

Abstract
Pangenomes—the cumulative set of genes encoded by a population or species—arise from the interplay of horizontal gene transfer, drift, and selection. The balance of these forces in shaping pangenomes has been debated, and studies to date focused on ancient evolutionary time scales have suggested that pangenomes generally confer niche adaptation to their bacterial hosts. To shed light on pangenome evolution on shorter evolutionary time scales, we inferred the selective pressures acting on mobile genes within individual human microbiomes from 176 Fiji islanders. We mapped metagenomic sequence reads to a set of known mobile genes to identify single nucleotide variants (SNVs) and calculated population genetic metrics to infer deviations from a neutral evolutionary model. We found that mobile gene sequence evolution varied more by gene family than by human social attributes, such as household or village. Patterns of mobile gene sequence evolution could be qualitatively recapitulated with a simple evolutionary simulation without the need to invoke the adaptive value of mobile genes to either bacterial or human hosts. These results stand in contrast with the apparent adaptive value of pangenomes over longer evolutionary time scales. In general, the most highly mobile genes (i.e., those present in more distinct bacterial host genomes) tend to have higher metagenomic read coverage and an excess of low-frequency SNVs, consistent with their rapid spread across multiple bacterial species in the gut. However, a subset of mobile genes—including those involved in defense mechanisms and secondary metabolism—showed a contrasting signature of intermediate-frequency SNVs, indicating species-specific selective pressures or negative frequency-dependent selection on these genes. Together, our evolutionary models and population genetic data show that gene-specific selective pressures predominate over human or bacterial host-specific pressures during the relatively short time scales of a human lifetime.
Keywords: pangenome, evolution, mobile genes, horizontal gene transfer, human gut microbiome, evolutionary simulations, population genetics
Introduction
Significance
It has recently been debated to what extent pangenomes, the total set of genes encoded by a species, are adaptive or shaped by neutral evolution. Based on a data set of mobile genes and metagenomes from bacteria in the human gut microbiome, we reframe this debate and find that gene sequence evolution can qualitatively be recapitulated by an evolutionary model in which mobile genes maximize their own replication but provide little adaptive benefit to their bacterial host genomes. We infer divergent regimes of natural selection acting on genes of different cellular functions within individual gut microbiomes. These results suggest that, at least on time scales of a human lifetime, selection acts on individual genes, but not in a way that is necessarily adaptive to microbial host cell fitness.
Human gut microbial communities (microbiomes) impact diverse aspects of human health, such as food digestion, nutritional uptake, immunity, and inflammation (Brito et al. 2016; Valdes et al. 2018). The gut microbiome is shaped by both ecological factors, such as shifts in species abundance or strain replacements, and evolutionary forces, such as mutation, horizontal gene transfer (HGT), drift, and selection (Garud and Pollard 2020). In particular, microbes in the gut dynamically and frequently exchange genetic material through HGT (Vos et al. 2015), resulting in pangenomes—defined as the total set of genes observed across all sampled members of a species or population—which are often much larger than an individual genome size (Sela et al. 2016; McInerney et al. 2017; Jiang et al. 2019). Horizontally transferred (mobile) genes can contribute to environmental adaptation, notably through the propagation of antibiotic resistance (Jiang et al. 2019). However, there are contexts in which pangenome evolution could be driven more by drift than by selection. For instance, the evolution of endosymbionts or intracellular pathogens, which have small effective population sizes, is generally driven by drift and gene loss, resulting in small pangenomes (Giovannoni et al. 2014). In contrast, selection seems to play a bigger role in free-living microbes, like hydrothermal vent bacteria (Bobay and Ochman 2018; Moulana et al. 2020). Whether pangenome evolution is mainly driven by selection (an adaptive model) or drift (a nonadaptive or neutral model) is a question that has generated active debate (Sela et al. 2016; Andreani et al. 2017; McInerney et al. 2017; Bobay and Ochman 2018).
Pangenome studies to date have focused on relatively long evolutionary time scales. For example, a model in which gene gain by HGT is predominantly adaptive to prokaryotic genomes provides a good fit to a data set of 707 distantly related genomes from the NCBI database (Sela et al. 2016). In that model, gene gain and loss maintain genome size equilibrium and have opposite fitness effects. Based on a synthesis of this model with additional population genomic data, McInerney et al. (2017) argued that mobile genes generally provide niche adaptation to their bacterial host genomes, and thus pangenomes can be considered an adaptive feature. A key piece of evidence supporting this conclusion is that bacteria with large effective population sizes (Ne) tend to have larger genomes that have gained many mobile genes (Sela et al. 2016). Since the effects of natural selection (relative to drift) are stronger when Ne is large, this suggests that mobile gene acquisition tends to be of adaptive value to the bacterial host genome. Similarly, Bobay and Ochman (2018) found that Ne correlates positively with pangenome size for most of the 153 prokaryotic species they analyzed. Another study of a similar NCBI data set revealed a positive correlation between genome fluidity (a measure of gene turnover in the pangenome) and synonymous nucleotide diversity, a proxy for the molecular clock (Andreani et al. 2017). Although this does not exclude a role for selection, the observation is most parsimoniously explained by a neutral model, in which mobile genes are gained and lost randomly over time. To reconcile these findings (Bobay and Ochman 2018), proposed a nearly neutral model of drift-selection balance. It assumes that most accessory genes in the pangenome are slightly beneficial, such that they can be considered neutral when Ne is small, but they can escape the effects of drift and spread when the selective coefficient s exceeds 1/Ne. This more nuanced model is likely more realistic than an artificial duality between either selection or drift alone.
Resolving the balance of evolutionary forces influencing pangenomes also depends on the biological scale or unit of evolution. For example, the consequences of selection at the level of single genes, genomes (i.e., the bacterial hosts of mobile genes) or humans (i.e., the hosts of microbiomes) could yield different patterns. The studies mentioned above focused on adaptation at the whole-genome level, but the selection also acts at the level of individual genes (Takeuchi et al. 2015; Shapiro 2017; Moulana et al. 2020). Mobile genes in particular may have their own Ne, which could be distinct from the Ne of the species as a whole (Shapiro 2017). For example, there is a distinct class of mobile genes, including phage and other “selfish” elements that have effectively instantaneous HGT rates (Wolf et al. 2016). Other mobile genes may provide rapid adaptive value to their microbial hosts, such as in the gut microbiome of humans with different diets or lifestyles (Brito et al. 2016). Therefore, some mobile genes appear to be selected to favor their own (“selfish”) replication, whereas others may provide benefits to their bacterial or even human hosts (Hehemann et al. 2010).
All the studies above investigated pangenome evolution among distantly related genomes over relatively ancient time scales. Yet selective pressures might differ on recent and shorter evolutionary time scales, such as within local populations of bacteria over dozens rather than millions of years. However, a targeted investigation of the population genetics of mobile genes on short time scales is still missing. To fill this gap, we used a data set of 37,853 mobile genes involved in recent HGT events in the human gut (Brito et al. 2016). HGT is known to occur frequently within individual human gut microbiomes (Smillie et al. 2011; Yaffe and Relman 2020; Groussin et al. 2021), making it an ideal system in which to study mobile gene evolution over short time scales. We mapped metagenomic reads from a cohort of 176 Fiji islander gut microbiomes to the set of mobile genes. From these mapped metagenomic reads, which sample multiple microbial populations within each person’s gut, we identified single nucleotide variants (SNVs) segregating within microbiomes, from which we calculated population genetic metrics (dN/dS and Tajima’s D) that contain information about the evolutionary and demographic histories of mobile genes. Our approach is thus gene-focused rather than species-focused because mobile genes are likely to inhabit multiple prokaryotic species (Shapiro 2017). By mapping metagenomic reads to mobile genes, we thus include mutations (SNVs) that occur in one or more bacterial species and evolve within the time frame of a single human lifespan. In contrast to studies over longer evolutionary time scales, which have concluded that gene acquisition is generally adaptive to bacterial hosts, we find that many aspects of mobile gene molecular evolution on shorter time scales can be explained without invoking adaptive benefits to the bacterial (or human) host. However, a small subset of genes with distinct functions shows dramatically different signatures of molecular evolution, suggesting that selection acts at the level of gene function. Therefore, selection acting at the level of individual gene function—rather than bacterial host genome adaptation—might predominate over shorter “human” time scales.
