

Abstract
Small molecules that selectively bind to the pseudokinase JH2 domain over the JH1 kinase domain of JAK2 kinase are sought. Virtual screening led to the purchase of 17 compounds among which 9 were found to bind to V617F JAK2 JH2 with affinities of 40 – 300 μM in a fluorogenic assay. Ten analogues were then purchased yielding 9 additional active compounds. Aminoanilinyltriazine 22 was particularly notable as it shows no detectable binding to JAK2 JH1, and it has a 65-μM dissociation constant Kd with V617F JAK2 JH2. A crystal structure for 22 in complex with wild-type JAK2 JH2 was obtained to elucidate the binding mode. Additional de novo design led to the synthesis of 19 analogues of 22 with the most potent being 33n with Kd values of 2–3 μM for WT and V617F JAK2 JH2, and with 16-fold selectivity relative to binding with WT JAK2 JH1.
Keywords: JAK2 kinase, Pseudokinase domain, JH2/JH1 selectivity, Kinase inhibitors
Graphical Abstract
Irregularities in JAK-STAT signaling pathways are known to cause numerous forms of cancer [1]. The key components of the pathways are Janus kinases (JAKs), the signal transducer and activator of transcription factors (STATs), and cytokine receptors such as erythropoietin or interleukin receptors. Binding of cytokines causes an intracellular signaling cascade featuring phosphorylations by JAKs (JAK1–3 and TYK2), which are multidomain proteins with a C-terminal kinase domain (JH1) and an adjacent pseudo-kinase domain (JH2). Though JH2 domains have an ATP-binding site, they lack the characteristic catalytic aspartate, and they play an essential regulatory role for the JH1 kinase activity. Of particular interest is a single-point mutation, V617F, in the JAK2 JH2 domain, which is responsible for ca. 70% of myeloproliferative neoplasms (MPNs) [2–5]. These conditions including polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF) arise from over-production of one or more types of blood cells in bone marrow.
Studies have shown that the hyperactivation caused by the V617F mutation can be reversed by proximal mutations in the JH2 binding site and C helix [6,7]. This coupled with the desire to avoid undesirable side effects associated with inhibition of normal JAK2 JH1 kinase activity, such as thrombocytopenia and anemia, has suggested a small-molecule therapeutic strategy featuring selective binding to the ATP site of JAK2 JH2 [6,8,9]. As an important step in this direction, we have sought to understand the structural features required to achieve strong binding to JAK2 JH2 and simultaneously weak or no binding to the JAK2 JH1domain [10–12].
An initial screen of kinase inhibitors led to identification of JNJ7706621 (1) as a strong JAK2 JH2 binder, however, it has a similar ca. 0.5-μM affinity for the JAK2 JH1 ATP-site using a fluorescence polarization assay [10]. Through computer-aided design and crystallography it was possible to evolve this lead into a series of arylamidotriazoles showing improved binding and selectivity with members such as 2 with Kd values of 0.3 – 0.6 μM with JAK2 JH2 and 6 – 42 μM with JAK2 JH1 [12].
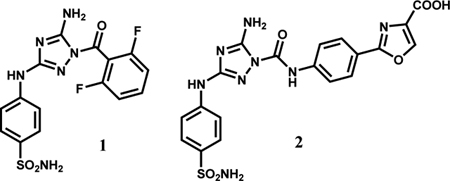
The present work reports on the discovery of an alternative series of JAK2 binding molecules that feature an indoloxytriazine core. This series arose from a virtual screen followed by lead refinement driven by computer-aided design and protein crystallography. Significant challenges are associated with direction of the right-hand substituent as in 2 into the polar ATP-site to achieve JH2/JH1 selectivity and strong binding.
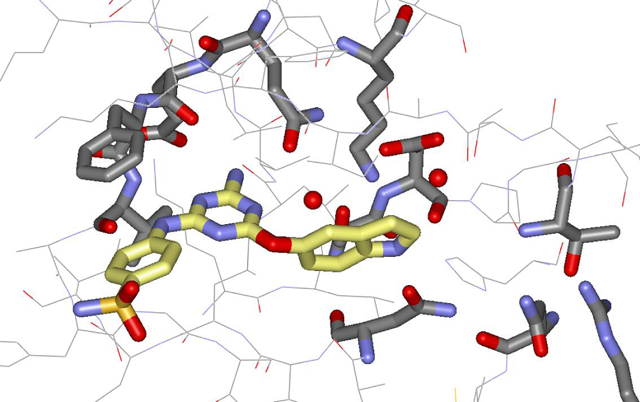
To begin, virtual screening was applied to find molecules that bind to the ATP site in the JH2 domain (Fig. 1). A crystal structure of JAK2 JH2 mutant V617F bound to ATP (PDBID: 4FVR [13]) was used for the protein target. The virtual screen was run using Glide SP [14] with the drug-like subset of the ZINC12 Database, which included more than 10 million compounds (10,637,968) with desirable ADME (absorption, distribution, metabolism and excretion) features [15]. Three hydrogen-bond constraints with Gln626, Glu627 and Val629 were included to provide strong binding to the hinge area.
