

Abstract
CD11c is a canonical dendritic cell (DC) marker with poorly defined functions in the immune system. Here, we found that blocking CD11c on human peripheral blood mononuclear cell‐derived DCs (MoDCs) inhibited the proliferation of CD4+ T cells and the differentiation into IFN‐γ‐producing T helper 1 (Th1) cells, which were critical in acute graft‐versus‐host disease (aGVHD) pathogenesis. Using allogeneic bone marrow transplantation (allo‐BMT) murine models, we consistently found that CD11c‐deficient recipient mice had alleviated aGVHD symptoms for the decreased IFN‐γ‐expressing CD4+ Th1 cells and CD8+ T cells. Transcriptional analysis showed that CD11c participated in several immune regulation functions including maintaining antigen presentation of APCs. CD11c‐deficient bone marrow‐derived DCs (BMDCs) impaired the antigen presentation function in coculture assay. Mechanistically, CD11c interacted with MHCII and Hsp90 and participated in the phosphorylation of Akt and Erk1/2 in DCs after multiple inflammatory stimulations. Therefore, CD11c played crucial roles in triggering aGVHD and might serve as a potential target for the prevention and treatment of aGVHD.
Keywords: acute graft‐versus‐host disease, bone marrow transplantation, CD11c
CD11c‐deficient recipient mice had alleviated aGVHD symptoms for the decreased IFN‐γ‐expressing CD4+ Th1 cells and CD8+ T cells during mice allo‐BMT. CD11c participated in triggering aGVHD and maintaining antigen presentation function of APCs. Mechanistically, CD11c interacted with MHCII and Hsp90 and participated in the phosphorylation of Akt and Erk1/2 in DCs after multiple inflammatory stimulations.

Abbreviations
- aGVHD
acute graft‐versus‐host disease
- allo‐BMT
allogeneic bone marrow transplantation
- allo‐HSCT
allogeneic haematopoietic stem cell transplantation
- APCs
antigen‐presenting cells
- BMDCs
bone marrow‐derived DCs
- CFSE
carboxyfluorescein diacetate succinimidyl ester
- Co‐IP
coimmunoprecipitation
- DCs
dendritic cells
- ECM
extracellular matrix
- FBS
fetal bovine serum
- GO
gene ontology
- GSEA
gene set enrichment analysis
- GVL
graft versus leukaemia
- Hsp
heat‐shock protein
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MHC
major histocompatibility complex
- MoDCs
mononuclear cell‐derived DCs
- NK
natural killer
- p‐Akt
phosphorylated Akt
- PBS
phosphate‐buffered saline
- p‐Erk1/2
phosphorylated Erk1/2
- TBI
total body irradiation
- Th1
T helper 1
- Treg
regulatory T
INTRODUCTION
Integrins, a large group of adhesion receptors that consist of α and β subunit to form heterodimers, are constitutively expressed on different kinds of cells and facilitate cell–cell and cell–extracellular matrix (ECM) adhesion and modulate a broad spectrum of processes, such as cell proliferation, differentiation and organ development [1]. In addition to its role in adhesion to the ECM, the same integrin dimer has different ligand specificities in different cell types due to associations with different transmembrane receptors [2, 3. β2 integrins, which consist of 4 subunits, that is CD11a, CD11b, CD11c and CD11d, paired with the common β2 chain CD18, play important roles in regulating immune responses [4], including immune cell recruitment, migration, cellular interactions and downstream cell signalling [4, 5.
Acute graft‐versus‐host disease (aGVHD) remains a life‐threatening complication that has limited the efficacy and application of allogeneic haematopoietic stem cell transplantation (allo‐HSCT) [6]. Therefore, it would be meaningful to obtain a better understanding of the induction and pathogenic mechanisms of aGVHD. Based on current knowledge of its mechanism, aGVHD is initiated when alloreactive T cells are primed by host antigen‐presenting cells (APCs) to undergo clonal expansion and maturation [7]. DCs are professional APCs that can efficiently take up both self‐antigens and exogenous antigens, digest them into peptides and present them to T cells in the context of major histocompatibility complex (MHC) class I and class II molecules [8]. In addition to directly presenting antigen locally, DCs can migrate from peripheral tissues to secondary lymphoid organs, where they activate naive T cells, thereby triggering an adaptive immune response [9]. Recipient APCs have been shown to be pivotal in MHC‐mismatched GVHD, and their depletion by alloreactive natural killer (NK) cells diminishes GVHD [10]. Recent studies found that anti‐CD83 Ab could deplete activated CD83+ human DCs and inhibited the expansion of CD4+ T cells [11], thereby preventing human PBMC‐induced aGVHD in SCID mouse recipients [12].
Previous study has found that the activation status of blood CD11c+ DC is highly associated with aGVHD [13]. To reduce the ability of CD11c+ recipient cells in stimulating the proliferation and interferon‐gamma expression of allogeneic T cells can alleviate GVHD [14]. These studies verified the role of CD11c+ DCs in triggering aGVHD and indicated that CD11c molecule may play an important part in this process.
CD11c is mainly expressed on dendritic cells (DCs) and is well recognized as a canonical DC marker. CD11c was initially reported to bind iC3b, matrix proteins such as fibrinogen and collagen and the Ig superfamily cell adhesion molecule ICAM‐1/2 and VCAM‐1, which is responsible for the recruitment of monocytes, macrophages and DCs in many physiological and pathological processes [15]. Deficiency of CD11c in mice results in reduction in adipose tissue T cells, which do not express CD11c, indicated a potential role of CD11c in macrophage/DC T‐cell interactions [16]. A recent study showed that CD11c and its partner Itgb2 were required for DC capture by CD47‐deficient cells dependent on the integrin signalling adaptor Talin1 and played a crucial role in DC antigen uptake and activation [17]. However, the precise function and mechanism of CD11c in the communication between DCs and other immune cells are still poorly defined. Regarding CD11c activation, a previous study revealed that the phosphorylation of Ser‐1158/Thr‐758 was necessary [18] for its downstream functions. Even though there have been several reports on integrin coactivity receptors, such as Toll‐like receptor [19], FcγR, IFNR and TNFR, and cytoplasmic signalling cascades including the FAK signalling pathway, PI3K‐MAPK cascade [20] and Ras‐Erk pathway [21], the coactivity receptors and signalling pathways specific to CD11c remain unknown. Meanwhile, how CD11c maintains DC function and whether CD11c on DCs is involved in the process of aGVHD remain to be clarified.
