

Abstract
Energetically inefficient inter-organ substrate shuttles are proposed contributors to cachexia-related weight loss. Here we examined glycolytic pathway metabolites, enzyme activity, and transport proteins in skeletal muscle, liver and tumors of mice with cachexia-related weight loss induced by colon-26 cancer cells. Skeletal muscle of cachexic mice had increased [L-lactate]/[pyruvate], LDH activity, and lactate transporter MCT1. Cachexic livers also showed increased MCT1. This is consistent with the proposal that the rate of muscle-derived lactate shuttling to liver for use in gluconeogenesis is increased i.e. an increased Cori cycle flux in weight-losing cachexic mice. A second shuttle between liver and tumor may also contribute to disrupted energy balance and weight loss. We found increased high-affinity glucose transporter GLUT1 in tumors, suggesting active glucose uptake, tumor MCT1 detection and decreased intratumor [L-lactate]/[pyruvate], implying increased lactate efflux and/or intratumor lactate oxidation. Last, high [L-lactate]/[pyruvate] and MCT1 in cachexic muscle provides a potential muscle-derived lactate supply for the tumor (a “reverse Warburg effect”), supporting tumor growth and consequent cachexia. Our findings suggest several substrate shuttles among liver, skeletal muscle and tumor contribute to metabolic disruption and weight loss. Therapies that aim to normalize dysregulated substrate shuttling among energy-regulating tissues may alleviate unintended weight loss in cancer cachexia.
Keywords: Warburg effect, Lactate, Cori cycle, Colon-26, Glucose transporter, Monocarboxylate transporter, Energy Metabolism
Introduction
Severe weight loss, muscle atrophy and frailty are critical paraneoplastic manifestations of cancer, termed cachexia1. Cachexia occurs in roughly half of all cancer patients, and in up to 80% of patients with advanced disease2,3; it is especially prevalent in cancers of the lung, colon, stomach, and pancreas4. Cachexia adversely affects patient outcomes in number of significant ways, including lowered treatment efficacy5, increased toxicity6, greater hospitalization cost and management of adverse events7,8, and reduced survival9. Overall, cachexia is responsible for 20-30% of cancer-related deaths2. Because cachexia impairs quality and quantity of life, and reversal of cachexia has the potential to increase survival10, therapies that mitigate cachexia are urgently needed. Currently, there are no effective treatments for cancer cachexia despite the significant impact of cachexia expressed by patients and their caregivers11. Developing an advanced understanding of mechanisms of metabolic disturbance that cause cachexia is important as it may guide efforts to develop therapeutic strategies that improve quality-of-life, response to clinical treatment, and survival in the growing number of patients fighting cancer annually.
Unraveling the mechanisms of metabolic disturbance that lead to cachexia are complicated by the systemic nature of the disease. Although emphasis is often placed on skeletal muscle pathology, cachexia involves altered function of multiple organs systems, particularly those regulating energy metabolism such as heart, adipose and liver among others12–15. The liver has gained renewed interest in cancer cachexia, with evidence suggesting that it is closely involved in cachexia-associated weight loss and muscle atrophy. For instance, the liver participates in the systemic acute phase response by synthesizing acute phase proteins such as albumin, fibrinogen and C-reactive protein among others. To support acute phase protein synthesis, amino acids are mobilized and released from skeletal muscle, contributing to tumor-induced muscle loss16. Specific targeting of hepatic metabolism can alleviate weight loss and muscle atrophy17, lending support for the liver as an important site for targeted therapeutic strategies.
While the features of cachexia are multi-faceted, involuntary weight loss is one of the most visibly alarming and conspicuous aspects of cachexia. Weight loss in cachexia appears to be linked with dysregulation of hepatic metabolism through several mechanisms. In cachexic mice, liver mitochondria are pathologically uncoupled, possibly due to increased proton leak across the mitochondrial inner membrane18. Uncoupling and proton leak in mitochondria is a significant physiological event because it accounts for a considerable proportion of basal metabolic rate19. Uncoupling is metabolically inefficient and may contribute to increased energy expenditure and unintended weight loss in cachexia. In addition, weight loss in cancer cachexia is frequently attributed to the Warburg effect (a high rate of tumor glycolysis and lactate production in the presence of oxygen) and lactate shuttling between the liver and tumor12–14. This shuttling of lactate constitutes a Cori cycle, similar to that identified between skeletal muscle and liver. Such intra-organ substrate shuttling is energetically inefficient because the production of lactate from glycolysis in the tumor nets 2 ATP, while exported lactate shuttled into the liver for use as a gluconeogenic substrate incurs a cost of 6 ATP. The hepatic glucose generated and released presumably supplies the glycolytic needs of the tumor, creating a futile substrate cycle.
Tissue-specific alterations in energy metabolism are routinely used to infer an association of cachexia-related weight loss with inter-organ substrate flux. Mice bearing human pancreatic cancer cells with high glycolytic activity (i.e. high glucose consumption and lactate production in culture media) showed evidence of cachexia such as weight loss, fat depletion, and muscle proteolysis20. These features were not present in mice bearing pancreatic cancer cells with low glycolytic activity, implying that the Warburg effect in cancer cells is independently associated with the induction of cachexia20. Recently, tissue-specific analysis of the proteome and metabolome were profiled in cancer cachexia21–23. Proteins and metabolites are downstream effectors and provide important clues about potential disease mechanisms. Proteome-wide expression analysis of liver tissue from mice with cancer cachexia revealed differential expression of several proteins that point to altered lactate metabolism and transport in the cachexic liver, including increased LDH-A chain, and increased lactate transporter MCT121. In a metabolome analysis of liver and plasma from mice with cancer cachexia, circulating glucose was decreased while lactate concentration was unchanged, suggesting increased consumption of glucose by peripheral tissues23. In the liver, glucose and glycogen were depleted, and lactate tended to be lower in cachexia compared with controls. Overall, this analysis supported derangements in glycolytic metabolism and ongoing inter-organ substrate shuttling among peripheral organs impacted by tumor-induced cachexia.
Advancing a holistic understanding of tissue-specific metabolic abnormalities in cancer cachexia should consider glycolytic pathway regulation concurrently in the tumor and peripheral organs impacted by cachexia-inducing tumor load. Candidate mechanisms meriting closer examination include transport proteins regulating glycolytic metabolite flux, of which the glucose and monocarboxylate transporter families feature prominently in cancer but have been given limited attention in cachexia24,25. Therefore, the purpose of this investigation was to examine glycolytic pathway metabolites, enzyme activity, and transporter proteins in liver, skeletal muscle, and tumor from mice with varying degrees of cancer cachexia severity, and their relationships with cachexia-related weight loss.