Figure 1.

Virtual screening protocol.
The best-ranked ca. 6400 compounds were redocked with Glide XP [16], and the ca. 1000 best-scoring complexes were visualized. After additional considerations of potential metabolic labilities, synthetic ease of preparing analogues, and novelty, 17 of the docking hits were purchased. Nine (3, 6, 9, 11–15, 19) of the purchased compounds did show binding to JAK2 JH2 V617F with Kd values of 40–300 μM, and five also showed affinity for wild-type (WT) JAK2 JH2 with Kd values of 92–300 μM (Table S1). Furthermore, only three compounds, 5, 9, and 15, exhibited binding with JAK2 JH1. These initial binding affinities were obtained through a fluorescence polarization (FP) assay in which a fluorescently labelled ATP molecule, BODIPY-ATP, was displaced from the JH2 ATP-binding site [10]. The purchase of ten analogues of 13, 20–29, which were found by substructure search, provided 9 additional active compounds for V617F JAK2 JH2 (Table S1).
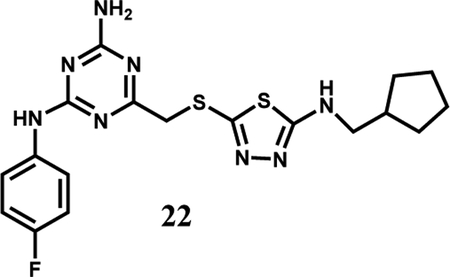
Compound 22 was particularly notable as it shows good selectivity with no binding to JAK2 JH1 and Kd values of 149 and 65 μM with WT and V617F JAK2 JH2, respectively. A crystal structure for the WT complex was obtained (Fig. 2A) and confirmed the four expected hydrogen bonds with Gln626, Glu627, and Val629. The thiadiazole-containing appendage, however, bends outward towards the solvent and the electron density for the terminal cyclopentylmethyl group could not be resolved. In order to achieve both selectivity and stronger binding, modifications to grow the ligand