Abstract
Despite the low overall prevalence of individual rare diseases, cochlear dysfunction leading to hearing loss represents a symptom in a large proportion. The aim of this work was to provide a clear overview of rare cochlear diseases, taking into account the embryonic development of the cochlea and the systematic presentation of the different disorders. Although rapid biotechnological and bioinformatic advances may facilitate the diagnosis of a rare disease, an interdisciplinary exchange is often required to raise the suspicion of a rare disease. It is important to recognize that the phenotype of rare inner ear diseases can vary greatly not only in non-syndromic but also in syndromic hearing disorders. Finally, it becomes clear that the phenotype of the individual rare diseases cannot be determined exclusively by classical genetics even in monogenetic disorders.
Key words: rare diseases, cochlea, embryology of the inner ear, cochlear malformations
Introduction
The cochlea is a highly complex microsystem. In its completely developed stage, it consists of the spiral ganglion (neuronal tissue supported by satellite cells), the organ of Corti (neuroepithelium for sensory perception), the stria vascularis (highly vascularized epithelium that is responsible for ion transportation), and the otic capsule (specialized bony tissue). Although the cochlea was initially considered an immune privileged organ, the immune system (endolymphatic sac) also contributes to the hearing process. Subsets of tissues in the cochlea are also found in other organ systems. For example, the brain, as well as cranial and peripheral nerves have a comparable network structure of neurons and glial cells; a complex and highly structured sensory epithelium such as the organ of Corti is also found in the retina; the stria vascularis and the renal corpuscles are both metabolically highly active tissues that maintain ionic balance. These structural similarities of the cochlea and other tissues are reflected in the clinical observation that many diseases of the inner ear may also affect other organ systems.
Diseases of the cochlea are usually associated with hearing loss. Even if the degree of hearing loss can be well assessed subjectively and objectively, the actual cause of hearing loss remains unknown in most cases. Frequently, rare diseases that affect cochlea are often undiagnosed and represent a particular challenge because many are unknown to most general practitioners and specialists.
Amongst diseases affecting the cochlea, Many diseases of the cochlea are classified as rare (see Table). In recent years, modern molecular biological procedures could define the cause and pathophysiology of most rare diseases. Investigating rare diseases on a molecular level led to the identification of novel mechanisms underlying the pathophysiology of cochlear dysfunction and leading to the discovery of new therapeutic interventions.
The present article summarizes all rare diseases to the authors known where primary involvement of the cochlea resulting in sensorineural hearing loss is present (summarized in Table 1 ). Further rare diseases occurring primarily in other organ systems of the field of oto-rhino-laryngology that may also affect the cochlea are described in other articles of this publication (Weiss NM, Rare diseases of the middle ear and the lateral skull base; Scherl C, Rare diseases of the head and neck part III: salivary glands and facial nerve; Dlugaiczyk J, Rare diseases of the vestibular labyrinth: of zebras, chameleons, and wolves in sheep’s clothing). The table organizes the diseases based on their pathophysiology or pathogenesis and gives a systematic overview.
Table 1 Rare diseases of the cochlea.
| Name | Cause | Gene | Inheritance | Incidence | Therapy | Symptoms | Annotations |
|---|---|---|---|---|---|---|---|
| Autoimmune-mediated inner ear diseases | |||||||
| Cogan syndrome | Autoantibody-mediated (?) vasculitis with systemic manifestation 127 | - | - | About 300 cases worldwide 127 | Corticosteroids, cyclophosphamide, methotrexate, mycophenolate mofetil, azathioprine, infliximab 127 | Non-syphilitic interstitial keratitis (IK) with audiovestibular Menière-like symptoms 127 ; typical and atypical types are described: in atypical types, the eye involvement manifests with non-IK inflammatory ocular symptoms | Max. 2 years between the affection of both organs (eye and inner ear) 127 ; is considered as vasculitis 117 |
| Muckle-Wells syndrome | Excessive release of IL1beta 128 | NLRP3 128 | aut. dom. 128 | 1–3:1,000,000 | Anakinra 128 | Fever, skin rash, musculo-skeletal symptoms and conjunctivitis. Progressive sensorineural hearing loss and kidney failure 128 | Belongs to the group of CAPS (cryopyrin-associated periodic syndrome); Muckle-Wells syndrome, FCAS (familial cold auto-inflammatory syndrome) and NOMID (neonatal onset multisystem inflammatory disorder) have a common causative gene defect (NLRP3) |
| Neonatal onset multisystem inflammatory disease (NOMID) | Excessive release of IL1beta 129 | CIAS1/NLRP3 129 | aut. dom. 129 | Very rare, 100 cases have been described worldwide 129 | Anakinra 129 | Skin rash, chronic meningitis, fever, joint inflammation 129 | |
| Relapsing polychondritis | Autoimmune-mediated inflammation of cartilage 130 | Multifactorial etiology 130 | 1:285,000 130 | Glucocorticoids 130 | Cartilage inflammation, uveitis, vasculitis, hearing loss in 50%, vertigo 130 | ||
| Vogt-Koyanagi-Harada disease | T cell mediated destruction of melanin-containing tissue 131 | - | - | 1:400,000 131 | Glucocorticoids 131 | Uveitis, alopecia, meningism 131 | |
| Vascular | |||||||
| Behçet’s syndrome | Vasculitis, HLA-B51-associated (?) | - | - | Regional differences, 1:100,000 in Germany | Symptom-based, steroids, non-steroidal antiphlogistics 132 | Recurrent oral aphthae, genital ulcers, eye and skin lesions 132 | Kidneys and peripheral nerve system are very rarely affected |
| Eosinophilic granulomatosis with polyangiits (‘EGPA; formerly: Churg-Strauss syndrome) | Allergic granulomatosis with polyangiitis, antinuclear cytoplasmic antibody-associated vasculitis 133 134 | - | - | 2.4:1,000,000 | High-dose glucocorticoids, cyclophosphamide, zafirlukast (leukotriene antagonist) 134 135 , mepolizumab (anti-interleukin-5 antibody) 136 | Blood eosinophilia, heart failure, allergic rhinitis, asthma, vasculitis with involvement of the skin, heart, lung, gastrointestinal tract, neural system 133 134 136 | Triphasic disease, manifestation in the inner ear in the 3 rd phase is rarely observed 133 , EGPA is classified as ANCA-associated vasculitis among minor vessel vasculitis |
| Generalized arterial calcification in infants | Calcium deposits in the arteries 137 | ENPP1, ABCC6 137 | Aut. rec. 137 | 1:391,000 137 | Bisphosphonates 137 | Heart failure, stroke, pseudoxanthoma elasticum 137 | Also conductive hearing loss 137 |
| Hereditary hemorrhagic telangiectasia (Weber-Osler-Rendu disease) | Vascular dysplasia, arterio-venous fistulas/malformations 138 | Chromosomes 9q and 12q 138 | Aut. dom 138 | 1–2:100,000 138 | Vascular malformation of multiple organs (kidney, gastrointestinal tract, liver, lung, brain), recurrent epistaxis as most frequent symptom | ||
| Kawasaki disease | Necrotizing vasculitis 139 140 | Unknown, corona virus (?) | >300,000 cases have been described worldwide 139 | Intravenous application of immunoglobulins, aspirin | Fever, skin rash, conjunctivitis, cardiac complications | Appears nearly exclusively in children | |
| Norrie disease | Disorder of the angiogenesis of eye and inner ear 141 | NDP 141 | X-linked | More than 400 cases have been described worldwide | Retinal detachment and progressive hearing loss 141 | ||
| Susac’s syndrome | CD8 T cell-mediated autoimmune-microangiopathic endotheliopathy 142 | - | - | Slightly more than 300 cases have been described worldwide 142 | Anti-platelet medicine, anti-coagulants, immunosuppressive treatment with e.g., cyclophosphamide, intravenous immunoglobulins, mycophenolate mofetil, azathioprine, methotrexate, natalizumab 142 | Visual field loss, visual loss, neurological symptoms, cephalgia 142 | |
| Granulomatosis with polyangiitis (GPA; formerly: Wegener’s granulomatosis) | Autoimmune vasculitis 143 | - | - | 1:6,400 143 | Glucocorticoids, rituximab 143 | Sinusitis, tracheal stenosis, kidney failure, pneumonia, mastoiditis 143 | ANCA+ 143 |
| Malformations | |||||||
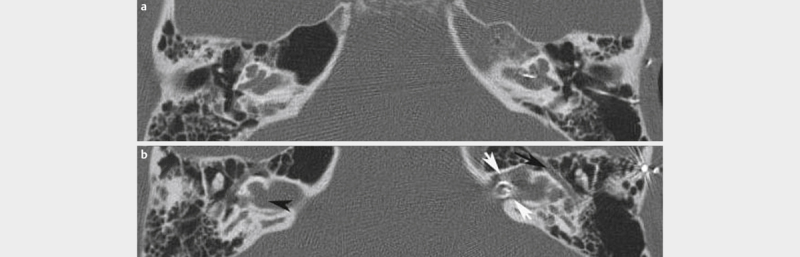
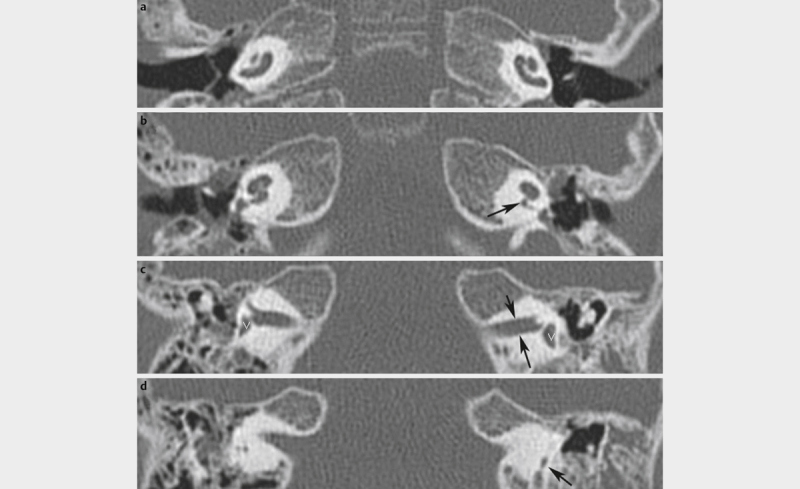
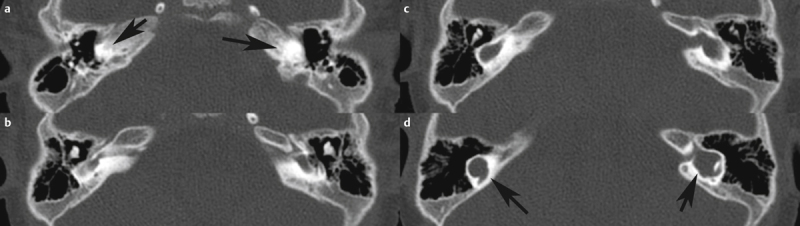
| Labyrinthine aplasia | Complete aplasia, when the development is disturbed before or at the beginning of the 3 rd week of gestation; may also be thalidomide-induced 144 145 | Increased risk in cases of consanguine parents 146 | 2% of all inner ear malformations | ABI | Facial nerve paresis or weakness 146 | Synonym: Michel deformity, Michel aplasia, bony canal of the internal carotid artery may be missing 146 | |
| Otocyst deformity | Developmental arrest in the 3 rd week of gestation 146 | Increased risk in cases of consanguine parents | 1% of all inner ear malformations | ABI | Congenital deafness, possible facial nerve palsy or weakness 146 | Synonym: common cavity (according to Jackler) | |
| Cochlear aplasia | 5% of all inner ear malformations 100 | CI, ABI 100 | Congenital deafness | Speech understanding with CI is possible 100 | |||
| Cochlear hypoplasia (isolated) | Frequently occurs in BOR syndrome 108 | 13% of all inner ear malformations 108 | CI | Deafness, high-grade hearing loss | Broad spectrum, nearly normal form up to a small basal cyst 108 | ||
| Complete aplasia of the semicircular canals | (includes hypoplastic cochlea and small vestibule with saccule) 147 | Often associated with CHARGE; single case reports with e.g., Wildervanck, Noonan, Goldenhar, or VACTERL* 148 | 16% of all inner ear malformations | CI | Deafness, high-grade hearing loss 147 | Main criterion of CHARGE | |
| Incomplete partition type 2 (IPT2) | 21% of all inner ear malformations 149 | CI | Deafness, high-grade hearing loss | Mondini malformation in the actual sense 149 | |||
| Incomplete partition type 1 (IPT1) | FOXF2 150 | 9% of all inner ear malformations 149 | CI | Deafness, high-grade hearing loss | Synonym: common cavity (E. Cock); cystic cochleovestibular malformation, figure-8 deformity | ||
| X-linked deafness (inner ear malformation called IPT3) | Different mutations of POU3F4 89 | POU3F4 | 3% of all inner ear malformations | Hearing aids, CI 151 | Variable hearing loss | Synonym: gusher, IPT3 – even if it is not in line with other incomplete partitions; often associated with hamartoma of the tuber cinereum 92 | |
| Modiolus aplasia | 1% of all inner ear malformations 110 | ||||||
| Hypoplasia or aplasia of the vestibulocochlear nerve or isolated of the cochlear nerve | Mostly combined with severe inner ear malformations 110 | ||||||
| * VACTERL describes an association of congenital malformations that has at least three of these symptoms: esophageal atresia, kidney malformation, heart defect, vertebral defects, anorectal malformations, and radial extremity malformations | |||||||
| Chromosomal | |||||||
| 3p deletion syndrome | Deletion of the short arm of chromosome 3 152 153 | Chromosome 3 152 153 | De novo 152 153 | Very rare | Microcephaly, triangular shape of the face, flat occiput, hypertelorism, polydactyly, cryptorchidism, renal and cardiac defects 152 153 | ||
| 10p deletion syndrome | Deletion of the short arm of chromosome 10 154 | Chromosome 10 154 | Very rare, about 50 cases are known 135 | Craniofacial malformations, growth disorders, congenital heart defects, hypoparathyroidism, immunodeficiency, mental retardation 154 | Haploinsufficiency 10p15 causes also HDR1 syndrome 155 | ||
| Cri-du-chat syndrome, 5p deletion syndrome | Deletion of the short arm of chromosome 5 156 | Haploinsufficiency of various genes, e.g., TERT, MARCH6, CTNND2, and SLC6A3 on chromosome 5 156 | De novo 156 | 1:15,000–1:50,000 156 | High-frequency cry (cri-du-chat), microcephaly, facial dysmorphia, delayed speech acquisition, mental disability 156 | Most frequent chromosomal defect 156 , neural hearing loss 157 | |
| DiGeorge anomaly 154 , chromosome 22q11.2 deletion | Haploinsufficiency of DiGeorge syndrome critical region gene 2 (DGCR2) 155 | DGCR2, centromere deletion of chromosome 10, 22q11.2 deletion 154 158 | 1:4,000 158 | Allogenic thymus tissue transplantation 159 | Thymus aplasia, congenital developmental disorder, T cell deficiency, hypocalcaemia, cardiovascular malformation, facial dysmorphia 154 | ||
| Cat eye syndrome, Schmid-Fraccaro syndrome | Anomaly of chromosome 22, 22 160 161 | Chromosome 22 160 161 | Aut. dom. (160, 161] | 1:100,000 160 161 | Symptom-based, experimental: GNE-886, selective inhibitor of the cat eye syndrome chromosome region candidate 2 bromodomain 162 | Coloboma, anal atresia, heard defects, preauricular tags 160 161 | |
| Mosaic trisomy 9 | Partial trisomy 163 164 165 | Chromosome 9 163 164 165 | Growth retardation, muscular weakness, mental disability, microcephaly, micrognathia, characteristic palpebral fissures, skeletal anomalies, microphthalmia, cleft palate, hydrocephalus 163 164 165 | Partly very mild courses that remain undiagnosed 163 | |||
| Mosaic trisomy 22 | Partial trisomy 166 167 | Chromosome 22 166 167 | Mental disability, growth disorders, failure to thrive, craniofacial asymmetry, microcephaly, brachycephaly, hypoplasia of the midface, preauricular tags, flat nose, micrognathia, cleft palate 166 167 | Overlapping with cat eye syndrome? | |||
| Pallister-Killian mosaic syndrome | Chromosome duplication (12p) 168 | 150 cases worldwide 168 | Muscular hypotonia and telecanthus 168 | ||||
| Smith-Magenis syndrome 169 | 17p11.2 deletion 169 | RAI1 169 | 1:15,000 169 | Brachycephaly, broad square-shaped face, hypotonia, sleep disorder, self-injury 169 | Initially conductive hearing loss, the progressive sensorineural hearing loss at the age of 10 years | ||
| Trichorhinophalangeal syndrome type II | Chromothripsis, chromosome deletion (q8) 170 | TRPS1, EXT1 170 | <60 worldwide 170 | Thin hair, short stature 170 | Langer-Giedion syndrome 170 | ||
| Metabolic diseases | |||||||
| Acyl-Co-A dehydrogenase deficiency (Schindler syndrome) | Lactate acidosis, mitochondrial disease of the complex I concerning the respiratory chain 171 | ACAD9 171 172 | Aut. rec. 171 | Very rare, 24 patient from 12 families have been described up to 2016 172 | Riboflavin substitution is effective in some patients 171 172 | Neurological, muscular, hepatic, and cardiac manifestation 171 172 | |
| Alpha galactosidase deficiency (Fabry’s disease) | Lysosomal storage disease, glycol-sphingolipid catabolism 173 | X-chromosomal | 1:40,000–1:117,000 | Agalsidase beta (enzyme substitution) 174 | Progressive kidney disease, cardiomyopathy, cerebrovascular complications, neuropathic pains, apoplexy 173 | Atrophy of the organ of Corti, stria vascularis, and the spiral ligament in 2 post-mortem analyses 173 | |
| Alpha mannosidosis | Deficiency of the lysosomal alpha D mannosidase | MAN2B1 | Aut. rec. | 1:500,000 135 | Velmanase alpha (Lamzede®) by Chiesi 175 176 | Recurrent infections, muscular weakness, skeletal and facial deformities, ataxia, hepatosplenomegaly, hydrocephalus, macroglossia, prognatism, strabism, hyperopia or myopia; immune deficiency, hypersomnia, psychiatric diseases, mental disability 135 177 | Lysosomal storage disease; different subtypes, severity and age at disease onset 135 177 |
| Biotinidase deficiency | Disorder of all mitochondrial caboxylases 178 | BTD 178 | Aut. rec. 178 | 1:50,000 178 | Biotin substitution | Seizures, muscular weakness, ataxia, developmental delay, visual loss, alopecia, skin rash 178 | Neuromyelitis optica spectrum disorders (NMOSDs) Holocarboxylase synthetase (HCLS) deficiency; incidence of 1:200,000 |
| Brown-Vialetto-van-Laere syndrome (riboflavin transporter deficiency) | Deficiency of riboflavin transporter proteins | SLC52A2, SLC52A3{179] | Aut. rec. 179 | Less than 100 cases are known 179 | Riboflavin substitution 180 | Progressive pontobulbar paralysis, respiratory insufficiency, muscular weakness, facial nerve palsy, ptosis, dysphagia, and ataxia 179 | |
| Camurti-Engelmann syndrome, diaphyseal hyperostosis or sclerosis | Permanent activity of the transforming growth factor beta 1 causing increased bone density and reduced fat and muscle tissue 181 182 183 184 | TGFB1 181 182 183 184 | Aut. dom. 181 182 183 184 | >300 cases are described worldwide 185 | Experimental approaches with TGF beta receptor antagonists 185 | Hyperostosis of the long bones, diffuse thickening of the skull base, ophthalmopathy, cephalgia, vasculopathy, pains, muscular weakness 181 182 183 184 | Craniotubular bone disease, progressive stenosis of the internal auditory canal 183 186 |
| Chanarin-Dorman syndrome | Abhydrolase deficiency and lacking activation of fat triglyceride lipase 187 | ABHD5 187 | Aut. rec. | More than 128 known cases | Symptom-based, fat-free diet | Congenital ichthyosiform erythroderma, hypothyroidism, neurological symptoms, liver function disorder, cataract, ectropion 187 | Neutral lipid storage disease with ichthyosis |
| Craniometaphyseal dysplasia | Inhibition of the regulated bone remodeling by extracellular pyrophosphate accumulation 188 | ANKH, GJA1 | Aut. rec. or aut. dom. | Very rare | Symptom-based | Hypertelorism, dolichocephaly, proptosis, prominent mandible, thickening of the skull bone, retarded dentition 188 189 190 | |
| Familial hypophosphatemia | Phosphate loss due to increased secretion of the phosphaturic hormone fibroblast growth factor 23 191 | X-chrom., more rarely aut. rec., aut. dom. 191 192 | 3:100,000 for X-linked | Symptom-based, phosphate and vitamin D | Rickets, abnormal gait, deformity of the lower extremities, retarded growth, dental abscesses 191 | ||
| Farber lipogranulomatosis | Lysosomal storage disease, acid ceramidase deficiency 193 194 | ASAH1 193 194 | Aut. rec. 193 194 | 201 cases were known in 2018 | Symptom-based | Subcutaneous nodes, deformed joints, progressive hoarseness, special types of muscle atrophy and progressive myoclonal epilepsy 193 194 | |
| Fibrodysplasia ossificans progressive | Heterotopic ossifications 195 196 | ACVR1/ALK2 195 196 | Aut. dom. 195 196 | 1:2,000,000 195 196 | Symptom-based 195 196 | Missing nails, progressive heterotopic ossification, hypoplasia of the brainstem, cognitive and motor developmental disorders | |
| Fibrous dysplasia (Jaffe-Lichtenstein syndrome) | Disorder of the osteogenesis due to overproduction of cAMP, phosphorylation CREB and activation of cAMP-depending protein kinase (PKA) 135 | GNAS | Non-hereditary | Unknown | Bisphosphonates | Exchange of normal bone and bone marrow with fibrous connective tissue and immature trabecular bone 135 | Often conductive but sometimes also sensorineural hearing loss 135 |
| Kernicterus | Deposit of unconjugated bilirubin 135 | Sporadic | Often occurs in premature births | Hyperbilirubinemia 135 | Neural and central hearing loss 135 | ||
| Congenital disease of the glycosylation | Defect biosynthesis of glycanes | Several genes 197 | Aut. rec., rarely x-linked | Less than 100 cases per type 198 | Symptom-based, mannose or D galactose supplementation 198 | Multisystem manifestation, neurological symptoms, mental disability, cardiomyopathy, edema, facial deformities 198 | More than 130 types are described 198 , defect N, O, and combined N and O glycosylation as well as lipid glycosylation 199 |
| Leigh syndrome (infantile necrotizing encephalopathy) | Congenital lactate acidosis, pyruvate dehydrogenase deficiency | PDHA, pyruvate dehydrogenase (E1) a subunit 200 201 | X-linked 200 201 | 1:40,000–70,000 | High-dose thiamine substitution 200 201 | Peripheral neuropathies, chorea, Parkinson-like symptoms, cognitive deficits, necrotic lesions in the brain, hypertrophic cardiomyopathy 200 201 | |
| Mucopolysaccharidosis type 1 (formerly: Hurler or Scheie syndrome) | Lysosomal storage disease 202 | NEU1 202 | Aut. rec. 202 | 1:42,000,000 202 | Ataxia, myoclonus, progressive visual loss 202 | ||
| Mucopolysaccharidosis type II (Hunter) | IDS 203 | X-linked, recessive 203 | 0.5–1:100,000 203 | Symptom-based, enzyme substitution therapy | Coarse facial features, skeletal deformities and stiff joints, growth retardation with hyposomia, impairment of respiration and heart including diffuse valvulopathy, inguinal and umbilical hernia, hepatosplenomegaly, neurological involvement in at least two third of the cases, adeno-tonsillar hypertrophy, obstructive sleep apnea, retinal degeneration 203 | Lysosomal storage disease | |
| Niemann-Pick-C syndrome 204 | Lysosomal storage disease; disturbed cholesterol and fatty acid transport 204 | NPC 1; NPC 2 204 | Aut. rec. | 1:100,000–250,000 204 | Progressive neurodegeneration, hepatomegaly 204 | Mild to high-grade hearing loss, also neuropathy | |
| NGLY1 deficiency | Inability to remove N-glycan 205 | NGLY1 205 | Aut. rec. | <63 patients worldwide | - | Neuropathy, corneal ulcerations, dystonia | |
| Oculo-auriculo-vertebral dysplasia (Goldenhar syndrome) 206 | Unknown | - | 1:30,000–1:40,000 206 | Unilateral malformation of cheekbones, jaw, mouth, ears, eyes, and/or vertebrae 206 | Part of Goldenhar syndrome, aural atresia, internal auditory canal may be malformed 206 | ||
| Primary distal renal tubular acidosis (distal RTA, type) | Proton pump subunit B1 is also expressed in the stria vascularis 207 ; some patients also have an enlarged vestibular aqueduct | ATP6V1B1;ATP6V0A4 207 | Aut. rec. | - | Correction of metabolic deficits 207 | Metabolic acidosis and osteomalacia 207 | Hearing loss is variable and is often not regressive under alkali therapy |
| Pompe disease (glycogenosis type 2) | Glycogen deposits in muscles 208 | GAA 208 | Aut. rec. 208 | 1:40,000 208 | Congenital or progressive muscular weakness, respiratory insufficiency 208 | Mild hearing loss, possible stapedius muscle weakness 208 | |
| Refsum disease | Failure of metabolism of phytanic acid 209 210 211 | PHXH; PEX7 209 210 211 | Aut. rec. | 1:1,000,000 209 210 211 | Retinitis pigmentosa, ichthyosis, anosmia 209 210 211 | Onset of the symptoms at an age of 10–20 years; mild to high-grade hearing loss, also neuropathy 209 210 211 , Bamiou et al. | |
| Rogers syndrome; thiamin responsive megaloblastic anemia | Thiamin pyrophosphokinase deficiency 212 213 ; highly-affine thiamin transporter | SLC19A2 212 213 | Aut. rec. 212 213 | Less than 80 cases are known 175 | Thiamin substitution 212 213 | Diabetes mellitus, megaloblastic anemia 212 213 | Thiamin pyrophosphokinase=highly affine thiamin transporter |
| Schindler syndrome | Lysosomal storage disease 214 | NAGA 214 | Aut. rec. 214 | <1:200,000 214 | Progressive neurodegeneration with hypotonia and telangiectasias in the adult type 214 | ||
| Keratoses and ichthyoses | |||||||
| Autosomal recessive congenital ichthyosis | Non-syndromic keratin disorder due to mutation of genes that regulate the keratinocyte differentiation | Different, e.g., TGM1, ALOXE3, ALOX12B, PNPLA1, and CERS2 215 | Aut. rec. | 1:100,000 | Symptom-based | Heat intolerance, pruritus, growth disorders, visual disorders 215 | Different types, syndromic types are e.g., KID |
| De Sanctis-Cacchione syndrome | Xeroderma pigmentosum, severe DNA reparation disorder (defective nucleotide excision reparation) | XPA or ERCC2/XSD 216 | Aut. rec. 216 217 | About 200 cases are known | Symptom-based | Cutaneous photosensitivity, microcephaly, mental disability, hyposomia, hypogonadism, spasm, peripheral neuropathy 216 217 | |
| Harlequin ichthyosis | Hyperkeratosis with defective keratinocyte transmembranous lipid transporter protein and disorder of the lipid transport to the stratum corneum 218 | ABCA12 218 | Aut. rec. | 1:500,000 218 | Symptom-based | Thickened yellowish skin with fissures, ectropium, eclabium, round open mouth, missing scalp hair as well as cilia and eyebrows 218 | Severest type of congenital ichthyosis |
| Keratosis-ichthyosis deafness syndrome (KID) | Connexin-26 disorder 219 | GJB2 219 | Sporadic, also aut. dom. and rec. cases are known 219 | Less than 100 cases are known 219 | Erthrokeratodermic follicular hyperkeratosis, psoriasisiform or verrucous plaques, palmoplantar keratodermatosis, conjunctivitis, hypotrichosis 219 | Characteristic triad: ichthyosiform erthrodermatosis, high-grade sensorineural hearing loss, vascularizing keratitis 219 | |
| Hereditary palmoplantar keratosis (PKK) | Connexin-26-related change of the Cx43 gap junctions (increased semicanal activity) 220 | GJB2 (Cx26-H73R, und Cx26-S183F) 220 | Aut. dom. or mitoch. 175 | Very rare,<1:1,000,000 175 | Palmar and plantar hyperkeratosis 220 | ||
| Trichothiodystrophy | Nucleotide excision reparation | ERCC2, ERCC3, TTDA, TTDN1, GTF2E2 | Aut. rec. | 1:1,000,000 | Dermal ichthyosis, mental and growth retardation, hypogonadism 221 | Variable manifestations, BIDS (brittle hair, impaired intelligence, decreased fertility, and short stature), IBIDS (with ichthyosis), PIBIDS (with photosensitivity), or Tay syndrome 221 | |