Methods
Animals
Ten-week-old Balb/c males (Envigo) were randomly assigned to receive either an injection of sterile PBS or cachexia-inducing colon-26 (C26) cancer cells. Tissues were collected from C26 tumor bearing mice between days 14-21 after injection of cancer cells, and classified by the degree of weight loss in accordance with previous literature evaluating mechanisms of cancer cachexia pathology16,26. This design yielded four experimental groups, including PBS injected mice that are non-tumor bearing and weight-stable (WS, n=4); C26 tumor bearing mice that had palpable tumors but did not undergo weight loss and classified as weight-stable (TB-WS, n=6); C26 tumor bearing mice that underwent 10% weight loss and classified with moderate cachexia (Mod, n=7); and C26 mice with ≥20% weight loss, categorized with severe cachexia (Sev, n=6). To determine weight loss for each mouse, the percentage change was calculated between carcass weight and body weight recorded on the day C26 cells were injected. Mice were individually housed, provided ad libitum water and food (5L0D, protein 29% fat 13%, carbohydrate 58%, PicoLab Laboratory Rodent Diet), and maintained on a 12:12 hr light:dark cycle. Approval by the Institutional Animal Care and Use Committee (#A16-39) was obtained before any experiments were conducted. Animal characteristics, myofiber size, mitochondrial function, and hepatic proteome analysis from this cohort of mice has been reported previously18,21. All data reported here have not been published.
Culture and injection of colon-26 cancer cells
Cells were cultured in a humidified incubator with 5% CO2 using completed media consisting of RPMI 1640 with 1% penicillin/streptomycin (vol/vol) and 10% FBS (vol/vol). Cancer cells were obtained commercially from CLS Cell Lines Service (Eppelheim, Germany). Culture media was replaced with fresh media every two to three days. For harvesting, cells were incubated with trypsin (0.05%, Gibco) and pelleted by centrifugation, the supernatant removed and remaining pellet resuspended in PBS. Cells were identified and counted with a hemocytometer by trypan blue staining and light microscopy. For mice assigned to tumor bearing groups, cells were injected subcutaneously in the upper back with a suspension containing 1 million cells, while non-tumor bearing mice received an equivalent volume of sterile PBS27,28.
Tissue collection
Euthanasia was performed by overdose with an intraperitoneal injection of ketamine/xylazine cocktail (300/30 mg/kg) during a four-hour time window beginning at 10:00 am. This timeframe ensured consistency in the timing of tissue collection. Body weight was monitored to reach the desired 10% and 20% weight loss in moderate and severe cachexia. Final tumor mass at euthanasia was anticipated at roughly 1g with moderate cachexia and target weight loss of 10%. For severely cachexic mice with 20% weight loss, final tumor mass of approximately 2g was projected based on our prior work in this model as well as reports by other users of C26 mice28–30. These outcomes were considered during routine surveillance because body weight measurements made before euthanasia would be confounded by tumor weight. During routine monitoring, a mouse showing ~7% weight loss would be euthanized in anticipation of tumor mass accounting for ~3-4% of body weight (1g tumor mass would represent ~3-4% of body weight in 25g Balb/c male). This allowed target weight loss of 10% for the moderate cachexia group (Mod), with tissue obtained on day 14 (n=4), 15 (n=1), 17 (n=1), and 21 (n=1). For TB-WS mice, tissue was collected on day 14 (n=1), 20 (n=2), and 21 (n=3). For severely cachexic mice (Sev, n=6), tissue was collected 21 days after tumor cell injection. Mice were not food deprived overnight or immediately prior to tissue collection. Collected tissue samples were weighed, sectioned, snap frozen and stored at −80°C for biochemical and immunoassays.
Tissue homogenate preparation
Liver, skeletal muscle, and tumor homogenates were prepared as previously described18. The gastrocnemius was used for assays involving skeletal muscle. Following animal experiments, all three tissues were rapidly excised, and rinsed with the ice-cold homogenization buffer (215 mM mannitol, 75 mM sucrose, 0.1% BSA, 20 mM HEPES, and 1 mM EGTA; pH adjusted to 7.2 with KOH). For tissue homogenate preparation, each tissue portion was homogenized using a manual Potter-Elvehjem tissue grinder containing 1 ml of ice-cold homogenization buffer. The tissue homogenate was stored immediately at −80°C until further analysis for biochemical assays and Western blotting. The protein concentration of tissue homogenates were quantified by BCA protein assay kit (Pierce, Rockford, IL, USA).
Glycolytic metabolites and enzyme activity
All biochemical assays in tissue homogenates were carried out using commercially available kits (Abcam Inc., Cambridge, MA) and followed the manufacturer’s instructions. The tissue metabolites including L-lactate (cat# ab65330) and pyruvate (cat# ab65342) were measured using fluorometric assay kits, and the ratio of L-lactate to pyruvate was then calculated and analyzed. Concentrations of NAD+ and NADH were measured by fluorometric assay kit (cat # ab176723), and the ratio of NAD+/NADH analyzed. Lactate dehydrogenase activity (LDH: cat# ab197000) was measured by fluorometric assay kit, and pyruvate dehydrogenase activity (PDH: cat# ab109902) was determined kinetically at 450 nm using monoclonal antibody pre-coated microplate colorimetric assay kit. Sample readings were normalized to protein concentration of the appropriate sample homogenate. A BioTek Synergy HTX multi-mode 96-well microplate reader (Winooski, VT) was used for fluorescence assays at Ex/Em = 530/590 nm.
Western blotting
To prepare samples for Western blotting, 30 μg of each protein sample was mixed with Laemmli sample buffer (cat# 1610747, Bio-Rad) containing 2-mercaptoethanol and then electrophoresed in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), using 4–20% Criterion™ TGX™ Precast gels (cat# 5671095, Bio-Rad). The gel was run at 140 V current for 100 min in Tris/Glycine/SDS buffer, pH8.3 (cat# 1610772, Bio-Rad). Subsequently, the separated proteins were transferred onto a PVDF membrane (0.2 μm size) at 75 V current for 90 min using ice-cold Tris/Glycine buffer, pH 8.3 (cat# 1610771, Bio-Rad) containing 20% methanol. Following electrotransfer, the PVDF membrane was blocked with 5% nonfat dry milk in TBST buffer, pH 7.4 containing 0.1% Tween-20 (Tris-buffered saline, cat# 35108649, Quality Biological) for 1 hour at room temperature. The membrane was then incubated with primary antibodies against mouse polyclonal to monocarboxylic acid transporter 1 (MCT1, 1: 2500 dilution, cat# ab90582, Abcam Inc.), rabbit polyclonal to glucose transporter 1 (GLUT1, 1:1000 dilution, cat# NB110-39113, Novus Biologicals), and rabbit polyclonal to glucose transporter 2 (GLUT2, 1:1000 dilution, cat# NBP2-22218, Novus Biologicals) at 4°C in 5% milk overnight. A rabbit monoclonal to GAPDH (1:5000 dilution, cat# 5174, Cell Signaling Inc.) was used as an internal protein loading control. After overnight incubation, the membrane was washed 3 times with TBST, and then incubated with corresponding horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hour period at room temperature. Subsequently, the membrane was washed 3 times in TBST and then incubated for 5 minutes in an enhanced chemiluminescent (ECL) horseradish peroxidase (HRP) substrate solution (cat# PI34580, Thermo Fisher) for protein detection. Reactive bands were identified using ChemiDocTM XRS+ imager with Image Lab™ software (Bio-Rad). The band density was quantified using ImageJ (NIH) software.