to the right towards Thr555 and Arg715 were desired (Fig. 2B) [12]. Modeling of complexes with WT JH2 using MCPRO [17] suggested that replacement of the right-hand substituent with substituted phenoxy groups might suffice, and benefits were also expected from replacing the fluorine atom in 22 with a cyano or sulfamyl group, as in 1 and 2 [12].
Figure 2.
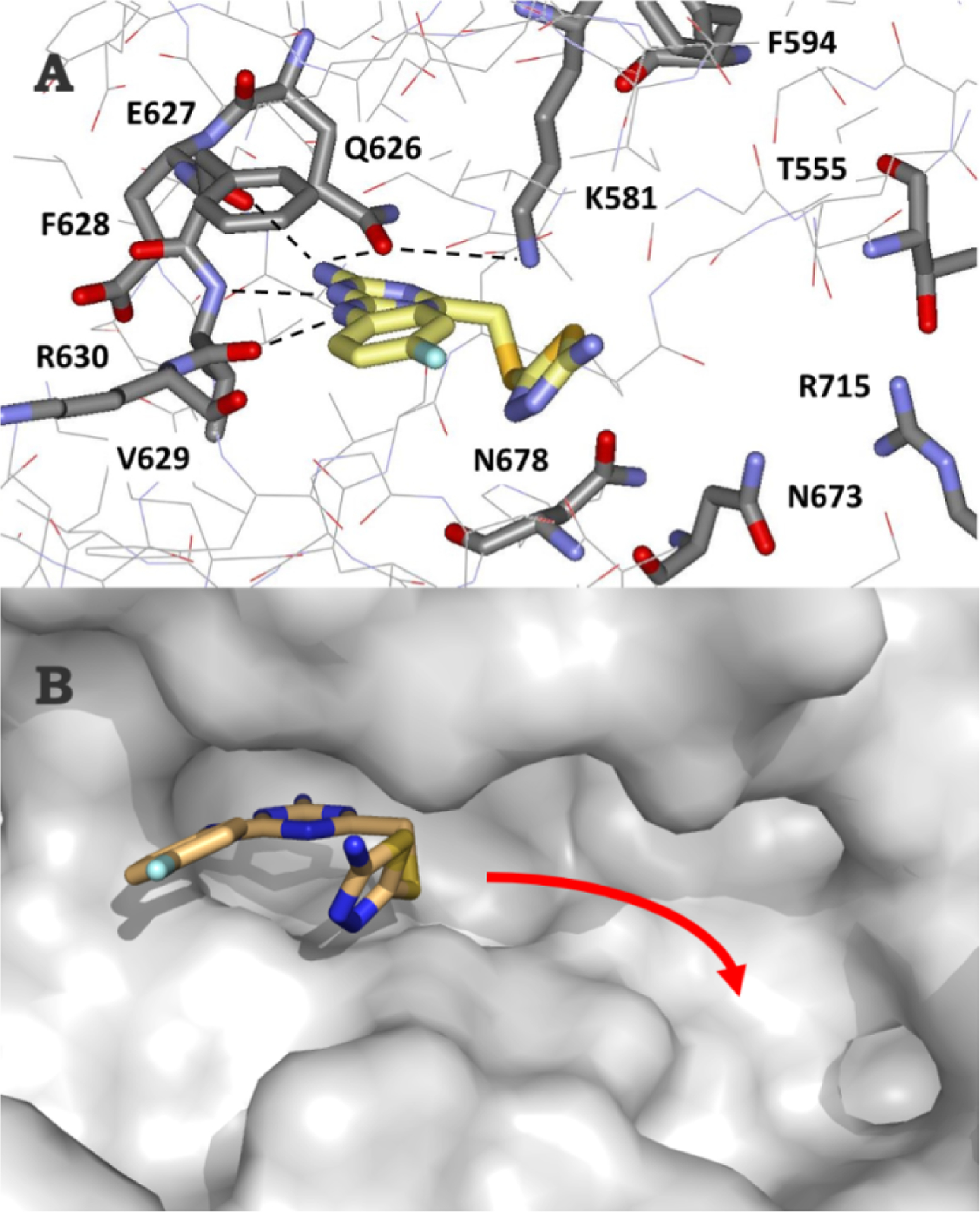
(A) Rendering from the 1.86-Å crystal structure of 22 with wild-type JAK2 JH2 (PDB ID 7JYQ). Orientation of 22, with hydrogen bonds in the hinge area dashed. (B) Desired direction of growth in the ATP-binding pocket is indicated with the red arrow.
Thus, alkylamino, phenoxy analogues 33a–m of 22 were prepared, as summarized in Scheme 1 and detailed in the ESI. The syntheses featured sequential SNAr introduction of the anilinyl and aryloxy groups starting from cyanuric chloride or the aminodichlorotriazine. The new compounds reported here are summarized in Table 1. Binding affinities were determined using the updated FP assay with a tracer derived from 1 [11, 12]. The three protein domains, WT JAK2 JH2, V617F JAK2 JH2, and WT JAK2 JH1, were expressed and purified as previously described [10,12].
Scheme 1.
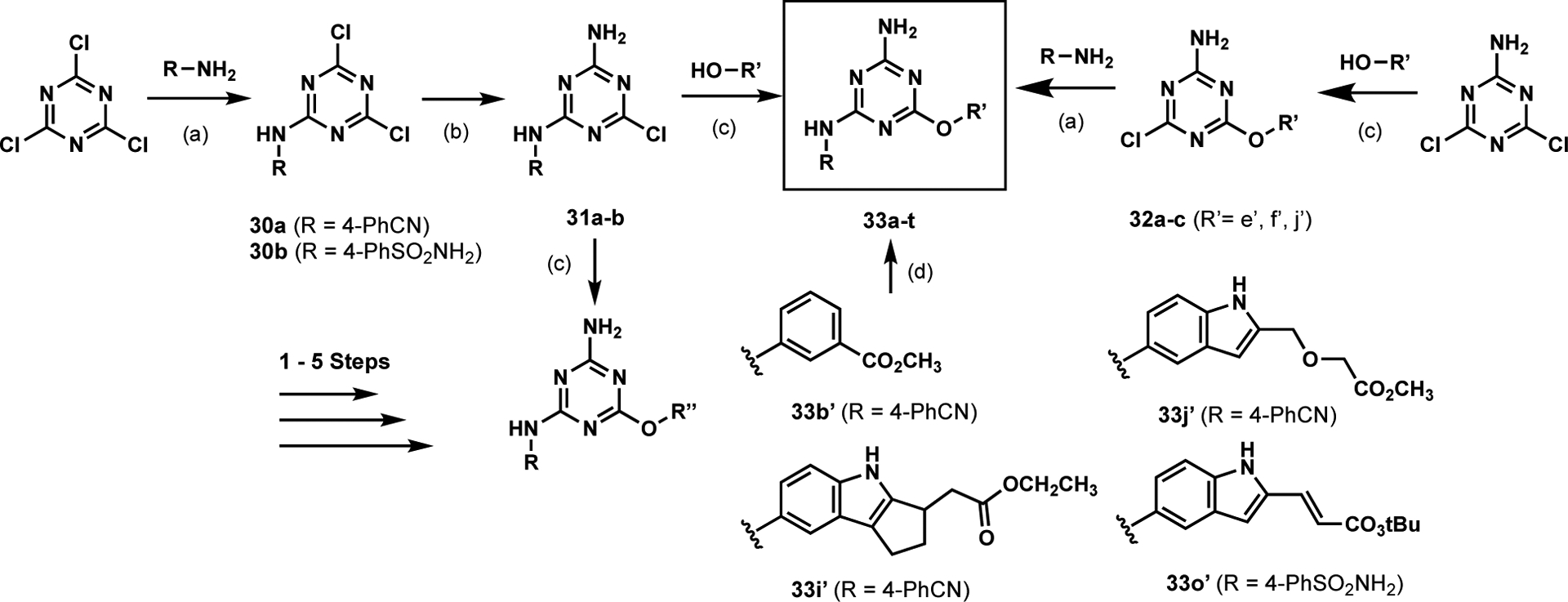
General synthetic scheme for anilinylaryloxytriazines.a
a Reagents and conditions: (a) K2CO3, Acetone, 0 → 23 °C; or K2CO3, DMF, 70 °C, 10–86 % yield (b) NH4OH (28%), Acetone, 0 → 23 °C, 71–85 % yield (c) K2CO3, Acetone, 0 → 23 °C; or K2CO3, DMF, 60–70 °C, 12.5–68 % yield (d) 2M NaOH (aq) or TFA, THF or EtOH, or dioxane, or CH2Cl2.