| Syndromes | |||||||
| Alström syndrome | Ciliopathy 222 | ALMS1 222 | Aut rec. 222 | 1–9:100,000 222 | CI | Photoreceptor dystrophy, obesity, type-2 diabetes, hyperlipidemia, acanthosis nigricans, hypogonadism, renal, pulmonary, and hepatic dysfunction, dilatative cardiomyopathy 222 | |
| Arts syndrome | Deficiency of phosphoribosyl pyrophosphate synthetase 1 223 | PRPS1 223 | X-linked | Very rare | Ataxia, mental retardation, hypotension, opticus atrophy, peripheral neuropathy 223 | ||
| Barakat syndrome | Developmental disorder of the parathyroid, kidney, and inner ear 135 | GATA3 224 | Aut. dom. | 180 patients worldwide 224 | Symptom-based | Hypoparathyroidism, deafness, and kidney diseases; variably phenotypes are possible 224 225 | |
| Bardet-Biedl syndrome | Cilipathy 135 226 | 21 different genes 226 | Aut. rec. | About 1:150,000 227 | Symptom-based; experimental gene therapeutic approaches 228 | Obesity, pigmental retinopathy, kidney disease, anosmia, hypogonadism, situs inversus 226 227 228 | Incidence is higher in regions with frequent consanguinity 227 |
| Bartter and Gitelman syndrome | Channelopathy 229 | Several, e.g., KCNJ1, NKCC, NCCT, BSND, ROMK, IBS, CLCNKB, SLC12A1 SLC12A3 229 230 | Aut. rec. 229 | Hypokaliemia, hypochloremic metabolic alkalosis, polyuria, polydipsia | Bartter type 1–4, Gitelman (SLC12A3) as mild, late onset type | ||
| Björnstad syndrome | Chaperonopathy, disorder of the ATPasis and lack of mitochondrial complex III 231 | BCS1L 231 | Aut. rec. and aut. dom. 231 | Extremely rare 231 | Pilli torti 231 | Disorder of the mitochondrial respirasome | |
| Branchio-oculo-facial syndrome (BOFS) | Disorder of the retinoic acid-induced transcription factor AP-2 alpha and thus of the regulation of eye, face, skin, neural tube, and kidney morphogenesis 232 | TFAP2A 232 233 | Aut. dom. 232 233 | <1:1,000,000 175 | Symptom-based | Low birth weight and growth and growth retardation, branchial skin alterations (hemangioma-like manifestation at the neck and behind the ears), microphthalmia, ptosis, cataract, dacryocystitis, characteristic facial changes (wide philtrum, cleft lip and palate, flat broad nose, deformed auricles 232 233 | Hearing loss may be conductive, sensorineural or mixed; clinical overlapping with BOR syndrome 232 233 |
| Branchio-oto-renal syndrome (BOR) | Disorder of the renal formation and the otic placode 235 236 , mild cochlear hypoplasia, second most frequent malformation | EYA1 (40% of the patients with clinical symptoms), SIX1, SIX5 (genes of the EYA-DACH-SIX-PAX pathways) 234 235 236 | Aut. dom. 234 236 | 1:40,000 234 236 | Symptom-based | Cervical or preauricular branchial fistula, hypoplasia, dysplasia, or agenesis of the kidneys, aplasia of the 8th cranial nerve 234 236 | Variable presentation and severity 235 , radiologically cochlear hypoplasia 236 |
| Boudhina-Yedes-Khiari syndrome | Neuro-cutaneous disease 237 | - | Aut. rec. 237 | 3 patients worldwide 237 | Symptom-based | Growth retardation, microcephaly, mental retardation, epilepsy and skin lesions 237 | |
| Carpenter syndrome, acrocephalopolysyndactyly type II | Mutation of the guanosin triphosphatase (GTPases) 238 239 240 | RAB23 238 239 240 | Aut. rec. 238 239 240 | Extremely rare 175 , about 40 cases are known | Symptom-based | Craniosynostosis, craniofacial malformations, polysyndactyly, obesity, mental disability, hypogonadism 238 239 240 | RAB23=Ras-associated binding protein 23; negative regulator of sonic hedgehog and fibroblast growth factor signalling pathway 238 |
| CHARGE syndrome, Hall-Hittner syndrome | Neurocristopathy, dysregulated gene expression and development of the neural crest, dysregulation of the neural crest stem cells, dysregulation of the alternative splicing (spliceosomopathy) 98 241 | CHD7 heterocygotic mutation 8q12 98 as well as newly identified genes: PUF60, EP300, RERE, KMT2D and KDM6A 241 | Aut. dom. (97% de novo) 98 241 | 0.1–1:10,000 135 | Symptom-based, CI | Variable expression of the symptoms, visual loss, cardiac anomalies, skeletal, oronasal, gastrointestinal, and genitourinary malformations, growth disorders, craniofacial malformations, anosmia, facial nerve palsy, immune deficiency 241 Diagnostic criteria 98 : typical: 2 or 3 main and 2 minor criteria; main criteria: coloboma (ocular), choanal atresia/stenosis, hypo-/aplasia of the semicircular canals; minor criteria: rhomb encephalic dysfunction (brainstem and cranial nerve anomalies), hypothalamo-pituitary dysfunction, malformation of the internal and/or external auditory canal, mediastinal organs (heart, esophagus), intellectual weakness | CHARGE= C oloboma of the eye, H eart defects, A tresia of choanae, R etardation of growth, G enital abnormalities, E ar anomalies; overlapping with Kallmann, Kabuki, 22q11.2 and Nager syndromes as well as with Guion-Almeida mandibulofacial dysostosis 241 ; the missing of the semicircular canals is highly predictive for CHD7 mutation |
| Cockayne syndrome, Neill-Dingwal syndrome | Delayed DNA reparation after UV light exposure, mitochondrial changes 242 243 | ERCC8, ERCC6 242 | Aut. rec. | 1:250,000 242 | Symptom-based | Hyposomia, cerebral and retinal atrophy, joint contractures, photosensitivity and wrinkled skin, atherosclerosis and vasculopathy, high blood pressure, stroke and cardiac infarction, peripheral neuropathy 242 | 3 types |
| Coffin-Lowry syndrome | Growth factor regulated serin-threonin-protein kinase 244 245 | RSK2; Locus Xp22.2 244 245 | X-linked 244 245 | >100 cases are known 244 245 | Symptom-based | Severe mental disability, hyposomia, hypertelorism, prominent front, anteverted nostrils, thick fingers with slim tips, kyphoscoliosis 244 245 | |
| Coffin-Siris syndrome | Mutations of the BRG-1 associated factor (BAF) complex, cell growth, division, replication, and differentiation as well as in DNA reparation 246 247 | ARID1A, ARID1B, SMARCA4, SMARCB1, SMARCE1, SOX11 246 247 | Aut. dom. and aut. rec. 246 247 | About 100 cases are known 246 247 | Symptom-based | Cognitive and developmental disorder, hypoplastic phalanxes and little finger nails, hirsutism, ptosis, cataract, strabism, hypospadias 246 247 | |
| Cornelia-de-Lange syndrome | Cohesinopathy, disorder of the chromatid cohesion and thus mitosis, disorder of the regulation of the transcription 248 249 250 | SMC1A, SMC3, RAD21 or HDAC8 248 249 250 | Aut. dom. or X-linked | More than 400 cases are known | Symptom-based | Facial dysmorphy (arched eyebrows with synophrys, long philtrum, thin lips, hairy front), prenatal and postnatal growth retardation, cognitive impairment, gastrointestinal malformations, congenital cardiac anomalies and malformed extremities 248 249 250 | |
| Curschmann-Batten-Steinert syndrome, myotonal dystrophy | Gene defect resulting in splicing defects of the pre-mRNAs of multiple genes | DMPK (type I), CNBP (type II); both loci adjacent to the DFNA18 locus 251 252 253 | Aut. dom. | Myotonia, muscular atrophy, insulin resistance, cardiac arrhythmia, cataract, cognition disorder and mental disability | Two types are known, also subclinically cochlear damage without hearing loss | ||
| Donnai-Barrow syndrome | Occulo-auditory syndrome 254 255 | LRP2 254 255 | Aut rec. 254 255 | <50 patients worldwide 254 255 | Symptom-based | Diaphragmatic hernia, exophthalmos, missing corpus callosum, myopia, proteinuria 254 255 | |
| DOOR syndrome | Unknown | TBC1D24 256 , SMARCB1 257 | Aut. rec. 256 257 | About 50 cases are known 257 | Symptom-based | Onychodystrophy, osteodystrophy, retardation, seizures 256 257 | DOOR=deafness, onychodystrophie, osteodystrophie, retardierung 256 257 |
| Ehlers-Danlos syndrome | Disorder of the collagen biosynthesis 258 259 | B4GALT7, B3GALT6, SLC39A13 and others 259 | Aut. rec. | 1:5,000 258 | Symptom-based | Skin and joint hyperlaxity, spondylodysplasia, kyphoscoliosis, aneurysms and ruptures of arteries, osteopenia/osteoporosis 258 259 | Different subtypes are known |
| Fountain syndrome | unknown | Unknown | Aut. rec. | Extremely rare | Symptom-based | Mental retardation, erythematous swelling of the face, skeletal changes 260 | |
| Freeman-Burian (Sheldon) syndrome | Distal arthrogryposis, multiple contractures 261 | MYH3 261 | Sporadic, aut. dom. 261 | About 100 cases are known | Symptom-based | Microstomia, pursed lips just as for siffling, H- or V-shaped chin defect, prominent nasolabial fold and major contractures of 2 or more body regions, typically hands and feet 261 | Craniofacial syndrome |
| Hajdu-Cheney syndrome | Disorder of the intercellular notch signaling pathway 262 | NOTCH2 262 | Aut. dom. | More than 80 cases are known 262 | Symptom-based | Craniofacial anomalies, cardiovascular disease, kidney cysts 262 | Craniofacial syndrome |
| HOXA1 syndrome | Developmental disorders of the head, the neural system, the inner ear, and the vestibular system 263 | HOXA1 263 | Aut. rec. 263 | Extremely rare 263 | Bilateral duane syndrome, cerebrovascular and cardiovascular malformations, autism, variable phenotypes are possible 263 | Mixed hearing loss | |
| Hutchinson-Gilford-Progerie syndrome | Reduced subcutaneous fat, aberrant lamin A production 264 | LMNA 264 | de novo, Aut. dom. 264 | 1:4,000,000 264 | Osteolysis, delayed eruption and loss of milk teeth, abnormal skin pigmentation, alopecia, osteoporosis, severe atherosclerosis, nightly lagophthalmos 264 | ||
| Johanson-Blizzard syndrome | Defect of ubiquitin protein ligase E3 components N-rekognin1 (UBR1) and thus disturbed ubiquitination and degradation of ubiquitin-associated proteins, disorder of the cell proliferation, differentiation, and apoptosis 265 | UBR1 265 | Aut. rec. 265 | Very rare, about 70 cases are known 265 | Exocrine pancreas insufficiency, hypoplasia of the nasal alae, oligodontia, skull defects, cognitive disorder, hyposomia, hypothyroidism, microcephaly, intrauterine growth disorder, congenital heart defect, urogenital and anorectal malformations kidney anomaly, late-onset diabetes mellitus 265 | ||
| Juberg-Marsidi syndrome | Disorder of the E3 ubiquitin ligase regulating the key factors such as p53 and Mcl1 266 | HUWE1 266 | X-linked, rec. 266 | 6 families are known 266 | Symptom-based | Mental retardation, growth disorder, hypogonadism, hypertelorism, microcephaly 266 | Hearing loss, possibly due to recurrent otitis media? |
| Kabuki syndrome | Disturbed histones-lysine methylation and chromatin remodeling 267 | KMT2D, sometimes also KDM6A 267 | X-linked 267 | 1:32,000–86,000 267 | Newborn hypotonia, nutritional problems in infants and toddlers, postnatal growth disorders, skeletal anomalies, disorders of the immune system, endocrine anomalies and congenital malformations of the heart, kidney, and palate 267 | ||
| Kallmann syndrome | Defect development of gonadotropin-releasing hormone secreting neurons and absence of puberty 268 | SOX10, KAL1, FGFR1, FGF8, FGF17, CHD7 and others 268 | X-linked, aut. rec. or aut. dom. | Very rare | Hormone replacement therapy | Hypogonadotropic hypogonadism with anosmia, cleft lip and palate, renal agenesis, short metacarpal bones, synkinesis, movement disorders of the eyes, cerebellar ataxia, and scoliosis 268 | Incidence of the congenital hypogonadotropic hypogonadism syndrome: 1:50,000 |
| Kearns-Sayre syndrome | Mitochondiopathy, disorder of the oxidative phosphorylation 269 | Deletion of the mitochondrial DNA 269 | de novo, rarely X-linked 269 | 1.6:100,000 269 | Chronic progressive external ophthalmoplegia, retinopathia pigmentosa, disorders of stimuli conduction, endocrine involvement, weakness of non-ocular muscles, encephalopathy 269 270 | Characteristic triad: disease onset before the age of 20, chronic-progressive external ophthalmoplegia, retinopathia pigmentosa 269 | |
| Klippel-Feil syndrome | Congenital synostosis 271 | Different, GDF6, GDF3, MEOX1 | Sporadic, sometimes aut. rec. or aut. dom. | 1:40,000 271 | Congenital malformation of the spine, extraskeletal manifestation with urogenital and cardiovascular anomalies, neural tube defects and cleft palate 271 272 | ||
| Kniest dysplasia | Type II collagenosis 273 | COL2A1 273 | Aut. dom. | Rare, exact incidence is unknown 273 | Short torso and extremities, kyphoscoliosis and craniofacial anomalies 273 | ||
| LADD syndrome | FGFR2, FGFR3, FGF10 274 | Very rare, less than 30 cases are known 274 | Hypoplasia/aplasia of the lacrimal glands/duct, hypoplasia/aplasia of the salivary glands, dental anomalies, malformation of the ears and fingers 274 | ||||
| Landau-Kleffner syndrome | Acquired epileptiform aphasia 275 | Unclear, mutation of GRIN2A, RELN, BSN, EPHB2, and NID2 have been described 275 | No data in the literature 275 | About 1:1,000,000 275 | Anticonvulsants, steroids, adrenocorticotropic hormone replacement diet, immunoglobulins 275 | Epileptic seizures, regression of speech, aggressive and hyperactive behavior 275 | Abnormal EEG, autism spectrum disorders |
| Noonan syndrome (formerly: LEOPARD syndrome) | RAS/MAPK disorders, developmental disorder of the neural crest due to mutations of the “non-receptor protein tyrosine phosphatase” SHP2 276 | PTPN11, RAF, and BRAF 276 | Aut. dom. | About 200 cases are known 276 | Lentigines, abnormal ECG, ocular hypertelorism, pulmonary valve stenoses, micrognathia, growth retardation 276 | ||
| Levy-Yeboa syndrome | KCNQ1 and KCNE3 277 | Aut. rec. | 1 family with 3 siblings has been described 277 | Congenital myopathy, recurrent secretory diarrhea, epidermolysis bullosa, microcephaly 277 | |||
| Marshall syndrome | Disturbed ectodermal development 278 | Coll11A1 278 | Aut. dom. | <1:1,000,000 | Facial dysmorphia, hypoplasia of the nasal bone and frontal sinus, skeletal anomalies 278 | Progressive hearing loss | |
| Maternally Inherited Leigh Syndrome (MILS) and NARP syndrome | Mitochondrial disease 279 | MTATP6 279 | Maternal 279 | 1:12,000–1:40,000 | Neuropathy, ataxia, retinitis pigmentosa 279 | MILS 90% Mt DNA mutated; NARP 70–80% Mt DNA mutated; Rawle et al. | |
| Mayer-Rokitansky-Küster-Hauser syndrome | Inhibition malformation of Müller’s ducts | - | - | 1:4,000–1:5000 280 | Ovarian and uterine agenesis, renal dysplasia 280 | ||
| McCune-Albright syndrome | Overproduction of growth factors and hormones 281 | GNAS 281 | Mosaic 281 | 1:100,000–1:1,000,000 | Fibrous dysplasia, Café-au-lait stains, pituitary dysfunction 281 | ||
| MELAS syndrome | Mitochondrial disease 282 283 | MT-TL1 and further mitochondrial DNA mutations 282 283 | Maternal | Myopathy, encephalopathy and stroke-like episodes, lactate acidosis 282 283 | Pathological changes in the stria vascularis, confirmed post mortem | ||
| MERRF | Mitochondrial disease 284 | MT-TK 284 | Maternal 284 | Myoclonus, epileptic seizures, ataxia, muscular weakness and dementia, hyposomia, degeneration of the optic nerve, peripheral neuropathy, cardiomyopathy 284 | |||
| Moebius syndrome | Disorder of the brainstem development 285 | REV3L, PLXND1 285 | De novo | 1:250,000 285 | Facial nerve palsy as well as paresis of other cranial nerves | Hearing loss in about 10% of the patients | |
| Myhre syndrome | “gain of function” mutation, excessive TGF beta signaling 286 | SMAD4 286 | Aut. dom. 286 | <1:1,000,000 | Microcephaly, midfacial hypoplasia, prognathia and blepharophimosis, hyposomia 286 | Enlarged vestibular aqueduct as most frequent radiological finding | |
| Otosponylomegaepiphyseal dysplasia, OSMED syndrome | Collagen defect 287 | COLL11A2 287 | Aut. rec. 287 | <1:1,000,000 287 | Enlarged epiphyses, skeletal dysplasia with relatively short extremities, vertebral anomalies 287 | High-frequency hearing loss, Pierre Robin sequence | |
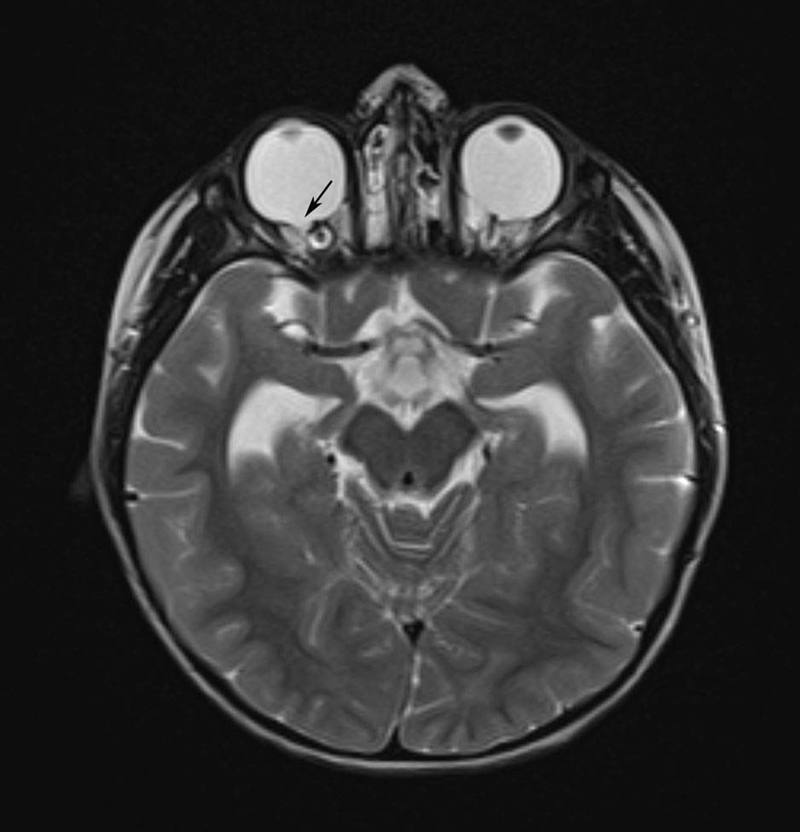
| Pendred syndrome | Partial disorder of the pendrin, an anion exchanger | SLC26A4, FOX11, KCNJ10 288 | Aut. rec. 288 | 7:100,000 288 | Anomalies of the temporal bone with dilated vestibular aqueduct and sometimes hypoplastic cochlea, vertigo, euthyroid goiter 288 289 | Also hypothyroid coursed | |
| Pfeiffer syndrome | Prolonged FGF signaling 290 | FGFR1, FGFR2 290 | Aut. dom. 290 | 1:100,000 290 | Craniosynostoses 290 | Conductive hearing loss and sometimes also inner ear hearing loss | |
| PIGA syndrome | Glycosyl phosphatidylinositol deficiency 291 | PIGA 291 | X-linked 291 | Rare 291 | Infantile spasms, epilepsy, mental retardation, cerebral lesions 291 | ||
| Primary ciliary dyskinesia (Kartagener syndrome) | DNAI1, DNAH5 DNAH11, CCDC39, CCDC40 292 | Aut. rec. 292 | 1:16,000 292 | Daily cough, chronic infection of the airways, situs inversus, asplenia, infertility 292 | Otitis media, inner ear hearing loss (30%) | ||
| Jervell-Lange-Nielsen syndrome | Ion channel mutation 293 | KCNE1 oder KCNQ1, SCN5A 293 | Aut. rec. or aut. dom. | 1:2,000–1:44,500 | Defibrillator, beta blocker, CI | Cardiac arrhythmia, prolonged QT interval 293 | |
| Rieger-Axenfeld syndrome | Irido-dental dysplasia 294 | FOXC1 8294] | Aut. dom. | 1–9:1,000,000 | Malformations of the face, teeth, umbilicum, and skeleton, congenital heart defect 294 | ||
| Russel-Silver syndrome | Methylation disorder/imprinting disorder 295 | - | Aut. dom., Aut. rec. 295 | 1:15,000 295 | Developmental retardation, hyposomia, clinodactyly, hypoglycemia, scoliosis 295 | ||
| Schinzel-Giedion syndrome | “Gain of function” mutation 296 | SETBP1 296 | Aut. dom. 296 | <1:1,000,000 296 | Facial dysmorphia, hydronephrosis, severe developmental delay, mental retardation as well as genital and cardiac anomalies, increased incidence of neuroepithelial dysplasia 8296] | ||
| Senior Løken syndrome | Ciliopathy 297 298 | More than 10 genes 298 | Aut. rec. 297 | 1:1,000,000 297 | Symptom-based | Nephronopthysis, retinopathy, diabetes insipidus, cerebellar ataxia, hepatic fibrosis 297 298 | |
| SeSAME syndrome | Disturbed development of the brain, kidneys, and stria vascularis 299 | KCNJ10 299 | Aut. rec. 299 | <1:100,000 299 | Epilepsy, ataxia, and electrolyte disorder 299 | Synonym: EAST syndrome | |
| Sotos syndrome | Mutation of the histon methyltransferase 300 | NSD1 300 | Aut. dom. 300 | 1:10,000 300 | Long, narrow face, high front, red cheeks and small pointed chin, ADHS, hypotension, excessive growth 300 | Also conductive hearing loss 300 | |
| STAR syndrome | Unknown 301 | FAM58A (CCNQ) 301 | X-linked 301 | Rare 301 | Telecanthus, syndactyly, renal and anogenital malformations 301 | ||
| Tietz syndrome | Disturbed development of melanocytes 302 | MITF 302 | Aut. dom. | <50 patients worldwide | CI | Hypopigmentation and high-grade hearing loss 302 | Also in Waardenburg syndrome |
| Townes-Brocks syndrome | Malformation of the cilia 303 | SALL1 303 | Aut. dom. 303 | - | Anal atresia, dysplastic ears and malformation of the thumbs 303 | ||
| Usher syndrome | Degeneration of hair cells and photoreceptors 304 | Ush 1: MYOVIIA, CDH23, PCDH15, SANS Ush2: ADGRV1, WHRN Ush3: CLRN1 288 304 | Aut. rec. 304 | 3:100,000 304 | CI | Ush 1: congenital hearing and balance disorder, visual loss before puberty Ush 2: congenital hearing loss, visual loss after puberty Ush 3: progressive hearing loss, variable visual loss and balance disorder 288 304 | 10% of all hearing disorders in children |
| Vici syndrome | Global developmental disorder 305 | EPG5 305 | Aut. rec. 305 | 100 patients worldwide 305 | Symptom-based 305 | Agenesis of the corpus callosum, cataract, oculocutaneous hypopigmentation, combined immune deficiency 305 | Disorder of autophagy |
| Waardenburg syndrome | Disturbed development of melanocytes | Pax 3 (type I, III) MITF, SNAI2 (type II) Sox10, EDN3, EDNRB (type IV) 288 306 | Aut. dom. (I, III) Aut. rec. (II, IV) | 1:40,000 | CI 307 | Hypopigmentation and hearing loss (type II)+dystopia canthorum (type I)+malformations of the upper extremities (type III)+Hirschsprung’s disease (type IV) 288 306 | Asymmetric and variable hearing loss |
| Wolfram syndrome, DIDMOAD | Mitochondrial disorder due to disturbed calcium homeostasis and stress in the endoplasmatic reticulum 308 309 | WFS2, WFS2 308 309 | Aut. rec. 308 309 | 1:55,000 308 309 | Insuline | D iabetes i nsipidus, d iabetes m ellitus, o pticus a trophy and d eafness (DIDMOAD) | Progressive hearing loss as of childhood, neurological symptoms |
| Zellweger spectrum diseases | Peroxisomal functional loss 310 | Pex 1,6,10 310 | Aut. rec. 310 | 1:50,000 310 | Bile acid 310 | Flat face, renal and hepatic insufficiency, retinitis pigmentosa 310 | Auditory neuropathy, infant-refsum disease |
| Neural/central | |||||||
| Alternating hemiplegia of childhood (AHC), Weber syndrome, medial medullar syndrome | Channelopathy, alpha3 subunit of Na+/K+ATPase (exclusively expressed in neurons of the CNS) | ATP1A3 | De novo mutations, rarely aut. dom. | 1:1,000,000 135 | Symptom-based | Episodes of weakness or paresis, choreoathetosis, dystonia, dyspnea, ataxia, dysfunction of the autonomous neural system, psychomotor regression, episodic nystagmus 135 | |
| Arnold-Chiari malformation | Syringomyelia, developmental disorder of the brainstem and the upper medulla 311 | Unknown | Unknown | Unknown | Symptom-based | Occipital cephalgia, diplopy, photophobia, spina bifida, meningocephaloceles, dysphagia, dysarthria, sleep apnea 311 | Type 0-VI |
| Autosomal dominant hereditary ataxia | Spinocerebellar degeneration, different types are known 312 | Different genes | Aut. dom., X-linked | 1–5:100,000 | Symptom-based | Ataxia, disturbed hand-eye coordination, speech disorders, nystagmus, diplopia, cognitive impairment, opticus atrophy, retinitis pigmentosa, opthalmoplegia, diabetes, cardiac and skeletal diseases 312 | |
| Canavan-Van Bogaert-Bertrand | Leukodystropia, asparto-acylase enzyme deficiency and accumulation of N-acetyl aspartate acid 313 , diffuse spongiform of the white brain substance, dys- and demyelination 314 | ASPA 313 314 | Aut. rec. 313 314 | 1:100,000, more frequently in Ashkenasim 175 | Symtpom-based, experimental gene and cell therapy, experimental approaches tested in humans with lithium | Macrocephaly, muscular weakness, dysphagia, seizures, nasal regurgitation, opticus atophy, severe progressive psychomotor retardation | Auditory neuropathy 314 , cases without auditory neuropathy and post mortem confirmation of hair cell loss 315 , congenital, infantile, and juvenile types are described |
| CAPOS/CAOS | Channelopathy, alpha3 subunit of Na+/K+ATPase (exclusively expressed in neurons of the CNS) | ATP1A3 c.2452G>A 316 | De novo mutation and aut. dom. 316 | <1:1,000,000 175 , slightly more than 40 patients are described in the literature 317 | Symptom-based | Cerebellar ataxia, areflexia, pes cavus, opticus atrophy 316 318 | |
| Charco-Marie-Tooth neuropathy | CMT with hearing loss 319 | Different genes: ABDH12; AIFM1; DNMT1; PRPS1; PTRH2 319 | Aut. rec., dom., X-linked 319 | 1:3,300 319 | Symptom-based 319 | Progressive neuropathy, muscular weakness, paralysis of the vocal folds, retinitis pigmentosa and cataracts, mental disability with dementia 319 | 80 genes; classification according to the genotype; mild to severe hearing loss, “hidden hearing loss” |
| (Stilling-Türk-)Duane syndrome | Cranial dysinnervation 320 321 | CHN1, MAFB, HOXA1, CDH2 320 321 | Aut. dom. and aut. rec. 320 321 | 1:1,000 320 321 | Symptom-based | Limited horizontal eye movement, abducens hypoplasia, skeletal, auricular, ocular, neural, and renal anomalies 320 321 | 3 types are known, sometimes conductive hearing loss 321 |
| Hereditary sensory neuropathy | Axonal atrophy and degeneration of the sensory neurons, disturbed sphingo-lipid synthesis [322m 323] | SPTLC1 322 323 | Aut. dom. 322 323 | 2:1,000,000 | Loss of distal sensorics, painless injuries, skin ulcer, bone infections, partly severe infections requiring amputations of toes or feet 322 323 , dementia | Extensive microglia activation that may also be classified as inflammatory or metabolic | |
| Superficial siderosis | Hemosiderin deposit as consequence of recurrent bleeding in the subarachnoidal space | - | - | 1:1,000,000 324 | Deferiprone, CI 324 | Progressive bilateral hearing loss, ataxia, vestibular dysfunction, myelopathy with pyramidal signs 324 | |
| Others | |||||||
| Cochlear dehiscence (“Third window” syndrome) | Dilated cochlear and vestibular aqueduct, bone dehiscence 325 | Surgery | Pseudo conductive hearing loss, vertigo (noise- or Valsalva-induced), autophonia 325 | ||||
| Intralabyrinthine schwannoma | Neoplasm | 1:100,000 326 | Surgery 327 , CI 328 | Hearing loss, slowly progressive or fluctuating, vertigo, unsteady gait 327 | |||
| Progressive myoclonal epilepsy | Group of disorders with common symptoms; comprises genetic diseases, mitochondrial diseases, and metabolic syndromes 329 | - | - | - | - | Myoclonus, epilepsy, neurodegeneration 329 |
In depth knowledge of the embryonic development of the cochlea allows for a better characterization of rare cochlear diseases. This is important especially for malformations and syndromic hearing loss. Common molecular principlesthe embryonic development of the cochlea are shared with other organs such as the heart, kidneys, and eyes. These common principles are revealed especially in syndromic cases.