Statistical Analysis
All data are expressed as mean ± SEM. GraphPad Prism 7.04 software was used for statistical analyses. For comparisons, one-way ANOVA followed by Tukey’s multiple comparisons test was performed to compare differences between groups. The significance threshold was set at P < 0.05.
Results
Altered [lactate]/[pyruvate] in liver, tumor and muscle of cachexic mice
To assess how major metabolites of the glycolytic pathway are altered in cachexia, we assayed pyruvate, and L-lactate concentrations in liver, tumor and muscle homogenates, and analyzed the ratio of lactate to pyruvate (Fig. 1). In the liver and tumor, [lactate]/[pyruvate] was decreased in moderate and severe cachexia (Fig. 1A). In contrast, skeletal muscle showed a general increase in [lactate]/[pyruvate] in cachexia. Together, [lactate]/[pyruvate] ratio was significantly reduced in liver and tumor of moderate and severe cachexic mice, while it tended to rise in skeletal muscle (P=0.113-0.128 vs. TB-WS) (Fig. 1A). Collectively this suggests increased glycolytic flux in muscle, with net accumulation of lactate due to impairment of downstream oxidative pathways in cachexic skeletal muscle (e.g. inhibition of pyruvate dehydrogenase complex or, as reported previously, reduced mitochondrial respiratory capacity18), but with increased rates of lactate oxidation in liver and tumor.
Figure 1. Glycolytic pathway metabolites in liver, skeletal muscle and tumors of mice with cancer cachexia.
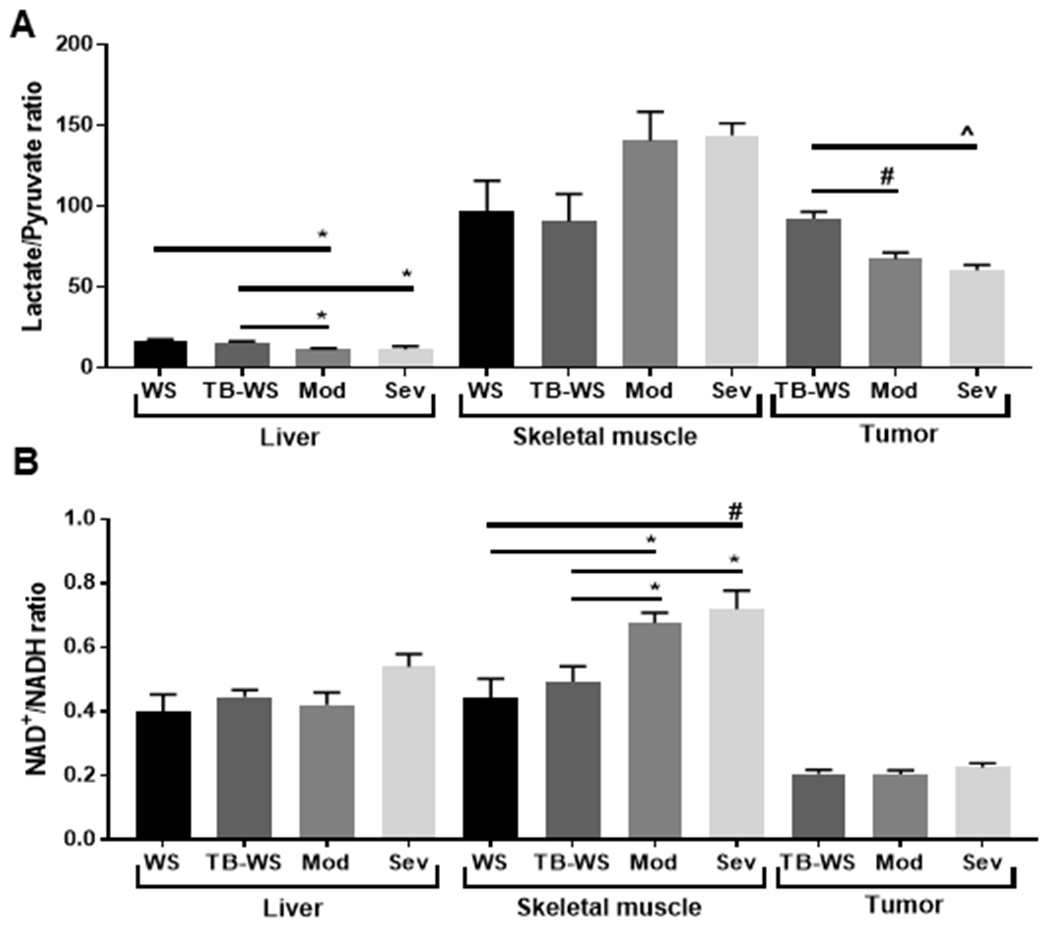
(A) Ratio of L-lactate to pyruvate. An increased ratio suggests respiratory limitations. (B) Ratio of NAD+/NADH, an indicator of the Warburg effect and glycolytic demand. Metabolites were measured in tissue lysates by fluorometry. Data shown as mean±SE. WS=PBS-injected weight-stable mice (n=4); TB-WS=colon-26 (C26) tumor bearing mice that are weight-stable (n=6); Mod=C26 mice with moderate cachexia (10% weight loss) (n=7); Sev=C26 mice with severe cachexia (20% weight loss) (n=6). Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test. Comparisons were made between groups within the same tissue type. P<0.05*; 0.01#; 0.001^.
NAD+/NADH is unaffected in liver and tumor, but increased in cachexic muscle
We next assayed NAD+ and NADH, the oxidized and reduced forms of NAD, and then analyzed the ratio of NAD+/NADH. NADH is generated during the catabolism of glucose and its production is increased with increased glycolytic flux31,32. NAD+ is formed when pyruvate is reduced to lactate, and this regenerated NAD+ is a substrate for G3P dehydrogenase higher in glycolysis31. A high NAD+/NADH ratio is indicative of the Warburg effect and reflects high glycolytic demand32. NAD+/NADH was not altered in liver and tumor tissue (Fig. 1B), suggesting that high rate glycolysis is not occurring in cachexia-inducing C26 tumors or the cachexic liver. In skeletal muscle, NAD+/NADH was significantly greater in cachexic muscle than in weight-stable animals (Fig. 1B). This is consistent with high rates of lactate production and regenerated NAD+ to support a high glycolytic demand.
Unaltered enzyme activity of LDH in cachexic liver and tumor despite reduced [lactate]/[pyruvate]
To determine whether altered [lactate]/[pyruvate] related to enzyme activity, we measured LDH and PDH activities in tissue homogenates. Muscle LDH activity significantly increased across cachexia severity (Fig. 2A). These increases in muscle LDH activity are consistent with the greater muscle [lactate]/[pyruvate] in cachexic mice (Figs. 1A). Muscle PDH also generally increased across tumor bearing mice, but this only reached statistical significance in moderate cachexia compared to saline controls (Fig. 2B). The tendency toward greater [lactate]/[pyruvate] in cachexic muscle (Fig. 1A), coupled with increased LDH activity, implies high glycolytic flux and lactate production that outstrips the rate of pyruvate transport across the mitochondrial membrane to supply the TCA cycle. In tumors, LDH activity was not altered (Fig. 2A), despite the observed reduction in tumor [lactate]/[pyruvate] in cachexic mice (Fig. 1A). Activity of tumor PDH decreased in severe cachexia only (Fig. 2B) also despite lower [lactate]/[pyruvate] (Fig. 1A). These findings are consistent with increased lactate tumor clearance (e.g. increased efflux) and/or lactate oxidation and intramitochondrial transport - potentially to support increased production of TCA cycle intermediates required for anabolism (a “reverse Warburg effect”). In liver, neither LDH nor PDH activity were affected by cachexia (Fig. 2A,B), despite lower liver [lactate]/[pyruvate] in cachexic mice (Fig. 1A). Similar to tumors, unchanged hepatic LDH suggests another mechanism is needed to account for reduced [lactate]/[pyruvate] in the liver, such as enhanced efflux or high gluconeogenic flux.
