination of hotspots leads to time-dependent inhibition of MraY. Dissecting the functional role of each hotspot would provide the key to building desired properties into MraY inhibitors.
Because all known effective MraY inhibitors contain a nucleoside base, which assumes a similar orientation in each MraY-inhibitor structure, it is unclear whether the uracil moiety can be replaced with a novel pharmacophore. Nucleosides or nucleotide inhibitors are notoriously difficult to synthesize and they also present cell permeability challenges. Can targeting HSs 1-4 and not the uridine pocket be sufficient for selective inhibition of MraY? Is the uridine moiety critical for stabilizing the inhibitor-bound conformation of MraY or can it be replaced with another chemotype? These types of mechanistic questions can be systematically addressed using a structure-based hotspot framework to understand MraY inhibition.
Acknowledgments
This work was supported by the National Institutes of Health (R01GM120594 to S.-Y. L.). We thank S. Ichikawa and J. Hong for our long-term and successful collaboration. We thank Ben Chung, J. Yoo, and A. Kuk for their contribution to structural and functional studies of MraY and GPT.
Footnotes
Declarations of interest: none
References
- 1.Lewis K, Platforms for antibiotic discovery. Nat Rev Drug Discov, 2013. 12(5): p. 371–87. [DOI] [PubMed] [Google Scholar]
- 2.McDonald LA, et al. , Structures of the muraymycins, novel peptidoglycan biosynthesis inhibitors. J Am Chem Soc, 2002. 124(35): p. 10260–1. [DOI] [PubMed] [Google Scholar]
- 3.Tanino T, et al. , Synthesis and Biological Evaluation of Muraymycin Analogues Active against Anti-Drug-Resistant Bacteria. ACS Medicinal Chemistry Letters, 2010. 1(6): p. 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isono F and Inukai M, Mureidomycin A, a new inhibitor of bacterial peptidoglycan synthesis. Antimicrob Agents Chemother, 1991. 35(2): p. 234–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isono F, et al. , Mureidomycins A-D, novel peptidylnucleoside antibiotics with spheroplast forming activity. III. Biological properties. J Antibiot (Tokyo), 1989. 42(5): p. 674–9. [DOI] [PubMed] [Google Scholar]
- 6.Isono F, Kodama K, and Inukai M, Susceptibility of Pseudomonas species to the novel antibiotics mureidomycins. Antimicrob Agents Chemother, 1992. 36(5): p. 1024–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi H, et al. , Capuramycin, a new nucleoside antibiotic. Taxonomy, fermentation, isolation and characterization. J Antibiot (Tokyo), 1986. 39(8): p. 1047–53. [DOI] [PubMed] [Google Scholar]
- 8.Reddy VM, Einck L, and Nacy CA, In vitro antimycobacterial activities of capuramycin analogues. Antimicrob Agents Chemother, 2008. 52(2): p. 719–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikonenko BV, et al. , Activity of SQ641, a capuramycin analog, in a murine model of tuberculosis. Antimicrob Agents Chemother, 2009. 53(7): p. 3138–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nikonenko B, et al. , Therapeutic efficacy of SQ641-NE against Mycobacterium tuberculosis. Antimicrob Agents Chemother, 2014. 58(1): p. 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore JH 2nd, et al. , Treatment of Clostridium difficile infection using SQ641, a capuramycin analogue, increases post-treatment survival and improves clinical measures of disease in a murine model. J Antimicrob Chemother, 2016. 71(5): p. 1300–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bogatcheva E, et al. , Chemical modification of capuramycins to enhance antibacterial activity. J Antimicrob Chemother, 2011. 66(3): p. 578–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isono K, et al. , Liposidomycins: novel nucleoside antibiotics which inhibit bacterial peptidoglycan synthesis. J Antibiot (Tokyo), 1985. 38(11): p. 1617–21. [DOI] [PubMed] [Google Scholar]