We illustrate the complexity of the evaluation of rare diseases with clinical examples. For example, the role of interdisciplinary and even international collaborations for the diagnosis of the rare vascular disease called Susac’s syndrome is emphasized. It further becomes obvious how difficult and long the way may be for patients to get a definite diagnostic assessment. Further examples (CHARGE syndrome, X-linked deafness) also illustrate the importance of interdisciplinary approaches, in particular with regard to imaging in the context of hearing restoration with cochlear implants.
The majority of the rare diseases show a high variability of their phenotype despite having the same genetic defect. This makes the classic approach of symptom-based diagnostics difficult. On the other hand, overlapping phenotypic patterns can be found for certain diseases even if different genetic defects are present. These observations emphasize the importance of electrophysiology, imaging, and in particular modern molecular diagnostics including proteome analysis that might be the basis for advances in oto-rhino-laryngology.
1 Embryonic Development and Morphology of the Cochlea
Accruing knowledge about the molecular evolution of its phenotypic development is a fundamental component of understanding an organ system. Knowing the molecular mechanisms leading to the development of the inner ear may contribute to better characterization and classification of rare diseases and malformations. In the following paragraphs, the embryonic development of the inner ear will be described.
Different tissue layers lead to the development of different portions of the inner ear. The membranous labyrinth is derived from the otic vesicle, which invaginates from the ectoderm whereas the bony labyrinth derives from the mesenchyme.
Beside morphogenesis, the formation of the inner ear also requires the specification of cellular fate. Morphogenesis of the inner ear is initiated from a flat thickening of the ectoderm leading to the development of the fluid filled spaces of the labyrinth and the cochlea. Specification of the cellular fate means the development of neurons, sensory cells as well as numerous non-sensory cells of the inner ear. A multitude of genes and thus induced biochemical processes contribute to the development that have a highly complex spatiotemporal expression pattern. The exact mechanisms playing a role in this context are only incompletely understood until now.
1.1 Morphogenesis of the inner ear
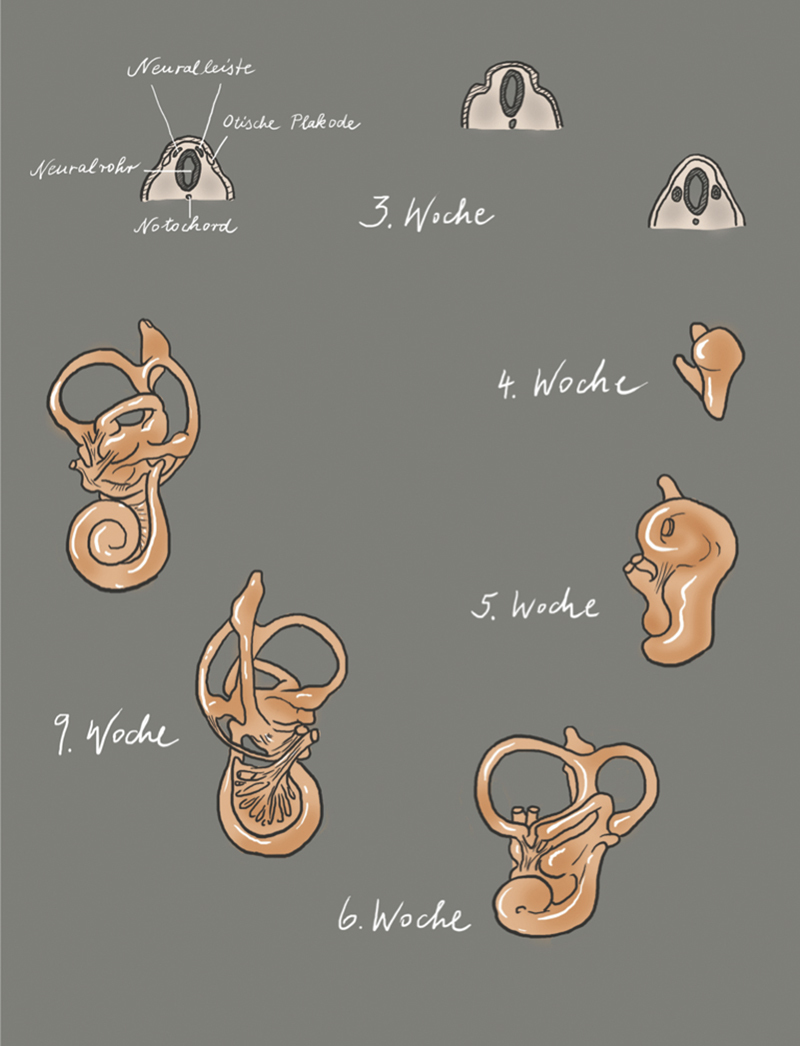
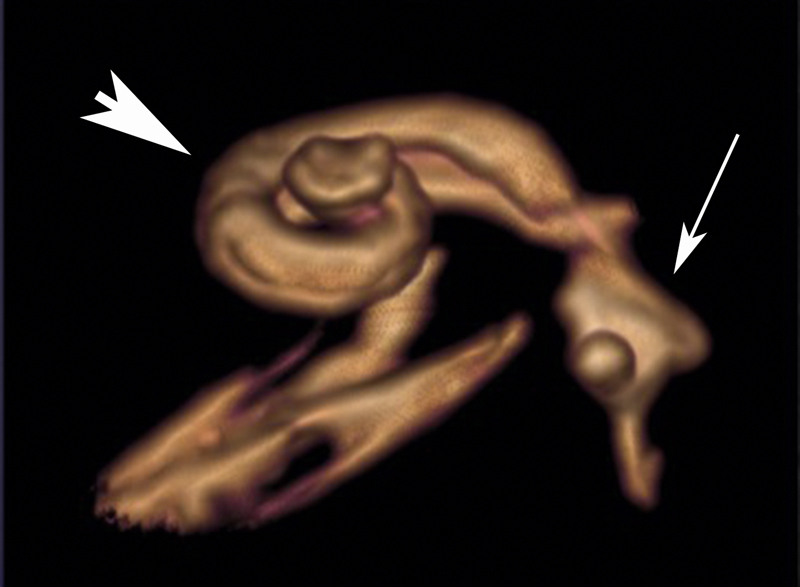
Most cell types forming the inner ear of adults have their developmental origin in the otic placode. The otic placode is the first step of inner ear development ( Fig. 1 ; 3 rd week, on the left). It is a thickening of the ectoderm, lateral to the rhombencephalon, that by invagination into the underlying mesenchyme forms a vesicular structure ( Fig. 1 ; 3 rd week), the otocyst (also known as the otic vesicle) 1 ( Fig. 1 ; 3 rd week, on the right). The otocyst is divided into a vestibular and a cochlear part ( Fig. 1 ; 4 th week). The vestibular compartment of the membranous labyrinth develops from the dorso-lateral part of the otic vesicle and the cochlear structures including the saccule develops from the ventromedial part 2 .
Fig. 1.
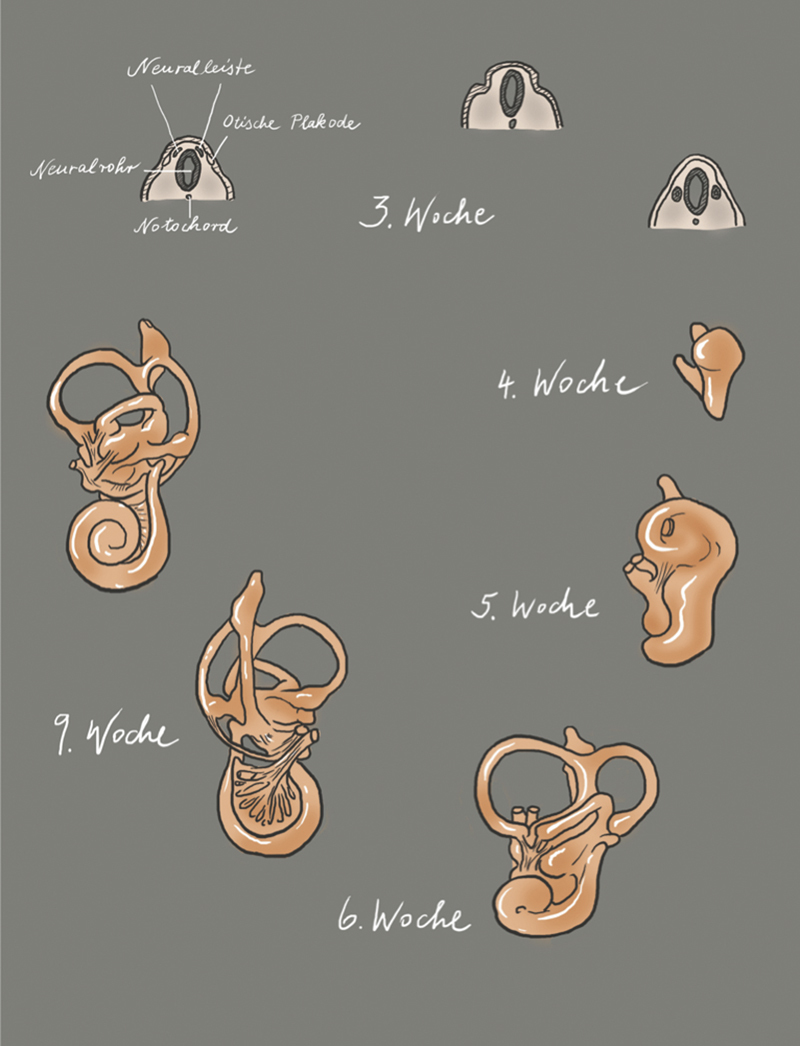
Illustration of the morphogenesis of the inner ear; modified according to Gray’s Anatomy, 41 st edition, 2016 8 , and according to Cummings, 7 th Edition, 2020 9 ; copyright Elsevier.
The developing cochlear part extends to a tubular structure, called cochlear duct ( Fig. 1 ; 5 th week). During growth, the cochlear duct forms a spiral and in the 8 th week of embryonic development, the 2.5 turns are complete ( Fig. 1 ; 6 th and 9 th week). In the 9 th embryonic week, the organ of Corti develops. At the beginning, it appears as an arrangement of polygonal cells equipped with a kinocilium and numerous microvilli on their surface 3 . The microvilli disappear within the next weeks and are replaced by stereocilia 4 that develop first on the inner and later on the outer sensory hair cells. The formation of stereocilia runs from the cochlear base to the apical area. It is the first morphological sign of sensory hair cell differentiation. Next, the arrangement of the inner and outer sensory hair cells becomes obvious. While one single row of inner sensory hair cells develops, the number of rows of outer sensory hair cells may vary between 3 and 4. In parallel, i. e., between the 9 th and 13 th embryonic week, the tectorial membrane develops that covers the organ of Corti. The binding of the tectorial membrane to the stereocilia seems to develop from an initially immature stage, characterized by loose binding, to a more mature stage where a firm connection between the outer sensory hair cells and the stereocilia is formed. Up to the 15 th week, the organ of Corti appears as a solid cell mass that is covered by a thin tectorial membrane.
All sensory hair cells develop a row of stereocilia, however, the inner sensory hair cells seem to be more mature than the outer ones with a characteristic U-shape in the configuration of the stereocilia. At the 22 nd week, this process is completed, the stereocilia have matured and show the same distribution pattern as in adults.
At the end of the 11 th embryonic week, the cochlear duct is surrounded by cartilage and fluid-filled spaces develop that form the scalae tympani and vestibuli by the 15 th week. In the 18 th week, the tunnel of Corti (also known as internal tunnel, cuniculus internus) has already developed from the solid cell mass of the organ of Corti (named after the Italian anatomist Alfonso Giacomo Gaspare Corti, 1822–1876). Also, Nuel’s space (also known as middle tunnel, cuniculus medius; named after the Belgian physician Jean Pierre Nuel, 1847–1920) is fully developed and due to regression of Kölliker’s organ (named after the German anatomist and physiologist Rudolf Albert von Kölliker, 1817–1905), the tectorial membrane is freed. Kölliker’s organ is a structure (greater epithelial ridge) that transitorily develops in the cochlea 5 . It consists of pillar like supporting cells releasing ATP. This binds to the ionotropic purinergic receptors (P2X receptors) of the inner sensory hair cells and leads to depolarization and calcium inflow. This process imitates the effect of depolarization by sound and leads to periodic excitation of the spiral ganglion cells during development. In addition, other trials could show that Ca 2+ spikes in neonatal inner sensory hair cells induce excitatory postsynaptic currents in the afferent dendrites of the spiral ganglia cells 6 . After maturation of the cochlea and onset of the hearing capacity, this ATP induced intrinsic activity of the inner sensory hair cells disappears. It is assumed that this spontaneous activity of the inner sensory hair cells and fibers of the auditory nerve is crucial for the neuronal survival of the cochlear nucleus in humans before hearing onset, for the correct interconnection of the auditory pathway, and for the formation/refining of the tonotopy in the auditory nuclei. In other words, this means that peripheral, non-sensory cells that are in Kölliker’s organ are responsible for the maturation of the auditory pathway 7 .
Between the 20 th and 22 nd fetal week, the cochlear duct is already manifestly longer with a larger diameter; the stria vascularis has developed its characteristic 3 cell layers, and the tectorial membrane is well developed. Afterwards, the outer pillar cells and the outer sensory hair cells extend step by step, and Deiters’ and Hensen’s supporting cells develop. At the end of the 2 nd trimester, the cochlea already has a mature appearance while the synapses of the efferent brainstem fibers are not yet fully developed.
The cochlear nerve develops from a group of cells (neuroblasts) that derive from the medial part of the ear epithelium and pass into the underlying mesenchyme. They form the VIII th (vestibulocochlear) ganglion leading to the development of the 8 th cranial nerve 10 . The ganglion cells, out of which the auditory part of the VIII th nerve will develop, organize around the modiolus to form the spiral ganglion. Axons that develop from these ganglion cells migrate centrally to the brain and peripherally to the organ of Corti. During the 5 th /6 th week of gestation, the axons first form synapses with the brainstem neurons. The dendrites only reach the basal turn of the cochlea at the end of the 9 th embryonic week and form contacts with the developing sensory hair cells between the 10 th and 12 th embryonic week 11 . By the end of the 12 th week of gestation, the development of the classic afferent synapses between neurons and sensory hair cells is initiated. Presynaptic bodies surrounded with vesicles can first be observed at the base of the inner sensory hair cell, with similar findings in the outer hair cells by the 13 th week. By the 14 th fetal week, basal to apical synapse formation is fully completed while the myelin-forming Schwann cells cannot be detected until the 15 th fetal week. At this early stage, the outer sensory hair cells are exclusively innervated by afferent neurons. In humans, the formation of axo-somatic synapses with the efferent system only takes place with the onset of the cochlear function, i. e., around the 20 th week. This observation seems to reflect an evolutionary process. At the beginning of synapse formation, the outer hair cells only function to transmit auditory signals to the brain. With the efferent interconnection at the end of maturation of the organ of Corti, the outer hair cells begin to function in their role as cochlear amplifiers. This process is more pronounced in the basal and middle regions of the cochlea compared to the apical areas leaving the apical outer hair cells innervated by mainly afferent rather than efferent neurons. Looking at ciliogenesis during this time period, it becomes obvious that the apical area of the cochlea remains in an immature stage so that the cochlea should be correctly subdivided into a basal and an apical part based on the embryonic development. As of the 22 nd fetal week, myelination within the cochlea and thin myelin sheaths are already visible 12 . In the 24 th fetal week, the extension of the myelin sheaths to the exit of the nerve from the temporal bone is revealed. After this time, myelination is performed by oligodendrocytes that have already settled at the nerve. Central myelination, however, has not occurred at this point 13 .
Between the 7 th and 8 th embryonic week, the auditory nuclei and pathways are already fully developed. The neurons of the brainstem containing information of the immature axons of the hearing nerve can be identified at the border of the brainstem as cochlear nuclei. A subset of these crosses in the brainstem and projects its extensions more centrally into the contralateral superior olivary nucleus 14 . The remaining neurons extend to the lateral lemniscus and from there into the inferior colliculus. The medial geniculate body can be identified in the 8 th embryonic week and is innervated by the axons from the inferior colliculus. Between the 9 th and 13 th week, only growth rather than structural change is observed in the brainstem. However, the neurons of the brainstem are still very small and immature even if the nuclei are relatively well developed. In the course of the second trimester, not only the neurons increase in size but also develop cytoplasm and cell organelles. By the end of the 24 th fetal week, more and more cytofilaments are present in the auditory neurons. Also, in the axons of the brainstem neurons, accelerated maturation is observed in the second trimester. Neurofilament that can only be detected in few neurons of the cochlear nerve at the end of the 16 th week is clearly visible at the end of the second trimester as bundled fascicles in the within the cochlear nerve and the brainstem.
1.2 Molecular biology of the embryonic development
The otic placode is one of the craniofacial placodes from which several structures develop (e.g., inner ear, the olfactory epithelium, neurons of different cranial sensory ganglia, eye lens). All these placodes develop in the pre-placodal region that is characterized by the expression of a common set of transcription factors (Six1, Eya2, and Foxi3) 15 . The otic vesicle (otocyst) develops from the pre-placodal region at the level of rhombomeres 5 and 6, influenced by the FGF signaling pathway 15 . The transcription factors Pax2 and Pax8 are markers of the otic vesicle. Gene expression profiles within the otic vesicle, in the adjacent tissue of the developing otic vesicle, within the borders between otic and adjacent tissue as well as within the borders of the compartments into which the developing otic vesicle may be divided schematically seem to be responsible for the orientation of the inner ear ( Figs. 2 and 3 ). The rhombencephalon is arranged in segments that are called rhombomeres. Each rhombomere is able to express specific genes. The otic placode is adjacent to rhombomeres 5 and 6 so that this area seems to play a crucial role in the axial organization as well as specification of the fate of inner ear cells. Mouse mutants with defects of the rhombencephalon in the area of the rhombomeres 5 and 6, where the border between these rhombomeres is maintained, show normal formation of the inner ear. The border between both rhombomeres corresponds nearly exactly to the midline of the otic vesicle and is possibly responsible for the specification of the otocyst cells into the anterior and posterior compartments. Since both rhombomeres develop very early, they may influence the development of the anterior and posterior otocyst by means of different signals 16 . Signal transmission between the cells of the otocyst and the cells of the rhombencephalon, is enabled by the Eph/ephrin system 17 . While the cells of the rhombomere 6 express high concentrations of the ligands of ephrin B2 and B3, a high density of the receptors EphA4, A7, B2, and B3 are found in rhombomere 5 18 19 . This means that the postero-medial cells of the otocyst that are in direct contact with rhombomere 6 receive ephrin-mediated signals, whereas cells adjacent to rhombomere 5 do not. Probably these signals are directly transmitted to the dorsal pole of the otic vesicle where the cells of the otic placode are in direct contact with the cells from the neural tube because no delimitation by a basal lamina is present. This would mean right from the beginning that distinct compartments form in the developing otocyst, i.e., the antero-medial and the postero-medial compartment 16 . These compartments are responsible for the organization of the cells and the inner ear specific development and orientation of the organ. They are characterized by a specific gene expression profile, and define and delineate the cell fate. This means that on one hand the cells in the respective compartments define the location and structure of the cochlea and the semicircular canals, the utricle, saccule as well as endolymphatic duct, on the other hand the mixing of the cells of different lines is not possible ( Fig. 3 ). Furthermore, probably the gene expression within the compartment decides which sensory organ (organ of Corti, crista or macula) develops. Soluble factors and cell surface molecules could then influence the cells along the border between the compartments, but only those that are directly adjacent to the border. Their diffusion seems to be possible but only in a radius of very few hundred micrometers. These factors may be for example morphogens, which would mean that elongating structures are induced along the border zone between two compartments while morphologically localized organs such as the crista may only develop at the border between three compartments. Thus, an exact location may be specified.
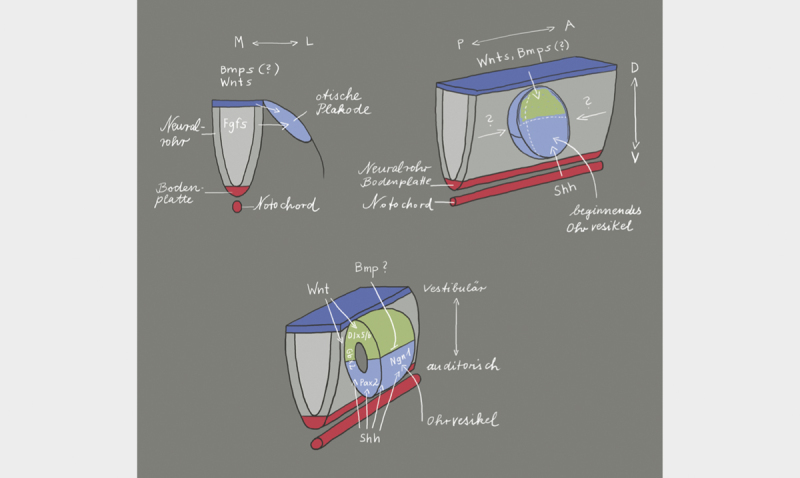
Fig. 2.
