

SUMMARY
Negative elongation factor (NELF) is a critical transcriptional regulator that stabilizes paused RNA polymerase to permit rapid gene expression changes in response to environmental cues. Although NELF is essential for embryonic development, its role in adult stem cells remains unclear. In this study, through a muscle-stem-cell-specific deletion, we showed that NELF is required for efficient muscle regeneration and stem cell pool replenishment. In mechanistic studies using PRO-seq, single-cell trajectory analyses and myofiber cultures revealed that NELF works at a specific stage of regeneration whereby it modulates p53 signaling to permit massive expansion of muscle progenitors. Strikingly, transplantation experiments indicated that these progenitors are also necessary for stem cell pool repopulation, implying that they are able to return to quiescence. Thus, we identified a critical role for NELF in the expansion of muscle progenitors in response to injury and revealed that progenitors returning to quiescence are major contributors to the stem cell pool repopulation.
Graphical abstract
In brief
NELF is a general transcription regulator thought to contribute ubiquitously to gene expression. Robinson et al. demonstrate that NELF plays a highly specific role in muscle regeneration, where it integrates extracellular signaling to control the number of muscle progenitor cells available for myofiber repair and stem cell niche repopulation.
INTRODUCTION
The ability of somatic stem cells to rapidly modify gene expression is essential for the recovery of damaged tissue. In the case of skeletal muscles, a tissue resident adult stem cell (MuSC), termed the satellite cell, is responsible for mediating the response to muscle injury (Aziz et al., 2012; Mauro, 1961). MuSC-mediated regeneration is a tightly coordinated process that requires properly timed responses to cellular signals to ensure efficient repair of myofibers and repopulation of the stem cell pool (Wosczyna and Rando, 2018). These include signals from the damaged muscle fibers (Hindi et al., 2012) as well as supporting cell populations including macrophages (Arnold et al., 2007), endothelial cells (Verma et al., 2018), and fibro-adipogenic progenitors (Joe et al., 2010). Recent studies, which focused on the isolation of RNA from quiescent cells, have revealed that MuSCs undergo a drastic change in gene expression in response to muscle injury that can be detected within 30 min of myofiber damage (Machado et al., 2017; van Velthoven et al., 2017). Although some insight into the signaling pathways utilized for cross-talk between these different cell populations has been elucidated (Wosczyna and Rando, 2018), we lack an understanding of the mechanisms used by MuSCs to integrate these signals and rapidly alter gene expression.
Promoter-proximal pausing is an important transcriptional regulatory process used to regulate RNA polymerase II (RNAPII) processivity (Adelman and Lis, 2012). Specifically, it was shown that after recruitment and initiation of transcription, RNAPII pauses approximately 25–50 bp downstream of the transcription start site (Luse, 2013). Although pause release is subsequently induced through the recruitment of P-TEFb in response to signals from the environment, the paused RNAPII is prone to arrest or premature termination (Adelman et al., 2005; Elrod et al., 2019). To prevent this, the negative elongation factor (NELF) complex is recruited to stabilize paused RNAPII (Henriques et al., 2013) through impeding RNAPII interactions with positive mediators of transcription elongation (Vos et al., 2018) and facilitating transcription re-initiation (Boettiger and Levine, 2009; Fant et al., 2020; Gilchrist and Adelman, 2010). As such, the loss of NELF leads to reduced RNAPII accumulation and transcription termination within the proximal-promoter region of its target genes (Core and Adelman, 2019). Initially identified on heat shock genes, transcriptional regulation through pausing is widely used at developmental genes for modulating transcriptional output in response to environmental stimuli (Muse et al., 2007; Zeitlinger et al., 2007).
Consistent with the importance of NELF in transcriptional regulation, targeted removal of NELF is lethal during the embryonic stage (Amleh et al., 2009). Interestingly, it has been shown that NELF regulates the spontaneous differentiation of cultured embryonic stem cells (Williams et al., 2015). However, we currently do not know whether NELF is involved in somatic stem cell-mediated tissue regeneration in vivo. In this study, using a MuSC-specific deletion of NELF-B, we showed that NELF is required for efficient regeneration of muscle tissue, whereby it mediates expansion of the muscle progenitor cell population in response to injury. Furthermore, these results led to the unanticipated finding that the expanded muscle progenitors can return to quiescence and play a major role in repopulating the stem cell pool.
RESULTS
NELF is required for the efficient regeneration of muscle after injury
The integrity and function of the NELF complex is dependent on all four of its constituent subunits—NELF-A, NELF-B, NELF-C/D, and NELF-E (Narita et al., 2003). To study the role of NELF-mediated transcript stabilization in MuSC-mediated muscle regeneration, we generated an inducible MuSC-specific deletion of NELF-B by crossing mice harboring a floxed NELF-B allele (Gupte et al., 2013) with mice expressing CreER from the Pax7 locus as a bicistronic transcript (Murphy et al., 2011). These mice were further crossed to integrate a ROSA26-lox-Stop-lox TdTomato (TdT) allele that yielded the desired NELF-BscKO mice that can monitor the efficiency of tamoxifen-induced excision by CreER (Figure 1A). Western blot analysis of MuSCs isolated from the tamoxifen-induced mice showed a 90% reduction in NELF-B protein (Figure 1B).
Figure 1. NELF is required for efficient MuSC-mediated myofiber repair after injury.

(A) Schematic representation of the recombination strategy. Tamoxifen administered in vivo activates CreERT2 to excise floxed genomic sequences. This is followed with intramuscular injection(s) of cardiotoxin to induce skeletal muscle damage.
(B) Western blot of a MuSCs whole cell extract isolated from WT (Pax7CreER/TdTscKO) and NELF-BscKO (NELF-BscKOPax7CreERTdTscKO) mice shows a 90% deletion efficiency of NELF-B.
(C) Hematoxylin and eosin stain of TA muscle cross-sections at 28 days post-injury (CTX), scale bar = 1 mm. The weight of the regenerated TA is presented relative to that of the undamaged contralateral leg and shows a reduction in size in the NELF-BscKO mice compared with WT controls (±SE, p-value < 0.01, n = 3).
(D) Magnified view of the regenerated and undamaged TA cross-sections shown in (C) demonstrate a reduced minimal Feret’s myofiber diameter in the regenerated NELF-BscKO myofibers (±SE, p-value < 0.0001, n = 3), scale bar = 50 μm.
(E) Single myofibers isolated from the regenerated EDL at 28 days post-injury were stained with DAPI to visualize nuclei from NELF-BscKO (n = 50 myofibers from 3 biological replicates) and WT (n = 57 myofibers from 3 biological replicates) mice, scale bar = 50 μm. Internuclear spacing is measured as the distance between the center of adjacent nuclei, whereas the myonuclei count was determined over the length of the myofiber (±SE, p-value < 0.0001).
(F) The relative abundance of newly regenerated myofibers in which the centrally located nucleus is captured in a 10 μm cross-section was calculated with tissues shown in (C) (±SE, p-value < 0.0001, n = 3).
Quiescent MuSCs are continuously activated where they fuse into myofibers to assure muscle homeostasis (Pawlikowski et al., 2015). Surprisingly, we observed that the loss of NELF-B caused no decrease in the number of MuSCs (marked by the transcription factor Pax7) when assayed 8 weeks after the deletion (Figures S1A and S1B). Similarly, histological examination of the tibialis anterior (TA) muscle showed no obvious pathology (Figure S1C). Thus, NELF is not required to maintain the quiescent state of MuSCs or to ensure muscle homeostasis.
To determine if NELF contributes to the regenerative capacity of MuSCs, we induced injury in the TA muscle using cardiotoxin (CTX). Twenty-eight days after the injury, the regenerated TA muscles of the NELF-BscKO mice showed myofiber hypotrophy with a 40% reduction in diameter and a ~50% reduction in weight compared with the wild-type (WT) mice (Figures 1C and 1D). Hematoxylin and eosin (H&E) staining of tissue cross-sections showed that MuSCs lacking NELF regenerated the injured TA muscle with a normal number of myofibers (Figure S1D), despite the significantly reduced diameter (Figure 1D). This reduced diameter in NELF-BscKO mice was already observed after 7 days of regeneration (Figures S1E and S1F). To address the origin of the myofiber defect, we examined the possibility that fewer nuclei could be incorporated into the newly regenerated fibers. Indeed, myofibers isolated from the extensor digitorum longus (EDL) muscle 28 days after injury showed a doubling of the spacing between the nuclei, which resulted from a reduced number of nuclei incorporated into the newly regenerated myofibers (Figure 1E). In the muscle cross-sections, this increased nucleus spacing significantly reduced the number of visible, centrally localized nuclei in the newly regenerated fibers (Figure 1F). Taken together, these results suggest that the muscle hypotrophy phenotype results from a reduced number of myonuclei per muscle fiber.
Loss of NELF induces premature differentiation of proliferating MuSCs
To better understand the mechanism underlying the regeneration defect in NELF-BscKO mice, we next sought to identify the specific stage(s) that is affected by NELF-B deletion during regeneration. Using TdT fluorescence to visualize MuSCs that have undergone Cre-mediated deletion of NELF-B (see Figure 1A), we examined the ability of MuSCs to transition from the quiescent to the activated state. Given that MuSCs undergo a first round of cell division 28 to 40 h after muscle injury (Rodgers et al., 2014; Siegel et al., 2011), we performed in vivo pulse labeling by injecting the deoxynucleotide analog, EdU, at 28 h and then sacrificing the mice at 40 h post-injury. The loss of NELF-B did not alter the number of MuSCs undergoing the first round of cell division within 40 h (Figures 2A and S2A). In an analogous experiment, mice were administered EdU at 48 h post-injury and were characterized for TA regeneration after 7 days. Again, the loss of NELF showed no effect on the number of myonuclei labeled with EdU (Figure S2B). This suggests that NELF is not required for MuSC activation.
Figure 2. NELF is required for the massive expansion of muscle progenitors in response to injury.

(A) FACS-acquired quantification of EdU+ MuSCs (identified as the TdT+/ITGA7+ population) derived from the skeletal muscle 40 h post-injury (±SE, n = 4).
(B) FACS-acquired quantification of EdU+ MuSCs (identified as the TdT+/ITGA7+ population) derived from the skeletal muscle 72 h post-injury to monitor in vivo myoblast proliferation (±SE, p-value < 0.0001, n = 5).
(C) Single myofibers isolated from the EDL muscle from NELF-BscKO (n = 47 from 3 biological replicates) or WT (n = 46 from 3 biological replicates) mice were cultured (0 or 72 h) and stained with antibodies as indicated, scale bar = 50 μm. Quantification of TdT+, EdU+, and Myog+ cells are shown (±SE, *p-value < 0.0001).
(D) Primary myoblasts isolated from NELF-BscKO and WT controls were plated at high density and induced to undergo terminal differentiation, scale bar = 100 μm. The differentiation index was calculated as the percentage of nuclei present in multinucleated myotubes (±SE, n = 3).