Results and Discussion
Gene Mobility Correlates Positively but Not Strongly with Metagenomic Coverage
To study pangenome evolution on time scales on the order of a human lifespan, we used an existing collection of mobile genes identified in 387 isolate genomes from the Human Microbiome Project (HMP) and 180 single-cell genomes from the Fiji Community Microbiome Project (FijiCOMP). Selected single-cell genomes came from 31 different genera and had less than 10% contamination as assessed by CheckM (Parks et al. 2015; Brito et al. 2016). The mobile genes were identified as genomic regions (≥500 bp) with >99% nucleotide identity over the whole gene length that was shared between distantly related reference or single-cell bacterial genomes (<97% identity in 16S rRNA), suggesting that HGT occurred within an individual human gut microbiome (Brito et al. 2016). Ribosomal genes, which tend to be highly conserved, were excluded from this set of mobile genes as they could represent false-positive HGT events (Brito et al. 2016). This procedure is strict, yielding likely true positive HGT events, at the expense of many false negatives (Smillie et al. 2011; Brito et al. 2016). We considered only genes with at least 10X metagenomic sequence coverage, and only metagenomes with at least 500 genes passing this coverage threshold. These filters yielded a total of 7,990 mobile genes out of the 37,853 genes present in the original data set, and 175 out of 176 metagenomes, each from a different person from Fiji. We operationally defined gene mobility as the number of single-cell genomes in which a mobile gene was found. Gene mobility ranged from 1 to 16 species (mean = 2.73, standard deviation = 2.42; supplementary fig. S1, Supplementary Material online) and is probably an underestimate of the true HGT rate because it was calculated from a limited sample (180 genomes) of the diversity in Fijian islanders’ gut. This could also be explained by small or incomplete assemblies of the single-cell genomes. Nonetheless, this data set allows us to assess the balance of evolutionary forces in the pangenome on short time scales.
We first asked whether our mobility metric behaves as expected in quantifying the spread of mobile genes in the gut. Assuming that genes with higher mobility will occur in more species, we expect them to be more deeply covered by metagenomic sequence reads. Consistent with this expectation, we found that a gene’s mobility is positively correlated with its depth of metagenomic read coverage (fig. 1, supplementary tables S1A and C, Supplementary Material online). The expectation of a positive correlation is not guaranteed because some mobile genes, such as selfish elements, have deleterious effects (Vogan and Higgs 2011) and can be subject to negative frequency-dependent selection (Takeuchi et al. 2015; Corander et al. 2017; Domingo-Sananes and McInerney 2019) such that they are carried only by a fraction of individuals within a species, even if prevalent across species. The correlation between gene mobility and coverage is significantly positive in 167 out of 175 gut metagenomes (false discovery rate [FDR]-adjusted P < 2.2 × 10−16), but the adjusted R2 and slope values are relatively modest (fig. 1, supplementary fig. S2, Supplementary Material online). Varying selective pressures across mobile genes (e.g., deleterious effects and negative frequency-dependent selection) might be responsible for reducing the scaling between gene mobility and coverage, but not enough to flatten the relationship completely. We conclude that gene mobility, even if estimated from a relatively small sample of 180 gut bacterial genomes, behaves approximately as expected: generally leading to higher gene copy numbers.
Fig. 1.
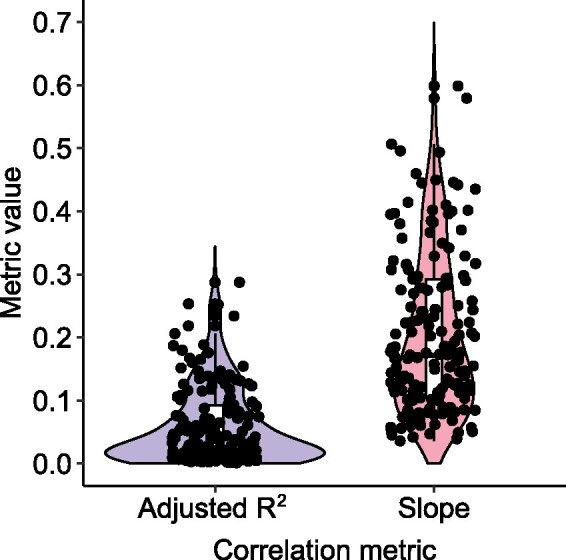
The correlation between gene mobility and metagenomic sequencing coverage is positive but widely variable. The boxplots and violin plots show the distributions of adjusted R2 values (blue) and slopes (red) across samples (individuals from Fiji) for the correlation between coverage (average depth per site) and gene mobility. The black dots represent the 167 samples (out of 175 tested) in which the correlation is significant (t-test, FDR-adjusted P < 0.05). Examples of this correlation in four randomly selected samples are shown in supplementary figure S2, Supplementary Material online.
Estimating Population Genetic Metrics from Metagenomic Data
The relationship between metagenomic coverage and gene mobility is generally positive but varies substantially across individuals (fig. 1). We therefore sought to ask whether this variation could be explained by either gene-specific factors, such as gene mobility and COG functional categories (Tatusov et al. 2000), or by human host-specific factors, such as age, diet, and social networks. Such factors are known to influence the patterns of mobile gene presence/absence across bacterial genomes (Takeuchi et al. 2015) and human hosts (Yatsunenko et al. 2012; Brito et al. 2016; Zhernakova et al. 2016; Garud and Pollard 2020), yet it is unknown how they influence the molecular evolution of mobile genes. To study molecular evolution, we mapped metagenomic reads to mobile genes to call SNVs segregating within and among bacterial species in the gut microbiome. We quantified mobile gene sequence evolution using four population genetic metrics that detect selection and capture deviations from a neutral evolutionary model:
, the nucleotide diversity calculated from the average number of pairwise nucleotide differences among metagenomic reads,
, the nucleotide diversity calculated from the normalized number of segregating/polymorphic sites in metagenomic reads,
Tajima’s D, the normalized difference between and , and
dN/dS, the ratio of nonsynonymous to synonymous substitution rates, measuring selective constraints at the protein level.
We note that our estimate of dN/dS, based on mapping metagenomic reads that could come from the same or different species, is a mixture of within-species polymorphism (often called pN/pS) and between-species divergence (dN/dS), but we refer to this hybrid metric as dN/dS for simplicity. We further note that and are two different estimators of the population mutation rate, = 2 Neµ, where µ is the mutation rate and Ne is the effective population size. is more sensitive to intermediate-frequency mutations whereas is more sensitive to low-frequency mutations. The difference between the two estimators is captured by Tajima’s D, with D < 0 indicating more low-frequency mutations than expected under a standard neutral model with no selection and a constant population size (Tajima 1989). Negative values of D can be the result of a population expansion, purifying selection, or a very recent selective sweep. Conversely, positive values of D indicate more intermediate-frequency mutations than expected under a neutral model (supplementary fig. S3, Supplementary Material online), due to population contraction, balancing selection, or negative frequency-dependent selection.
These population genetic metrics were calculated for each mobile gene in each sample by mapping metagenomic reads and calling SNVs after applying a 10X sequencing coverage filter (Materials and Methods). Consistent with previous estimates across multiple kingdoms of life (Koonin and Wolf 2010), we observe that and distributions across samples span 3 to 4 orders of magnitude (supplementary fig. S4, Supplementary Material online). Also consistent with previous estimates in bacteria over different time scales (Sela et al. 2016; Gardon et al. 2020; Garud and Pollard 2020), dN/dS tends to be less than one, suggesting the predominance of purifying selection at the protein level (supplementary fig. S4, Supplementary Material online). Our estimates of these population genetic metrics from metagenomic data are thus within an expected range.
Population Genetic Metrics Vary More across Mobile Genes Than across Human Host Attributes
With these metrics in hand, we asked whether mobile gene evolution is mainly driven by microbial- or human host-specific selective pressures. To do so, we determined whether population genetic metrics varied more across gene families or across individual human hosts. We first compared distributions of pairwise differences for each metric using the Kolmogorov–Smirnov test (KS test) and found much greater variation between genes than between individuals (fig. 2 and supplementary fig. S4, Supplementary Material online). This result indicates that, on short time scales, the selective pressures quantified by the four metrics may be less affected by person-specific factors, such as lifestyle or social networks, than by gene functions within a microbial cell. In other words, although some mobile genes may enable adaptations to personalized factors such as diet (Brito et al. 2016), sequence evolution is modestly affected by these factors on short time scales (within an individual). In contrast, population genetic metrics vary substantially more across genes, suggesting that selective pressures act predominantly at the level of gene function.
