

Abstract
A novel proteasome deubiquitinase inhibitor, VLX1570, has been highlighted as a promising therapeutic agent mainly for lymphoid neoplasms and solid tumors. We examined in vitro effects of VLX1570 on eight myeloid and three lymphoid leukemia cell lines. From cell culture studies, 10 out of 11 cell lines except K562 were found to be susceptible to VLX1570 treatment and it inhibited cell growth mainly by apoptosis. Next, to identify the signaling pathways associated with apoptosis, we performed gene expression profiling using HL‐60 with or without 50 nmol/L of VLX1570 for 3 hours and demonstrated that VLX1570 induced the genetic pathway involved in “heat shock transcription factor 1 (HSF1) activation”, “HSF1 dependent transactivation”, and “Regulation of HSF1 mediated heat shock response”. VLX1570 increased the amount of high molecular weight polyubiquitinated proteins and the expression of HSP70 as the result of the suppression of ubiquitin proteasome system, the expression of heme oxygenase‐1, and the amount of phosphorylation in JNK and p38 associated with the generation of reactive oxygen species (ROS) induced apoptosis and the amount of phosphorylation in eIF2α, inducing the expression of ATF4 and endoplasmic reticulum (ER) stress dependent apoptosis protein, CHOP, and the amount of phosphorylation slightly in IRE1α, leading to increased expression of XBP‐1s in leukemia cell lines. In the present study, we demonstrate that VLX1570 induces apoptosis and exerts a potential anti‐leukemic effect through the generation of ROS and induction of ER stress in leukemia cell lines.
Keywords: acute myeloid leukemia, proteasome deubiquitinase, VLX1570, reactive oxygen species, endoplasmic reticulum stress
VLX1570 induces apoptosis mainly through reactive oxygen (ROS) stress signaling and unfolded protein response pathway under endoplasmic reticulum (ER) stress. Regarding ER stress, VLX1570 activated the protein kinase RNA‐like endoplasmic reticulum kinase signaling pathway, leading to ER stress‐dependent apoptosis. As another pathway, excess accumulation of polyubiquitinated proteins on the mitochondria induces ROS generation, followed by phosphorylation of JNK and p38, leading to caspases‐dependent apoptosis.
1. INTRODUCTION
Acute myeloid leukemia (AML) is characterized by clonal expansion of myeloid blasts in the peripheral blood and bone marrow.1 The prognosis of patients with AML has been improved by advances in hematopoietic stem cell transplantation (HSCT).2, 3 Dombret and Gardin reported that more than 80% of younger patients with AML reach complete remission with 5‐year overall survival up to 40%.4 On the other hand, it is important for aged patients ineligible for HSCT to establish new treatment strategies for AML.5
Intracellular excessive accumulation of misfolded or damaged proteins reduces cell survival. The ubiquitin proteasome system (UPS) is a selective procedure to degrade unnecessary proteins, distinct from the autophagy‐lysosome pathway.6, 7 Misfolded or damaged proteins are marked by ubiquitin chains and degraded in the 20S core particle of proteasome. UPS routinely contributes to cellular processes including cell cycle progression and apoptosis, whereas perturbation of UPS leads to development of inflammatory disorders, neurodegeneration, and cancer.8 Since turnover of protein synthesis in cancer cells is rapid as compared with those in normal cells, the role of UPS is further required.9 A 20S proteasome inhibitor, bortezomib, and carfilzomib have been reported to exert high sensitivity to various malignant cells and are currently used as the first‐line therapy for patients with multiple myeloma.10, 11
The cellular redox state is important as a decision factor of apoptosis.12 Generation of reactive oxygen species (ROS) is harmful for cells to cause oxidative stress induced apoptosis and serves the role of controlling apoptosis signaling pathways as the second messenger.13 Previous reports showed that UPS inhibition by bortezomib induces apoptosis through ROS generation and endoplasmic reticulum (ER) stress induction.14
Recently, the resistance to bortezomib has been recognized as a serious clinical problem. Different mechanisms have been described for bortezomib resistance, including mutations in the PSMB5 subunit and overexpression of this subunit.15 Proteasome deubiquitinases (DUBs) are the enzymes which remove ubiquitin chains for proper proteasome degradation of polyubiquitinated substrates.16 Novel proteasome DUBs inhibitors, b‐AP15 and VLX1570 (a derivative of b‐AP15), predominantly target ubiquitin‐specific protease 14 (USP14) and interfere with the proteasomal degradation by inhibiting the activities of the proteasome DUBs, such as USP14 and ubiquitin C‐terminal hydrolase L5 (UCHL5) localized on the 19S proteasome.17, 18 The antiproliferative effect of these agents has been reported on several solid tumors19, 20 and lymphoid malignancies.21, 22, 23, 24, 25 These agents lead to a similar pattern to the expression of genes induced by 20S proteasome inhibitors26 and show high susceptibility to bortezomib‐resistant cells.27, 28
In this study, we investigated the effect of VLX1570 on the growth of myeloid/lymphoid leukemia cell lines and demonstrated that it suppresses cell proliferation by inducing apoptosis through ROS generation and ER stress induction.
2. MATERIALS AND METHODS
2.1. Reagents
VLX1570, purchased from Selleck Chemicals, was dissolved in dimethyl sulfoxide and stored at −80℃ with being protected from light. N‐acetyl cysteine (NAC) and N,N’‐dimethylthiourea (DMTU) were purchased from Sigma‐Aldrich and mannitol from FUJIFILM Wako Pure Chemical Corporation.
2.2. Cell lines and culture
A myelodysplastic syndromes (MDS)‐derived cell line, MDS92, was established from the bone marrow of a patient with MDS. The MDS‐L cell line was established as a blastic subline from the parental MDS92.29, 30, 31 MDS‐L and F‐36P were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 U/mL IL‐3. As human leukemia cell lines, myeloid cell lines HL‐60, MOLM13, MV4‐11 and OCI‐AML3, a monocytic cell line THP‐1, a chronic myelogenous leukemia‐derived cell line K562, T lymphoblastic cell lines MOLT‐4 and Jurkat, and a histiocytic lymphoma cell line U937 were used in this study. The morphological assessment was done with May–Gruenwald Giemsa‐stained cytospin slides.
2.3. Cell growth assay and MTT assay
Cell growth was assessed by counting living cells after trypan blue staining. Cell suspensions were plated into 96‐well plates in the presence of the drug or solvent alone, incubated as above at 37℃ for 24‐48 hours, and analyzed by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT) assay.
2.4. Apoptosis assay
Apoptosis was examined using an Annexin V Apoptosis Detection Kit (BD Pharmingen) and all samples were analyzed with FACSCalibur flow cytometer and CellQuest software (Becton Dickinson).
2.5. Cell cycle analysis
Cells were fixed with 70% methanol for 30 minutes and treated with 2 mg/mL ribonuclease A (Nacalai Tesque) for 30 minutes at 37℃, then with 50 μg/mL propidium iodide (PI; Sigma) for a further 30 minutes at room temperature.