In this study, we focused on the function of CD11c molecule in aGVHD, including its role in the interaction with T cells and antigen presentation function. The effect of CD11c deletion on aGVHD was explored using Itgax gene‐knockout mice. In an allogeneic bone marrow transplantation (allo‐BMT) murine model, CD11c‐deficient recipient mice showed decreased aGVHD severity and prolonged overall survival, which were due to the decreased expansion of alloreactive CD4+ T cells early after allo‐BMT and the decreased production of inflammatory cytokines in target organs. Blocking CD11c on human peripheral blood mononuclear cell‐derived DCs (MoDCs) impaired the proliferation of allogeneic CD4+ T and the differentiation of IFN‐γ‐producing T helper 1 (Th1) cells in vitro. Functional analysis demonstrated critical roles of CD11c in maintaining antigen presentation of DCs. Mechanistic study showed that CD11c interacted with MHCII and Hsp90 and participated in the phosphorylation of Akt and Erk1/2 in DCs after multiple inflammatory stimulations. Altogether, these results demonstrated that CD11c played an important role in maintaining DC functions and participated in triggering aGVHD.
RESULTS
Blocking CD11c on human MoDCs inhibited allogeneic CD4+ T‐cell proliferation and Th1 cell differentiation
Previous studies demonstrated that host‐derived DCs are important to elicit allogeneic T‐cell responses and trigger aGVHD [22]. The reduced adipose tissue T cells in CD11c‐deleted mice indicated a potential role of CD11c in macrophage/DC T‐cell interactions [16]. Therefore, we determined whether and how CD11c on DCs may be involved in induction of aGVHD. We first evaluated the function of CD11c on human MoDCs in the proliferation and activation of allogeneic T cells in vitro, which mimics activated donor T cells in patients with GVHD. Mature human MoDCs were pretreated with CD11c blocking antibody (clone 3.9) or control antibody and then cocultured with allogeneic CD4+ T cells or CD8+ T cells for 3 days. As shown in Figure 1, blocking CD11c molecule on human MoDCs significantly decreased the percentages of activated CD4+ CD69+ T cells (Figure 1a,b), CD4+ IFN‐γ+ T cells (Figure 1c,d) and proliferated CD4+ T cells (Figure 1e,f) compared with that in their self‐control groups pretreated with CD11c control antibody. Even though CD11c blockade significantly reduced the expression of IFN‐γ by CD4+ T cells (Figure 1c,d), it did not influence the expression of IL‐4 or IL‐17 (Figure S1A,B). The activation, proliferation and IFN‐γ expression of CD8+ T cells were not significantly affected after CD11c blockade (Figure S1C–E). In addition, blocking CD11c on human MoDCs did not affect the percentages of CD4+ FoxP3+ regulatory T (Treg) cells or effect memory T cells, including CD4+ CD44+ CD62L− T cells or CD4+ CD44+ CD62L− T cells (Figure S1F–I). These results indicated that CD11c on DCs critically participated in alloreactive CD4+ T‐cell proliferation and T helper 1 (Th1) cell differentiation. We hypothesized that CD11c expressed on residual DCs in recipients could promote donor CD4+ T‐cell proliferation and IFN‐γ‐producing Th1 cell differentiation, triggering or aggravating aGVHD.
FIGURE 1.
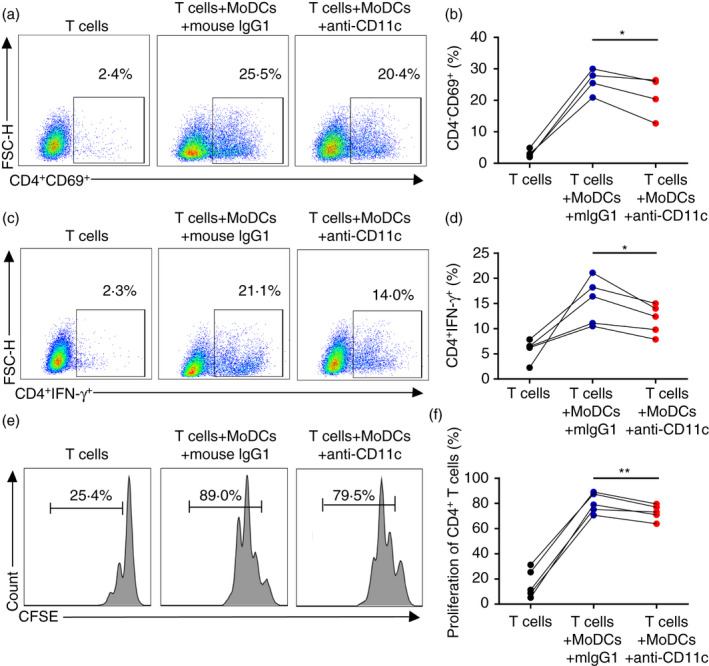
Blocking CD11c on human MoDCs inhibited allogeneic CD4+ T‐cell proliferation and Th1 cell differentiation. (a, c, e) Representative flow cytometric analysis of human CD4+ CD69+ T‐cell (a), CD4+ IFN‐γ+ T‐cell (c) and CFSE‐labelled CD4+ T‐cell (e) percentages, cocultured with allogeneic MoDCs for 3 days pretreated with CD11c blocking antibody or CD11c control antibody. (b, d, f) Percentages of human CD4+ CD69+ T cells (b), CD4+ IFN‐γ+ T cells (d) and proliferative CD4+ T cells (f), cocultured with allogeneic MoDCs for 3 days pretreated with CD11c blocking antibody or CD11c control antibody. Results are presented as mean ± SD, *P < 0·05, **P < 0·01, ***P < 0·001, by paired‐samples t‐test (b, d, f)
CD11c‐deficient recipient mice showed alleviated AGVHD symptoms
To investigate the role of recipient CD11c in aGVHD, Itgax gene‐knockout mice (CD11c−/−) were employed. We established a MHC‐mismatched [BALB/c mice to C57BL/6WT or CD11c−/− mice, C57BL/6WT mice to C57BL/6WT mice were used as autotransplantation control and lethally irradiated C57BL/6WT mice and CD11c−/− mice without transplantation were used as total body irradiation (TBI) control] BMT model to explore the function of recipient CD11c in aGVHD. Compared with WT recipient mice, CD11c−/− recipient mice had significantly alleviated aGVHD symptoms, reflected by lower body weight loss (Figure 2a), improved survival (Figure 2b) and lower clinical aGVHD scores (Figure 2c) after allo‐BMT. Pathologic analysis of the recipient lung, liver, intestine and colon also showed reduction in aGVHD severity in CD11c−/− recipient mice (detailed pathological description was in figure legend) (Figure 2d). Consistently, the percentages of IFN‐γ‐producing CD4+ Th1 cells and CD8+ T cells were dramatically decreased in the spleen and lymph node and slightly decreased in the lung in CD11c−/− recipient mice (Figure 2e–h). However, the percentages of IL‐4‐ and IL‐17‐producing CD4+ T cells or CD8+ T cells were almost unchanged compared with WT recipient mice (Figure S2A–D). Altogether, CD11c−/− recipient mice had significantly improved aGVHD symptoms and reduced death (within 30 days) early after allo‐BMT, supporting the hypothesis that the CD11c molecule and its related APCs in recipients play a major role in the early and middle stages of aGVHD after transplantation. Previous studies revealed that host DCs, which were activated during preparative conditioning for allo‐HSCT, presented host antigens to prime donor CD4+ and CD8+ T cells and promoted their proliferation and differentiation into alloreactive effector cells [23]. Therefore, we simultaneously tested the proliferation of donor T cells after allo‐BMT and found that the proliferation of donor CD4+ T cells was significantly decreased in CD11c−/− recipient mice at 3 days after allo‐BMT (Figure 2i,j). Reductions in T‐cell proliferation and activation, with decreased IFN‐γ‐expressing CD4+ Th1 cells and CD8+ T cells, may explain the alleviated aGVHD observed in CD11c−/− recipient mice.