Fig. 2.

Mobile gene evolution is more variable across genes than across human hosts. Each panel shows the distribution of the variation of population genetic metrics among samples (red) or among gene families (black) through the distribution of log10(DKS) statistics. The DKS statistic from the KS test measures the maximal distance between a pair of cumulative distributions—in this case, across either samples or genes. Panels a, b, c, and d represent the variation of , , Tajima’s D, and dN/dS, respectively. We downsampled the 37,853 genes to the same size as the number of samples to make them comparable and repeated the downsampling 999 times. This figure presents the result for 999 replicate downsamplings of 175 genes and shows that there is more variation across genes than across samples/individuals for all the population genetics metrics (KS test, P < 2.2 × 10−16). See supplementary figure S4, Supplementary Material online for example distributions across genes and samples.
To further assess the evidence that person-specific factors have weaker effects than gene-specific factors on mobile gene sequence evolution, we used a linear regression where the continuous response variable is one of the population genetic metrics and the qualitative/categorical explanatory variable is a human host attribute (Materials and Methods). Because the statistical significance of such an analysis is affected by sample size, we selected mobile genes with less than 30% missing values across the 172 samples for which metadata were available, for a total of 1,333 tested genes. Human host age and sex did not show any significant effects on mobile gene sequence evolution. However, a person’s household or village significantly influenced the evolution of a relatively small percentage of mobile genes (7.84–15.2% of the 1,333 tested genes; fig. 3a). In this small subset of significant genes, the correlations between population genetic metrics and household (adjusted R2 ∼0.30 to ∼0.85) were stronger than correlations with village (adjusted R2 < 0.30). These results varied somewhat depending on the value of the missing value filter. For example, using a stringent filter, the village of origin had a significant influence on dN/dS in up to 20% of genes (supplementary fig. S5, Supplementary Material online). The small set of genes significantly influenced by household and village could be representative of very specific family/village selective pressures such as diet. Annotations of these genes show that they are involved in a set of functions involved in carbohydrates, lipids, secondary metabolites and ions transport or metabolism, and potential antibiotic resistance through ABC-type multidrug transporter system (supplementary table S2, Supplementary Material online). Some of these functions are similar to those identified by Brito et al. (2016) as differentially abundant among villages. Therefore, although village- or household-specific selective pressures do not explain much of the variation in population genetic metrics across genes, we cannot exclude specific instances in which social networks or lifestyles drive the evolution of few mobile genes over short time scales.
Fig. 3.

Gene function explains more variation in mobile gene sequence evolution than human host attributes. (a) Adjusted R2 values for the categorical regressions between population genetic metrics (color-coded) and human host attributes. We only considered genes with at least 10X coverage in a sample, and we also required that mobile gene should have less than 30% missing values across samples, for a total of 1,333 genes included in this analysis. The five strongest and most prevalent correlations between population genetics metrics and human host factors are shown (FDR-adjusted P < 0.05). Not shown are village significantly correlated with Tajima’s D (0.75%) and dN/dS (0%), and household significantly correlated with (0.38%). Human host age and sex did not show any significant effects on mobile gene sequence evolution. Each black point represents a mobile gene for which the categorical regression is significant. The percentage of significant genes out of the total number of genes tested is indicated in parentheses along the x-axis. For dN/dS, the sample size was reduced to n = 255 genes because an additional filter requiring mutations to be seen in the least five metagenomic reads was applied before computing dN/dS, which can other be sensitive to sequencing errors (Materials and Methods). (b) Adjusted R2 values of the categorical regressions between a population genetic metric and the gene family. Each black point represents a sample for which the categorical regression is significant. The percentage of significant samples out of the total number of samples tested is indicated in parenthesis along the x-axis. Only 172 out of 175 samples for which metadata were available are included in this analysis. We only considered genes with at least 10X coverage in a sample. We only included genes with a gene family annotation and required that each gene family be represented by at least two genes. Finally, we only included genes present in 70% or more of the samples (less than 30% missing values), for a total of 512 genes.
Although human host factors seem to have relatively little effect on the sequence evolution of most mobile genes on short time scales, selective pressures at the level of the genes might be more important. Indeed, we observed a higher variation of population genetics metrics between genes than between samples (fig. 2), which could be explained by gene attributes such as their cellular function. To test this hypothesis, we used linear regressions between population genetics metrics and gene families based on the following set of conditions:
the gene should have at least 10X coverage to limit the impact of sequencing errors and increase confidence in variant calling,
the gene should have an available gene family annotation, which is the explanatory variable of the regression. Gene family annotations come from COG, KEGG, TIGRFAM, PFAM, or dbCAN databases (Brito et al. 2016),
the COG family should be represented by at least two genes within the data set to avoid low sample sizes, and
the mobile gene should have less than 30% missing values across samples, for a total of 512 tested genes.
In contrast to human factors, gene functions defined by COG families explained much of the variation in mobile gene sequence evolution across the majority of tested samples. For , , and Tajima’s D, COG families explained from ∼10% to ∼60% of the variance in more than 88% of the samples (FDR-adjusted P < 0.05; fig. 3b). For dN/dS, COG families explained from ∼26% to ∼83% of the variance in 52.3% of samples. To ensure that the less prevalent effect of host factors was robust to differential sampling of genes (n = 512) and individuals (n = 172), we downsampled 999 times the set of tested genes to n = 172 and repeated the regressions for each subset of randomly selected genes. After downsampling, host village and household significantly correlated with mobile gene sequence evolution for ∼1–28% of tested genes compared with 52.3–97.1% of tested samples for COG family (fig. 3a and supplementary fig. S6, Supplementary Material online), which confirms the less prevalent effect of host factors compared with gene functions. A remaining caveat is that the explanatory power and the reproducibility of the correlation (i.e., the proportion of samples for which the correlation is significant at an FDR-adjusted P < 0.05) depends on the balance between sample size (number of genes) and the stringency of the data quality filters (supplementary fig. S7, Supplementary Material online). Indeed, as the filter stringency increases, only the most prevalent mobile genes are included and the correlation R2 tends to increase. However, the sample size and the reproducibility of the correlation tend to decrease (supplementary fig. S7, Supplementary Material online). For instance, as the stringency of the missing value filter increases, fewer samples show significant correlations between Tajima’s D and COG function, going from 88.4% of samples when a gene can be absent in at most 30% of samples to 40.1% significant when a gene can be absent in at most 10% of samples (supplementary fig. S7, Supplementary Material online). Although the correlation strength depends on the filter stringency, the median adjusted R2 of the correlation is always higher than 20% and can reach up to ∼60%. Altogether, these results suggest that COG functions appear to explain much of the short-term molecular evolution of a subset of mobile genes, which is much larger than the subset of genes that are significantly influenced by human host factors.
Higher Gene Mobility Is Associated with Low-Frequency SNVs in the Gut Microbiome
In addition to gene- or environment-specific selective pressures, the rate of HGT is also expected to affect mobile gene molecular evolution, as it allows genes to spread across different species, possibly altering their population size and thus the efficacy of selection (Vos et al. 2015; Shapiro 2017). To first order, each human host represents a distinct short-term evolutionary trial. Thus, to study the influence of HGT rate on molecular evolution within each of the human guts sampled, we correlated gene mobility with the population genetic metrics described above: dN/dS, , , and Tajima’s D.
Using this regression approach, we first observed that the correlation between dN/dS and gene mobility was significant and positive in 147 out of 175 samples (supplementary table S3D, Supplementary Material online and fig. 4a), but with a low average adjusted R2 of 0.03 (SD = 0.02). This pattern is robust whether or not we include gene length and coverage as covariates in linear regressions to control for the effect of sequencing artifacts (supplementary fig. S8, Supplementary Material online). This correlation could be explained by the fixation of slightly deleterious nonsynonymous mutations in the early stage of a population expansion (Parsch et al. 2009) as might be the case when mobile genes are spreading across species on short time scales. Alternatively, this could also be explained by slightly increasing positive or relaxed purifying selection with increasing gene mobility, but we refrain from drawing strong conclusions due to the weak R2 values.