2.6. Immunoblotting analysis
Cell lysates of all 11 cell lines were prepared in lysis buffer containing 50 mmol/L Tris‐HCL, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton X‐100, 0.05%, 1 mmol/L phenylmethylsulfonyl fluoride and 1 mmol/L Na3VO4. The lysates were separated by SDS‐polyacrylamide gel electrophoresis and immunoblotting analysis was performed as previously described.32
Primary antibodies were obtained from Santa Cruz Biotechnology (bcl‐2, Bcl‐x S/L), Cell Signaling Technology (cleaved‐PARP [cPARP], XBP‐1s, CHOP, ATF4, eIF2α, phospho‐eIF2α [S51], IREα, JNK, phospho‐JNK [T183/Y185], cleaved caspase‐3, −6, −8 and −9), Sigma‐Aldrich (HSP70), Merck Millipore (anti‐ubiquitin K48 [Apu2]), New England Biolabs Japan Inc. (p38, phospho‐p38 [Thr180/ Tyr182]), BD Transduction Laboratories (BiP/GRP78), Invitrogen (phospho‐IRE1α(Ser724)), and BioAcademia (ATF6). Horse‐radish peroxidase‐conjugated mouse and rabbit antibodies were from GE Healthcare Life Sciences.
For detection of the expression of XBP‐1s, ATF4, and CHOP, the cells were lysed with the lysis buffer and incubated for 20 minutes on ice. After eliminating supernatant including cytosolic component by centrifugation at 18 000 g for 20min at 4℃, we prepared protein lysate extracted from the pellet including the nuclear component by heating for 10 minutes at 100℃ with sample buffer.
2.7. Gene expression profiling and gene set enrichment analysis
Gene expression profiling of HL‐60 cells was carried out in three independent experiments. Treated cells were harvested after 3 hours of treatment with 50 nmol/L of VLX1570. Total RNA was extracted with an RNeasy Mini Kit (Qiagen), converted to cDNA and amplified with a GeneChip WT Terminal Labeling and Controls Kit (Affymetrix). The fragmentation, the labeling, and the hybridization of cDNA were treated with a GeneChip Hybridization, Wash, and Stain Kit (Affymetrix). Chips were scanned with a GeneChip Scanner 3000 7G System (Affymetrix).
Gene set enrichment analysis (GSEA) (Broad Institute Cambridge) was performed using gene expression profiling data and by handling the GSEA software.33 The whole expression change in the gene sets was defined as statistically significant if both the false discovery rate (FDR) q values and the familywise error rate (FWER) P values were less than .25.
2.8. Measurement of ROS production
To measure ROS levels, cells were stained with 0.1 µmol/L of a redox sensitive dye CM‐H2DCFDA (Thermo Fisher Scientific) and 10 µmol/L of hydroxyphenyl fluorescein (HPF) (Goryo Chemical) to detect highly ROS and 0.2 µmol/L of HYDROPTM (Goryo Chemical), which is a specific probe for the detection of intracellular H2O2 in PBS, for 30 minutes at 37℃.34, 35 Cells were washed and resuspended in PBS.
ROS production was analyzed using a FACSCalibur flow cytometer and CellQuest software (Becton Dickinson).
2.9. Assay of the mitochondrial membrane potential
The mitochondrial membrane potential of cells with or without 100 nM of VLX1570 was estimated by flow cytometry using rhodamine 123 staining. Cells were cultured with indicated concentrations of VLX1570 for 24 hours and stained with 1 μmol/L of rhodamine 123 for 30 minutes at 37℃. After the staining, cells were washed by PBS with 1% BSA.36
The decrease in mitochondrial membrane potential (loss of ΔΨ) was calculated as follows: the mean fluorescence intensity (MFI) of VLX1570‐treated samples minus that of a cell‐only sample treated with VLX1570 divided by the MFI of control samples (VLX1570 untreated) minus that of a cell‐only sample untreated with VLX1570.
2.10. Statistical analyses
All results are shown as the mean values with ranges. Comparisons between the groups were done using the Dunnett's and Scheffe's tests. Differences were considered statistically significant if P values were less than .05. These analyses were carried out using SPSS for Windows version 14.0.
3. RESULTS
3.1. VLX1570 inhibits the proliferation of leukemia cell lines and mainly induces apoptosis
To determine whether VLX1570 inhibits leukemia cell growth, we treated 11 cell lines including eight myeloid (HL‐60, MOLM13, MV4‐11, OCI‐AML3, THP‐1, K562, MDS‐L, F‐36P) and three lymphoid cell lines (MOLT‐4, Jurkat, U937) with VLX1570 for 0‐48 hours and examined the half maximal inhibitory concentration (IC50) of VLX1570 in all cell lines with MTT assay (Figure 1A). The IC50 value of VLX1570 in each cell line is indicated in Table 1. We found that 10 cell lines, not including K562, were susceptible to VLX1570 (Table 1). Sensitivity to VLX1570 in myeloid cell lines except K562 was similar to that of lymphoid cell lines. Manual cell counting nearly corresponded to the results of the MTT assay (data not shown).
FIGURE 1.
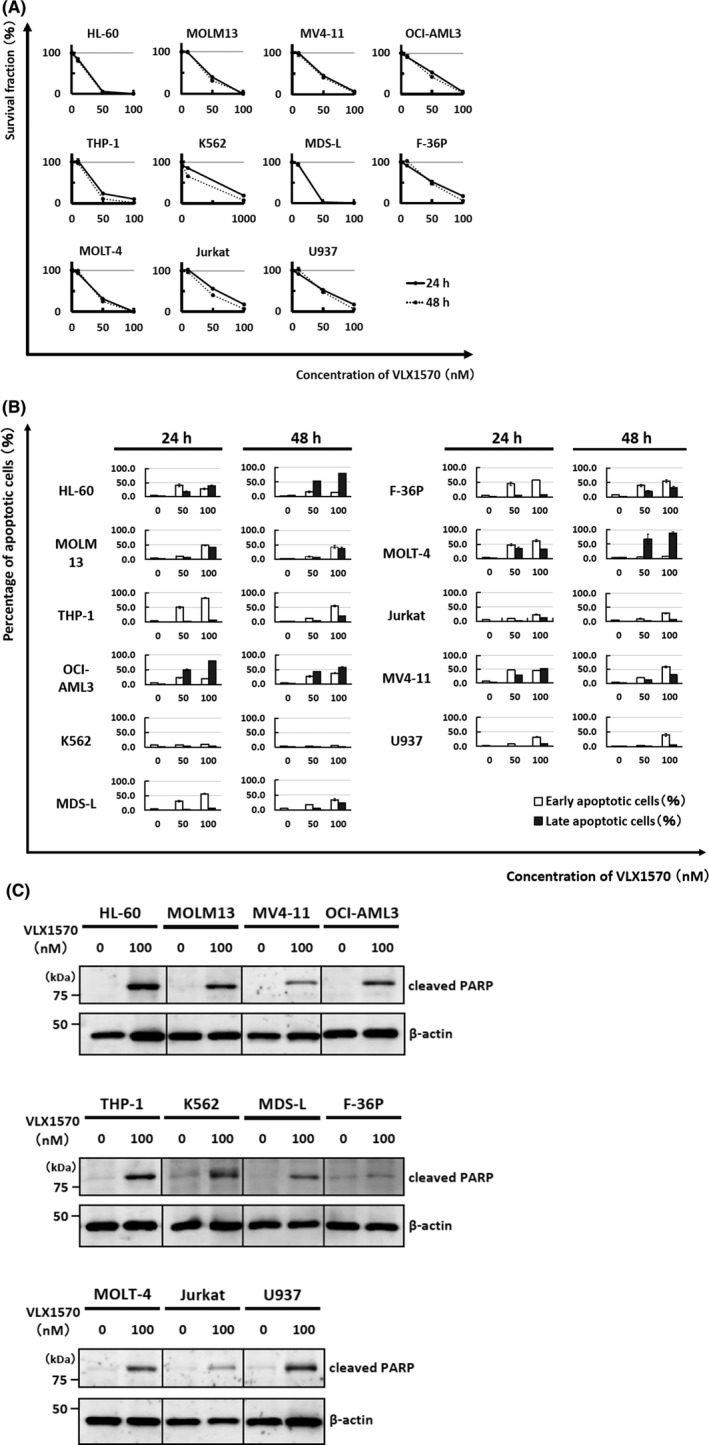
VLX1570 inhibits the proliferation of leukemia cells and induces apoptosis in most cell lines. (A) Eleven cell lines, including eight myeloid leukemia cell lines (HL‐60, MDS‐L, F‐36P, THP‐1, MOLM13, OCI‐AML3, MV4‐11, K562) and three lymphoid cell lines (MOLT‐4, Jurkat, U937), were cultured with VLX1570 (0‐100 nmol/L) for the indicated times. The cell count was measured by MTT assay. The value without VLX1570 was adjusted to 100%. The data represent the mean values with SD from five independent experiments. Solid line, 24 h; dotted line, 48 h. (B) These cell lines were cultured with different concentrations of VLX1570 (0‐100 nmol/L) for the indicated times and apoptosis was assessed by flow cytometry using annexin V‐FITC/PI staining. The single‐positive fraction for annexin V‐FITC (white bar) implies early apoptosis and the double‐positive fraction for annexin V‐FITC/PI (black bar) implies late apoptosis. The data represent the mean values with SD from three independent experiments. (C) Cell lysates of each cell line treated with or without 100 nmol/L of VLX1570 for 12 h were analyzed by immunoblotting analysis for detection of cleaved PARP (cPARP). The amount of β‐actin is shown as a loading control
TABLE 1.