Figure 2. LDH activity is unaffected in liver and tumor despite reduced lactate, but increased in cachexic skeletal muscle.
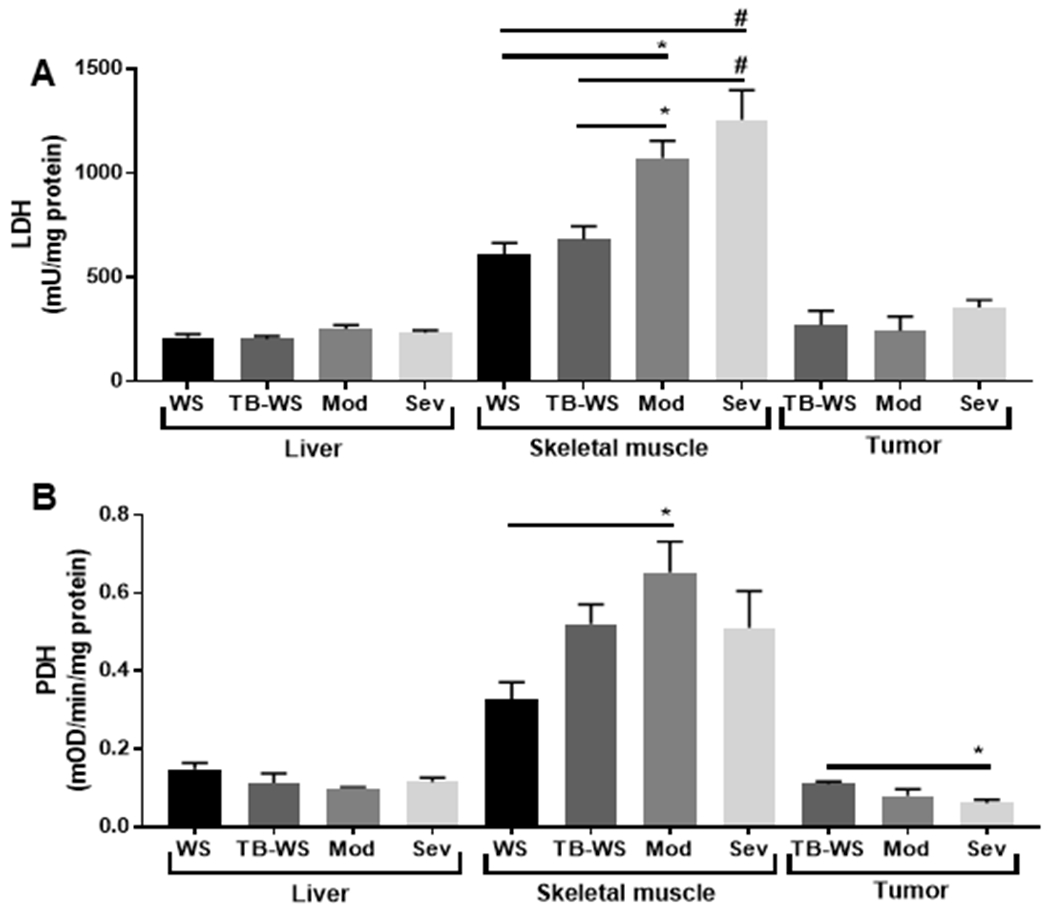
(A) Lactate dehydrogenase (LDH) activity measured in tissue lysates by fluorometry. (B) Pyruvate dehydrogenase (PDH) activity measured in tissue lysates by colorimetry. Data shown as mean±SE. WS=PBS-injected weight-stable mice (n=4); TB-WS=colon-26 (C26) tumor bearing mice that are weight-stable (n=6); Mod=C26 mice with moderate cachexia (10% weight loss) (n=7); Sev=C26 mice with severe cachexia (20% weight loss) (n=6). Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test. Comparisons were made between groups within the same tissue type. P<0.05*; 0.01#.
Metabolite transporter expression is altered in liver, tumor and muscle of cachexic mice
[Lactate]/[pyruvate] was significantly lowered in liver and tumor of cachexic mice (Figs. 1A). Because there were no changes LDH activity (Fig. 2A), we sought to establish the expression of monocarboxylate transporter-1 (MCT1), a major member of the monocarboxylate transporter family that regulates influx and efflux of lactate. In liver, MCT1 expression was significantly increased in moderate and severe cachexia compared to weight-stable mice (Fig. 3A–B), with no differences between the two cachexic groups. This induction of liver MCT1 suggests that active export or import of lactate in hepatocytes is a general feature of cancer cachexia. Although it has been suggested that the liver can function as a lactate producing organ under certain conditions33, import by MCT1 and subsequent oxidation is most probable given its established role as a major site of lactate consumption via gluconeogenesis.
Figure 3. Increased lactate transporter MCT1 but unaltered glucose transporters GLUT1 or GLUT2 in cancer cachexic liver.
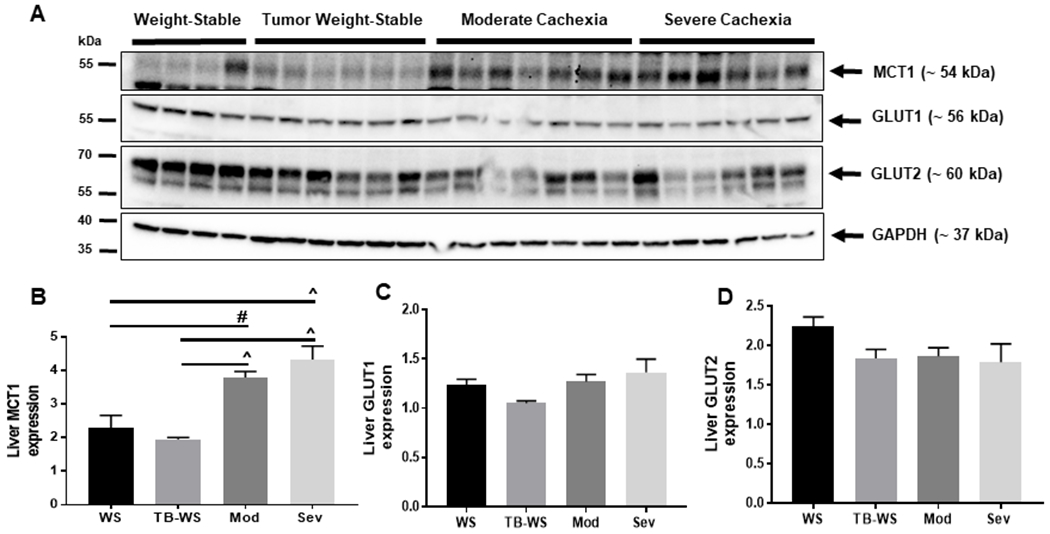
(A) Blots of monocarboxylate and glucose transporters in liver tissue lysates from moderate and severely cachexic mice. (B) Monocarboxylate transporter-1 (MCT1). (C) Glucose transporter-1 (GLUT1). (D) Glucose transporter-2 (GLUT2). Data shown as mean±SE. WS=PBS-injected weight-stable mice (n=4); TB-WS=colon-26 (C26) tumor bearing mice that are weight-stable (n=6); Mod=C26 mice with moderate cachexia (10% weight loss) (n=7); Sev=C26 mice with severe cachexia (20% weight loss) (n=6). Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test. P<0.01#; 0.001^.