FIGURE 2.
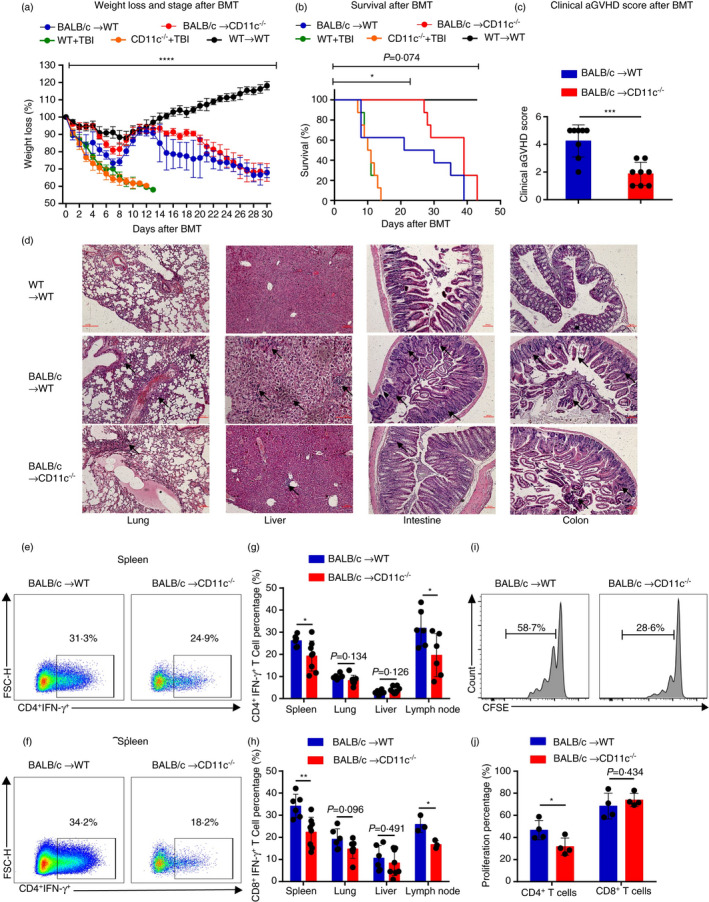
CD11c‐deficient recipient mice had alleviated aGVHD symptoms. (a–h) Lethally irradiated CD11c−/− mice or WT mice were transplanted with 1 × 107 BM cells and 2 × 107 splenic cells from BALB/c mice. (a, b) Lethally irradiated WT mice were transplanted with 1 × 107 BM cells and 2 × 107 splenic cells from WT mice. Lethally irradiated WT mice and CD11c−/− mice without infusion. (a, b) Body weight curves (n = 4–8) (a) and overall survival (n = 4–8) (b) are depicted. (c) Clinical aGVHD scores of recipients CD11c−/− mice and WT mice 14 days after BMT. (d) Representative haematoxylin‐ and eosin‐stained sections of the lung, liver, intestine and colon from recipient CD11c−/− mice and WT mice and autotransplantation WT mice (scale bar, 100 μm). The arrows were used to indicate the representative areas. Lung GVHD showed bronchiolitis with lymphohistiocytic infiltration. The marked epithelial damage was also observed. Hepatic GVHD showed an extensive lobular hepatitis with intense lymphocytic inflammation of sinusoids and portal spaces, and hepatocellular necrosis. Gut GVHD showed numerous lymphocytes infiltrated the glands within the oedematous mucosa. Apoptosis was widespread, along with segmental disruption of the glands. The degenerative colonic crypts were observed in colon. (e, f) Representative flow cytometric analysis of CD4+ IFN‐γ+ T‐cell (e) and CD8+IFN‐γ+ T‐cell (f) percentages in recipient mice in spleen 14 days after BMT. (g, h) Percentages of CD4+ IFN‐γ+ T cells (n = 6–8) (g) and CD8+ IFN‐γ+ T cells (n = 3–8) (h) in recipient mice in spleen, lung, liver and lymph node 14 days after BMT. (i, j) Lethally irradiated CD11c−/− mice and WT mice were transplanted with 1 × 107 T‐cell‐deleted (TCD) BM cells and 5 × 106 CFSE‐labelled CD3+ T cells sorted from spleens of BALB/c mice (n = 4). (i) Representative flow cytometric analysis of CFSE‐labelled CD4+ T cell in recipient mice in spleen 3 days after BMT. (j) Percentages of proliferative donor CD4+ T cell and CD8+ T cells in spleen 3 days after BMT. Results are presented as mean ± SD, *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001, Student's t‐test (c, g, h, j), two‐way ANOVA (a) and log‐rank (Kaplan–Meier method) test (b). All data are representative of three independent experiments
CD11c deleted impaired several immune regulation functions in APCS by transcriptional analysis
To explore the potential molecular mechanism by which CD11c molecule regulates the functions of APCs, we further profiled the transcriptome sequencing of APCs sorted from the splenocytes of CD11c−/− and WT mice. Remarkably, CD11c‐deficient APCs had extensive transcriptional changes, among 29 136 gene transcripts, 1352 transcripts with a ≥2 or ≤−2‐fold change in transcription were detected. Gene ontology (GO) enrichment analysis was employed to identify the main functions impaired in the absence of CD11c. As summarized in Figure 3a, regulation of T‐cell activation and lymphocyte/mononuclear cell proliferation were decreased with CD11c deletion, consistent with the impaired function in CD11c−/− recipient mice during allo‐BMT. Network analysis revealed genes that participate in the main impaired functions mentioned above, such as Ccl2, which contributes to T‐cell activation and leucocyte adhesion (Figure 3b). In addition, pathway enrichment analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database and gene set enrichment analysis (GSEA) were performed. Pathways involved in cytokine–cytokine receptor interactions, MAPK signalling pathway and Th1/2 cell and Th17 cell differentiation were significantly downregulated in CD11c‐deficient APCs (Figure 3c), and the related molecules are shown in the network in Figure 3d. Based on previous studies on integrin‐related signalling pathways, including the PI3K‐MAPK, JAK‐STAT, FAK‐RAS and AKT‐NFκB signalling pathways, GSEA was employed to determine their involvement in the function of CD11c‐deficient APCs. Strikingly, the MAPK signalling pathway, AKT signalling pathway (Figure S3), IFN‐γ‐related signalling pathway and antigen receptor‐mediated signalling pathway were associated with the CD11c molecule with significantly high confidence levels (Figure 3e).
FIGURE 3.
