

Abstract
The gut microbiome is shaped by diet and influences host metabolism, but these links are complex and can be unique to each individual. We performed deep metagenomic sequencing of >1,100 gut microbiomes from individuals with detailed long-term diet information, as well as hundreds of fasting and same-meal postprandial cardiometabolic blood marker measurements. We found strong associations between microbes and specific nutrients, foods, food groups, and general dietary indices, driven especially by the presence and diversity of healthy and plant-based foods. Microbial biomarkers of obesity were reproducible across cohorts, and blood markers of cardiovascular disease and impaired glucose tolerance were more strongly associated with microbiome structure. While some microbes such as Prevotella copri and Blastocystis spp., were indicators of favorable postprandial glucose metabolism, several species were more directly predictive of postprandial triglycerides and C-peptide. The panel of intestinal species associated with healthy dietary habits overlapped with those associated with favourable cardiometabolic and postprandial markers, indicating our large-scale resource can potentially stratify the gut microbiome into generalizable health levels among individuals without clinically manifest disease.
Introduction
Dietary contributions to health and chronic conditions, such as obesity, metabolic syndrome, cancer and cardiovascular disease, are of universal importance. Obesity and associated mortality/morbidity have risen dramatically over the past decades1, with the gut microbiome implicated as one of several potentially causal human-environment interactions2,3. Surprisingly, the details of the microbiome’s role in obesity and cardiometabolic health have proven difficult to define reproducibly in large human populations4, likely due to the complexity of habitual diets, the difficulty of measuring them at scale and disentangling them from other lifestyle variables5,6, and the personalized nature of the microbiome7.
To overcome these challenges, we launched the Personalised Responses to Dietary Composition Trial (PREDICT 1) of diet-microbiome interactions in metabolic health8. PREDICT 1 included >1,000 participants profiled pre- and post- standardised dietary challenges using intensive in-clinic biometric and blood measures, habitual dietary data collection, continuous glucose monitoring, and stool metagenomics. The study was inspired by previous large-scale diet-microbiome interaction profiles, identifying gut microbiome configurations and microbial taxa associated with postprandial glucose responses9,10, obesity-associated biometrics such as body mass index (BMI) and adiposity11–13, and blood lipids and inflammatory markers14,15.
Results
Large metagenomically-profiled cohorts with rich clinical, cardiometabolic, and dietary information
PREDICT 18,16 is an intervention study of diet-microbiome-cardiometabolic interactions (see Methods), including a discovery cohort in the UK (n=1,002) and a validation population in the US (n=100). We collected demographic information, habitual diet data, cardiometabolic blood biomarkers, and postprandial responses to standardized test meals in the clinic and in free-living settings8,16 (Fig. 1A). At-home stool collection yielded 1,098 baseline and 105 follow-up microbiome samples (+14 days) that were all shotgun sequenced, and then taxonomically and functionally profiled (Fig. 1A, Methods).
Fig. 1: The PREDICT 1 study associates gut microbiome structure with habitual diet and blood cardiometabolic markers.
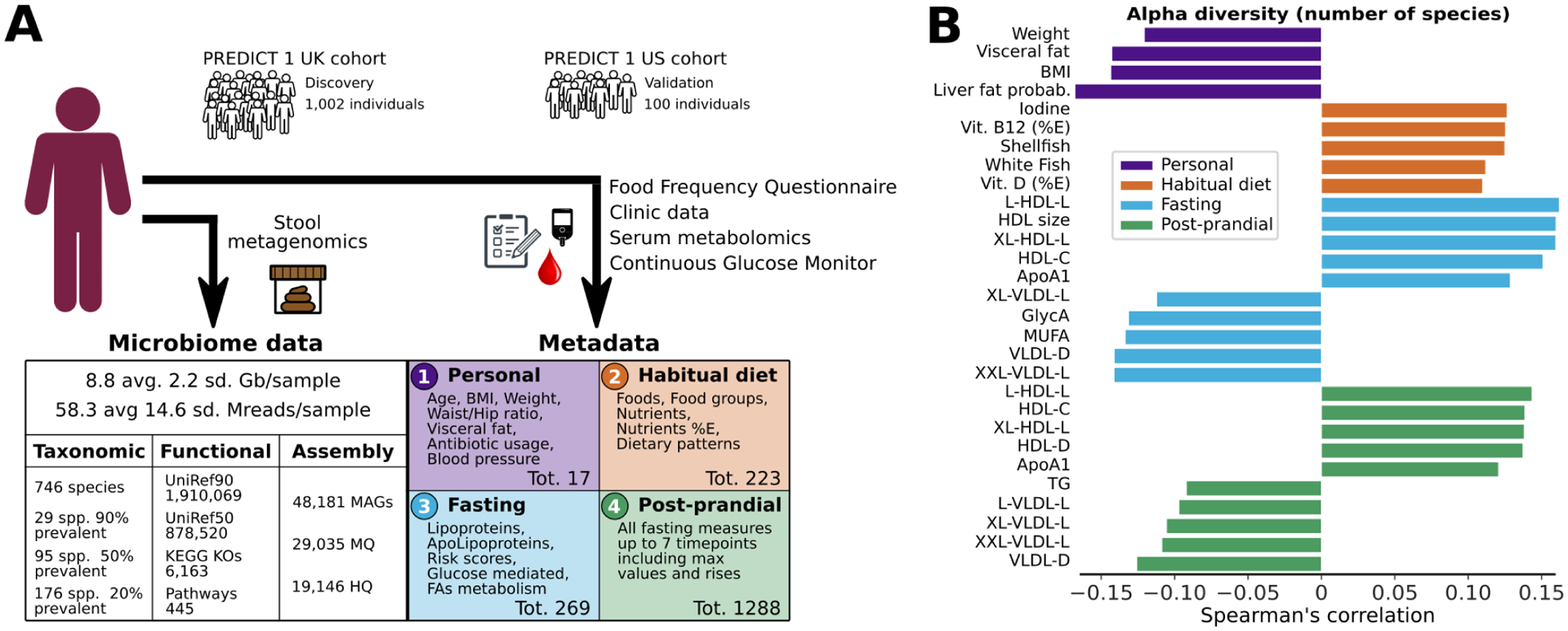
(A) The PREDICT 1 study assessed the gut microbiome of 1,098 volunteers from the UK and US via metagenomic sequencing of stool samples. Phenotypic data obtained through in-person assessment, blood/biospecimen collection, and the return of validated study questionnaires queried a range of relevant host/environmental factors including (1) personal characteristics, such as age, BMI, and estimated visceral fat; (2) habitual dietary intake using semi-quantitative food frequency questionnaires (FFQs); (3) fasting; and (4) postprandial cardiometabolic blood and inflammatory markers, total lipid and lipoprotein concentrations, lipoprotein particle sizes, apolipoproteins, derived metabolic risk scores, glycaemic-mediated metabolites, and metabolites related to fatty acid metabolism. (B) Overall microbiome alpha diversity, estimated as the total number of confidently identified microbial species in a given sample (richness), was correlated with HDL-D (positive) and estimated hepatic steatosis (negative). The five strongest positive and negative Spearman’s correlations with q<0.05 are reported for each of the four categories. Top species based on Shannon diversity are reported in Extended Data Fig. 1A and all correlations are reported in Supplementary Table 1.
Microbial diversity and composition are linked with diet and fasting and postprandial biomarkers
We first leveraged a unique subpopulation of 480 monozygotic and dizygotic twins and confirmed that host genetics influences microbiome composition only to a limited extent17. Indeed, twin-pair microbiome similarity remained substantially lower than intra-subject longitudinal similarity (day 0 vs. day 14, p<1e-12, Extended Data Fig. 1B), a testament to the personalized nature of the gut microbiome attributable to non-genetic factors (Extended Data Fig. 1C–D).
We then investigated overall intra-sample (alpha) microbiome diversity as a broad summary statistic of microbiome structure and found it was significantly associated (q<0.05) in 56 of the 295 tested correlations with personal characteristics, habitual diet, and metabolic indices (Fig. 1B, Supplementary Table 1A). BMI, visceral fat measurements, and probability of fatty liver (using a validated prediction model18) were inversely associated with species richness. Among clinical circulating measures, High-Density Lipoprotein cholesterol (HDL-C) was positively correlated with species richness. Emerging cardiometabolic biomarkers19 that are not routinely used clinically, including lipoprotein particle size (diameter, “-D”), and GlycA (inflammatory biomarker; glycoprotein acetyls), were also associated (positively or negatively) with microbiome richness. These results associating simple indicators such as microbiome richness to cardiometabolic health indicators and diet, motivated our more detailed investigations of specific gut microbiome components.
Diversity of healthy plant-based foods in habitual diet shapes gut microbiome composition
We assessed links between habitual diet and the microbiome using random forest (RF) models, each trained on quantitative microbiome features to predict each dietary variable from food frequency questionnaires (FFQs) (Methods). The performance of the models was quantified with ROC AUCs for classification and with correlation for regression (Methods). Several foods and food groups exceeded 0.15 median Spearman’s correlation over bootstrap folds (denoted as “ρ”) between predicted and FFQ-estimated values (14.5%) and AUC>0.65 (10.8%; Fig. 2A). The strongest association was for coffee (instant or ground) (ρ=0.43, AUC=0.8), with dose-dependent effects and validated in the US cohort (Fig. 2D). Tighter microbiome links were found for energy-adjusted nutrients (Fig. 2A) with almost one-third (Supplementary Table 2) showing correlations above 0.3.
Fig. 2: Food quality, regardless of source, is linked to overall and feature-level composition of the gut microbiome.
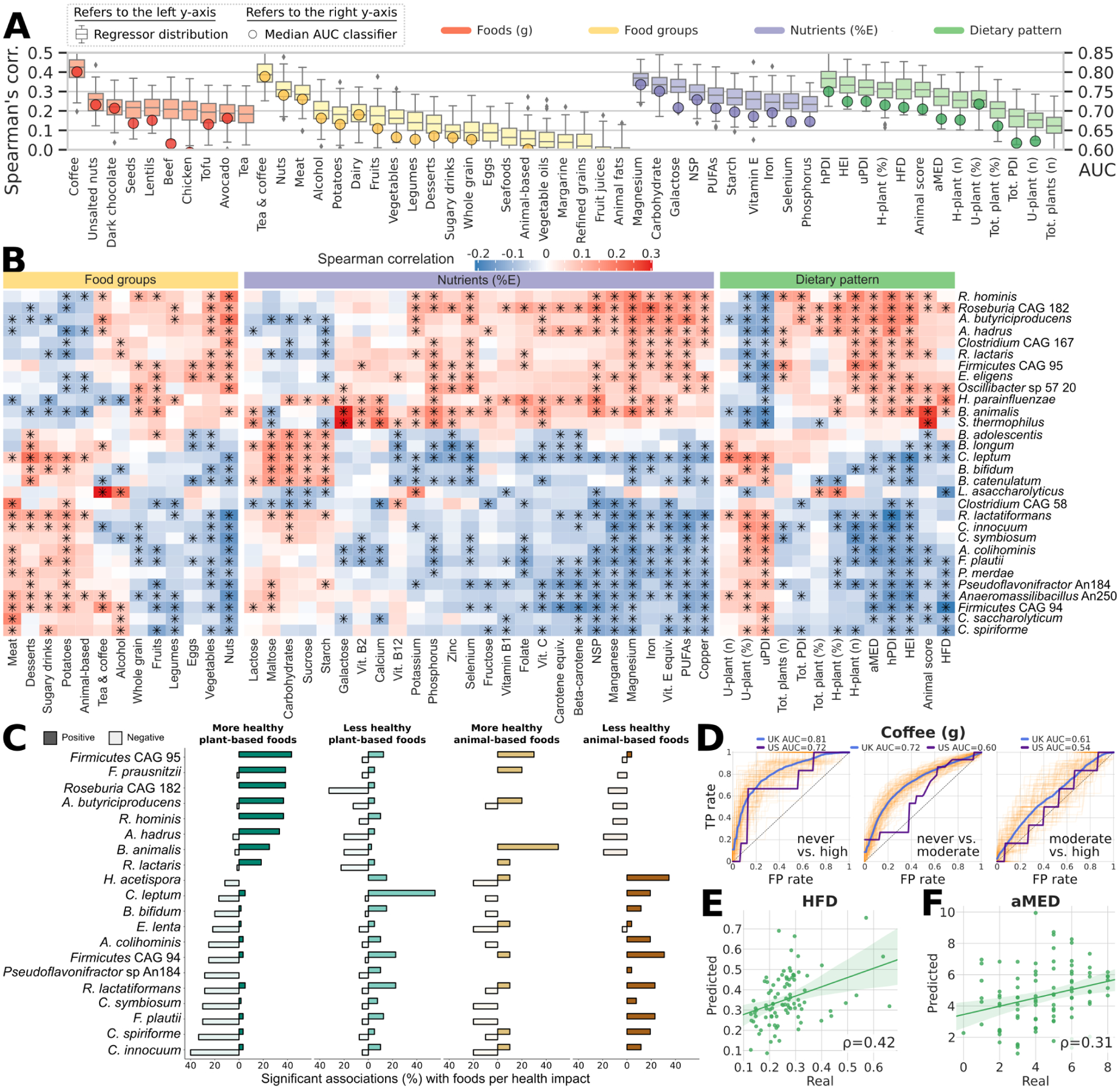
(A) Specific components of habitual diet comprising foods, nutrients, and dietary indices are linked to the composition of the gut microbiome with variable strengths as estimated by machine learning regression and classification models. Boxplots report the correlation between the real value of each component and the value predicted by regression models across 100 training/testing folds (Methods). Circles denote median area-under-the-curve (AUC) values across 100 folds for a corresponding binary classifier between the highest and lowest quartiles (Methods). (B) Single Spearman’s correlations adjusted for BMI and age between microbial species and components of habitual diet with asterisks denoting significant associations (FDR q<0.2). The 30 microbial species with the highest number of significant associations across habitual diet categories are reported. All indices of dietary patterns are reported, whereas only food groups and nutrients (energy-adjusted) with at least 7 associations among the top 30 microbial species are reported (NSP: non‐starch polysaccharides). Rows and columns are hierarchically clustered (complete linkage, Euclidean distance). Full heatmaps of foods and unadjusted nutrients are reported in Extended Data Fig. 2, and the full set of correlations is available in Supplementary Table 5. (C) Number of significant positive and negative associations (Spearman’s correlation p<0.2) between foods and taxa categorized by more and less healthy plant-based foods and more and less healthy animal-based foods according to the PDI. Taxa shown are the 20 species with the highest total number of significant associations regardless of category. (D) The association between the gut microbiome and coffee consumption in UK participants is dose-dependent, i.e. stronger when assessing heavy (e.g. >4 cups/d) vs. never drinkers, and was validated in the US cohort when applying the UK model. The reported ROC curves represent the performance of the classifier at varying classification thresholds with respect to the True Positive Rate (i.e. recall) and the False Positive Rate (i.e. precision). (E-F) Among general dietary patterns and indices, the Healthy Food Diversity index (HFD) and the Alternate Mediterranean Diet score (aMED) were validated in the US cohort, thus showing consistency between the two populations on these two important dietary indices. Other validations of the UK model applied to the US cohort are reported in Extended Data Fig. 3.
We then summarized constituent foods into dietary indices (Supplementary Table 2), including the Healthy Food Diversity index (incorporating dietary diversity and food quality, HFD)20, the Healthy/Unhealthy Plant-based Dietary Indices (considering quality and quantity of plant-based foods, hPDI/uPDI), Healthy Eating Index (extent of alignment with dietary guidelines, HEI)21 and the Alternate Mediterranean Diet score (aMED)22, all of which are associated with reduced risk of chronic disease22–27. We demonstrated tight correlations between microbial composition and the HFD, hPDI/uPDI and HEI in the UK (ρ between 0.31 and 0.37, Fig. 2A) and the results were consistent in the US validation cohort, with ρ reaching 0.42 for HFD and 0.31 for aMED (Fig. 2E–F and Extended Data Fig. 3), highlighting the relationship between the microbiome and health-associated dietary patterns.
Microbial species segregate into groups associated with more healthy and less healthy plant- and animal-based foods
We proceeded to identify the specific microbial taxa most responsible for these diet-based community associations (Fig. 2B). After adjusting for age and BMI, we found 42 species (24% of those at >20% prevalence) significantly correlated with at least five dietary exposures (q<0.2, Supplementary Table 5). This included expected associations (Extended Data Fig. 2), such as enrichment of the probiotic taxa Bifidobacterium animalis28 and Streptococcus thermophilus with greater full-fat yogurt consumption (ρ=0.22 for both). The strongest food/microbe association was between the recently characterized butyrate-producing Lawsonibacter asaccharolyticus29 and coffee consumption (Fig. 2B). However, due to the low resolution of FFQ data, the complexity of dietary patterns, nutrient-nutrient interactions, and clustering of ‘healthy’/’less-healthy’ food items, it is challenging to disentangle the independent associations of single foods with microbial species.