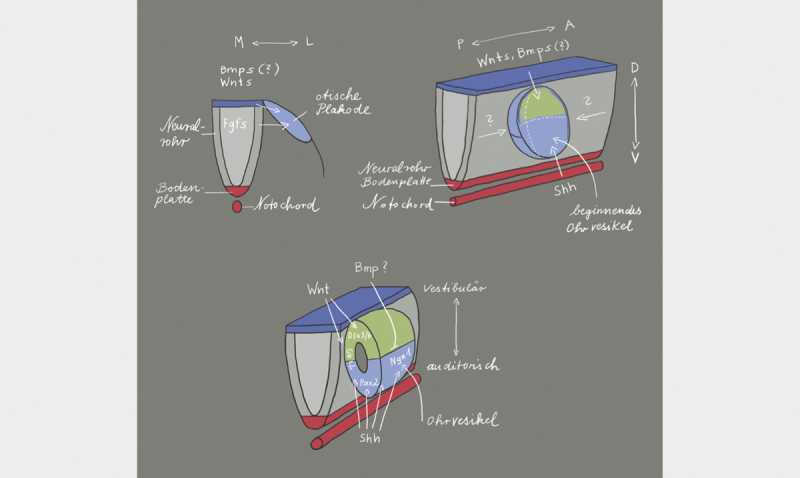
The location of formation of the otic placode along the body axis is defined via the expression of fibroblast growth factors (FGF) from the neural tube 20 . The release of FGF in the periotic mesoderm shortly before the development of the otic placode 21 leads to the expression of several transcription factors that are necessary for the development of the inner ear 22 23 . The orientation of the antero-posterior axis starts with the expression of FGF10, lunatic fringe (Lfng), delta 1, neurogenin1 (Ngn1), and neuronal differentiation factor (NeuroD1) in the anterior region of the invaginating otic placode. This gene expression pattern is limited to the anterior region of the otocyst. This limitation is mediated by Tbx1 that is exclusively expressed in the posterior part of the otocyst. The dorso-ventral axis depends on the WNT and SHH expression in the rhomb encephalon. WNT is expressed in the dorsal area and leads to upregulation of Dlx5, Dlx6, Hmx2, and Gbx2. These genes are responsible for the development of vestibular structures in the dorsal region of the otocyst. On the other hand, there is the expression of SHH from the notochord that determines the fate (auditory) of the cells in the ventral part of the otocyst by regulating the expression of the transcription factors Pax2, Ngn1, Lfng, NeuroD1, Sox2, and Six1. BMP (bone morphogenetic protein) and SHH inhibit each other so that BMP assumes a significant role in the morphogenesis of the inner ear. Illustration modified according to 24 25 .
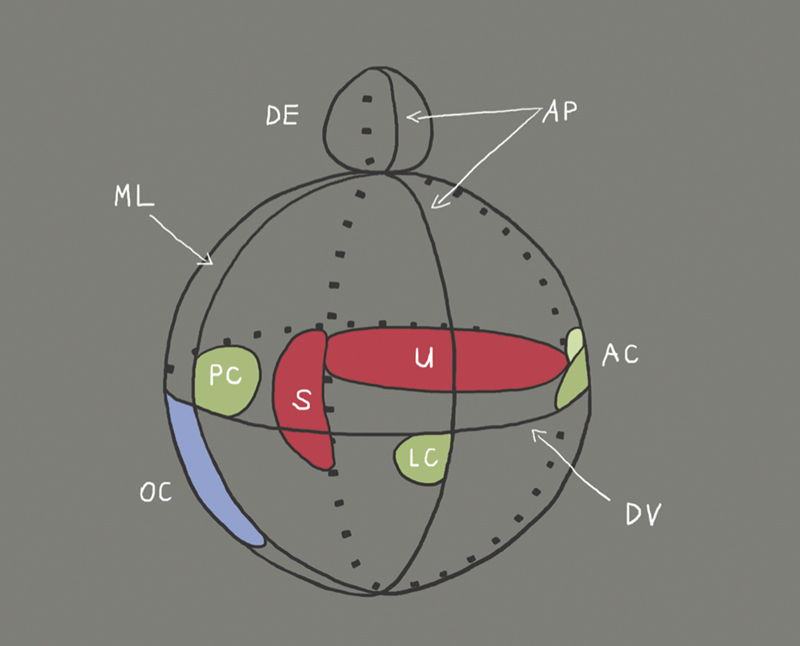
Fig. 3.
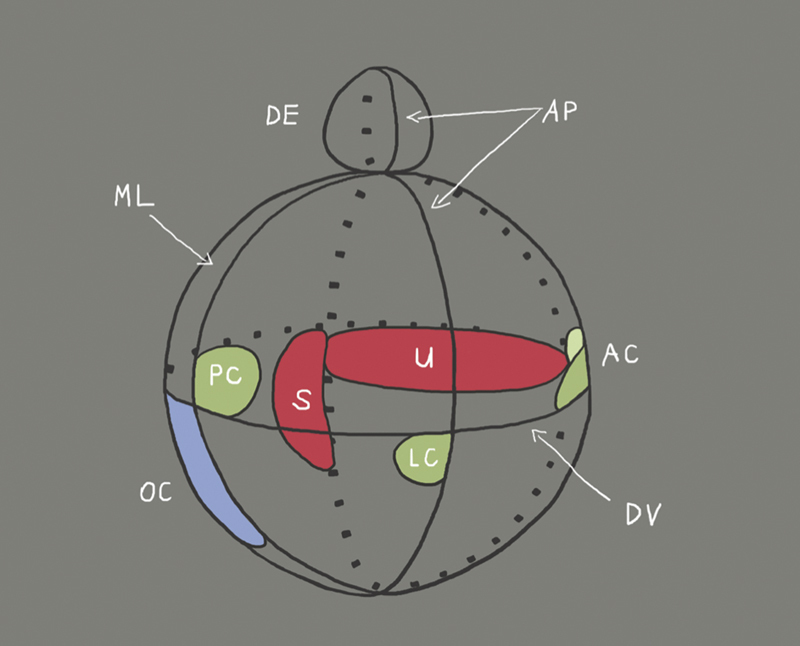
Illustration of the compartments of the developing inner ear and location of the different organs (organ of Corti: OC; saccule: S; utricle: U; endolymphatic duct: ED; cristae of the semicircular canals: AC, PC and LC) as well as the axes (AP: antero-posterior; DV: dorso-ventral; ML: medio-lateral). The orientation in the antero-posterior axis takes place before the orientation in dorso-ventral direction 26 . The dorso-ventral axis is not defined until the formation of the otocyst 27 . The axial specification already starts with formation of the otic placode and depends on factors ( Fig. 2 ) that are expressed by rhombomeres 5 and 6 of the rhombencephalon. As soon as a rhombomere is rotated in ovo along the dorso-ventral axis, the expression of the ventral genes Lfng, NeuroD1, and Six1 (see Table, BOR syndrome) is shifted into the dorsal regional of the otocyst, whereas the expression of dorsal genes like Gbx2 is inhibited. This means that by rotation of the rhombomeres ventral areas of the rhombencephalon may transform ventral areas of the otocyst into dorsal otic tissue 26 . The formation of organs of the inner ear after the stage of otocyst depends on the expression of Gata3 (see table, Bakarat-HDR syndrome), Eya1 (see table, BOR syndrome), and FGF3/8 (see table, Kallmann syndrome, which was shown in investigations of Gata3, Eya1, and FGF3/8 deficient mice 28 29 30 . SHH (see Table, incomplete partition and Carpenter syndrome) as well as Pax2 are cochlear genes because mutations in these genes allow only the formation of a short, straight cochlear duct. Gbx2, Hmx2, Hmx3, and WNT are considered as vestibular genes because a defect of one of these genes leads to morphological defects of the saccule, utricle, or the semicircular canals (illustration modified according to Brigande et al., 2000 16 ).
1.3 Development of sensory hair cells
Different soluble factors are necessary for the induction of the otic placode: FGF from the mesoderm and the neuroectoderm, SHH from the notochord and the base plate of the neural tube, WNT from the rhombencephalon as well as BMP from the ectoderm. The anterior area of the otocyst where the neurosensory cells of the inner ear develop is characterized by the expression of Lfng, Sox2, and Eya1 31 while Tbx1 and Lmx1a are expressed in the dorsal part in the non-sensory region. Within the prosensory region, the proneuronal transcription factor Neurog1 is upregulated at an early stage. It is not only necessary for the formation of neurons but it also contributes essentially to the development of the sensory epithelium including the sensory hair cells 32 . It belongs to the basic helix-loop-helix (bHLH) transcription factors and is expressed together with Neurod1 by proliferating progenitor cells. The bHLH gene Atoh1 (atonal-homolog-1)/Math1 (murine atonal homolog 1)/Hath1 (human atonal homolog1) is necessary for the development of sensory hair cells 33 34 . Furthermore, the POU domain transcription factor POU4f3, the zinc finger transcription factor Gfi1, and the homeodomain factor Barhl1 are needed for the maintenance and formation of sensory hair cells 32 . The singular and highly specific pattern of cell alignment in the sensory epithelium of the inner ear where sensory hair cells and supporting cells are arranged alternatingly allows the assumption that local cell communication mechanisms override predetermined cell specification. The notch-signaling pathway regulates the determination of the cellular fate in numerous organ systems 35 . During the development of the inner ear, notch (see Table; Hajdu-Cheney syndrome) is initially distributed in the entire epithelium. With the differentiation of the sensory hair cells, however, its expression is limited to the supporting cells. Delta 1 and Jagged2, however, are synthesized by the sensory hair cells of the murine cochlea about one day after the onset of Math1 expression 36 . Hereby, the cells that are supposed to develop into sensory hair cells express Jagged1, which increases the notch activity in the neighboring cells and thus forces them to adopt another cell type (the one of supporting cell). This process is called lateral inhibition.
The formation of the apical mechanosensory region (the site where the stereocilia are located) plays a vital role for the function of the sensory hair cells. The stereocilia of the sensory hair cells are packed with actin and other cytoskeletal proteins. They are organized in V-shaped ascending rows. The organization is uniform throughout the entire organ of Corti while the tip of the V always points into the periphery of the cochlear duct and is essential for correct function 37 . In the context of orientation of the sensory hair cell bundles, the WNT/planar cell polarity-signaling pathway plays a crucial role. The asymmetric distribution of the planar cell polarity proteins of frizzled (Fzd), disheveled (Dvl), Van Gogh (Vangl), and prickle (Pk) leads to polarization of the sensory hair cells. It could be shown that Fzd and Dvl proteins form a complex on one side of the cell whereas Vangl and Pk arrange at the contralateral side 38 . It is assumed that the subunit of the kinesin II motor complex, Kif3a, regulates the organization of the sensory hair cells. In Kif3a mouse mutants, the kinocilium is missing, the cochlear duct is shortened, and the shape of the sensory hair cell bundles seems to be flattened 39 . Disorders of the cilia, so-called ciliopathies, are characteristic for Bardet-Biedl and Senior-Løken syndrome ( Table 1 ).
1.4 Development of spiral ganglion cells
During the development of the central nervous system, basic helix-loop-helix (bHLH) transcription factors are responsible for the specification of cells, whereas lateral inhibition by the delta/notch system inhibits neuronal differentiation in neighboring cells by activating the inhibitory effector genes Hes and Hey 40 . Several soluble factors such as WNT (wingless), FGF, BMP, and SHH (sonic hedgehog) induce neuronal progenitor cells 41 expressing proneuronal genes and allow the change of fate to glia formation via activation of the COUP-TF/II transcription factor 42 . These basal patterns of embryonic development of the nervous system can also be observed in the context of ontogenesis of the inner ear. It can be assumed that the entire otocyst is able to form neuroblasts. Already shortly after invagination of the otic placode, a delamination of neuroblasts occurs from the anterior and ventral region, that is called neurosensory domain, and out of which the VIII. cranial ganglion (also known as cochleovestibular ganglion) develops 15 . For differentiation of spiral ganglia cells, the proneuronal bHLH transcription factor neurogenin1 (Ngn1) is needed. After an initial overexpression of Ngn1, an upregulation of Neurod1 as well as delta and notch genes occurs within the developing spiral ganglion cells while Ngn1 itself is downregulated 43 . The expression of Gata3 also seems to play a crucial role for the formation of neurons, in particular in the context of central connection. However, this process could not be fully clarified up to now because Gata3 contributes to the development of the inner ear already at an early stage in embryonic development and a systemic deletion of Gata3 leads to impaired formation of the inner ear 44 . The development of neurons further depends on the expression of POU4f1 (formerly called Brn3a) 45 . During their development, the spiral ganglion cells migrate from the cochlear duct to the spiral canal of the modiolus (Rosenthal’s canal). They reach their postmitotic phase already in the cochlear duct in a baso-apical gradient, i. e., first the neurons of the basal and middle cochlear parts exit from the cell cycle, later the ones of the apical parts. The outgrowing dendrites arrange and retract in a way that the inner and outer sensory hair cells are innervated according to a certain pattern. This process seems to be regulated via the G protein RhoA-GTP and the rho-associated GTP binding proteins Rnd2 and Rnd3. It could be shown that ectopic sensory hair cells are able to form connections with spiral ganglion neurons 46 so that it may be expected that the sensory hair cells attract the dendrites of the neurons. This might be an explanation for the fact, why patients with severe malformations of the inner ear (e. g., incomplete partition) might nonetheless benefit from cochlea implantation.
Interestingly, the region developing into the organ of Corti expresses neurotropic factors before the differentiation of sensory hair cells so that developing neurites grow into the organ of Corti even if the formation of sensory hair cells does not occur 47 . However, if they reach the habenula perforata, they need a stimulus originating from the sensory hair cells so that they can continue to grow in direction of the greater epithelial ridge or the sensory hair cells 48 . These factors may be semaphorin/neurophilin1, Eph/ephrin as well as Slit/Robo. Their expression defines a path along which the dendrites may grow out 49 . Morphogens such as WNT and SHHs are expressed so that a dorso-ventral gradient is formed that is necessary for the development of the cochlea 24 . Furthermore, a significant role for direction-oriented outgrowth of the axons is attributed to WNT and SHH. In order to allow outgrowth of the neurites, WNT and SHH are required together with the growth factors FGF and BMP 48 . Type I and type II spiral ganglion neurons forward stimuli from the inner and outer sensory hair cells in a central direction. It is still unknown when this innervation pattern develops that is clearly seen in adults. However, this process seems to be closely related to the peripherin expression in type II spiral ganglia neurons 50 . Another protein, Prox 1 required for the coordination of the outgrowth of neurites of type II neurons is expressed in the spiral ganglion cells but possibly also in the supporting cells, along which the afferent fibers are expected to grow 51 .
The expression of the neurotrophins BDNF and NT3 as well as their receptors NTRK2 and NTRK3 regulate the survival and the outgrowth of the developing spiral ganglion neurons. In the developing cochlea, a BDNF-NT3 gradient is formed from apical into basal direction 52 . If BDNF is deleted, a normal cochlea develops with a reduced neuronal population of about 7–15%. However, if NT3 is missing, the complete innervation of the basal cochlea is missing and is reduced in the middle turn 48 . Deletion of neurotrophin expression during development not only alters neuronal development but also results in a clearly shorter cochlear duct and disorganized rows of sensory hair cells similar to Neurod1 deficient mice 43 .
1.5 Regenerative factors
Although the development of the inner ear is a highly complex process that may be influenced by multiple factors, a targeted modulation of single signaling pathways can be an approach for regenerative therapies. The REGAIN trial ( RE generation of hair cells with a GA mma secretase IN hibitor) aims at treating patients with mild to moderate hearing loss by inhibiting the notch-signaling pathway. In this context, the notch inhibitor LY3056480 is used to stimulate the regeneration of sensory hair cells in the inner ear that are lost with increasing age 53 . Furthermore, the discovery of WNT-reactive progenitor cells in the murine cochlea being positive for LGR5 54 has revealed that hair cell regeneration is fostered by blocking the notch signal 55 and is possible also in the adult cochlea of mammals 56 . In the context of human embryonic development of the inner ear, the expression of LGR5 increases from the 8 th to 12 th week of gestation 57 . During this time, also the development of the organ of Corti takes place. Interestingly, the LGR5 protein complex that was found in the apical poles of the sensory epithelium of the cochlea is limited to the sensory hair cells as of the 12 th week of gestation 57 . Current studies show that in particular proteoglycans of the extracellular matrix contribute to the development of sensory hair cells and spiral ganglion neurons by up- or downregulating certain genes 58 .
1.6 Embryonic development principles – correlation with other organ systems
For proper organogenesis, organ-specific genes are upregulated during embryonic development while genes that contribute to cell division and general morphogenesis are downregulated 59 . At later stages, genes that encode organ-specific functions are upregulated 59 . Multi-species and longitudinal gene expression analyses show a high overlap of the transcriptome over the entire embryonic development period and in particular of the brain, cerebellum, liver, kidney, testis, and ovaries 59 .
Next generation sequencing analyses reveal that there are specific groups of genes responsible for organ development, for crosstalk and interaction between the organs (organ pattern genes) as well as house keeping genes that mainly coordinate metabolism 60 . The specific biological properties of these organ pattern genes may possibly give hints to new biomarkers or therapeutic targets for precise and effective prognosis and treatment of complex and in particular of rare diseases 60 . They might even explain how a gene defect leads to the involvement of different organ systems.
The principles of embryonic development of the cochlea and the auditory nerve show that the single developmental steps are complex and depend on multiple factors. This becomes obvious especially in the context of malformations, which, in contrast to initial assumptions, do not reflect the result of developmental arrest at certain stages. Different genes and gene families are responsible for the development of the inner ear and many of these regulate the organogenesis of other systems too. For example, forkhead box transcription factors play a role in the development of different organ systems by regulation and post-translational modification of different genes such as Neurod and Sox2 61 . Sox2 as well as BMP, WNT, and FGF are involved in cardiogenesis 62 , otogenesis 63 , skeletogenesis 64 as well as retinal development 65 . Another group of evolutionarily conserved factors that are involved in the development of numerous organs (eyes, kidneys, heart, muscles, and inner ear) are the Eya (eyes absent) molecules 66 . In particular, the carboxyl domain is conserved in the Eya molecules that binds Six (sine oculis) proteins 66 . Eya4 mutations are for example the cause for a rare autosomal-dominantly inherited hearing disorder that is sometimes associated with dilatative cardiomyopathy 66 .
2 Non-syndromic Hearing Disorders
Non-syndromic hearing loss (NSHL) is inherited in an autosomal-recessive (about 80%) but also autosomal-dominant (about 15%), X-linked (2–5%) as well as maternal/mitochondrial (1%) fashion. Some but not all known mutations are listed in Table 1 . Despite our understanding of these mutation and their inheritance patterns, a complete understanding of non-syndromic hearing disorders is a major challenge due to the extreme clinical and genetic heterogeneity 67 .
Nearly 100 genes have already been identified that may lead to non-syndromic hearing disorders. A comprehensive overview of the genes that have been identified as of 2015 is found in the publication of Vona et al. 67 . A significant gain in knowledge about the function of the mature cochlea results from the study of genes that are mutated in cases of non-syndromic hearing disorders. The signaling pathways that are involved in rare as well as in non-syndromic diseases allow the identification of new pathophysiological processes that lead to hearing loss. Variations of the genotype-phenotype correlation are known in the context of mutations of single genes; and despite autosomal-dominant inheritance patterns, siblings may have hearing loss of various severities 67 . Such variations also became apparent in syndromic diseases. Sometimes entire organ systems are not affected (incomplete or reduced penetrance) even if the same gene mutation is found 68 . Secondary genetic factors or environmental factors might be responsible for the variations (epigenetics). Already in 1941, statistical methods were applied to prove the existence of modifier genes in Huntington’s chorea so that the concept of dominant modifier and suppressor genes was made responsible for the high degree of variability 69 . In fact, 2 loci for modifier genes have been identified that might cause the variability of certain hearing disorders 67 .
In spite of the variations (e. g., the onset of hearing loss may occur very variably even with the same gene mutation), autosomal-dominant hearing disorders often show a characteristic audio profile 70 that may be helpful for diagnosis. Despite the remarkable progress in the identification of a multitude of gene defects resulting in hearing loss, the mechanistic understanding in particular of non-syndromic hearing disorders does not suffice to allow clarification of the pathophysiology or the development of effective therapies.
The advent of modern molecular biological methods in combination with artificial intelligence and machine learning will will bring about individualized precision healthcare for patients with hearing loss. In addition, preventive measures, e.g., control of structural, mutational, and epigenetic changes of the hearing disorder, are possible. This means that beside classic, meanwhile widely distributed and available omics technologies and imaging procedures for confirmation of the diagnosis, intensive diagnostics have to be developed to identify e. g., posttranslational changes, virus load or the inflammasome 71 . Screening technologies that include evaluation of protein-protein interactions 72 also integrate miRNA assessment in the perilymph 73 as well as analyses of model cells or patient cells 74 will allow real personalized medicine.
3 Malformations of the Cochlea
Before the era of CT imaging, reports about inner ear malformations were based on post-mortem examinations; and first pioneers such as Carlo Mondini (1729–1803) described changes like a missing apical turn and an enlarged vestibular aqueduct that is known as Mondini dysplasia 75 . Eugene Michel (1819–1883), the German otolaryngologist Arno Scheide (1864–1837), Gustav Alexander (1873–1932) as well as the Swiss neurologist Paul Robert Bing (1878–1956) lent their names for characteristic malformations of the inner ear 76 . In the context of post-mortem investigations, characteristic inner ear malformations of the bony (20%) and the membranous labyrinth (80%) have been assessed. In 1974, the American otologist Harold Frederick Schuknecht (1917–1996) published his standard reference work about ear pathologies by listing beside malformations all other diseases concerning the cochlea based on histological examinations of a large temporal bone collection.
Based on polytomography and sometimes CT scans, Jackler and his colleagues Luxford and House published the first classification of congenital malformations of the cochlea and the labyrinth 77 in 1987 that is still clinically used today.
Nearly 100 malformed inner ears were classified as follows:
Complete aplasia (Michel aplasia)
Common cavity
Cochlear aplasia with normally developed labyrinth
Cochlear hypoplasia
Incomplete partition (small cochlea with incomplete or missing interscalar septum; normal or malformed semicircular canals)
The hitherto revolutionary aspect of this classification was the concept of the embryonic developmental arrest to explain the different types of malformations 76 . However, the absence of inner ear development in different stages of embryonic development as pathomechanism of malformations can only explain some of the observed changes. Even Jackler already mentioned in his work that malformations indicate a disturbed rather than an absent development 77 . The malformations were listed according to the week of gestation when the embryonic development seemed to be disturbed. Jackler and his colleagues substantiated this assumption with the similarity of polytomographic findings and the (illustrated) developmental stages according to Streeter 78 . The series starts with the labyrinthine aplasia (Michel deformity, 3 rd week of gestation), followed by the common cavity in the 4 th week of gestation, cochlear aplasia in the 5 th week of gestation, and severe and mild cochlear hypoplasia in the early and late 6 th week of gestation. The incomplete partition, classic Mondini malformation, is the chronologically last malformation in the 7 th week of gestation. The classification performed by Jackler et al. differentiates further a group A with missing or malformed cochlea (complete labyrinthine aplasia, cochlear aplasia, cochlear hypoplasia, incomplete partition, and common cavity) and a group B with normal cochlea (enlarged vestibular aqueduct, exactly defined dysplasia of the semicircular canals consisting of an enlarged vestibulum and a short but dilated lateral semicircular canal). Sennaroglu and colleagues enhanced the Jackler classification with regard to the surgical anatomy for cochlear implantation 79 . The incomplete partition type I (IPT1) describes a severe type with missing partitioning of the entire cochlea with conspicuously enlarged vestibule and undetectable modiolus. The outer borders of the cochlea are coarse and often bloated. An accompanying malformation of the vestibule and the semicircular canals may be expected while an enlarged vestibular aqueduct is not found. Clear delimitation of the common cavity is not possible according to the used definitions. The wide transition from the cochlea to the vestibule characterizes also IPT1, which is reflected in the synonymous term of cystic cochleovestibular malformation 80 81 82 . The common cavity has been described as malformation with a common cavity of the cochlea and the vestibule that are connected via a wide transition. This definition that was originally coined by the histological report of Edward Cock from 1838 83 is not sufficient for a clear delimitation against incomplete partition type I. The result is an unclear use in the literature. Similar to the enlarged vestibular aqueduct that may be found in numerous other malformations as accompanying symptom, the modiolus is regularly not defined by CT scan in its typical form in cases of X-linked deafness and IPT1. Today, the malformation originally described by Mondini is understood as the incomplete partition type 2.
The aspect of the classification away from the developmental arrest theory to multifactorial genetic defects is illustrated in a review article about cochlear implantation in children with cochleovestibular malformations 84 . According to Papsin et al., the genetic coding of the murine otocyst ( Figs. 2 and 3 ) mostly excludes the hypothesis of developmental arrest of a single development pathway 84 . Instead, possible multiple distinct pathways of the inner ear development are described. The identification of the genetic signature of single malformations and the correlation with radiological findings crucially changes the understanding of the pathogenesis in this field.
3.1 X-linked deafness DFN3, Gusher-associated
The hearing loss defined as X-linked deafness shows a characteristic CT scan (incomplete partition type 3, IPT3) with a widely open connection between the cochlea and the internal auditory canal. The fundus of the internal auditory canal is dilated ( Fig. 4 ). The modiolus and the cribiform plate are missing so that a direct transition of the perilymph of the inner ear and the liquor in the subarachnoidal space is found. Further characteristics are the corkscrew-like appearance of the cochlea, dilated nerve canals of the facial nerve and the posterior ampullary nerve from the inferior vestibular nerve (in the singular canal) as well as coarse protrusions at the vestibule. Also a small, mostly completely bone surrounded endolymphatic sac is observed.
Fig. 4.
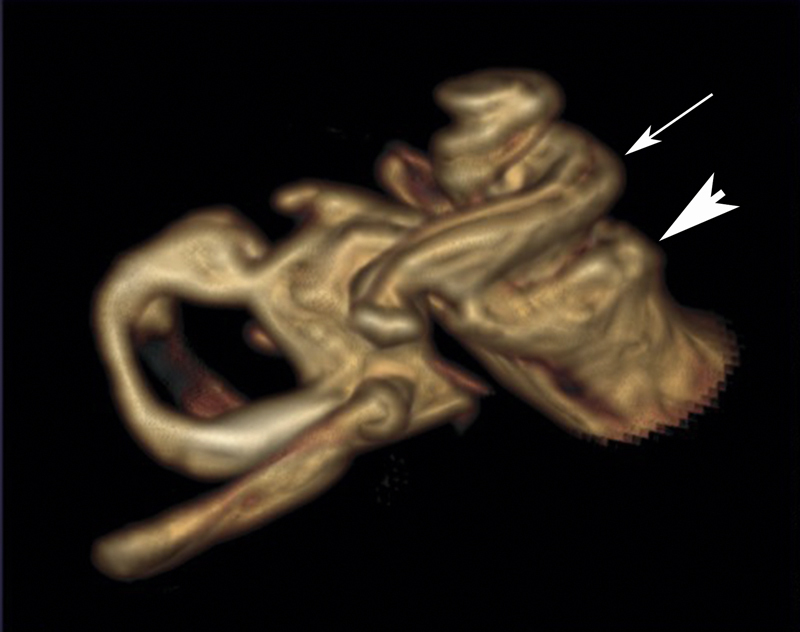
Volume rendering from the T2 dataset of an MRI of a patients with X-linked deafness. A typical corkscrew-like picture of the cochlea (white arrow pointing to the basal turn). The fundus (arrowhead) of the internal auditory canal is dilated.