We next observed that 153 out of 175 samples had a stronger and significant positive correlation between and gene mobility (linear regression with FDR-adjusted P < 0.05; mean adjusted R2 = 0.06; SD = 0.04; supplementary table S3B, Supplementary Material online and fig. 4a). This is consistent with a model in which mobile genes accumulate SNVs that remain at low frequency (as measured by which is sensitive to these low-frequency mutations) as they spread across species. This pattern is reproducible in most samples, but it is less robust to the potential effects of sequencing artifacts than other observed patterns (supplementary fig. S8, Supplementary Material online). We also observed that , which is more sensitive to intermediate-frequency mutations, decreases with gene mobility (supplementary table S3A, Supplementary Material online and fig. 4a). Among samples in which versus gene mobility regression results were significant (157 out of 175 samples with FDR-adjusted P < 0.05), ∼97% of them exhibited this negative correlation (mean adjusted R2 = 0.08; SD = 0.04). As a result, Tajima’s D (which reflects the difference between and ) is significantly negatively correlated with gene mobility in 149 out of 175 samples (fig. 4a and supplementary table S3C, Supplementary Material online). Even if the R2 values are modest, we note that the trends are highly repeatable across samples even when we control for the effect of sequencing artifacts (supplementary fig. S8, Supplementary Material online). Reasons for the relatively low R2 values could include noise in the gene mobility metric (based on a small sample of genomes) and/or variable selective pressures across genes. There are several reasons for this enrichment of low-frequency SNVs (resulting in lower Tajima’s D values) in more highly mobile genes, including purifying selection keeping deleterious mutations at low frequency, recovery of new polymorphism after a recent selective sweep, or population expansion. Overall, these results suggest that HGT can spread genes across species faster than SNVs are able to rise to high frequency.
Fig. 4.

Gene mobility is negatively correlated with Tajima's D and positively correlated with dN/dS in real and simulated microbiomes. (a) Real data from Fiji. The heatmap shows the slope of a regression model in which either , , Tajima’s D, or dN/dS is the response variable and gene mobility is the explanatory variable (across samples). Regression P-values were obtained through a t-test. The heatmap contains nonsignificant regressions results after FDR correction for multiple testing (black), negative significant correlations (red), and positive significant correlations (blue). Data standardization was performed before each regression to respect the t-test’s assumption of normality, and t-tests were concordant with nonparametric tests (Materials and Methods). Heatmap rows and columns were clustered with Euclidean distance and complete linkage clustering. (b) Representation of simulation events over two generations. In the first generation, a gene gain event occurs through HGT. Gene gain is represented by the transfer of gene from a donor to a recipient cell and increases the genome size of the recipient. The probability of future gene gain or loss events (Pgain and Ploss, respectively) is determined by the difference between the current genome size of the cell (x) and the equilibrium genome size (x0). At equilibrium, the probability of gene gain and loss is the same by definition (Pgain = Ploss). An increase of genome size until it exceeds the equilibrium point (x > x0) leads to gene loss being more likely than gain (Pgain < Ploss). Gene gain also increases the fitness (f > fWT) of the recipient cell based on the selection coefficient of the transferred gene (sgain). In the model, each gene has its own selective coefficient drawn from an exponential distribution exp(λ) with an expected value of 1/λ. Gene gain is either slightly beneficial or neutral in this model and has the opposite fitness effect of gene loss, which is slightly deleterious or neutral (-sgain = sloss where sgain ≥ 0). Gene loss decreases the genome size of the target cell and in case this decrease leads to a smaller genome size than equilibrium, the probability of gene gain becomes higher than the probability of gene loss (Pgain > Ploss). Gene loss also decreases the fitness of the target cell (f < fWT) based on the selection coefficient of the lost gene (sloss). Finally, as represented in the second generation, mutations can also occur and change the fitness of the cell based on a selective coefficient (smutation) which is drawn from a distribution (Materials and Methods). (c) Simulated data. The heatmap shows the slope of a regression model in which either , , Tajima’s D, or dN/dS is the response variable and gene mobility is the explanatory variable (across simulation replicates). Simulations with different parameters for HGT rate and or distributions of selective coefficients (s ∼ exp(λ)) are color-coded (n = 10 replicates per simulation). Heatmap rows and columns were clustered with Euclidean distance and complete linkage clustering.
Simple Evolutionary Simulations Recapitulate the Observed Effects of HGT on Mobile Gene Sequence Evolution
To better understand potential mechanisms underlying the relationship between gene mobility and sequence evolution observed in the Fiji microbiome data, we implemented the explicit simulation of HGT and sequence evolution in SodaPop, a forward evolutionary simulation toolkit (Gauthier et al. 2019), which we updated to include HGT. Similar to Sela et al. (2016), gene gain and loss are constrained to maintain genome size equilibrium and to have opposite fitness effects (fig. 4b). SodaPop simulates protein sequence evolution with the distribution of fitness effects of mutations derived from biophysics-based protein fitness landscapes (Gauthier et al. 2019). Briefly, we simulated a Wright–Fisher process for asexual populations with 10 bacterial species. Each simulation included 5,000 cells in total, divided into 10 species, and ran for 105 generations. Each gene has an explicit DNA sequence, where each site can be mutated to another nucleotide at a defined mutation rate (10−7 mutations per site per generation). These mutations include synonymous changes that are assumed to be neutral, and nonsynonymous changes with a distribution of fitness effects of which 30% are lethal and 70% are drawn from a normal distribution N (µ = −0.02, σ = 0.01) (Eyre-Walker and Keightley 2007) (Materials and Methods). Microbial genomes also experience HGT events, with explicit gene gain and loss events. The rates of these two events are updated at each generation for each cell to maintain an equilibrium around the genome size x0, set to 500 genes (fig. 4b) (Sela et al. 2016). Genomes larger than x0 are prone to gene loss, but genomes smaller than x0 are prone to gene gain. We also modeled gene gain and loss selection coefficients, specific to each gene and drawn from an exponential distribution with parameter λ (Materials and Methods). We kept simulated population sizes small due to memory limitations. To make sure this limitation does not cause excessive drift (e.g., the accumulation of deleterious mutations leading to extinction, also known as Muller’s Ratchet (Bachtrog and Gordo 2004)) we forced species relative abundances to remain constant. We also set a relatively high mutation rate of 1 × 10−7 mutations per site per generation to compensate for the small population sizes and to ensure that enough mutations were generated in a reasonable number of generations. Genome size equilibrium was reached for every simulation, indicating robustness to variable starting conditions (supplementary fig. S9, Supplementary Material online). Altogether, this model allows us to test if the relationships between gene mobility and population genetic metrics observed in the real data can be observed under varying rates of HGT and the adaptive benefit of acquired genes.
We found that the simulation could recapitulate the major features observed in the real Fiji microbiome data without requiring that mobile genes provide adaptive value to a human host or to its bacterial genome. First, the simulations can recapitulate the shape of the observed distribution of gene mobility (supplementary fig. S1, Supplementary Material online). A caveat is that simulations are far from including all the complexity of the gut microbiome that is the number of species, population structures, and other features not simulated, and the distributions were only compared for one illustrative set of input parameters (supplementary fig. S1, Supplementary Material online). Thus, we do not claim that our model can provide a precise quantitative description of gene mobility in the gut microbiome, but rather that it can recapitulate the major qualitative features.
Second, the simulations recover the positive correlation between gene mobility and census population size (metagenomic coverage) observed in the real data (fig. 1). The positive correlation was always stronger in the simulations (mean adjusted R2 of 0.705 across all parameter settings, standard deviation = 0.190) compared with the real data (mean adjusted R2 of 0.057 across all parameter settings, standard deviation = 0.062). This suggests that factors not included in the model, such as negative frequency-dependent selection and noise in the gene mobility metric, reduced the strength of the correlation in the real data. The positive correlation was stronger in simulations with relatively low HGT rates but was largely unaffected by whether HGT events were neutral or adaptive to microbial host cell fitness (supplementary table S4, Supplementary Material online). This suggests that relatively high HGT rates could also explain the weaker correlation between gene mobility and coverage observed in the real data.