At a broader level, we found clear segregation of species (Fig. 2B) into two distinct clusters with either more healthy plant-based foods (e.g. spinach, seeds, tomatoes, broccoli) or with less healthy plant-based (e.g. juices, sweetened beverages, refined grains) and animal-based foods, as defined by the PDI30 (Supplementary Table 4). Taxa linked to healthy plant-based foods (Fig. 2B–C, Extended Data Fig. 2) mostly included butyrate producers, such as Roseburia hominis, Agathobaculum butyriciproducens, Faecalibacterium prausnitzii, and Anaerostipes hadrus, as well as uncultivated species, predicted to have this metabolic capability (Roseburia CAG182 and Firmicutes CAG95). Clades correlating with several ‘less-healthy’ plant-based and animal-based foods included several Clostridium species (Clostridium innocuum, C. symbiosum, C. spiroforme, C. leptum, C. saccharolyticum). The segregation of species according to animal-based ‘healthy’ foods (e.g. eggs, white and oily fish) or animal-based ‘less-healthy’ foods (e.g. meat pies, bacon, dairy desserts) using a novel categorisation (Methods), was also distinct and overlapping with taxa signatures for ‘healthy’ and ‘less-healthy’ plant foods (Fig. 2C and Extended Data Fig. 2). The few foods not fitting into the ‘healthy’ cluster despite being ‘healthy plant’ foods, were (ultra) processed foods31 (e.g. sauces, baked beans; Extended Data Fig. 2). This emphasises the importance of food quality (e.g. highly processed vs. unprocessed), food source (e.g. plant vs. animal), and food type (i.e. not all plant foods are healthy) both in overall health and in microbiome ecology.
Poorly characterized microbes drive the strongest microbiome-habitual diet associations
Many of the strongest microbial associations with diet occurred with only recently-isolated or still uncultured taxa including five species defined using co-abundance gene groups (CAGs) from metagenomics32. Among indices, the hPDI significantly correlated with 60 of the 176 prevalent species, highlighting the impact of dietary diversity and quality on gut microbial responsiveness. Among other dietary indices and nutrients, we observed general concordance with the two sets of microbes associated with healthy and less-healthy foods. A greater animal-based food score (definition in Supplementary Table 4), was associated with the ‘healthy’ cluster, suggesting that a diet rich in healthier animal-based foods is associated with the more favourable diet-microbiome signature, although this may also reflect an overall healthier dietary pattern. The healthy and unhealthy PDI, which differentially affect disease risk25,30 also had distinct clusters, again emphasising the oversimplification of conventional plant and animal-based food groupings. The taxa with the highest correlations in the two clusters are Firmicutes CAG95 and Clostridium leptum for healthy and unhealthy diet, respectively. The lack or paucity of cultivated representatives for these two taxa may explain why these links were previously overlooked9,12. The US validation cohort generally confirmed these associations despite its smaller sample size: among the subset of derived pattern/index scores shared between the UK and US cohorts, of the 54 associations that were significant both in the UK cohort (FDR q<0.2) and in the US cohort (p<0.05), 70.4% were concordant.
Microbial indicators of obesity are reproducible across varied populations
Microbiome links to obesity have attracted much interest although results have varied in human populations3,4. Our machine learning (ML) approach (Methods) found visceral fat to be more strongly linked to gut microbial composition than BMI33, a finding again validated in US participants (Fig. 3A). Some obesity-associated taxa were also associated with poor dietary patterns after controlling for BMI (e.g. Clostridium CAG58, Flavonifractor plautii), whereas markers of lower visceral fat mass (e.g. Faecalibacterium prausnitzii) were more strongly linked to healthier foods and patterns of intake, illustrating that diet and obesity microbiome signatures overlap but are not identical (Fig. 3B).
Fig. 3: Random forest machine learning models trained on microbial or functional profiles are capable of predicting obesity phenotypic markers, even on independent cohorts.
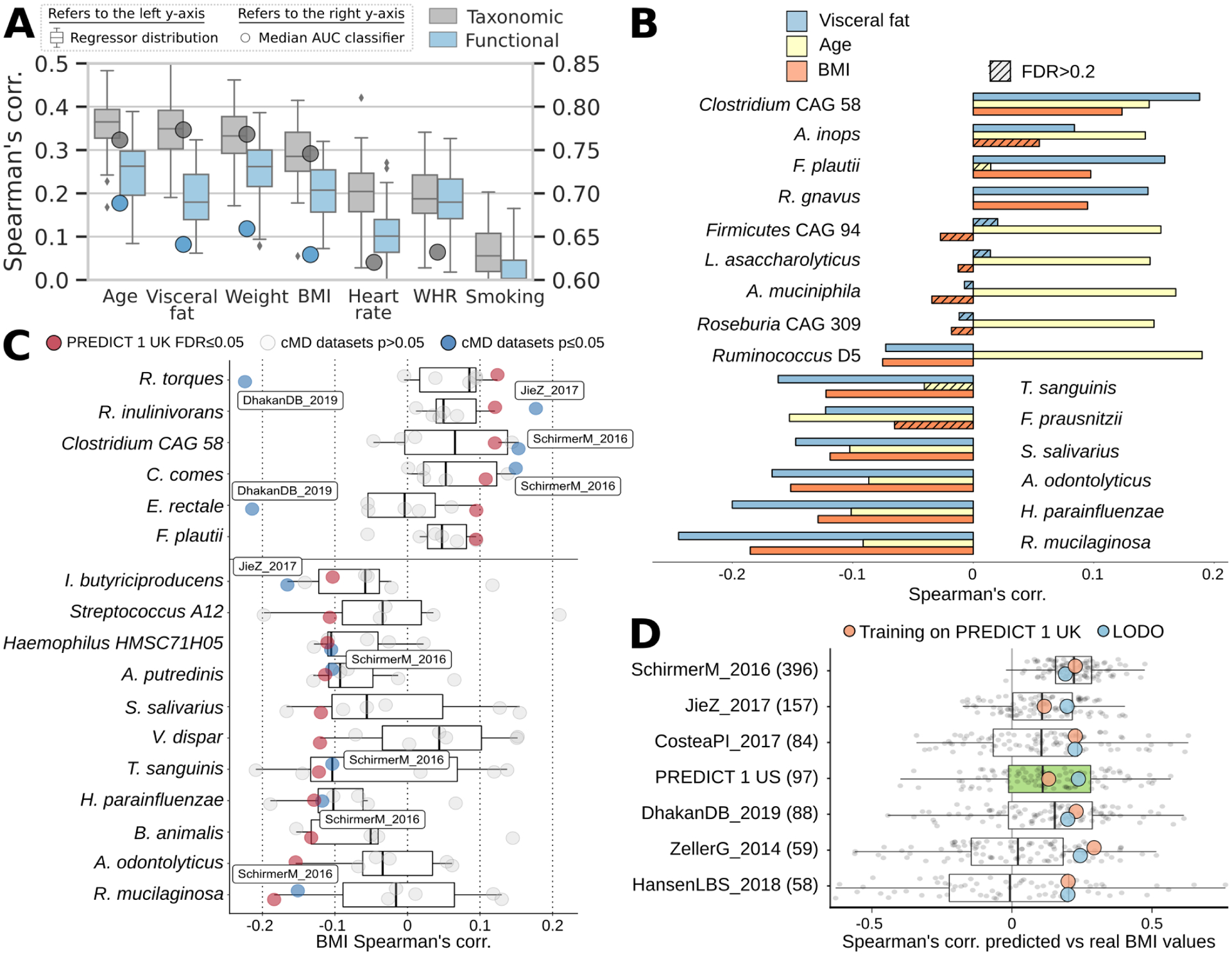
(A) Whole-microbiome machine learning models can assess personal factors with RF regression (boxplots and left-side y-axis) using only taxonomic or functional (i.e. pathway) microbiome features. Classification models (circles and right-side y-axis) exceed AUC 0.65 except for waist-to-hip ratio (WHR) and smoking. (B) We observed the highest correlations between the relative abundance of microbial species and age, BMI, and visceral fat. The link between microbial features and visceral fat was of greater effect and more often significant than with traditional BMI. (C) Using several independent datasets34 we confirmed correlations between single microbial species and BMI with blue points denoting significant associations at p<0.05. (D) The machine learning model for BMI trained on PREDICT 1 data is reproducible in several external datasets (Extended Data Fig. 5), achieving correlations with true values exceeding those obtained in cross-validation of a single given dataset in five of seven cases. When the PREDICT 1 microbiome model is expanded to include other datasets (excluding those ones used for testing, i.e. leave-one-dataset-out/LODO approach) the performance remains comparable, confirming the generalizability of the PREDICT 1 model on obesity-related indicators.
Microbiome models to predict BMI in the UK cohort were further validated in six independent datasets available in curatedMetagenomicData34 (Methods). Despite inter-population differences11,35, the UK model improved cohort-specific cross-validation accuracy in the majority of cases, on par with the leave-one-dataset-out approach (Fig. 3D). Of the 17 species surpassing q<0.05, three had an (absolute) ρ>0.1 in the US cohort and two of these were concordant with those in the UK cohort (Fig. 3C). Across harmonized independent datasets, all but two median associations were consistent with the PREDICT 1 UK signatures, and 12 of the 14 were concordant despite different sample collection and DNA extraction methods.
Fasting cardiometabolic markers associated with specific microbiome structures
To explore the connections between the gut microbiome and cardiometabolic health, we developed and evaluated microbiome-based ML models for each selected clinical and emerging cardiometabolic biomarker. We found modest concordance between microbiome models and several traditional clinical fasting cardiometabolic biomarkers (Fig. 4A) including blood pressure, lipids (TG, TC, HDL-C, LDL-C), fasting glucose, and glycosylated haemoglobin (HbA1c%) as well as a clinical prediction score estimating latent 10-year risk of heart disease (ASCVD score)36.
Fig. 4: Fasting and postprandial cardiometabolic responses to standardized test meals associated with the microbiome.
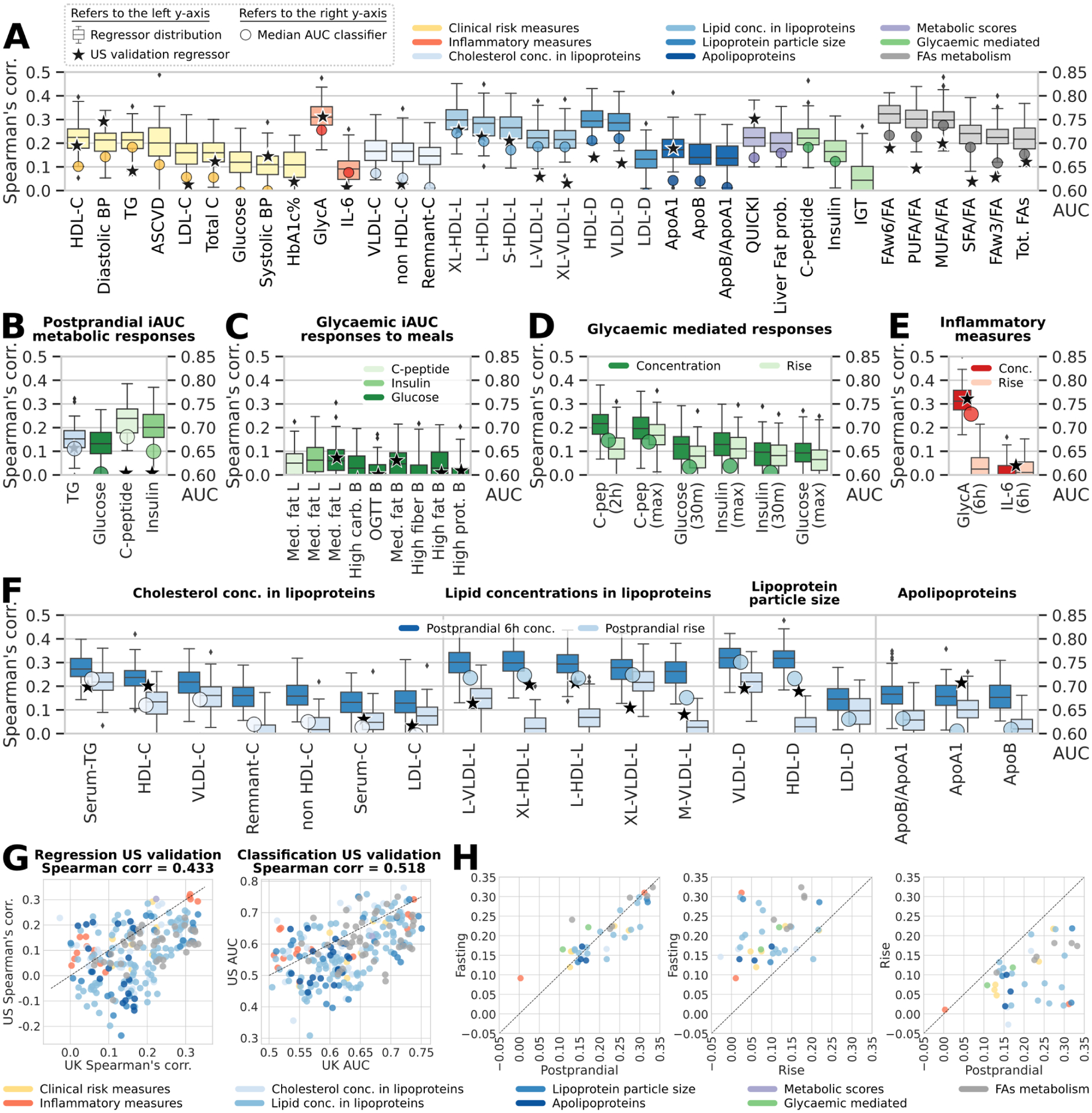
(A) The strongest observed links according to correlation of the predicted versus collected measures between the gut microbiome and fasting metabolic blood markers. For measures of lipid concentration in lipoproteins, we report the five strongest correlations only. Indices are grouped in nine distinct categories, and boxplots report the correlation between the prediction of RF regression models trained on microbial taxa or pathway abundances across 100 training/testing folds and stars report regressor performance when trained on the UK cohort and evaluated on the independent US validation cohort (left-side y-axis). Circles denote AUC values for RF classification (right-side y-axis) (B-F) Performance of our microbiome-based ML-model in estimating postprandial absolute levels and postprandial increases in cardiometabolic markers. Stars denote regression model results in our US validation cohort for postprandial measurements (not rises; Extended Data Fig. 4B-C). (B) RF regression and classification performance in predicting postprandial metabolic responses for clinic Meal 1 (breakfast) measured as iAUC at 6h for triglycerides and iAUC at 2h for glucose, C-peptide, and insulin. (C) Glycaemic-mediated postprandial iAUCs at 2h for the other meals (Supplementary Table 7), and (D) glycaemic-mediated markers absolute levels vs. rise. (E) Postprandial inflammatory measures (concentration and rise). (F) Postprandial lipoproteins measures (6h concentration and rise). (G) Overall agreement between RF regression and classification tasks for UK models applied to the independent US cohort. (H) RF microbiome-based model performance with postprandial changes (concentrations and rise) in lipoprotein concentration, composition, and size. Fasting and postprandial performance indices (correlation of the regressors’ outputs) were more tightly linked to gut community structure than were their corresponding postprandial rises.
For other blood biomarkers (Fig. 1A), we found stronger correlations between the microbiome and an inflammatory surrogate (GlycA, Fig. 4A), circulating polyunsaturated fatty acids (PUFA) (both omega-6 [FAω6/FA] and total PUFA [PUFA/FA] to total fatty acid ratios, ρ=0.3 and 0.32, respectively), as well as emerging lipid measures linked to host health, including HDL and VLDL particle size (-D, ρ=0.29 for both) and the lipid content of lipoprotein subfractions (including XL-HDL-L and L-HDL-L, ρ=0.3 and 0.28 respectively). GlycA and VLDL-D are associated with increased risk for metabolic syndrome, CVD, and T2D, whereas HDL-D and its lipid constituents, omega-6, and total PUFA have inverse associations37,38. Similarly, the majority of glycaemic indicators such as insulin and C-peptide were also coupled to human gut microbiome composition (ρ=0.17 and 0.22, respectively), as well as derived predictors of insulin sensitivity (QUICKI, ρ=0.22)39 and hepatic steatosis (Liver Fat Probability, ρ=0.2).
Species-based predictors proved more accurate than pathway abundance profiles (Extended Data Fig. 4A), consistent with other reports40. Our primary findings were generally replicated in the US cohort (Fig. 4A), corroborating the existence of a strong, previously overlooked link between the gut microbiome and surrogates of cardiometabolic health.