Clinically, X-linked deafness presents as severe mixed conductive and sensorineural hearing loss in both ears. In 1971, Nance et al. described first a congenital fixation of the stapes footplate in X-chromosomal combined hearing loss and gusher 85 . The open connection to the cerebrospinal fluid leads to leakage during stapes mobilization, so-called gusher. Female carriers are described, however, only a low number of anatomical conspicuities are found in the CT scans and the hearing loss is clearly milder 86 .
In some cases, the provision with hearing aids is sufficient, but often cochlear implantation is indicated. Surgically, the insertion of the electrode array is a particular challenge because it may easily reach into the internal auditory canal due to the open connection ( Fig. 5 ). Intraoperative cone beam computed tomography is a very useful tool in this context. Speech understanding after cochlear implantation is good and comparable to patients without inner ear malformation 87 88 .
Fig. 5.
Patient with X-linked deafness. Axial CT scan of the temporal bone in two levels a A widely open connection between the internal auditory canal and the basal turn is revealed as well as a missing modiolus and missing lamina cribrosa (black arrow). b On the left side, the broad canal of the facial nerve is well displayed (black arrowhead). After cochlea implantation, malposition of the left-sided inserted CI electrode array in the internal auditory canal (white arrows) is observed.
In 1995, Kok et al. were the first to describe the location of the mutation on the gene POU3F4 89 . Since then, more than 63 causal mutations on the POU3F4 gene (DFNX2) have been reported, among them 44 point mutations and various deletions 90 . POU3F4 is expressed in the developing neural tube and later especially in the areas of the brain, supraoptically and paraventricularly in the hypothalamus nuclei. Already in 1982, Myhre et al. reported about a recessive X-linked disorder with congenital deafness and hypogonadism 91 . The patients of our own department have hamartomas of the hypothalamus in more than 90% of the cases that are characterized by hypogonadotrophic hypogonadism in contrast to the usual clinics in the context of hamartomas that are associated with laughing fits and precocious puberty 92 . Siddiqui et al. described hypothalamic malformations in patients with X-linked deafness and IPT3 so that these may be classified as syndromic malformations 93 .
X-linked deafness with an identified gene defect in POU3F4 is not the only hearing disorder associated with mutations identified on the X chromosome. Altogether, they make up about 1–2% of syndromic and non-syndromic hearing disorders. Up to now, 6 loci and 5 genes have been identified for non-syndromic hearing loss and at least 15 for syndromic hearing loss 94 , among them Norrie syndrome, Cornelia-de-Lange syndrome, Fabry syndrome, Alport syndrome, STAR syndrome, PIGA syndrome, and X-linked adrenoleukodystrophy, just to mention a few syndromes that are all considered as rare diseases.
3.2 Complete aplasia of the semicircular canals and CHARGE syndrome
The complete aplasia of the semicircular canals is the main criterion of CHARGE syndrome, which may also occur as isolated symptom. It has also been described in combination with Wildervanck, Noonan, Goldenhar, or VACTERL syndrome 95 96 . The first description was made in 1979 independently by Hall and von Hittner so that the disease is known under the name of Hall-Hittner syndrome. The acronym CHARGE, however, was suggested by Pagon et al. in 1981 97 : C oloboma, H eart defects, choanal A tresia, R etardataion, G enitourinary and E ar abnormalities. The criteria applied today were suggested by Blake et al. in 1998 and revised by Verloes et al. in 2005 98 . Verloes emphasized in particular the three Cs as main criteria: coloboma, choanal atresia, and hypo-/aplasia of the semicircular canals. Depending on the number of the fulfilled criteria, the difference is made between typical, partial, and atypical CHARGE syndrome. However, only the presence of a few criteria is required for the diagnosis. The phenotypes in CHARGE syndrome are protean. Regarding the main criteria, ear malformation is observed in 95–100%, 90% occurring in the inner ear 99 , followed by coloboma in 90% of the cases and neural malformations especially of the facial nerve in 50–90%, depending on the literature. The diagnosis of complete aplasia of the semicircular canals (SCC aplasia) is made based on imaging by means of computed tomography. Among the inner ear malformations, it represents a particularity because the phylogenetically older part of the labyrinth is missing. Accordingly, severe genetic alterations are present that become obvious due to the described multitude of associated malformations of the eye, midline structures of the facial skull, mediastinal malformations (cardiac and esophageal ones, possible thymus aplasia), and malformations of the efferent urinary pathways and genitals that all belong to different non-adjacent embryonic territories. CT scan and MRI of the temporal bone is the imaging technique of choice for existing hearing disorders in order to exclude possible inner ear malformations. CT scan of the temporal bone reveals the complete aplasia of the semicircular canals including the utricle ( Fig. 6 ). The visible vestibule is small, mostly comma-shaped, and contains only the saccule belonging to the inferior part ( Fig. 7 ). The cochlear mostly has a reduced number of turns (hypoplasia). Missing separation of the scalae may be an accompanying finding. In frequent neural hypo- or aplasias, also the internal auditory canal is narrow. In MRI, the depiction of the nerves is performed by means of high-resolution T2 sequence. The facial nerve and the vestibulocochlear nerve may be completely missing (4% of the cases); however, mostly a facial nerve is found and the hypo- and aplasia concerns more frequently a part of the vestibular nerve than of the cochlear nerve. Another accompanying malformation in the area of the temporal bone is a persisting petrosquamous sinus in up to 80% of the cases. It is a surgical challenge in the context of cochlear implantation like a missing oval or round window and a small middle ear. An aberrant course of the facial nerve in the middle ear can also be observed. In rare cases, the petrosquamous sinus may perform the venous drainage of the intracranial space from the transverse sinus. Furthermore, in a small percentage the venous exit occurs via the postglenoid foramen dorsal of the temporomandibular joint. In these cases, only a very small jugular foramen is found.
Fig. 6.
Complete aplasia of the semicircular canals. Axial CT scan of the temporal bone with complete aplasia of the semicircular canals. a Bilateral hypoplastic cochlea. b On the right, a normal width of the cochlear aperture is found, on the left, the aperture is severely narrowed (arrow) – hypo- or aplasia of the cochlear nerve can already be assumed but MRI has to provide the evidence. c Narrow internal auditory canals (black arrows) are typical such as the bilateral comma-shaped vestibule (white V). d The semicircular canals cannot be displayed, the vestibular aqueduct (arrow) is the only narrow structure.
Fig. 7.
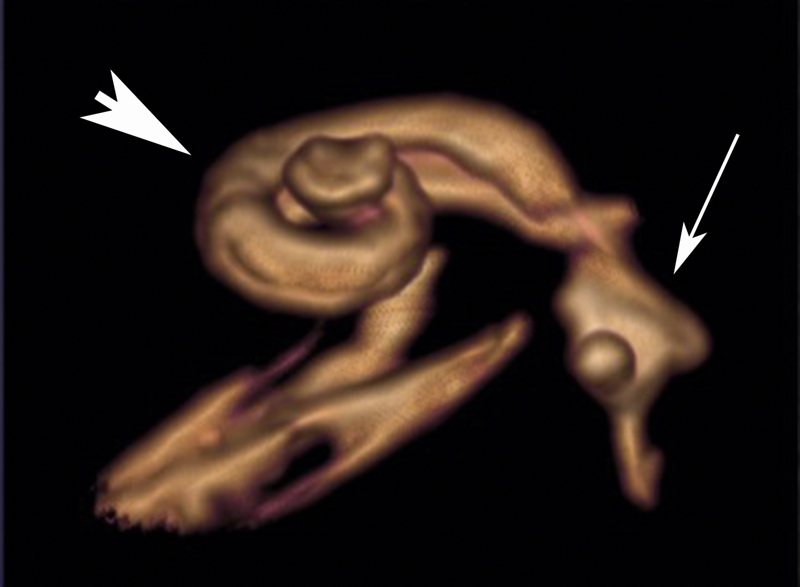
Aplasia of the semicircular canals. Volume rendering from the T2 dataset of MRI of a patient with complete aplasia of the semicircular canals and CHARGE syndrome. The cochlea (arrowhead) is hypoplastic and the vestibule (arrow) contains only the saccule. The semicircular canals are not developed.
In some cases, also coloboma of the eye is directly seen in the imaging. It is another of the three Cs, the main criteria according to Verloes of 2005. Coloboma is a congenital cleft of the iris, lens, and ocular fundus. In the last mentioned case, it can be made visible in tomography as protrusion of the ocular bulb around or beside the optic nerve ( Fig. 8 ). Accompanying microphthalmia may be observed. The third C can also be diagnosed through CT scanning: choanal atresia, which may be present unilaterally or bilaterally as well as bony or only as fibrous closure. However, especially in cases of bilateral appearance it becomes obvious already at birth and has been treated at the time of assessing the appropriateness for cochlear implantation.
Fig. 8.
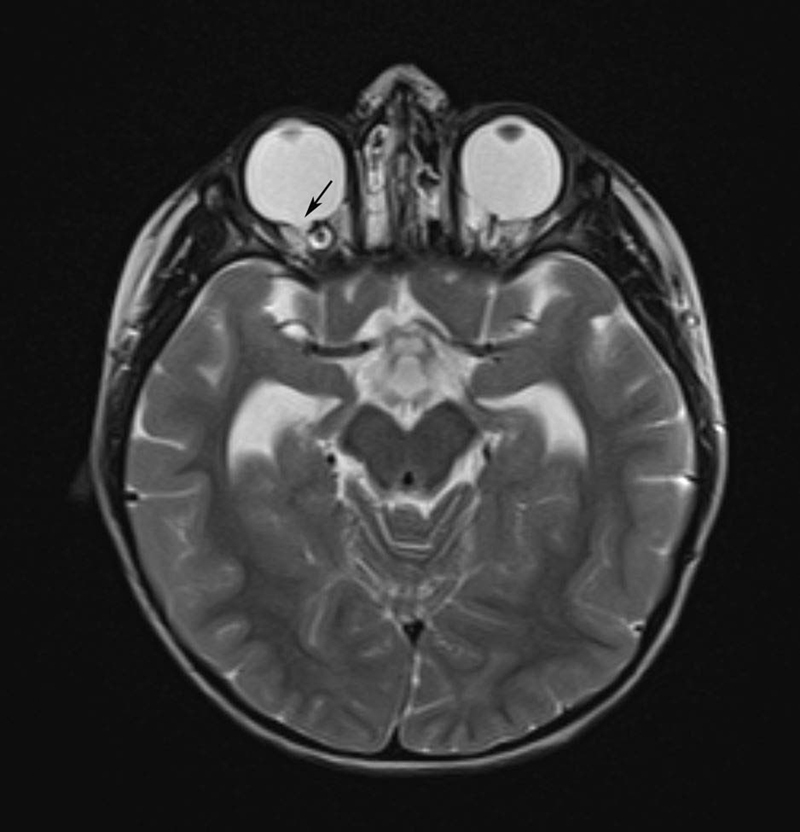
T2 weighted axial MRI of a child with CHARGE syndrome in the context of preliminary cochlea implant examination. Coloboma is found at the right (black arrow) and left eye.
Data on the incidence of accompanying hearing disorder in CHARGE vary between 50% for severe hearing disorder and 90% for deafness. In our own patient population with exclusively patients with complete aplasia of the semicircular canals, nearly all cases have severe hearing loss or deafness. If the cochlear nerve is present, cochlear implantation is the therapy of choice. Implantation may represent a particular challenge because the accompanying malformations complicate the access to the cochlea. Therefore, careful assessment of the CT scan should be performed in order to identify the optimal access. Speech understanding after CI has a broad spectrum depending on the abilities of the patients with possible simultaneous retardation, blindness and other disabilities. Overall, a benefit is reported, independently from the severity of CHARGE. About half of the implanted children use speech as communication way one year after implantation 97 .
The genetic cause of CHARGE syndrome is a mutation of the CHD7 that leads to functional loss. Patients who meet the diagnostic criteria of CHARGE syndrome have a mutation in up to 95%. Most mutations are singular and equally distributed over the coding region of CHD7. More than 500 different pathological changes have been described up to now. 75% of them are frameshift or nonsense mutations. Nearly all mutations develop de novo; but also familial accumulation has been described. In such CHARGE families, a broad spectrum of clinical manifestations is observed with parents suffering from very mild symptoms that barely meet the criteria up to the full spectrum in the children.
Regarding the manifestation of CHARGE syndrome, overlapping with Kallmann syndrome is found as well as 22q11.2 deletion. Anomalies of the olfactory nerve are found in Kallmann as well as in CHARGE syndromes. Immune defects are frequent in 22q11.2 deletion, but they may also appear in CHARGE. As in 22q11.2 deletion, thymus aplasia may be observed. In cases of middle ear infections a thymus aplasia must be taken into consideration in children with CHARGE because middle ear infections are not always due only to anatomical circumstances.
3.3 Cochlear implantation in cases of cochlear malformations
Hearing rehabilitation has a high success rate not only in patients with normally developed anatomical labyrinth displayed in CT scans but also in patients having a bone malformation revealed in the context of cochlea implant examination (about 20%) 100 . The care for these patients represents a particular challenge. Complications e. g., caused by an aberrant course of the facial nerve or the increased risk of meningitis may occur very frequently when anomalies of the cochlea are found 79 84 101 102 103 . Hence, an intensive evaluation of every cochlear implant candidate is crucial, in particular of children, in experienced centres. A series of different descriptions of inner ear malformations is found in the literature and the same term may be interpreted in quite various ways, depending on the author. Already in the 1990ies, Lenarz and colleagues and Sennaroglu et al. in 2017 requested a standardized description of malformations 79 103 in order not only to allow comparisons and knowledge exchange, but especially to establish guidelines for patient care.
A standardization taking into account the anatomy was pursued by Jackler in the 20 th century and by Sennaroglu in 2002 as well as numerous other authors. Not only the entire spectrum of malformations was described in this context but also single and partly rare subgroups. The complete partitions 1 and 2 were completed by atypical cases, e.g., IPT2 (former Mondini in the classic sense) without enlarged vestibular aqueduct 104 ; or IPT1 that has exactly this enlargement 104 . The course of the facial nerve in the temporal bone has been investigated in detail 105 . Cochlear hypoplasia was classified more specifically 106 107 108 . All this reflects the enormous possibilities to combine genetic factors in order to provide this highly complex spectrum of different malformations. However, several malformations occur more frequently than others and they are worth being accurately classified.
Taking the example of the term common cavity, it becomes clear how differently the term is used and how incompletely the development of inner ear malformations is understood despite advances in genetics and imaging. In the literature, the term of“common cavity is used for different types of malformations, at least three fundamentally different groups are included: 1) With the term of common cavity, Jackler described an otocyst that still bears the predisposition of the cochlea, the vestibule, and the semicircular canals, i. e., a malformation at a very early developmental level. 2) Other publications use the term in the sense that the cochlea and the vestibule, both coarsely developed, are non-partitioned and dilated and form a common cavity, i. e., a broad transition is found between both. At the fundus of the internal auditory canal, the cochlear and the vestibular nerve can generally not be differentiated. In the American literature, sometimes the term of cystic cochleovestibular anomaly ( Fig. 8 deformity) is applied 109 . At the same time, the malformation has no clear delimitation criteria with regard to incomplete partition 1, where also the cochlea is dilated and non-partitioned and the vestibule is severely stumpy. An arbitrary definition could possibly be the width of the transition, which has no functional consequences for cochlea implantation. 3) Sennaroglu indicates that cochlear aplasia cannot always be delimited of common cavity in all cases 79 . He describes an oval malformation that is located mainly dorsal to the internal auditory canal and definitely has neural connections (in contrast to the otocyst deformities described by Jackler that does not need them). Both, cochlear aplasia as well as common cavity may have rudimentary or partly well-developed semicircular canals. Thus, the common cavity cannot be differentiated from cochlear aplasia in CT scans. Only the criterion of sclerotic areas ventral/inferior to the internal auditory canal that often exists in cases of cochlear aplasia is not applied for differentiation ( Fig. 9 ). However, based on own observations, this sclerotic area is present in nearly all cochlear aplasias. This overlapping makes differentiation rather difficult; decisions for cochlear implantation should be made based on the evidence by MRI of the internal auditory canal and the nerves. This example shows how important tomography with multiple levels of the labyrinth are to assess the respective malformations. If only one layer is shown, as it is often found in publications, the criteria that are necessary for evaluating a malformation cannot always be understood. With regard to the individual assessment and therapy, 3-dimensional reconstruction and the individual adaptation of the electrode that is possible in this way represents a logical consequence 106 .
Fig. 9.
Patient with cochlear aplasia. a The black arrows show the sclerotic area of the otic capsule where normally the cochlea is found. b and c show further dorsally located areas that comprise the dysplastic vestibule that is marked in d with black arrows on both sides.
Publications are available that report about speech understanding after cochlear implantation in cases of cochlear aplasia 100 . Those cochlear aplasias are called common cavity by other authors. The presence of a spiral ganglion that can be stimulated cannot be assessed in CT scans. A combination of CT morphology and neural predisposition as it appears in MRI is more suitable to predict the possible benefit of cochlear implantation. These techniques are applied since long time in cochlear implant candidacy evaluation. Giesemann et al. have established the classification based on the severity of associated neural malformations in 2012 110 . It becomes clear that severe malformations regularly have an entire spectrum of different nerve aplasias with certain probabilities. The resolution limit of MRI is crucial and further procedures such as the promontory test and BERA should be applied as well. It is still one of the open questions in the field of healthcare regarding malformations, up to which level hypoplasia of the cochlear nerve may lead to successful implantations.
The Table summarizes examples of a classification that includes the clinically most important malformations that may be well differentiated by imaging morphology in relation to the presence of a cochlear nerve 110 .
4 Susac’s Syndrome
Susac’s syndrome defines an autoimmune microangiopathic endotheliopathy that leads to closure of the precapillary arterioles of the brain, the retina, and the inner ear 111 . The neuroophthalmologist John O. Susac (1940–2012) lent his name for this disease. A systematic review article from 2013 summarizes the data of all cases of this rare disease that had been described until then 111 and defines criteria based on which this disease may be diagnosed that is characterized by multifaceted phenotypes 112 . The majority (nearly 80%) of the patients are females. Since autoimmune diseases are observed more frequently in women, this fact supports the possible autoimmune origin of Susac’s syndrome 111 . Even if the characteristics of the disease are clearly defined, the diagnosis is often difficult and is associated with a long way for the patients. Severe neuropsychological deficits, visual field failures, and hearing loss but also unspecific symptoms like cephalgia may occur.
The majority of these patients initially develop neurological symptoms so that the disease is often misdiagnosed as multiple sclerosis. Within 2 years, about 85% of the patients develop the characteristic triad 111 . Also, non-classic symptoms have been described in patients with Susac’s syndrome. A recently published paper that retrospectively investigated the hearing loss of the well-known painter Francisco Goya (1746–1828) with the background of current knowledge assumes an uncharacteristic manifestation of Susac’s syndrome even if the painter fortunately had no significant disorder of his visual field 113 . An otologic manifestation of syphilis, other vasculitis diseases such as Churg-Strauss or autoimmune diseases like Cogan syndrome may cause symptoms that are similar to Susac’s syndrome. One characteristic of the disease is the hearing loss of low frequencies, initially unilateral and reversible, later also bilateral and persistent 111 114 . In rare cases, the hearing loss manifests as the first symptom, even years before the disease is diagnosed 115 . Fluorescence angiography and tone audiometry should be applied as early as possible in order to confirm the suspected diagnosis 111 . Multiple disseminated lesions, in particular snowball-like changes in the area of the corpus callosum and a leptomeningeal enhancement are characteristic cranial changes in MRI 116 . An early and aggressive treatment in particular in cases of neurological manifestation leads to a favorable prognosis.
An interdisciplinary approach and close communication between neurologists, ophthalmologists, neuroradiologists, and otolaryngologists is vital and accelerates the confirmed diagnosis. The low frequency hearing loss, as it can appear as initial sign of Susac’s syndrome, may also suggest Menière’s disease, low frequency type of sudden sensorineural hearing loss, or intra-cochlear schwannoma 117 . With a prevalence of about 0.2%, also Menière’s disease is considered as rare disease of the inner ear and is often assumed in patients presenting with low frequency hearing loss and vertigo. The symptoms of vertigo alone or in combination with low frequency hearing loss, however, may also occur in patients with Susac’s syndrome. Therefore it is possible that Susac’s syndrome remains unidentified and the patients are treated for Menière’s disease with steroids. Patients who are initially diagnosed with low frequency type of sudden hearing loss may suffer from Susac’s syndrome. With this background, an ophthalmological and neurological consultation of all patients with assumed sudden hearing loss or Menière’s disease seems to be reasonable.
The pathophysiology of Susac’s syndrome is caused by an occlusion of the lumen of minor vessels 114 . It is hypothesized that the underlying cause is binding by antibodies against vascular endothelial cells or T cell mediated swelling of vascular endothelial cells. Furthermore, MRI studies have revealed a disturbed microvascular blood-brain barrier in Susac’s syndrome that was caused by inflammatory changes of the vascular wall 118 .
5 Hearing Loss and Microcirculation
Normal inner ear function is predicated on a perfectly functioning microcirculation of the vessels of the labyrinthine artery that enters the organ via the internal auditory canal. The presence of a disturbed microcirculation in the inner ear is suspected in several diseases. The inner ear supply is based on an end artery, i. e. there are no anastomoses with other vessels that could take over the organ supply in cases of occlusion. The labyrinthine artery originates from the anterior inferior cerebellar artery and divides into 3 main branches in the inner ear: the anterior vestibular artery, the vestibulocochlear artery, and the cochlear artery 119 . A disorder of the microvascular blood-labyrinth barrier is also assumed in Menière’s disease, based on an increased gadolinium uptake in affected inner ears in MRI trials 120 . Disturbance of the blood-labyrinth barrier is assumed to be more severe in sudden sensorineural hearing loss 120 . Post mortem analyses of patients who had suffered from Menière’s disease reveal an increased expression of inducible nitrogen monoxide synthase, damage of the vascular endothelial cells, degeneration of the perivascular basal membrane and extracellular matrix, and loss of the blood-labyrinth barrier. These changes are consistent with increased oxidative stress 120 . Even more interestingly, similar molecular processes (release of pro-inflammatory cytokines and endothelial and mitochondrial dysregulation as well as oxidative stress) have been identified as basic and common sign of multiple organ failure 121 so that these processes must rather be considered as the result of a series of insults and damages and not as causes. At the molecular level, patients suffering from Menière’s disease reveal an upregulation of cochlin as well as downregulation of collagen IV and laminin-beta 122 . To date, post-mortem investigations of patients having suffering from Susac’s syndrome, have not confirmed these findings. The classic vertigo attacks that are reported by patients with Menière’s disease are not observed in Susac’s syndrome. Nonetheless, from an otolaryngological point of view, acute or fluctuating, or (intermittently) progressive sensorineural hearing loss restricted to the apical cochlear region based on an endolymphatic cochlear hydrops is one of the most important differential diagnoses of Susac’s syndrome besides Cogan syndrome.
To discover molecular markers in the blood or other body fluids for certain inner ear diseases, such as cochlin in Menière’s disease, might be one of the ways to lead us into modern oto-rhino-laryngology. Initial approaches of perilymph analyses in patients performed during surgical inner ear interventions have allowed the identification of numerous proteins that could not be detected in the liquor or plasma 123 . Furthermore, inflammatory marker proteins were identified in human perilymph 71 so that establishing an inflammasome profile of the perilymph may give hints about the pathomechanisms of certain diseases. In particular, it may be a valuable method to characterize rare inner ear diseases. Even if the perilymph collection during cochlea implantation does not influence the residual hearing of the patients 123 , i. e. possible additional damage of the inner ear seems to be very improbable, perilymph collection as minimally invasive intervention under local anesthesia in cases of significant residual hearing has not been proposed until now. The characterization of the perilymph of a “normal” cochlea is rather difficult because up to now only examinations have been performed in patients who suffered from other diseases of the neural system, e. g., meningiomas 124 .
Another development in the era of Big Data, artificial intelligence, and machine learning is the availability of databases ideally providing the complete data worldwide in specific consortiums of patients who suffer from a certain (rare) disease. Even as contact points for patients to retrieve information about competence centers and support groups, disease-specific consortiums are highly valuable especially in cases of rare diseases. With regard to Susac’s syndrome, such activities are coordinated via the European Susac Consortium (EuSaC; http://www.eusac.net).
6 Rare Diseases of the Cochlea: Outlook and Conclusion
The inner ear is anatomically and histologically a highly complex organ consisting of different tissue types. Developmental disorders and pathophysiological processes occurring during life may affect all tissue types of the inner ear and lead to hearing loss. Even if hearing loss is the most frequently observed degenerative sensorineural disease from a statistical point of view with 16% of affected Europeans, hearing loss is also an important component of many rare diseases. In particular in pediatric patients, these diseases may be overlooked. Even if the diagnostics of many rare diseases is nowadays facilitated by accessing bioinformatics databases and analysis software, an interdisciplinary examination (e. g., neurology, cardiology, nephrology, rheumatology, ophthalmology as well as otorhinolaryngology) is obligatory because of the highly variable phenotypes.
Due to their rare occurrence, rare diseases are often neglected in teaching and education of young physicians because the probability to encounter such cases in daily routine is rather low. However, if a combination of disorders (e. g., eye-inner ear, musculoskeletal system-inner ear, heart-kidney-inner ear, inner ear-thyroid, inner ear-gonads) is observed, it becomes clear that they may in fact occur more frequently. However, it also becomes clear how valuable the clinical discussion of the rare disease of an organ system is beside the embryonic development in order to understand the organ with its structure, its functions, and its diseases.