Third, we assessed whether the simulations could reproduce the observed correlations between population genetics metrics and gene mobility. Simulations recapitulated most of the observed effects of HGT on nucleotide diversity in real data. Specifically, Tajima’s D correlates negatively with gene mobility in simulations, with a median adjusted R2 of 0.32 (mean = 0.23; SD = 0.13) compared with a median adjusted R2 of 0.01 (mean = 0.01; SD = 0.01) in the real data and reproducible across all the simulations compared with ∼85% of the samples in the real data (fig. 4; supplementary tables S3C and S5C, Supplementary Material online). The variation in this correlation is explained more by the HGT rate than by fitness effects (i.e., neutral or adaptive selective coefficients on gene gain/loss). This can be seen in the heatmap, in which simulations cluster by HGT rate rather than by selective coefficients (fig. 4c). Along the same lines, we performed a K-S test on the slopes of the regression between Tajima’s D and mobility and observed that this slope varies more by HGT rate than by selective coefficients (supplementary fig. S10, Supplementary Material online). Simulations also predict that dN/dS correlates positively but weakly with mobility, but this is only significant at intermediate HGT rates (supplementary fig. S11, Supplementary Material online). A similar pattern is observed in the real data, in which dN/dS correlates weakly with mobility (fig. 4a). This effect could be due to slightly deleterious nonsynonymous mutations spreading by HGT before they can be purged by the selection, but further work will be needed to test this thoroughly and to exclude the competing hypothesis of positive selection. Overall, real human microbiome data are recapitulated by our simple evolutionary model, which includes only selection for a stable genome size, without the need to invoke adaptive advantage of mobile genes to their bacterial genomes, or to include any human host factors whatsoever.
A Subset of Gene Functions Experiences a Divergent Regime of Natural Selection
Having established that Tajima’s D correlates negatively with gene mobility whereas coverage tends to correlate positively both in simulations and in the real data set, we sought to determine if these general trends apply equally to all gene families. Although the trends are significant across samples, the large variations observed across genes (fig. 2 and supplementary fig. S4, Supplementary Material online) could be due to gene-specific evolutionary regimes. To test this hypothesis, we used linear mixed models with gene mobility as a predictor of either Tajima’s D or coverage as a response variable, while controlling for variations across gut microbiome samples like alpha or beta diversity, and allowing the response to vary across COG categories (Materials and Methods). This analysis was performed on genes with at least 10X coverage and available COG annotations (n = 3,608 mobile genes).
As expected, based on the overall positive relationship observed (fig. 1), coverage and gene mobility are positively and significantly correlated across most COG categories (fig. 5a). COG category X (mobilome, prophages, and transposons) stood out as the strongest contributor to this positive relationship, consistent with this signal being driven by genes with the highest mobility. Removing sample identity or COG category from the linear mixed models significantly decreased the model fit, suggesting that they both contribute substantially to explaining variation in the coverage-mobility and Tajima’s D-mobility relationships (supplementary tables S1A and B, Supplementary Material online). We confirmed that Tajima’s D is negatively correlated with gene mobility (fig. 5a), as observed in the regression analysis (fig. 4a), and the same relationship holds even when mobile genes are binned according to bacterial host range. Regardless of whether mobile genes are shared within a genus, across genera of the same phylum, or across phyla, gene mobility is positively correlated with coverage and negatively with Tajima’s D, and there is no apparent trend according with increasing host range (supplementary fig. S12, Supplementary Material online). We also considered whether community alpha diversity (Shannon entropy) or beta diversity (the first principal component of Bray–Curtis dissimilarity between samples) within individual microbiomes could affect these results. However, adding these diversity metrics to the mixed models did not change the sign of the correlations, nor did they significantly improve the model fits (Likelihood ratio test P > 0.05; Materials and Methods). Therefore, the negative correlation between Tajima’s D and gene mobility appears to be rather general and robust to factors such as host range and microbiome diversity.
Fig. 5.

Gene mobility regressions reveal a minority of genes with distinct signals of selection. (a) Linear mixed model regression slopes per COG category. This figure illustrates COG categories regression slopes for the linear mixed models “” and “” with “” and “” being considered as random effects. The asterisks at the tip of each bar indicate the significance of the simple linear regressions “” and “,” respectively, for the associated COG category (*Significant; N.S., not significant; Cutoff: FDR-adjusted P < 0.05). (b) Schematic of evolutionary scenarios. Scenario 1 represents the situation in which mobile genes Tajima’s D is negatively correlated with gene mobility because HGT is faster than fixation of mutant alleles (red stars). Scenario 2 represents the situation in which Tajima’s D correlates positively with mobility. These genes maintain intermediate frequency mutations (blue stars) despite being frequently transferred to new species. Note that the gene copies (dots or stars) illustrated here could come from members of the same or different species in the microbiome.
Certain functional categories of genes deviated from the general negative correlation between D and mobility. The COG categories for which Tajima’s D is positively and significantly correlated with mobility include P (Inorganic ion transport and metabolism), I (Lipid transport and metabolism), Q (Secondary metabolites biosynthesis, transport, and catabolism), V (Defense mechanisms), and O (Posttranslational modification, protein turnover, chaperones), representing ∼30% of gene families (supplementary fig. S13, Supplementary Material online). There are several explanations for why these gene families maintain or accumulate more intermediate-frequency SNVs (i.e., an increase in Tajima’s D) while being transferred to many new species (fig. 5b). The first explanation is a population contraction, or in this context, a reduction of the number of gene copies across species. However, this is unlikely to be the case for these genes because their coverage (a proxy for their relative abundance) increases with mobility. The second explanation is that these genes could be subject to species-specific selective pressures that push mutations to fixation in some species but not in others, resulting in intermediate SNV frequencies in the bulk metagenome. The third potential explanation is that negative frequency-dependent selection, which is thought to be an important force shaping pangenome evolution (Cordero and Polz 2014; Domingo-Sananes and McInerney 2019), is acting on these genes, within species, between species, or both. Thus, the last two scenarios, which rely on the presence of distinct selective pressures on these subsets of genes, most likely explain how some mobile genes can maintain or accumulate intermediate-frequency SNVs as they spread across species.
Conclusion
To date, pangenome evolution has been studied primarily over long evolutionary time scales by comparing relatively distantly related genomes (McInerney et al. 2017). These studies have largely concluded, although with some debate (Andreani et al. 2017; Shapiro 2017) that pangenomes are predominantly adaptive—that selection plays a bigger role in pangenome evolution than drift. Here we have refocused the study of pangenome evolution to shorter time scales that is within individual gut microbiomes in which gene transfer events likely occurred within a human lifespan (Brito et al. 2016; Groussin et al. 2021). Based on microbiome data from a Fiji cohort, we found that mobile gene sequence evolution is more influenced by selective pressures at the level of gene function than at the human host level. Of course, there were many unmeasured human host factors that could impose selective pressures that we were unable to study, and certain mobile genes do indeed provide adaptive benefits to humans (Hehemann et al. 2010; Brito et al. 2016). However, evolutionary simulation results showed that mobile genes need not provide any special adaptive value to their human host or microbial genomes in order to recapitulate the qualitative patterns of molecular evolution observed in the real data. We therefore conclude that the bulk of mobile gene evolution within the gut microbiome can be explained by selective pressures acting at the level of individual genes rather than human or bacterial hosts.
The observed patterns of molecular evolution based on population genetic metrics provide clues about the balance of evolutionary forces acting on mobile genes in microbiomes within a human lifespan. We found that most genes accumulate low-frequency mutations as they spread within and between bacterial species. One interpretation of this result is that most mobile genes are under purifying selection to maintain a conserved function, even as they spread across species, such that most mutations are deleterious and kept at low frequency. Another nonexclusive interpretation is that low-frequency mutations could become enriched during the rapid spread of a gene, before mutations are able to rise to a higher frequency. This pattern of rapid spread is strongest in phages and transposons (COG category X), which are known to have extremely high rates of transfer across genomes (Wolf et al. 2016). In contrast, a minority of genes involved in specific cellular functions, such as defense mechanisms (COG category V), accumulate intermediate-frequency mutations as they spread across species, possibly due to negative frequency-dependent selection within species and/or fixation of beneficial mutations within some species but not others. Further investigation is needed to explore the nature of these variable selective pressures across genes.