IC50 (nmol/L) of VLX1570 in leukemia cell lines
| IC50 (nmol/L) | 24 h | 48 h |
|---|---|---|
| HL‐60 | 20.5 ± 0.3 | 19.0 ± 0.8 |
| MOLM13 | 38.0 ± 1.0 | 32.2 ± 0.6 |
| MV4‐11 | 43.1 ± 0.2 | 37.9 ± 2.1 |
| OCI‐AML3 | 52.2 ± 0.8 | 38.4 ± 2.1 |
| THP‐1 | 29.1 ± 1.7 | 24.0 ± 0.6 |
| K562 | 340.8 ± 15.1 | 184.7 ± 8.6 |
| MDS‐L | 21.8 ± 0.5 | 21.4 ± 0.6 |
| F‐36P | 52.4 ± 1.1 | 46.3 ± 2.8 |
| MOLT‐4 | 30.6 ± 0.9 | 28.9 ± 0.3 |
| Jurkat | 55.9 ± 2.8 | 37.9 ± 1.5 |
| U937 | 38.2 ± 1.2 | 31.4 ± 2.1 |
Next, we found that VLX1570 induced apoptosis in 10 cell lines, not including K562 (Figure 1B), and confirmed VLX1570‐induced apoptosis morphologically (data not shown). The amount of cleaved PARP (cPARP), a marker of undergoing apoptosis, was increased after VLX1570 treatment regardless of variable degrees in 11 cell lines (Figure 1C). The expression of Bcl‐2 family members (Bcl‐2, Bcl‐x S/L) did not change by immunoblotting analysis (data not shown).
The amount of LC3‐II protein, an autophagosome membrane‐bound form, was not increased in HL‐60, OCI‐AML3, MDS‐L, and K562 after up to 500 nmol/L of VLX1570 treatment (Figure S1), and therefore autophagy might not be involved in this treatment process.
To determine whether VLX1570 affects the cell cycle, we performed the cell cycle analysis of VLX1570‐treated cells (HL‐60, MV4‐11, OCI‐AML3, THP‐1, K562, MDS‐L, F‐36P, and MOLT‐4) by flow cytometry, but we did not find any significant deviation of the cell cycle pattern except for F‐36P, which showed the increase in G2/M fraction. The sub‐G1 fraction implying apoptotic cell fraction was 6.1 ± 0.1% and 10.0 ± 0.9% in control F‐36P cells and VLX1570 (100 nmol/L)‐treated cells, respectively.
3.2. VLX1570 suppresses leukemia cell growth through inhibition of proteasome ubiquitin processing
To determine whether UPS inhibition by VLX1570 contributes to growth suppression of leukemia cells, we examined the amount of high molecular weight polyubiquitinated proteins in the cells treated with or without VLX1570. VLX1570 increased the amount of phosphorylated Lys‐48 in all cell lines (Figure 2A). Since UPS inhibition promotes the induction of chaperon to counteract the accumulation of misfolded proteins, we analyzed the amount of HSP70 in the cells treated with or without VLX1570 and found that VLX1570 increased the expression of HSP70 in most leukemia cell lines (Figure 2B).
FIGURE 2.
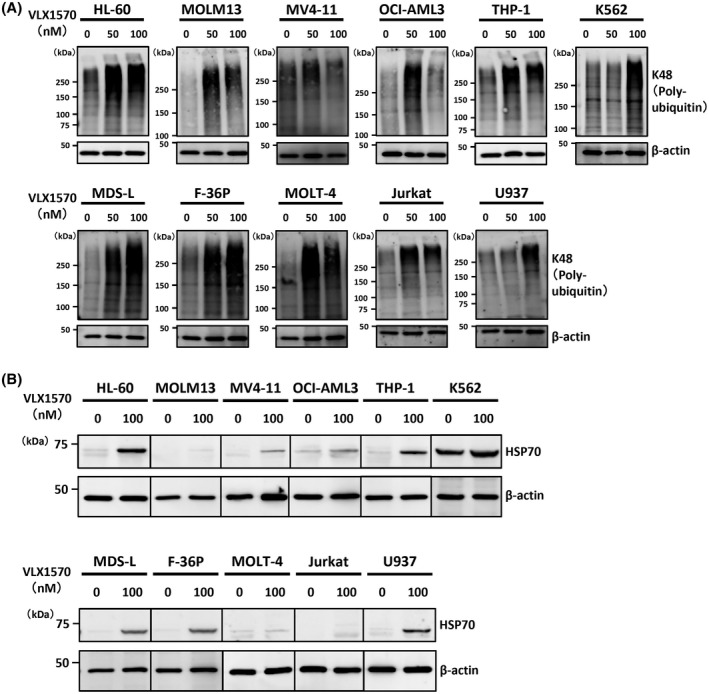
The antiproliferative effect is related closely to the inhibition of proteasome ubiquitin processing by VLX1570. The cells were treated with or without 50 or 100 nmol/L of VLX1570 for 6 h and cell lysates were analyzed by immunoblotting analysis for detection of high molecular weight polyubiquitinated proteins (A) and HSP70 (B). The amount of β‐actin is shown as a loading control
3.3. VLX1570 possibly affects the expression of the gene sets associated with heat shock transcription factor 1 activation
To further explore the action mechanisms of VLX1570, the molecular pathways regulated by VLX1570 were explored using gene expression profiling of HL‐60 cells (TP53‐mutant type) treated with or without VLX1570. Genes whose expression changed by more than 1.5 fold or <0.66 following the treatment were defined as the regulated genes. We performed GSEA and found that some of the most significantly affected GSEA sets were “HSF1 activation” (FDR q‐val 0.000; FWER p‐val 0.000), “HSF1‐dependent transactivation” (FDR q‐val 0.000; FWER p‐val 0.000) and “Regulation of HSF1‐mediated heat shock response” (FDR q‐val 0.000; FWER p‐val 0.000) (Figure 3A,B). We performed the same analysis using the OCI‐AML3 cell line (TP53‐wild type) and confirmed that the profiles of the most significantly affected GSEA sets in OCI‐AML cells were similar to those in HL‐60 (Figure S2).