The gut microbiome is a better predictor of postprandial triglycerides and insulin concentrations than of glucose levels
Fasting blood assays are standard for research and clinical investigations; however, individuals consume multiple mixed-nutrient meals throughout the day and spend most of their waking hours in the postprandial state, resulting in repeated elevations in circulating TG, glucose and related metabolites8. Whilst postprandial glucose responses may, in part, be predicted by the gut microbiome 9, ‘real-life’ variations in both postprandial lipid and glucose-mediated metabolites have not been explored. We assessed them by considering the overall magnitude of the response by iAUC, peak concentrations, and change from fasting (i.e. rise).
Firstly, we measured postprandial TG, glucose, C-peptide, insulin, and circulating metabolite concentrations at regular intervals (0–6h) in the clinic after two sequential test meals (890 kcal, 50g fat and 85g carb at 0h [breakfast] and 500 kcal, 22g fat and 71g carb at 4h [lunch]; Fig. 4B–C). Notably, we found that postprandial TG (0–6h iAUC), insulin, and C-peptide (both 0–2h iAUC) responses were more strongly associated with the gut microbiome (ρ=0.15, 0.2, 0.24, respectively; AUCs >0.65) compared with postprandial glucose (0–2h iAUC) responses (ρ=0.13 and AUC 0.6, Fig. 4B), findings replicated in our US cohort (Fig. 4B–G). We also measured glucose concentrations during the 13-day at-home period16 following isocaloric standardized meals, with different macronutrient compositions (Supplementary Table 3). However, contrary to our clinic meal responses (Fig. 4B) and previous work9, the glucose 0–2h iAUCs following these meals did not achieve high correlations with the microbiome (all ρ<0.07 and AUC<0.59, Fig. 4C). Whilst this may be dependent on meal composition and the effect of multiple meals consumed after stool collection, these results suggest that the microbiome is a stronger predictor of postprandial lipaemia than glycaemia.
Postprandial rises in lipid- and glucose-mediated measures are differentially predicted by the microbiome compared with fasting levels
Postprandial measures depend both on the corresponding fasting levels and the meal-induced rise. Therefore, we compared the differential prediction accuracy of the gut microbiome for fasting levels, postprandial (peak) total levels, and postprandial rises (Fig. 4H). For lipid and glucose-mediated (clinic day) measures, despite a similar strength of association between peak (6h), magnitude (iAUC) and fasting TG concentrations, the rise (6–0h) was not similarly correlated (Fig. 4A–E–F). In contrast, the microbiome associations with glycaemic measures were comparable between fasting, peak, and rise (Fig. 4A–D).
Of particular interest were the lipoprotein subfraction concentrations, composition, and size (Extended Data Fig. 4B-C), which are remodelled postprandially into potentially atherogenic lipoproteins (e.g. Large VLDL particles and TG-enriched LDL, and HDL particles)41. These particles were predicted at comparable accuracy for both fasting and postprandial peak 6h concentrations (Fig. 4A–F–H), and notably, HDL-D and VLDL-D achieve modestly stronger correlations (ρ=0.32 for both) postprandially (Fig. 4F). However, as with TG, we found that the microbiome was substantially less predictive for the postprandial rise in all lipid metabolite measures compared with fasting and postprandial peak concentration (Fig. 4A–F–H). For example, HDL-D is closely associated with gut microbial composition at fasting and 6h postprandially (ρ=0.29 and 0.32; AUC 0.71 and 0.72 respectively; Fig. 4A–F–H), but not with the rise (Fig. 4F). These differential associations suggest that the microbiome may influence postprandial lipid-mediated measures via effects on fasting measures.
Distinct microbial signatures discriminate between positive and negative metabolic health indices under fasting conditions
Motivated by the observed potential of the gut microbiome to predict the fasting and postprandial levels of circulating metabolic markers, we next assessed the microbiome features driving these associations. Among three general risk indices of cardiovascular health (ASCVD, liver fat probability, and insulin sensitivity or QUICKI, Fig. 4A), we found six species significantly and concordantly correlated with all three (negatively or positively, p<0.05), hinting at a global underlying microbial signature of improved metabolic health. These taxa included Clostridium CAG58 (higher cardiometabolic risk) and Haemophilus parainfluenzae (lower risk) that we had previously linked with healthy and less-healthy dietary habits (Fig. 2B).
We found similarly distinct separations between two opposing clearly defined clusters of species either positively or negatively correlated with fasting cardiometabolic measures (Fig. 5A–B). Species correlated with positive markers included some prevalent taxa generally regarded as healthy (F. prausnitzii) but also eight uncultivated and under-characterized bacteria. The positive cluster included many distinct genera, pointing at a rich functional diversity. In contrast, the cluster negatively correlated with positive markers included eight Clostridium species and the recurrent negatively connotated R. gnavus and F. plautii. Large HDL particles (and their lipid compositions, Extended Data Figs. 6 and 7), which have strong inverse associations with cardiometabolic outcomes38, were associated with the healthy cluster. Conversely, lipoproteins associated with increased risk of CVD and T2D (VLDL of all sizes and lipid composition) and atherogenicity42 (S-LDL, M-HDL and S-HDL TG), were associated with the less-healthy cluster (Extended Data Figs. 6 and 7).
Fig. 5: Species-level segregation into healthy and unhealthy microbial signatures of fasting and postprandial cardiometabolic markers.
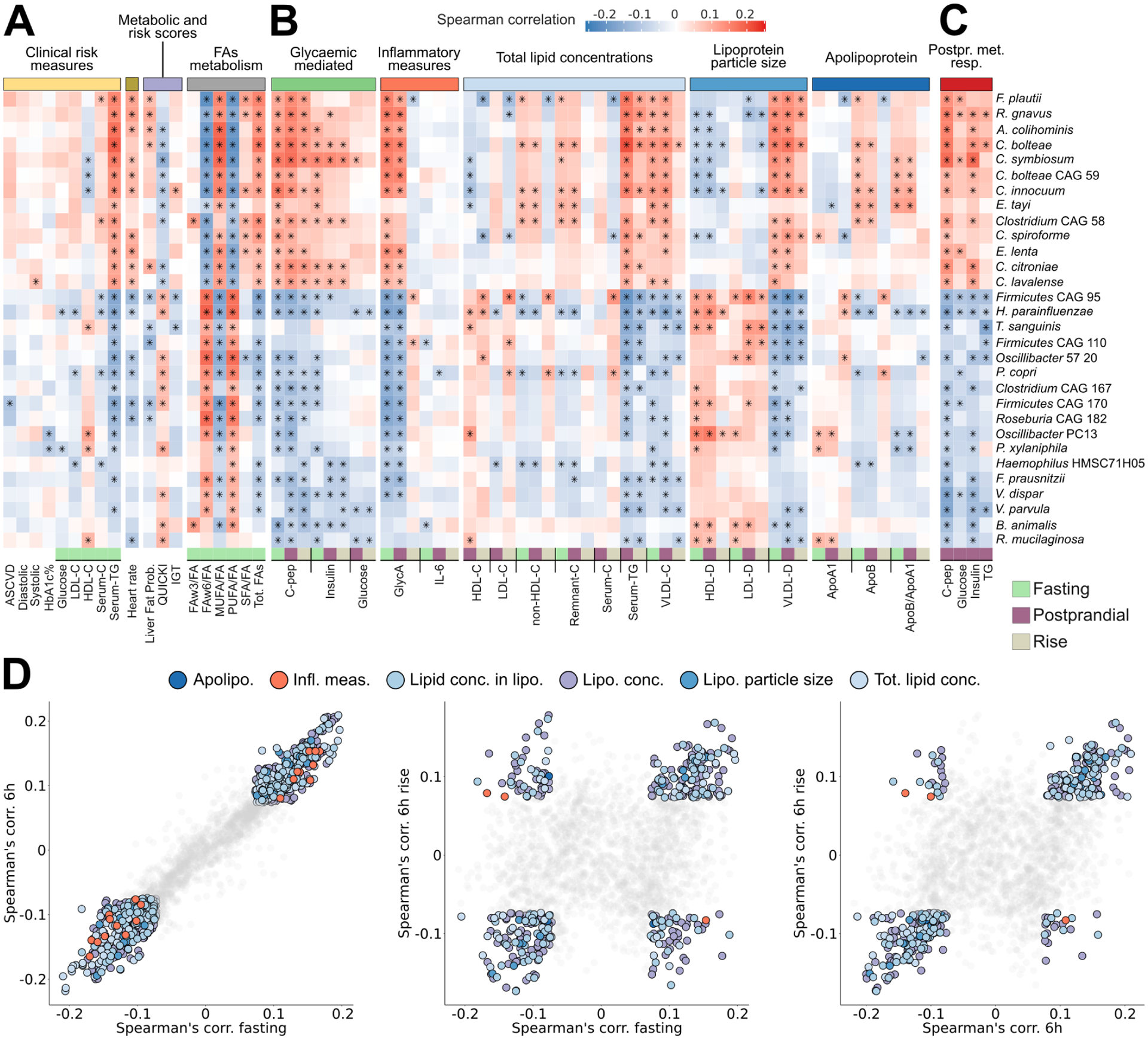
(A) Associations (Spearman’s correlation, q<0.2 marked with stars) between single microbial species and fasting clinical risk measures and (B) glycaemic, inflammatory, and lipaemic indices. (C) Correlation between microbial species and the iAUC for glucose and C-peptide estimations based on clinical measurements before and after standardized meals. The 30 species with the highest number of significant correlations with distinct fasting and postprandial indices are shown. Rows are hierarchically clustered (complete linkage, Euclidean distance). (D) Microbe-metabolite correlations are very consistent when evaluated for fasting versus postprandial (6h) conditions (left panel). Associations with postprandial variations (rise) conversely often show opposing relationships, with several species positively correlated with fasting measures being negatively correlated with postprandial variation of the same metabolite (or vice versa, central panel). This was mitigated somewhat when comparing absolute postprandial responses with rise (right panel).
Due to the lack of endogenous production of these fatty acids, circulating omega-6 and total polyunsaturated fatty acids (PUFA) that closely reflect dietary intake43 and are linked to a reduced risk of chronic disease38, were associated here with the healthy cluster (Fig. 5A and Supplementary Table 5). In contrast, circulating monounsaturated fatty acids (MUFA), which (unlike dietary MUFA) have been linked to increased risk of chronic disease38, were associated with the unhealthy cluster, with an under-characterized Firmicutes species (CAG 170) and Clostridium bolteae responsible for the strongest negative and positive associations respectively.
Both favourable and unfavourable microbial signatures of metabolic health were maintained under postprandial conditions
Links between postprandial levels of cardiometabolic and inflammatory measures corresponded with the segregation of healthful vs. detrimental taxa observed under fasting conditions (Fig. 5B and Extended Data Figs. 6 and 7). Notably, fasting and postprandial GlycA were strongly linked with the microbiome (47 species significantly correlated at 6 hours and 64 at fasting), substantially exceeding IL-6 (5 and 16 significant associations, Fig. 5B). C. boltae and R. gnavus were the most correlated with increased fasting and postprandial inflammation, whereas H. parainfluenzae and Firmicutes bacterium CAG95 were the strongest associations with reduced GlycA levels. VLDL lipoprotein subfractions (markers of adverse cardiometabolic effects) were also consistently associated with the less-healthy cluster both at fasting and postprandially.
Postprandial rises, rather than absolute postprandial levels, were in some cases uncoupled from the microbial associations with fasting markers (Fig. 5D). For example, the change in GlycA (Fig. 5B) was differentially associated with clusters compared to fasting and postprandial levels (especially for F. plautii, Firmicutes CAG95, and Firmicutes CAG110); likely due to the small reduction in GlycA postprandially. Other immunological markers and some lipid and cholesterol levels paralleled this behaviour (Extended Data Fig. 6), possibly reflecting postprandial lipoprotein remodelling44.
We observed the same “favourable” vs. “unfavourable” clustering of microbiome features when analyzing microbial pathways and gene families (Extended Data Fig. 8) supporting taxa segregation by their underlying biochemical activities. The strengths of microbe-blood marker associations were confirmed by RF feature relevance analysis (Extended Data Fig. 9) and, importantly, confirmed in the US cohort. For the 209 microbe-index correlations that were significant both in the UK cohort (q<0.2) and in the US cohort (p<0.05) the concordance in the sign of the correlation reached 88.7% for the associations in fasting conditions and 96.1% postprandially.
Prevotella copri diversity and Blastocystis presence are markers of improved postprandial glucose responses
Some ecologically unusual microbes hypothesized to have population-scale health effects solely based on their presence or absence appeared in our microbial signatures45. Among them, Prevotella copri45,46 had conflicting previous accounts for its role in glucose homeostasis47,48 possibly due to subspecies diversity49,50. Our data find P. copri to be associated with beneficial cardiometabolic markers, being negatively correlated with estimated visceral fat (ρ=−0.11, p=0.0006), fasting VLDL-D (ρ=−0.08, p=0.011), and fasting GlycA (ρ=−0.14, p<0.0001) among others (Supplementary Table 5). While almost no diet indices were associated with P. copri, postprandial rises in glucose (ρ=−0.11, p<0.001) and polyunsaturated/omega-6 fatty acids (ρ=0.15 and 0.14, respectively, and p<0.001) were top-scoring correlations for this bacterium and were stronger than corresponding fasting and postprandial levels, in contrast with what we observed for the overall microbiome (Fig. 4A–B). P. copri is present in at least one of its subtypes49 in 29.8% of the PREDICT 1 individuals, and P. copri-carriers had lower C-peptide (−9.2%, p=0.002), insulin (−14%, p=0.006), and TG levels (−3.2%, p=0.003) compared to P. copri-negative individuals (Extended Data Fig. 10 and Supplementary Table 8). Similarly, postprandial glucose after breakfast was significantly less pronounced in individuals with P. copri (−20.4% glucose iAUC at 2h, p=0.002, Extended Data Fig. 10C), and visceral fat was significantly lower (−12.5%, p=0.005, Extended Data Fig. 10A). This set of diverse associations supports that the presence of P. copri in the gut microbiome could be beneficial in glucose homeostasis and host metabolism.
Blastocystis is a unicellular eukaryotic parasite increasingly regarded as a commensal member of the gut microbiome51–53. It shares with P. copri a limited prevalence in Western-lifestyle populations53 and a high abundance when present. We found evidence that Blastocystis-positive individuals (25.7% in our cohort) also have favourable glucose homeostasis and lower estimated visceral fat (−15.7% glucose iAUC, −22.1% visceral fat, p<0.002, Extended Data Fig. 10). The latter confirms that Blastocystis is less prevalent in individuals with high BMI, as previously suggested53. Interestingly, the effect of the simultaneous presence of P. copri and Blastocystis (12% of the individuals) appears to further promote healthier metabolic function. Visceral fat is 17.3% lower on average (p<0.005, Supplementary Table 8) for individuals positive for both P. copri and Blastocystis compared to individuals with only one or the other and 23.3% lower (p=8.9E-6) compared with individuals lacking both.
A clear microbial signature of cardiometabolic health levels consistent across diet, obesity indicators, and cardiometabolic risks
We observed above a consistent set of microbial species that were strongly linked to (1) food indices reflecting different levels of “healthy” diets, (2) indicators of obesity and cardiometabolic health, (3) fasting circulating metabolites connected with cardiometabolic risk, and (4) postprandial responses. To test the consistency of such a signature, we selected representative cardiometabolic “health” indicators from each category and ranked microbial species based on their correlation coefficient. We found remarkable agreement among microbes associated with different positive or negative indicators of cardiometabolic health (Fig. 6, Supplementary Table 9).
Fig. 6: The panel of 30 species showing the strongest overall correlations with a selection of markers of nutritional and cardiometabolic health.