After completing a first cell division, MuSCs undergo rapid expansion to permit repair of the damaged muscle (Singh and Dilworth, 2013). To measure expansion of the MuSC population, mice were pulse labeled with EdU by i.p. injection at 60 h and then sacrificed at 72 h post-injury. Interestingly, we observed that MuSCs from NELF-BscKO mice displayed a large decrease in their proliferation capacity in vivo (Figure 2B). Similarly, when EdU was administered to mice 72 h post-injury, we observed a reduced number of myofibers containing EdU-labeled, centrally located nuclei when measured after 7 days of regeneration (Figure S2B). This suggests that in the absence of NELF, some of the MuSCs had already begun exiting the cell cycle and committed to terminal differentiation within the first 72 h of regeneration. Characterization of MuSC proliferation on ex vivo cultured myofibers showed a significant reduction in the incorporation of EdU in MuSCs that lacked NELF-B, resulting in fewer total MuSCs per fiber at 72 h post-isolation (Figure 2C). Similarly, we observed that NELF-B depletion (Figures S2C and S2D) resulted in a significant reduction in the number of MuSCs progressing through the cell cycle, which resulted in impaired expansion of the population in vitro (Figures S2E and S2F). These results suggest that in the absence of NELF-B, the muscle progenitor population cannot fully expand to repair the damaged myofibers completely.
The last step of muscle regeneration encompasses cell cycle exit and fusion of MuSCs to form multinucleated myofibers. The histological analysis of myofibers in the regenerated TA muscle suggested that MuSCs are able to efficiently undergo terminal differentiation and fuse into myofibers (Figure 1D). Consistent with the in vivo findings, MuSCs associated with myofiber explants were able to initiate differentiation as indicated by expression of the myogenic marker, Myog (Figure 2C). In fact, a higher percentage of the TdT+ MuSCs were positive for Myog in the absence of NELF at 72 h post-isolation suggesting an accelerated differentiation (Figure 2C). Finally, cultured MuSCs lacking NELF-B were able to efficiently form multinucleated myotubes (Figure 2D). Thus, the primary muscle regeneration defect in NELF-BscKO mice results from a decreased expansion of the MuSC population, which led to a reduced myofiber size.
Ablation of NELF-B leads to a depletion of the MuSC pool after muscle injury
After muscle injury, the pool of MuSCs must be replenished to ensure that the regenerative capacities are maintained throughout life. To determine whether NELF contributes to the replenishment of the MuSCs pool, we used a serial injury model in which the muscle was first injured and then allowed to regenerate for 28 days before being subjected to a second injury (Figure 3A). If the MuSC pool was efficiently repopulated after the first injury, the muscle will fully recover from the second injury. However, a defect in MuSC pool repopulation will prevent recovery from the second injury. Although we observed that the WT mice were able to efficiently recover from consecutive injuries, the NELF-BscKO mice showed a devastating inability to regenerate the TA muscle after the second injury (Figure 3B). More specifically, the TA muscle showed a ~70% decrease in mass with a visible pathology of interstitial cells outnumbering the regenerated myofibers (Figures 3B and 3C). In further support of a defect in MuSC pool replenishment in the NELF-BscKO mice, we observed a diminution of MuSCs (Pax7+) associated with the regenerated myofibers when measured at either 7 or 28 days post-injury (Figures 3D, S3A, and S3B). The inability of NELF-BscKO MuSCs to repopulate the niche was not due to reduced viability, as the isolated TdT+ cells sorted using FACS did not show increased cell death (Figure S3C) nor did they show the presence of activated caspases (Figure S3D). Thus, in the absence of NELF-B, MuSCs do not repopulate the stem cell pool sufficiently to allow for regeneration after successive muscle injuries.
Figure 3. Loss of NELF in the regenerating muscle leads to a depletion of the MuSC pool.

(A) Experimental schematic showing the sequential injury approach where mice that had undergone tamoxifen-induced recombination were subjected to two rounds of CTX treatment at 28-day intervals.
(B) Hematoxylin and eosin stain of the regenerated TA muscle after the repeat injury (±SE, p-value< 0.001, n = 3) was used to visualize regeneration and the weight of the regenerated muscle was determined relative to the uninjured contralateral TA muscle (±SE, n = 3), scale bar = 1 mm.
(C) Magnification of the hematoxylin and eosin staining for the repeat-injured TA from (B) showing fibrosis and interstitial cells, scale bar = 50 μm.
(D) Immunofluorescence characterization of MuSCs (Pax7+) in the regenerated TA muscle 7 or 28 days after injury (±SE, p-value < 0.001, n = 4), scale bar = 25 μm.
Transplanted donor NELF-BscKO MuSCs can replenish the stem cell pool in uninjured recipient muscle
The inability of NELF-BscKO MuSCs to repopulate the niche presents the possibility that NELF may be required for activated MuSCs to return to a quiescent state. To directly address this possibility, 12,000 TdT-labeled MuSCs from WT or NELF-BscKO mice were transplanted into the contralateral TA muscles of recipient mice (Feige and Rudnicki, 2020), which had been irradiated to deplete endogenous MuSCs.
We first examined whether early activated MuSCs from uninjured NELF-BscKO mice maintained their ability to return to quiescence for stem cell pool repopulation on healthy myofibers. By transplanting MuSCs isolated from either WT or NELF-BscKO mice, we confirmed a proliferation defect because MuSCs from the NELF-BscKO mice gave rise to less TdT+ myofibers in homeostatic muscles (Figures 4A, S4A, and S4B). By characterizing the ability of the transplanted MuSCs to repopulate the niche, we observed that all the TdT+ MuSCs were associated with TdT+ myofibers regardless of their genotype (Figure 4A). As such, niche repopulation was scored as a percentage of TdT+ myofibers associated with TdT+ MuSCs. Despite the fact that the proliferation defect resulted in NELF-BscKO MuSCs being incorporated into fewer myofibers, we observed that MuSCs from both WT and NELF-BscKO mice contributed to niche repopulation on these TdT+ myofibers with similar efficiencies (Figure 4A). The expression of CalcR (Baghdadi et al., 2018) and absence of Ki-67 confirmed that the transplanted MuSCs from both the WT and NELF-BscKO mice had returned to the quiescent state (Figures 4B and S4C). This suggests that in the absence of injury, MuSCs do not require NELF to return to quiescence. We next explored the possibility that the regenerative environment might lead to changes specifically in the NELF-BscKO MuSCs that prevented their return to quiescence. MuSCs were isolated from WT or NELF-BscKO mice 40 h after muscle injury, allowing the cells to fully activate and enter the cell cycle before transplantation. Again, we observed that MuSCs from WT and NELF-BscKO mice were able to contribute both to myofiber homeostasis (Figures 4C, S4B, and S4D) and niche repopulation (Figure 4B). These results suggest that NELF-B is not required for MuSCs to return to quiescence when a sufficient number of progenitors are available to ensure a healthy myofiber environment.
Figure 4. NELF is not required for muscle progenitors to return to quiescence and repopulated the MuSC niche.

Engraftment experiments were performed using MuSCs derived from WT and NELF-BscKO into recipient mice. In all instances, 12,000 donor MuSCs (TdT+ ITGA7+) derived from WT or NELF-BscKOs were transplanted into NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice, which were irradiated to deplete the endogenous MuSC population. The TA muscles were recovered 28 days after transplantation and were subjected to immunofluorescence analysis using the antibodies indicated. The niche replenishment ratio was calculated by normalizing the number of donor MuSCs (TdT+ Pax7+) to the number of TdT+ myofibers.
(A) Freshly isolated MuSCs from the healthy muscle of WT and NELF-BscKO donors were transplanted into uninjured, irradiated recipient NSG muscle(±SE, n = 5), scale bar = 50 μm.
(B) The ability of transplanted MuSCs to return to the quiescent state was examined through co-staining of TdT+ cells in the niche with Pax7 and CalcR, or Pax7 and Ki67 (see Figure S4F) (±SE, n = 3), scale bar = 50 μm.
(C) Activated MuSCs isolated from the regenerating muscle of WT and NELF-BscKO donors (40 h after CTX injection) into uninjured, irradiated recipient NSG muscle (±SE, n = 3), scale bar = 50 μm.
(D) Freshly isolated MuSCs from the healthy muscle of WT and NELF-BscKO donors transplanted into irradiated, recipient NSG muscle at 48 h after CTX damage, scale bar = 50 μm. A paired, two-tailed t test was used to compare the contralateral-matched TA muscles for TdT+ donor cell contribution with the Pax7+ MuSC niche (±SE, p-value < 0.05, n = 3).
Finally, we examined whether MuSCs freshly isolated from an uninjured donor muscle could repopulate the niche of regenerated myofibers of an injured recipient mouse. Consistent with an inability to expand the muscle progenitor population, MuSCs from NELF-BscKO mice gave rise to a significantly smaller number of regenerated myofibers compared with their WT counterparts (Figures S4B and S4E). Most importantly, we observed that the freshly isolated NELF-BscKO MuSCs that efficiently contributed to repopulating the niche of an uninjured muscle (Figure 4A), were unable to repopulate the MuSC pool when injected into a regenerating muscle (Figure 4D). Thus, NELF is required for MuSCs to re-establish the quiescent state in a regenerative signaling environment. Taken together, our results strongly support a mechanism whereby depletion of the MuSC pool in NELF-BscKO mice after injury is caused by reduced muscle progenitor expansion that limits the number of undifferentiated cells available for return to quiescence, rather than by an active role of NELF in re-establishing the quiescent state of MuSCs.
NELF promotes the efficient expansion of muscle progenitor cells
To determine how loss of NELF affects the progression of MuSCs along the continuum of muscle regeneration, single cell RNA-sequencing (scRNA-seq) analysis was performed on purified TdT-labeled mononuclear cells isolated from regenerating muscle tissue 3 days after injury. Louvain clustering identified 16 distinct myogenic populations that were present in both the WT and NELF-BscKO cell populations (Figure 5A). The myogenic trajectory of the combined WT and NELF-BscKO cell populations was modeled based on differential gene expression using the PAGA algorithm where Pax7 expression was set as the common anchor (Figures 5B, 5C, and S5A). Independent plotting of the WT or NELF-BscKO cells on the trajectory (Figure 5D) clearly demonstrated that WT cells (clusters 2, 7, and 9) accumulated within distinct clusters compared with NELF-BscKO cells (clusters 0 and 8). We next used gene expression analysis (Figure 5C) to classify the clusters into 4 representative groups: (1) quiescence (cluster 13); (2) proliferating cells (clusters 2, 3, 4, 5, 7, 9, 11); (3) early differentiation (clusters 0, 8); and (4) cells committed to differentiation (clusters 1, 6, 12, 15)(Figures 5C and 5D). Measuring the number of WT and NELF-BscKO MuSCs in each of these populations revealed that WT cells (60%) were more likely to proliferate than cells from the NELF-BscKO (50%) mice (Figures 5D and 5E). Reciprocally, WT cells (39%) were underrepresented in early or late differentiation compared with NELF-BscKO cells (47%) (Figure 5E). Although the increase in the number of cells undergoing differentiation in the NELF-BscKO may not seem drastic, the numbers should be viewed as a snapshot in time where more and more cells will cease to proliferate through each round of the cell cycle, and can explain the significantly reduced myofiber size after 28 days. When focusing on the clusters that were enriched in either the WT (clusters 2, 7, and 9) or NELF-BscKO (clusters 0 and 8) cells, Gene Ontology (GO) analysis revealed that the NELF-BscKO cells showed decreased expression of genes involved in cell cycle progression in all five clusters (Figures 5F and 5G). This was accompanied by an increased expression of genes involved in cell adhesion and muscle contraction (Figures 5F and 5G) suggesting that MuSCs from NELF-BscKO mice had progressed toward differentiation. Looking at signaling pathways that were disrupted in the NELF-BscKO cells, the two overrepresented progenitor clusters (0 and 8) showed altered expression of genes involved in the p53 signaling pathway, including ccng1 and cdkn1a (Figures 5F, 5G, S5B, and S5C). Thus, our scRNA-seq data provide strong support for a model whereby NELF contributes to the expansion of the muscle progenitor pool by preventing cells from undergoing premature differentiation.