The human genome project and the high throughput sequencing methods that are now available as well as analyses of the proteome, transcriptome, epigenome, metabolome as well as microbiome offer the chance already today to sharpen disease profiles. Different disorders having similar symptoms (phenotypes) may be different on a molecular level and have to be treated in different ways (e. g., mitochondrial disease versus lysosomal storage diseases). It is well understood that genes controlling several signals and chromosomal anomalies that lead to the simultaneous loss of several genes cause broad-spectrum effects and severe manifestations. For example, the original classification system of the Charcot-Marie-Tooth syndrome reveals how clinical thinking about rare diseases changed during the last years. Initially based on phenotypes, the classification system was completed on the basis of progression and physiological measurements until finally the current classification based on the genotype (currently more than 80 genes) was established. Even if it could be shown that disease-specific genes generally tend to be expressed in a limited number of tissues, it is still unclear how tissue-specific expression patterns of disease genes correlate with their pathological manifestations. Proteome analyses reveal that most gene products assume their function often in combination as complexes of several different proteins 125 . This might explain why mutations of different proteins lead to a similar phenotype. Recent approaches show that a tissue-specific overexpression of genes in the medulla, the dorsal ganglion, and the skeletal muscles coding certain protein complexes correlate with the pathological manifestation of Charcot-Marie-Tooth syndrome 125 . The cellular components that are affected are the telomere regions of the chromosomes; and the biological processes that are disturbed belong to the mechanosensory system 125 . Those classification systems also mean that several biological processes that were unknown may lead to hearing loss: basal membrane/collagen defects, overexpression of growth factors (e. g., TGF beta/interleukins) as well as disorders of the melanocytes, autophagy, and methylation. How this additional knowledge may be used in order to establish new treatment methods especially for patients who suffer from rare diseases, will have to be investigated in future trials.
The example of Susac’s syndrome shows that not only other inner ear diseases have to be considered in the differential diagnosis, but also ophthalmological or neurological diseases. It is important to think of an (possibly initial) audiological manifestation of a rare disease if sensorineural hearing loss has an unknown origin. Interdisciplinary diagnostic assessment may be helpful to detect occult symptoms and to early find a correct diagnosis. How hearing loss may already be diagnosed in these cases before it manifests, will be a prognostically relevant question. Patients with subjectively undisturbed hearing at rest may have difficulties in understanding speech in noise (also known as synaptopathy or hidden hearing loss, which may be present as early symptom of progressive neurodegeneration). Often this disorder remains undetected because the patients are not aware of it; and with routinely applied procedures, it cannot be identified. Targeted testing of speech understanding in noise can lead to a correct indication. This would allow an early introduction of therapy in diseases such as Susac’s syndrome. The identification of patients suffering from hidden hearing loss 126 is finally also relevant because currently 3 clinical trials investigate new therapies for treatment with promising results.
Numerous molecular and cell physiological processes are the basis for hearing loss especially in the context of rare diseases. The future of (also merely symptomatic) treatment of inner ear diseases, that are often rare diseases, may benefit from the early identification of molecular disorders.
Footnotes
Interessenkonflikt Die Autorinnen/Autoren geben an, dass kein Interessenkonflikt besteht.
Literatur
- 1.O’Rahilly R. The timing and sequence of events in the development of the human eye and ear during the embryonic period proper. Anat Embryol (Berl) 1983;168:87–99. doi: 10.1007/BF00305401. [DOI] [PubMed] [Google Scholar]
- 2.Som P M, Curtin H D, Liu K et al. Current Embryology of the Temporal Bone, Part II: the Middle and External Ears, the Statoacoustic and Facial Nerves, and When Things Go Developmentally Wrong. Neurographics. 2016;6:332–349. doi: 10.3174/ng.5160174. [DOI] [Google Scholar]
- 3.Lavigne-Rebillard M, Pujol R. Surface Aspects of the Developing Human Organ of Corti. Acta Otolaryngol. 1987;104:43–50. doi: 10.3109/00016488709124975. [DOI] [PubMed] [Google Scholar]
- 4.Dabdoub A, Donohue M J, Brennan A et al. Wnt signaling mediates reorientation of outer hair cell stereociliary bundles in the mammalian cochlea. Development. 2003;130:2375–2384. doi: 10.1242/dev.00448. [DOI] [PubMed] [Google Scholar]
- 5.Tritsch N X, Zhang Y X, Ellis-Davies G et al. ATP-induced morphological changes in supporting cells of the developing cochlea. Purinergic Signal. 2010;6:155–166. doi: 10.1007/s11302-010-9189-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fettiplace R. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2017. Hair Cell Transduction, Tuning, and Synaptic Transmission in the Mammalian Cochlea. In: Comprehensive Physiology; pp. 1197–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tritsch N X, Yi E, Gale J E et al. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- 8.Standring S. Gray’s anatomy: the anatomical basis of clinical practice. 2016.
- 9.Flint P, Haughey B, Lund V Cummings Otolaryngology Head and Neck Surgery. 2020.
- 10.Gibaja A, Aburto M R, Pulido S et al. TGFβ2-induced senescence during early inner ear development. Sci Rep. 2019;9:1–13. doi: 10.1038/s41598-019-42040-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pujol R, Lavigne-Rebillard M. Early stages of innervation and sensory cell differentiation in the human fetal organ of Corti. Acta Otolaryngol Suppl. 1985;423:43–50. doi: 10.3109/00016488509122911. [DOI] [PubMed] [Google Scholar]
- 12.Lavigne-Rebillard M, Pujol R. Hair Cell Innervation in the Fetal Human Cochlea. Acta Otolaryngol. 1988;105:398–402. doi: 10.3109/00016488809119492. [DOI] [PubMed] [Google Scholar]
- 13.Moore J K, Linthicum F H. Myelination of the Human Auditory Nerve: Different Time Courses for Schwann Celland Glial Myelin. Ann Otol Rhinol Laryngol. 2001;110:655–661. doi: 10.1177/000348940111000711. [DOI] [PubMed] [Google Scholar]
- 14.Moore J K. Organization of the human superior olivary complex. Microsc Res Tech. 2000;51:403–412. doi: 10.1002/1097-0029(20001115)51:4<403::AID-JEMT8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 15.Brown R, Groves A K. Hear, hear for notch: Control of cell fates in the inner ear by notch signaling. Biomolecules. 2020;10:1–18. doi: 10.3390/biom10030370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brigande J V, Kiernan aE, Gao X et al. Molecular genetics of pattern formation in the inner ear: do compartment boundaries play a role? Proc Natl Acad Sci U S A. 2000;97:11700–11706. doi: 10.1073/pnas.97.22.11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Q, Mellitzer G, Robinson V et al. In vivo cell sorting in complementary segmental domains mediated by Eph receptors and ephrins. Nature. 1999;399:267–271. doi: 10.1038/20452. [DOI] [PubMed] [Google Scholar]
- 18.Bergemann A D, Zhang L, Chiang M K et al. Ephrin-B3, a ligand for the receptor EphB3, expressed at the midline of the developing neural tube. Oncogene. 1998;16:471–480. doi: 10.1038/sj.onc.1201557. [DOI] [PubMed] [Google Scholar]
- 19.Gale N W, Flenniken A, Compton D C et al. Elk-L3, a novel transmembrane ligand for the Eph family of receptor tyrosine kinases, expressed in embryonic floor plate, roof plate and hindbrain segments. Im Internet http://www.ncbi.nlm.nih.gov/pubmed/8808709. Oncogene. 1996;13:1343–1352. [PubMed] [Google Scholar]
- 20.Wright T J, Hatch E P, Karabagli H et al. Expression of mouse fibroblast growth factor and fibroblast growth factor receptor genes during early inner ear development. Dev Dyn. 2003;228:267–272. doi: 10.1002/dvdy.10362. [DOI] [PubMed] [Google Scholar]
- 21.Sai X, Ladher R K. Early steps in inner ear development: Induction and morphogenesis of the otic placode. Front Pharmacol. 2015;6:1–8. doi: 10.3389/fphar.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bouchard M, de Caprona D, Busslinger M et al. Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev Biol. 2010;10:89. doi: 10.1186/1471-213X-10-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chatterjee S, Kraus P, Lufkin T. A symphony of inner ear developmental control genes. BMC Genet. 2010;11:68. doi: 10.1186/1471-2156-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riccomagno M M. Wnt-dependent regulation of inner ear morphogenesis is balanced by the opposing and supporting roles of Shh. Genes Dev. 2005;19:1612–1623. doi: 10.1101/gad.1303905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bok J, Brunet L J, Howard O et al. Role of hindbrain in inner ear morphogenesis: analysis of Noggin knockout mice. Dev Biol. 2007;311:69–78. doi: 10.1016/j.ydbio.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bok J, Chang W, Wu D K. Patterning and morphogenesis of the vertebrate inner ear. Int J Dev Biol. 2007;51:521–533. doi: 10.1387/ijdb.072381jb. [DOI] [PubMed] [Google Scholar]
- 27.Wu D K, Nunes F D, Choo D. Axial specification for sensory organs versus non-sensory structures of the chicken inner ear. Im Internet: http://www.ncbi.nlm.nih.gov/pubmed/9389659. Development. 1998;125:11–20. doi: 10.1242/dev.125.1.11. [DOI] [PubMed] [Google Scholar]
- 28.Duncan J S, Lim K-C, Engel J D et al. Limited inner ear morphogenesis and neurosensory development are possible in the absence of GATA3. Int J Dev Biol. 2011;55:297–303. doi: 10.1387/ijdb.103178jd. [DOI] [PubMed] [Google Scholar]
- 29.Zou D, Silvius D, Rodrigo-Blomqvist S et al. Eya1 regulates the growth of otic epithelium and interacts with Pax2 during the development of all sensory areas in the inner ear. Dev Biol. 2006;298:430–441. doi: 10.1016/j.ydbio.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maroon H, Walshe J, Mahmood R et al. Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Im Internet: http://www.ncbi.nlm.nih.gov/pubmed/11959820. Development. 2002;129:2099–2108. doi: 10.1242/dev.129.9.2099. [DOI] [PubMed] [Google Scholar]
- 31.Kiernan A E, Pelling A L, Leung KK H et al. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- 32.Fritzsch B, Beisel KW K, Hansen La. The molecular basis of neurosensory cell formation in ear development: a blueprint for hair cell and sensory neuron regeneration? Bioessays. 2006;28:1181–1193. doi: 10.1002/bies.20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fritzsch B, Matei Va, Nichols D H et al. Atoh1 null mice show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev Dyn. 2005;233:570–583. doi: 10.1002/dvdy.20370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bermingham Na, Hassan Ba, Price S D et al. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- 35.Artavanis-Tsakonas S, Muskavitch MA T. Notch: the past, the present, and the future. Curr Top Dev Biol. 2010;92:1–29. doi: 10.1016/S0070-2153(10)92001-2. [DOI] [PubMed] [Google Scholar]
- 36.Bryant J, Goodyear R J, Richardson G P.Sensory organ development in the inner ear: molecular and cellular mechanisms Br Med Bull 20026339–57.Im Internethttp://www.ncbi.nlm.nih.gov/pubmed/12324383 [DOI] [PubMed] [Google Scholar]
- 37.Petit C, Richardson G P. Linking genes underlying deafness to hair-bundle development and function. Nat Neurosci. 2009;12:703–710. doi: 10.1038/nn.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin H, Copley C O, Goodrich L V et al. Comparison of phenotypes between different vangl2 mutants demonstrates dominant effects of the Looptail mutation during hair cell development. PLoS One. 2012;7:e31988. doi: 10.1371/journal.pone.0031988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sipe C W, Lu X. Kif3a regulates planar polarization of auditory hair cells through both ciliary and non-ciliary mechanisms. Development. 2011;138:3441–3449. doi: 10.1242/dev.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.García-Bellido A, De Celis J F. The complex tale of the achaete-scute complex: A paradigmatic case in the analysis of gene organization and function during development. Genetics. 2009;182:631–639. doi: 10.1534/genetics.109.104083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaspard N, Vanderhaeghen P. Mechanisms of neural specification from embryonic stem cells. Curr Opin Neurobiol. 2010;20:37–43. doi: 10.1016/j.conb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 42.Naka H, Nakamura S, Shimazaki T et al. Requirement for COUP-TFI and II in the temporal specification of neural stem cells in CNS development. Nat Neurosci. 2008;11:1014–1023. doi: 10.1038/nn.2168. [DOI] [PubMed] [Google Scholar]
- 43.Jahan I, Pan N, Kersigo J et al. Neurod1 suppresses hair cell differentiation in ear ganglia and regulates hair cell subtype development in the cochlea. PLoS One. 2010;5:e11661. doi: 10.1371/journal.pone.0011661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karis A, Pata I, van Doorninck J H et al. Transcription factor GATA-3 alters pathway selection of olivocochlear neurons and affects morphogenesis of the ear. J Comp Neurol. 2001;429:615–630. doi: 10.1002/1096-9861(20010122)429:4<615::aid-cne8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 45.Huang E J, Liu W, Fritzsch B et al. Brn3a is a transcriptional regulator of soma size, target field innervation and axon pathfinding of inner ear sensory neurons. Im Internet http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2710107&tool=pmcentrez&rendertype=abstract. Development. 2001;128:2421–2432. doi: 10.1242/dev.128.13.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jahan I, Pan N, Elliott K L et al. The quest for restoring hearing: Understanding ear development more completely. BioEssays. 2015;37:1016–1027. doi: 10.1002/bies.201500044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan N, Jahan I, Kersigo J et al. Conditional deletion of Atoh1 using Pax2-Cre results in viable mice without differentiated cochlear hair cells that have lost most of the organ of Corti. Hear Res. 2011;275:66–80. doi: 10.1016/j.heares.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang T, Kersigo J, Jahan I et al. The molecular basis of making spiral ganglion neurons and connecting them to hair cells of the organ of Corti. Hear Res. 2011;278:21–33. doi: 10.1016/j.heares.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fekete D M, Campero A M. Axon guidance in the inner ear. Int J Dev Biol. 2007;51:549–556. doi: 10.1387/ijdb.072341df. [DOI] [PubMed] [Google Scholar]
- 50.Barclay M, Julien J-P, Ryan A F et al. Type III intermediate filament peripherin inhibits neuritogenesis in type II spiral ganglion neurons in vitro. Neurosci Lett. 2010;478:51–55. doi: 10.1016/j.neulet.2010.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fritzsch B, Dillard M, Lavado A et al. Canal cristae growth and fiber extension to the outer hair cells of the mouse ear require Prox1 activity. PLoS One. 2010;5:1–12. doi: 10.1371/journal.pone.0009377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fariñas I, Jones K R, Tessarollo L et al. Spatial shaping of cochlear innervation by temporally regulated neurotrophin expression. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2710117&tool=pmcentrez&rendertype=abstract. J Neurosci. 2001;21:6170–6180. doi: 10.1523/JNEUROSCI.21-16-06170.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samarajeewa A, Jacques B E, Dabdoub A. Therapeutic Potential of Wnt and Notch Signaling and Epigenetic Regulation in Mammalian Sensory Hair Cell Regeneration. Mol Ther. 2019;27:904–911. doi: 10.1016/j.ymthe.2019.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chai R, Kuo B, Wang T et al. Wnt signaling induces proliferation of sensory precursors in the postnatal mouse cochlea. Proc Natl Acad Sci U S A. 2012;109:8167–8172. doi: 10.1073/pnas.1202774109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bramhall N F, Shi F, Arnold K et al. Lgr5-positive supporting cells generate new hair cells in the postnatal cochlea. Stem Cell Reports. 2014;2:311–322. doi: 10.1016/j.stemcr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McLean W J, Yin X, Lu L et al. Clonal Expansion of Lgr5-Positive Cells from Mammalian Cochlea and High-Purity Generation of Sensory Hair Cells. Cell Rep. 2017;18:1917–1929. doi: 10.1016/j.celrep.2017.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson Chacko L, Sergi C, Eberharter T et al. Early appearance of key transcription factors influence the spatiotemporal development of the human inner ear. Cell Tissue Res. 2020;379:459–471. doi: 10.1007/s00441-019-03115-6. [DOI] [PubMed] [Google Scholar]
- 58.Miwa T, Ohta K, Ito N et al. Tsukushi is essential for the development of the inner ear. Mol Brain. 2020;13:1–11. doi: 10.1186/s13041-020-00570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cardoso-Moreira M, Halbert J, Valloton D et al. Gene expression across mammalian organ development. Nature. 2019;571:505–509. doi: 10.1038/s41586-019-1338-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qin Y, Pan J, Cai M et al. Pattern Genes Suggest Functional Connectivity of Organs. Sci Rep. 2016;6:1–7. doi: 10.1038/srep26501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lam EW F, Brosens J J, Gomes A R et al. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13:482–495. doi: 10.1038/nrc3539. [DOI] [PubMed] [Google Scholar]
- 62.Stefanovic S, Abboud N, Désilets S et al. Interplay of Oct4 with Sox2 and Sox17: A molecular switch from stem cell pluripotency to specifying a cardiac fate. J Cell Biol. 2009;186:665–673. doi: 10.1083/jcb.200901040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Som P M, Curtin H D, Liu K et al. Current Embryology of the Temporal Bone, Part I: the Inner Ear. Neurographics. 2016;6:250–265. doi: 10.3174/ng.4160166. [DOI] [Google Scholar]
- 64.Lefebvre V. Roles and regulation of SOX transcription factors in skeletogenesis. Curr Top Dev Biol. 2019;133:171–193. doi: 10.1016/bs.ctdb.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heavner W E, Andoniadou C L, Pevny L H. Establishment of the neurogenic boundary of the mouse retina requires cooperation of SOX2 and WNT signaling. Neural Dev. 2014:9. doi: 10.1186/1749-8104-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, Sewell W F, Kim S D et al. Eya4 regulation of Na+/K+-ATPase in required for sensory system. development in zebrafish. Development. 2008;135:3425–3434. doi: 10.1242/dev.012237. [DOI] [PubMed] [Google Scholar]
- 67.Vona B, Nanda I, Hofrichter MA H et al. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol Cell Probes. 2015;29:260–270. doi: 10.1016/j.mcp.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 68.Girirajan S, Eichler E E. Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet. 2010;19:R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Génin E, Feingold J, Clerget-Darpoux F. Identifying modifier genes of monogenic disease: strategies and difficulties. Hum Genet. 2008;124:357–368. doi: 10.1007/s00439-008-0560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hildebrand M S, DeLuca A P, Taylor K R et al. A contemporary review of AudioGene audioprofiling: A machine-based candidate gene prediction tool for autosomal dominant nonsyndromic hearing loss. Laryngoscope. 2009;119:2211–2215. doi: 10.1002/lary.20664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Warnecke A, Prenzler N K, Schmitt H et al. Defining the Inflammatory Microenvironment in the Human Cochlea by Perilymph Analysis: Toward Liquid Biopsy of the Cochlea. Front Neurol. 2019;10:1–10. doi: 10.3389/fneur.2019.00665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yue Q, Stahl F, Plettenburg O et al. The Noncompetitive Effect of Gambogic Acid Displaces Fluorescence-Labeled ATP but Requires ATP for Binding to Hsp90/HtpG. Biochemistry. 2018;57:2601–2605. doi: 10.1021/acs.biochem.8b00155. [DOI] [PubMed] [Google Scholar]
- 73.Shew M, Warnecke A, Lenarz T et al. Feasibility of microRNA profiling in human inner ear perilymph. Neuroreport. 2018;29:894–901. doi: 10.1097/WNR.0000000000001049. [DOI] [PubMed] [Google Scholar]
- 74.Wang H, Stahl F, Scheper T et al. Microarray-based screening system identifies temperature-controlled activity of Connexin 26 that is distorted by mutations. Sci Rep. 2019;9:13543. doi: 10.1038/s41598-019-49423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mondini C. Minor works of Carlo Mondini: the anatomical section of a boy born deaf. Am J Otol. 1997;18:288–293. [PubMed] [Google Scholar]
- 76.Brotto D, Uberti A, Manara R. From Mondini to the latest inner ear malformations’ classifications: an historical and critical review. Hear Balanc Commun. 2019;17:241–248. doi: 10.1080/21695717.2019.1663041. [DOI] [Google Scholar]
- 77.Jackler R K, Luxford W M, House W F. Congenital malformations of the inner ear: A classification based on embryo genesis. Laryngoscope. 1987;97:2–14. doi: 10.1002/lary.5540971301. [DOI] [PubMed] [Google Scholar]
- 78.Streeter G L.Developmental horizons in human embryos; a review of the histogenesis of cartilage and bone Contrib Embryol 194933149–168.Im Internethttp://www.ncbi.nlm.nih.gov/pubmed/18144445 [PubMed] [Google Scholar]
- 79.Sennaroğlu L, Bajin M D. Classification and current management of inner ear malformations. Balkan Med J. 2017;34:397–411. doi: 10.4274/balkanmedj.2017.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sennaroglu L, Saatci I. Unpartitioned Versus Incompletely Partitioned Cochleae: Radiologic Differentiation. Otol Neurotol. 2004;25:520–529. doi: 10.1097/00129492-200407000-00020. [DOI] [PubMed] [Google Scholar]
- 81.Giesemann A, Götz F, Lanfermann H. Fehlbildungen des Innenohrs - Diagnostik und Einteilung in CT und MRT. Radiol up2date. 2013;13:201–218. doi: 10.1055/s-0033-1344189. [DOI] [Google Scholar]
- 82.Phelps P D, Michaels L. The Common Cavity Congenital Deformity of the Inner Ear. ORL. 1995;57:228–231. doi: 10.1159/000276746. [DOI] [PubMed] [Google Scholar]
- 83.Cock E. A contribution to the pathology of congenital deafness. Guys Hosp Rep. 1838.
- 84.Papsin B C. Cochlear implantation in children with anomalous cochleovestibular anatomy. Laryngoscope. 2005;115:1–26. doi: 10.1097/00005537-200501001-00001. [DOI] [PubMed] [Google Scholar]
- 85.Nance W E, Setleff R, McLeod A et al. X-linked mixed deafness with congenital fixation of the stapedial footplate and perilymphatic gusher. Im Internet http://www.ncbi.nlm.nih.gov/pubmed/5173351. Birth Defects Orig Artic Ser. 1971;07:64–69. [PubMed] [Google Scholar]
- 86.Phelps P D, Reardon W, Pembrey M et al. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology. 1991;33:326–330. doi: 10.1007/BF00587816. [DOI] [PubMed] [Google Scholar]
- 87.Kang W S, Shim B S, Lee K S. Audiologic performance after cochlear implantation in children with X-linked deafness: Comparison with deaf children with a normal inner ear structure. Otol Neurotol. 2013;34:544–548. doi: 10.1097/MAO.0b013e3182839864. [DOI] [PubMed] [Google Scholar]
- 88.Smith J D, El-Kashlan N, Darr OA F et al. Systematic Review of Outcomes After Cochlear Implantation in Children With X-Linked Deafness-2. Otolaryngol – Head Neck Surg (United States) 2020:1–8. doi: 10.1177/0194599820932138. [DOI] [PubMed] [Google Scholar]
- 89.de Kok Y J, van der Maarel S M, Bitner-Glindzicz M et al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science. 1995;267:685–688. doi: 10.1126/science.7839145. [DOI] [PubMed] [Google Scholar]
- 90.Pollak A, Lechowicz U, Kȩdra A et al. Novel and De Novo mutations extend association of POU3F4 with distinct clinical and radiological phenotype of hearing loss. PLoS One. 2016;11:1–13. doi: 10.1371/journal.pone.0166618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Myhre S A, Ruvalcaba RH A, Kelley V C. Congenital deafness and hypogonadism: a new X-linked recessive disorder. Clin Genet. 2008;22:299–307. doi: 10.1111/j.1399-0004.1982.tb01843.x. [DOI] [PubMed] [Google Scholar]
- 92.Giesemann A, Hartmann H, Franke D Hamartome in Kombination mit X-chromosomaler Taubheit zeigen keine Epilepsie und keine Pubertas praecox. In: Clinical Neuroradiology. 2013.
- 93.Siddiqui A, D’Amico A, Colafati G S et al. Hypothalamic malformations in patients with X-linked deafness and incomplete partition type 3. Neuroradiology. 2019;61:949–952. doi: 10.1007/s00234-019-02230-z. [DOI] [PubMed] [Google Scholar]
- 94.Corvino V, Apisa P, Malesci R et al. X-Linked Sensorineural Hearing Loss: A Literature Review. Curr Genomics. 2017;19:327–338. doi: 10.2174/1389202919666171218163046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Satar B, Mukherji S K, Telian S A. Congenital Aplasia of the Semicircular Canals. Otol Neurotol. 2003;24:437–446. doi: 10.1097/00129492-200305000-00014. [DOI] [PubMed] [Google Scholar]
- 96.Lanson B G, Green J E, Roland J T et al. Cochlear implantation in Children with CHARGE syndrome: therapeutic decisions and outcomes. Laryngoscope. 2007;117:1260–1266. doi: 10.1097/MLG.0b013e31806009c9. [DOI] [PubMed] [Google Scholar]
- 97.Pagon R A, Graham J M, Zonana J et al. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99:223–227. doi: 10.1016/s0022-3476(81)80454-4. [DOI] [PubMed] [Google Scholar]
- 98.Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A:306–308. doi: 10.1002/ajmg.a.30559. [DOI] [PubMed] [Google Scholar]
- 99.Hsu P, Ma A, Wilson M et al. CHARGE syndrome: A review. J Paediatr Child Health. 2014;50:504–511. doi: 10.1111/jpc.12497. [DOI] [PubMed] [Google Scholar]
- 100.Kontorinis G, Goetz F, Giourgas A et al. Aplasia of the cochlea: Radiologic assessment and options for hearing rehabilitation. Otol Neurotol. 2013;34:1253–1260. doi: 10.1097/MAO.0b013e318291c48f. [DOI] [PubMed] [Google Scholar]
- 101.Phelps P D. Cochlear Implants For Congenital Deformities. J Laryngol Otol. 1992;106:967–970. doi: 10.1017/S0022215100121486. [DOI] [PubMed] [Google Scholar]
- 102.Dahm M C, Weber B P, Lenarz T. Cochlear implantation in a Mondini malformation of the inner ear and the management of perilymphatic gusher. Adv Otorhinolaryngol. 1995;50:66–71. doi: 10.1159/000424437. [DOI] [PubMed] [Google Scholar]
- 103.Weber B P, Lenarz T, Hartrampf R et al. Cochlear implantation in children with malformation of the cochlea. Adv Otorhinolaryngol. 1995;50:59–65. doi: 10.1159/000424436. [DOI] [PubMed] [Google Scholar]
- 104.Kontorinis G, Goetz F, Giourgas A et al. Radiological diagnosis of incomplete partition type I versus type II: significance for cochlear implantation. Eur Radiol. 2012;22:525–532. doi: 10.1007/s00330-011-2301-5. [DOI] [PubMed] [Google Scholar]
- 105.Sennaroğlu L, Tahir E. A Novel Classification: Anomalous Routes of the Facial Nerve in Relation to Inner Ear Malformations. Laryngoscope. 2020:1–8. doi: 10.1002/lary.28596. [DOI] [PubMed] [Google Scholar]
- 106.Halawani R T, Dhanasingh A. New Classification of Cochlear Hypoplasia Type Malformation: Relevance in Cochlear Implantation. J Int Adv Otol. 2020;16:153–157. doi: 10.5152/iao.2020.7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cinar B C, Batuk M O, Tahir E et al. Audiologic and radiologic findings in cochlear hypoplasia. Auris Nasus Larynx. 2017;44:655–663. doi: 10.1016/j.anl.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 108.Giesemann A M, Goetz F, Neuburger J et al. Appearance of hypoplastic cochleae in CT and MRI: A new subclassification. Neuroradiology. 2011;53:49–61. doi: 10.1007/s00234-010-0777-3. [DOI] [PubMed] [Google Scholar]
- 109.Harnsberger HR. Diagnostic Imaging Head and Neck. 2004.