- 14.Kimura K.-i., et al. , Liposidomycin C Inhibits Phospho-N-acetylmuramyl-pentapeptide Transferase in Peptidoglycan Synthesis of Escherichia coliY-10. Agricultural and Biological Chemistry, 2014. 53(7): p. 1811–1815. [Google Scholar]
- 15.Igarashi M, et al. , Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J Antibiot (Tokyo), 2003. 56(6): p. 580–3. [DOI] [PubMed] [Google Scholar]
- 16.Tanino T, et al. , Mechanistic Analysis of Muraymycin Analogues: A Guide to the Design of MraY Inhibitors. Journal of Medicinal Chemistry, 2011. 54(24): p. 8421–8439. [DOI] [PubMed] [Google Scholar]
- 17.Brandish PE, et al. , Modes of action of tunicamycin, liposidomycin B, and mureidomycin A: inhibition of phospho-N-acetylmuramyl-pentapeptide translocase from Escherichia coli. Antimicrob Agents Chemother, 1996. 40(7): p. 1640–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muramatsu Y, Ishii MM, and Inukai M, Studies on novel bacterial translocase I inhibitors, A-500359s. II. Biological activities of A-500359 A, C, D and G. J Antibiot (Tokyo), 2003. 56(3): p. 253–8. [DOI] [PubMed] [Google Scholar]
- 19.Brandish PE, et al. , Slow binding inhibition of phospho-N-acetylmuramyl-pentapeptide-translocase (Escherichia coli) by mureidomycin A. J Biol Chem, 1996. 271(13): p. 7609–14. [DOI] [PubMed] [Google Scholar]
- 20.Yamashita A, et al. , Muraymycins, novel peptidoglycan biosynthesis inhibitors: synthesis and SAR of their analogues. Bioorg Med Chem Lett, 2003. 13(19): p. 3345–50. [DOI] [PubMed] [Google Scholar]
- 21.Takeoka Y, et al. , Expansion of Antibacterial Spectrum of Muraymycins toward Pseudomonas aeruginosa. ACS Med Chem Lett, 2014. 5(5): p. 556–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirano S, Ichikawa S, and Matsuda A, Synthesis of caprazamycin analogues and their structure--activity relationship for antibacterial activity. J Org Chem, 2008. 73(2): p. 569–77. [DOI] [PubMed] [Google Scholar]
- 23.Spork AP, et al. , Lead structures for new antibacterials: stereocontrolled synthesis of a bioactive muraymycin analogue. Chemistry, 2014. 20(47): p. 15292–7. [DOI] [PubMed] [Google Scholar]
- 24.Cui Z, et al. , Antibacterial Muraymycins from Mutant Strains of Streptomyces sp. NRRL 30471. J Nat Prod, 2018. 81(4): p. 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichikawa S, et al. , Carbacaprazamycins: Chemically Stable Analogues of the Caprazamycin Nucleoside Antibiotics. ACS Infectious Diseases, 2015. 1(4): p. 151–156. [DOI] [PubMed] [Google Scholar]
- 26.Hotoda H, et al. , Synthesis and antimycobacterial activity of capuramycin analogues. Part 1: substitution of the azepan-2-one moiety of capuramycin. Bioorganic & Medicinal Chemistry Letters, 2003. 13(17): p. 2829–2832. [DOI] [PubMed] [Google Scholar]
- 27.Hotoda H, et al. , Synthesis and antimycobacterial activity of capuramycin analogues. Part 2: acylated derivatives of capuramycin-related compounds. Bioorganic & Medicinal Chemistry Letters, 2003. 13(17): p. 2833–2836. [DOI] [PubMed] [Google Scholar]
- 28.Koga T, et al. , Activity of capuramycin analogues against Mycobacterium tuberculosis, Mycobacterium avium and Mycobacterium intracellulare in vitro and in vivo. J Antimicrob Chemother, 2004. 54(4): p. 755–60. [DOI] [PubMed] [Google Scholar]
- 29.Dubuisson T, et al. , In vitro antimicrobial activities of capuramycin analogues against non-tuberculous mycobacteria. J Antimicrob Chemother, 2010. 65(12): p. 2590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duksin D and Mahoney WC, Relationship of the structure and biological activity of the natural homologues of tunicamycin. J Biol Chem, 1982. 257(6): p. 3105–9. [PubMed] [Google Scholar]