Figure 5. Single-cell transcriptome analysis of muscle progenitors in regeneration.

Myogenic progenitor cells (TdT+, ITGA7+) were isolated from CTX injured hind-limb muscles at 72 h post-injury, sorted based on fluorescence, and subjected to scRNA-Seq using the 10x genomic platform.
(A) Myogenic cells were clustered using the PAGA algorithm, which identified 16 different Louvain clusters on combined cell analysis for NELF-BscKO and WT samples.
(B) Pseudotime trajectory re-construction using the PAGA algorithm shows the cell trajectory across 16 clusters from the Pax7+ MuSCs (blue), through to the Myog+ myocytes (red).
(C) Dotplot analysis of the 16 clusters on selected genes shows differences in Louvain cluster gene signatures.
(D) Density mapping of WT and NELF-BscKO myoblasts on the PAGA trajectory reveals differential population occupancy in clusters 0 and 8 (up in NELF-BscKO) as well as clusters 2, 7, and 9 (down in NELF-BscKO).
(E) Cells were classified into different myogenic cell states based on global gene signatures representing quiescence (cluster 13), proliferation (cluster 2,3,4,5,7,9,11), early differentiation (cluster 0,8), and late differentiation (cluster 1,6,12,15).
(F and G) Gene Ontology analysis was performed on differentially expressed genes identified in the 5 clusters that were either underrepresented (F) or overrepresented (G) in NELF-BscKO mice. Representative upregulated (blue) and downregulated (red) GO terms are shown as a function of Log10(p-value).
To elucidate the mechanism underlying NELF-mediated expansion of muscle progenitor cells, we next performed bulk RNA-seq analysis on mononuclear TdT-labeled cells isolated from either regenerating muscle or MuSCs cultured in vitro (Figures 6 and S6). A time point of 48 h post-injury was chosen based on our observations that NELF-BscKO MuSCs began to display a proliferation defect between 40 and 72 h post-injury (Figures 2B and 2C). Differential gene expression analysis (adjusted p value < 0.01) showed that loss of NELF-B resulted in the down-regulation of 342 genes, whereas 305 genes were upregulated (Figure 6A). Consistent with the phenotypic observation of premature differentiation, loss of NELF-B resulted in the up-regulation of transcripts encoding key genes involved in muscle differentiation (including Myog, Mef2c, Mylpf), ECM receptor interactions, as well as mitogen-activated protein kinase (MAPK) signaling (Figures 6A and S6A). Loss of NELF-B in cultured muscle progenitors similarly showed an increase in the expression of genes involved in muscle differentiation and MAPK signaling while also identifying negative regulation of TGF-β receptor signaling (Figure 6B). Looking at genes downregulated in the absence of NELF, those encoding proteins of the extracellular matrix or controlling cell proliferation were highly enriched in data sets obtained both in vivo and in vitro (Figures 6A and 6B). Interesting, TdT+ cells isolated from regenerating muscle of NELF-BscKO mice 120 h post-injury showed increased expression of genes involved in the cell cycle suggesting that, at this later time point, the remaining undifferentiated muscle progenitors are trying to compensate for the inability to expand the progenitor population (Figure S6B). Overall, these findings are consistent with a mechanism whereby NELF controls the expansion of muscle progenitors by preventing cells from initiating differentiation.
Figure 6. Transcriptome analysis of muscle progenitors in regeneration.

RNA-seq analysis was performed on fluorescence-sorted NELF-BscKO and WT myogenic progenitors (TdT+ ITGA7+) using and adjusted p-value < 0.01 as a cutoff at different timepoints, represented as volcano plots. Gene Ontology analysis performed on differentially expressed genes shows terms that are upregulated and downregulated in NELF-BscKO samples as a function of Log10(p-value). Terms are represented based on their origin from biological process (blue), cell component (red), or Kegg pathway (black).
(A) Myogenic progenitors freshly isolated from skeletal muscle at 48 h post-injury show 305 upregulated and 343 downregulated genes.
(B) In vitro cultured myoblasts show 441 downregulated genes and 102 upregulated genes.
(C) Precision Run-On sequencing mapped onto RNA-sequencing results show that downregulated genes in the NELF-BscKO mice have decreased nascent transcript levels at the promoter-proximal region as well as the gene body (p-value = 0.006). Genes upregulated in NELF-BscKO mice show no significant change in nascent transcript levels compared with WT controls (p-value = 0.77).
(D) A representative UCSC browser track showing the reduced gene expression of SerpinF1 for RNA-seq performed at 48-h post-injury on RNA collected from cultured myoblasts. PRO-seq shows a loss of nascent transcript emerging from the transcription start site (TSS) upon a NELF-BscKO, and Cut&Tag analysis showing a loss of NELF-E binding at the promoter region upon a NELF-BscKO and a reduced occupancy of RNAPII (S5P).
Because the loss of NELF-B can contribute both directly and indirectly to changes in gene expression, we next sought to identify NELF target genes across the genome. Using an antibody directed against NELF-E, we observed that the NELF complex was associated with RNAPII at the pause site across active genes in cultured muscle progenitors (Figure S6C). To determine how the loss of NELF was altering gene expression at these targets, we analyzed nascent transcription in proliferating progenitors using precision run-on (PRO)-seq. Consistent with a role for NELF in stabilizing paused RNAPII, we observe that genes downregulated in NELF-BscKO progenitors showed a significant decrease in the PRO-seq signal poised at the promoter before entering the gene (Figure 6C – left panel) and within the gene body (Figure 6C – right panel). The loss of PRO-seq signal was accompanied by a decrease in the occupancy of engaged RNAPII (S5P) and H3K4me3 accumulation at these genes (Figure S6C). These findings indicate that NELF stabilizes paused RNAPII against termination at these genes in MuSCs/muscle progenitors to increase their rate of transcription. Looking at genes that were upregulated upon loss of NELF, we observed a limited increase in RNAPII transcription at the gene body, but this effect was not statistically significant (Figure 6C – middle and right panels). Thus, in agreement with work in cell culture (Aoi et al., 2020; Core and Adelman, 2019), our results suggest that NELF functions to stabilize paused RNAPII in regenerating muscle progenitors.
PEDF and p53 signaling are important mediators of NELF-dependent expansion of muscle progenitor cells
We next wanted to know how NELF contributes to the expansion of muscle progenitors. GO analysis suggested that the loss of NELF-B in MuSCs led to altered expression of multiple cell signaling pathways as well as proteins regulating cell cycle progression (Figures 6A and 6B). Among the NELF target genes that also showed altered expression in both bulk RNA-seq (in vivo and in vitro) and scRNA-seq experiments, SerpinF1 and Ccng1 were identified as potential mediators of the effects of NELF on muscle progenitor expansion. The SerpinF1 gene encodes the pigment-epithelium derived factor (PEDF) protein that has been shown to have mitogenic effects in various stem cell populations (Ho et al., 2015, 2019). Loss of NELF led to reduced levels of engaged RNAPII S5P with a concomitant reduction in the number of nascent transcripts across the SerpinF1 gene (Figure 6D). Similar transcriptional profiles were observed for the NELF target gene Ccng1 (Figure S6D), which encodes the cyclin G1 component of the p53 signaling pathway that acts to maintain proliferation of stem cells by facilitating the degradation of the cell cycle check-point protein p53 (Gordon et al., 2018). Thus, we have identified the SerpinF1/PEDF and p53 signaling as two important pathways directly modulated by NELF in proliferating muscle progenitors.
Finally, we examined whether the SerpinF1/PEDF and p53 signaling were primary mediators of the regulatory effects of NELF on muscle progenitor expansion during regeneration. For this purpose, the p53 inhibitor pifithrin-α was used to mimic the effects of cyclin G1 on ensuring reduced p53 activity in cells. We first examined the ability of the p53 inhibition to rescue the NELF phenotype in vivo. Mice were administered three doses of pifithrin-α (or vehicle) at 24-h intervals starting at 40 h (40, 64, and 88 h) post-CTX injury, and then sacrificed after 7 days of regeneration. Histological examination of the muscle fibers showed that transient inhibition of p53 function during the time period when the muscle progenitor population undergoes rapid expansion resulted in improved myofiber diameter in NELF-BscKO mice 7 days after injury (Figures 7A and S7A). Importantly, we observed that transient inhibition of p53 signaling led to a significant recovery of Pax7+ MuSCs associated with muscle fibers at 7 days post-injury (Figures 7B, S7B, and S7C). To determine whether this improved myofiber size and niche repopulation was due to expansion of the muscle progenitors, we measured the ability of pifithrin-α to rescue progenitor proliferation in culture. Although progenitors from the NELF-B null mice showed reduced EdU incorporation compared with their WT littermates, supplementing the culture media of NELF-BscKO muscle progenitors with pifithrin-α was sufficient to rescue their proliferation defect (Figure 7C). These findings show that NELF-dependent inhibition of p53 activity allows expansion of the progenitor cell population to efficiently repair myofibers and self-renewal of the MuSC population in vivo. Finally, because PEDF has previously been shown to downregulate p53 expression in mesenchymal stem cells (Ding et al., 2017), we examined the ability of recombinant PEDF protein to restore proliferation of NELF-BscKO progenitors. Similar to pifithrin-α, we observed that the addition of PEDF protein to the culture media was also able to rescue the proliferation defect in NELF-null MuSCs (Figure 7C). Taken together, these results identified p53 and PEDF signaling as key MuSC regulatory axes modulated by NELF to control expansion of the progenitor population during muscle regeneration.
Figure 7. Rescue experiments and proposed model explaining role of NELF in myogenesis.

(A) Pifithrin-α (25 μM or vehicle) in Captisol (20% w/v in saline) was administered to mice by intraperitoneal injections (2 mg/kg of pifithrin-α) at 40, 64, and 88 h after CTX injury of the TA muscle. Following regeneration (7 days), histological analysis of regenerating muscle with hematoxylin and eosin staining was used to visualize the regeneration (±SE, p-value < 0.01, n = 3 with 500 myofiber measurements per biological replicate), scale bar = 50 μm.
(B) Immunofluorescent characterization of the Pax7+ population in the regenerated TA cross-sections after pifithrin-α treatment (±SE, **p-value < 0.01 or ***p-value < 0.001, n = 3), scale bar 50 μm.