FIGURE 3.
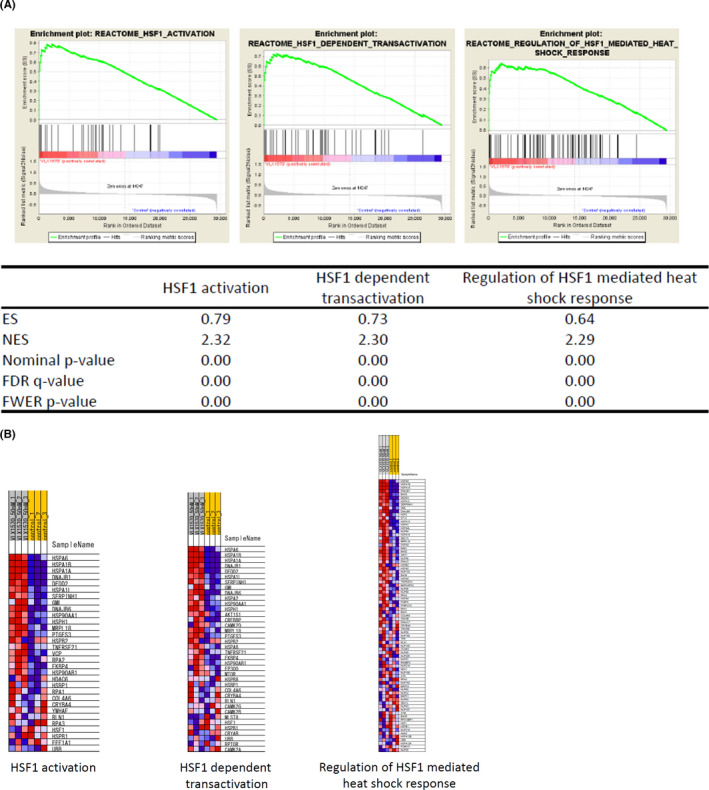
GSEA revealed the activation of the gene sets associated with HSF activation in VLX1570‐treated HL‐60 cells. (A) VLX1570 (50 nmol/L)‐treated or untreated HL‐60 cells were harvested at 3 h. Gene expression profiling was examined in triplicate experiments and obtained data were used for GSEA. The gene set “HSF1 activation”, “HSF1‐dependent transactivation”, and “Regulation of HSF1‐mediated heat shock response” were strongly upregulated by VLX1570 treatment and several statistical values are also presented. (B) Heat map presentation of affected genes included in the gene set is shown in the triplicate experiments (VLX1570 50 nmol/L: 50 nmol/L of VLX1570‐treated; control: untreated)
Taken together, gene expression profiling suggested that VLX1570 induced the genetic pathway involved in HSF1 activation irrespective of TP53 status.
3.4. VLX1570‐induced ROS generation causes apoptosis
Since the expression of heme oxygenase‐1 (HO‐1) mRNA significantly was found to be increased from a result of gene expression profiling using VLX1570‐treated HL‐60 cells, we determined whether VLX1570 actually increases the expression of HO‐1 protein in myeloid cells (MDS‐L and HL‐60) and lymphoid cells (MOLT‐4 and Jurkat). VLX1570 induced strong expression of HO‐1 in three cell lines, but not MOLT‐4 (Figure 4A). This finding prompted us to examine the association between high expression of HO‐1 and induction of oxidative stress. We demonstrated by flow cytometry using a redox‐sensitive dye CM‐H2DCFDA that exposure of HL‐60 cells to VLX1570 triggered a rapid burst of ROS and it was suppressed by antioxidant agent, NAC, which is a precursor of reduced glutathione (GSH) and attenuates oxidative stress (Figure 4B). Next, we examined whether NAC protects AML cells from ROS‐induced apoptosis. The annexin V‐FITC/PI staining revealed that NAC prevented VLX1570 from inducing apoptosis in HL‐60 and MV4‐11 (Figure 4C). It showed a similar tendency to OCI‐AML3 cells (data not shown.).
FIGURE 4.
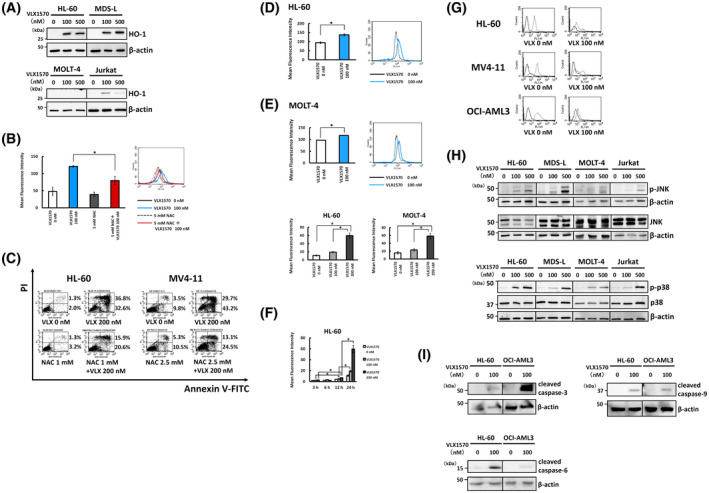
VLX1570 induces ROS generation. (A) HL‐60, MDS‐L, MOLT‐4, and Jurkat cells were treated with indicated doses of VLX1570 for 6 h and the cell lysates were analyzed by immunoblotting analysis for detection of HO‐1. (B) HL‐60 cells were treated with or without 100 nmol/L of VLX1570 or 5 mmol/L of NAC for 3 h and stained with 0.1 μmol/L of CM‐H2DCFDA for 30 min at 37℃. ROS levels were determined by flow cytometry. (C) HL‐60 and MV4‐11 cells were treated with VLX1570 in the absence or presence of NAC for 24 h. The treated cells were stained by Annexin V‐FITC/PI and analyzed by flow cytometry. (D) HL‐60 and MOLT‐4 cells were treated with or without 100 nmol/L of VLX1570 for 3 h and stained with 10 μmol/L of HPF for 30 min at 37℃. ROS levels were determined by flow cytometry. (E) HL‐60 and MOLT‐4 cells were treated with or without indicated doses of VLX1570 for 24 h and stained with 0.2 μmol/L of HYDROPTM for 30 min at 37℃. ROS levels were determined by flow cytometry. (F) HL‐60 and MOLT‐4 cells were treated with or without indicated doses of VLX1570 for the indicated times and stained with 0.2 μmol/L of HYDROPTM for 30 min at 37℃. ROS levels were determined by flow cytometry. (G) HL‐60, MV4‐11, and OCI‐AML3 cells were exposed to 100 nmol/L of VLX1570 for 24 h. Mitochondrial membrane potential was detected by rhodamine 123 staining and flow cytometry. Solid line, negative control; dotted line, cells treated with rhodamine 123. (H) HL‐60, MDS‐L, MOLT‐4, and Jurkat cells were treated with indicated doses of VLX1570 for 3 h and the cell lysates were analyzed by immunoblotting analysis for detection of JNK, phospho‐JNK, p38, and phospho‐p38. β‐actin was used as a loading control. (I) HL‐60 and OCI‐AML3 cells were treated with indicated doses of VLX1570 for 12 h and the cell lysates were analyzed by immunoblotting analysis for detection of cleaved caspase‐3, ‐6, and ‐9. β‐actin was used as a loading control. In B, D, E, F, statistical differences were evaluated and presented by asterisks if P values were less than .05
As CM‐H2DCFDA was reported to be inadequate for detection of specific ROS,37 we attempted to detect highly ROS including ·OH and ONOO‐ using HPF in HL‐60, OCI‐AML3 cells, and MOLT‐4 treated with or without VLX1570 and found that exposure of HL‐60 cells and MOLT‐4 to VLX1570 significantly increased the generation of highly ROS (Figure 4D). Next, we determined whether an ·OH specific inhibitor, mannitol, suppresses VLX1570‐induced apoptosis, but mannitol failed to protect HL‐60 cells from VLX1570‐induced apoptosis (data not shown).