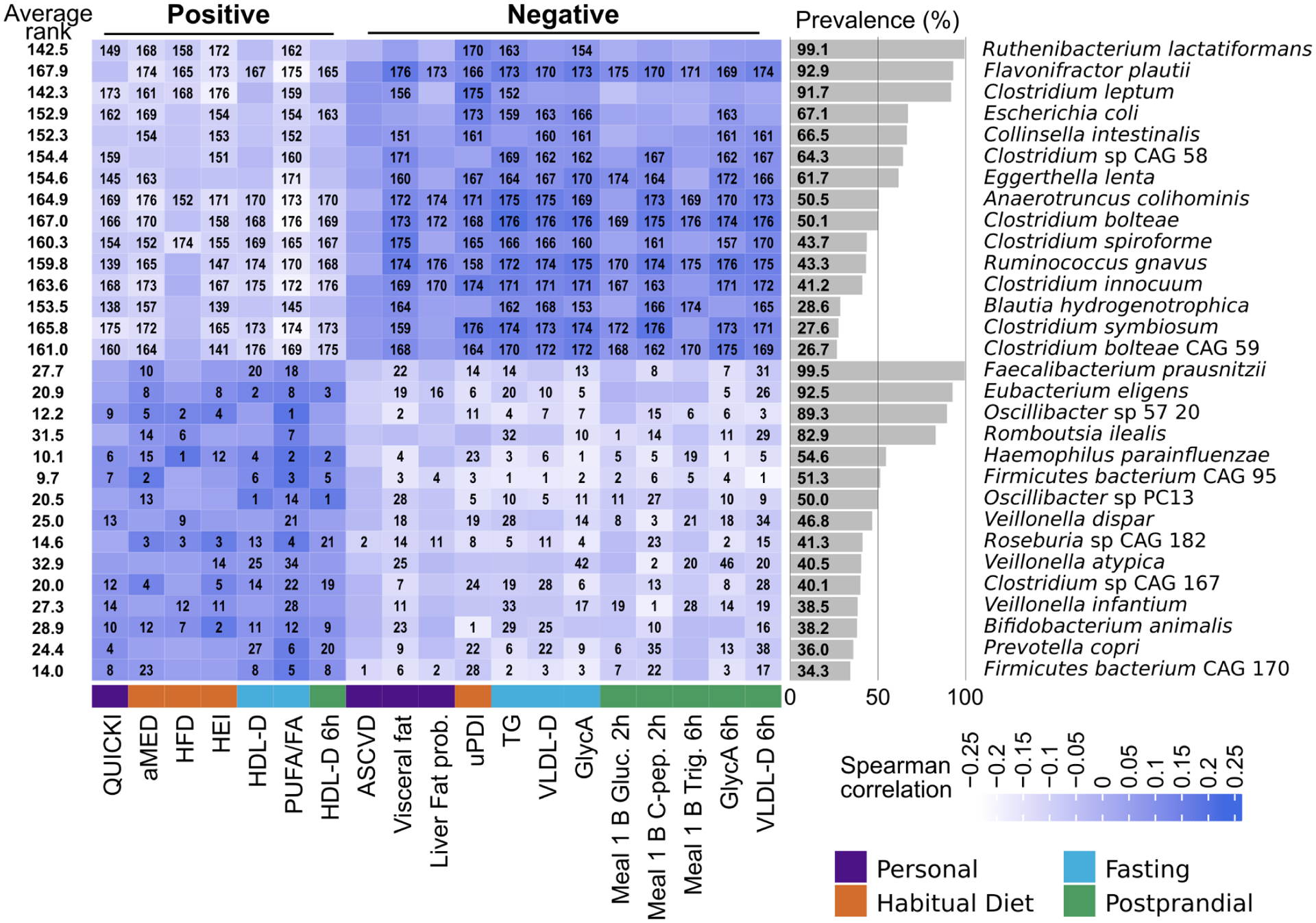
The 30 species with the highest and lowest average ranks with diverse positive and negative cardiometabolic health and healthy diet indicators, respectively, are shown here. The rank of each microbe’s correlation with individual indicators is written within cells when significant (p<0.05). For each of the main categories of indices, we selected up to five representative markers (for “Personal” we considered only four as the remaining were highly correlated with visceral fat or not relevant in this context). Indices can be considered “positive” and “negative” depending on whether higher or lower values are a proxy for more or less healthy conditions.
In particular, Firmicutes CAG95 is the uncultivated species with the most beneficial score. Of the “health”-associated microbial species only F. prausnitzii, and - partially - P. copri were already convincingly linked with health in previous investigations54. The beneficial signature also included E. eligens and H. parainfluenzae without previous clear roles in health, and additional species without cultivated representatives such as Roseburia CAG182, Oscillibacter sp 57_20, Firmicutes bacterium CAG170, Oscillibacter sp PC13, Clostridium sp CAG167, and Roseburia CAG182. Species conversely consistent with indicators of poor overall health (Fig. 6) included the already discussed set of Clostridia (C. spiroforme, C. bolteae CAG59, C. bolteae, Clostridium CAG58, C. symbiosum, C. innocuum, C. leptum) and the mucolytic microbe R. gnavus and F. plautii, again previously found to be associated with disease conditions55,56. Overall, this set of 30 species serves as a marker of overall good or poor cardiometabolic health and dietary patterns in non-diseased human hosts.
Discussion
PREDICT 1 represents the first diet-microbiome study to identify both individual components of the microbiome and an overall gut microbial signature associated with multiple measures of dietary intake and cardiometabolic health. These signatures reproduced across UK and US populations, across multiple previously-published study populations, and for multiple dietary and health indicators. Notably, microbiome signatures grouped both microbiome and dietary components into health-associated and anti-associated clusters, the latter in agreement with dietary quality and diversity scores20,57. The diversity of a healthy diet (HFD and PDI) was particularly predictable by the microbiome, surpassing other indices including the Mediterranean diet previously linked with microbiome composition58. The segregation of favourable and unfavourable microbial clusters according to the heterogeneity of the food source (healthy or unhealthy animal or plant), quality (processed vs unprocessed), and dietary patterns highlights the importance of looking beyond nutrients and single foods in diet-microbiome research. The substantially greater detail and consistency in our results relative to prior diet-microbiome work9,11–13,15 may be due to the quality in the dietary recording, metagenomic profiling, and the large sample size. However, given the limitations of FFQ dietary data, future diet-microbiome studies would benefit further from higher resolution dietary assessment methodologies such as weighed food record data.
Several aspects of the consistent gut microbiome signatures across diet, obesity, and cardiometabolic health measures are striking for their potential novel epidemiology and microbial biochemistry. A surprising proportion of diet- or health-associated taxa in these results are largely uncharacterized or represented solely by metagenomic assemblies5. Other microbes found here to have dietary or cardiometabolic associations, such as Prevotella spp. or Blastocystis spp., have been characterized in greater biochemical detail, but their population structure in the human microbiome have only recently begun to be appreciated49,53. The latter in particular may be only one of many examples of non-bacterial microbiome members not amenable to most current high-throughput approaches, but with unexpected and potentially key positive roles in humans.
Likewise, these new contributions of the gut microbiome to human dietary responses may help to explain some of the heterogeneity seen among previous population studies4,9,59. First, diet-microbiome-blood marker associations were overall strongest with respect to circulating lipid levels relative to glycemic indices. It is possible that the relative contribution of gut microbes is higher for circulating lipid levels than carbohydrate derivatives, through either direct processes or indirectly through gastrointestinal or systemic bile acid signalling60. Alternatively, host metabolism may play a greater role in circulating glucose and insulin levels relative to microbial bioactivity. The lipoprotein features most closely associated with the microbiome (such as L-HDL-L) are also more strongly associated with cardiovascular risk compared with typically measured lipids (e.g. TC, HDL-C, LDL-C), suggesting that their utility as clinical biomarkers or as targets for beneficial gut microbiome manipulation warrants further investigation.
Overall, this is the first study to identify a shared diet-metabolic-health microbial signature, segregating favourable and unfavourable taxa with multiple measures of both dietary intake and cardiometabolic health. These results will aid as a resource both in utilization of the gut microbiome as a biomarker for cardiometabolic risk and in strategies for reshaping the microbiome to improve personalized dietary health.
Methods
The PREDICT 1 study
The PREDICT 1 clinical trial (NCT03479866) aimed to quantify and predict individual variations in metabolic responses to standardised meals. We integrated data from a cohort of twins and unrelated adults from the UK to explore genetic, metabolic, microbiome composition, meal composition, and meal context data to distinguish predictors of individual responses to meals. We then validated these predictions in an independent cohort of adults from the US. The trial was a single-arm, single-blinded intervention study that commenced in June 2018 and completed in May 2019.
For full protocol, see Berry et al., 202016. In brief; 1,002 generally healthy adults from the United Kingdom (UK; non-twins, and identical [monozygotic; MZ] and non-identical [dizygotic; DZ] twins) and 100 healthy adults from the United States (US; non-twins; validation cohort) were enrolled into the study (see Berry et al.8 for eligibility criteria) and completed baseline clinic measurements. The study consisted of a 1-day clinical visit at baseline followed by a 13-day at-home period. At baseline (Day 1), participants arrived fasted and were given a standardised metabolic challenge meal for breakfast (0h; 86g carbohydrate, 53g fat) and lunch (4h; 71g carbohydrate, 22g fat). Fasting and postprandial (9 timepoints; 0–6h) venous blood was collected to determine serum concentrations of glucose, triglycerides (TG), insulin, C-peptide (as a surrogate for insulin), and metabolomics (NMR). Stool samples, anthropometry, and a questionnaire querying habitual diet, lifestyle and medical health were obtained at baseline. During the home-phase (Days 2–14), participants consumed standardised test meals in duplicate varying in sequence and in macronutrient composition, while wearing digital devices to continuously monitor their blood glucose (continuous glucose monitor; CGM), physical activity, and sleep. Capillary blood was collected using dried blood spot cards, during the clinic visit and at home, to analyze fasting and postprandial concentrations of TG and C-peptide. Participants were supported throughout the study with reminders and communication from study staff delivered through the ZOE study app. A second stool sample was collected at home by participants following completion of the study, and all devices and samples were mailed back to study staff. To monitor compliance, all test meals consumed by participants were logged in the Zoe app (with an accompanying picture) and reviewed in real-time by the study nutritionists. Only test meals that were consumed according to the standardised meal protocol (outlined in Berry et al. 20208) were included in the analysis.
The recruitment criteria, meal intervention challenges, outcome variables, and sample collection and analysis procedures relevant to this paper are described elsewhere8,16. The trial was approved in the UK by the Research Ethics Committee and Integrated Research Application System (IRAS 236407) and in the US by the Partners Healthcare Institutional Review Board (IRB 2018P002078). The core characteristics of study participants at baseline were not significantly different between UK and US cohorts8.
Overview of microbiome sequencing and profiling
We performed deep shotgun metagenomic sequencing (mean 8.8±2.2 gigabases/sample) in stool samples from a total of 1,098 PREDICT 1 participants (UK n=1,001; US n=97). From a random subset of these participants (n=105), we additionally sequenced faecal metagenomes from a second stool sample collected 14 days after the first collection (Fig. 1A) for a total of 1,168 metagenomes. Computational analysis was performed using the bioBakery suite of tools61 to obtain species-level microbial abundances for the 769 taxa identified using the newly updated MetaPhlAn 2.96 tool62, functional potential profiling of >1.91 M microbial gene families, 445 KEGG pathways with HUMAnN 2.063, and reconstruction of 48,181 metagenome-assembled genomes (MAGs) of medium or high-quality using our validated pipeline5, which includes assembly with MegaHIT64, binning with MetaBAT265, and quality-control with CheckM66.
Microbiome sample collection
Participants were mailed a pre-visit study pack with a stool collection kit and relevant questionnaires and asked to collect an at-home stool sample at two timepoints (one prior to their in-person clinical visit on day 0 and the next at the conclusion of their home-phase, day 14). Those who did not collect a sample prior to their in-person, baseline visit completed the collection as soon as possible during the home-phase. Baseline samples in the UK were collected using the EasySampler collection kit (ALPCO, NH, US), whereas post-study samples, as well as the entirety of the US collection was conducted using the Fecotainer collection kit (Excretas Medical BV, Enschede, the Netherlands). For baseline samples, one fresh unfixed sample was deposited into a sterile universal collection container (Sarstedt, Australia, Cat #L0263-10) and one into a tube containing DNA/RNA Shield buffer (Zymo Research, CA, US, Cat #R1101). Samples were stored at ambient temperature until returned to the study staff. Follow-up samples were collected similarly, but only sampled into a DNA/RNA Shield buffer tube and sent by standard mail to study staff. Upon receipt in the laboratory, samples were homogenized, aliquoted, and stored at −80°C in Qiagen PowerBeads 1.5 mL tubes (Qiagen, Germany). This sample collection procedure was tested and validated internally comparing different storage conditions (fresh, frozen, buffer), different DNA extraction kits (PowerSoilPro, FastDNA, ProtocolQ, Zymo), and different sequencing technologies (16S rRNA, shotgun metagenomics, and arrays), data not shown.
DNA extraction and sequencing
DNA was isolated by QIAGEN Genomic Services using DNeasy® 96 PowerSoil® Pro from all Day 0 (baseline) DNA/RNA shield fixed microbiome samples. A random subset of Day 14 (end of at-home phase) samples (n=105) were also extracted. Optical density measurement was done using Spectrophotometer Quantification (Tecan Infinite 200). Before library preparation and sequencing, the quality and quantity of the samples were assessed using the Fragment Analyzer (Agilent Technologies, Inc.) according to manufacturer’s guidelines. Samples with a high-quality DNA profile were further processed. The NEBNext® Ultra II FS DNA module (cat# NEB #E7810S/L) was used for DNA fragmentation, end-repair, and A-tailing. For adapter ligation, the NEBNext® Ultra II Ligation module (cat# NEB #E7595S/L) was used. The quality and yield after sample preparation were measured with the Fragment Analyzer. The size of the resulting product was consistent with the expected size of approximately 500–700 bp. Libraries were sequenced for 300 bp paired-end reads using the Illumina NovaSeq6000 platform according to manufacturer’s protocols. 1.1 nM library was used for flow cell loading. NovaSeq control software NCS v1.5 was used. Image analysis, base calling, and the quality check was performed with the Illumina data analysis pipeline RTA3.3.5 and Bcl2fastq v2.20.
Metagenome quality control and pre-processing
All sequenced metagenomes were QCed using the pre-processing pipeline as implemented in https://github.com/SegataLab/preprocessing. Pre-processing consists of three main steps: (1) read-level quality control; (2) screening of contaminants, i.e. host sequences; and (3) split and sorting of cleaned reads. Initial quality control involves the removal of low-quality reads (quality score <Q20), fragmented short reads (<75 bp), and reads with >2 ambiguous nucleotides. Contaminant DNA was identified using Bowtie 2 (Langmead and Salzberg 2012) using the --sensitive-local parameter, allowing confident removal of the phiX174 Illumina spike-in and human-associated reads (hg19). Sorting and splitting allowed for the creation of standard forward, reverse, and unpaired reads output files for each metagenome.
Microbiome taxonomic and functional potential profiling
The metagenomic analysis was performed following the general guidelines67 and relying on the bioBakery computational environment61. The taxonomic profiling and quantification of organisms’ relative abundances of all metagenomic samples have been quantified using MetaPhlAn 3.062. The updated species-specific database of markers was built using 99,237 reference genomes representing 16,797 species retrieved from Genbank (January 2019). From this set of reference genomes, we extracted a total of 1,132,166 markers used to profile 13,393 species. This set of species included also 83 species defined by the co-abundant gene (CAG) groups approach32 that were very genetically distinct from species represented by isolate genomes and for which the use of unique marker genes limited the potential issues of using metagenomic assemblies reconstructed over multiple samples. Compared to the previous version of the MetaPhlAn2 database (mpa_v20_m200), the updated database is able to profile 7,116 more species. Metagenomes were mapped internally in MetaPhlAn 3.0 against the marker genes database with BowTie2 version 2.3.4.3 with the parameter “very-sensitive”. The resulting alignments were filtered to remove reads aligned with a MAPQ value <5, representing an estimated probability of the likelihood of the alignments.
For estimating the microbiome species richness of an individual from the taxonomic profiles of PREDICT 1 participants, we computed two alpha diversity measures: the number of species found in the microbiome (“observed richness”), and the Shannon entropy estimation. We did not perform rarefaction prior to alpha diversity calculations because of the low standard deviation in sequencing depths and the verified missing correlation between metadata of interest and sequencing depth. Microbiome dissimilarity between participants (beta diversity) was computed using the Bray-Curtis dissimilarity on microbiome taxonomic profiles.
Functional potential analysis of the metagenomic samples was performed using HUMAnN2 (version 0.11.2 and UniRef database release 2014–07)63 that computed pathway profiles and gene-family abundances.
Metagenomic assembly
Metagenomic samples were processed to obtain metagenome-assembled genomes (MAGs) following the procedure we used elsewhere 5. In brief, we used MEGAHIT (version 1.2.9)64 with parameters “--k-max 127” for assembly and assembled contigs ≥1.5kb were considered for the binning step performed using MetaBAT2 (version 2.14)65 with parameters: “-m 1500 --unbinned”. Quality control of the obtained MAGs was performed using CheckM (version 1.0.18)66 using default parameters. High-quality and medium-quality microbial genomes were integrated into the existing database of >150,000 human MAGs.
Collection and processing of habitual diet information
Habitual diet information was collected using food frequency questionnaires (FFQ). For the UK, the European Prospective Investigation into Cancer and Nutrition (EPIC) FFQ was used and in the US, the Harvard semi-quantitative FFQ was used.
For the UK, we used an adapted 131-item EPIC FFQ that was developed and validated against pre-established nutrient biomarkers for the EPIC Norfolk68. The questionnaire captured average intakes in the past year. UK nutrient intakes were determined using FETA software to calculate macro- and micro- nutrient data69. 16 additional foods in the modified FFQ were reviewed by two dietitians and one nutritionist who manually matched food items to corresponding foods within McCance and Widdowson’s 7th Edition, with portions allocated according to the Food Standards Agency 3rd Edition ‘Food Portion Sizes’. US participants completed the Harvard 2007 Grid 131-item FFQ previously validated against two-week dietary records70. Nutrient intakes were estimated using the Harvard Nutrient Database. Submitted FFQs were excluded if greater than 10 food items were left unanswered, or if the total energy intake estimate derived from FFQ as a ratio of the subject’s estimated basal metabolic rate (determined by the Harris-Benedict equation71) was more than two standard deviations outside the mean of this ratio (<0.52 or >2.58).