(C) Myoblasts were isolated from induced WT and NELF-BscKO mice and treated in culture with PEDF (500 ng/mL) or pifithrin-α (25 μM). Proliferating cells were then pulsed with EdU (10 μM) for 4 h (n = 3) scale bar = 100 μm.
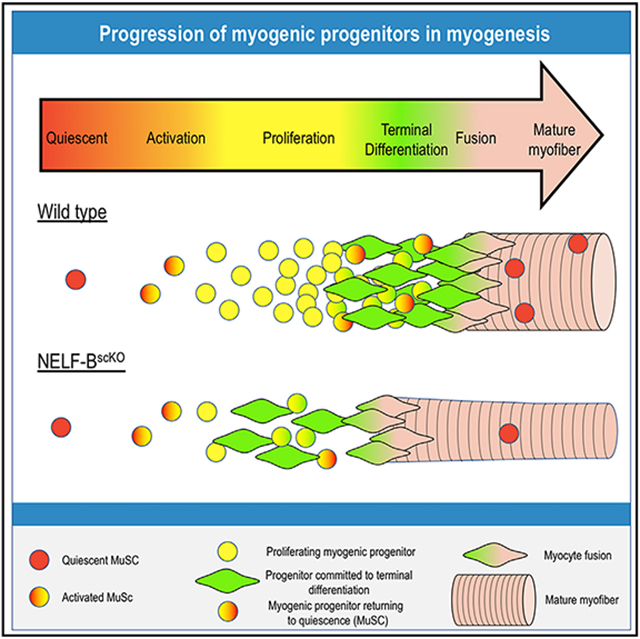
(D) Proposed model for the implication of NELF in skeletal muscle regeneration. When healthy skeletal muscle is injured (i), MuSCs are activated and beginning to undergo population expansion (ii). Reduced proliferation upon a NELF-BscKO results in lower myogenic progenitor cell presence than in WT conditions (iii). As regeneration progresses (iv), myogenic progenitors will either commit to terminal differentiation to form new skeletal muscle, or return to quiescence to repopulate the stem cell niche (v).
DISCUSSION
NELF has been extensively studied at the mechanistic level and has been shown to be essential for stabilizing paused RNAPII (Core and Adelman, 2019). However, the role of transcriptional pausing in regulating in vivo cellular processes remains poorly defined. Here, we showed that NELF plays an essential role in muscle regeneration and repopulation of the stem cell pool. Through systematic dissection of the stages of myofiber regeneration after injury, we found that NELF regulated the PEDF and p53 signaling to modulate the dramatic expansion of muscle progenitors required for the production of sufficient myoblasts to reconstruct the muscle. Unexpectedly, we also found that NELF-dependent expansion of progenitors is required for repopulation of the MuSC niche and provide evidence that the majority of the MuSCs present on the regenerated myofiber are derived from muscle progenitors that have returned to a quiescent state. Therefore, our work identified a new role for NELF in inducing muscle progenitor cell proliferation in response to injury and repopulation of the MuSC pool.
Proximal-promoter pausing is an important transcriptional control mechanism shared across all cell types (Day et al., 2016). RNAPII pausing has been shown to occur at genes involved in various processes including development (Muse et al., 2007; Zeitlinger et al., 2007), cell cycle progression (Day et al., 2016), metabolism (Zhu et al., 2014), and cell signaling (Williams et al., 2015). In addition, embryos lacking NELF-B do not mature beyond day E5 (Amleh et al., 2009), suggesting that RNAPII pausing is essential for early development. Given that transcriptional pausing is broadly used to regulate gene expression, our finding that NELF function in regeneration is restricted to muscle progenitor expansion was unexpected. Dissecting the regenerative process, we have shown that NELF acts to ensure the efficient transcription of key signaling genes that transduce signals from the extracellular environment to expand the muscle progenitor pool by preventing differentiation. Specifically, we found that the PEDF and p53 signaling pathways are important targets directly regulated by NELF to modulate the expansion of muscle progenitors. Previous studies have shown that p53 signaling contributes to myoblast differentiation through its ability to induce cell cycle exit (Porrello et al., 2000). Our studies identified the p53 pathway component Ccng1 as a direct target of NELF whose expression is decreased in progenitors lacking NELF-B. In parallel, our scRNA-seq studies identified the p53 target gene Cdkn1a (p21cip1) as a gene upregulated in populations that are differentially enriched for NELF-BscKO cells. This is interesting because cyclin G1 is an important cyclin that acts to modulate p53 levels in the cells by co-regulating the activity of the ubiquitin ligase MDM2 (Gordon et al., 2018). In the absence of cyclin G1, MDM2 is dephosphorylated and no longer targets p53 for degradation. Accumulation of p53 in the cell activates target genes such as Cdkn1a/p21 to block cell cycle progression and prepare the cells for differentiation (See Figure S7D). The ability of p53 inhibition to partially rescue the proliferation defect in NELF-BscKO MuSCs to improve fiber diameter and increase MuSCs numbers clearly establishes p53 signaling as an important NELF target in muscle regeneration. We similarly showed that PEDF is an important target of NELF that regulates progenitor expansion. Consistent with the importance for PEDF in MuSC function, previous studies have shown that PEDF has strong mitogen activity that stimulates the proliferation of muscle progenitors in vitro (Ho et al., 2015). Furthermore, it was found that a synthetic PEDF-derived short peptide can enhance the growth of regenerating myofibers through stimulation of MuSC expansion (Ho et al., 2015). Our ability to rescue the proliferation defect in NELF-null muscle progenitors using recombinant PEDF affirms the importance of this mitogen as an important contributor to the expansion of the muscle progenitor pool. Thus, our results point to an essential role for NELF in allowing muscle progenitors to respond to signals from the environment to ensure progenitor cell expansion. Remarkably, NELF-regulated genes include not only signaling molecules (and downstream transducers of the signals) but also extracellular matrix proteins (collagens and fibronectin) that stabilize receptors at the membrane and matrix metalloproteinases that activate/inactivate receptors and ligands. This suggests that cell-to-cell communication between muscle progenitors and non-muscle cells may also be regulated through proximal pausing. Finally, it is interesting to note that NELF and the process of promoter-proximal pausing emerged during evolution of the early metazoans in parallel with proteins possessing tyrosine kinase signaling domains, collagen-like domains, and/or laminin domains (King et al., 2008). Taken together, our results revealed a role for NELF-dependent transcriptional pausing in enhancing communication between muscle progenitors and their environment.
Using high-resolution nascent RNA assays, we confirmed that NELF-affected genes such as SerpinF1 and Ccng1 were directly targeted at the transcriptional level. We found that genes with decreased expression upon loss of NELF-B displayed significantly reduced promoter-proximal RNAPII and less polymerase released into the gene bodies. This finding is consistent with a role for NELF in stabilizing paused RNAPII (Henriques et al., 2013; Vos et al., 2018) and suggests that destabilized RNAPII at this set of promoters is particularly susceptible to termination. Intriguingly, recent work implicates an interplay between NELF and the Integrator termination complex at such genes (Elrod et al., 2019; Stadelmayer et al., 2014). The Integrator complex subunit, IntS11, possesses endonuclease activity that cleaves nascent transcripts to attenuate transcription (Elrod et al., 2019; Tatomer et al., 2019). Thus, it is possible that the Integrator complex may be selectively recruited to a subset of NELF target genes in proliferating progenitor cells. Nonetheless, determining why the disruption of NELF affects productive elongation levels at discrete subsets of genes and why functional NELF targets display such exquisite cell-type specificity remain active areas of research (Core and Adelman, 2019).
Previous lineage tracing experiments have shown that naive MuSCs that reside on myofibers undergo asymmetric cell division within 40 h of injury to produce one naive MuSC that will repopulate the stem cell pool and one poised MuSC that will become committed to the muscle progenitor lineage (Feige et al., 2018; Gurevich et al., 2016; Kuang et al., 2007). However, naive MuSCs represent only 5% to 10% of quiescent MuSCs on a myofiber (Kuang et al., 2007), suggesting that additional mechanisms could contribute to replenishing the MuSC population. Our results showed that expansion of the muscle progenitor cell population is necessary to replenish the MuSC pool on regenerated myofibers, and that cycling MuSCs can return to the quiescent state. These findings are interesting in light of recent work showing that not all of the muscle progenitors are destined to undergo differentiation, but instead will frequently undergo asymmetric cell division to produce one Myog+ myocyte that is committed to differentiate, and one muscle progenitor that would be available to replenish the MuSC pool (Evano et al., 2020). Thus, we propose that in parallel with asymmetric cell division that occurs to repopulate the niche, a second mechanism is in place that allows muscle progenitors that do not differentiate to return to quiescence for niche repopulation (see model in Figure 7D). Consistent with this model, Chakkalakal et al. have previously identified two populations of MuSCs in vivo—one that divides frequently and one that that only undergoes rare cell divisions (Chakkalakal et al., 2014). Furthermore, in vivo EdU incorporation experiments at specific time points after muscle injury have shown that 75% of quiescent MuSCs that reside on myofibers have extensively proliferated between the time of injury and their homing in the myofiber niche (Pawlikowski et al., 2019). Thus, we propose that progenitor amplification and return to quiescence is a major mechanism underlying MuSC niche repopulation upon injury, and that it occurs in parallel with asymmetric division of naive MuSC. Our findings that muscle progenitors are major drivers of muscle regeneration and that niche repopulation has important therapeutic implications for the treatment of myopathies, such as Duchenne Muscular Dystrophy, suggesting that NELF-dependent genes and pathways could be targeted by novel therapies for expansion of the muscle progenitor population to prevent MuSC exhaustion.
Interestingly, the phenotype we have observed in NELF-BscKO mice is reminiscent of defects identified in muscle aging with reduction in both the myofiber size and MuSC numbers (Brack and Muñoz-Cánoves, 2016). Although it has been shown that age-related muscle hypotrophy is due to reduced expansion of the muscle progenitors (Bernet et al., 2014), it has been less clear what gives rise to the reduction in MuSC numbers. Our results suggest that a deficiency in muscle progenitor expansion may also contribute to a decrease in the number of MuSCs in aged muscles. This opens the possibility that targeting pathways that modulating muscle progenitor expansion may also serve the purpose of preventing age-related loss of MuSCs.
In conclusion, we demonstrated that NELF-mediated RNAPII pausing is a key transcriptional control mechanism used to modulate expansion of the muscle progenitor population in response to muscle injury cues. Furthermore, we identified a need for NELF to repopulate the MuSC pool. This need for NELF to repopulate the niche led to the unexpected finding that the majority of the MuSCs associated with a regenerated fiber are derived from the expanding myoblast population that fail to differentiate and instead return to quiescence.
Limitations of the study
There are several limitations in our study. First, our transcriptomic analysis identified p53 signaling as a key pathway regulated by NELF to modulate the expansion of the muscle progenitors. Although this was corroborated by small molecule inhibitor studies, as well as the demonstration that the p53 target gene, p21, was upregulated in the NELF-BscKO progenitors by western blot, we did not test how the p53 protein levels changed upon loss of NELF-B. Second, we did not obtain a full rescue of the NELF-BscKO muscle regeneration defect upon transient inhibition of p53 function. A GO analysis of NELF-regulated genes identified several different signaling pathways that are disrupted upon depletion of the NELF complex. We expect that several of these additional pathways contribute to the NELF-Bscko phenotype, and that future studies will be required to elucidate their contribution to the expansion of MuSCs after muscle injury.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. F. Jeffrey Dilworth (jdilworth@ohri.ca)
Materials availability
This study did not generate new unique reagents.