Pangenome evolution is the product of a fine balance between drift and selection, which can shift depending on the time scale and level of biological organization. In the gut microbiome of a single person, the time scale of evolution may be too short to easily resolve the balance between drift and selection. Indeed, on very short time scales during which mutations could still be segregating and HGT likely occurs more rapidly than point mutation (Levade et al. 2017; Yaffe and Relman 2020), slightly adaptive genes that have been recently transferred could be largely influenced by drift because of their initially small Ne, such that their adaptive value could be effectively detected only over long time scales. This is supported by our relatively short-term simulations, in which the HGT rate (which increases the census population size and thus Ne) was found to be a major determinant of sequence evolution, whereas gene-specific selection coefficients were not. These findings are broadly consistent with a drift-barrier model of evolution (Bobay and Ochman 2018; Gardon et al. 2020), which could be explored in additional genomic data sets and simulations. We suggest that future work on pangenome evolution should ask what factors control shifts in the drift-selection balance and its interplay with species ecology (Ne, species lifestyle, etc.) and gene ecology (i.e., gene function, to what extent are genes selfish or cooperative within a genome, etc.). Rather than settling for a binary distinction between an adaptive or neutral model, the relative strengths of drift and selection should be considered at varying evolutionary time scales and levels of biological organization, from gene to genome to community.
Materials and Methods
Population Genetics of Fijian Gut Microbiome Mobile Genes
The Fiji Community Microbiome project provides open access to metagenomes from the gut microbiomes of 176 individuals. For each of these individuals, we mapped metagenomic sequence reads to a set of 37,853 mobile genes previously defined as follows from bacterial whole-genome sequences from the HMP and FijiCOMP. To be considered mobile, pairs of genes had to be identified in ≥500 bp fragments that shared >99% nucleotide identity over the whole fragment length between reference or single-cell assembled genomes with <97% identity in the 16S rRNA gene (Brito et al. 2016). Metadata about the single-cell assemblies are available in the following link: http://fijicomp.bme.cornell.edu/data/Singlecellassemblies.xlsx. This procedure selects nearly identical genes present in distinct species or genera as candidates for very recent HGT, likely within an individual gut microbiome (Smillie et al. 2011; Brito et al. 2016). As ribosomal genes can be highly conserved between species without being horizontally transferred, inferred HGT events involving from ribosomal genes were excluded. For the metagenomic read mapping, only reads that aligned with 99% identity across ≥50% of their own length were considered (Brito et al. 2016). From the mappings, we used Anvi’o to report SNVs (–min-coverage-for-variability 10 –min-contig-length 50) (Eren et al. 2015), followed by a pipeline to compute population genetics metrics (, , dN/dS, and Tajima’s D) based on the SNVs. The pipeline scripts are available at https://github.com/arnaud00013/Fiji_Mobile_Gene_Specific_PopGen_scripts. The Anvi’o SNV calling module (Eren et al. 2015) has the advantage of being fast and simple to use, can be executed in parallel (High-Performance Computing), and has filters to control minimum gene coverage or mutation frequency. For each sample mapping, a gene was retained if its mean site depth was ≥10. Only one sample was excluded for having less than 500 genes passing the site depth filter, reducing the sample size to 175 metagenomes. Among all samples, 7,990 unique genes were conserved after applying the site depth filter. Finally, mobile gene COG annotations, available in the FijiCOMP data (http://fijicomp.bme.cornell.edu//), were used to define two levels of gene functions: COG gene family (which is more specific), and COG category (which is more general).
Detecting Selection by dN/dS
dN/dS is the nonsynonymous to synonymous mutations per site ratio. Different methods have been developed to estimate dN/dS with the common purpose of inferring selection in protein-coding genes. More precisely, dN/dS can detect purifying selection (dN/dS < 1), neutral evolution (dN/dS ≈ 1), and positive selection (dN/dS > 1). Because we are working with metagenomic gene variants, we defined our own estimator of dN/dS:
| (1) |
where Nbnsm is the number of nonsynonymous mutations (SNVs), Nbnss is the number of nonsynonymous sites, Nbsm is the number of synonymous mutations (SNVs), and Nbss is the number of synonymous sites.
Measuring Mobile Genes Nucleotide Diversity at Metagenomic Level
Because mobile genes are by definition present in multiple species, we calculated population genetic metrics based on all reads from a metagenome that map to a particular mobile gene. Based on these mapped reads, we calculated Tajima’s D (Tajima 1989), which measures the difference between average per site pairwise nucleotide differences () and the normalized number of polymorphic sites (
| (2) |
where the denotes the expected sampling variance of . For each sample, we estimated mobile gene nucleotide diversity from sequence variants detected in the mapping between metagenomic reads and mobile gene reference sequence from FijiCOMP as follows:
| (3) |
where n is the gene length, ci is the depth of the site i of the gene, is the choose() function, which calculates the number of pairs of reads in a set of size ci and Nb_reads_pwdiff is the number of pairwise nucleotide differences, and
| (4) |
where S in the number of segregating sites and a1 is a normalizing factor that represents the sample size (n):
| (5) |
Usually, Tajima’s D is estimated from a multiple alignments between gene alleles. The sample size used to estimate the normalizing factor a1 is the number of alleles. Here we use the average depth of coverage at polymorphic sites as an estimator of the sample size n.
Effect of Gene Mobility on Metagenomic Coverage
We operationally defined gene mobility as the number of single-cell genomes in which a mobile gene was found and tested if this metric behaves as expected in explaining gene frequencies in metagenomes. More precisely, we correlated gene mobility with metagenomic coverage with the expectation that more mobile genes occur in multiple species and should thus be more deeply covered by metagenomic sequence reads. Metadata on the 180 single-cell genomes used to estimate gene mobility are available at http://fijicomp.bme.cornell.edu/data/Singlecellassemblies.xlsx. Linear regression analyses and t-tests were calculated using the R function “summary.lm()” (R Core Team 2019). Data standardization was performed before each regression to respect the t-test’s assumption of normality. The distributions of the significant correlations adjusted R2 obtained from the t-tests converged with the ones from a nonparametric permutational ANOVA (K-S test P > 0.05) (Anderson 2001; Wheeler and Torchiano 2016). The results from the t-tests on the standardized data are reported instead of the permutational ANOVA on the raw data because standardization removes units and thus facilitates comparison between real and simulated data. Finally, we adjusted the regression P-values using the FDR correction for type I error after multiple testing implemented in the p.adjust() function of the R package “stats” (Benjamini and Hochberg 1995; R Core Team 2019).
Assessing Variation in Sequence Evolution across Genes and Across Individuals
To determine whether mobile gene evolution is driven more by gene-specific factors or by human host attributes, we first compared the variation of mobile genes nucleotide diversity (and other population genetic metric described above) across genes versus across samples through the KS test. The KS test involves a statistic D, which measures the maximal distance between a pair of cumulative distributions. We downsampled the mobile genes to the same size as the number of samples to avoid the potential bias due to different sized data sets and repeated this for a total of 999 resamples. We performed this series of KS test with the function ks.test() from the R package “stats” (R Core Team 2019).
Gene Function and Human Host (Individual) Attributes as Predictors of Mobile Genes Evolution
To determine whether mobile gene evolution is driven more by gene function or human host attributes, we performed linear regressions between a continuous response variable and a qualitative/categorical explanatory variable, which we will refer as a factor. Regressions between a quantitative continuous variable, for example Tajima’s D, and a factor, for example gene function family, require transforming the factor as it cannot be integrated into a regression equation in its original form (R Core Team 2019). We therefore used the R contrast function “constr.sum()” to transform factors (R Core Team 2019). This transformation allows the regression coefficients to represent how each level/state of the factor differs. Then, we assess the significance of the regression model with a permutational ANOVA (Anderson 2001). This test makes random permutations of the response variable between the different groups/levels of the factor and estimates the P as the proportion of permutations with an F-statistic greater than or equal to that observed in the real (unpermuted) data. This test is implemented in the R library “lmPerm” (v.2.1.0) (R Core Team 2019). We also adjusted the regression P-values using the FDR correction implemented in the p.adjust() function of the R package “stats” (Benjamini and Hochberg 1995; R Core Team 2019).
For the correlations between human host attributes and population genetic metrics, we focused on 172 samples with available metadata. Metadata about these samples were extracted from Brito et al. (2016) and NCBI accession numbers of the corresponding stool metagenomes are publicly available at http://fijicomp.bme.cornell.edu//data/FijiCOMPmetagenomicsamples.xlsx. Mobile genes selected for this analysis needed to respect the following conditions: 1) the gene should have at least 10X coverage to limit the impact of sequencing errors and 2) mobile gene should have less than 30% missing values across samples, for a total of 1,333 tested genes.