We then determined the expression of glucose transport members GLUT1 and GLUT2. GLUT1 is ubiquitously expressed and found in most cell types where it mediates basal glucose uptake due to its low Km and high affinity for glucose34. Induction of GLUT1 occurs in response to cellular stress, such as glucose deprivation, to increase glucose transport. GLUT2 is low affinity (higher Km) and high capacity, abundantly expressed in liver, and allows bidirectional flux of glucose35. Cachexia had no effect on expression of either GLUT1 or GLUT2 in the liver (Figs. 3C,D), suggesting that glucose transport is not altered in the cachexic liver, and that glucose flux is adequately supported by these transporters.
Skeletal muscle is also major site of glycolytic metabolism and substrate shuttling, known for its participation in the Cori cycle in which muscular production of lactate is released and taken up by liver for gluconeogenesis. In cachexic muscle, [lactate]/[pyruvate] was high (Fig. 1A), and increased MCT1 would be expected to accommodate muscular release into circulation for shuttling to other organs. We found muscle MCT1 expression was significantly increased in all C26 tumor-bearing groups compared to saline controls (Figs. 4A,B), implying increased lactate efflux from muscle in response to tumor burden. Muscle GLUT1 expression was significantly decreased in severe cachexia compared to weight-stable mice (Fig. 4C). Because muscle GLUT1 is known for glucose uptake under basal conditions, this suggests limited GLUT1-mediated glucose transport into skeletal muscle of tumor-bearing mice. We also cannot exclude compensatory uptake by other transporters abundantly expressed in muscle such as GLUT4. Interestingly, muscle GLUT2, an isoform not abundantly expressed in skeletal muscle, was significantly increased in weight-stable and moderately cachexic C26 mice compared to saline controls, but unchanged in severe cachexia (Fig. 4D).
Figure 4. Increased MCT1 protein expression in skeletal muscle of cachexic mice.
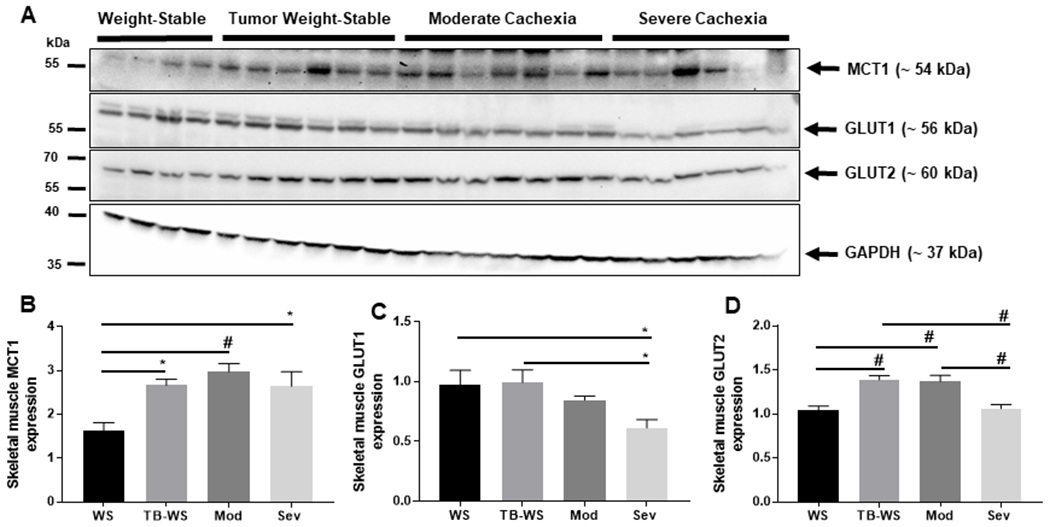
(A) Blots of monocarboxylate and glucose transporters in colon-26 skeletal muscle lysates from moderate and severely cachexic mice. (B) Monocarboxylate transporter-1 (MCT1). (C) Glucose transporter-1 (GLUT1). (D) Glucose transporter-2 (GLUT2). Data shown as mean±SE. WS=PBS-injected weight-stable mice (n=4); TB-WS=colon-26 (C26) tumor bearing mice that are weight-stable (n=6); Mod=C26 mice with moderate cachexia (10% weight loss) (n=7); Sev=C26 mice with severe cachexia (20% weight loss) (n=6). Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test. P<0.05*; 0.01#.
In contrast to liver, tumor MCT1 expression was not altered by cachexia (Fig. 5A–B), implying that bulk uptake or release of lactate is not occurring to a significant extent through MCT1 in cachexia-inducing C26 tumors. Alternatively, basal levels of tumor MCT1 may be sufficient to accommodate the required lactate transport in transformed cells. We also cannot rule out the possibility that transport is mediated by other MCT members induced in malignant tissues such as MCT2 and MCT425,36. In tumors, GLUT1 expression was significantly increased in mice with severe cachexia (Fig. 5C). This induction of GLUT1 suggests adaptation by the tumor to facilitate glucose uptake, possibly sourced from hepatic gluconeogenesis. In contrast, tumor GLUT2 significantly decreased in severe cachexia (Fig. 5D). Together this suggests enhanced glucose transport into the cachexia-inducing C26 tumor through a GLUT-1 dependent mechanism.
Figure 5. Increased tumor expression of high affinity glucose transporter GLUT1 but unaltered MCT1 in cancer cachexia.