Data and code availability
The datasets generated during this study are available at the Gene Expression Omnibus as the following DOIsq
The accession number for the RNA-seq and ChIP-Seq datasets reported in this paper is GEO: GSE150280
The accession number for the PRO-seq datasets reported in this paper is GEO: GSE149766
Analysized transcriptomic data are available on Mendeley Data: https://dx.doi.org/10.17632/r4tj555pc7.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal models
All animal experiments were performed in accordance with the recommendations of the University of Ottawa Animal Care Facility, and the guidelines published by the Canadian Council on the Animal Care (CCAC). Experimental mice used in the study were in early adult stage (6 – 10 weeks of age), with equal repetitions performed between males and females. The exception to this falls within NSG recipient mice for animal studies, which were performed in mice 6 months of age (middle adulthood). Animal strains used for experiments are indicated in the key resources table. These strains were used to generate combined experimental strains, which include the NELF-Bfl/flPax7CreER, NELF-Bfl/flPax7CreERTdTfl/fl, and Pax7CreERTdTfl/fl strains described. Generating combined strains was performed through breeding of homozygous parental strains (F0) to generate a heterozygote population (F1), which was crossed amongst itself to generate the desired homozygous strain (F2). Genotyping was performed throughout to assure the desired genotype (see key resources table). Harem breeding in accordance with animal care protocols consisted of housing 2 females with 1 male. Mice of the same sex, from a same litter and of the same verified genotype were caged together (maximum of 5 mice per cage), in accordance with our animal care protocols. Engraftment experiments were performed double-blinded until results were quantified and identities revealed.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Pax7 | Developmental Studies Hybridoma Bank | Name : PAX7; RRID: AB_2299243 |
| Mouse monoclonal anti-myogenin | Santa Cruz Biotechnology | Cat# sc-12732; RRID: AB_627980 |
| Mouse monoclonal anti-NELF-E | Santa Cruz Biotechnology | Cat# sc-377052; RRID: AB_2847957 |
| Rabbit polyclonal anti-NELF-E | ProteinTech | Cat# 10705-1-AP; RRID: AB_513966 |
| Rat monoclonal anti-Alpha-integrin 7 (R2F2 clone) | AbLab | Cat# 67-0010-05 |
| Rabbit monoclonal anti-NELF-B | Cell Signaling Technology | Cat# 14894S; RRID: AB_2798637 |
| Rabbit monoclonal anti-alpha-tubulin | Cell Signaling Technology | Cat# 2125S; RRID: AB_2619646 |
| Rabbit polyclonal anti-calcitonin receptor | Abcam | Cat# ab11042; RRID: AB_297696 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Rabbit polyclonal anti-H3K4me3 | Millipore Sigma | Cat# 07-473; RRID: AB_1977252 |
| Goat polyclonal anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 647 | ThermoFisher Scientific | Cat# A-21235; RRID: AB_2535804 |
| Goat polyclonal anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 546 | ThermoFisher Scientific | Cat# A-11030; RRID: AB_144695 |
| Goat polyclonal anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 | ThermoFisher Scientific | Cat# A-11001; RRID: AB_2534069 |
| Goat polyclonal anti-Rabbig IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 647 | ThermoFisher Scientific | Cat# A-21244; RRID: AB_2535812 |
| Goat polyclonal anti-Rabbig IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 546 | ThermoFisher Scientific | Cat# A-11010; RRID: AB_2534077 |
| Goat polyclonal anti-Rabbig IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 | ThermoFisher Scientific | Cat# A-11034; RRID: AB_2576217 |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase from Clostridium histolyticum | Millipore Sigma | Cat# C0130-1G; |
| HyClone Dulbecco’s Modified Eagles Medium | ThermoFisher Scientific | Cat# SH3024301 |
| Ham’s F10 medium | Wisent bioproducts | Cat# 318-050-CL |
| Fibroblast growth factor basic protein, human recombinant | Millipore sigma | Cat# GF003AF-MG |
| Penicillin-Streptomycin Solution | Wisent bioproducts | Cat# 450-201-EL |
| HyClone Bovine Growth Serum | ThermoFisher Scientific | Cat# SH3054103 |
| Fluorescence Mounting Medium | Agilent | Part# S302380-2 |
| M.O.M. (Mouse on Mouse) Blocking Reagent | Vector Laboratories | Product# VECTMKB22131 |
| Dispase II (neutral protease, grade II) | Millipore Sigma | Cat# 4942078001 |
| Matrigel Membrane Matrix | Corning | Cat# 354234 |
| Red Blood Cell Lysing Buffer Hybri-Max | Millipore Sigma | Cat# R7757 |
| DAPI | Millipore Sigma | Cat# D9542 |
| Horse serum | Millipore Sigma | Cat# H1138 |
| Insulin solution from bovine pancreas | Millipore Sigma | Cat# I0516 |
| Holo-Transferrin bovine | Millipore Sigma | Cat# T1283 |
| NucleoSpin RNA Plus XS, micro kit for RNA purification with DNA removal column | Macherey-Nagel | Ref740990 |
| 2-Methylbutane | Millipore sigma | Cat# 320404 |
| Fisherbrand Superfrost Plus Microscope Slides | ThermoFisher Scientific | Cat# 12-550-15 |
| Falcon Cell Strainers (100 μm) | VWR | Cat# CA21008-950 |
| Falcon Cell Strainers (40 μm) | VWR | Cat# CA21008-949 |
| Cardiotoxin | Latoxan | Cat# L8102 |
| Premium Frozen Section Compound | VWR | Cat# 95057-838 |
| PureLink RNA Mini Kit | ThermoFisher Scientific | Cat# 12183020 |
| EdU, DNA synthesis monitoring probe | Abcam | Cat# ab146186 |
| KAPA Stranded mRNA-seq kit | Roche | Cat# 07962169001 |
| Captisol | Captisol | Cat# RC-0C7-100 |
| Recombinant human PEDF protein | Abcam | Cat# Ab56289 |
| Pifithrin-α | Millipore Sigma | Cat# P4359; Cas#63208-82-2 |
| Digitonin | Millipore Sigma | Cat# D141 |
| Concanavalin A-coated magnetic beads | Bangs Laboratories | Cat# BP531 |
| Wheat Germ Agglutinin, Texas Red-X Conjugate | ThermoFisher Scientific | Cat# W21405 |
| Wheat Germ Agglutinin, Alexa Fluor 488 Conjugate | ThermoFisher Scientific | Cat# W11261 |
| Critical commercial assays | ||
| Click-iT EdU cell proliferation kit for imaging, Alexa Fluor 488 dye | ThermoFisher Scientific | Cat# C10337 |
| FAM FLICA Poly Caspase Kit | Bio-Rad | Product# ICT091 |
| NELFB TaqMan Gene Expression Assay | ThermoFisher Scientific | Cat# 4331182; AssayID : Mm00480507_m1 |
| GAPDH TaqMan Gene Expression Assay | ThermoFisher Scientific | Cat# 4331182; AssayID : Mm_99999915_g1 |
| Deposited data | ||
| RNA-Seq Data | GEO | GSE150280 |
| Cut&Tag Data | GEO | GSE150280 |
| PRO-Seq Data | GEO | GSE149766 |
| Differential Gene Expression Analysis and Gene Ontology | Mendeley | https://dx.doi.org/10.17632/r4tj555pc7.1 |
| Experimental models: organisms/strains | ||
| Mouse : C57BL/6N-Nelfbtm1.1Ehs/J | Karen Adelman Lab (now available at The Jackson Laboratory) | JAX 033115 |
| Mouse : B6.Cg-Pax7tm1(cre/ERT2)Gaka/J | The Jackson Laboratory | JAX 017763 |
| Mouse : B6.Cg-Gt(ROSA) 26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX 007909 |
| Mouse : NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | JAX 005557 |
| Oligonucleotides | ||
| Pax7CreER WT primers : Fwd : GCTGCTGTTGATTACCTGGC; Rev : CTGCACTGAGACAGGACCG | Murphy et al., 2011 | N/A |
| Pax7CreER Mutant primers : Fwd : GCTGCTGTTGATTACCTGGC; Rev : CAAAAGACGGCAATATGGTG | Murphy et al., 2011 | N/A |
| NELF-B 3’ sequencing primers : Fwd : TGCCTGATGTAGGCACTAGGAGTT; Rev : AAGCCAGTTCCAGGACACCCAGG | Gupte et al., 2013 | N/A |
| NELF-B 5’ sequencing primers : Fwd: ATGTAGGTGCTGGAACTGAACCCA; Rev: TGCTGTCACACTTGGCAATGATGC | Gupte et al., 2013 | N/A |
| TdTomato WT sequencing primers : Fwd : AAGGGAGCTGCAGTGGAGTA; Rev : CCGAAAATCTGTGGGAAGTC | Madisen et al., 2010 | N/A |
| TdTomato Mut sequencing primers : Fwd : GGCATTAAAGCAGCGTATCC; Rev : CTGTTCCTGTACGGCATGG | Madisen et al., 2010 | N/A |
| Software and algorithms | ||
| FIJI | Schindelin et al., 2012; Preibisch et al., 2009 | https://imagej.net/Fiji |
| Seurat v3.0 | Stuart et al., 2019 | https://satijalab.org/seurat/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| bamCoverage | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html |
| SEACR | Meers et al., 2019 | https://seacr.fredhutch.org/ |
| BEDTools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| ChIPseeker | Yu et al., 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
Primary cell cultures
Primary cell cultures used for ex vivo experimentation were derived from adult mice according to protocols described below. Experiments were performed with combined populations of male and female cells, isolated from adult mice (6- 10 weeks). Cells were cultured in a humid sterile environment at 37°C and 5%CO2. Cell authentication was performed by monitoring for TdT expression, and genotype verification of adult mice used to derive cell cultures.
METHOD DETAILS
Phenotypic methods
In vivo muscle regeneration
Animals were subject to four intraperitoneal (IP) tamoxifen injection (100 μL at 10 mg/mL in corn oil) at 24h intervals followed by a 72h recovery period. Mice were anesthetized with isoflurane (2% in oxygen), and skeletal muscle damaged through intra-muscular injection of cardiotoxin (10 μM in saline, 50 μL per muscle group) in the designated muscle group (TA, EDL, gastrocnemius, bicep posterior). Buprenorphine was concurrently administered (0.1 mg/kg) for pain management. Muscle regeneration periods were provided to experiment-specific endpoint. Re-injury experiments were performed through repeating the above steps at the specified time point. Upon experimental endpoint, mice were euthanized with CO2 asphyxiation and cervical dislocation.