The following dietary indices were calculated as described below and according to categorisation listed in Supplementary Tables 2 and 4.
Healthy Food Diversity Index.
The Healthy Food Diversity (HFD) index considers the number, distribution, and health value of consumed foods. To obtain this index, food frequency questionnaire foods were first aggregated into 15 food groups according to the HFD20. Health values were then derived from the German Nutrition Society (DGE) dietary guidelines (https://www.dge.de/en/) and the weight of each food group was multiplied by its corresponding health value (hv). Scores were divided by the maximum (hv=0.26) to bind values between 0–1 before multiplication with the Berry-Index. The original HFD was used instead of the US-HFD for the following reasons: the original HFD gives greater emphasis to plant-based foods and less to meat than the US-HFD which would more closely align with hypothesised microbiome-plant food/fibre interactions, and converting UK g/serving to US volume measures (as required for the US-HFD) would introduce additional error to the FFQ estimates.
Healthy Eating Index 2010.
The Healthy Eating Index 2010 (HEI-2010)21 assesses to which extent an individual’s food intake aligns with the Dietary Guidelines for Americans 201072, developed by the US Department of Agriculture. These guidelines cover a total of twelve food groups and nutrients. The HEI has 9 adequacy (encouraged) and 3 moderation (discouraged) components; firstly a density approach is used to set per 1000 calories; and secondly least-restrictive standards are employed, i.e., those that are easiest to achieve among recommendations that vary by energy level, sex, and/or age. Total fruits, whole fruits, total vegetables, greens and beans, whole grains, dairy (lean portion only), total protein foods (lean portion of meat only), seafood and plant proteins and fatty acids (PUFAs + MUFAs/SFAs) are considered adequate, whereas refined grains, sodium and empty calories (considered added sugars, solid fats, and alcohol above 13g per 1000kcal) are considered detrimental and should be consumed in moderation. The index ranged from 0 (not in agreement with the guidelines) to 100 (completely in agreement with guidelines).
The plant-based diet index.
Three versions of the plant-based diet index30 were considered: the original plant-based diet index (PDI), the healthy plant-based index (h-PDI), and the unhealthy plant-based index (u-PDI). Eighteen food groups (amalgamated from the FFQ food groups; Supplementary Table 2) were assigned either positive or reverse scores after segregation into quintiles, as outlined in Supplementary Table 430. Participants with an intake above the highest quintile for the positive score received a score of 5. Those below the lowest quintile intake received a score of 1. A reverse value was applied for the reverse scores. The scores for each participant were summed to create the final score. For the PDI, a positive score was applied to the “healthy” and “less-healthy”/“unhealthy” plant foods, and a reverse score applied to the animal-based foods. For the h-PDI, positive scores were applied to the “healthy” plant foods, and a reverse score to the “less-healthy”/“unhealthy” plant foods and the animal-based foods. For the u-PDI, a positive score was applied to the “less-healthy”/“unhealthy” plant foods and a reverse score applied to the “healthy” plant foods and the animal-based foods.
Animal score.
The animal-based score categorised animal foods into “healthy” and “less-healthy”/“unhealthy” categories according to previous epidemiological studies73–81. A similar approach to the PDI scoring was applied to the animal-based food groups, with either a positive (“healthy”) or reverse (“less-healthy”/“unhealthy”) quintile scoring (Supplementary Tables 2 and 4).
The aMED score.
Adherence to the aMED diet was calculated by following the method outlined by Fung et al. 22. Nine food/nutrient categories were included (Supplementary Table 4) and the score ranged from 0 to 9 (“least” to “most” Mediterranean). To form groups, weekly intake frequencies were first multiplied for assigned foods by the amount in grams per serving and then divided by 7 to determine grams per day. Next, food gram amounts were summed to make the final category total. For all food categories as well as the fatty acid intake ratio, the median intake of each category was calculated. A score of 0 (no aMED) or 1 (aMED) was given for each category depending on whether the participant was above or below the median intake. For alcohol intake, a range was used for score assignment: females: 5–25 g/d; males:10–50 g/d were assigned a score of 1, while those above or below this range were assigned a score of 0. Finally, the aMED was then generated by summation of each category score.
Food groups.
For individual analyses of food groups-microbe interaction, food groups were formed by aggregation of FFQ foods into the 18 PDI food groups plus margarine and alcohol (Supplementary Table 4).
Percentage of plants within diet:
The percentage of plants within diet was calculated as weight in grams of plant foods within total weight (g) of diet after adjustment of FFQ foods into quantities (g) per week.
Number of plant foods.
For the number of plant foods, each plant food item within the FFQ above the value of 0g was allocated a score of 1 and summed for each participant. For the total number of plants and the number of “healthy” and “unhealthy” plants, FFQ food items were allocated into groups according to the PDI food groupings.
Collection and processing of fasting and postprandial markers
Venous blood samples were collected as outlined in the accompanying protocol paper82. In brief, participants were cannulated, and venous blood was collected at fasting (prior to a test breakfast) and at 9 timepoints postprandially (15, 30, 60, 120, 180, 240, 270, 300, and 360 minutes). Plasma glucose and serum C-peptide and insulin were measured at all timepoints. Serum TG was measured at hourly intervals, and serum metabolomics (NMR by Nightingale Health, Helsinki, Finland, using the 2020 platform) at 0, 4 and 6h. Fasting samples were analysed for lipid profile, thyroid-stimulating hormone, alanine aminotransferase, liver function panel, and complete blood count (CBC) analysis.
Continuous glucose monitoring on day 2–14 was measured every 15 minutes using Freestyle Libre Pro continuous glucose monitors (Abbott, Abbott Park, IL, US), fitted on the upper, non-dominant arm at participants’ baseline clinical visit. Given the CGM device requires time to calibrate once fitted to a participant, CGM data collected 12 hours and onwards after activating the device was used for analysis.
Dry blood spot analysis of TG and C-peptide was completed by participants on the first 4 days of the home-phase while consuming test meals. The timepoints were dependent on the test meal as described elsewhere8,82. Test cards were stored in aluminum sachets with desiccant once completed and placed in the refrigerator at the end of the study day or until participants mailed them back to the study site. DBS cards were frozen at −80 °C upon receipt in the laboratory until being shipped to Vitas for analysis (Vitas Analytical Services, Oslo, Norway).
Specific timepoints and increments for TG, glucose, insulin, and C-peptide were selected for the current analysis to reflect the different pathophysiological processes for each measure as described in our protocol82. The incremental area under the postprandial TG (0–6h), glucose (0–2h), and insulin (0–2h) curves (iAUC) were computed using the trapezium rule83.
Detailed descriptions of sample collection, processing and analysis have been reported in8,16.
Machine learning
The machine learning (ML) framework employed is based on the scikit-learn Python package84. The ML algorithms used for the prediction and classification of personal, habitual diet, fasting, and postprandial metadata are based on Random Forest (RF) regressor and classification. We selected RF-based methods a priori as it has been repeatedly shown to be particularly suitable and robust to the statistical challenges inherent to microbiome abundance data40,85. For both the regression and classification tasks, a cross-validation approach was implemented, based on 100 bootstrap iterations and an 80/20 random split of training and testing folds. To specifically avoid overfitting as a result of our twin population and their shared factors, we removed any twin from the training fold if their twin was present in the test fold.
For the regression task, we trained an RF regressor to learn the feature to predict, and simple linear regression to calibrate the output for the test folds on the range of values in the training folds. From the scikit-learn package, we used the RandomForestRegressor with “n_estimators=1000, criterion=‘mse’, max_features=‘sqrt’” parameters and LinearRegression with default parameters. For the classification task, we divided the continuous features into two classes: the top and bottom quartiles. From the scikit-learn package we used the RandomForestClassifier function with “n_estimators=1000, max_features=‘sqrt” parameters.
We used RF classification and regression on both species-level taxonomic relative abundance and functional potential profiles. For taxonomic abundances, we used the species-level relative abundances as estimated by MetaPhlAn 3.0 (see above normalized using the arcsin-sqrt transformation for compositional data). For functional profiles, we considered both raw relative abundance estimates of single microbial gene families, as well as pathway-level relative abundance as provided by HUMAnN2.
As an additional control, we verified that when randomly swapping the target labels or values (classification and regression, respectively), the performances were reflecting a random prediction, hence an AUC very close to 0.5 and a non-significant correlation between the real and predicted values approaching 0.
Statistical analysis
Spearman’s correlations (reported with “ρ” in the text), have been computed using the cor.test from the stats R package and a modified version of the pcor.test from the ppcor package (available at http://www.yilab.gatech.edu/pcor.R) that permits to control for a set of covariates rather than single ones, respectively. Correlations and p-values were computed for each couple of metadata and species, and p-values were corrected using FDR through the Benjamini-Hochberg procedure, which are reported in the text as q-values. We considered significant correlations with a q<0.2. Significant species have been selected by ranking them according to their number of significant associations for the panel of metadata considered, and then the top thirty unique species are considered for each panel of metadata. In the heatmaps for partial correlations, the asterisk indicates that the correlation index for the corresponding species-metadata pair is significant at FDR≤0.2.
The contribution of metadata variables to microbiota community variation was determined by distance-based redundancy analysis (dbRDA) on species-level Bray-Curtis dissimilarity and Aitchison distance with the capscale function in the vegan R package86. Correction for multiple testing (Benjamini–Hochberg, FDR) was applied and significance was defined at FDR <0.1. The cumulative contribution of metadata variables or metadata categories was determined by forward model selection on dbRDA (stepwise dbRDA) with the ordiR2step function in vegan, with variables that showed a significant contribution to microbiota community variation in the previous step. Because of the high consistency between the two distance functions, we performed the cumulative distribution analysis using the Bray-Curtis dissimilarity. Only metadata variables with <15% missing data and without high collinearity with other variables (Spearman’s rho <0.8) were used as input in the stepwise model.
Data validation on the US cohort and on the cMD datasets
As independent validation, we considered the publically available datasets collected in the curatedMetagenomicData version 1.16.0 R package (cMD)34. Of the 57 datasets available we selected those that have samples with the following characteristics: (1) gut samples collected from healthy adult individuals at first collection (“days_from_first_collection”=0 or NA), (2) samples with age and BMI data available and BMI interquartile range (IQR) of these samples between 3.5 and 7.5 (± 2 with respect to the PREDICT 1 UK IQR of 5.5, Extended Data Fig. 5). For each dataset with samples meeting the above criteria, only datasets with at least 50 samples were considered: CosteaPI_201787 (84 samples out of 279), DhakanDB_201988 (88 samples out of 110), HansenLBS_201889 (58 samples out of 208), JieZ_201790 (157 samples 385), SchirmerM_201614 (396 samples out of 471), and ZellerG_201491 (59 samples out of 199).
We used the previously selected validation datasets from cMD in two analyses: one based on machine learning to verify the reproducibility of the ML model we trained using the PREDICT 1 UK samples, and the second to verify the species-level correlations found in the PREDICT 1 UK cohort. For the first task, we applied a regression algorithm to predict BMI and age. Three different cross-validation approaches were used. First, using each dataset independently in 100 bootstrap iterations and an 80/20 random split of training and testing folds. Second, one more iteration was performed using the PREDICT 1 UK dataset as training fold and each dataset as testing fold. Third, a final prediction was made using Leave-One-Dataset-Out cross-validation (LODO), meaning that all datasets (PREDICT 1 UK, PREDICT 1 UK, and the cMD datasets) were considered together and each validation dataset was successively used as the test fold while all others were used for training. An additional validation performed using the cMD datasets was done by applying a pairwise Spearman correlation for each species in each cMD dataset against BMI and age. For each correlation we selected the top associated species in PREDICT 1 UK (FDR q<=0.05) and reported their correlation in cMD. For those species found also in the PREDICT 1 US, we reported their correlation as well.
Extended Data
Extended Data Fig. 1. Alpha diversity linked with personal factors, habitual diet, fasting, and postprandial markers.
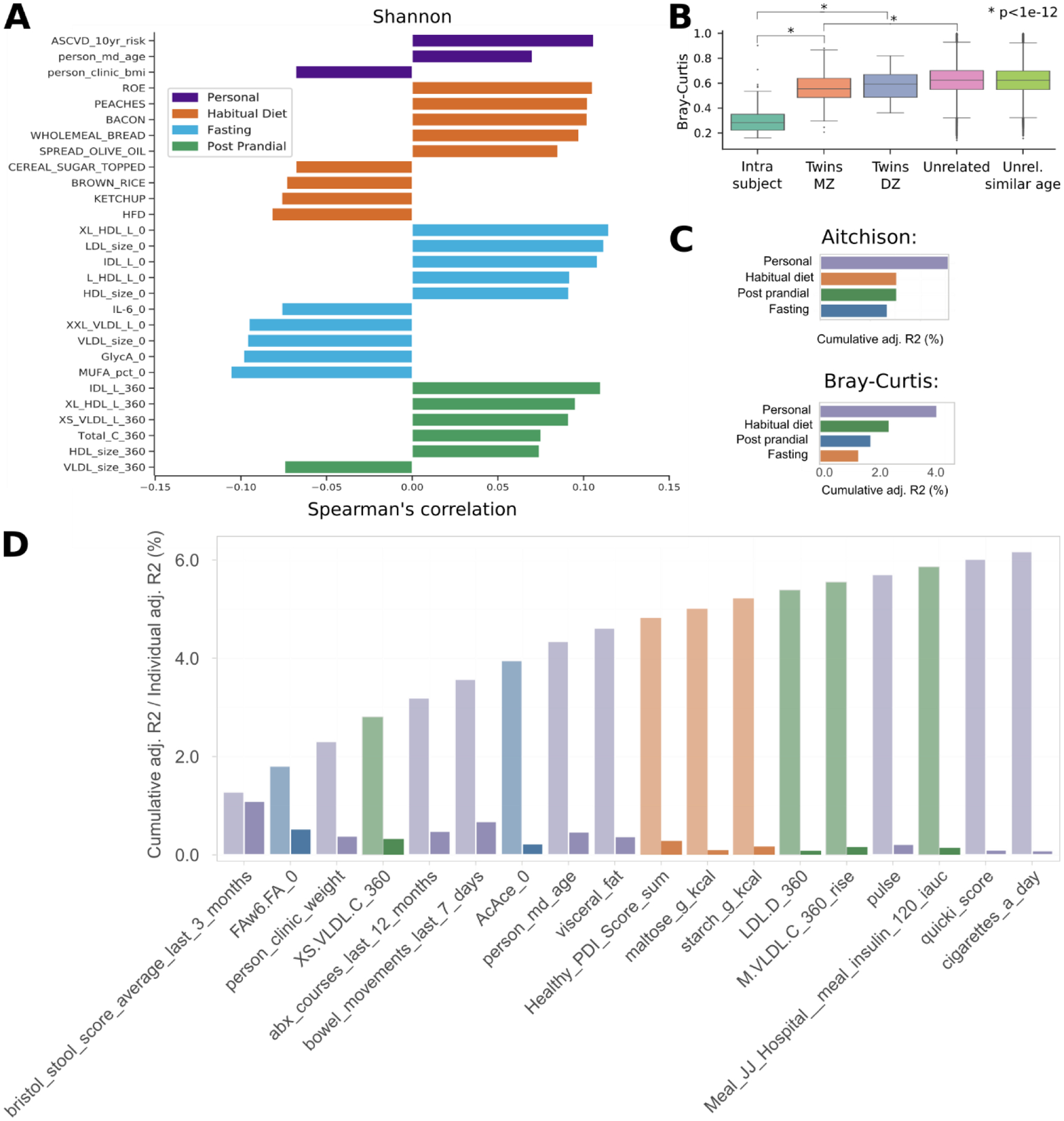
a, Microbiome alpha diversity computed using the Shannon index correlated markers from the four categories: personal, habitual diet, fasting, and post-prandial. Reported are the five strongest positive and negative Spearman correlations for each category with p < 0.05. All correlations and p-values available in the Supplementary Table 1. b, Inter-sample microbiome distances (beta-diversity) were substantially lower, that is closer, among samples from the same individuals (two weeks apart) compared to those amongst different individuals. Gut microbial communities in monozygotic twins were slightly more similar than in dizygotic twins (Mann–Whitney U test two-sided p = 0.06), which, in turn, were more similar than unrelated individuals (p < 1e-12), even after adjusting for age (p < 1e-12). c, After excluding twin status (that is non-twin, vs. mono vs. dizygotic twins) from the model, personal factors still accounted for the greatest proportion of variance explained in overall microbial diversity, followed by dietary habits, fasting and postprandial cardiometabolic blood markers (by cumulative stepwise dbRDA). d, Cumulative (left bars) contributions and individual (right bars) contributions for each metadata variable based on Bray-Curtis dissimilarity. Box plots show first and third quartiles (boxes) and the median (middle line), whiskers extends up-to 1.5× the interquartile range.