- 31.Faye L and Chrispeels MJ, Apparent Inhibition of beta-Fructosidase Secretion by Tunicamycin May Be Explained by Breakdown of the Unglycosylated Protein during Secretion. Plant Physiol, 1989. 89(3): p. 845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zeng Y and Elbein AD, UDP-N-acetylglucosamine:dolichyl-phosphate N-acetylglucosamine-1-phosphate transferase is amplified in tunicamycin-resistant soybean cells. Eur J Biochem, 1995. 233(2): p. 458–66. [DOI] [PubMed] [Google Scholar]
- 33.Koizumi N, et al. , Overexpression of a gene that encodes the first enzyme in the biosynthesis of asparagine-linked glycans makes plants resistant to tunicamycin and obviates the tunicamycin-induced unfolded protein response. Plant Physiol, 1999. 121(2): p. 353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oslowski CM and Urano F, Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol, 2011. 490: p. 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura K, et al. , Selective inhibition of the bacterial peptidoglycan biosynthesis by the new types of liposidomycins. J Antibiot (Tokyo), 1998. 51(12): p. 1099–104. [DOI] [PubMed] [Google Scholar]
- 36.Inukai M, Isono F, and Takatsuki A, Selective inhibition of the bacterial translocase reaction in peptidoglycan synthesis by mureidomycins. Antimicrob Agents Chemother, 1993. 37(5): p. 980–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chung BC, et al. , Structural insights into inhibition of lipid I production in bacterial cell wall synthesis. Nature, 2016. 533(7604): p. 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hakulinen JK, et al. , MraY-antibiotic complex reveals details of tunicamycin mode of action. Nat Chem Biol, 2017. 13(3): p. 265–267. [DOI] [PubMed] [Google Scholar]
- 39.Mashalidis EH, et al. , Chemical logic of MraY inhibition by antibacterial nucleoside natural products. Nat Commun, 2019. 10(1): p. 2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoo J, et al. , GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat Struct Mol Biol, 2018. 25(3): p. 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong YY, et al. , Structures of DPAGT1 Explain Glycosylation Disease Mechanisms and Advance TB Antibiotic Design. Cell, 2018. 175(4): p. 1045–1058 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung BC, et al. , Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science, 2013. 341(6149): p. 1012–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Dabbagh B, et al. , Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry, 2008. 47(34): p. 8919–28. [DOI] [PubMed] [Google Scholar]
- 44.Anderson MS, Eveland SS, and Price NP, Conserved cytoplasmic motifs that distinguish sub-groups of the polyprenol phosphate:N-acetylhexosamine-1-phosphate transferase family. FEMS Microbiol Lett, 2000. 191(2): p. 169–75. [DOI] [PubMed] [Google Scholar]
- 45.Inukai M, et al. , Mureidomycins A-D, novel peptidylnucleoside antibiotics with spheroplast forming activity. I. Taxonomy, fermentation, isolation and physicochemical properties. J Antibiot (Tokyo), 1989. 42(5): p. 662–6. [DOI] [PubMed] [Google Scholar]
- 46.Isono F, et al. , Mureidomycins A-D, novel peptidylnucleoside antibiotics with spheroplast forming activity. II. Structural elucidation. J Antibiot (Tokyo), 1989. 42(5): p. 667–73. [DOI] [PubMed] [Google Scholar]
- 47.Lemoine RC, Magon A, and Hecker SJ, Synthesis of base-modified dihydropacidamycins. Bioorg Med Chem Lett, 2002. 12(7): p. 1121–3. [DOI] [PubMed] [Google Scholar]
- 48.Wiegmann D, Koppermann S, and Ducho C, Aminoribosylated Analogues of Muraymycin Nucleoside Antibiotics. Molecules, 2018. 23(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heib A, et al. , Muraymycin Nucleoside Antibiotics: Structure-Activity Relationship for Variations in the Nucleoside Unit. Molecules, 2019. 25(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dini C, et al. , Synthesis of the nucleoside moiety of liposidomycins: elucidation of the pharmacophore of this family of MraY inhibitors. Bioorg Med Chem Lett, 2000. 10(16): p. 1839–43. [DOI] [PubMed] [Google Scholar]