Table 1.
Compounds (33) and their experimental binding affinities (Kd) using the fluorescence polarization (FP) assay [11].
| Kd (μM) | |||||
|---|---|---|---|---|---|
| Compound | R | R’ | JAK2 JH1 | JAK2 JH2 WT | JAK2 JH2 V617F |
| 33a | a | a’ | 73.4 ± 20.3 | ||
| 33b | a | b’ | 39.6 ± 3.2 | ||
| 33c | a | c’ | 40.7 ± 4.9 | ||
| 33d | a | d’ | 43.8 ± 12.2 | ||
| 33e | a | f’ | 17.4 ± 9.1 | ||
| 33f | a | g’ | 10.7 ± 1.9 | ||
| 33g | a | h’ | 34.9 ± 1.5 | ||
| 33h | a | J’ | 47.4 ± 6.9 | ||
| 33i | a | k’ | 14.0± 1.0 | ||
| 33j | a | l’ | ND (15% @ 50 μM) | 5.9± 0.7 | 4.3± 0.1 |
| 33k | b | h’ | 20.9 ± 9.2 | ||
| 33l | b | i’ | 31.6± 5.7 | ||
| 33m | b | J’ | ND (16% @ 50 μM) | 18.3 ± 3.3 | 8.0± 0.8 |
| 33n | b | l’ | 41.1 ± 7.8 | 2.6 ± 0.1 | 2.4 ± 0.4 |
| 33o | b | m’ | 18.0± 2.7 | 3.3± 1.2 | 2.0± 0.1 |
| 33p | c | J’ | 53.5 ± 17.9 | ||
| 33q | d | J’ | ND (15% @ 50 μM) | ||
| 33r | f | e’ | 122.7 ± 22.0 | ||
| 33s | f | f’ | 54.7 ± 12.2 | ||
| 33t | f | J’ | 23.2 ± 1.4 | ||
| JNJ7706621 (1) | 0.671 ± 0.175 | 0.456 ± 0.124 | 0.599 ± 0.087 | ||
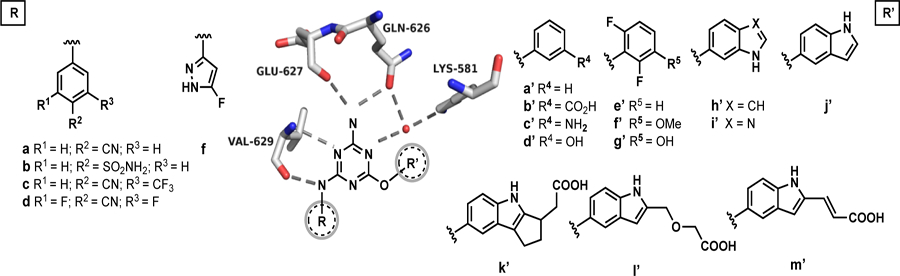
The SAR was initially explored for the p-cyanoanilinyl analogues 33a–f. The parent phenoxy compound 33a showed improved binding to WT JAK2 JH2 with a Kd of 73 μM, and addition of a meta carboxylate, amino, or hydroxy group brought a two-fold benefit to ca. 40 μM (33b–d, Table 1). Fortunately, it was possible to obtain a crystal structure for the complex of 33b with WT JAK2 JH2 (Fig. 3A); however, it revealed that the phenoxy substituent is again directed outward with one oxygen atom of the m-carboxylate group engaged in three hydrogen bonds with the backbone nitrogen (2.79 Å) and hydroxyl oxygen (2.84 Å) of Ser633 and with the hydroxyl oxygen (2.95 Å) of Thr636. This suggested modification of 33d to increase the acidity of the hydroxyl group by addition of two ortho fluorine atoms yielding 33f. A lower Kd of 10.7 μM did result, though the expectation was that the substituent remained directed outward, as in Figure 3A.