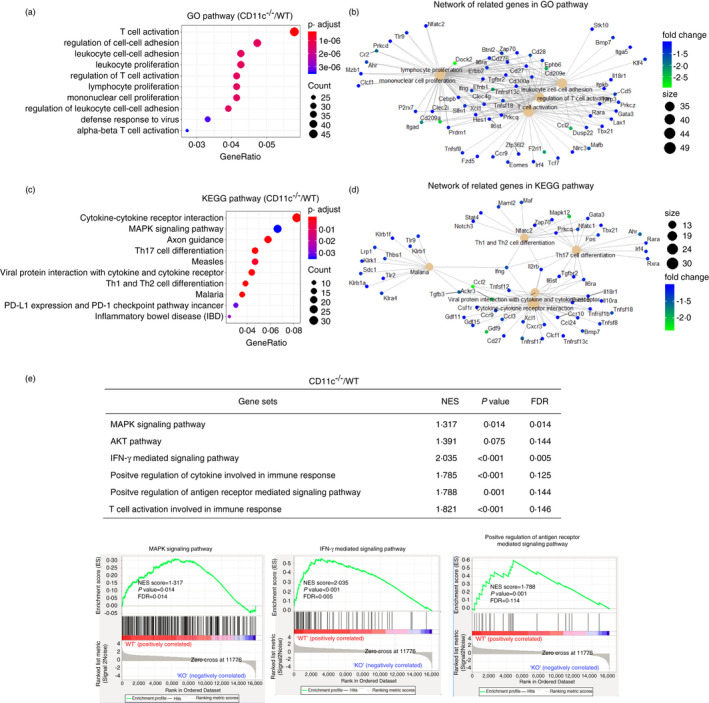
CD11c deleted impaired several immune regulation functions in APCs by transcriptional analysis. (a, c) Dot graph shows GO pathways (a) and KEGG pathways (c) downregulated in CD11c‐deficient APCs compared with WT APCs. Dot size relates to the number of differentially expressed genes. (b, d) Network displayed the related genes involved in the main pathways. (e) Gene pathways that were differentially expressed in APCs from CD11c−/− mice and WT mice according to GSEA. Gene sets were considered statistically significant at P‐value <0·05. Data were analysed with R3.3.5 software and GSEA
CD11c deficiency impaired antigen presentation function of DCS
Based on the transcriptional analysis, the antigen presentation function of APCs during allo‐BMT might be impaired when CD11c was deleted. To understand how CD11c regulates T cells in alloresponses, we determined the expression of the MHCII molecule and costimulatory molecules in splenocytes from CD11c−/− and WT mice. Results showed that the expression of MHCII molecule and the percentage of MHCII+ cells were significantly reduced while the levels of CD40, CD80 and CD86 remained comparable in the splenocytes from CD11c−/− and WT mice (Figure 4a–c). The MHCI and MHCII proteins play a pivotal role in the adaptive branch of the immune system. Both classes of proteins are responsible for presenting peptides on the cell surface for their recognition by T cells. MHCI complexes presented on nucleated cells were shown to be recognized by cytotoxic CD8+ T cells, while the presentation of MHCII on APCs regularly activates CD4+ T cells, leading to the co‐ordination and regulation of effector cells [24]. Therefore, we further studied the function of CD11c in antigen presentation by OVA immunization and found that CD4+ T cells sorted from OT‐II transgenic mice had a lower proliferation rate after CD11c‐deficient bone marrow‐derived DC (BMDC) stimulation in vitro, as reflected by significantly decreased carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution (Figure 4d,e). These results demonstrated that CD11c might act as a vital molecule in maintaining the antigen presentation function of DCs during allo‐BMT.
FIGURE 4.
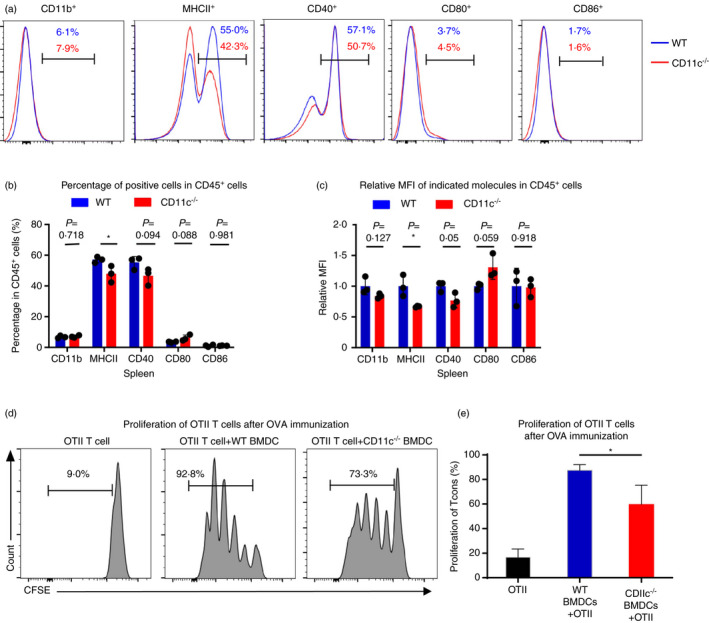
CD11c deficiency could impair the ability of antigen presentation function of DCs. (a) Representative flow cytometric analysis of the percentage of CD11b+, MHCII+, CD40+ and CD80+ in CD45+ cells in spleen in CD11c−/− mice or WT mice. (b) Percentages of cells expressing CD11b, MHCII, CD40, CD80 and CD86 in CD45+ cells in spleen in CD11c−/− mice or WT mice (n = 3). (c) Relative MFI (mean fluorescence intensity) of CD11b, MHCII, CD40, CD80 and CD86 in respective positive cells in spleen in CD11c−/− mice or WT mice (n = 3). (d) Representative flow cytometric analysis of CFSE‐labelled CD4+ T cells sorted from OT‐II transgenic mice cocultured with BMDCs from CD11c−/− mice or WT mice for 3 days pretreated with OVA for 30 min. (e) Percentages of proliferative CD4+ T cells sorted from OT‐II transgenic mice cocultured with BMDCs from CD11c−/− mice or WT mice for 3 days. Results are presented as mean ± SD, *P < 0·05, Student's t‐test (b, c, e). All data are representative of two‐three independent experiments
CD11c interacted with MHCII and Hsp90 and participated in the phosphorylation of AKT and Erk1/2 in DCS after multiple inflammatory stimulations
To investigate the underlying molecule mechanisms by which CD11c maintains the functions of antigen presentation in DCs, we pulled down proteins that interact with CD11c in DC2.4 cells. Mass spectrometry analysis showed that the top‐most related molecules were members of the heat‐shock protein (Hsp) family, especially Hsp90 (Figure 5a). We next verified these results by coimmunoprecipitation (Co‐IP) and Western blotting of DC2.4 cells stimulated with IFN‐γ, TNF‐α and LPS at different concentrations for different lengths of time. IFN‐γ and LPS could promote the interaction of CD11c with MHCII and Hsp90, while TNF‐α could promote the interaction of CD11c with MHCII but not Hsp90. Phosphorylated Erk1/2 (p‐Erk1/2) and phosphorylated Akt (p‐Akt), which are downstream of CD11c, were enhanced under inflammatory stimulation (Figure 5b–d), which supported the results of transcriptional analysis of CD11c‐related signalling pathways by GSEA and KEGG analysis. Furthermore, we analysed the colocalization of CD11c/MHCII, CD11c/Hsp90 and MHCII/Hsp90 in regions of interest under stimulation with IFN‐γ, TNF‐α and LPS using immunofluorescence confocal microscopy (Figure 5e) with Pearson's correlation analysis. Consistent with the Co‐IP results, IFN‐γ significantly increased the colocalization of CD11c/MHCII, CD11c/Hsp90 and MHCII/Hsp90 (Figure 5f–h). Notably, the TNF‐α‐treated group showed a lower colocalization score for MHCII/Hsp90 than the IFN‐γ‐ and LPS‐treated groups (Figure 5f). Studies showed that treatment of mouse APCs with a pharmacological Hsp90 inhibitor inhibited the MHCII‐mediated presentation of endocytosed and cytosolic proteins and synthetic peptides to specific T cells [25]. Therefore, CD11c interacted with MHCII and Hsp90 and participated in the phosphorylation of Akt and Erk1/2 in DCs after multiple inflammatory stimulations, which could be the potential molecule mechanisms by which CD11c maintains the functions of antigen presentation in DCs.