- 51.Dini C, et al. , Synthesis of analogues of the O-beta-D-ribofuranosyl nucleoside moiety of liposidomycins. Part 1: contribution of the amino group and the uracil moiety upon the inhibition of MraY. Bioorg Med Chem Lett, 2001. 11(4): p. 529–31. [DOI] [PubMed] [Google Scholar]
- 52.Chatterjee S, et al. , Napsamycins, new Pseudomonas active antibiotics of the mureidomycin family from Streptomyces sp. HIL Y-82,11372. J Antibiot (Tokyo), 1994. 47(5): p. 595–8. [DOI] [PubMed] [Google Scholar]
- 53.Gruschow S, et al. , New pacidamycin antibiotics through precursor-directed biosynthesis. Chembiochem, 2009. 10(2): p. 355–60. [DOI] [PubMed] [Google Scholar]
- 54.Spork AP, et al. , Analogues of Muraymycin Nucleoside Antibiotics with Epimeric Uridine-Derived Core Structures. Molecules, 2018. 23(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muramatsu Y, et al. , Studies on novel bacterial translocase I inhibitors, A-500359s. III. Deaminocaprolactam derivatives of capuramycin: A-500359 E, F, H; M-1 and M-2. J Antibiot (Tokyo), 2003. 56(3): p. 259–67. [DOI] [PubMed] [Google Scholar]
- 56.Kimura K, et al. , New types of liposidomycins that inhibit bacterial peptidoglycan synthesis and are produced by Streptomyces. I. Producing organism and medium components. J Antibiot (Tokyo), 1998. 51(7): p. 640–6. [DOI] [PubMed] [Google Scholar]
- 57.Kimura K, et al. , New types of liposidomycins that inhibit bacterial peptidoglycan synthesis and are produced by Streptomyces. II. Isolation and structure elucidation. J Antibiot (Tokyo), 1998. 51(7): p. 647–54. [DOI] [PubMed] [Google Scholar]
- 58.Esumi Y, et al. , New types of liposidomycins produced by Streptomyces that inhibit bacterial peptidoglycan synthesis. Structure elucidation of fatty acid components by tandem mass spectrometry. J Antibiot (Tokyo), 1999. 52(3): p. 281–7. [DOI] [PubMed] [Google Scholar]
- 59.Hirano S, Ichikawa S, and Matsuda A, Structure-activity relationship of truncated analogs of caprazamycins as potential anti-tuberculosis agents. Bioorg Med Chem, 2008. 16(9): p. 5123–33. [DOI] [PubMed] [Google Scholar]
- 60.Kimura K.-i. and Bugg TDH, Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Natural Product Reports, 2003. 20(2): p. 252–273. [DOI] [PubMed] [Google Scholar]
- 61.Howard NI and Bugg TDH, Synthesis and activity of 5′-Uridinyl dipeptide analogues mimicking the amino terminal peptide chain of nucleoside antibiotic mureidomycin A. Bioorganic & Medicinal Chemistry, 2003. 11(14): p. 3083–3099. [DOI] [PubMed] [Google Scholar]
- 62.A Gentle C, et al. , Structure–function studies on nucleoside antibiotic mureidomycin A: synthesis of 5′-functionalised uridine models. Journal of the Chemical Society, Perkin Transactions 1, 1999(10): p. 1287–1294. [Google Scholar]
- 63.Wang R, et al. , A search for pyrophosphate mimics for the development of substrates and inhibitors of glycosyltransferases. Bioorg Med Chem, 1997. 5(4): p. 661–72. [DOI] [PubMed] [Google Scholar]
- 64.Price NP and Momany FA, Modeling bacterial UDP-HexNAc: polyprenol-P HexNAc-1-P transferases. Glycobiology, 2005. 15(9): p. 29R–42R. [DOI] [PubMed] [Google Scholar]
- 65.Xu L, et al. , Conformational analysis of chirally deuterated tunicamycin as an active site probe of UDP-N-acetylhexosamine:polyprenol-P N-acetylhexosamine-1-P translocases. Biochemistry, 2004. 43(42): p. 13248–55. [DOI] [PubMed] [Google Scholar]
- 66.Lehrman MA, A family of UDP-GlcNAc/MurNAc: polyisoprenol-P GlcNAc/MurNAc-1-P transferases. Glycobiology, 1994. 4(6): p. 768–71. [DOI] [PubMed] [Google Scholar]
- 67.Hering J, et al. , Structural basis for selective inhibition of antibacterial target MraY, a membrane-bound enzyme involved in peptidoglycan synthesis. Drug Discov Today, 2018. 23(7): p. 1426–1435. [DOI] [PubMed] [Google Scholar]