As for linear regressions between population genetics metrics and gene families, we selected genes based on the following set of conditions: 1) the gene should have at least 10X coverage to limit the impact of sequencing errors, 2) the gene should have available gene family annotations, which come from COG, KEGG, TIGRFAM, PFAM, or dbCAN databases (Brito et al. 2016), 3) the gene family should be represented by at least two genes within the data set, and 4) the mobile gene should have less than 30% missing values across samples, for a total of 512 tested genes. The first two filters are the basic requirements for doing these regressions analyses. However, the 3rd and 4th filters were chosen, respectively, to avoid the effects of small sample size for COG families that are underrepresented in the data set and to handle missing values caused by gene absence across sample or genes with low coverage in gut metagenomes.
Effect of HGT on Sequence Evolution
To determine the impact of HGT on mobile gene sequence evolution, multiple linear regressions were performed. In these multiple linear regressions, coverage, Gene Mobility—the number of species in which a mobile gene has been identified when looking for HGT events—and gene length were the explanatory variables and the various population genetic metrics were the response variables. We used the lm() function in R to remove collinearity with QR-decomposition/Gram-Schmidt orthogonalization. Thus, it is possible to assess the effect of Gene Mobility on each population genetics metrics while controlling for the effect of potential confounders like coverage and gene length. The significance of the multiple linear regression was evaluated with the F-test of the R function summary.lm() (R Core Team 2019). For each response variable Y tested (, , dN/dS, and Tajima’s D), there are two regression models:
| (6) |
| (7) |
The asterisk represents the fact that the regression controls for the effects of coverage and gene length, which increases the chance of observing sequencing errors. The adjusted R2 of a correlation represents the proportion of variable Y variance that is explained by the regression model with a correction for the number of explanatory parameters included in the model (k) and the sample size (n):
| (8) |
where SSres is the residual sum of squares and SStotal is the fitted data sum of squares. The type of correlation (positive or negative) can be determined by the regression coefficient. The reproducibility of the regressions was measured by the number of samples in which the correlation is significant.
For the simple linear regression, P-values were obtained from a t-test computed by the R function summary.lm() (R Core Team 2019). We adjusted the regression P-values using the FDR correction implemented in the p.adjust() function of the R package “stats” (Benjamini and Hochberg 1995; R Core Team 2019). Data standardization was performed before each regression to respect the t-test’s assumption of normality. The distributions of adjusted R2 values obtained from the t-tests converged with those from a nonparametric permutational ANOVA (K-S test P-value > 0.05) (Anderson 2001; Wheeler and Torchiano 2016). The results from the t-tests on standardized data are reported.
Variation across COG Categories
To assess how the relationships between gene mobility and Tajima’s D or coverage varied across COG categories, we considered 22 COG categories (Tatusov et al. 2000). We then used linear mixed models, through the R package lme4, to study the effect of gene mobility on coverage and Tajima’s D across COG categories (Bates et al. 2015). A linear mixed model allows to build a linear model between the response variable and the fixed effects while controlling for random effects. In the regression model, fixed effects are explanatory variables for which we want to know the relationship with the response variable. Random effects are grouping factors that explain the random variance of the relationship between the response variable and the fixed effects across a finite number of different groups. To control for random effects, the algorithm builds a linear model for each group. In the two regression models, the variables “COG category” and “Sample” were included as random effects:
| (9) |
| (10) |
Data were normalized using the Box-Cox transformation to ensure the condition of residual normality was accounted for before building the linear mixed models. We only used the 99.6% of Tajima’s D values that were negative and thus inversed their sign before applying Box-Cox transformation, which only works with positive values. We then performed the linear mixed model regression “” and inversed the sign of its slope.
We included human “Sample” as a random effect in the models to control for differences in alpha and beta diversity, and other sample-specific effects. As an additional test, we specifically included the microbial community alpha or beta diversity in the linear mixed models. To do so, we used Metaphlan2 (Segata et al. 2012) to estimate the relative abundances of named prokaryotic species in each metagenome (supplementary table S6, Supplementary Material online). We estimated the alpha diversity using Shannon entropy and performed principal component analysis on the Bray–Curtis dissimilarity matrix across samples (Legendre and Legendre 2012). We then added alpha diversity and the first principal component (PC1) of the Bray–Curtis matrix, which we used as a proxy of beta diversity, to the linear mixed models as follows:
| (11) |
| (12) |
We then removed “alpha” and “PC1_beta” one at a time and used the R function anova() to perform a likelihood ratio test between each linear mixed model and their nested model. This allowed us to test the significance of alpha and beta diversity in the models (Crainiceanu and Ruppert 2004; R Core Team 2019). The likelihood ratio test compares the likelihood of a nested model to the likelihood of the full linear mixed model, with the assumption that the test statistic follows a Chi-square distribution. Thus, we can create each nested model by the removal of a single variable from the full linear mixed model and assess the significance of this variable using a P-value from the Chi-square distribution (Crainiceanu and Ruppert 2004). We repeated the same procedure for “COG category” and “Sample” to test their significance in the models.
Effect of Mobile Gene Host Range
We divided the set of 7,990 mobile genes with enough coverage (≥10x) and prevalence (≤30% missing values) in the Fiji metagenomes in three categories of host range: shared within a single genus (n = 2,275 mobile genes), shared between genera of a single phylum (n = 874), and shared between phyla (n = 4,841). We then performed the linear mixed model regressions with “” as a random effect to determine the relationships Tajima’s D-mobility and coverage-mobility while controlling for random variations across genes with different host range:
| (13) |
| (14) |
Simulation of Pangenome Evolution
We simulated Sela, Wolf, and Koonin’s prokaryotic genome size evolution model with few changes, using the SodaPop simulation tool (Sela et al. 2016; Gauthier et al. 2019). In this model, the selective advantage of gene gain that is the advantage of having x + 1 genes instead of x genes, depends on the genome size, which is measured by the number of genes in the genome (x). Selection coefficients for gene loss have the opposite sign as gene gain; thus, gene gain is slightly beneficial while gene loss is slightly deleterious (Sela et al. 2016). The selection coefficient of gene gain and gene loss can thus be described by the following formula:
| (15) |
where is the selection coefficient of gene gain through HGT, “” is a constant input parameter of the simulation that allows to improve the fit of the linear expression with the real data, “” is a constant input parameter that represents the benefit or cost associated with the gain of a single gene, x represents genome size (number of genes), and is the selection coefficient of gene loss. We modified this formula to simulate a model where each gene has its own constant selective advantage regardless of genome size (x). To do so, we only needed to set the condition b = 0. This change allowed us to reproduce the shape of gene mobility distribution in simulation (supplementary fig. S1, Supplementary Material online). In this case:
| (16) |
where ∼ Exp(λ), λ is an input parameter of the simulation, and 1/λ represents the expected value of the exponential distribution of selection coefficients.
In the model, genome size (x) influences gene gain rate and gene loss rate. Indeed, the more genome size increases, the more gene gain rate decreases, and the more gene loss rates increase to find an equilibrium around a certain genome size x0. Therefore, when genome size (x) is smaller than genome size at equilibrium (x0), the cell has a higher probability of gene gain than loss. To consider the stochastic component of evolution, the cells and genes that are involved in each gain or loss events are randomly selected. Also, the number of gain or loss events is drawn from a Poisson distribution with the gain and loss rates as follows:
| (17) |
| (18) |
where is the gain rate that is the number of gene gain events per generation, is the loss rate that is the number of gene loss events per generation, and r’, s’, and are simulation input parameters that allow to tune the gain and loss rates.