Extended Data Fig. 2. Species-level correlation with single foods.
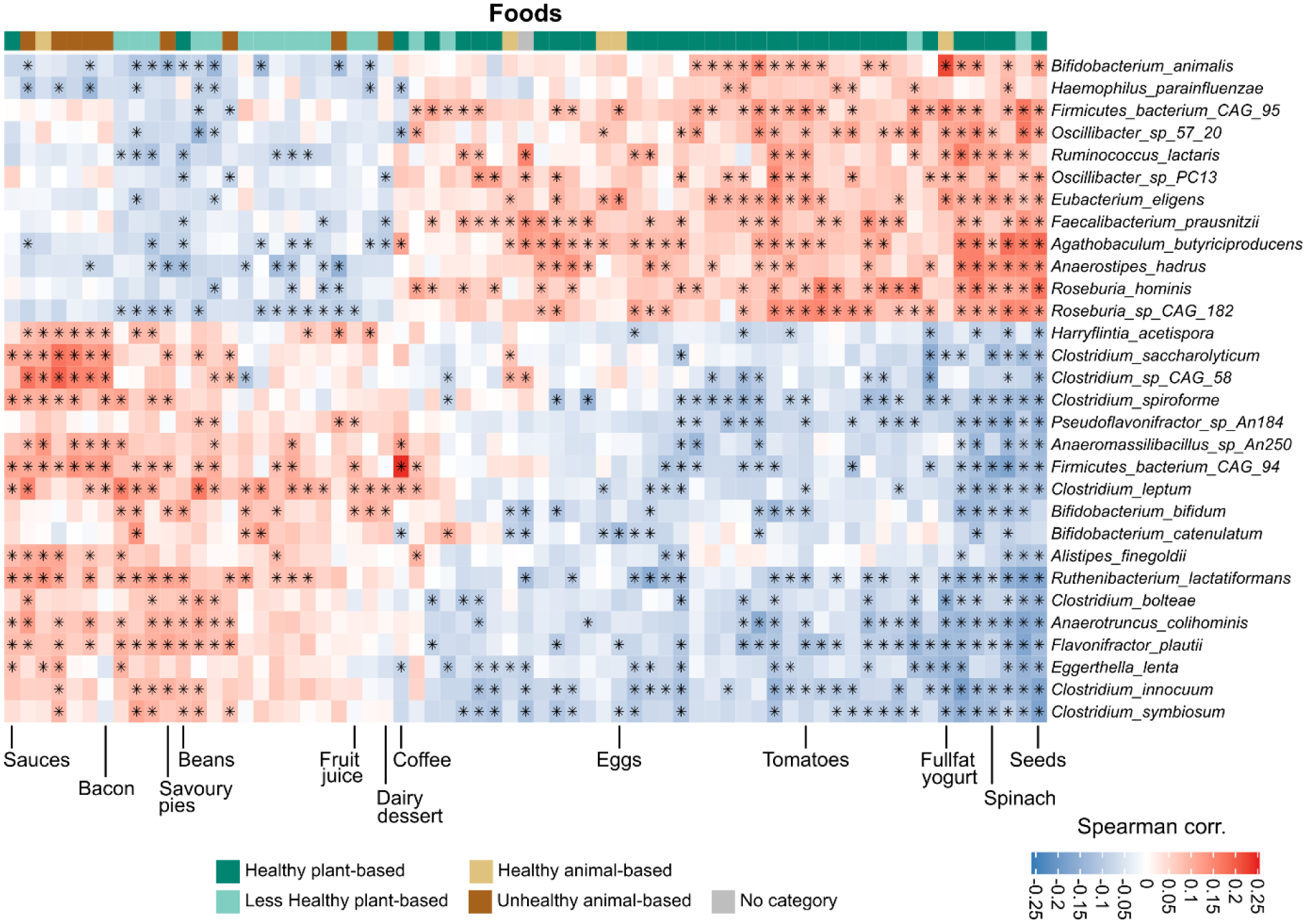
The figure shows the species-level correlations (Spearman) with single food quantities as estimated from the food frequency questionnaires. Only foods with at least 5 significant associations (q-value≤0.2) are displayed. Species are sorted by the number of significant associations, and the top 30 are reported in the figure.
Extended Data Fig. 3. Top foods, food groups, nutrients, and dietary patterns validated in the PREDICT 1 US cohort.
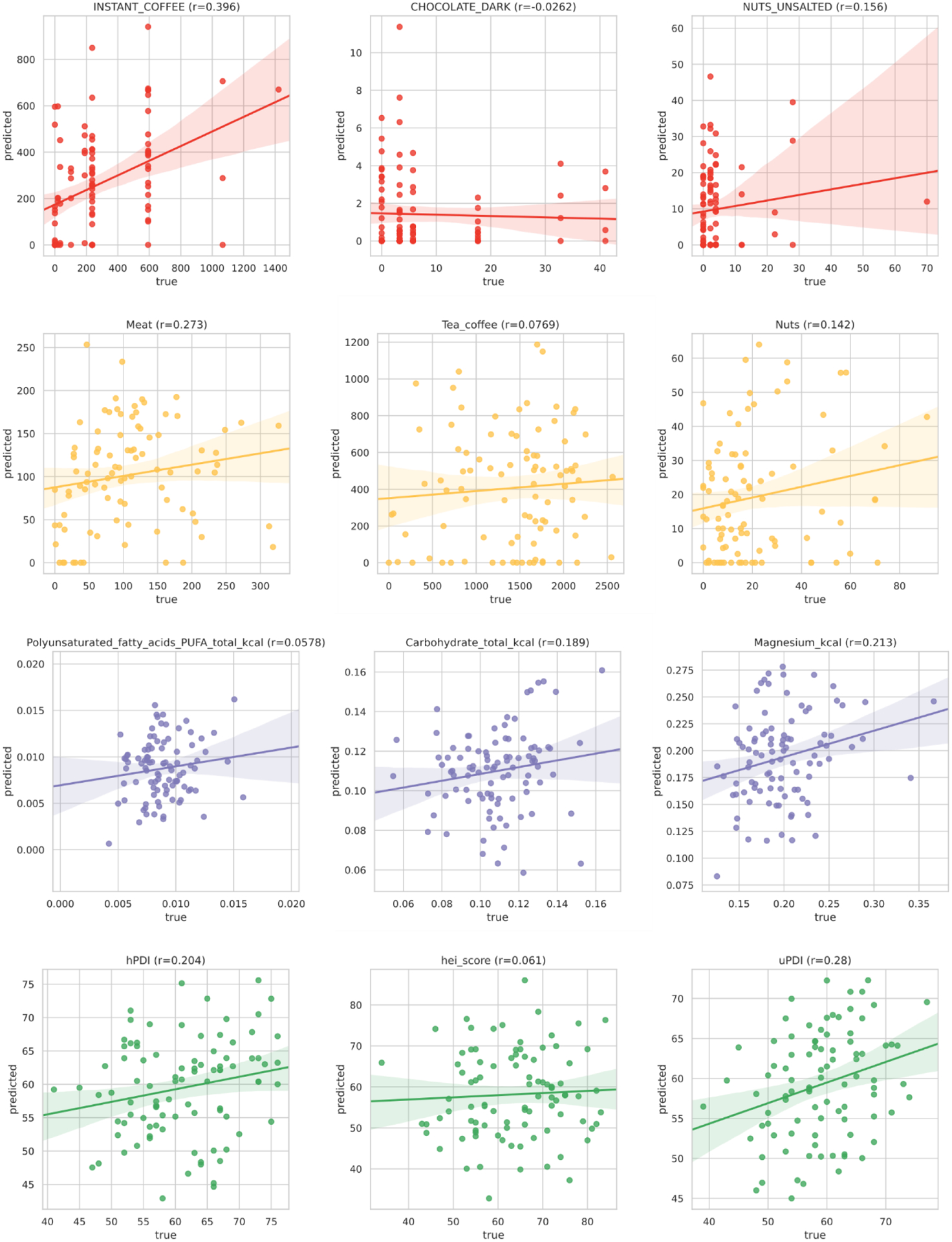
The application of the RF regression model trained on the PREDICT 1 UK cohort on the PREDICT 1 US participants, validating the associations with food-related variables found in the PREDICT 1 UK.
Extended Data Fig. 4. Performance for random Forest regression and classification on microbiome functional potential in predicting fasting measurements, total cholesterol and triglycerides in different lipoproteins.
The figure shows the performance of both RF regression and classification tasks trained on microbiome gene families profiles in predicting (a) the fasting measurements presented in Fig. 4a, sorted as in Fig. 4a. b, Predicting performances of the total cholesterol and (c) of triglycerides in different sizes of lipoproteins. For each lipoprotein, we considered its concentration values at both fasting and postprandial (6 h), and also the difference (rise) between the post-prandial concentration and the fasting one. Box plots show the distribution of the Spearman correlations (left axis) between real and predicted values using RF regression. Box plots show first and third quartiles (boxes) and the median (middle line), whiskers extends up-to 1.5× the interquartile range. Circles show the median AUC (right axis) of RF classification in predicting the bottom quartile of the distribution vs. the top quartile.
Extended Data Fig. 5. Distributions of BMI in each curatedMetagenomicData dataset.
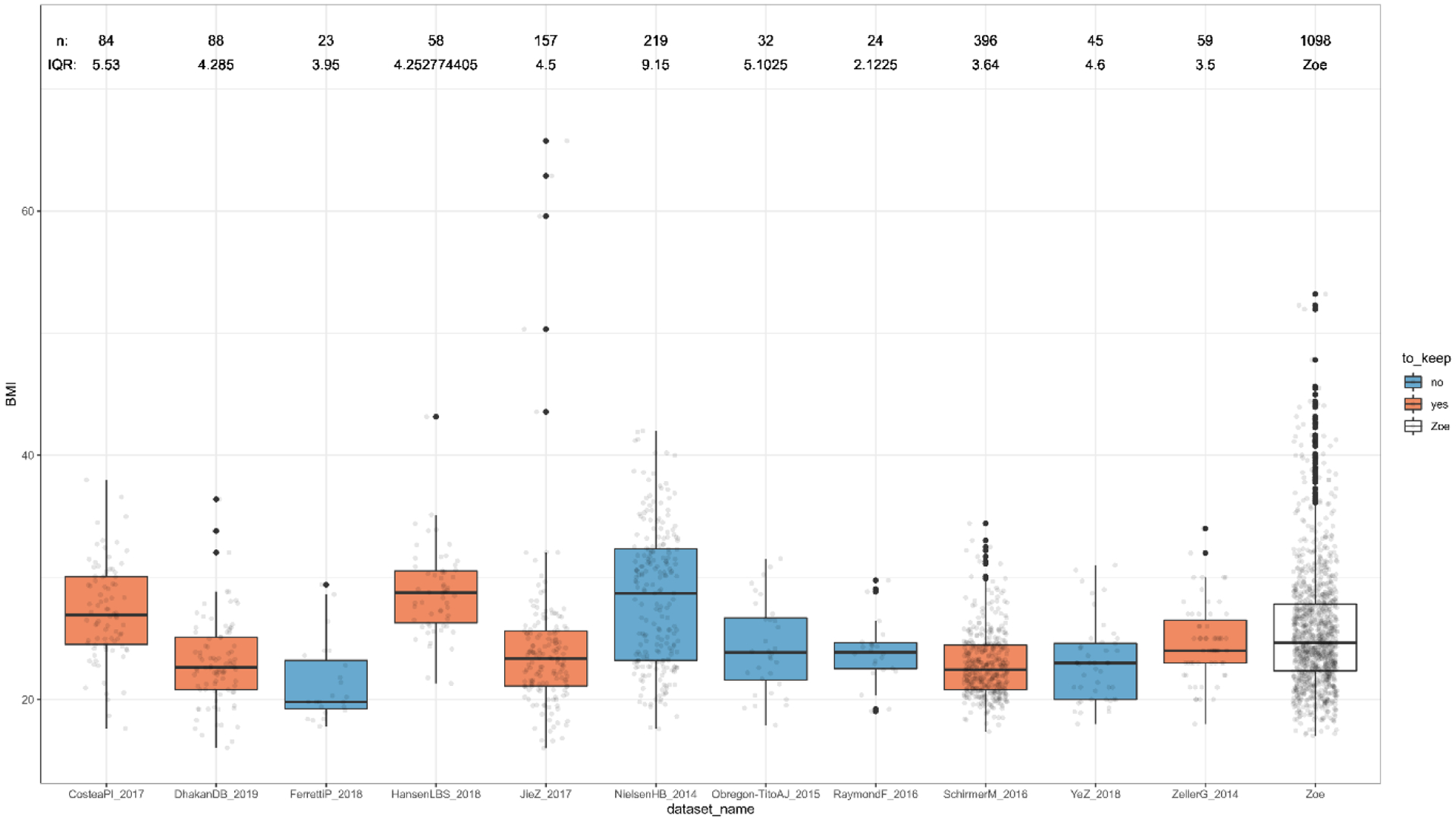
The figure shows the distributions of BMI values for the datasets available in curatedMetagenomicData. This was used to further select those datasets with a comparable range of values (interquartile range between 3.5 and 7.5) as the one in the PREDICT 1 UK dataset (IQR of 5.5), to be used as validation datasets for the associations found. Box plots show first and third quartiles (boxes) and the median (middle line), whiskers extends up-to 1.5× the interquartile range.
Extended Data Fig. 6. Pairwise partial Spearman correlations between bacterial species and total lipids and cholesterol in lipoproteins.
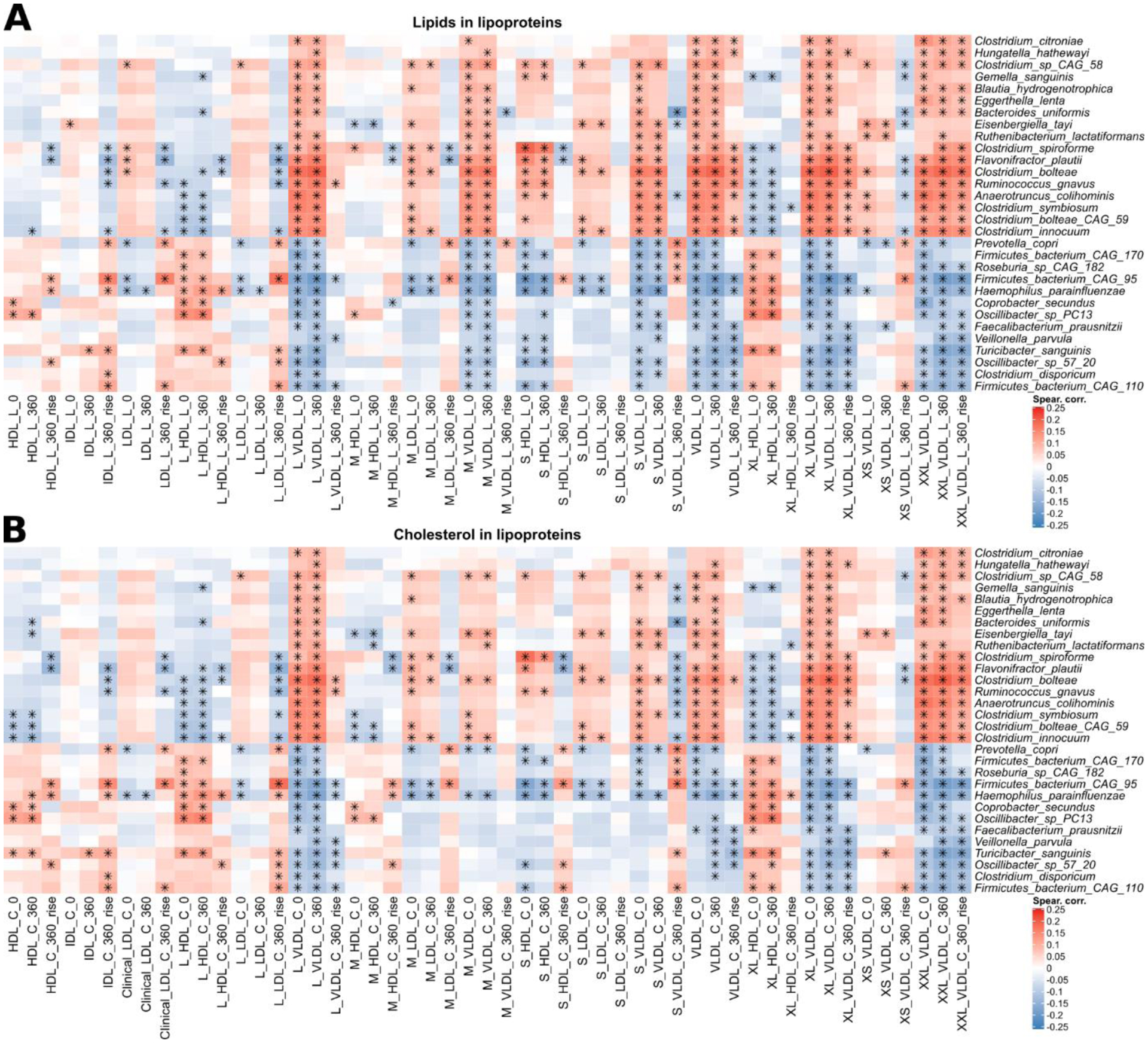
a, The heatmap shows the species-level correlations with total lipids in lipoprotein variables at fasting, post-prandial (6 h), and the difference (rise) between the postprandial and fasting concentrations. The 30 species with the highest number of significant associations (FDR ≤ 0.2) are shown. The asterisk indicates a significant correlation between species and metadata variable using a t-test two-sided, corrected with FDR with q < 0.2. b, The heatmap shows the species-level correlations with total cholesterol in lipoprotein variables at fasting, post-prandial (6 h), and the difference (rise) between the postprandial and fasting concentrations. The 30 species with the highest number of significant associations (FDR ≤ 0.2) are shown. The asterisk indicates a significant correlation between species and metadata variable using a t-test two-sided, corrected with FDR with q < 0.2. All correlations, p-values, and q-values are available in the Supplementary Table 6.