Figure 3.
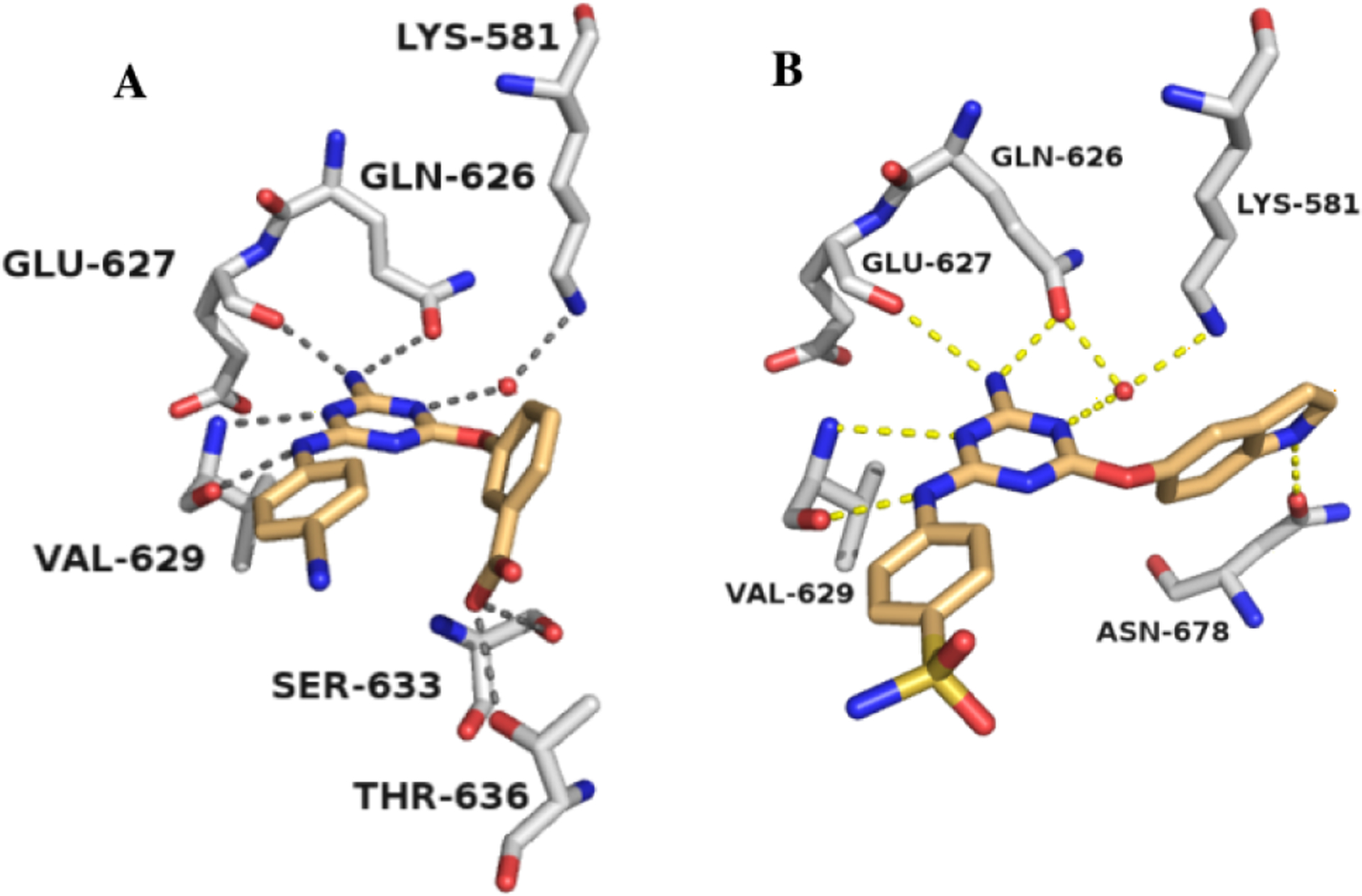
Renderings from the (A) 2.16-Å crystal structure of 33b (PDB ID 7JYO) and (B) 2.02-Å crystal structure of 33m (PDB ID 6XJK) with wild type JAK2 JH2.
Modeling then considered bicyclic aryloxy possibilities, which led to the notion that a 5-indoloxy substituent might be directed inward to form a hydrogen bond between the indole NH and the sidechain carbonyl group of Asn678. Thus, the cyano and sulfamyl alternatives 33h and 33m were synthesized and yielded Kd values of 47 and 18 μM, respectively. Importantly, the structural prediction was confirmed by a crystal structure for 33m (Fig. 3B). The isomeric indole 33k and benzimidazole 33l are less potent, though they are also expected to benefit from cation-π interactions with Lys581 [18].