In vivo EdU pulse
In vivo EdU pulse experiments were adapted from (Shea et al., 2010). First, EdU was dissolved in sterile saline (10 mg/mL) and administered to mice by IP-injection at a designated timepoint as follows. For EdU labeling of actively dividing cells using FACS analysis (Figures 2A and 2B), in vivo EdU was administered at either 28h post-injury followed with a 12h incubation to label activating satellite cells, or at 66h post-injury followed with a 6h incubation to label actively dividing myogenic precursors. For Edu pulse-label on regenerating skeletal muscle, EdU was administered with a single IP-injection at 48h or 72h post-injury, and allowed to regenerate for 7d post-injury.
Muscle perfusion
To maintain the TdTomato fluorescence signal, mice were perfused prior to muscle processing. For this, mice were euthanized with sodium pentobarbital (200 mg/Kg bodyweight) without cervical dislocation. Once no reflex could be detected, mice were exsanguinated by cardiac perfusion with 25 mL chilled PBS, then 50 mL chilled PFA (4% w/V, 4°C), both delivered at a flow rate of 5 mL/min. Harvested TA muscle was post-fixed in 4% PFA (w/V) overnight (4°C) followed by overnight incubation at 4°C over a two-layer sucrose gradient (15% w/V layered on 30% w/V sucrose in PBS). Muscle was embedded in Frozen Section Compound, and frozen in isopentane cooled in liquid-nitrogen.
Isolation of tibialis anterior (TA)
Isolation of TA muscle was modified from (Le Moal et al., 2018). Upon experimental endpoint, mice were euthanized as described above, and TA muscles harvested from the hindlimbs, submerged in a tinfoil cup containing Frozen Section Compound, then frozen in isopentane cooled in liquid nitrogen. For long-term storage, muscle was stored at −80°C. Skeletal muscle was processed by sectioning on a cryostat cooled to −20°C until the widest segment of the muscle was reached. From there, muscle was sectioned at 10 μm thickness and sections collected on positively-charged microscope slides. Slides were either immediately processed or placed at −80°C for long-term storage.
In vivo rescue of regeneration with pifithrin-α
Mice were induced with tamoxifen to induce Cre-mediated recombination prior to cardiotoxin injury of the TA muscle as described above. At 40h, 64h, and 88h post-injury, mice were administered an intraperitoneal injection of either the p53 inhibitor pifithrin-α (0.04mg dissolved in 50 μL of 20% captisol/PBS solution (v/v) or an untreated control (50 μL of 20% captisol/PBS solution (w/v) dilute in saline). Mice were sacrificed at 7-days post cardiotoxin-injury to assess regeneration of the TA as described above.
Single myofiber isolation
Single myofiber isolation from Extensor Digitorum Longus (EDL) muscle was performed using established protocols (Brun et al., 2018). EDL muscles were carefully harvested from a euthanized mouse and immediately placed in sterile PBS pre-warmed to 37°C. Paired EDL muscles from each mouse were processed together. EDLs were washed twice in pre-warmed PBS, then placed in digestion medium (2 mg/mL Collagenase Type 1 in DMEM high-glucose with sodium pyruvate). EDLs were gently swirled at 15 min intervals for 1.5h, then the digestion quenched through addition of primary growth medium (Ham’s F10 medium, 20% Bovine Growth Serum, 1% pen/strep, Fibroblast Growth Factor 2.5 ng/μL). Fibers were allowed to settle at the bottom of the plate (5 min), and the medium replaced with fresh primary growth medium. Primary growth medium was changed at 24h intervals up to the endpoint of the fiber culture. For EdU-pulsed experiments, one volume of 2X EdU solution (20 μM in primary growth medium) was added to one volume of fibers in growth medium, and incubated (37°C, 5% CO2) for the desired incubation period. Upon culturing endpoint, fibers were fixed (4%PFA, 20 min, RT) and stored in PBS.
Primary myoblast isolation
Primary myoblast isolation was carried out as described in (Hindi et al., 2017). First, skeletal muscle was dissected from hindlimbs of euthanized mice (TA, EDL, quadriceps, gastrocnemius, bicep posterior) and adipose tissue and tendons trimmed away. Skeletal muscle was minced with dissection scissors to small pieces (approx. 1 mm3), then washed twice in PBS. Muscle was enzymatically digested in digestion medium (1 mg/mL Type 1 collagenase, 1 U/mL Dispase II in Ham’s F10 medium, no serum) for 1h with gentle swirling at 15 min intervals (37°C, 5%CO2). On ice, digested muscle tissue was suspended in two volumes of chilled PBS, passed through a 40 μm strainer, and the mononuclear cells were recovered as a pellet by centrifugation (150 x g, 10 min, 4°C). For cell culturing, cells were preplated on an uncoated tissue culture dish for 3h, then the supernatant collected and plated on a Matrigel coated dish (20 μL Matrigel resuspended in 1 mL DMEM High-glucose medium, swirled on a 10 cm dish, and allowed to solidify for 1h at 37°C°, 5% CO2). For FACS sorting of MuSCs, cell pellets were resuspended in 350 μL Red Blood Cell lysis solution, then immediately resuspended in 10 mL FACS buffer (10% Bovine Growth serum in PBS, 3mM EDTA), and pelleted (350 x g, 4°C, 10 min). Cell pellets were resuspended in 1 mL FACS buffer, incubated with 647nm-fluorophore tagged alpha-integrin 7 (20 min, 4°C), washed, and exposed to Sytox 488 cell viability dye, then passed through a 50 μm cell strainer. Cells were stored on ice in a polystyrene tube while awaiting FACS sorting.
Differentiation of cultures myoblasts
Primary myoblasts were expanded while maintaining low cell confluence to avoid cell-cell contact on Matrigel-coated dish. Upon sufficient population expansion, myoblasts were trypsinized, resuspended in primary growth medium, and the cell concentration quantified using a Countess II Automated Cell Counter. Cells were plated on a Matrigel-coated dish at a density of 650 cells / mm2 (650,000 cells per well in a 6-well dish), allowed to adhere for 1h, then changed to a low-serum differentiation medium (2% horse serum, 0.1% insulin, 0.1% transferrin, 1% pen/strep in DMEM high-glucose with sodium pyruvate). Differentiation was allowed to proceed for 48h (37°C, 5% CO2) without any changes to the medium.
NSG mouse engraftment preparation
Allograft transplantation experiments were performed according to previously established protocols (Feige and Rudnicki, 2020) with minor modifications. NSG mice (Shultz et al., 2005) were anesthetized with isoflurane (2% in oxygen) and both the endogenous MuSC population in the hindlimbs incapacitated through 8 Gy irradiation, delivered to both hindlimb at 0.89 Gy/min (X-rad 320, Precision X-ray), while the rest of the body was protected with a lead shield. If damaged TA muscle was experimentally required, cardiotoxin (50 μL, 10 μM) was immediately administered to both TAs of the NSG recipient mice, with buprenorphine (0.1 mg/Kg bodyweight) for pain management. Mice were provided a 48h recovery period, then each TA was injected with 12,000 MuSCs derived from either WT or NELF-BscKO donor mice. A 21-day regeneration period was provided, then mice euthanized and perfused as previously described.
Primary myoblast drug treatments
Primary myoblasts were isolated from WT or NELF-BscKO mice and plated at low confluence in Matrigel-coated 12-well plates. Primary growth medium was refreshed with primary growth medium containing either PEDF (500 ng/mL), pifithrin-α (27 μM), or untreated primary growth medium, and provided a 4-hour incubation period. Subsequently, 1 volume of untreated primary growth medium, or primary growth medium containing the same concentrations of PEDF and pifithrin-α as previously described was spiked with EdU (20 μM) and added to the corresponding wells for a 4 h incubation period. Myoblasts were subsequently fixed with PFA (4% (w/v) in PBS) and treated for subsequent immunofluorescent characterization (see ‘immunofluorescent staining of cell cultures’, below).
Apoptosis screening assay
Primary myoblasts were isolated and expanded in culture as previously described. Apoptosis screening was performed using an activated capsase screening kit, following the manufacturer’s protocol (FAM-FLICA poly caspase kit, Bio-Rad Antibodies, ICT091). A positive control was prepared alongside experimental samples by adding staurosporine (5 μM in DMSO) for 3 h to the culture medium of WT myoblasts.
Immunofluorescent characterization
Myofiber immunofluorescent staining
Isolation of myofibers was performed according to protocols described in (Brun et al., 2018). Fixed fibers were permeabilized (5% Triton X-100 vol/vol in 100 mM glycine in TBS), reacted with EdU Click-It chemistry according to manufacturer protocol, pre-blocked in Goat anti-mouse IgG (100 μg/mL in TBS), then blocked for 1h (5% goat serum, 2% BSA in TBST). Fibers were subsequently incubated with primary antibody (overnight, 4°C) according to the specified dilution (key resources table), then with secondary antibody dilute in TBST, counterstained with DAPI (0.2 mg/mL in PBS), and mounted with fluorescent mounting media. Immunofluorescence images were captured using a Zeiss Z1 inverted epifluorescent microscope.
Immunofluorescent staining of muscle cross-sections
Slides containing muscle cross-sections were thawed to room temperature, washed (PBS), then fixed with PFA (4% w/v, 10 min). Antigen retrieval was performed (heat-induced epitope retrieval, 98°C°, 10 min), then slides moved to PBS (RT), and permeabilized (0.5% Triton X-100 V/V, 100mM glycine, in TBS) for 10 min. An optional EdU Click-it reaction was performed at this step according to the manufacturer protocol, then tissues washed (TBS), and blocked (1h, RT, 5% Goat serum V/V, 2% BSA w/V in TBST with 1:40 Mouse-on-mouse Ig blocking reagent). Samples were incubated with primary antibody (4°C, overnight), washed (TBS), and secondary antibody dilute in TBST was added (1h, RT). DAPI counterstaining followed (0.2 mg/mL in TBS), and mounted with fluorescent mounting media. Where applicable, the TdTomato fluorescence signal was preserved through PFA perfusion (described above) and did not require any signal rescue. Immunofluorescence images were captured using a Zeiss Z1 inverted epifluorescent microscope.
Immunofluorescent staining of cell cultures
Cell cultures were fixed in PFA (4% w/V, 10 min, RT), washed (PBS), then permeabilized (5% Triton X-100 vol/vol, 100 mM glycine, 20 min, RT). If cells were pre-incubated with EdU, the EdU Click-It reaction was performed according to the manufacturer’s protocol; otherwise, this step was omitted. Cells were blocked with blocking buffer (5% goat serum V/V, 2% BSA w/V, in TBST) for 1h, then incubated with primary antibody diluted in blocking buffer (4°C, overnight) according to the dilution in the key resources table. Cells were washed (TBS), incubated with secondary antibody dilute in TBS (1h, RT), counterstained with DAPI, and resuspended in TBS. Cells maintained in culture dish during staining were covered with PBS and immediately imaged. Immunofluorescence images were captured using a Zeiss Z1 inverted epifluorescent microscope.
Real-time quantitative PCR
A subset population of FACS isolated MuSCs derived from the donor mice for engraftment experiments was set aside, and RNA isolated using a NucleoSpin RNA Plus XS micro kit for RNA purification kit (Macherey-Nagel) according to the manufacturer protocol. Reverse transcription was performed to generate a cDNA library using random primers. Quantitative PCR was performed using Taqman gene expression assays according to the manufacturer protocol.