Extended Data Fig. 7. Species-level correlations with triglycerides in lipoproteins.
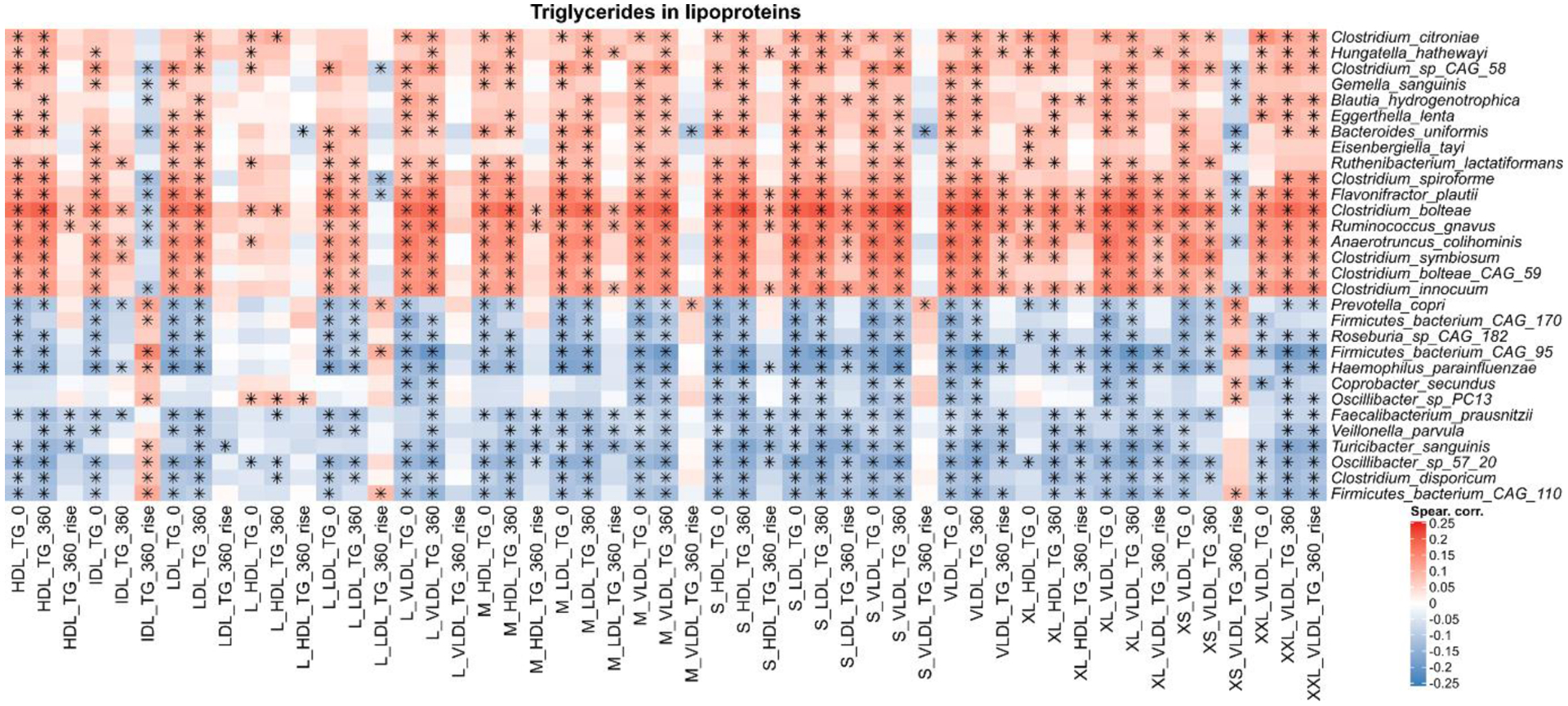
The heatmap shows the species-level correlations with triglycerides in lipoprotein variables at fasting, post-prandial (6 h), and the difference (rise) between the postprandial and fasting concentrations. The 30 species with the highest number of significant associations (FDR ≤ 0.2) are shown. The asterisk indicates a significant correlation between species and metadata variable using a t-test two-sided, corrected with FDR with q < 0.2. All correlations, p-values, and q-values are available in the Supplementary Table 6.
Extended Data Fig. 8. Pairwise partial Spearman correlations between bacterial gene families and pathway abundances with clinical and metabolic risk scores, glycaemic and inflammatory measures, and lipoproteins.
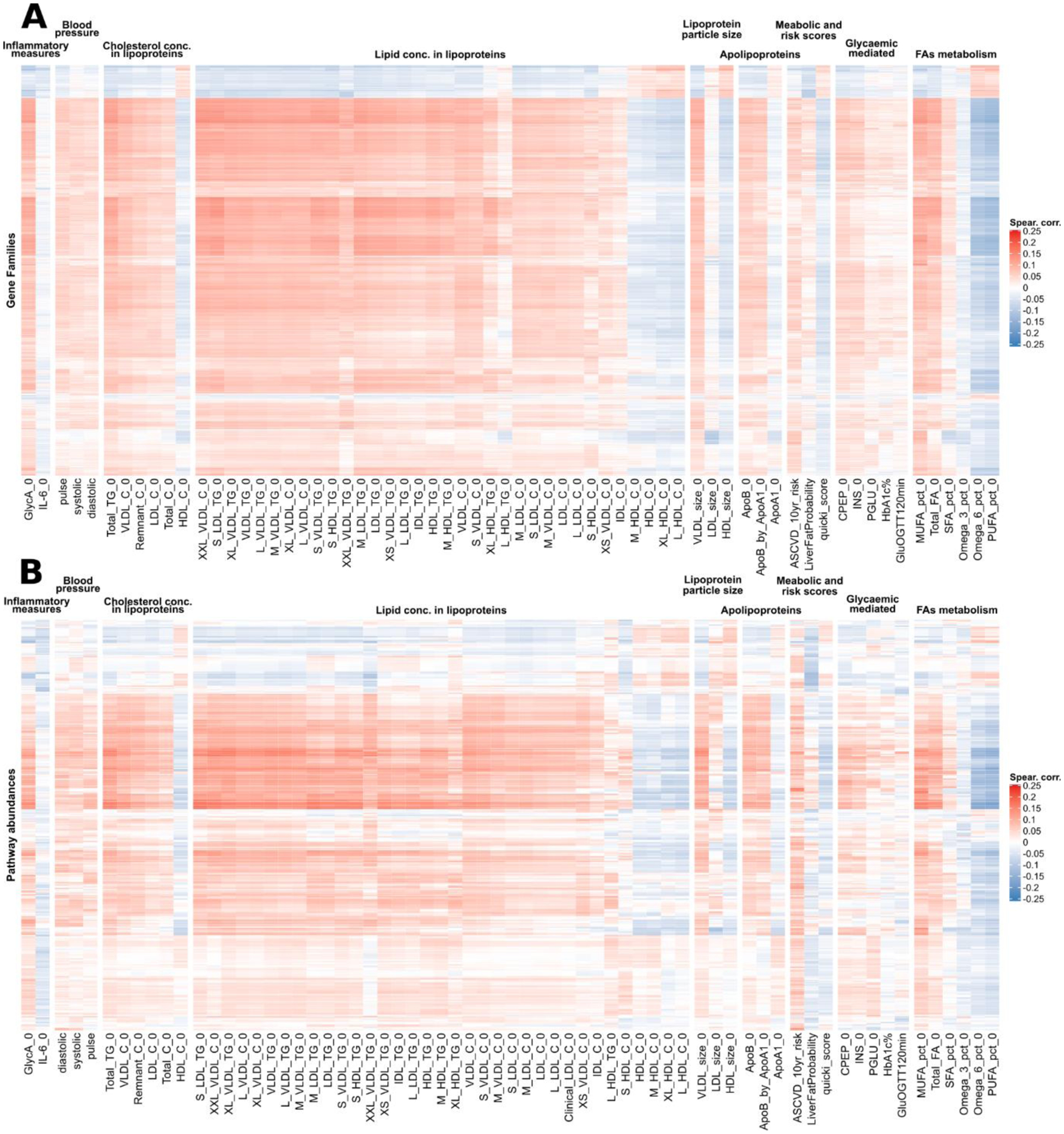
a, The heatmap shows gene families correlations with the set of metadata presented in Fig. 5a–c reporting the top 2,000 genes selected among those with at least 20% prevalence on their number of significant correlations (q < 0.2). Gene families’ correlations are showing the same clusters as the species-level correlations in Fig. 5a–c. b, The heatmap shows pathway abundances correlations with the set of metadata presented in Fig. 5a–c reporting all the pathways at 20% prevalence (349 in total). Pathway abundances correlations are showing the same cluster structure as the species-level correlations in Fig. 5a–c.
Extended Data Fig. 9. Concordance of Random Forest scores with species-level partial correlations.
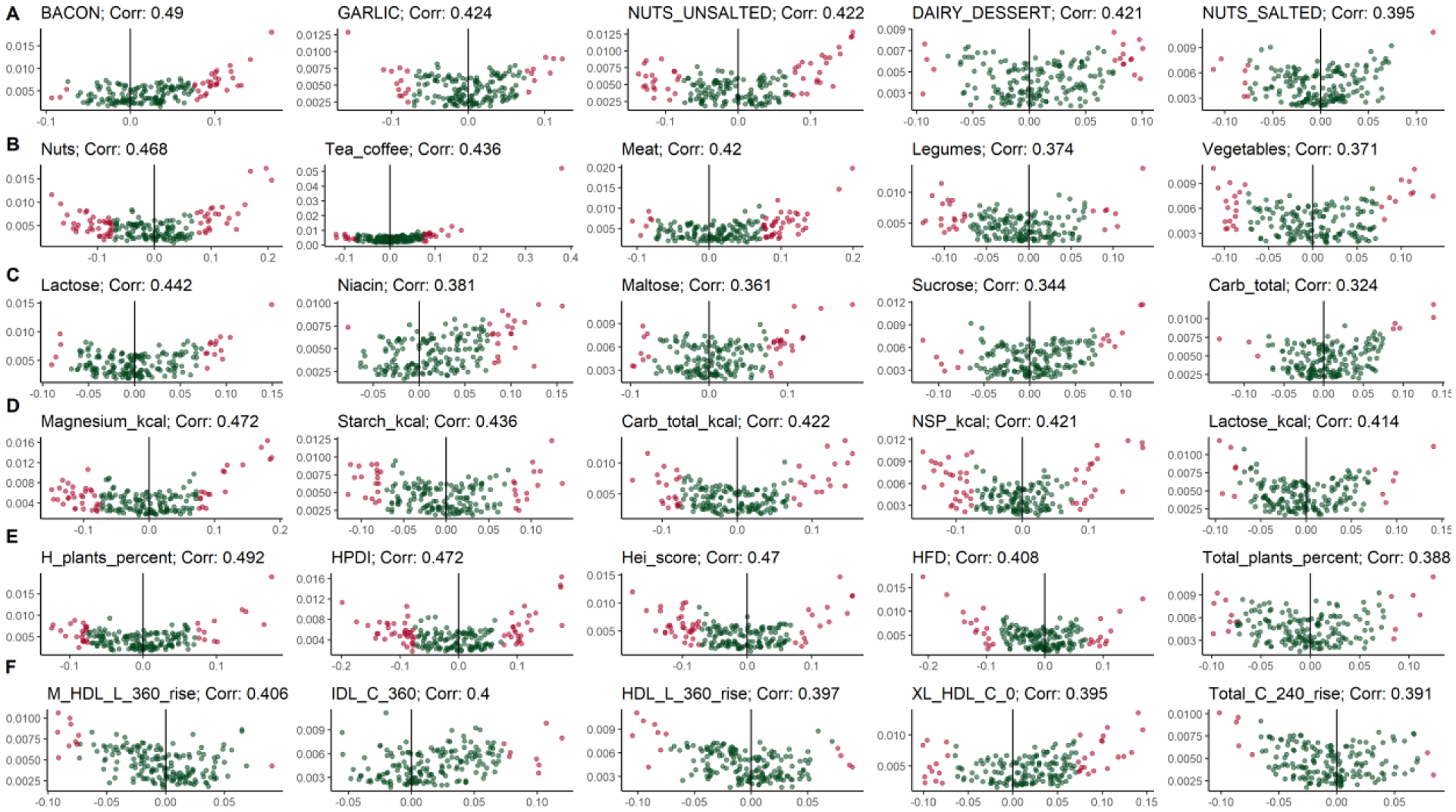
Volcano plots of the scores assigned to each species by Random Forest and their partial correlation, showing an overall concordance between the two independent approaches. We considered the top 5 metadata variables for the six metadata categories: a, Foods, bacon (g) (corr. 0.49), garlic (g) (corr. 0.424), unsalted nuts (g) (0.422), dairy dessert (g) (corr. 0.421), salted nuts (g) (corr. 0.395). b, Food groups, nuts (corr. 0.468), tea and coffee (corr. 0.436), meat (corr. 0.42), legumes (corr. 0.374), vegetables (corr. 0.371). c, Nutrients, lactose (corr. 0.442), niacin (corr. 0.381), maltose (corr. 0.361), sucrose (corr. 0.344), total carbohydrates (corr. 0.324). d, Nutrients normalized by daily energy intake, magnesium (corr. 0.472), starch (corr. 0.436), total carbohydrates (corr. 0.422), non-starch polysaccharides (NSP) (corr. 0.421), lactose (corr. 0.414). e, Dietary patterns, healthy plant percentage (corr. 0.492), healthy PDI (corr. 0.472), hei score (corr. 0.47), HFD (corr. 0.408), total plants percentage (0.388). f, Lipoproteins, M-HDL-L 6 h rise (corr. 0.406), IDL-C 6 h (corr. 0.4), HDL-L 6 h rise (corr. 0.397), XL-HDL-C 0 h (corr. 0.395), Total Cholesterol 4 h rise (corr. 0.391).
Extended Data Fig. 10. Prevotella copri and/or Blastocystis presence are indicators of a more favourable postprandial glucose response to meals.