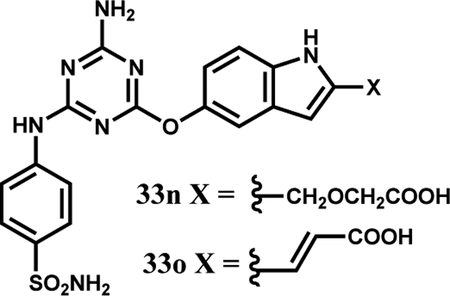
The next step was to further extend the indoles by attachments at C2 to terminate in a carboxylic acid group as in 2 (Fig. 4). This was realized with 33i, 33j, 33n, and 33o, which did yield the strongest binding molecules at 2–3 μM for WT and V617F JAK2 JH2. The 9- and 17-fold selectivities of 33n and 33o for binding the V617F JH2 domain over WT JH1 were also gratifying (Table 1). The right-hand-sides of 33n and 33o were synthesized from the common intermediate 37 (Scheme 2), which arose from DIBAL-reduction of the C2- ethyl ester after TBS-protection of the C5-hydroxyl group. Rh(II)-catalyzed etherification of 37 followed by TBAF-deprotection of the silyl group yielded ester 34, while ester 35 was prepared by oxidation of 37 to the aldehyde, Wittig reaction, and deprotection. The two hydroxyesters underwent SNAr coupling with anilinylchlorotriazines to yield 33n directly (the ester hydrolyzed during coupling) and the ester of 33o, which upon hydrolysis provided the corresponding acrylic acid.
Figure 4.
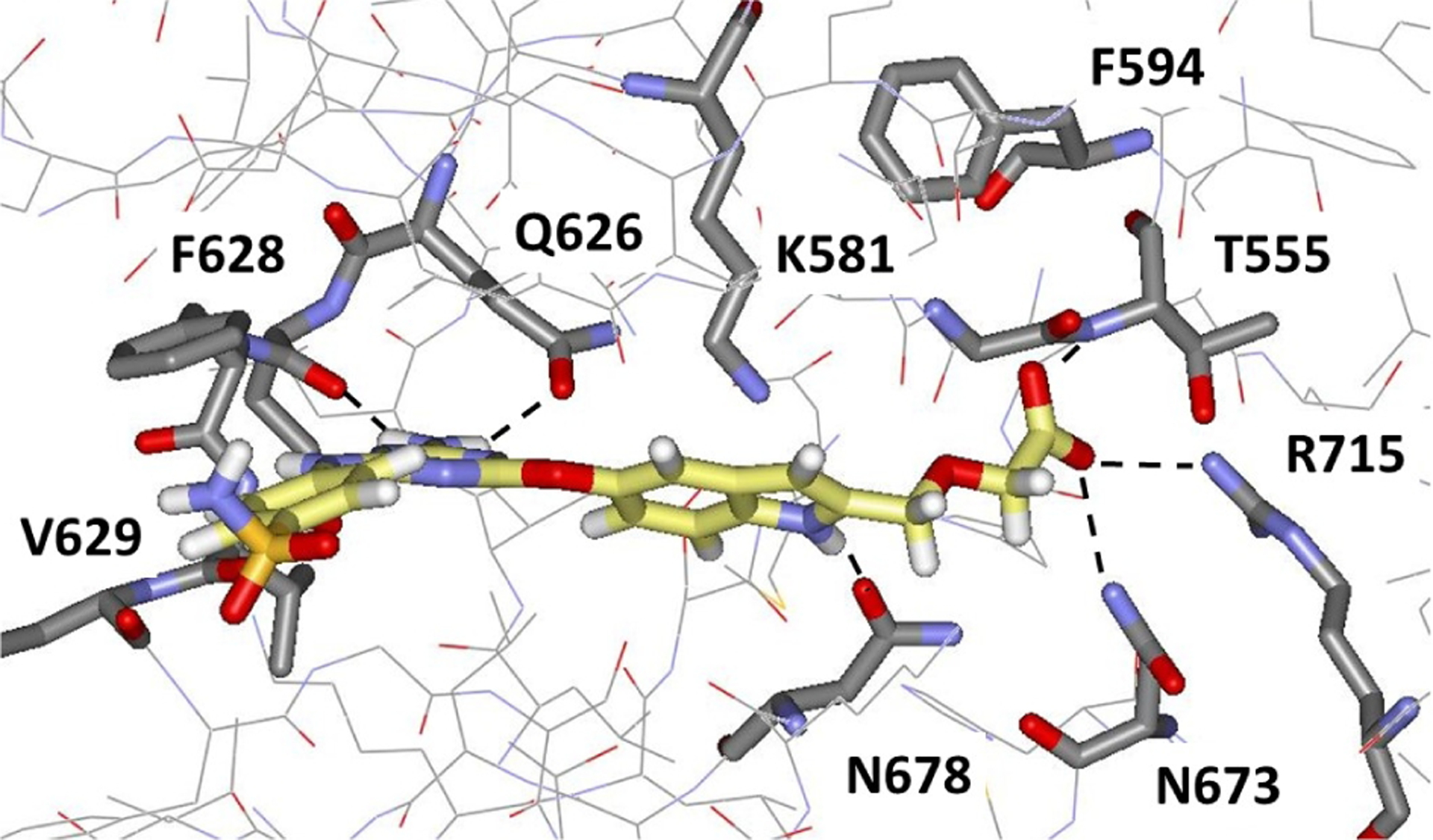
Computed structure using MCPRO [17] for 33n with WT JAK2 JH2 illustrating the expected eastward extension towards Arg715.