We implemented this model in the SodaPop software, which simulates a Wright–Fischer process for asexual populations (Gauthier et al. 2019). In SodaPop, the mutation model is equivalent to Jukes–Cantor in which all single nucleotides occur at the same constant rate (Jukes and Cantor 1969). We also implemented a distribution of nonsynonymous mutation fitness effect in which 30% of mutations are lethal, as previously reported in the literature (Eyre-Walker and Keightley 2007), and 70% are drawn from a normal distribution, N (µ = −0.02, σ = 0.01). Synonymous mutations are all considered neutral unless the user provides data on species codon usage and the related fitness effects. SodaPop also offers flexibility in the initial setup of the simulation (Gauthier et al. 2019). We created scripts to facilitate the creation of the simulation starting conditions (https://github.com/arnaud00013/SodaPop/tree/Sodapop-pev/tools). The scripts allow to define each species abundance, gene content, and to define the genes that are mobile (https://github.com/arnaud00013/SodaPop/blob/Sodapop-pev/tools/Setup_SodaPop_with_PEV.py). Mobile genes can be transferred and lost whereas core genes and accessory genes (defined at the start of the simulation) can only be lost. For each set of simulations sharing the same input parameters, we ran 10 replicates. Each simulation included 5,000 cells, 10 species, 500 genes per cells at equilibrium, and a simulation time of 105 generations and a timestep of 104 generations to save simulation data. Population size is small in simulation because of hardware memory limitations. To avoid undesirable effects, like Muller’s Ratchet, we maintained species abundance constant. We also established a relatively high mutation rate on the order of 10−7 mutations per site per generation to compensate for small population sizes. Genome size equilibrium was reached for every simulation and the model is thus robust to the initial conditions (supplementary fig. S9, Supplementary Material online). The software is available on GitHub at: https://github.com/arnaud00013/SodaPop.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors would like to thank Compute Canada for allocated resources, and Louis-Marie Bobay and Gavin Douglas for constructive comments. B.J.S. was supported by aNatural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant. A.W.R.S. acknowledges funding from a Canada Research Chair Tier 2 and an NSERC Discovery Grant. A.N. was supported by an Fonds de Recherche du Quebec-Nature & Technologie (FRQNT) scholarship.
Data Availability
All metagenomic data used in this study is described at: http://fijicomp.bme.cornell.edu/data/FijiCOMPmetagenomicsamples.xlsx
All computer code used in this study is available at: https://github.com/arnaud00013/Fiji_Mobile_Gene_Specific_PopGen_scripts and https://github.com/arnaud00013/SodaPop
Literature Cited
- Anderson MJ.2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26:32–46. [Google Scholar]
- Andreani NA, Hesse E, Vos M.. 2017. Prokaryote genome fluidity is dependent on effective population size. ISME J. 11(7):1719–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Gordo I.. 2004. Adaptive evolution of asexual populations under Muller's ratchet. Evolution 58(7):1403–1413. [DOI] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, Walker S.. 2015. Fitting linear mixed-effects models using lme4. J Stat Soft. 67(1):1–48. [Google Scholar]
- Benjamini Y, Hochberg Y.. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Ser B. 57:289–300. [Google Scholar]
- Bobay LM, Ochman H.. 2018. Factors driving effective population size and pan-genome evolution in bacteria. BMC Evol Biol. 18(1):153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito IL, et al. 2016. Mobile genes in the human microbiome are structured from global to individual scales. Nature 535(7612):435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, et al. 2017. Frequency-dependent selection in vaccine-associated pneumococcal population dynamics. Nat Ecol Evol. 1(12):1950–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero OX, Polz MF.. 2014. Explaining microbial genomic diversity in light of evolutionary ecology. Nat Rev Microbiol. 12(4):263–273. [DOI] [PubMed] [Google Scholar]
- Crainiceanu C, Ruppert D.. 2004. Likelihood ratio tests in linear mixed models with one variance component. J R Stat Soc B. 66(1):165–185. [Google Scholar]
- Domingo-Sananes MR, McInerney JO.. 2019. Selection-based model of prokaryote pangenomes. bioRxiv 782573. doi: 10.1101/782573. [DOI] [Google Scholar]
- Eren AM, et al. 2015. Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3:e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Keightley PD.. 2007. The distribution of fitness effects of new mutations. Nat Rev Genet. 8(8):610–618. [DOI] [PubMed] [Google Scholar]
- Gardon H, Biderre-Petit C, Jouan-Dufournel I, Bronner G.. 2020. A drift-barrier model drives the genomic landscape of a structured bacterial population. Mol Ecol. doi: 10.1111/mec.15628. [DOI] [PubMed] [Google Scholar]
- Garud NR, Pollard KS.. 2020. Population genetics in the human microbiome. Trends Genet. 36(1):53–67. [DOI] [PubMed] [Google Scholar]
- Gauthier L, Di Franco R, Serohijos AWR.. 2019. SodaPop: a forward simulation suite for the evolutionary dynamics of asexual populations on protein fitness landscapes. Bioinformatics 35(20):4053–4062. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, Cameron Thrash J, Temperton B.. 2014. Implications of streamlining theory for microbial ecology. ISME J. 8(8):1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groussin M, et al. 2021. Elevated rates of horizontal gene transfer in the industrialized human microbiome. Cell 184(8):2053–2067.e18. [DOI] [PubMed] [Google Scholar]
- Hehemann JH, et al. 2010. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464(7290):908–912. [DOI] [PubMed] [Google Scholar]
- Jiang X, Hall AB, Xavier RJ, Alm EJ.. 2019. Comprehensive analysis of chromosomal mobile genetic elements in the gut microbiome reveals phylum-level niche-adaptive gene pools. PLoS One 14(12):e0223680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes TH, Cantor CR.. 1969. Evolution of protein molecules. In: Elsevier, editor. Mammalian protein metabolism. New York: Academic Press. p. 21–132. [Google Scholar]
- Koonin EV, Wolf YI.. 2010. Constraints and plasticity in genome and molecular-phenome evolution. Nat Rev Genet. 11(7):487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P, Legendre L.. 2012. Numerical ecology. Amsterdam (The Netherlands): Elsevier. [Google Scholar]
- Levade I, et al. 2017. Vibrio cholerae genomic diversity within and between patients. Microb Genom. 3(12):e000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney JO, McNally A, O'Connell MJ.. 2017. Why prokaryotes have pangenomes. Nat Microbiol. 2:17040. [DOI] [PubMed] [Google Scholar]
- Moulana A, Anderson RE, Fortunato CS, Huber JA.. 2020. Selection is a significant driver of gene gain and loss in the pangenome of the bacterial genus Sulfurovum in geographically distinct deep-sea hydrothermal vents. mSystems. 5(2):e00673–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW.. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25(7):1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsch J, Zhang Z, Baines JF.. 2009. The influence of demography and weak selection on the McDonald-Kreitman test: an empirical study in Drosophila. Mol Biol Evol. 26(3):691–698. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2019. A language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing. [Google Scholar]
- Segata N, et al. 2012. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 9(8):811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela I, Wolf YI, Koonin EV.. 2016. Theory of prokaryotic genome evolution. Proc Natl Acad Sci U S A. 113(41):11399–11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro BJ.2017. The population genetics of pangenomes. Nat Microbiol. 2(12):1574. [DOI] [PubMed] [Google Scholar]
- Smillie CS, et al. 2011. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480(7376):241–244. [DOI] [PubMed] [Google Scholar]
- Tajima F.1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi N, Cordero OX, Koonin EV, Kaneko K.. 2015. Gene-specific selective sweeps in bacteria and archaea caused by negative frequency-dependent selection. BMC Biol. 13:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV.. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28(1):33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes AM, Walter J, Segal E, Spector TD.. 2018. Role of the gut microbiota in nutrition and health. BMJ 361:k2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogan AA, Higgs PG.. 2011. The advantages and disadvantages of horizontal gene transfer and the emergence of the first species. Biol Direct. 6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos M, Hesselman MC, Te Beek TA, van Passel MWJ, Eyre-Walker A.. 2015. Rates of lateral gene transfer in prokaryotes: high but why? Trends Microbiol. 23(10):598–605. [DOI] [PubMed] [Google Scholar]
- Wheeler B, Torchiano M.. 2016. Package ‘lmPem’: permutation tests for linear models. 2.1.0 ed. Available from: https://cran.r-project.org/web/packages/lmPerm/index.html.
- Wolf YI, Makarova KS, Lobkovsky AE, Koonin EV.. 2016. Two fundamentally different classes of microbial genes. Nat Microbiol. 2:16208. [DOI] [PubMed] [Google Scholar]
- Yaffe E, Relman DA.. 2020. Tracking microbial evolution in the human gut using Hi-C reveals extensive horizontal gene transfer, persistence and adaptation. Nat Microbiol. 5(2):343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, et al. 2012. Human gut microbiome viewed across age and geography. Nature 486(7402):222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhernakova A, et al. ; LifeLines cohort study. 2016. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352(6285):565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All metagenomic data used in this study is described at: http://fijicomp.bme.cornell.edu/data/FijiCOMPmetagenomicsamples.xlsx
All computer code used in this study is available at: https://github.com/arnaud00013/Fiji_Mobile_Gene_Specific_PopGen_scripts and https://github.com/arnaud00013/SodaPop