FIGURE 5.
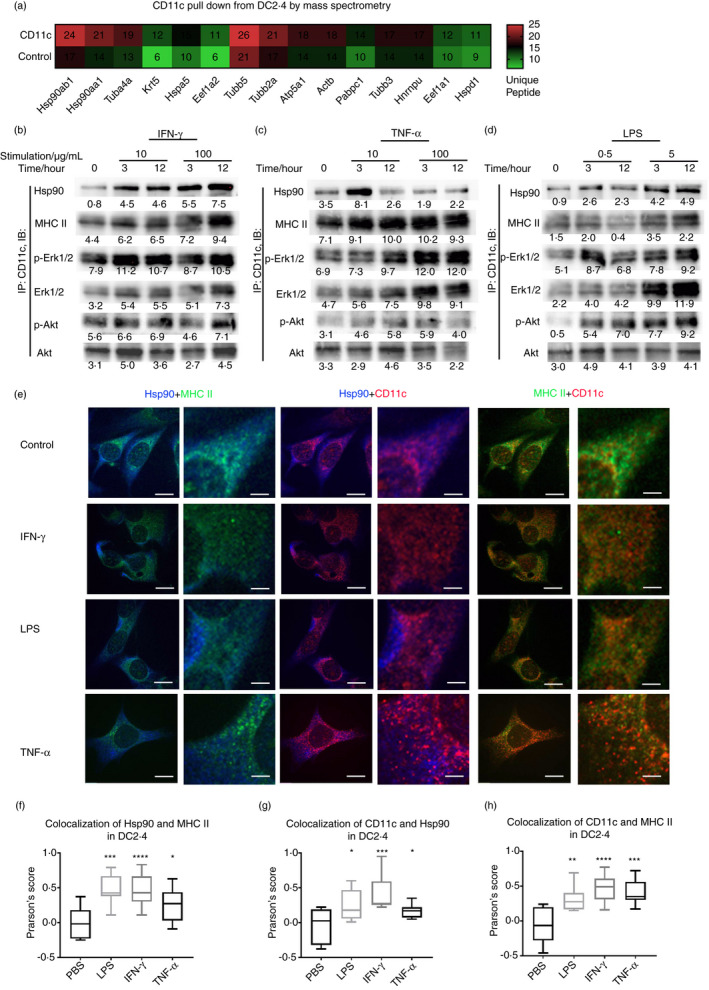
The downstream molecules interacted with CD11c after multiple inflammatory stimulations. (a) Proteins pulled down by CD11c from DC2.4 cells were analysed by mass spectrometry and ranked by the amount of unique peptides. (b–d) After incubation with 10 ng/ml IFN‐γ, 100 ng/ml IFN‐γ (b), 10 ng/mL TNF‐α, 100 ng/mL TNF‐α (c), 0.5 µg/ml LPS and 5ug/ml LPS (d) for 0, 3 or 12 h, DC2.4 cells were collected, coimmunoprecipitation was performed by CD11c antibody, and Hsp90, MHCII, phosphorylated Akt, total Akt, phosphorylated Erk1/2 and total Erk1/2 levels were detected by Western blotting. (e) Representative Hsp90, MHCII and CD11c confocal microscopy in DC2.4 cells treated with IFN‐γ, LPS and TNF‐α. Hsp90 was labelled in blue, MHCII in green and CD11c in red, bar = 50 μm. (f–h) Colocalization of Hsp90/MHCII (f), CD11c/Hsp90 (g) and CD11c/MHCII (h) of DC2.4 cells treated with IFN‐γ, LPS and TNF‐α, calculated regions of interest by Pearson's Score System (NIH) using ImageJ–Coloc2 with Pearson's Score Algorithm (https://imagej.net/Colocalization_Analysis)
DISCUSSION
CD11c, acts as one of the β2 integrin family, is well known as a surface marker of DCs. Evidence suggests that leucocyte recruitment to tissues is dependent on β2 integrins [26]. Functional β2 integrins are important in the formation of the immunological synapse between APCs and T cells [4]. The weak binding of a T lymphocyte to its specific antigen on the surface of an APC triggers intracellular signalling pathways in the T cell that activate its β2 integrins. The activated integrins then enable the T cell to adhere strongly to the APCs so that it remains in contact long enough to become stimulated fully.
At present, the understanding of the pathogenesis of aGVHD includes the activation of APCs, alloactivation of donor T cells and organ destruction by alloreactive T cells [27]. Importantly, alloreactive donor T cells can attack neoplastic cells, mediating the graft‐versus‐leukaemia (GVL) effect [28, 29, and also attack normal cells, causing GVHD [30, 31, 32. A central goal of allo‐HSCT research has been to develop methods to minimize GVHD while preserving T‐cell‐mediated GVL activity and immune reconstitution. Towards this end, substantial research has been devoted towards understanding how donor alloreactive T cells are activated, migrate to tissues and mediate end‐organ damage [32, 33. Previously studies have found that early T‐cell activation could be important, especially as the immediate post‐transplantation period is particularly conducive to T‐cell activation when host‐derived APCs are still present and functional [34]. Host APCs and donor T cells are critical for the induction and development of GVHD. APCs provided an essential checkpoint that controls the initiation of GVHD [7, 35. Conditioning induces tissue damage and releases a storm of cytokines that promote the maturation and activation of APCs and further amplification of donor T cells [7, 35, 36. Donor T cells are the best‐studied effectors of aGVHD, and most prophylaxis and treatment strategies are aimed at attenuating their function. T cells must receive adequate costimulation from APCs to become activated and acquire effector functions [31]. Many costimulatory ligand/receptor interactions and their involvement in aGVHD have been described [38]. The decreased expression of CD11c, CD40 and CD86 on host DCs was one of the mechanisms of minimizing aGVHD [39].