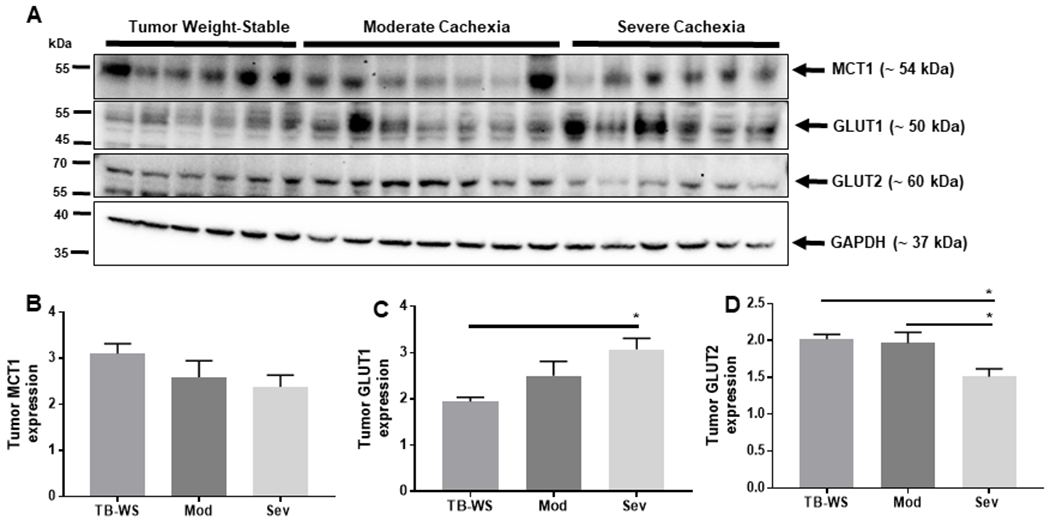
(A) Blots of monocarboxylate and glucose transporters in colon-26 tumor tissue lysates from moderate and severely cachexic mice. (B) Monocarboxylate transporter-1 (MCT1) was detected in colon-26 tumors, but not differentially affected by cachexia. (C) Glucose transporter-1 (GLUT1). (D) Glucose transporter-2 (GLUT2). Data shown as mean±SE. WS=PBS-injected weight-stable mice (n=4); TB-WS=colon-26 (C26) tumor bearing mice that are weight-stable (n=6); Mod=C26 mice with moderate cachexia (10% weight loss) (n=7); Sev=C26 mice with severe cachexia (20% weight loss) (n=6). Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test. P<0.05*.
Discussion
This study examined glycolytic pathway metabolites, enzyme activity, and transport protein expression in liver, muscle, and tumor from mice with cancer cachexia. We provide evidence of altered glycolytic and lactate metabolites, enzyme activity, and transporter protein expression in all three tissues during cachexia. Changes in these three energy-regulating tissues suggest ongoing substrate shuttling that may contribute to tumor growth, energetic inefficiency and therefore, unintended weight loss in cachexia (Fig. 6).
Figure 6. Hypothesized substrate shuttles among liver, skeletal muscle and tumor that may contribute to cachexia-associated weight loss.
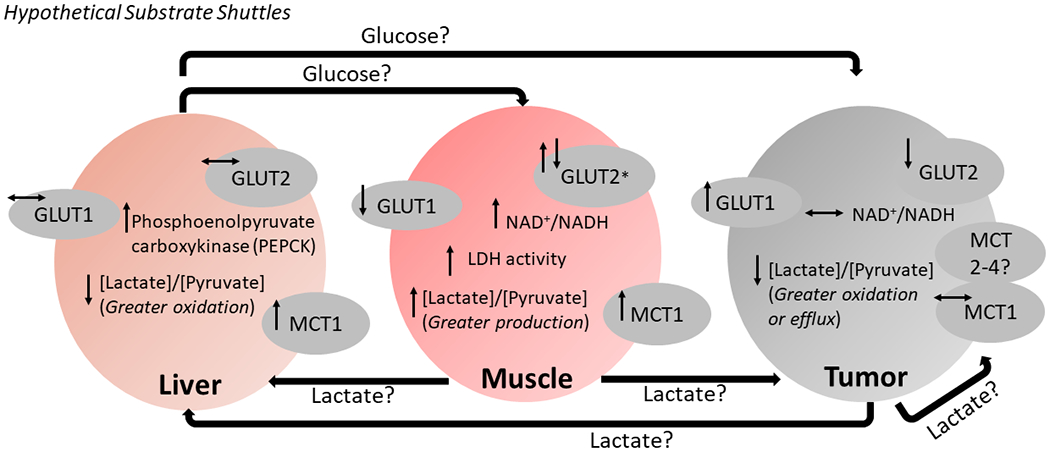
The classical Cori cycle that involves shuttling of lactate and glucose between skeletal muscle and liver appears to be active in mice with cancer cachexia. Increased lactate dehydrogenase (LDH) activity, [L-lactate]/[pyruvate], and lactate transporter MCT1 is supportive of enhanced lactate production by cachexic skeletal muscle (2 ATP yield), release into circulation and shuttling to the liver. Livers of cachexic mice had increased MCT1, suggesting lactate uptake into the liver for use in gluconeogenesis (6 ATP cost). In our prior proteome analysis, cachexic livers showed increased phosphoenolpyruvate carboxykinase (PEPCK) in this same cohort of tumor bearing mice21. PEPCK catalyzes a rate-limiting step of gluconeogenesis, therefore, increased PEPCK suggests maintained drive for hepatic gluconeogenesis. Glucose generated by gluconeogenesis can be used as energy by muscle, however decreased glucose transporter expression and ongoing accumulation of glycolytic metabolites may impair uptake and use. Another form of Cori cycle may involve lactate release from the tumor, shuttling to the liver, gluconeogenesis, and shuttling of glucose for use by the tumor. Both muscle-liver and tumor-liver shuttles would be energetically costly, with persistent shuttling possibly leading to unintended weight loss in cachexia. Another possible inter-organ substrate exchange could occur between muscle and tumor, with high production and release of muscular lactate shuttled to the energetically demanding tumor. Thus, muscle-derived lactate may serve as an important source of energy for tumors that induce subsequent cachexia. (↑, ↓) Arrows adjacent to a given metabolite or transport protein represent statistically significant comparisons for moderate and/or severe cachexia vs. weight-stable mice (PBS injected or tumor bearing). *Muscle GLUT2 initially increased in tumor-bearing weight stable and moderately cachexic mice compared to saline controls, but decreased in severe cachexia back to saline control levels.
The Cori Cycle described in 1929 by Gerty Cori and Carl Cori37, explains how lactate produced by skeletal muscle is shuttled to the liver and used as a gluconeogenic substrate. Glucose generated from hepatic gluconeogenesis can then be used for skeletal muscle bioenergetics. This cycle is energetically inefficient due to the cost of hepatic gluconeogenesis (6 ATP) relative to energy yield by production of lactate from glycolysis in muscle (2 ATP), but allows muscle activity to be maintained in conditions of extreme energetic stress. Our data are consistent with this Cori cycle between skeletal muscle and liver in weight-losing cachexic mice. Concurrently increased [lactate]/[pyruvate] and LDH activity occurred only in muscle of cachexic mice and not any other tissues (Figs. 1–2), along with reduced mitochondrial oxidative capacity18 implying high production of muscular lactate that is released to serve as a gluconeogenic substrate in the liver. Significantly increased expression of the lactate transporter MCT1 in cachexic muscle is supportive of this inference (Fig. 4). In the liver, cachexic mice had increased lactate transporter MCT1 (Fig. 3) and decreased [lactate]/[pyruvate] (Fig. 1) but no change in glucose transporters GLUT1 or GLUT2 (Fig. 3). These hepatic events suggest active uptake of lactate as a preferred substrate, oxidation into pyruvate and conversion to glucose by gluconeogenesis, with glucose export accommodated by the basal function of GLUT transporters. Ongoing hepatic gluconeogenesis is supported by our prior proteome analysis of cachexic livers from this same cohort of mice21. The differentially expressed protein list revealed that liver phosphoenolpyruvate carboxykinase (PEPCK) was increased in all C26 tumor bearing mice (both weight-stable and cachexic) compared to PBS injected controls (fold-change 1.56-2.41)21. PEPCK catalyzes an irreversible step of gluconeogenesis, therefore increased PEPCK implies sustained drive for hepatic gluconeogenesis associated with a C26 tumor load. Collectively, these data imply that the classical Cori cycle between muscle and liver may be ongoing in mice with cachexia, and that persistent substrate shuttling between these organs may at least partly contribute to systemic energy balance disruption and unintended weight loss in cachexia. We note that in our mitochondrial phenotyping experiments from this same cohort of mice, oxidative phosphorylation was impaired in cachexic skeletal muscle18. Defective oxidative metabolism likely contributes to the high muscular [lactate]/[pyruvate] reported in the present work (Fig. 1). It would also compound energetic stress experienced by cachexic muscle due to an inability to fully catabolize glucose by aerobic glycolysis, when supplied by hepatic gluconeogenesis during energetically inefficient substrate shuttling.