We also attempted to detect H2O2 using HYDROPTM in HL‐60, MDS‐L, OCI‐AML3, and MOLT‐4 cells treated with or without VLX1570. Exposure of HL‐60 and MOLT‐4 cells except for MDS‐L and OCI‐AML3 to VLX1570 significantly increased the generation of H2O2 (Figure 4E, F). Because MDS‐L and OCI‐AML3 were found to originally produce a lot of H2O2 as compared with HL‐60 and MOLT‐4 cells by HYDROPTM assay (data not shown), it may be hard to detect subtle changes in the generation of H2O2 in VLX1570‐treated cells. Next, we determined whether a H2O2 specific inhibitor, DMTU, suppresses VLX1570‐induced apoptosis, but DMTU treatment did not protect HL‐60 cells from apoptosis.
As above described, we detected fine alteration of ROS generation in the cells treated with VLX1570 using some ROS‐sensitive probes, but we could not demonstrate the direct relationship between ROS induction and cell death from the study using specific ROS scavengers.
We also examined whether VLX1570 treatment leads to the loss of mitochondrial membrane potential using rhodamine 123 and demonstrated that VLX1570 increased the proportion of cells with loss of ΔΨ in three myeloid cell lines (HL‐60, MV4‐11, and OCI‐AML3) (Figure 4G). We calculated the decrease in MFI of mitochondrial membrane potential after VLX1570 treatment as described in the Materials and methods section. The relative ratios of MFI in HL‐60, MV4‐11, and OCI‐AML3 decreased to 0.34, 0.42, and 0.15, respectively, after VLX1570 treatment.
To identify the signaling pathway involved in ROS generation and apoptosis, we examined the degree of phosphorylation in p38 and JNK in myeloid cells (MDS‐L and HL‐60) and lymphoid cells (MOLT‐4 and Jurkat). As shown in Figure 4H, the phosphorylation of JNK and p38 increased at 3 hours after VLX1570 treatment in four cell lines except for the absence of robust JNK phosphorylation in MOLT‐4.
We attempted to examine the expression of cleaved caspases‐3, ‐6, ‐8, and ‐9 using HL‐60 and OCI‐AML3 treated with or without VLX1570 for 12 hours. Exposure of these cells to 100 nmol/L of VLX1570 increased the expression of cleaved caspases‐3, ‐6, and ‐9, but not cleaved caspase‐8 (Figure 4I).
3.5. VLX1570 induces apoptosis through ER stress in leukemia cell lines
UPS inhibition by bortezomib, b‐AP15 or VLX1570 has been reported to induce unfolded protein response and to activate protein kinase RNA‐like endoplasmic reticulum kinase (PERK), IRE1, and ATF6 mediated ER stress signaling in cancer cells.7, 9, 21, 22, 27 First, we examined the degree of phosphorylation of eIF2α and IRE1α in myeloid cells (HL60 and MDS‐L) and lymphoid cells (MOLT‐4 and Jurkat) after VLX1570 treatment (Figure 5A, B). VLX1570 clearly increased phosphorylation of eIF2α, whereas it slightly increased phosphorylation of IRE1α in four cell lines (Figure 5A, B). Regarding the PERK signaling pathway, VLX1570 increased the expression of the downstream molecules, ATF4 and CHOP, indicating the induction of ER stress‐dependent apoptosis (Figure 5A). On the other hand, regarding the IRE1 signaling pathway, VLX1570 also increased the expression of the downstream molecule, the spliced form of the X‐box binding protein 1 (XBP‐1s), which promotes transcriptional activity of the gene involved in ER‐associated degradation (ERAD), ER chaperon, and ER membrane synthesis (Figure 5B).
FIGURE 5.