Deep-sequencing sample preparation
Single-cell RNA sequencing
Sample preparation for single-cell RNA sequencing were modified from protocols described in (Giordani et al., 2019). Regenerating TdT+ cells were isolated from mice at 72h post-injury and purified by FACS as described above. TdT+ cells were concentrated to approximately 600 cells / μL, and their concentrations verified by quantification on a hemocytometer. Cell viability was monitored to assure viability remained above 85%, then 2,000 cells were prepared for each sample according to the 10X platform manufacturing guidelines. Once complete, samples were sequenced on an Illumina NextSeq 500 Platform with a read depth of 50,000 reads per cell.
RNA-sequencing
TdT+ myogenic cells for in vivo RNA-seq regeneration studies were isolated and purified from mice at either 48h or 120h post-injury as described above. RNA was collected using a PureLink RNA mini kit according to the manufacturer’s protocol. Total RNA (340 ng) underwent rRNA depletion, and cDNA library prepared using a KAPA stranded mRNA sequencing kit according to the manufacturer protocol. Sequencing was performed on an Illumina HiSeq 2500 platform.
Precision run-on sequencing
MuSCs were isolated from NELF-BscKO and WT mice as described above for primary myoblast culturing techniques. After 48h of colony expansion, myoblasts were exposed to 4-hydroxytamoxifen (5 μM in primary growth medium) for 72h, refreshing the primary growth medium and 4-OHT at 24h intervals. Primary myoblasts were maintained at low confluence to avoid cell-cell contacts. Cell permeabilization was performed as follows. Primary myoblasts were detached with trypsin, collected in chilled (4°C) PBS with 10% BGS, and immediately quantified. Using 10x106 cells per replicate, myoblasts were centrifuged (150 x g, 10 min, 4°C), resuspended in wash buffer (10 mM Tris-HCl pH 8, 10 mM KCl, 250 mM sucrose, 5 mM MgCl2, 0.5 mM DTT, 10% glycerol), and passed through a 40 μm strainer. Samples were permeabilized with permeabilization buffer (wash buffer supplemented with 0.1% Igepal) for 90s on a nutator, then immediately centrifuged (100 x g, 5min, 4°C). Permeabilized myoblasts were resuspended in freezing buffer (50 mM Tris HCl pH 8.3, 40% glycerol, 5 mM MgCl2, 0.5 mM DTT) to a final concentration of 10 x 106 cells / mL. Nuclear run-on was performed according to the run-on assay described in (Mahat et al., 2016).
Genome-wide localization using CUT&Tag
Cleavage Under Target and Tagmentation (CUT&Tag) was performed as originally described in the protocol (Kaya-Okur et al., 2019). Briefly, primary myoblasts were exposed to 4-hydroxytamoxifen (5 μM in primary growth medium) for 72h, refreshing the primary growth medium and 4-OHT at 24h intervals. Recovered myoblasts were then immobilized on Concanavalin A-coated paramagnetic beads and permeabilized with digitonin. Cells were incubated overnight with the corresponding antibodies: Nelf-E (Proteintech, Cat#10705-1-AP + Nelf-E(F-9) Santa Cruz, Cat#SC – 377052, mixed 1:1 ratio); RNAPII S5P (D9N5I - Cell Signaling Cat #13523); and H3K4me3 (EMD Millipore, Cat# 07-473). Tethered cells were washed thoroughly to remove unbound antibody, and then with pA-Tn5 for 60 min. Tn5 transposase mediated tagmentation was then initiated by the addition of MgCl2. Cellular DNA was extracted using Phenol/Chloroform/IsoamyI alcohol and then precipitated. Tagmented DNA libraries were generated as previously described (Kaya-Okur et al., 2019), and sequenced on an Illumina HiSeq 2500 platform.
Deep-sequencing analysis
No new code was generated for sequencing dataset analysis. All code used was obtained from previously published peer-reviewed journals indicated below.
Single-cell RNA sequencing
Single-cell RNA-sequencing data were analyzed on the 10X cellranger platform using v2.1.1 to generate a gene expression matrix. This was analyzed using Seurat V3.0 (Stuart et al., 2019; Butler et al., 2018) to remove non-myogenic populations and perform clustering occupancy analysis. Myogenic populations were identified and exported for further use on PAGA (partition-based graph abstraction) on Scanpy v1.4. Various trajectories were constructed using ForceAtlas2 embedding as previously described (Jacomy et al., 2014).
RNA-sequencing analysis
RNA-seq reads were aligned using STAR v2.6.0a, counts summarized using FeatureCounts v1.6.1, and differential gene expression identified with DESeq2 v1.26.0. Significant genes were identified with a p-value cutoff < 0.01. DotPlots were constructed using ggplot2, and Volcano Plots with EnhancedVolcano functions on R version 3.6.3. In all instances, gene ontology analysis was performed using DAVID 6.8 (Huang et al., 2009a, 2009b), and values represented as DotPlots.
CUT&Tag analysis
High-throughput sequencing of CUT&Tag samples were processed by first trimming adapters from raw reads with Cutadapt 2.6 (Martin, 2011). The trimmed reads were aligned to the mouse genome (mm10) with Bowtie2 (v2.3.4.1) using the following parameters “—local –very-sensitive-local –no-unal –no-mixed –no-discordant –phred33 -I 10 -X 700” (Langmead and Salzberg, 2012). Samtools v1.10 was used for post-alignment processing i.e. duplicates were marked and removed; non-uniquely aligned reads were filtered and only reads mapped in proper pairs were retained for analysis.
Spike-in normalized genomic coverage was calculated using bamCoverage (deeptools v3.3.2) (Ramírez et al., 2016). For the normalization, trimmed reads were aligned to spike-in DNA with Bowtie2 (v2.3.4.1) and the resulting number of total mapped fragments was used to calculate the per-sample scaling factors. For each sample, this was done by dividing the min(total-mapped-fragments) by the total-mapped-fragments for that sample. Additionally, bamCoverage was used to generate bigWig tracks for visualization as well as normalized bedGraphs for downstream peak-calling.
SEACR was used to call “stringent” peaks for each spike-in normalized bedgraph target file (Meers et al., 2019). In the presence of an IgG control, the control data were used to calculate the empirical thresholds and conversely, in the absence of a control, top 5% of enriched regions were selected as peaks by AUC. ENCODE Blacklist regions (Amemiya et al, 2019) were filtered from peaks using BEDTools v2.27.1 (Quinlan and Hall, 2010) and nearest-gene annotations were obtained with ChIPseeker v1.24.0 (Yu et al., 2015).
Signal intensity heatmaps for the Cut&Tag libraries were generated using deeptools v3.3.2 (Ramírez et al., 2016). Using both normalized bigwigs and the M25 mouse gene annotation file from Gencode, ComputeMatrix was used with the scale-regions mode (–beforeRegionStartLength 1000, –regionBodyLength 5000, –afterRegionStartLength 1000). PlotHeatmap was used with the resulting matrix to plot the signal distribution.
PRO-sequencing analysis
FASTQ read pairs were trimmed to 41bp per mate, and read pairs with a minimum average base quality score of 20 were retained. Read pairs were then further trimmed using cutadapt 1.14 to remove adapter sequences and low-quality 3’ bases (–match-read-wild-cards -m 20 -q 10).
R1 reads were then aligned to the spike genome index (dm3) using Bowtie 1.2.2 (-v 2 -p 6 –best –un) (Langmead and Salzberg, 2012), with those reads not mapping to the spike genome serving as input to the reference, i.e. primary, genome alignment step (again using the same Bowtie 1.2.2 options). Reads mapping to the mm10 reference genome were then sorted, via samtools 1.3.1 (-n), and subsequently converted to bedGraph format. Because PRO-seq reveals the position of the RNA 3’ end, the “+” and “−“ strands were swapped to generate bedGraphs representing 3’ end positions. The bedGraphs were ultimately depth-normalized only, merged within conditions, and used to generate bigWig files binned at 10bp. TSS-centric read count matrices were calculated over a window of +/− 2kb with 25bp bins.
For direct comparison to PRO-Seq data, RNA-sequencing analysis was performed on proliferating primary myoblasts. FASTQ read pairs were aligned to a GRCm38 cDNA index and quantified using kallisto 0.45.1 (-b 30 –rf-stranded –genomebam –gtf –chromosomes -t 6). Replicate samples belonging to the same condition were merged and depth-normalized, and bigWigs were then generated with 10bp binning. Differentially expressed genes were determined via a combination of tximport v1.12.3 and DESeq2 v1.24.0, using a padj < 0.01 threshold.
To integrate the differentially expressed genes, determined via RNA-seq, with the PRO-seq analyses, all wild type R2 reads (i.e. 5’ end) were aligned (in the same manner as 3’ end R1 reads) to identify putative transcription start sites using TSScall (https://github.com/lavenderca/TSScall, –set_read_threshold 8 –annotation_join_distance 500 –annotation_search_window 200). A single, dominant TSS was then assigned to each gene based on having the highest number of reads within a +/− 150 interval relative to each called TSS. Dominant TSS-centric 3’ read count matrices were then intersected with lists of up- and down-regulated gene lists from the RNA-seq analyses to visualize TSS-proximal profiles. Gene body read counts (+250-2250 relative to dominant TSS’s) were similarly intersected with the RNA-seq results and statistics were computed via Wilcoxon test.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless stated in the manuscript, all values are reported as the mean where error bars represent SEM. Statistical significance was identified among comparisons having a p-value below 0.01 in transcriptome data. For all other experiments *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Statistical quantification was performed on the statistical graphing software GraphPad Prism v7.0.
Supplementary Material
Highlights.
Muscle progenitors are a major source of muscle stem cells on regenerated myofibers
Loss of NELF in regeneration causes muscle stem cell pool depletion
NELF transduces signals that modulate the expansion of the muscle progenitor population
NELF-mediated expansion of progenitors occurs through regulation of p53 activity
ACKNOWLEDGMENTS
We thank Lynn Megeney for insightful discussions and comments on the manuscript; Caroline Brun, Peter Feige, Alex Lin, and Marie-Claude Sincennes for helpful advice; Odile Neyret for Cut&Tag and RNA-seq library sequencing; Kathy Sheikheleslamy for scRNA-seq library sequencing; Fernando Ortiz for assistance with FACS; and the HMS Nascent transcriptomics core for PRO-seq; Magid Fallahi for ensure breeding of the mouse colony; and Lifang Li for technical help. This work was funded by the Canadian Institutes of Health Research: FDN-143330 to F.J.D., MOP-343603 to M.B.; and Harvard Medical School Startup Funds to K.A. D.C.L.R. held studentships from the CIHR, OGS, Mitacs, and QEIIST training programs. G.M.N. holds an NIH Biomedical Informatics and Data Science Research Training Program (BIRT) grant - T15LM007092.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.devcel.2021.02.025.