It is well known that DCs are the most important APCs, which we found that the blockade of CD11c on human MoDCs could significantly reduce the proliferation of CD4+ T cells and the percentage of IFN‐γ‐producing Th1 cells. CD11c molecule involves in regulating the immune response via impacting the antigen presentation functions of DCs and plays a key role in activating alloreactive T cells. Moreover, CD11c‐deficient APCs had reprogrammed functions revealed by transcriptional analysis, such as its downregulation of T‐cell activation and lymphocyte cell proliferation. In an aGVHD model, CD11c‐deficient recipient mice exhibited an impaired ability to expand donor CD4+ T cells at the early stage after allo‐BMT and a reduced infiltration of inflammatory cytokines in aGVHD‐target organs, alleviating aGVHD symptoms and decreasing GVHD‐associated mortality. Therefore, CD11c may act as a therapeutic target in aGVHD. Targeting CD11c molecule in receipts early after allo‐HSCT, instead of depleting DCs, may decrease DC‐mediated donor T‐cell activation and further alleviate aGVHD.
MHCII‐expressing intestinal epithelial cells functioned as APCs to prime donor CD4+ T cells in vivo and induced lethal aGVHD [40]. We found that CD11c deficiency impaired the expression of MHCII and CD11c could interact with MHCII under inflammatory stimulation, which might participate in donor CD4+ T‐cell activation and trigger aGVHD. Proteins in the Hsp family are well‐known chaperones during antigen digestion and processing. A recent study suggested that Hsp90 activity plays a major role in MHCII‐mediated antigen presentation pathways and implicated IFN‐inducible Hsp90‐independent mechanisms [41]. Pharmacological inhibition of Hsp90 could decrease GVHD‐associated mortality without impairing the GVL effect [42, 43. Mechanistically, we demonstrated that CD11c interacted with Hsp90 and MHCII under inflammatory stimulation and maintained the phosphorylation of Akt and Erk1/2 in DCs.
Our study revealed the crucial role of CD11c molecule in the triggering aGVHD during allo‐BMT. CD11c expressed on DCs could promote the proliferation and differentiation of CD4+ T cells via maintaining the expression of MHCII. Under multiple inflammatory stimulations, CD11c could interact with MHCII and Hsp90 and participate in the phosphorylation of Akt and Erk1/2 in DCs. In an aGVHD model, CD11c‐deficient recipient mice had milder symptoms and less inflammatory cell infiltration. Therefore, CD11c is not only an important marker of DCs but also a signalling molecule vital for maintaining the normal interaction between DCs and T cells. Altogether, we suggested that therapeutic targeting of the CD11c molecule holds enormous potential as a treatment option for aGVHD and other immune‐ or inflammation‐related diseases.
MATERIALS AND METHODS
Mice
All animals were maintained in specific pathogen‐free barrier facilities and used in accordance with protocols approved by the Institutional Animal Care and Use Committee at the Institute of Hematology, Chinese Academy of Medical Sciences. C57BL/6 (H‐2b), BALB/c (H‐2d) and CD45.1+ mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). CD11c−/− mice were generated as previously described [44], and the colony was maintained in the animal facility of the Institute of Hematology, Chinese Academy of Medical Sciences. All mice used were 6–8 weeks old unless otherwise noted. The sample sizes for the various animal experiments were chosen based on prior data generated in the laboratory.
Acute GVHD murine models
Recipient C57BL/6WT and CD11c−/− mice (8–10 weeks old) were lethally irradiated with 800 cGy split into two doses on day 0. Irradiated recipient mice were then intravenously injected with 1 × 107 BM cells with 2 × 107 splenic cells from BALB/c mice. Lethally irradiated C57BL/6WT recipient mice intravenously injected with 1 × 107 BM cells with 2 × 107 splenic cells from C57BL/6WT mice were used as autotransplantation control group while lethally irradiated C57BL/6WT recipient mice and CD11c−/− recipient mice were used as TBI control group. The BM and splenic cell suspensions had been prepared using leg bones and spleens, respectively. The secretion of inflammatory cytokines by T cells in the spleen, lung and liver was analysed by flow cytometry 14 days after transplantation. Recipient mouse survival, body weight and clinical score were monitored until the mouse died or up to 50 days. Animals were given water supplemented with trimethoprim/sulphamethoxazole for 2 weeks following BMT.
Cell purification and flow cytometry
To analyse cell surface markers, single‐cell suspensions were prepared from the spleen, lymph node, lung, BM and liver, and stained with the indicated antibodies according to the instructions of the manufacturers. Cell surface staining was mostly performed at 4° for 40 min. For intracellular cytokine staining, cells were stimulated for 4–5 h with phorbol myristate acetate (50 ng/ml) and ionomycin (500 ng/ml). APCs were purified by negative selection by removing T cells using CD3‐negative isolation kit (Stem Cell) first and then sorted as CD11b+ MHCII+ cells by excluding B cells (B220+) and neutrophils (CD11b+ Ly6G+) with a FACSAria III (BD Biosciences). The sorted populations were more than 98% pure and used for RNA extraction with TRIzol reagent (Invitrogen). The antibodies were obtained from eBioscience, Biolegend, BD Biosciences and Invitrogen. Flow cytometry data were acquired on a FACSCanto II (BD Biosciences) and analysed with FlowJo software (FlowJo LLC, Ashland, Oregon). RNA transcriptome sequencing data sets were analysed with R3.3.5 software and assessed by GSEA.
Culture with GM‐CSF
A total of 5–10 × 106 BM cells per well were cultured in tissue culture‐treated 12‐well plates in 2 ml of RPMI 1640 medium containing 2 mM l‐glutamine, 100 μg/ml penicillin, 100 μg/ml streptomycin (all from Gibco), 100 ng/ml GM‐CSF (PeproTech) and 50 ng/ml IL‐4 (Peprotech) at 37° in 5% CO2 for 6 days. Half of the medium was removed every two days, and new medium was added, GM‐CSF and IL‐4 concentrations were maintained throughout medium changes. On day 6, cells were harvested and cultured in 2 ml of complete medium with GM‐CSF, IL‐4 and TNF‐α (100 ng/ml, PeproTech) for another two days. On day 8, the cells were harvested for the following experiments.
T‐cell proliferation and activation in vitro
A total of 2 × 105 BMDCs were treated with 0·5 μg/mL OVA323‐339 peptide (Sigma) in 100 μl for 30 min at 37° in 5% CO2 in RPMI 1640 containing 2 mM l‐glutamine, 100 μg/ml penicillin and 100 μg/ml streptomycin (all from Gibco) in 96‐well round‐bottom plates. CD4+ T cells from the spleens and lymph nodes of OT‐II transgenic mice were sorted by magnetic separation using a mouse CD4‐negative isolation kit (Stem Cell) according to the manufacturer's instructions. Enriched CD4+ T cells were 98% pure, as analysed by a FACSCanto II (BD Biosciences). Then, 2 × 105 CFSE‐labelled CD4+ T cells were added to BMDCs and cultured at 37° in 5% CO2. After 3–4 days of culture, responding CD4+ T cells were assessed for CFSE dilution by flow cytometry.