Scheme 2.
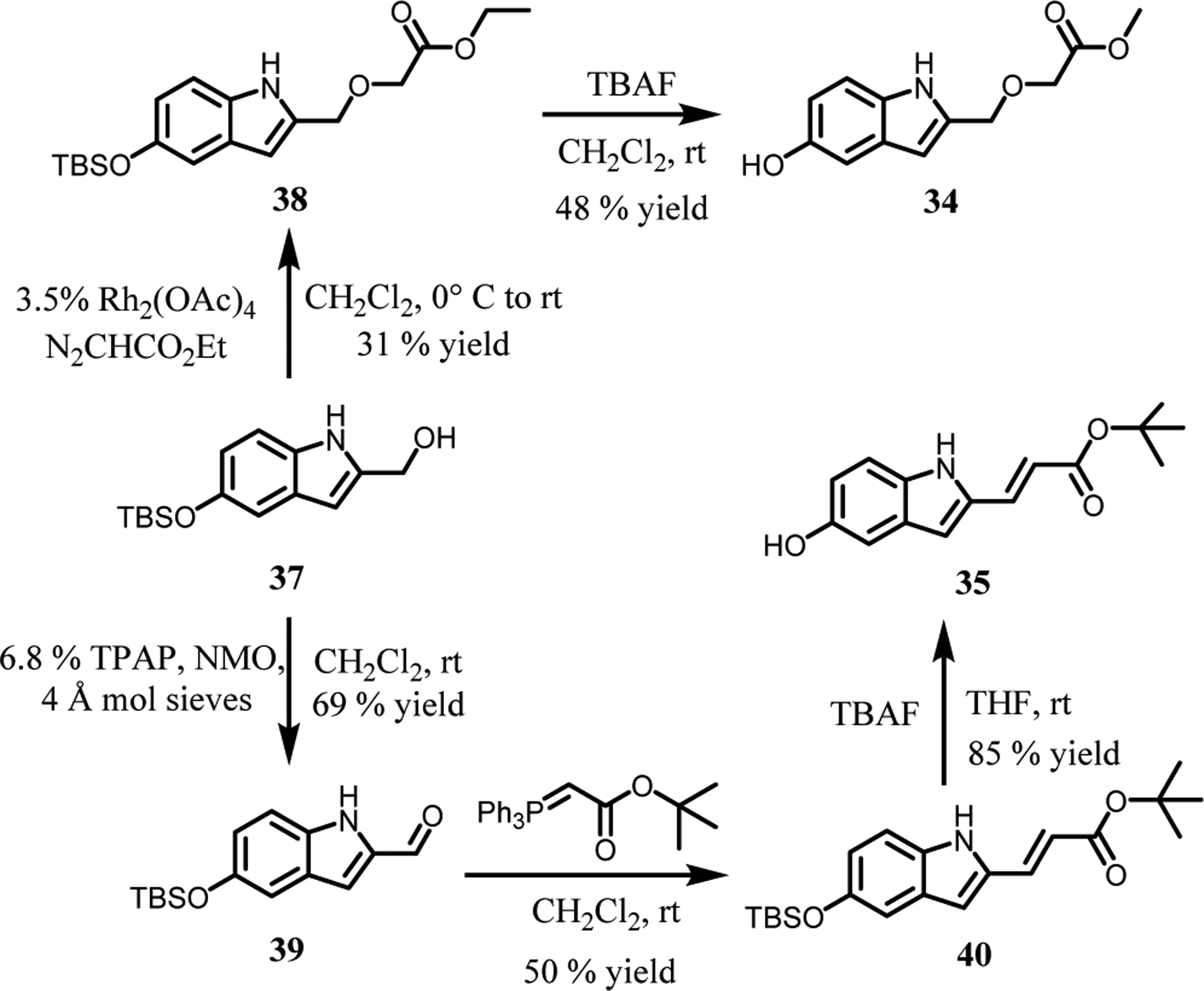
Synthetic scheme for substituted indoles.
Finally, some modifications of the anilinyl group were considered with 33p–t, but they did not yield clear binding boosts. The near-equipotency for 33t and 33m, however, does provide the 3-amino-5-fluoropyrazole group as a promising p-sulfamylaniline surrogate. Additional studies with 33n, 33o, and analogues are ongoing. This includes investigation of prodrug strategies to increase permeability for potential testing in cell assays to seek compounds that selectively reduce the hyperactivity of V617F JAK2.