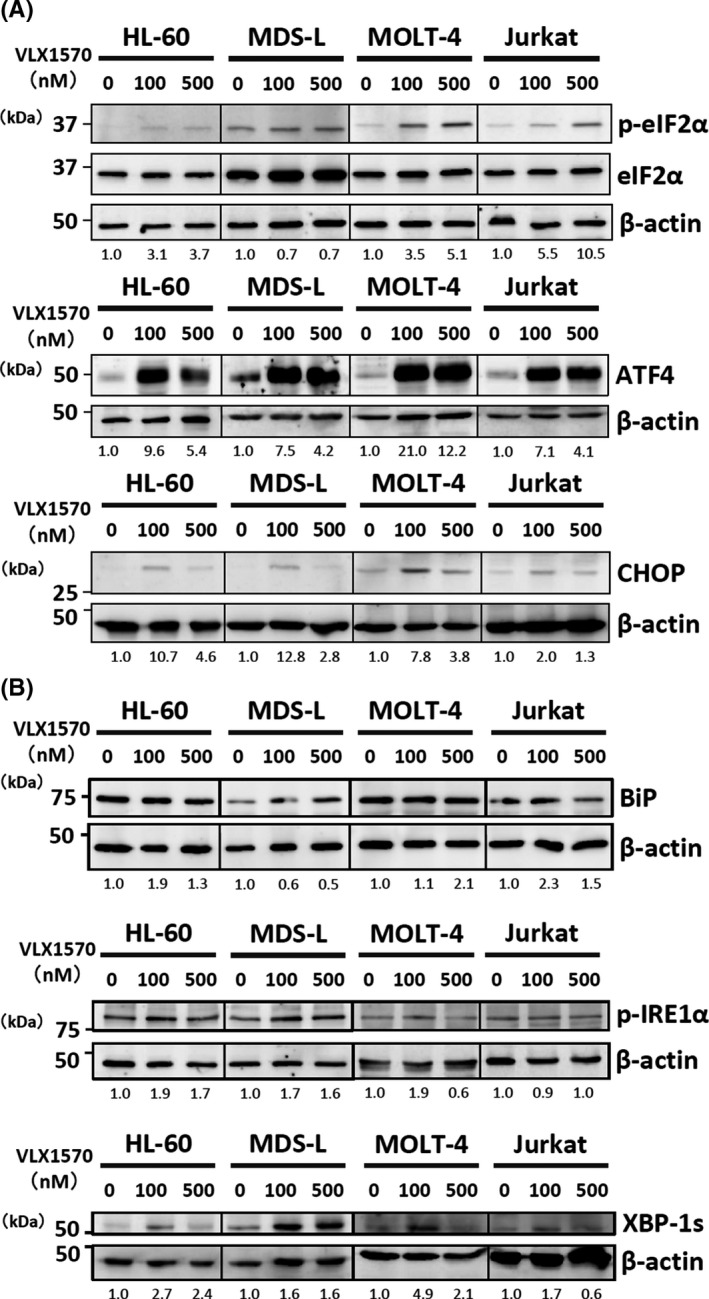
VLX1570 exerted an antiproliferative effect on leukemia cells by apoptosis involved in ER stress induction. (A), (B) HL‐60, MDS‐L, MOLT‐4 and Jurkat cells were treated with 0‐500 nmol/L of VLX1570 for 3 h and the cell lysates were analyzed by immunoblotting analysis for detection of XBP‐1s, CHOP, ATF4, BiP, eIF2α, phospho‐eIF2α, and phospho‐IRE1α. For detection of the expression of XBP‐1s, ATF4, and CHOP, the nuclear fraction was extracted as described in the Materials and methods section. β‐actin was used as a loading control. The number of protein bands was measured by densitometry and the ratio was adjusted as 1.0 in the untreated sample and the changes of the ratio of treated samples relative to untreated sample were indicated
ATF6 is expressed as a 90‐kDa protein (p90ATF6) in ER under physiological conditions, whereas in ER‐stressed cells activated ATF6 is transported to the Golgi apparatus and cleaved to a 50‐kDa protein (p50ATF6) by two Golgi‐resident proteases, site‐1 and site‐2 protease, followed by translocation to the nucleus.38 We examined the expression of p90 and p50ATF6 in HL‐60 and OCI‐AML3 treated with or without VLX1570 but found that only the expression of p90ATF6 was detected, suggesting unlikely the contribution of ATF6 to VLX1570‐induced ER stress.
4. DISCUSSION
In this study, we investigated the effect of a novel proteasome DUB inhibitor, VLX1570, on 11 myeloid/lymphoid leukemia cell lines and demonstrated that it suppressed cell proliferation by inducing apoptosis through ROS generation and ER stress induction. Some study groups have previously analyzed the relationship between UPS inhibition and the antiproliferative effect by a proteasome DUB inhibitor in various cancer cells. In hematopoietic tumors, the effects of VLX1570 on lymphoid malignancies have been reported. Mazurkiewicz et al reported that VLX1570 suppressed the growth of acute lymphoblastic leukemia (ALL) cells, induced the chaperone HSP70, the oxidative stress marker HO‐1, and a cell cycle regulator p21, and exerted an additive or synergistic antiproliferative effect by combination with L‐asparaginase.21 Wang et al reported that VLX1570 showed selectivity for USP14 and induced apoptosis on myeloma cells.23 Paulus et al reported that VLX1570 facilitated the antiproliferative effect by inhibition of USP14 and UCHL5 simultaneously.22 Likewise, VLX1570 has an antitumor activity in solid tumors.19, 20 b‐AP15 has also exerted an antiproliferative effect on various tumor models as well as VLX1570.7, 27, 39, 40, 41, 42
As shown in Figure 1A, all leukemia cell lines except K562 revealed a susceptibility to VLX1570. Although the reason why K562 was resistant to VLX1570 remains unclear, it might be related to our data that exposure of K562 to VLX1570 did not induce the expression of HO‐1. There are two mechanisms including UPS and autophagy‐lysosome systems to degrade unnecessary proteins.43 Since these two systems are mutual, even if UPS activity is impaired by VLX1570, the degradation of unnecessary proteins is maintained by the autophagy‐lysosome system, leading to drug resistance. Vogel et al reported that simultaneous inhibition of DUBs and autophagy synergistically suppressed the growth of breast cancer cells.42 In our study, the amount of LC3‐II was not increased after up to 100 nmol/L of VLX1570 treatment (Figure S1), and therefore autophagy might not be involved in this treatment process.
As described in Figure 1, we did not find any significant deviation of the cell cycle pattern except for F‐36P, which showed the increase in G2/M fraction at 12 hours. Therefore, we further performed cell cycle analysis in HL‐60 and OCI‐AML3 treated with various doses of VLX1570 for 3 or 6 hours, but VLX1570 did not significantly affect the pattern of cell cycle. The reason why VLX1570 did not alter the cell cycle despite phosphorylation of p38 and JNK remains unclear. As a possible explanation, VLX1570 quite promptly induces apoptosis in leukemia cells as compared with other agents, and therefore it may be hard to detect the apparent alteration of the cell cycle pattern.
A DUB inhibitor showed a promising effect on bortezomib‐resistant cancer cells.15 Zang et al reported that whereas similar expression profiles were induced by b‐AP15 and bortezomib in colon cancer cells, b‐AP15 induced stronger expression of chaperones and stronger accumulation of polyubiquitinated proteins.44, 45 Likewise, in our study VLX1570 induced stronger expression of HSP70 and stronger accumulation of polyubiquitinated proteins in leukemia cells (Figure 2A, B).
VLX1570 and b‐AP15 were previously reported to induce apoptosis through ROS generation.21, 45 We found strong expression of HO‐1 from gene expression profiling of VLX1570‐treated HL‐60 cells (Figure 3A,B) and confirmed that VLX1570 increased the expression of HO‐1 in HL‐60, MDS‐L, and Jurkat cells, indicating that ROS generation contributed to VLX1570‐induced apoptosis (Figure 4A). We indicated that exposure of leukemia cells to VLX1570 induced ROS generation (Figure 4B). NAC protected leukemia cells from VLX1570‐induced apoptosis (Figure 4C). Exceptionally, as VLX1570 did not induce the expression of HO‐1 in MOLT‐4 cells, VLX1570‐induced apoptosis in MOLT‐4 cells might not be associated with ROS‐related oxidative stress.
We further measured highly ROS and H2O2 in VLX1570‐treated cells using ROS specific probes including HRP and HYDROPTM. Although we detected fine alteration of these specific ROS, we could not demonstrate the direct relationship between ROS induction and cell death (Figure 4D–F). Proteasome inhibition by VLX1570 also promoted ROS generation, mitochondrial damage (Figure 4G), and increased phosphorylation of JNK and p38 (Figure 4H) and the expression of cleaved caspase‐9 (Figure 4I). Although the origin of ROS remained unclear, Maharjan et al reported that treatment with a proteasome inhibitor, MG132, induced mitochondrial oxidation and accumulation of polyubiquitinated proteins in CHO cells.46 Zang et al demonstrated that elevated level of polyubiquitinated proteins was localized on the mitochondria in Hela cells treated with b‐AP15.44 In our study, excess accumulation of polyubiquitinated proteins on the mitochondria may induce ROS generation and subsequent apoptosis.