- 110.Giesemann A M, Kontorinis G, Jan Z et al. The vestibulocochlear nerve: Aplasia and hypoplasia in combination with inner ear malformations. Eur Radiol. 2012;22:519–524. doi: 10.1007/s00330-011-2287-z. [DOI] [PubMed] [Google Scholar]
- 111.Dörr J, Krautwald S, Wildemann B et al. Characteristics of Susac syndrome: A review of all reported cases. Nat Rev Neurol. 2013;9:307–316. doi: 10.1038/nrneurol.2013.82. [DOI] [PubMed] [Google Scholar]
- 112.Kleffner I, Dörr J, Ringelstein M et al. Diagnostic criteria for Susac syndrome. J Neurol Neurosurg Psychiatry. 2016;87:1287–1295. doi: 10.1136/jnnp-2016-314295. [DOI] [PubMed] [Google Scholar]
- 113.Hertzano R, Tomlinson J A, Mackowiak P A. Goya’s Lost Hearing: A Twenty-First Century Perspective on Its Cause, Effects and Possible Treatment. Am J Med Sci. 2019;357:275–279. doi: 10.1016/j.amjms.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 114.Schelenz D, Kleffner I, Tsiampalis N et al. Susac syndrome – interdisciplinary tracking of the chameleon: two different case reports. Ophthalmologe. 2020;117:369–375. doi: 10.1007/s00347-019-0926-y. [DOI] [PubMed] [Google Scholar]
- 115.Wang Y, Burkholder B, Newsome S D. Progressive sensorineural hearing loss many years preceding completion of Susac’s syndrome triad: A case report. Mult Scler Relat Disord. 2020;37:101436. doi: 10.1016/j.msard.2019.101436. [DOI] [PubMed] [Google Scholar]
- 116.Kleffner I, Duning T, Lohmann H et al. A brief review of Susac syndrome. J Neurol Sci. 2012;322:35–40. doi: 10.1016/j.jns.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 117.Plontke S K, Caye-Thomasen P, Strauss C Management of transmodiolar and transmacular cochleovestibular schwannomas with and without cochlear implantation. HNO. 2020. [DOI] [PMC free article] [PubMed]
- 118.Baskin J, Hardy T A, Law L Y et al. Black blood MRI: endotheliopathy of Susac syndrome unmasked. Neurol Sci. 2020:8–10. doi: 10.1007/s10072-020-04562-8. [DOI] [PubMed] [Google Scholar]
- 119.Mei X, Glueckert R, Schrott-Fischer A et al. Vascular Supply of the Human Spiral Ganglion: Novel Three-Dimensional Analysis Using Synchrotron Phase-Contrast Imaging and Histology. Sci Rep. 2020;10:5877. doi: 10.1038/s41598-020-62653-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ishiyama G, Wester J, Lopez I A et al. Oxidative stress in the blood labyrinthine barrier in the macula utricle of Meniere’s disease patients. Front Physiol. 2018;9:1–16. doi: 10.3389/fphys.2018.01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gu C, Qiao W, Wang L et al. Identification of genes and pathways associated with multiple organ dysfunction syndrome by microarray analysis. Mol Med Rep. 2018;18:31–40. doi: 10.3892/mmr.2018.8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ishiyama G, Lopez I A, Acuna D et al. Investigations of the Microvasculature of the Human Macula Utricle in Meniere’s Disease. Front Cell Neurosci. 2019;13:1–11. doi: 10.3389/fncel.2019.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schmitt H A, Pich A, Schröder A.Proteome Analysis of Human Perilymph using an Intraoperative Sampling Method. J Proteome Res 2017. acs.jproteome.6b00986 10.1021/acs.jproteome.6b00986 [DOI] [PubMed]
- 124.Lin H C, Ren Y, Lysaght A C et al. Proteome of normal human perilymph and perilymph from people with disabling vertigo. PLoS One. 2019;14:1–21. doi: 10.1371/journal.pone.0218292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lage K, Hansena N T, Karlberg E O et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci U S A. 2008;105:20870–20875. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Knipper M, Hofmeier B, Singer W et al. Differentiating cochlear synaptopathies into different hearing disorders. HNO. 2019;67:406–416. doi: 10.1007/s00106-019-0660-4. [DOI] [PubMed] [Google Scholar]
- 127.Espinoza G M, Wheeler J, Temprano K K et al. Cogan’s Syndrome: Clinical Presentations and Update on Treatment. Curr Allergy Asthma Rep. 2020;20:2–7. doi: 10.1007/s11882-020-00945-1. [DOI] [PubMed] [Google Scholar]
- 128.Kuemmerle-Deschner J B, Koitschev A, Ummenhofer K et al. Hearing loss in Muckle-Wells syndrome. Arthritis Rheum. 2013;65:824–831. doi: 10.1002/art.37810. [DOI] [PubMed] [Google Scholar]
- 129.Goldbach-Mansky R, Dailey N J, Canna S W et al. Neonatal-Onset Multisystem Inflammatory Disease Responsive to Interleukin-1β Inhibition. N Engl J Med. 2006;355:581–592. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bachor E, Blevins N H, Karmody C et al. Otologic manifestations of relapsing polychondritis. Auris Nasus Larynx. 2006;33:135–141. doi: 10.1016/j.anl.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 131.Noguchi Y, Nishio A, Takase H et al. Audiovestibular findings in patients with Vogt-Koyanagi-Harada disease. Acta Otolaryngol. 2014;134:339–344. doi: 10.3109/00016489.2013.868604. [DOI] [PubMed] [Google Scholar]
- 132.Kemal O, Anadolu Y, Boyvat A et al. Behçet Disease as a Cause of Hearing Loss: A Prospective, Placebo-Controlled Study of 29 Patients. Ear, Nose Throat J. 2013;92:112–120. doi: 10.1177/014556131309200309. [DOI] [PubMed] [Google Scholar]
- 133.Ovadia S, Dror I, Zubkov T et al. Churg-Strauss syndrome: A rare presentation with otological and pericardial manifestations: Case report and review of the literature. Clin Rheumatol. 2009;28:35–38. doi: 10.1007/s10067-009-1119-x. [DOI] [PubMed] [Google Scholar]
- 134.Moosig F, Holle J. Current treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) Z Rheumatol. 2019;78:333–338. doi: 10.1007/s00393-018-0580-9. [DOI] [PubMed] [Google Scholar]
- 135.rarediseases.orgIm Internet https://rarediseases.org/rare-diseases/alpha-mannosidosis/ Stand: 16.08.2020
- 136.Faverio P, Bonaiti G, Bini F et al. Mepolizumab as the first targeted treatment for eosinophilic granulomatosis with polyangiitis: A review of current evidence and potential place in therapy. Ther Clin Risk Manag. 2018;14:2385–2396. doi: 10.2147/TCRM.S159949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Brachet C, Mansbach A L, Clerckx A et al. Hearing Loss Is Part of the Clinical Picture of ENPP1 Loss of Function Mutation. Horm Res Paediatr. 2014;81:63–66. doi: 10.1159/000354661. [DOI] [PubMed] [Google Scholar]
- 138.Maher C O, Piepgras D G, Brown R D et al. Cerebrovascular manifestations in 321 cases of hereditary hemorrhagic telangiectasia. Stroke. 2001;32:877–882. doi: 10.1161/01.STR.32.4.877. [DOI] [PubMed] [Google Scholar]
- 139.Kim G B. Reality of Kawasaki disease epidemiology. Korean J Pediatr. 2019;62:292–296. doi: 10.3345/kjp.2019.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rahne T, Plontke S, Keyßer G. Vasculitis and the ear: A literature review. Curr Opin Rheumatol. 2020;32:47–52. doi: 10.1097/BOR.0000000000000665. [DOI] [PubMed] [Google Scholar]
- 141.Nadol J B, Eavey R D, Liberfarb R M et al. Histopathology of the ears, eyes, and brain in norrie’s disease (oculoacousticocerebral degeneration) Am J Otolaryngol. 1990;11:112–124. doi: 10.1016/0196-0709(90)90007-I. [DOI] [PubMed] [Google Scholar]
- 142.Gross C C, Meyer C, Bhatia U et al. CD8+ T cell-mediated endotheliopathy is a targetable mechanism of neuro-inflammation in Susac syndrome. Nat Commun. 2019:10. doi: 10.1038/s41467-019-13593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Naini A S, Ghorbani J, Elahi SM L et al. Otologic manifestations in patients with Wegener’s granulomatosis: A survey in 55 patients. Iran J Otorhinolaryngol. 2017;29:327–331. doi: 10.22038/ijorl.2017.25253.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Marsot-Dupuch K, Dominguez-Brito A, Ghasli K et al. CT and MR findings of Michel anomaly: Inner ear aplasia. Am J Neuroradiol. 1999;20:281–284. [PMC free article] [PubMed] [Google Scholar]
- 145.Daneshi A, Farhadi M, Asghari A et al. Three familial cases of Michel’s aplasia. Otol Neurotol. 2002;23:346–348. doi: 10.1097/00129492-200205000-00020. [DOI] [PubMed] [Google Scholar]
- 146.Giesemann A M, Goetz F, Neuburger J et al. From labyrinthine aplasia to otocyst deformity. Neuroradiology. 2010;52:147–154. doi: 10.1007/s00234-009-0601-0. [DOI] [PubMed] [Google Scholar]
- 147.Vesseur A C, Verbist B M, Westerlaan H E et al. CT findings of the temporal bone in CHARGE syndrome: aspects of importance in cochlear implant surgery. Eur Arch Oto-Rhino-Laryngology. 2016;273:4225–4240. doi: 10.1007/s00405-016-4141-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Giesemann A M, Goetz G F, Neuburger J et al. Persistent petrosquamosal sinus: High incidence in cases of complete aplasia of the semicircular canals. Radiology. 2011;259:825–833. doi: 10.1148/radiol.11101466. [DOI] [PubMed] [Google Scholar]
- 149.Sennaroglu L, Saatci I. A New Classification for Cochleovestibular Malformations. Laryngoscope. 2002;112:2230–2241. doi: 10.1097/00005537-200212000-00019. [DOI] [PubMed] [Google Scholar]
- 150.Bademci G, Abad C, Incesulu A et al. FOXF2 is required for cochlear development in humans and mice. Hum Mol Genet. 2019;28:1286–1297. doi: 10.1093/hmg/ddy431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Alballaa A, Aschendorff A, Arndt S et al. Incomplete partition type III“ – Langzeitergebnisse nach Cochleaimplantation. HNO. 2019;67:760–768. doi: 10.1007/s00106-019-00733-y. [DOI] [PubMed] [Google Scholar]
- 152.Kaur A, Khetarpal S. 3P Deletion Syndrome. Indian Pediatr. 2013;50:795–796. [PubMed] [Google Scholar]
- 153.Ţuţulan-Cuniţǎ A C, Papuc S M, Arghir A et al. 3p interstitial deletion: Novel case report and review. J Child Neurol. 2012;27:1062–1066. doi: 10.1177/0883073811431016. [DOI] [PubMed] [Google Scholar]
- 154.Lindstrand A, Malmgren H, Verri A et al. Molecular and clinical characterization of patients with overlapping 10p deletions. Am J Med Genet Part A. 2010;152:1233–1243. doi: 10.1002/ajmg.a.33366. [DOI] [PubMed] [Google Scholar]
- 155.Ohta S, Isojima T, Mizuno Y et al. Partial monosomy of 10p and duplication of another chromosome in two patients. Pediatr Int. 2017;59:99–102. doi: 10.1111/ped.13181. [DOI] [PubMed] [Google Scholar]
- 156.Corrêa T, Feltes B C, Riegel M. Integrated analysis of the critical region 5p15.3–p15.2 associated with cri-du-chat syndrome. Genet Mol Biol. 2019;42:186–196. doi: 10.1590/1678-4685-gmb-2018-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Swanepoel D. Auditory pathology in cri-du-chat (5p-) syndrome: Phenotypic evidence for auditory neuropathy. Clin Genet. 2007;72:369–373. doi: 10.1111/j.1399-0004.2007.00870.x. [DOI] [PubMed] [Google Scholar]
- 158.Du Q, de la Morena M T, van Oers NS C. The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front Genet. 2020;10:1–16. doi: 10.3389/fgene.2019.01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bhalla P, Wysocki C A, van Oers NS C. Molecular Insights Into the Causes of Human Thymic Hypoplasia With Animal Models. Front Immunol. 2020:11. doi: 10.3389/fimmu.2020.00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Romagna E S, Appel da Silva M C, Zanetti Ballardin P A. Schmid-Fraccaro Syndrome: Severe Neurologic Features. Pediatr Neurol. 2010;42:151–153. doi: 10.1016/j.pediatrneurol.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 161.Alamer L, Bassant S, Alhazmi R et al. Rare otologic presentation of cat eye syndrome. Ann Saudi Med. 2019;39:441–443. doi: 10.5144/0256-4947.2019.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Crawford T D, Audia J E, Bellon S et al. GNE-886: A Potent and Selective Inhibitor of the Cat Eye Syndrome Chromosome Region Candidate 2 Bromodomain (CECR2) ACS Med Chem Lett. 2017;8:737–741. doi: 10.1021/acsmedchemlett.7b00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pejcic L, Stankovic T, Ratkovic-Jankovic M et al. Clinical manifestations in trisomy 9 mosaicism. Turk J Pediatr. 2018;60:729–734. doi: 10.24953/turkjped.2018.06.015. [DOI] [PubMed] [Google Scholar]
- 164.Dhangar S, Korgaonkar S, Vundinti B R. Partial trisomy 9 (9pter->9q22.1) and partial monosomy 14 (14pter->14q11.2) due to paternal translocation t(9(q22.1;q11.2) in a case of Dysmorphic features. Intractable Rare Dis Res. 2019;8:72–77. doi: 10.5582/irdr.2019.01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lee C Y, Su H J, Cheng Y T et al. Detection of fetal trisomy 9 mosaicism by noninvasive prenatal testing through maternal plasma DNA sequencing. Taiwan J Obstet Gynecol. 2018;57:594–597. doi: 10.1016/j.tjog.2018.06.021. [DOI] [PubMed] [Google Scholar]
- 166.Thomas S, Parker M, Tan J et al. Ocular manifestations of mosaic trisomy 22: A case report and review of the literature. Ophthalmic Genet. 2004;25:53–56. doi: 10.1076/opge.25.1.53.29004. [DOI] [PubMed] [Google Scholar]
- 167.Abdelgadir D, Nowaczyk MJ M, Li C. Trisomy 22 Mosaicism and Normal Developmental Outcome: Report of Two Patients and Review of the Literature. Am J Med Genet Part A. 2013;161:1126–1131. doi: 10.1002/ajmg.a.35812. [DOI] [PubMed] [Google Scholar]
- 168.Schuster M, Hoppe U, Eysholdt U et al. Severe Hearing Loss in Pallister-Killian Syndrome. ORL. 2002;64:343–345. doi: 10.1159/000066080. [DOI] [PubMed] [Google Scholar]
- 169.Brendal M A, King K A, Zalewski C K et al. Auditory Phenotype of Smith–Magenis Syndrome. J Speech, Lang Hear Res. 2017;60:1076–1087. doi: 10.1044/2016_JSLHR-H-16-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lei M, Liang D, Yang Y et al. Long-read DNA sequencing fully characterized chromothripsis in a patient with Langer–Giedion syndrome and Cornelia de Lange syndrome-4. J Hum Genet. 2020;65:667–674. doi: 10.1038/s10038-020-0754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nouws J, Wibrand F, van den Brand M et al. A Patient with Complex I Deficiency Caused by a Novel ACAD9 Mutation Not Responding to Riboflavin Treatment. In: JIMD Reports. 2013:37–45. doi: 10.1007/8904_2013_242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dewulf J P, Barrea C, Vincent M F et al. Evidence of a wide spectrum of cardiac involvement due to ACAD9 mutations: Report on nine patients. Mol Genet Metab. 2016;118:185–189. doi: 10.1016/j.ymgme.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 173.Yazdanfard P D, Madsen C V, Nielsen L H et al. Significant hearing loss in Fabry disease: Study of the Danish nationwide cohort prior to treatment. PLoS One. 2019;14:e0225071. doi: 10.1371/journal.pone.0225071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Syed Haneef S A, George Priya Doss C. Personalized Pharmacoperones for Lysosomal Storage Disorder: Approach for Next-Generation Treatment. Adv Protein Chem Struct Biol. 2016;102:225–265. doi: 10.1016/bs.apcsb.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 175.orpha.net
- 176.Ärztezeitung.de Im Internethttps://www.aerztezeitung.de/Specials/Lamzede-erste-Enzymersatztherapie-fuer-Patienten-mit-Alpha-Mannosidose-255697.html; Stand: 16.08.2020
- 177.Lehalle D, Colombo R, O’Grady M et al. Hearing impairment as an early sign of alpha-mannosidosis in children with a mild phenotype: Report of seven new cases. Am J Med Genet Part A. 2019;179:1756–1763. doi: 10.1002/ajmg.a.61273. [DOI] [PubMed] [Google Scholar]
- 178.Canda E, Kalkan Uçar S, Çoker M. Biotinidase Deficiency: Prevalence, Impact And Management Strategies. Pediatr Heal Med Ther. 2020;11:127–133. doi: 10.2147/PHMT.S198656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jaeger B, Bosch A M. Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. J Inherit Metab Dis. 2016;39:559–564. doi: 10.1007/s10545-016-9924-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Garg M, Kulkarni S, Hegde A et al. Riboflavin treatment in genetically proven Brown–Vialetto–Van Laere syndrome. J Pediatr Neurosci. 2018;13:471. doi: 10.4103/JPN.JPN_131_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dhar S U, Taylor T, Trinh C et al. Cranio-meta-diaphyseal dysplasia: 25 Year follow-up and review of literature. Am J Med Genet Part A. 2010;152:2335–2338. doi: 10.1002/ajmg.a.33582. [DOI] [PubMed] [Google Scholar]
- 182.PLM Huygen, CWRJ Cremers, WIM Verhagen et al. Camurati-Engelmann disease presenting as „juvenile otosclerosis“. Int J Pediatr Otorhinolaryngol. 1996;37:129–141. doi: 10.1016/0165-5876(96)01392-4. [DOI] [PubMed] [Google Scholar]
- 183.Carlson M L, Beatty C W, Neff B A et al. Skull base manifestations of Camurati-Engelmann disease. Arch Otolaryngol - Head Neck Surg. 2010;136:566–575. doi: 10.1001/archoto.2010.68. [DOI] [PubMed] [Google Scholar]
- 184.Kim Y M, Kang E, Choi J H et al. Clinical characteristics and treatment outcomes in Camurati-Engelmann disease. Med (United States) 2018;97:1–6. doi: 10.1097/MD.0000000000010309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Qin Y, Tang S, Zhen G et al. Bone-targeted delivery of TGF-β type 1 receptor inhibitor rescues uncoupled bone remodeling in Camurati-Engelmann disease. Ann N Y Acad Sci. 2018;1433:29–40. doi: 10.1111/nyas.13941. [DOI] [PubMed] [Google Scholar]
- 186.Lenarz T. JG Neuro-otologic early symptoms of Camurati-Engelmann disease. Laryngol Rhinol Otol (Stuttg) 1983;62:463–467. [PubMed] [Google Scholar]
- 187.Louhichi N, Bahloul E, Marrakchi S et al. Thyroid involvement in Chanarin-Dorfman syndrome in adults in the largest series of patients carrying the same founder mutation in ABHD5 gene. Orphanet J Rare Dis. 2019;14:1–8. doi: 10.1186/s13023-019-1095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Faruqi T, Dhawan N, Bahl J Molecular, phenotypic aspects and therapeutic horizons of rare genetic bone disorders. Biomed Res Int. 2014. p. 2014. [DOI] [PMC free article] [PubMed]
- 189.Sun G H, Samy R N, Tinkle B T Imaging Case of the Month Craniometaphyseal Dysplasia-Induced Hearing Loss. 2011. pp. 9–10. [DOI] [PubMed]
- 190.Vasu C K, Rajendran V R, Regi George A N et al. Progressive facial disfigurement and deafness in craniometaphyseal dysplasia. Indian J Pediatr. 2006;73:1105. doi: 10.1007/bf02763055. [DOI] [PubMed] [Google Scholar]
- 191.Haffner D, Emma F, Eastwood D M et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15:435–455. doi: 10.1038/s41581-019-0152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Morava E, Kühnisch J, Drijvers J M et al. Autosomal recessive mental retardation, deafness, ankylosis, and mild hypophosphatemia associated with a novel ANKH mutation in a consanguineous family. J Clin Endocrinol Metab. 2011;96:189–198. doi: 10.1210/jc.2010-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kyriakou K, Lederer C W, Kleanthous M et al. Acid ceramidase depletion impairs neuronal survival and induces morphological defects in neurites associated with altered gene transcription and sphingolipid content. Int J Mol Sci. 2020;21:1–24. doi: 10.3390/ijms21051607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Yu FP S, Amintas S, Levade T et al. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis. 2018;13:1–19. doi: 10.1186/s13023-018-0845-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pignolo R J, Wang H, Kaplan F S. Fibrodysplasia Ossificans Progressiva (FOP): A Segmental Progeroid Syndrome. Front Endocrinol (Lausanne) 2020;10:1–8. doi: 10.3389/fendo.2019.00908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kaplan F S, Kobori J A, Orellana C et al. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva ( ACVR1 c.772G>A; R258G): A report of two patients. Am J Med Genet Part A. 2015;167:2265–2271. doi: 10.1002/ajmg.a.37205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Jaeken J, Vleugels W, Régal L et al. RFT1-CDG: Deafness as a novel feature of congenital disorders of glycosylation. J Inherit Metab Dis. 2009;32:335–338. doi: 10.1007/s10545-009-1297-3. [DOI] [PubMed] [Google Scholar]
- 198.Kościelak J. Congenital disorders of glycosylation. Handb Carbohydr Eng. 2005;6:99–140. doi: 10.21037/atm.2018.10.45. [DOI] [Google Scholar]
- 199.Mohamed M, Guillard M, Wortmann S B et al. Clinical and diagnostic approach in unsolved CDG patients with a type 2 transferrin pattern. Biochim Biophys Acta - Mol Basis Dis. 2011;1812:691–698. doi: 10.1016/j.bbadis.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 200.Sedel F, Challe G, Mayer J M et al. Thiamine responsive pyruvate dehydrogenase deficiency in an adult with peripheral neuropathy and optic neuropathy. J Neurol Neurosurg Psychiatry. 2008;79:846–847. doi: 10.1136/jnnp.2007.136630. [DOI] [PubMed] [Google Scholar]
- 201.Naito E, Ito M, Yokota I et al. Thiamine-responsive pyruvate dehydrogenase deficiency in two patients caused by a point mutation (F205L and L216F) within the thiamine pyrophosphate binding region. Biochim Biophys Acta – Mol Basis Dis. 2002;1588:79–84. doi: 10.1016/S0925-4439(02)00142-4. [DOI] [PubMed] [Google Scholar]
- 202.Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018;6:476. doi: 10.21037/atm.2018.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Dʹavanzo F, Rigon L, Zanetti A Mucopolysaccharidosis type II: One hundred years of research, diagnosis, and treatment. Int J Mol Sci. 2020. [DOI] [PMC free article] [PubMed]
- 204.King K A, Gordon-Salant S, Yanjanin N Auditory Phenotype of Niemann-Pick Disease, Type C1. Ear Hear. 2014. [DOI] [PMC free article] [PubMed]
- 205.Lipari Pinto P, Machado C, Janeiro P et al. Ngly1 deficiency—a rare congenital disorder of deglycosylation. JIMD Rep. 2020;53:2–9. doi: 10.1002/jmd2.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.RFM Rosa, da Silva A P, Goetze T B et al. Ear abnormalities in patients with oculo-auriculo-vertebral spectrum (Goldenhar syndrome) Braz J Otorhinolaryngol. 2011;77:455–460. doi: 10.1590/S1808-86942011000400008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Vargas-Poussou R, Houillier P, Le Pottier N et al. Genetic Investigation of Autosomal Recessive Distal Renal Tubular Acidosis: Evidence for Early Sensorineural Hearing Loss Associated with Mutations in the ATP6V0A4 Gene. J Am Soc Nephrol. 2006;17:1437–1443. doi: 10.1681/ASN.2005121305. [DOI] [PubMed] [Google Scholar]
- 208.Hanisch F, Rahne T, Plontke S K. Prevalence of hearing loss in patients with late-onset Pompe disease: Audiological and otological consequences. Int J Audiol. 2013;52:816–823. doi: 10.3109/14992027.2013.840932. [DOI] [PubMed] [Google Scholar]
- 209.Oysu C, Aslan I, Basaran B et al. The site of the hearing loss in Refsum’s disease. Int J Pediatr Otorhinolaryngol. 2001;61:129–134. doi: 10.1016/S0165-5876(01)00559-6. [DOI] [PubMed] [Google Scholar]
- 210.Vandana V P, Bindu P S, Nagappa M et al. Audiological findings in Infantile Refsum disease. Int J Pediatr Otorhinolaryngol. 2015;79:1366–1369. doi: 10.1016/j.ijporl.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 211.Bamiou D-E, Spraggs PR D, Gibberd F B et al. Hearing loss in adult Refsum’s disease. Clin Otolaryngol Allied Sci. 2003;28:227–230. doi: 10.1046/j.1365-2273.2003.00694.x. [DOI] [PubMed] [Google Scholar]
- 212.Liberman M C, Tartaglini E, Fleming J C et al. Deletion of SLC19A2, the high affinity thiamine transporter, causes selective inner hair cell loss and an auditory neuropathy phenotype. JARO - J Assoc Res Otolaryngol. 2006;7:211–217. doi: 10.1007/s10162-006-0035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Di Giaimo R, Riccio M, Santi S et al. Targeted disruption of Slc19a2, the gene encoding the high-affinity thiamin transporter Thtr-1, causes diabetes mellitus, sensorineural deafness and megaloblastosis in mice. Hum Mol Genet. 2002;11:2951–2960. doi: 10.1093/hmg/11.23.2951. [DOI] [PubMed] [Google Scholar]
- 214.Mohamed F E, Al Sorkhy M, Ghattas M A et al. A Novel Homozygous Missense Variant in the NAGA Gene with Extreme Intrafamilial Phenotypic Heterogeneity. J Mol Neurosci. 2020;70:45–55. doi: 10.1007/s12031-019-01398-6. [DOI] [PubMed] [Google Scholar]
- 215.Rodríguez-Pazos L, Ginarte M, Vega A et al. Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr. 2013;104:270–284. doi: 10.1016/j.adengl.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 216.Fekete R. Xeroderma pigmentosum/De Sanctis-Cacchione syndrome: Unusual cause of ataxia. Case Rep Neurol. 2014;6:83–87. doi: 10.1159/000362115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Rahbar Z, Naraghi M. De Sanctis-Cacchione syndrome: A case report and literature review. Int J Women’s Dermatology. 2015;1:136–139. doi: 10.1016/j.ijwd.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kale K, Ghonge N, Kaul A. Prenatal diagnosis of congenital harlequin ichthyosis with fetal MRI. Indian J Radiol Imaging. 2019;29:448. doi: 10.4103/ijri.IJRI_105_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cammarata-Scalisi F, Willoughby C E, Cárdenas Tadich A Clinical, etiopathogenic, and therapeutic aspects of KID syndrome. Dermatol Ther. 2020. [DOI] [PubMed]