Supplementary Material
Aberrant increase in JAK2 kinase activity causes myeloproliferative disorders
Selective binding to the JAK2 pseudokinase domain JH2 may be deactivating
Small molecules that selectively bind JH2 over the JH1 kinase domain are reported
The best binding molecules have an indoloxytriazine core
Acknowledgments
Gratitude is expressed to the U.S. National Institutes of Health (GM32136) for research support, to the Deutsche Forschungsgemeinschaft (DFG) for a postdoctoral fellowship to S.G.K. (KR 5228/1-1), and to the NIH for training support to D.E.P. (GM007324). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357 and also funded by the National Institutes of Health (P30 GM124165) and an NIH-ORIP HEI grant (S10OD021527). This research also used resources of the Yale Macromolecular X-ray Core Facility (NIH grant 1S10OD018007-01). This report is dedicated to Prof. Dale L. Boger with congratulations on his receipt of the Tetrahedron Prize
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing Interest
The authors declare no competing interests.
References
- [1].Hammarén HM, Virtanen AT, Abraham BG, Peussa H, Hubbard SR, Silvennoinen O, J. Allergy Clin. Immunol 143 (2019) 1549–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Avis T, Barthorpe A, Bignell G, Blow M, Brackenbury L, Buck G, Clegg S, Clements J, Cole J, Davies K, Edkins S, Gray K, Gorton M, O’Meara S, Halliday K, Harrison R, Haynes W, Hills K, Hunter C, Jones D, Kosmidou V, Laman R, Lugg R, Parker A, Perry J, Petty R, Small A, Solomon H, Stephens P, Stephens Y, Stevens C, Smith R, Tarpey P, Tofts C, Varian J, West S, Widaa S, Bamford S, Butler A, Dawson E, Dicks E, Edwards K, Forbes S, Greenman C, Hinton J, Menzies A, Raine K, Shepherd R, Teague J, Yates A, Wooster R, Futreal A, Stratton M, Green AR, Lancet 365 (2005) 1054–1061. [DOI] [PubMed] [Google Scholar]
- [3].James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, Garçon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker WA Nature 434 (2005) 1144–1148. [DOI] [PubMed] [Google Scholar]
- [4].Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, Tichelli A, Cazzola M, Skoda RC, N. Engl. J. Med 352 (2005) 1779–1790. [DOI] [PubMed] [Google Scholar]
- [5].Levine RL, Wadleigh M, Cools J, Eberts BL, Wernig G, Huntly BJP, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Fröhling S, Döhner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG, Cancer Cell 7387–397. [DOI] [PubMed] [Google Scholar]
- [6].Hammarén HM, Ungureanu D, Grisouard J, Skoda RC, Hubbard SR, Silvennoinen O, Proc. Natl. Acad. Sci. USA 112 (2015) 4642–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Leroy E, Balligand T, Pecquet C, Mounton C, Colau D, Shiau AK, Dusa A, Constantinescu SN, J. Allergy Clin. Immunol 144 (2019) 224–235. [DOI] [PubMed] [Google Scholar]
- [8].Leroy E, Constantinescu SN, Leukemia 31 (2017) 1023–1038. [DOI] [PubMed] [Google Scholar]
- [9].Kung JE, Jura N Nature Reviews 18 (2019) 501–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Puleo DE, Kucera K, Hammarén HM, Ungureanu D, Newton AS, Silvennoinen O, Jorgensen WL, Schlessinger J, ACS Med. Chem. Lett 8 (2017) 618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Newton AS, Deiana L, Puleo DE, Cisneros JA, Cutrona KJ, Schlessinger J, Jorgensen WL, ACS Med. Chem. Lett 8 (2017) 614–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liosi M-E, Krimmer SG, Newton AS, Dawson TK, Puleo DE, Cutrona KJ, Suzuki Y, Schlessinger J, Jorgensen WL, J. Med. Chem 63 (2020) 5324–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bandaranayake RM, Ungureanu D, Shan Y, Shaw DE, Silvennoinen O, Hubbard SR, Nat. Struct. Mol. Biol 19 (2012) 754–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS, J. Med. Chem 47 (2004) 1739–1749. [DOI] [PubMed] [Google Scholar]
- [15].Irwin JJ, Shoichet BK, J. Chem. Inf. Model 45 (2005) 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT, J. Med. Chem 49 (2006) 6177–6196. [DOI] [PubMed] [Google Scholar]
- [17].Jorgensen WL, Tirado-Rives J, J. Comput. Chem 26 (2005) 1689–1700. [DOI] [PubMed] [Google Scholar]
- [18].Turupcu A, Tirado-Rives J, Jorgensen WL, J. Chem. Theory Comput 16 (2020) 7184–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.