Based on the GSEA from gene expression profiling of HL‐60 and OCI‐AML3 cells treated with VLX1570, we particularly focused on three gene sets: “HSF1 activation” (FDR q‐val 0.000; FWER p‐val 0.000), “HSF1‐dependent transactivation” (FDR q‐val 0.000; FWER p‐val 0.000), and “regulation of HSF1‐mediated heat shock response” (FDR q‐val 0.000; FWER p‐val 0.000) (Figure 3A,B). HSF1 is induced after temperature stress and plays an important role in mediating several heat shock proteins, including HSP70 and HSP90. At steady states, HSF1 is bound to HSP70 and HSP90 as an inactive monomer, whereas under stress it is released as an active trimer and binds heat shock promoter elements (HSE), promoting the transcription of HSP‐family genes.47 As shown in Figure 2B, VLX1570 increased the expression of HSP70, indicating upregulation of heat shock response (HSR). Generally, the unfolded protein response (UPR) pathway is further activated and induces ER stress‐related apoptosis. BiP/GRP78 is an ER chaperone and induces the activation of PERK. Subsequently, phosphorylation of eIF2α, the expression of ATF4 and C/EBP homologous protein, CHOP is elevated. b‐AP15 and VLX1570 were reported to induce apoptosis through ER stress in cancer cells.7, 9, 21, 22, 27 Mazurkiewicz et al reported that VLX1570 increased the phosphorylation of eIF2α in ALL cells at higher concentrations than the doses which induced polyubiquitin accumulation and reduced cell viability in other lymphoid cells.21 In our study, the phosphorylation of eIF2α and the upregulation of ATF4 and CHOP expression were observed at the concentration close to IC50 in leukemia cell lines (Figure 5A).
As HL‐60 cells are deficient in TP53,48 we examined whether the stress reactions are different between HL‐60 and OCI‐AML3 (TP53‐wild type). Previous reports described a similar sensitivity of TP53‐wild type and TP53‐mutant cell lines to b‐AP15. Bazzaro et al indicated that IC50 of TP53‐wild type cell lines in the NCI60 panel to b‐AP15 is 0.18 μmol/L, and IC50 of TP53‐mutant cell lines is 0.15 μmol/L (P = .70).49 Didier et al reported that b‐AP15 has an antitumor effect irrespective of mutation status on BRAF, NRAS, and TP53.9 D'Arcy et al reported using isogenic clones of HCT‐116 that b‐AP15 induced apoptosis that was insensitive to overexpression of Bcl‐2 and disruption of TP53.
In our study, IC50 of TP53‐mutant cell lines (HL‐60 and MDS‐L) to VLX1570 was similar to that of TP53‐wild type cell lines (OCI‐AML3, MV4‐11 and MOLM13). Next, we compared the expression of ROS‐ and ER stress‐associated protein using TP53‐wild type cell line OCI‐AML3 and TP53‐mutant cell line HL‐60 treated with or without VLX1570. As a result, the exposure of OCI‐AML3 to VLX1570 increased phosphorylation of JNK and p38, and the expression of HO‐1, CHOP, and ATF4 as well as HL‐60 (Figure S3).
In the present study, we investigated the effect of a DUB inhibitor, VLX1570, by using leukemia cell lines and demonstrated that VLX1570 exerted an antiproliferative effect mainly by apoptosis derived from ROS generation and ER stress induction. Our data together with previous reports are summarized in Figure 6. Further study regarding the availability of VLX1570 is needed to establish a promising therapy for patients with intractable hematological malignancies.
FIGURE 6.
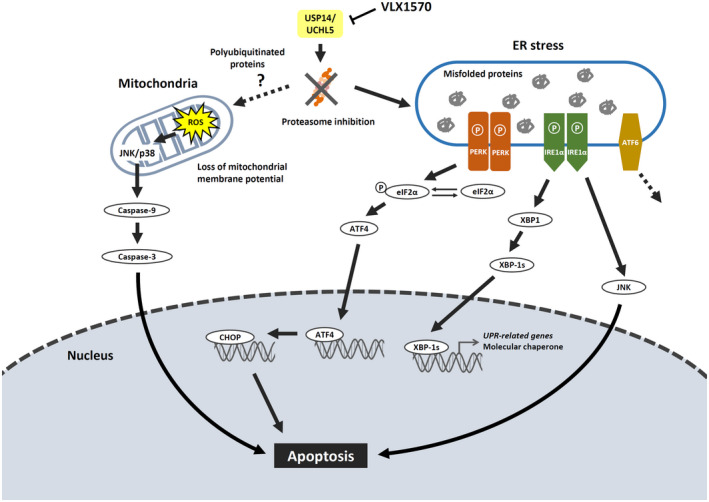
Proteasome inhibition by VLX1570 induces ROS generation and the activation of unfolded protein response under ER stress followed by apoptosis. VLX1570 induces apoptosis mainly through ROS stress signaling and the unfolded protein response pathway under ER stress. Regarding the PERK signaling pathway, VLX1570 increases the amount of phospho‐eIF2α and the expression of the downstream molecules, ATF4 and CHOP, induction of ER stress‐dependent apoptosis. As another pathway, excess accumulation of polyubiquitinated proteins on the mitochondria induces ROS generation, followed by phosphorylation of JNK and p38, leading to caspase‐3‐ and 9‐dependent apoptosis
DISCLOSURE
The authors have no conflict of interest to declare.
Supporting information
Fig S1
Fig S2
Fig S3
ACKNOWLEDGMENTS
This work was supported in part by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science, and in part by a Kawasaki Medical School project grant. The authors thank Ms Aki Kuyama for editorial assistance.
Kurozumi N, Tsujioka T, Ouchida M, et al. VLX1570 induces apoptosis through the generation of ROS and induction of ER stress on leukemia cell lines. Cancer Sci. 2021;112:3302–3313. 10.1111/cas.14982
Funding information
A Kawasaki Medical School project grant, a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science.