- 220.Shuja Z, Li L, Gupta S et al. Connexin26 mutations causing palmoplantar keratoderma and deafness interact with connexin43, modifying gap junction and hemichannel properties. J Invest Dermatol. 2016;136:225–235. doi: 10.1038/JID.2015.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yoon H K, Sargent M A, Prendiville J S et al. Cerebellar and cerebral atrophy in trichothiodystrophy. Pediatr Radiol. 2005;35:1019–1023. doi: 10.1007/s00247-005-1495-6. [DOI] [PubMed] [Google Scholar]
- 222.Valverde D, Alvarez-Satta M, Castro-Sánchez S. Alström syndrome: current perspectives. Appl Clin Genet. 2015. p. 171. [DOI] [PMC free article] [PubMed]
- 223.Mittal R, Patel K, Mittal J et al. Association of PRPS1 Mutations with Disease Phenotypes. Dis Markers. 2015;2015:1–7. doi: 10.1155/2015/127013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Barakat A J, Raygada M, Rennert O M. Barakat syndrome revisited. Am J Med Genet Part A. 2018;176:1341–1348. doi: 10.1002/ajmg.a.38693. [DOI] [PubMed] [Google Scholar]
- 225.Sheehan-Rooney K, Swartz M E, Zhao F et al. Ahsa1 and Hsp90 activity confers more severe craniofacial phenotypes in a zebrafish model of hypoparathyroidism, sensorineural deafness and renal dysplasia (HDR) DMM Dis Model Mech. 2013;6:1285–1291. doi: 10.1242/dmm.011965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Husson H, Bukanov N O, Moreno S et al. Correction of cilia structure and function alleviates multi-organ pathology in Bardet–Biedl syndrome mice. Hum Mol Genet. 2020;00:1–15. doi: 10.1093/hmg/ddaa138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.M’Hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of bardet-biedl syndrome. Mol Syndromol. 2014;5:51–56. doi: 10.1159/000357054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Datta P, Ruffcorn A, Seo S. Limited time window for retinal gene therapy in a preclinical model of ciliopathy. Hum Mol Genet. 2020;29:2337–2352. doi: 10.1093/hmg/ddaa124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gajendragadkar A, Bhamkar R. Antenatal Bartter′s syndrome with sensorineural deafness. Indian J Nephrol. 2009;19:23. doi: 10.4103/0971-4065.50677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Miyamura N, Matsumoto K, Taguchi T et al. Atypical Bartter Syndrome with Sensorineural Deafness with G47R Mutation of the β-Subunit for ClC-Ka and ClC-Kb Chloride Channels, Barttin. J Clin Endocrinol Metab. 2003;88:781–786. doi: 10.1210/jc.2002-021398. [DOI] [PubMed] [Google Scholar]
- 231.Zhang J, Duo L, Lin Z et al. Exome sequencing reveals novel BCS1L mutations in siblings with hearing loss and hypotrichosis. Gene. 2015;566:84–88. doi: 10.1016/j.gene.2015.04.039. [DOI] [PubMed] [Google Scholar]
- 232.Min J, Mao B, Wang Y et al. A Heterozygous Novel Mutation in TFAP2A Gene Causes Atypical Branchio-Oculo-Facial Syndrome With Isolated Coloboma of Choroid: A Case Report. Front Pediatr. 2020;8:1–6. doi: 10.3389/fped.2020.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Milunsky J M, Maher T A, Zhao G et al. TFAP2A Mutations Result in Branchio-Oculo-Facial Syndrome. Am J Hum Genet. 2008;82:1171–1177. doi: 10.1016/j.ajhg.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Klingbeil K D, Greenland C M, Arslan S et al. Novel EYA1 variants causing Branchio-oto-renal syndrome. Int J Pediatr Otorhinolaryngol. 2017;98:59–63. doi: 10.1016/j.ijporl.2017.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Shah A M, Krohn P, Baxi A B et al. Six1 proteins with human branchio-oto-renal mutations differentially affect cranial gene expression and otic development. DMM Dis Model Mech. 2020:13. doi: 10.1242/dmm.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hsu A, Desai N, Paldino M J. The Unwound Cochlea: A Specific Imaging Marker of Branchio-Oto-Renal Syndrome. AJNR Am J Neuroradiol. 2018;39:2345–2349. doi: 10.3174/ajnr.A5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Boudhina T, Yedes A, Khiari S et al. Familial syndrome combining short stature, microcephaly, mental deficiency, seizures, hearing loss, and skin lesions. A new syndrome. ediatr (Paris) 1990;37:399–403. [PubMed] [Google Scholar]
- 238.Hasan M R, Takatalo M, Ma H et al. RAB23 coordinates early osteogenesis by repressing FGF10-pERK1/2 and GLI1. Elife. 2020;9:1–26. doi: 10.7554/eLife.55829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tarhan E, Oğuz H, Şafak M A et al. The Carpenter syndrome phenotype. Int J Pediatr Otorhinolaryngol. 2004;68:353–357. doi: 10.1016/j.ijporl.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 240.Twigg SR F, Lloyd D, Jenkins D et al. Mutations in multidomain protein MEGF8 identify a carpenter syndrome subtype associated with defective lateralization. Am J Hum Genet. 2012;91:897–905. doi: 10.1016/j.ajhg.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Bérubé-Simard F A, Pilon N. Molecular dissection of CHARGE syndrome highlights the vulnerability of neural crest cells to problems with alternative splicing and other transcription-related processes. Transcription. 2019;10:21–28. doi: 10.1080/21541264.2018.1521213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Karikkineth A C, Scheibye-Knudsen M, Fivenson E et al. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res Rev. 2017;33:3–17. doi: 10.1016/j.arr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Morris D P, Alian W, Maessen H et al. Cochlear implantation in Cockayne syndrome: Our experience of two cases with different outcomes. Laryngoscope. 2007;117:939–943. doi: 10.1097/MLG.0b013e3180325106. [DOI] [PubMed] [Google Scholar]
- 244.Hanauer A, Young I D. Coffin-Lowry syndrome: Clinical and molecular features. J Med Genet. 2002;39:705–713. doi: 10.1136/jmg.39.10.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Rosanowski F, Eysholdt U. Late-Onset Sensorineural Hearing Loss in Coffin-Lowry Syndrome. 1998:224–226. doi: 10.1159/000027598. [DOI] [PubMed] [Google Scholar]
- 246.Vasileiou G, Vergarajauregui S, Endele S et al. Mutations in the BAF-Complex Subunit DPF2 Are Associated with Coffin-Siris Syndrome. Am J Hum Genet. 2018;102:468–479. doi: 10.1016/j.ajhg.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Schrier S A, Bodurtha J N, Burton B et al. The Coffin-Siris syndrome: A proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet Part A. 2012;158A:1865–1876. doi: 10.1002/ajmg.a.35415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sarogni P, Pallotta M M, Musio A. Cornelia de Lange syndrome: From molecular diagnosis to therapeutic approach. J Med Genet. 2020;57:289–295. doi: 10.1136/jmedgenet-2019-106277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Avagliano L, Parenti I, Grazioli P et al. Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin Genet. 2020;97:3–11. doi: 10.1111/cge.13674. [DOI] [PubMed] [Google Scholar]
- 250.Marchisio P, Selicorni A, Bianchini S et al. Audiological findings, genotype and clinical severity score in Cornelia de Lange syndrome. Int J Pediatr Otorhinolaryngol. 2014;78:1045–1048. doi: 10.1016/j.ijporl.2014.03.038. [DOI] [PubMed] [Google Scholar]
- 251.Kurkiewicz A, Cooper A, McIlwaine E et al. Towards development of a statistical framework to evaluate myotonic dystrophy type 1 mRNA biomarkers in the context of a clinical trial. PLoS One. 2020;15:1–19. doi: 10.1371/journal.pone.0231000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Van Vliet J, Tieleman A A, Van Engelen BG M et al. Hearing impairment in patients with myotonic dystrophy type 2. Neurology. 2018;90:e615–e622. doi: 10.1212/WNL.0000000000004963. [DOI] [PubMed] [Google Scholar]
- 253.Balatsouras D G, Felekis D, Panas M et al. Inner ear dysfunction in myotonic dystrophy type 1. Acta Neurol Scand. 2013;127:337–343. doi: 10.1111/ane.12020. [DOI] [PubMed] [Google Scholar]
- 254.Khalifa O, Al-Sahlawi Z, Imtiaz F et al. Variable expression pattern in Donnai-Barrow syndrome: Report of two novel LRP2 mutations and review of the literature. Eur J Med Genet. 2015;58:293–299. doi: 10.1016/j.ejmg.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 255.Kantarci S, Al-Gazali L, Hill R S et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39:957–959. doi: 10.1038/ng2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Campeau P M, Kasperaviciute D, Lu J T et al. The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol. 2014;13:44–58. doi: 10.1016/S1474-4422(13)70265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Campeau P M, Hennekam R C, Aftimos S et al. DOORS syndrome: Phenotype, genotype and comparison with coffin-siris syndrome. Am J Med Genet Part C Semin Med Genet. 2014;166:327–332. doi: 10.1002/ajmg.c.31412. [DOI] [PubMed] [Google Scholar]
- 258.Pradhan N, Shilawant J, Akkamahadevi C H et al. Ehlers-Danlos syndrome with huge bladder diverticulum in pregnancy – A rare and interesting case report. Eur J Obstet Gynecol Reprod Biol. 2020;250:231–234. doi: 10.1016/j.ejogrb.2020.05.001. [DOI] [PubMed] [Google Scholar]
- 259.Ritelli M, Dordoni C, Cinquina V et al. Expanding the clinical and mutational spectrum of B4GALT7-spondylodysplastic Ehlers-Danlos syndrome. Orphanet J Rare Dis. 2017;12:1–7. doi: 10.1186/s13023-017-0704-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Fryns J P. Fountain’s syndrome: Mental retardation, sensorineural deafness, skeletal abnormalities, and coarse face with full lips. J Med Genet. 1989;26:722–724. doi: 10.1136/jmg.26.11.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Poling M I, Dufresne C R, Chamberlain R L. Findings, Phenotypes, Diagnostic Accuracy, and Treatment in Freeman-Burian Syndrome. J Craniofac Surg. 2020;31:1063–1069. doi: 10.1097/SCS.0000000000006299. [DOI] [PubMed] [Google Scholar]
- 262.Regev M, Pode-Shakked B, Jacobson J M et al. Phenotype variability in Hajdu-Cheney syndrome. Eur J Med Genet. 2019;62:35–38. doi: 10.1016/j.ejmg.2018.04.015. [DOI] [PubMed] [Google Scholar]
- 263.Abu-Amero K K, Hagr AAl, Almomani M O et al. HOXA1 mutations are not commonly associated with non-syndromic deafness. Can J Neurol Sci. 2014;41:448–451. doi: 10.1017/S0317167100018473. [DOI] [PubMed] [Google Scholar]
- 264.Lai W F, Wong W T. Progress and trends in the development of therapies for Hutchinson–Gilford progeria syndrome. Aging Cell. 2020;19:1–17. doi: 10.1111/acel.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Liu S, Wang Z, Jiang J et al. Severe forms of Johanson-Blizzard syndrome caused by two novel compound heterozygous variants in UBR1: Clinical manifestations, imaging findings and molecular genetics. Pancreatology. 2020;20:562–568. doi: 10.1016/j.pan.2020.01.007. [DOI] [PubMed] [Google Scholar]
- 266.Friez M J, Brooks S S, Stevenson R E et al. HUWE1 mutations in Juberg-Marsidi and Brooks syndromes: The results of an X-chromosome exome sequencing study. BMJ Open. 2016;6:1–9. doi: 10.1136/bmjopen-2015-009537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Cuvertino S, Hartill V, Colyer A et al. A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet Med. 2020;22:867–877. doi: 10.1038/s41436-019-0743-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Stamou M I, Georgopoulos N A. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism. 2018;86:124–134. doi: 10.1016/j.metabol.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Finsterer J, Winklehner M, Stöllberger C et al. Unusual Phenotype and Disease Trajectory in Kearns–Sayre Syndrome. Case Rep Neurol Med. 2020;2020:1–6. doi: 10.1155/2020/7368527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Weidauer H, Lenarz T. Kearns-Sayre syndrome from the otorhinolaryngologic viewpoint. Laryngol Rhinol Otol (Stuttg) 1984;63:141–146. [PubMed] [Google Scholar]
- 271.Frikha R. Klippel-Feil syndrome: a review of the literature. Clin Dysmorphol. 2020;29:35–37. doi: 10.1097/MCD.0000000000000301. [DOI] [PubMed] [Google Scholar]
- 272.Mayer B, Lenarz T, Haels J. Cervically-induced symptoms of the Klippel-Feil syndrome. Laryngol Rhinol Otol (Stuttg) 1984;63:364–370. [PubMed] [Google Scholar]
- 273.Husain Q, Cho J, Neugarten J et al. Surgery of the head and neck in patient with Kniest dysplasia: Is wound healing an issue? Int J Pediatr Otorhinolaryngol. 2017;93:97–99. doi: 10.1016/j.ijporl.2016.12.025. [DOI] [PubMed] [Google Scholar]
- 274.Hey Ryu Y, Kyun Chae J, Kim J W et al. Lacrimo-auriculo-dento-digital syndrome: A novel mutation in a Korean family and review of literature. Mol Genet Genomic Med. 2020:1–11. doi: 10.1002/mgg3.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Muzio M R, Cascella M, Al Khalili Y.Landau Kleffner Syndrome 2020. Im Internethttp://www.ncbi.nlm.nih.gov/pubmed/31613525 [PubMed]
- 276.Kim J, Kim M R, Kim H J et al. LEOPARD syndrome with PTPN11 gene mutation showing six cardinal symptoms of LEOPARD. Ann Dermatol. 2011;23:232–235. doi: 10.5021/ad.2011.23.2.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Levy J, Chung W, Garzon M et al. Congenital myopathy, recurrent secretory diarrhea, bullous eruption of skin, microcephaly, and deafness: A new genetic syndrome? Am J Med Genet. 2003;116:20–25. doi: 10.1002/ajmg.a.10072. [DOI] [PubMed] [Google Scholar]
- 278.Griffith A J, Sprunger L K, Sirko-Osadsa D A et al. Marshall Syndrome Associated with a Splicing Defect at the COL11A1 Locus. Am J Hum Genet. 1998;62:816–823. doi: 10.1086/301789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Rawle M, Larner A. NARP Syndrome: A 20-Year Follow-Up. Case Rep Neurol. 2013;5:204–207. doi: 10.1159/000357518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Strubbe E H, CWRJ Cremers, Dikkers F G et al. Hearing loss and the Mayer-Rokitansky-Kuster-Hauser syndrome. Am J Otol. 1994;15:431–436. [PubMed] [Google Scholar]
- 281.Boyce A M, Collins M T. Fibrous Dysplasia/McCune-Albright Syndrome: A Rare, Mosaic Disease of Gα s Activation. Endocr Rev. 2020;41:345–370. doi: 10.1210/endrev/bnz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Di Stadio A, Pegoraro V, Giaretta L et al. Hearing impairment in MELAS: New prospective in clinical use of microRNA, a systematic review. Orphanet J Rare Dis. 2018;13:1–9. doi: 10.1186/s13023-018-0770-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Handzel O, Ungar O J, Lee D J et al. Temporal bone histopathology in MELAS syndrome. Laryngoscope Investig Otolaryngol. 2020;5:152–156. doi: 10.1002/lio2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Tsutsumi T, Nishida H, Noguchi Y et al. Audiological findings in patients with myoclonic epilepsy associated with ragged-red fibres. J Laryngol Otol. 2001:115. doi: 10.1258/0022215011909224. [DOI] [PubMed] [Google Scholar]
- 285.Picciolini O, Porro M, Cattaneo E et al. Moebius syndrome: clinical features, diagnosis, management and early intervention. Ital J Pediatr. 2016;42:56. doi: 10.1186/s13052-016-0256-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Gürsoy S, Hazan F, Öztürk T et al. Novel Ocular and Inner Ear Anomalies in a Patient with Myhre Syndrome. Mol Syndromol. 2020;10:339–343. doi: 10.1159/000504829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Melkoniemi M, Brunner H G, Manouvrier S et al. Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene. Am J Hum Genet. 2000;66:368–377. doi: 10.1086/302750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Koffler T, Ushakov K, Avraham K B. Genetics of Hearing Loss. Otolaryngol Clin North Am. 2015;48:1041–1061. doi: 10.1016/j.otc.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Gettelfinger J, Dahl J. Syndromic Hearing Loss: A Brief Review of Common Presentations and Genetics. J Pediatr Genet. 2018;07:1–8. doi: 10.1055/s-0037-1617454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Desai U, Rosen H, Mulliken J B et al. Audiologic Findings in Pfeiffer Syndrome. J Craniofac Surg. 2010;21:1411–1418. doi: 10.1097/SCS.0b013e3181ebcf58. [DOI] [PubMed] [Google Scholar]
- 291.Tarailo-Graovac M, Sinclair G, Stockler-Ipsiroglu S et al. The genotypic and phenotypic spectrum of PIGA deficiency. Orphanet J Rare Dis. 2015:10. doi: 10.1186/s13023-015-0243-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Knowles M R, Zariwala M, Leigh M. Primary Ciliary Dyskinesia. Clin Chest Med. 2016;37:449–461. doi: 10.1016/j.ccm.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Chang Q, Wang J, Li Q et al. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol Med. 2015;7:1077–1086. doi: 10.15252/emmm.201404929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Zhang Q, Liang D, Yue Y et al. Axenfeld-Rieger syndrome-associated mutants of the transcription factor FOXC1 abnormally regulate NKX2-5 in model zebrafish embryos. J Biol Chem. 2020;2:jbc.RA120.013287. doi: 10.1074/jbc.RA120.013287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Wakeling E L, Brioude F, Lokulo-Sodipe O et al. Diagnosis and management of Silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13:105–124. doi: 10.1038/nrendo.2016.138. [DOI] [PubMed] [Google Scholar]
- 296.Hoischen A, Van Bon BW M, Gilissen C et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 297.Tsang S H, Aycinena AR P, Sharma T. Ciliopathy: Senior-Løken Syndrome. 2018:175–178. doi: 10.1007/978-3-319-95046-4_34. [DOI] [PubMed] [Google Scholar]
- 298.Kaur A, Dhir S K, Goyal G et al. Senior loken syndrome. J Clin Diagnostic Res. 2016;10:SD03–SD04. doi: 10.7860/JCDR/2016/21832.8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Abdelhadi O, Iancu D, Stanescu H et al. EAST syndrome: Clinical, pathophysiological, and genetic aspects of mutations in KCNJ10. Rare Dis (Austin, Tex) 2016;4:e1195043. doi: 10.1080/21675511.2016.1195043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Gaudreau P, Zizak V, Gallagher T Q. The otolaryngologic manifestations of Sotos syndrome. Int J Pediatr Otorhinolaryngol. 2013;77:1861–1863. doi: 10.1016/j.ijporl.2013.08.029. [DOI] [PubMed] [Google Scholar]
- 301.Boczek N J, Kruisselbrink T, Cousin M A et al. Multigenerational pedigree with STAR syndrome: A novel FAM58A variant and expansion of the phenotype. Am J Med Genet Part A. 2017;173:1328–1333. doi: 10.1002/ajmg.a.38113. [DOI] [PubMed] [Google Scholar]
- 302.Smith S D, Kelley P M, Kenyon J B et al. Tietz syndrome (hypopigmentation/deafness) caused by mutation of MITF. J Med Genet. 2000;37:446–448. doi: 10.1136/jmg.37.6.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Liang Y, Shen D, Cai W. Two coding single nucleotide polymorphisms in the SALL1 gene in Townes-Brocks syndrome: a case report and review of the literature. J Pediatr Surg. 2008;43:391–393. doi: 10.1016/j.jpedsurg.2007.09.079. [DOI] [PubMed] [Google Scholar]
- 304.Géléoc GG S, El-Amraoui A. Disease mechanisms and gene therapy for Usher syndrome. Hear Res. 2020;394:107932. doi: 10.1016/j.heares.2020.107932. [DOI] [PubMed] [Google Scholar]
- 305.Hedberg-Oldfors C, Darin N, Oldfors A. Muscle pathology in Vici syndrome-A case study with a novel mutation in EPG5 and a summary of the literature. Neuromuscul Disord. 2017;27:771–776. doi: 10.1016/j.nmd.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 306.Song J, Feng Y, Acke F R et al. Hearing loss in Waardenburg syndrome: a systematic review. Clin Genet. 2016;89:416–425. doi: 10.1111/cge.12631. [DOI] [PubMed] [Google Scholar]
- 307.Kontorinis G, Lenarz T, Giourgas A et al. Outcomes and Special Considerations of Cochlear Implantation in Waardenburg Syndrome. Otol Neurotol. 2011;32:951–955. doi: 10.1097/MAO.0b013e31821b3ae3. [DOI] [PubMed] [Google Scholar]
- 308.La Morgia C, Maresca A, Amore G et al. Calcium mishandling in absence of primary mitochondrial dysfunction drives cellular pathology in Wolfram Syndrome. Sci Rep. 2020;10:1–15. doi: 10.1038/s41598-020-61735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Samara A, Rahn R, Neyman O et al. Developmental hypomyelination in Wolfram syndrome: New insights from neuroimaging and gene expression analyses. Orphanet J Rare Dis. 2019;14:1–14. doi: 10.1186/s13023-019-1260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Elumalai V, Pasrija D.Zellweger Syndrome 2020. Im Internethttp://www.ncbi.nlm.nih.gov/pubmed/32809511
- 311.Sclafani A P, DeDio R M, Hendrix R A.The Chiari-I malformation Ear Nose Throat J 199170208–212.Im Internethttp://www.ncbi.nlm.nih.gov/pubmed/1874153 [PubMed] [Google Scholar]
- 312.Barsottini O G, Pedroso J L, Martins C R et al. Deafness and Vestibulopathy in Cerebellar Diseases: a Practical Approach. Cerebellum. 2019;18:1011–1016. doi: 10.1007/s12311-019-01042-4. [DOI] [PubMed] [Google Scholar]
- 313.Bokhari M R, Samanta D, Bokhari SRA.Canavan Disease 2020. Im Internethttp://www.ncbi.nlm.nih.gov/pubmed/28613566
- 314.Roscoe R B, Elliott C, Zarros A et al. Non-genetic therapeutic approaches to Canavan disease. J Neurol Sci. 2016;366:116–124. doi: 10.1016/j.jns.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 315.Ishiyama G, Lopez I, Baloh R W et al. Canavan’s leukodystrophy is associated with defects in cochlear neurodevelopment and deafness. Neurology. 2003;60:1702–1704. doi: 10.1212/01.WNL.0000065893.60879.D3. [DOI] [PubMed] [Google Scholar]
- 316.Demos M K, Van Karnebeek CD M, Ross CJ D et al. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet J Rare Dis. 2014;9:1–9. doi: 10.1186/1750-1172-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Stenshorne I, Rasmussen M, Salvanos P et al. Fever-related ataxia: A case report of CAPOS syndrome. Cerebellum and Ataxias. 2019;6:3–7. doi: 10.1186/s40673-019-0096-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Rosewich H, Weise D, Ohlenbusch A et al. Phenotypic overlap of alternating hemiplegia of childhood and CAPOS syndrome. Neurology. 2014;83:861–863. doi: 10.1212/WNL.0000000000000735. [DOI] [PubMed] [Google Scholar]
- 319.Choi J E, Seok J M, Ahn J et al. Hidden hearing loss in patients with Charcot-Marie-Tooth disease type 1A. Sci Rep. 2018;8:10335. doi: 10.1038/s41598-018-28501-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Park J G, Tischfield M A, Nugent A A et al. Loss of MAFB Function in Humans and Mice Causes Duane Syndrome, Aberrant Extraocular Muscle Innervation, and Inner-Ear Defects. Am J Hum Genet. 2016;98:1220–1227. doi: 10.1016/j.ajhg.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Weir F W, Kreicher K L, Hatch J L et al. Audiologic and otologic phenotype in children with Duane’s Retraction Syndrome: A rare ophthalmologic disorder. Int J Pediatr Otorhinolaryngol. 2016;89:154–158. doi: 10.1016/j.ijporl.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 322.Auer-Grumbach M, Bode H, Pieber T R et al. Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur J Med Genet. 2013;56:266–269. doi: 10.1016/j.ejmg.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Hojo K, Kawamata T, Tanaka C et al. Inflammatory glial activation in the brain of a patient with hereditary sensory neuropathy type 1 with deafness and dementia. Neurosci Lett. 2004;367:340–343. doi: 10.1016/j.neulet.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 324.Tyler G K, Martin TP C, Baguley D M. Systematic review of outcome of cochlear implantation in superficial siderosis. Otol Neurotol. 2012;33:976–982. doi: 10.1097/MAO.0b013e3182565a46. [DOI] [PubMed] [Google Scholar]
- 325.Iversen M M, Rabbitt R D. Biomechanics of Third Window Syndrome. Front Neurol. 2020:11. doi: 10.3389/fneur.2020.00891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Marinelli J P, Lohse C M, Carlson M L. Incidence of Intralabyrinthine Schwannoma. Otol Neurotol. 2018;39:1191–1194. doi: 10.1097/MAO.0000000000001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Plontke S K, Rahne T, Pfister M et al. Intralabyrinthine schwannomas: Surgical management and hearing rehabilitation with cochlear implants. HNO. 2017;65:136–148. doi: 10.1007/s00106-017-0364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Plontke S K, Fröhlich L, Wagner L et al. How Much Cochlea Do You Need for Cochlear Implantation? Otol Neurotol. 2020;41:694–703. doi: 10.1097/MAO.0000000000002614. [DOI] [PubMed] [Google Scholar]
- 329.Orsini A, Valetto A, Bertini V et al. The best evidence for progressive myoclonic epilepsy: A pathway to precision therapy. Seizure. 2019;71:247–257. doi: 10.1016/j.seizure.2019.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]