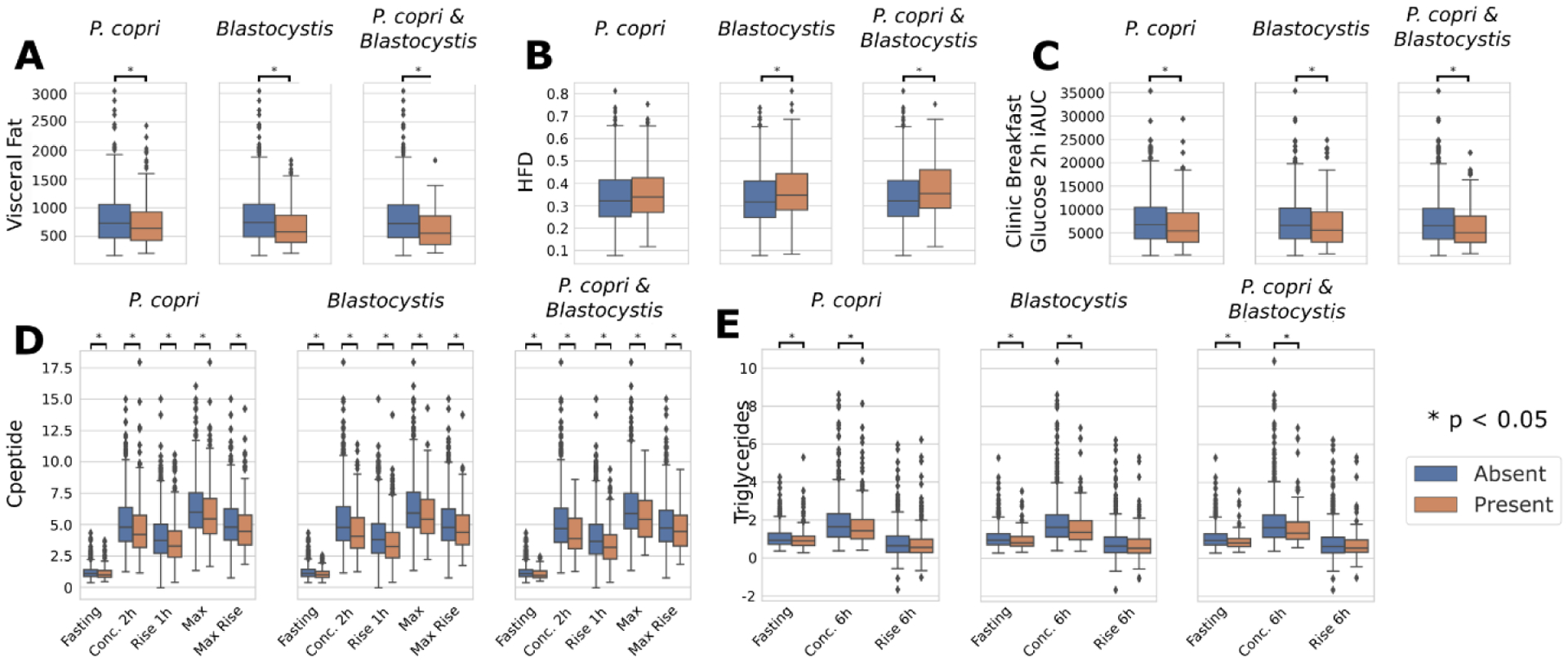
a–c, Differential analysis of visceral fat, HFD and glucose iAUC 2 h after standardised breakfast according to presence-absence of one and both of P. copri and Blastocystis. The analysis reveals that both these species are indicators of reduced visceral fat, good cholesterol and meal-driven increase of glucose. d,e, Differential analysis of C-peptide and triglycerides at different time points according to presence-absence of one and both of P. copri and Blastocystis. The distributions of the concentrations for C-peptide and triglycerides were typically lower when one or both are absent. An asterisk between two box plots represents a significant p-value (p < 0.05) according to the Mann-Whitney U test (two-sided, Supplementary Table 8). Box plots show first and third quartiles (boxes) and the median (middle line), whiskers extends up-to 1.5× the interquartile range. P-values are available in Supplementary Table 8.
Supplementary Material
Acknowledgements
We express our sincere thanks to the participants of the PREDICT 1 study. We thank Naeimeh Atabaki-Pasdar for generating the liver fat score. We thank the staff of Z. Global, the Department of Twin Research and Massachusetts General Hospital, and all the members of the Segata, Berry and Spector labs for their tireless work in contributing to the running of the study, data collection, and data processing. We thank Nightingale Health and Affinity Biomarker Labs for their support and analytical work.
Funding
This work was supported by Zoe Global Ltd and also received support from grants from the Wellcome Trust (212904/Z/18/Z) and the Medical Research Council (MRC)/British Heart Foundation Ancestry and Biological Informative Markers for Stratification of Hypertension (AIMHY; MR/M016560/1). The work was also supported by the European Research Council (ERC-STG project MetaPG) to NS; by MIUR ‘Futuro in Ricerca’ (grant No. RBFR13EWWI_001) to NS; by the European H2020 program (ONCOBIOME-825410 project and MASTER-818368 project) to NS; by the National Cancer Institute of the National Institutes of Health (1U01CA230551) to NS; and by the Premio Internazionale Lombardia e Ricerca 2019 to N.S. SEB was supported in part by a grant funded by the BBSRC (BB/NO12739/1). PWF was supported in part by grants from the European Research Council (CoG-2015_681742_NASCENT), Swedish Research Council, SSF (IRC15-0067) and Novo Nordisk Foundation. ATC was supported in part as a Stuart and Suzanne Steele MGH Research Scholar. TwinsUK is funded by the Wellcome Trust, Medical Research Council, European Union, Chronic Disease Research Foundation (CDRF), Zoe Global Ltd and the National Institute for Health Research (NIHR)-funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
Footnotes
Conflict of interest statement
TD Spector, SE Berry, AM Valdes, F Asnicar, PW Franks, C Huttenhower, and N Segata, are consultants to Zoe Global Ltd (“Zoe”). J Wolf, G Hadjigeorgiou, R Davies, J Capdevila, C Bonnett, R Hine, L Francis, F Giordano, and S Danzanvilliers are or have been employees of Zoe. Other authors have no conflict of interest to declare.
Data availability
Metagenomes are deposited in EBI ENA under accession number PRJEB39223. The non-metagenomic data used for analysis in this study are held by the department of Twin Research at King’s College London. The data can be released to bona fide researchers using our normal procedures overseen by the Wellcome Trust and its guidelines as part of our core funding. We receive around 100 requests per year for our datasets and have a meeting 3 times per month with independent members to assess proposals. The application is at https://twinsuk.ac.uk/resources-for-researchers/access-our-data/. This means that the data need to be anonymized and conform to GDPR standards.
References
- 1.Ng M et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 766–781 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown JM & Hazen SL Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol 16, 171–181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le Chatelier E et al. Richness of human gut microbiome correlates with metabolic markers. Nature 500, 541–546 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Sze MA & Schloss PD Looking for a Signal in the Noise: Revisiting Obesity and the Microbiome. MBio 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasolli E et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 176, 649–662.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yatsunenko T et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilbert JA et al. Current understanding of the human microbiome. Nat. Med 24, 392–400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berry S et al. Human postprandial responses to food and potential for precision nutrition. Nature Medicine (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeevi D et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell 163, 1079–1094 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Mendes-Soares H et al. Model of personalized postprandial glycemic response to food developed for an Israeli cohort predicts responses in Midwestern American individuals. Am. J. Clin. Nutr 110, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falony G et al. Population-level analysis of gut microbiome variation. Science 352, 560–564 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Zhernakova A et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thingholm LB et al. Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition. Cell Host Microbe 26, 252–264.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schirmer M et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 167, 1897 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Fu J et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res 117, 817–824 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berry S et al. Personalised REsponses to DIetary Composition Trial (PREDICT): an intervention study to determine inter-individual differences in postprandial response to foods. Protocol Exchange (2020). [Google Scholar]
- 17.Xie H et al. Shotgun Metagenomics of 250 Adult Twins Reveals Genetic and Environmental Impacts on the Gut Microbiome. Cell Syst 3, 572–584.e3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atabaki-Pasdar N et al. Predicting and elucidating the etiology of fatty liver disease: A machine learning modeling and validation study in the IMI DIRECT cohorts. PLoS Med 17, e1003149 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vojinovic D et al. Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat. Commun 10, 5813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vadiveloo M, Dixon LB, Mijanovich T, Elbel B & Parekh N Development and evaluation of the US Healthy Food Diversity index. Br. J. Nutr 112, 1562–1574 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Guenther PM et al. Update of the Healthy Eating Index: HEI-2010. J. Acad. Nutr. Diet 113, 569–580 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fung TT et al. Diet-quality scores and plasma concentrations of markers of inflammation and endothelial dysfunction--. Am. J. Clin. Nutr 82, 163–173 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Reedy J et al. Higher diet quality is associated with decreased risk of all-cause, cardiovascular disease, and cancer mortality among older adults. J. Nutr 144, 881–889 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitrou PN et al. Mediterranean dietary pattern and prediction of all-cause mortality in a US population: results from the NIH-AARP Diet and Health Study. Arch. Intern. Med 167, 2461–2468 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Satija A et al. Plant-Based Dietary Patterns and Incidence of Type 2 Diabetes in US Men and Women: Results from Three Prospective Cohort Studies. PLoS Med 13, e1002039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vadiveloo M, Parekh N & Mattei J Greater healthful food variety as measured by the US Healthy Food Diversity index is associated with lower odds of metabolic syndrome and its components in US adults. J. Nutr 145, 564–571 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Onvani S, Haghighatdoost F, Surkan PJ, Larijani B & Azadbakht L Adherence to the Healthy Eating Index and Alternative Healthy Eating Index dietary patterns and mortality from all causes, cardiovascular disease and cancer: a meta-analysis of observational studies. J. Hum. Nutr. Diet 30, 216–226 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Redondo-Useros N et al. Associations of Probiotic Fermented Milk (PFM) and Yogurt Consumption with Bifidobacterium and Lactobacillus Components of the Gut Microbiota in Healthy Adults. Nutrients 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakamoto M, Iino T, Yuki M & Ohkuma M Lawsonibacter asaccharolyticus gen. nov., sp. nov., a butyrate-producing bacterium isolated from human faeces. Int. J. Syst. Evol. Microbiol 68, 2074–2081 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Satija A et al. Healthful and Unhealthful Plant-Based Diets and the Risk of Coronary Heart Disease in U.S. Adults. J. Am. Coll. Cardiol 70, 411–422 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monteiro CA et al. The UN Decade of Nutrition, the NOVA food classification and the trouble with ultra-processing. Public Health Nutr 21, 5–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen HB et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol 32, 822–828 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Beaumont M et al. Heritable components of the human fecal microbiome are associated with visceral fat. Genome Biol 17, 189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasolli E et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods 14, 1023–1024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Truong DT, Tett A, Pasolli E, Huttenhower C & Segata N Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res 27, 626–638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Agostino R. B., Sr et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation 117, 743–753 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Kettunen J et al. Biomarker Glycoprotein Acetyls Is Associated With the Risk of a Wide Spectrum of Incident Diseases and Stratifies Mortality Risk in Angiography Patients. Circ Genom Precis Med 11, e002234 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Würtz P et al. Metabolite profiling and cardiovascular event risk: a prospective study of 3 population-based cohorts. Circulation 131, 774–785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hrebícek J, Janout V, Malincíková J, Horáková D & Cízek L Detection of insulin resistance by simple quantitative insulin sensitivity check index QUICKI for epidemiological assessment and prevention. J. Clin. Endocrinol. Metab 87, 144–147 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Thomas AM et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med 25, 667–678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wojczynski MK et al. High-fat meal effect on LDL, HDL, and VLDL particle size and number in the Genetics of Lipid-Lowering Drugs and Diet Network (GOLDN): an interventional study. Lipids Health Dis 10, 181 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skeggs JW & Morton RE LDL and HDL enriched in triglyceride promote abnormal cholesterol transport. J. Lipid Res 43, 1264–1274 (2002). [PubMed] [Google Scholar]
- 43.Hodson L, Skeaff CM & Fielding BA Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res 47, 348–380 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Cohn JS Postprandial lipemia: emerging evidence for atherogenicity of remnant lipoproteins. Can. J. Cardiol 14Suppl B, 18B–27B (1998). [PubMed] [Google Scholar]
- 45.Arumugam M et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kovatcheva-Datchary P et al. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab 22, 971–982 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Pedersen HK et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535, 376–381 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Tett A et al. The Prevotella copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations. Cell Host Microbe (2019) doi: 10.1016/j.chom.2019.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Filippis F et al. Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe 25, 444–453.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Clark CG, van der Giezen M, Alfellani MA & Stensvold CR Recent developments in Blastocystis research. Adv. Parasitol 82, 1–32 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Lukeš J, Stensvold CR, Jirků-Pomajbíková K & Wegener Parfrey L Are Human Intestinal Eukaryotes Beneficial or Commensals? PLoS Pathog 11, e1005039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beghini F et al. Large-scale comparative metagenomics of Blastocystis, a common member of the human gut microbiome. ISME J 11, 2848–2863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sokol H et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U. S. A 105, 16731–16736 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hall AB et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med 9, 103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valles-Colomer M et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat Microbiol 4, 623–632 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Kim H, Caulfield LE & Rebholz CM Healthy Plant-Based Diets Are Associated with Lower Risk of All-Cause Mortality in US Adults. J. Nutr 148, 624–631 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meslier V et al. Mediterranean diet intervention in overweight and obese subjects lowers plasma cholesterol and causes changes in the gut microbiome and metabolome independently of energy intake. Gut (2020) doi: 10.1136/gutjnl-2019-320438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kurilshikov A et al. Gut Microbial Associations to Plasma Metabolites Linked to Cardiovascular Phenotypes and Risk. Circ. Res 124, 1808–1820 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Ko C-W, Qu J, Black DD & Tso P Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol (2020) doi: 10.1038/s41575-019-0250-7. [DOI] [PubMed] [Google Scholar]
- 61.McIver LJ et al. bioBakery: a meta’omic analysis environment. Bioinformatics 34, 1235–1237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Truong DT et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Franzosa EA et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li D, Liu C-M, Luo R, Sadakane K & Lam T-W MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Kang DD et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Quince C, Walker AW, Simpson JT, Loman NJ & Segata N Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol 35, 833–844 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Bingham SA et al. Nutritional methods in the European Prospective Investigation of Cancer in Norfolk. Public Health Nutr 4, 847–858 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Mulligan AA et al. A new tool for converting food frequency questionnaire data into nutrient and food group values: FETA research methods and availability. BMJ Open 4, e004503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rimm EB et al. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am. J. Epidemiol 135, 1114–26; discussion 1127–36 (1992). [DOI] [PubMed] [Google Scholar]
- 71.Frankenfield DC, Muth ER & Rowe WA The Harris-Benedict studies of human basal metabolism: history and limitations. J. Am. Diet. Assoc 98, 439–445 (1998). [DOI] [PubMed] [Google Scholar]
- 72.McGuire S U.S. Department of Agriculture and U.S. Department of Health and Human Services, Dietary Guidelines for Americans, 2010 7th Edition, Washington, DC: U.S. Government Printing Office, January 2011. Adv. Nutr.2, 293–294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.WHO | Effect of trans-fatty acid intake on blood lipids and lipoproteins: a systematic review and meta-regression analysis. (2016).
- 74.Zhong VW et al. Associations of Dietary Cholesterol or Egg Consumption With Incident Cardiovascular Disease and Mortality. JAMA 321, 1081–1095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.de Souza RJ et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: systematic review and meta-analysis of observational studies. BMJ 351, h3978 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michaëlsson K et al. Milk intake and risk of mortality and fractures in women and men: cohort studies. BMJ 349, g6015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mazidi M et al. Consumption of dairy product and its association with total and cause specific mortality - A population-based cohort study and meta-analysis. Clin. Nutr 38, 2833–2845 (2019). [DOI] [PubMed] [Google Scholar]
- 78.Petsini F, Fragopoulou E & Antonopoulou S Fish consumption and cardiovascular disease related biomarkers: A review of clinical trials. Crit. Rev. Food Sci. Nutr 59, 2061–2071 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Rimm EB et al. Seafood Long-Chain n-3 Polyunsaturated Fatty Acids and Cardiovascular Disease: A Science Advisory From the American Heart Association. Circulation 138, e35–e47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim K et al. Role of Total, Red, Processed, and White Meat Consumption in Stroke Incidence and Mortality: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. J. Am. Heart Assoc 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Website, N. H. S. Dairy and alternatives in your diet. nhs.uk. https://www.nhs.uk/live-well/eat-well/milk-and-dairy-nutrition/.
- 82.Berry S et al. Personalised REsponses to DIetary Composition Trial (PREDICT): an intervention study to determine inter-individual differences in postprandial response to foods. Protocol Exchange (2020). [Google Scholar]
- 83.Matthews JN, Altman DG, Campbell MJ & Royston P Analysis of serial measurements in medical research. BMJ 300, 230–235 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pedregosa F et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830 (2011). [Google Scholar]
- 85.Pasolli E, Truong DT, Malik F, Waldron L & Segata N Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Comput. Biol 12, e1004977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oksanen J et al. Vegan: community ecology package. R package version 1.17–4 URLhttp://CRAN.R-project.org/package=vegan (2010).
- 87.Costea PI et al. Subspecies in the global human gut microbiome. Mol. Syst. Biol 13, 960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dhakan DB et al. The unique composition of Indian gut microbiome, gene catalogue, and associated fecal metabolome deciphered using multi-omics approaches. Gigascience 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hansen LBS et al. A low-gluten diet induces changes in the intestinal microbiome of healthy Danish adults. Nat. Commun 9, 4630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jie Z et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun 8, 845 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeller G et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol 10, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metagenomes are deposited in EBI ENA under accession number PRJEB39223. The non-metagenomic data used for analysis in this study are held by the department of Twin Research at King’s College London. The data can be released to bona fide researchers using our normal procedures overseen by the Wellcome Trust and its guidelines as part of our core funding. We receive around 100 requests per year for our datasets and have a meeting 3 times per month with independent members to assess proposals. The application is at https://twinsuk.ac.uk/resources-for-researchers/access-our-data/. This means that the data need to be anonymized and conform to GDPR standards.