A second form of Cori cycle involving tumor and liver is often described in mechanistic frameworks of cancer cachexia12–14,38,39. In this substrate cycle, lactate produced by high intratumor glycolytic flux is released and shuttled to the liver, where uptake and conversion by gluconeogenesis may further supply transformed tissues with glucose to meet high energy demand. We found that in tumors from cachexic mice, GLUT1 increased (Fig. 5), suggesting active glucose uptake and oxidation. No change in intratumor NAD+/NADH suggests that high rate glycolytic demand may not necessarily be a prominent feature of cachexia-inducing C26 tumors (Fig. 1B). We also report that tumor content of MCT1 was not affected during cachexia (Fig. 5B). This suggests several possibilities including: 1) limited lactate transport in tumors by MCT1 during cachexia; or 2) lactate transport is adequately supported by the basal function of MCT1 proteins already expressed in tumors. We also cannot exclude the reasonable possibility that lactate flux is mediated by other MCT family members known to be expressed in transformed tissues such as MCT4, which mostly exports lactate25,36,40. We did find decreased intratumor [lactate]/[pyruvate] in cachexic mice (Fig. 1), implying ongoing lactate oxidation to supply TCA cycle intermediates involved in anabolism in the tumor and/or clearance of lactate41. We note that increased tumor GLUT1 is associated with poor survival in patients42. Here, tumor GLUT1 showed a step-wise increase as cachexia worsened, with abundance greatest in severely cachexic mice (Fig. 5C). Elevated GLUT1 in transformed tissues may therefore be an informative marker that reflects ensuing cachexia, shortened survival and overall poor prognosis.
Other possible inter-organ substrate shuttling may be associated with cancer cachexia pathology (Fig. 6). These substrate shuttles may not constitute a cycle, but provide possible sources of energy for the tumor at the expense of the host, and thus contributes to tumor-induced cachexia. Compelling evidence suggests that tumors exhibit propensity to use lactate as a source of energy. In patients with non-small-cell lung cancer, infusion of radiolabeled lactate subsequently revealed a significant amount of labeled TCA cycle metabolites, indicating oxidation of lactate and entry into the TCA cycle in the tumor43. When MCT1 was deleted in tumors of mice, metabolites derived from labelled lactate were eliminated, supportive of active lactate uptake by tumors43. It was suggested therefore, that lactate rather than glucose may be the predominant source of TCA cycle precursors for some tumors43. In the present work, muscle [lactate]/[pyruvate] and MCT1 were significantly increased in cachexic mice (Figs. 1,4), implying high production and release of lactate by skeletal muscle. Given that lactate is a significant source of carbon for the TCA cycle, it is conceivable that a lactate shuttle exists between skeletal muscle and tumor, where muscular lactate is metabolized directly by the tumor in addition to the liver in the Cori cycle. In this instance, skeletal muscle-derived lactate may in fact be an important source of carbons for cachexia-inducing tumors at the expense of the host, which has been termed a “reverse Warburg effect”44. We noted that in our previous work, mitochondrial oxidative phosphorylation was impaired in skeletal muscle from the same cohort of cachexic mice reported here18. An enticing possibility exists in which therapeutic approaches that improve muscle oxidative phosphorylation may not only improve muscle function and mitigate weakness in patients, but also reduce excessive muscular lactate production, thereby potentially limiting tumor growth. Lastly, tumor-derived lactate may be released and shuttled to adjacent cancer cells, stromal cells, and other cells in the tumor microenvironment to support malignant growth40, and thus cachexia onset and/or progression.
In conclusion, this study examined tissue-specific alterations in glycolytic metabolism during tumor load and cachexia. We propose that several substrate shuttles among liver, skeletal muscle, and tumor may contribute to energy balance disruption and unintended weight loss in cancer cachexia. Liver and skeletal muscle demonstrated changes suggestive of an ongoing, energetically costly Cori cycle during cachexia that could contribute to weight loss. Furthermore, tumor tissue showed high metabolic demand as evidenced by upregulation of glucose transporters to harness energy precursors such as glucose as well as lactate (a “reverse Warburg effect”) from storage tissues (i.e. liver, skeletal muscle) for self-propagation; thus, contributing to cachexia in the host. Therapeutic strategies that seek to normalize abnormal metabolism and substrate shuttling among energy-regulating tissues may alleviate and slow involuntary weight loss in cancer cachexia. Candidates to consider are interventions that restrict nutrients for malignant tissues and inhibit intratumor hypermetabolism, thereby reducing the development of abnormal substrate cycling with other organs and mitigating unintended weight loss.