REFERENCES
- Adelman K, and Lis JT (2012). Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet 13, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman K, Marr MT, Werner J, Saunders A, Ni Z, Andrulis ED, and Lis JT (2005). Efficient release from promoter-proximal stall sites requires transcript cleavage factor TFIIS. Mol. Cell 17, 103–112. [DOI] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amleh A, Nair SJ, Sun J, Sutherland A, Hasty P, and Li R (2009). Mouse cofactor of BRCA1 (Cobra1) is required for early embryogenesis. PLoS One 4, e5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi Y, Smith ER, Shah AP, Rendleman EJ, Marshall SA, Woodfin AR, Chen FX, Shiekhattar R, and Shilatifard A (2020). NELF regulates a promoter-proximal step distinct from RNA Pol II pause-release. Mol. Cell 78, 261–274.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, and Chazaud B (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz A, Sebastian S, and Dilworth FJ (2012). The origin and fate of muscle satellite cells. Stem Cell Rev. Rep 8, 609–622. [DOI] [PubMed] [Google Scholar]
- Baghdadi MB, Castel D, Machado L, Fukada SI, Birk DE, Relaix F, Tajbakhsh S, and Mourikis P (2018). Reciprocal signalling by Notch-Collagen V-CALCR retains muscle stem cells in their niche. Nature 557, 714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, and Olwin BB (2014). p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med 20, 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, and Levine M (2009). Synchronous and stochastic patterns of gene activation in the Drosophila embryo. Science 325, 471–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, and Muñoz-Cánoves P (2016). The ins and outs of muscle stem cell aging. Skelet. Muscle 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun CE, Wang YX, and Rudnicki MA (2018). Single EDL myofiber isolation for analyses of quiescent and activated muscle stem cells. Methods Mol. Biol 1686, 149–159. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal JV, Christensen J, Xiang W, Tierney MT, Boscolo FS, Sacco A, and Brack AS (2014). Early forming label-retaining muscle stem cells require p27Kip1 for maintenance of the primitive state. Development 141, 1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L, and Adelman K (2019). Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev. 33, 960–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day DS, Zhang B, Stevens SM, Ferrari F, Larschan EN, Park PJ, and Pu WT (2016). Comprehensive analysis of promoter-proximal RNA polymerase II pausing across mammalian cell types. Genome Biol. 17, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding DC, Wen YT, and Tsai RK (2017). Pigment epithelium-derived factor from ARPE19 promotes proliferation and inhibits apoptosis of human umbilical mesenchymal stem cells in serum-free medium. Exp. Mol. Med 49, e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod ND, Henriques T, Huang KL, Tatomer DC, Wilusz JE, Wagner EJ, and Adelman K (2019). The integrator complex attenuates promoter-proximal transcription at protein-coding genes. Mol. Cell 76, 738–752.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evano B, Khalilian S, Le Carrou G, Almouzni G, and Tajbakhsh S (2020). Dynamics of asymmetric and symmetric divisions of muscle stem cells in vivo and on artificial niches. Cell Rep 30, 3195–3206.e7, 10.1016/j.celrep.2020.01.097. [DOI] [PubMed] [Google Scholar]
- Fant CB, Levandowski CB, Gupta K, Maas ZL, Moir J, Rubin JD, Sawyer A, Esbin MN, Rimel JK, Luyties O, et al. (2020). TFIID enables RNA polymerase II promoter-proximal pausing. Mol. Cell 78, 785–793.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige P, Brun CE, Ritso M, and Rudnicki MA (2018). Orienting muscle stem cells for regeneration in homeostasis, aging, and disease. Cell Stem Cell 23, 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige P, and Rudnicki MA (2020). Isolation of satellite cells and transplantation into mice for lineage tracing in muscle. Nat. Protoc 15, 1082–1097. [DOI] [PubMed] [Google Scholar]
- Gilchrist DA, and Adelman K (2010). Moonshine illuminates a developmental role for regulated transcription elongation. Dev. Cell 19, 9–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordani L, He GJ, Negroni E, Sakai H, Law JYC, Siu MM, Wan R, Corneau A, Tajbakhsh S, Cheung TH, and Le Grand F (2019). High-dimensional single-cell cartography reveals novel skeletal muscle-resident cell populations. Mol. Cell 74, 609–621.e6. [DOI] [PubMed] [Google Scholar]
- Gordon EM, Ravicz JR, Liu S, Chawla SP, and Hall FL (2018). Cell cycle checkpoint control: the cyclin G1/Mdm2/p53 axis emerges as a strategic target for broad-spectrum cancer gene therapy - a review of molecular mechanisms for oncologists. Mol. Clin. Oncol 9, 115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte R, Muse GW, Chinenov Y, Adelman K, and Rogatsky I (2013). Glucocorticoid receptor represses proinflammatory genes at distinct steps of the transcription cycle. Proc. Natl. Acad. Sci. USA 110, 14616–14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich DB, Nguyen PD, Siegel AL, Ehrlich OV, Sonntag C, Phan JM, Berger S, Ratnayake D, Hersey L, Berger J, et al. (2016). Asymmetric division of clonal muscle stem cells coordinates muscle regeneration in vivo. Science 353, aad9969. [DOI] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, and Adelman K (2013). Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol. Cell 52, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindi L, McMillan JD, Afroze D, Hindi SM, and Kumar A (2017). Isolation, culturing, and differentiation of primary myoblasts from skeletal muscle of adult mice. Bio Protoc. 7, e2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindi SM, Paul PK, Dahiya S, Mishra V, Bhatnagar S, Kuang S, Choi Y, and Kumar A (2012). Reciprocal interaction between TRAF6 and notch signaling regulates adult myofiber regeneration upon injury. Mol. Cell. Biol 32, 4833–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TC, Chiang YP, Chuang CK, Chen SL, Hsieh JW, Lan YW, and Tsao YP (2015). PEDF-derived peptide promotes skeletal muscle regeneration through its mitogenic effect on muscle progenitor cells. Am. J. Physiol. Cell Physiol 309, C159–C168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TC, Tsai SH, Yeh SI, Chen SL, Tung KY, Chien HY, Lu YC, Huang CH, and Tsao YP (2019). PEDF-derived peptide promotes tendon regeneration through its mitogenic effect on tendon stem/progenitor cells. Stem Cell Res. Ther 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jacomy M, Venturini T, Heymann S, and Bastian M (2014). ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the Gephi software. PLoS One 9, e98679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, and Rossi FM (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol 12, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, Fairclough S, Hellsten U, Isogai Y, Letunic I, et al. (2008). The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451, 783–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, and Rudnicki MA (2007). Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 129, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Moal E, Juban G, Bernard AS, Varga T, Policar C, Chazaud B, and Mounier R (2018). Macrophage-derived superoxide production and antioxidant response following skeletal muscle injury. Free Radic. Biol. Med 120, 33–40. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luse DS (2013). Promoter clearance by RNA polymerase II. Biochim. Biophys. Acta 1829, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado L, Esteves de Lima J, Fabre O, Proux C, Legendre R, Szegedi A, Varet H, Ingerslev LR, Barrès R, Relaix F, and Mourikis P (2017). In situ fixation redefines quiescence and early activation of skeletal muscle stem cells. Cell Rep. 21, 1982–1993. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12. [Google Scholar]
- Mauro A (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol 9, 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers MP, Tenenbaum D, and Henikoff S (2019). Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet. Chromatin 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, and Kardon G (2011). Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 138, 3625–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, and Adelman K (2007). RNA polymerase is poised for activation across the genome. Nat. Genet 39, 1507–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita T, Yamaguchi Y, Yano K, Sugimoto S, Chanarat S, Wada T, Kim DK, Hasegawa J, Omori M, Inukai N, et al. (2003). Human transcription elongation factor NELF: identification of novel subunits and reconstitution of the functionally active complex. Mol. Cell. Biol 23, 1863–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlikowski B, Betta ND, Antwine T, and Olwin BB (2019). Skeletal muscle stem cell self-renewal and differentiation kinetics revealed by EdU lineage tracing during regenaration. bioRxiv. 10.1101/627851. [DOI] [Google Scholar]
- Pawlikowski B, Pulliam C, Betta ND, Kardon G, and Olwin BB (2015). Pervasive satellite cell contribution to uninjured adult muscle fibers. Skelet. Muscle 5, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrello A, Cerone MA, Coen S, Gurtner A, Fontemaggi G, Cimino L, Piaggio G, Sacchi A, and Soddu S (2000). p53 regulates myogenesis by triggering the differentiation activity of pRb. J. Cell Biol 151, 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preibisch S, Saalfeld S, and Tomancak P (2009). Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai C-R, et al. (2014). mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature 510, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea KL, Xiang W, LaPorta VS, Licht JD, Keller C, Basson MA, and Brack AS (2010). Sprouty1 regulates reversible quiescence of a self-renewing adult muscle stem cell pool during regeneration. Cell Stem Cell 6, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. (2005). Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol 174, 6477–6489. [DOI] [PubMed] [Google Scholar]
- Siegel AL, Kuhlmann PK, and Cornelison DD (2011). Muscle satellite cell proliferation and association: new insights from myofiber time-lapse imaging. Skelet. Muscle 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, and Dilworth FJ (2013). Differential modulation of cell cycle progression distinguishes members of the myogenic regulatory factor family of transcription factors. FEBS J. 280, 3991–4003. [DOI] [PubMed] [Google Scholar]
- Stadelmayer B, Micas G, Gamot A, Martin P, Malirat N, Koval S, Raffel R, Sobhian B, Severac D, Rialle S, et al. (2014). Integrator complex regulates NELF-mediated RNA polymerase II pause/release and processivity at coding genes. Nat. Commun 5, 5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatomer DC, Elrod ND, Liang D, Xiao MS, Jiang JZ, Jonathan M, Huang KL, Wagner EJ, Cherry S, and Wilusz JE (2019). The Integrator complex cleaves nascent mRNAs to attenuate transcription. Genes Dev. 33, 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velthoven CTJ, de Morree A, Egner IM, Brett JO, and Rando TA (2017). Transcriptional profiling of quiescent muscle stem cells in vivo. Cell Rep. 21, 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma M, Asakura Y, Murakonda BSR, Pengo T, Latroche C, Chazaud B, McLoon LK, and Asakura A (2018). Muscle satellite cell cross-talk with a vascular niche maintains quiescence via VEGF and notch signaling. Cell Stem Cell 23, 530–543.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Urlaub H, and Cramer P (2018). Structure of paused transcription complex Pol II-DSIF-NELF. Nature 560, 601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LH, Fromm G, Gokey NG, Henriques T, Muse GW, Burkholder A, Fargo DC, Hu G, and Adelman K (2015). Pausing of RNA polymerase II regulates mammalian developmental potential through control of signaling networks. Mol. Cell 58, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosczyna MN, and Rando TA (2018). A muscle stem cell support group: coordinated cellular responses in muscle regeneration. Dev. Cell 46, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, and Young RA (2007). RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet 39, 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu HJ, Pan H, Cui Y, Wang XQ, Wang LJ, Li NS, Yang HB, and Gong FY (2014). The changes of serum glypican4 in obese patients with different glucose metabolism status. J. Clin. Endocrinol. Metab 99, E2697–E2701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at the Gene Expression Omnibus as the following DOIsq
The accession number for the RNA-seq and ChIP-Seq datasets reported in this paper is GEO: GSE150280
The accession number for the PRO-seq datasets reported in this paper is GEO: GSE149766
Analysized transcriptomic data are available on Mendeley Data: https://dx.doi.org/10.17632/r4tj555pc7.1