Coimmunoprecipitation and western blotting
DC2.4 cells were cultured in RPMI 1640 with IFN‐γ (10 ng/ml and 100 ng/ml), TNF‐α (10 ng/ml and 100 ng/ml) or LPS (0·5 µg/ml and 5 µg/ml) (all from PeproTech) for 3 h or 12 h at 37° in 5% CO2. Coimmunoprecipitation and Western blotting were carried out as previously described [45, 46 with antibodies against CD11c, Hsp90, MHCII, Akt/p‐Akt and Erk1/2/p‐Erk1/2 (Cell Signaling Technology).
Immunofluorescence microscopy
Cells were washed with phosphate‐buffered saline (PBS), fixed and permeabilized with cold (−20°) 50% methanol and 50% acetone, washed with PBS, blocked with 5% fetal bovine serum (FBS) in PBS for 1 h, washed with PBS and incubated with primary antibody (anti‐CD11c, 1:250; anti‐Hsp90, 1:250) in 3% FBS in PBS for 2 h. The cells were washed with PBS, incubated with secondary antibody conjugated with Alexa Fluor 647 or 405 for 1 h, washed with PBS and then incubated with anti‐MHCII antibody conjugated with Alexa Fluor 488. Images among the experiments were taken with a Nikon Ti‐E spinning disc confocal microscope (UltraVIEW VOX, PerkinElmer) using identical exposure times and scaled equivalently. Confocal images were analysed with ImageJ–Coloc2 software with Pearson's correlation analysis (https://imagej.net/Colocalization_Analysis).
Histological analyses
Samples were harvested from the lungs, livers, intestines and colons of transplanted recipients on day 14 after transplantation and immediately fixed in 10% formalin and washed with 70% ethanol. Samples were then embedded in paraffin, cut into 6‐μm thick sections and stained with H&E. The presented data are from individual GVHD target organs. All slides used for GVHD analysis were coded and read in a blinded fashion. Photomicrographs were taken at a 10× magnification.
Human T‐cell and DC coculture
Human T cells from different individuals were isolated from PBMCs by Ficoll density gradient centrifugation and sorted with a FACSAria III (BD Biosciences), the sorted populations were more than 98% pure. Human DCs were differentiated from monocytes isolated from PBMCs. The matured DCs were sorted with a FACSAria III (BD Biosciences), and the sorted populations were more than 98% pure. Purified human T cells from each individual were labelled with CFSE and separated into 3 parts, and then cultured alone or cocultured with mature allogeneic DCs pretreated with CD11c‐blocking antibody (anti‐human CD11c, clone 3.9, Biolegend) or cocultured with mature allogeneic DCs pretreated with control antibody (mouse IgG1, Biolegend) at a 2:1 ratio for an additional 3 days, respectively. The expression of IFN‐γ and CD69 and the CFSE dilution were analysed by flow cytometry (Canto II, BD Biosciences). All patients gave their written informed consent in accordance with the Declaration of Helsinki, and this work was approved by the Ethics Committee of the Institute of Hematology, Chinese Academy of Medical Sciences (Tianjin, China).
Statistics
Data were analysed using Prism 5.00 (GraphPad Software, San Diego, CA, USA) to calculate the statistical significance of differences in the mean values when two groups were compared and to determine P values. Survival curves were generated using the Kaplan–Meier method and compared using the log‐rank test. Student's t‐test, the paired t‐test or two‐way ANOVA was performed, depending on the number of compared groups. Differences for which P < 0·05 were considered statistically significant.
CONFLICT OF INTEREST
The authors declare no financial or commercial conflicts of interest.
AUTHOR CONTRIBUTIONS
Q.W., X.P. and E.J. designed the study. Q.W. performed the experiments and analysed the data. X.S. and X.P. helped with some experiments. X.P. helped to analyse some data. Y.H., M.W., D.Y., R.ZH., J.W., Q.M., W.ZH., A.P., Y.H. and S.F. contributed to case data collection and sample preparation. C.M.B. and H.W. provided some results about the phenotype of the CD11c−/− mice; Q.W. wrote the paper. H.W., X.P., X.F. and E.J. edited the paper. E.J. and X.P helped to obtain the funding for the research. E.J., X.F. and M.H. oversaw the project.
Supporting information
Figure S1
{kind=link}
Figure S2
{kind=link}
Figure S3
{kind=link}
ACKNOWLEDGMENTS
We thank Professor Ying Wang (Department of Immunology, Peking University) for editing the manuscript.
Funding information
This work was supported by grants from the National Natural Science Foundation of China (81670171 to E.J., 81601369 to X.P., 81870090 to X.F.), the Non‐profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019‐RC‐HL‐013 to X.F.) and the National Basic Research Program of China (2015CB964402 to E.J.).
Contributor Information
Xiaolei Pei, Email: peixiaolei@ihcams.ac.cn.
Xiaoming Feng, Email: fengxiaoming@ihcams.ac.cn.
Mingzhe Han, Email: mzhan@ihcams.ac.cn.
Erlie Jiang, Email: jiangerlie@163.com.