Supplementary Material
Acknowledgements
We extend our sincere thanks to Ms. Peggy Donnelly and Ms. Denise Merrill for administrative support. AVK is supported by the Transdisciplinary Research in Energetics and Cancer (TREC) Training Workshop R25CA203650 (PI: Melinda Irwin). HBR is supported by grants from NIH (R01HL15145, P50HD098593, R01DK122767, P2CHD086851) and the Tobacco Related Disease Research Program (T31IP1666).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Fearon K, Strasser F, Anker SD, et al. Definition and classification of cancer cachexia: an international consensus. The lancet oncology. 2011. [DOI] [PubMed] [Google Scholar]
- 2.von Haehling S, Anker SD. Cachexia as a major underestimated and unmet medical need: facts and numbers. J Cachexia Sarcopenia Muscle. 2010;1(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers-update 2014. J Cachexia Sarcopenia Muscle. 2014;5(4):261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vagnildhaug OM, Balstad TR, Almberg SS, et al. A cross-sectional study examining the prevalence of cachexia and areas of unmet need in patients with cancer. Support Care Cancer. 2018;26(6):1871–1880. [DOI] [PubMed] [Google Scholar]
- 5.Antoun S, Baracos VE, Birdsell L, Escudier B, Sawyer MB. Low body mass index and sarcopenia associated with dose-limiting toxicity of sorafenib in patients with renal cell carcinoma. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2010;21(8):1594–1598. [DOI] [PubMed] [Google Scholar]
- 6.Prado CM, Baracos VE, McCargar LJ, et al. Sarcopenia as a determinant of chemotherapy toxicity and time to tumor progression in metastatic breast cancer patients receiving capecitabine treatment. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(8):2920–2926. [DOI] [PubMed] [Google Scholar]
- 7.Arthur ST, Van Doren BA, Roy D, Noone JM, Zacherle E, Blanchette CM. Cachexia among US cancer patients. J Med Econ. 2016;19(9):874–880. [DOI] [PubMed] [Google Scholar]
- 8.Arthur ST, Noone JM, Van Doren BA, Roy D, Blanchette CM. One-year prevalence, comorbidities and cost of cachexia-related inpatient admissions in the USA. Drugs Context. 2014;3:212265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachmann J, Ketterer K, Marsch C, et al. Pancreatic cancer related cachexia: influence on metabolism and correlation to weight loss and pulmonary function. BMC cancer. 2009;9:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou X, Wang JL, Lu J, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142(4):531–543. [DOI] [PubMed] [Google Scholar]
- 11.Reid J, McKenna HP, Fitzsimons D, McCance TV. An exploration of the experience of cancer cachexia: what patients and their families want from healthcare professionals. European journal of cancer care. 2010;19(5):682–689. [DOI] [PubMed] [Google Scholar]
- 12.Hulmi JJ, Penna F, Pollanen N, et al. Muscle NAD(+) depletion and Serpina3n as molecular determinants of murine cancer cachexia-the effects of blocking myostatin and activins. Mol Metab. 2020;41:101046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. 2016;5:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manring H, Abreu E, Brotto L, Weisleder N, Brotto M. Novel excitation-contraction coupling related genes reveal aspects of muscle weakness beyond atrophy-new hopes for treatment of musculoskeletal diseases. Front Physiol. 2014;5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visavadiya NP, Pena GS, Khamoui AV. Mitochondrial dynamics and quality control are altered in a hepatic cell culture model of cancer cachexia. Mol Cell Biochem. 2020. [DOI] [PubMed] [Google Scholar]
- 16.Bonetto A, Aydogdu T, Kunzevitzky N, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PloS one. 2011;6(7):e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goncalves MD, Hwang SK, Pauli C, et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc Natl Acad Sci U S A. 2018;115(4):E743–E752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halle JL, Pena GS, Paez HG, et al. Tissue-specific dysregulation of mitochondrial respiratory capacity and coupling control in colon-26 tumor-induced cachexia. Am J Physiol Regul Integr Comp Physiol. 2019;317(1):R68–R82. [DOI] [PubMed] [Google Scholar]
- 19.Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda). 2011;26(3):192–205. [DOI] [PubMed] [Google Scholar]
- 20.Wang F, Liu H, Hu L, et al. The Warburg effect in human pancreatic cancer cells triggers cachexia in athymic mice carrying the cancer cells. BMC Cancer. 2018;18(1):360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khamoui AV, Tokmina-Roszyk D, Rossiter HB, Fields GB, Visavadiya NP. Hepatic proteome analysis reveals altered mitochondrial metabolism and suppressed acyl-CoA synthetase-1 in colon-26 tumor-induced cachexia. Physiol Genomics. 2020;52(5):203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunzke T, Buck A, Prade VM, et al. Derangements of amino acids in cachectic skeletal muscle are caused by mitochondrial dysfunction. J Cachexia Sarcopenia Muscle. 2020;11(1):226–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pin F, Barreto R, Couch ME, Bonetto A, O’Connell TM. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J Cachexia Sarcopenia Muscle. 2019;10(1):140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ancey PB, Contat C, Meylan E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. Febs J. 2018;285(16):2926–2943. [DOI] [PubMed] [Google Scholar]
- 25.Payen VL, Mina E, Van Hee VF, Porporato PE, Sonveaux P. Monocarboxylate transporters in cancer. Mol Metab. 2020;33:48–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White JP, Baynes JW, Welle SL, et al. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PloS one. 2011;6(9):e24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diffee GM, Kalfas K, Al-Majid S, McCarthy DO. Altered expression of skeletal muscle myosin isoforms in cancer cachexia. Am J Physiol Cell Physiol. 2002;283(5):C1376–1382. [DOI] [PubMed] [Google Scholar]
- 28.Khamoui AV, Park BS, Kim DH, et al. Aerobic and resistance training dependent skeletal muscle plasticity in the colon-26 murine model of cancer cachexia. Metabolism. 2016;65(5):685–698. [DOI] [PubMed] [Google Scholar]
- 29.Acharyya S, Ladner KJ, Nelsen LL, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. The Journal of clinical investigation. 2004;114(3):370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, Crawford D, Hutchinson KR, et al. Myocardial dysfunction in an animal model of cancer cachexia. Life sciences. 2011;88(9-10):406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alameddine AK, Conlin FT, Binnall BJ, Alameddine YA, Alameddine KO. How do cancer cells replenish their fuel supply? Cancer Rep (Hoboken). 2018;1(1):e1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moreira JD, Hamraz M, Abolhassani M, et al. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites. 2016;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phypers B, Pierce JT. Lactate physiology in health and disease. Continuing Education in Anaesthesia Critical Care & Pain. 2006;6(3):128–132. [Google Scholar]
- 34.Chadt A, Al-Hasani H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflugers Arch. 2020;472(9):1273–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leturque A, Brot-Laroche E, Le Gall M, Stolarczyk E, Tobin V. The role of GLUT2 in dietary sugar handling. J Physiol Biochem. 2005;61(4):529–537. [DOI] [PubMed] [Google Scholar]
- 36.Kim HK, Lee I, Bang H, et al. MCT4 Expression Is a Potential Therapeutic Target in Colorectal Cancer with Peritoneal Carcinomatosis. Mol Cancer Ther. 2018;17(4):838–848. [DOI] [PubMed] [Google Scholar]
- 37.Cori CF, Cori GT. Glycogen formation in the liver from d- and l-lactic acid. Journal of Biological Chemistry. 1929;81:389–403. [Google Scholar]
- 38.Woo JS, Hwang JH, Huang M, et al. Interaction between mitsugumin 29 and TRPC3 participates in regulating Ca(2+) transients in skeletal muscle. Biochem Biophys Res Commun. 2015;464(1):133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Correll RN, Lynch JM, Schips TG, et al. Mitsugumin 29 regulates t-tubule architecture in the failing heart. Sci Rep. 2017;7(1):5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodwin ML, Gladden LB, Nijsten MW, Jones KB. Lactate and cancer: revisiting the warburg effect in an era of lactate shuttling. Front Nutr. 2014;1:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab. 2020;2(2):127–129. [DOI] [PubMed] [Google Scholar]
- 42.Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol. 2005;202(3):654–662. [DOI] [PubMed] [Google Scholar]
- 43.Faubert B, Li KY, Cai L, et al. Lactate Metabolism in Human Lung Tumors. Cell. 2017;171(2):358–371 e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pavlides S, Whitaker-Menezes D, Castello-Cros R, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.