REFERENCES
- 1.Arber DA, Orazi A, Hasserjian RP, et al. In: Swerdlow SH, Campo E, Harris NL, eds. Introduction and overview of the classification of myeloid neoplasms: WHO classification of tumours of Haematopoietic and Lymphoid Tissues, Revised, 4th edn. IARC: Lyon; 2017:16‐27. [Google Scholar]
- 2.Koreth J, Schlenk R, Kopecky KJ, et al. Allogeneic stem cell transplantation for acute myeloid leukemia in first complete remission: systematic review and meta‐analysis of prospective clinical trials. JAMA. 2009;301:2349‐2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood. 2016;127:62‐70. [DOI] [PubMed] [Google Scholar]
- 4.Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016;127:53‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oran B, Weisdorf DJ. Survival for older patients with acute myeloid leukemia: a population‐based study. Haematologica. 2012;97:1916‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;11–20. [DOI] [PubMed] [Google Scholar]
- 7.Cai J, Xia X, Liao Y, et al. A novel deubiquitinase inhibitor b‐AP15 triggers apoptosis in both androgen receptor‐dependent and ‐independent prostate cancers. Oncotarget. 2017;8:63232‐63246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol. 2011;12:605‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Didier R, Mallavialle A, Ben Jouira R, et al. Targeting the Proteasome‐Associated Deubiquitinating Enzyme USP14 Impairs Melanoma Cell Survival and Overcomes Resistance to MAPK‐Targeting Therapies. Mol Cancer Ther. 2018;17:1416‐1429. [DOI] [PubMed] [Google Scholar]
- 10.Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high‐dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487‐2498. [DOI] [PubMed] [Google Scholar]
- 11.Varga C, Laubach J, Hideshima T, et al. Novel targeted agents in the treatment of multiple myeloma. Hematol Oncol Clin North Am. 2014;28:903‐925. [DOI] [PubMed] [Google Scholar]
- 12.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931‐947. [DOI] [PubMed] [Google Scholar]
- 13.Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1‐MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780(11):1325‐1336. [DOI] [PubMed] [Google Scholar]
- 14.Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS‐341 induces apoptosis through induction of endoplasmic reticulum stress‐reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 2004;24:9695‐9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niewerth D, Jansen G, Assaraf YG, Zweegman S, Kaspers GJ, Cloos J. Molecular basis of resistance to proteasome inhibitors in hematological malignancies. Drug Resist Updat. 2015;18:18‐35. [DOI] [PubMed] [Google Scholar]
- 16.Rechsteiner M, Hoffman L, Dubiel W. The multicatalytic and 26 S proteases. J Biol Chem. 1993;268:6065‐6068. [PubMed] [Google Scholar]
- 17.D'Arcy P, Brnjic S, Olofsson MH, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17:1636‐1640. [DOI] [PubMed] [Google Scholar]
- 18.Ward JA, Pinto‐Fernandez A, Cornelissen L, et al. Re‐evaluating the mechanism of action of α, β‐unsaturated carbonyl DUB inhibitors b‐AP15 and VLX1570: A paradigmatic example of unspecific protein cross‐linking with Michael acceptor motif‐containing drugs. J Med Chem. 2020;63:3756‐3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shukla N, Somwar R, Smith RS, et al. Proteasome addiction defined in Ewing sarcoma is effectively targeted by a novel class of 19S proteasome inhibitors. Cancer Res. 2016;76:4525‐4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vogel RI, Pulver T, Heilmann W, et al. USP14 is a predictor of recurrence in endometrial cancer and a molecular target for endometrial cancer treatment. Oncotarget. 2016;7:30962‐30976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazurkiewicz M, Hillert EK, Wang X, et al. Acute lymphoblastic leukemia cells are sensitive to disturbances in protein homeostasis induced by proteasome deubiquitinase inhibition. Oncotarget. 2017;8:21115‐21127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulus A, Akhtar S, Caulfield TR, et al. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib‐ or bortezomib‐resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016;6.e492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, Mazurkiewicz M, Hillert EK, et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin‐specific protease‐14 and induces apoptosis of multiple myeloma cells. Sci Rep. 2016;6:26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pellegrini P, Selvaraju K, Faustini E, et al. Induction of ER stress in acute lymphoblastic leukemia cells by the deubiquitinase inhibitor VLX1570. Int J Mol Sci. 2020;21:4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowinsky EK, Paner A, Berdeja JG, et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Invest New Drugs. 2020;38:1448‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Arcy P, Linder S. Proteasome deubiquitinases as novel targets for cancer therapy. Int J Biochem Cell Biol. 2012;44:1729‐1738. [DOI] [PubMed] [Google Scholar]
- 27.Tian Z, D'Arcy P, Wang X, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123:706‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mofers A, Perego P, Selvaraju K, et al. Analysis of determinants for in vitro resistance to the small molecule deubiquitinase inhibitor b‐AP15. PLoS One. 2019;14.e0223807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuoka A, Tochigi A, Kishimoto M, et al. Lenalidomide induces cell death in an MDS‐derived cell line with deletion of chromosome 5q by inhibition of cytokinesis. Leukemia. 2010;24:748‐755. [DOI] [PubMed] [Google Scholar]
- 30.Tsujioka T, Yokoi A, Uesugi M, et al. Effects of DNA methyltransferase inhibitors (DNMTIs) on MDS‐derived cell lines. Exp Hematol. 2013;41:189‐197. [DOI] [PubMed] [Google Scholar]
- 31.Kida JI, Tsujioka T, Suemori SI, et al. An MDS‐derived cell line and a series of its sublines serve as an in vitro model for the leukemic evolution of MDS. Leukemia. 2018;32:1846‐1850. [DOI] [PubMed] [Google Scholar]
- 32.Kitanaka A, Mano H, Conley ME, Campana D. Expression and activation of the nonreceptor tyrosine kinase Tec in human B cells. Blood. 1998;91:940‐948. [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem. 2003;278:3170‐3175. [DOI] [PubMed] [Google Scholar]
- 35.Tomita K, Kuwahara Y, Takashi Y, et al. Clinically relevant radioresistant cells exhibit resistance to H2O2 by decreasing internal H2O2 and lipid peroxidation. Tumour Biol. 2018;40:1010428318799250. [DOI] [PubMed] [Google Scholar]
- 36.Mehdipour M, Daghigh Kia H, Najafi A, Mohammadi H, Álvarez‐Rodriguez M. Effect of crocin and naringenin supplementation in cryopreservation medium on post‐thaw rooster sperm quality and expression of apoptosis associated genes. PLoS One. 2020;15.e0241105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oparka M, Walczak J, Malinska D, et al. Quantifying ROS levels using CM‐H2DCFDA and HyPer. Methods. 2016;109:3‐11. [DOI] [PubMed] [Google Scholar]
- 38.Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane‐bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355‐1364. [DOI] [PubMed] [Google Scholar]
- 39.Chitta K, Paulus A, Akhtar S, et al. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenström macroglobulinaemia tumour cells. Br J Haematol. 2015;169:377‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kropp KN, Maurer S, Rothfelder K, et al. The novel deubiquitinase inhibitor b‐AP15 induces direct and NK cell‐mediated antitumor effects in human mantle cell lymphoma. Cancer Immunol Immunother. 2018;67:935‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding Y, Chen X, Wang B, Yu B, Ge J. Deubiquitinase inhibitor b‐AP15 activates endoplasmic reticulum (ER) stress and inhibits Wnt/Notch1 signaling pathway leading to the reduction of cell survival in hepatocellular carcinoma cells. Eur J Pharmacol. 2018;825:10‐18. [DOI] [PubMed] [Google Scholar]
- 42.Vogel RI, Coughlin K, Scotti A, et al. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget. 2015;6:4159‐4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79‐87. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Pellegrini P, Saei AA, et al. The deubiquitinase inhibitor b‐AP15 induces strong proteotoxic stress and mitochondrial damage. Biochem Pharmacol. 2018;156:291‐301. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Espinosa B, Saei AA, D'Arcy P, Zubarev RA, Linder S. Oxidative stress induced by the deubiquitinase inhibitor b‐AP15 Is associated with mitochondrial impairment. Oxid Med Cell Longev. 2019;2019:1659468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maharjan S, Oku M, Tsuda M, Hoseki J, Sakai Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci Rep. 2014;4:5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089‐1115. [DOI] [PubMed] [Google Scholar]
- 48.Drexler HG. Myelocytic cell lines HL‐60. In: Drexler HG (Eds.), Leukemia‐Lymphoma Cell Lines, 2nd ed. Braunschweig: Academic Press; 2010:705‐706. [Google Scholar]
- 49.Bazzaro M, Linder S. Dienone compounds: targets and pharmacological responses. J Med Chem. 2020;63:15075‐15093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3