REFERENCES
- 1.Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res. 2010;339:269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kechagia JZ, Ivaska J, Roca‐Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. 2019;20:457–73. [DOI] [PubMed] [Google Scholar]
- 3.Anderson LR, Owens TW, Naylor MJ. Structural and mechanical functions of integrins. Biophys Rev. 2014;6:203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schittenhelm L, Hilkens CM, Morrison VL. β2 Integrins as regulators of dendritic cell, monocyte, and macrophage function. Front Immunol. 2017;8:1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison VL, James MJ, Grzes K, et al. Loss of beta2‐integrin‐mediated cytoskeletal linkage reprogrammes dendritic cells to a mature migratory phenotype. Nat Commun. 2014;5:5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teshima T, Reddy P, Zeiser R. Acute graft‐versus‐host disease: Novel biological insights. Biol Blood Marrow Transplant. 2016;22:11–6. [DOI] [PubMed] [Google Scholar]
- 7.Chakraverty R, Sykes M. The role of antigen‐presenting cells in triggering graft‐versus‐host disease and graft‐versus‐leukemia. Blood. 2007;110:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. [DOI] [PubMed] [Google Scholar]
- 9.Sallusto F, Lanzavecchia A. The instructive role of dendritic cells on T‐cell responses. Arthritis Res. 2002;4(Suppl 3):S127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teshima T, Ordemann R, Reddy P, et al. Acute graft‐versus‐host disease does not require alloantigen expression on host epithelium [published correction appears in Nat Med 2002 Sep; 8(9):1039]. Nat Med. 2002;8:575–81. [DOI] [PubMed] [Google Scholar]
- 11.Wong KY, Baron R, Seldon TA, et al. CD83 antibody inhibits human B cell responses to antigen as well as dendritic cell‐mediated CD4 T cell responses. J Immunol. 2018;200:3383–96. [DOI] [PubMed] [Google Scholar]
- 12.Seldon TA, Pryor R, Palkova A, et al. Immunosuppressive human anti‐CD83 monoclonal antibody depletion of activated dendritic cells in transplantation. Leukemia. 2016;30:692–700. [DOI] [PubMed] [Google Scholar]
- 13.Lau J, Sartor M, Bradstock KF, Vuckovic S, et al. Activated circulating dendritic cells after hematopoietic stem cell transplantation predict acute graft‐versus‐host disease. Transplantation. 2007;83:839–46. [DOI] [PubMed] [Google Scholar]
- 14.Fischer JC, Bscheider M, Göttert S, Thiele Orberg E, et al. Type I interferon signaling before hematopoietic stem cell transplantation lowers donor T cell activation via reduced allogenicity of recipient cells. Sci Rep. 2019;9:14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ting HJ, Stice JP, Schaff UY, et al. Triglyceride‐rich lipoproteins prime aortic endothelium for an enhanced inflammatory response to tumor necrosis factor‐alpha. Circ Res. 2007;100:381–90. [DOI] [PubMed] [Google Scholar]
- 16.Wu H, Perrard XD, Wang Q, et al. CD11c expression in adipose tissue and blood and its role in diet‐induced obesity. Arterioscler Thromb Vasc Biol. 2010;30:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Wu H, An J, et al. Critical role of integrin CD11c in splenic dendritic cell capture of missing‐self CD47 cells to induce adaptive immunity. Proc Natl Acad Sci USA. 2018;115:6786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uotila LM, Aatonen M, Gahmberg CG. Integrin CD11c/CD18 α‐chain phosphorylation is functionally important. J Biol Chem. 2013;288:33494–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerold G, Abu Ajaj K, Bienert M, et al. A Toll‐like receptor 2‐integrin beta3 complex senses bacterial lipopeptides via vitronectin. Nat Immunol. 2008;9:761–8. [DOI] [PubMed] [Google Scholar]
- 20.Klekotka PA, Santoro SA, Zutter MM. alpha 2 integrin subunit cytoplasmic domain‐dependent cellular migration requires p38 MAPK. J Biol Chem. 2001;276:9503–11. [DOI] [PubMed] [Google Scholar]
- 21.Fincham VJ, James M, Frame MC, Winder SJ. Active ERK/MAP kinase is targeted to newly forming cell‐matrix adhesions by integrin engagement and v‐Src. EMBO J. 2000;19:2911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H, Tian Y, Wang Y, et al. Dendritic cell regulation of Graft‐Vs.‐Host Disease: immunostimulation and tolerance. Front Immunol. 2019;10:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Germain RN. MHC‐dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–99. [DOI] [PubMed] [Google Scholar]
- 24.Wieczorek M, Abualrous ET, Sticht J, et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: conformational plasticity in antigen presentation. Front Immunol. 2017;8:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajagopal D, Bal V, Mayor S, et al. A role for the Hsp90 molecular chaperone family in antigen presentation to T lymphocytes via major histocompatibility complex class II molecules. Eur J Immunol. 2006;36:828–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lämmermann T, Bader BL, Monkley SJ, et al. Rapid leukocyte migration by integrin‐independent flowing and squeezing. Nature. 2008;453:51–5. [DOI] [PubMed] [Google Scholar]
- 27.Boieri M, Shah P, Dressel R, Inngjerdingen M. the role of animal models in the study of hematopoietic stem cell transplantation and GvHD: a historical overview. Front Immunol. 2016;7:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vincent K, Roy DC, Perreault C. Next‐generation leukemia immunotherapy. Blood. 2011;118:2951–9. [DOI] [PubMed] [Google Scholar]
- 29.Bleakley M, Riddell SR. Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol. 2011;89:396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blazar BR, Murphy WJ, Abedi M. Advances in graft‐versus‐host disease biology and therapy. Nat Rev Immunol. 2012;12:443–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holtan SG, Pasquini M, Weisdorf DJ. Acute graft‐versus‐host disease: a bench‐to‐bedside update. Blood. 2014;124:363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markey KA, MacDonald KP, Hill GR. The biology of graft‐versus‐host disease: experimental systems instructing clinical practice. Blood. 2014;124:354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi J, Ziga ED, Ritchey J, et al. IFNγR signaling mediates alloreactive T‐cell trafficking and GVHD. Blood. 2012;120:4093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson BE, McNiff JM, Jain D, et al. Distinct roles for donor‐ and host‐derived antigen‐presenting cells and costimulatory molecules in murine chronic graft‐versus‐host disease: requirements depend on target organ. Blood. 2005;105:2227–34. [DOI] [PubMed] [Google Scholar]
- 35.Socié G, Blazar BR. Acute graft‐versus‐host disease: from the bench to the bedside. Blood. 2009;114:4327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toubai T, Sun Y, Tawara I, et al. Ikaros‐Notch axis in host hematopoietic cells regulates experimental graft‐versus‐host disease. Blood. 2011;118:192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu L, Zhang Y, Luo G, Li Y. The roles of stem cell memory T cells in hematological malignancies. J Hematol Oncol. 2015;8:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Briones J, Novelli S, Sierra J. T‐cell costimulatory molecules in acute‐graft‐versus host disease: therapeutic implications. Bone Marrow Res. 2011;2011:976793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo X, Xu L, Li Y, Tan H. Notch pathway plays a novel and critical role in regulating responses of T and antigen‐presenting cells in aGVHD. Cell Biol Toxicol. 2017;33:169–81. [DOI] [PubMed] [Google Scholar]
- 40.Koyama M, Mukhopadhyay P, Schuster IS, et al. MHC Class II antigen presentation by the intestinal epithelium initiates Graft‐versus‐Host disease and is influenced by the microbiota. Immunity. 2019;51:885–98.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12‐CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–88. [DOI] [PubMed] [Google Scholar]
- 42.Joly AL, Deepti A, Seignez A, et al. The HSP90 inhibitor, 17AAG, protects the intestinal stem cell niche and inhibits graft versus host disease development. Oncogene. 2016;35:2948. [DOI] [PubMed] [Google Scholar]
- 43.Huang Q, He S, Tian Y, et al. Hsp90 inhibition destabilizes Ezh2 protein in alloreactive T cells and reduces graft‐versus‐host disease in mice. Blood. 2017;129:2737–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Gower RM, Wang H, et al. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pei X, Zheng D, She S, et al. Elevated expression levels of PC3‐Secreted Microprotein (PSMP) in prostate cancer associated with increased xenograft growth and modification of immune‐related microenvironment. Front Oncol. 2019;9:724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu X, Zelmer A, Wellmann S. Visualization of protein‐protein interaction in nuclear and cytoplasmic fractions by co‐immunoprecipitation and in situ proximity ligation assay. J Vis Exp. 2017;119:55218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3