

This review discusses how the control of proliferation and cell division by cell cycle regulators is also involved in the metabolic adaptive responses when cells need to produce energy. We describe how cell cycle regulators are involved in the control of metabolic pathways, in cell growth and the functionality of organelles including endolysosomes and mitochondria.
Keywords: cell cycle regulators, cell growth, endolysosomes, metabolism, mitochondria
Abstract
Adapting to changes in nutrient availability and environmental conditions is a fundamental property of cells. This adaptation requires a multi‐directional coordination between metabolism, growth, and the cell cycle regulators (consisting of the family of cyclin‐dependent kinases (CDKs), their regulatory subunits known as cyclins, CDK inhibitors, the retinoblastoma family members, and the E2F transcription factors). Deciphering the mechanisms accountable for this coordination is crucial for understanding various patho‐physiological processes. While it is well established that metabolism and growth affect cell division, this review will focus on recent observations that demonstrate how cell cycle regulators coordinate metabolism, cell cycle progression, and growth. We will discuss how the cell cycle regulators directly regulate metabolic enzymes and pathways and summarize their involvement in the endolysosomal pathway and in the functions and dynamics of mitochondria.
Abbreviations
- AA
amino acids
- AATYK1
Apoptosis‐associated tyrosine kinase 1
- AMPK
AMP‐activated protein kinase
- AP2M1
clathrin‐associated/assembly/adaptor protein medium 1
- APC/C
anaphase‐promoting complex/cyclosome
- CDK
cyclin‐dependent kinases
- CDK4
cyclin‐dependent kinase 4
- CIE
clathrin‐independent endocytosis
- CME
clathrin‐mediated endocytosis
- COX
cytochrome c oxidase
- DRP1
dynamin‐related protein 1
- EGFR
epidermal growth factor receptor
- ERC
endocytic recycling compartments
- EZH2
enhancer of zeste homolog 2
- FAO
fatty acid oxidation
- FBXO4
F‐Box protein 4
- FEME
fast endophilin‐mediated endocytosis
- GLS1
glutaminase 1
- GLUT
glucose transporter
- HKII
hexokinase II
- INK4
inhibitors of CDK4
- IRS2
insulin receptor substrate 2
- LC3
microtubule‐associated proteins 1A/1B light chain 3B
- LD
lipid droplets
- MAT1
ménage‐à‐trois 1
- MCOLN1
mucolipin 1
- MEF
mouse embryonic fibroblasts
- MFN
mitofusin
- mTORC1
mammalian target of rapamycin complex
- NRF
nuclear respiratory factor
- NRF1
nuclear respiratory factor 1
- OMM
outer mitochondrial membrane
- PDK
pyruvate dehydrogenase kinase
- PFKP
6‐phosphofructokinase
- PGC‐1α
eroxisome proliferator‐activated receptor gamma coactivator 1‐alpha
- PI3P
phosphatidylinositol‐3 phosphate
- PK
pyruvate kinase
- PKM2
pyruvate kinase M2
- pRB
pocket protein of the retinoblastoma
- RALA
ras‐like proto‐oncogene A
- RNR
ribonucleotide reductase
- ROS
reactive oxygen species
- TFEB
transcription factor EB
- TS
thymidylate synthase
- TSC2
tuberous sclerosis complex 2
- TXNIP
thioredoxin interacting protein
- VPS34
vacuolar protein sorting 34
Introduction
Cell cycle regulators are key factors for the control of proliferation and cell survival. They have been typically involved in several physio‐pathological processes, including development, tissue regeneration, or cancer [1]. The progression into the cell cycle is dependent on several families of proteins with specific functions in each phase of the cell cycle, ultimately leading to cell division, which is the final step of this process. The cyclins‐CDKs holoenzymes, the retinoblastoma family of pocket protein (pRB, p130, p107), the E2F transcription factors (E2F1‐8), and the cyclin‐dependent kinases (CDK)‐inhibitor families are key effectors of the cell cycle. The participation of these proteins in the control of proliferation and cell division has been extensively studied and is reviewed elsewhere [1, 2, 3]. Here, we will discuss how this pathway is also involved in the metabolic adaptive response of the cells triggered by growth factors and the energetic need of the cells.
In particular, we will focus on how cell cycle regulators control metabolic pathways, cellular growth, and the functionality of organelles such as endolysosomes and mitochondria.
The multi‐directional coordination of the cell division machinery, metabolism, and cell growth (the accumulation of mass) is essential for cell division [4] (Fig. 1). Determining the mechanisms accountable for this coordination is crucial for understanding various disease states such as cancer or physiological processes such as differentiation and aging [5]. Hence, it is important to understand the molecular mechanisms determining the cell cycle–metabolism–growth interface [6]. While it was broad consensus that metabolism is driving the cell division cycle, it is becoming increasingly clear that also the cell division machinery is coordinating metabolism with the cell cycle and growth, thereby ensuring that nutritional demands of cells are met [5, 6].
Fig. 1.
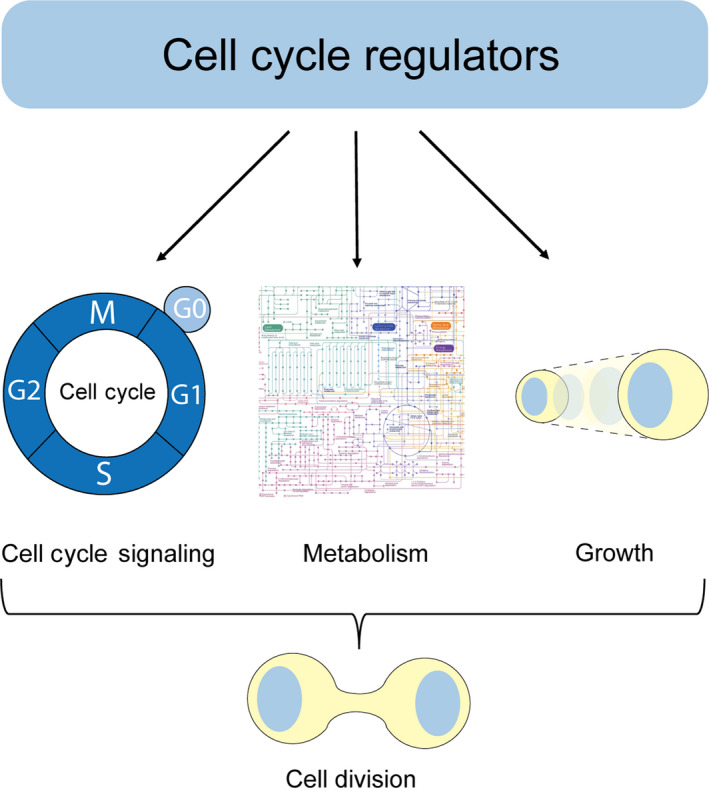
Multiple roles of cell cycle regulators in the regulation of cell cycle signaling, metabolism, and growth. Upon proper stimuli, cell cycle regulators trigger progression through the cell cycle. This is accompanied by an adaptive metabolic response and regulation of cellular growth, ultimately leading to cell division.
Pioneering work in Saccharomyces cerevisiae in the 1970s identified that cell division cycle mutants, which arrested in different cell cycle phases, continued cell growth. This led to the understanding that metabolism and growth control cell cycle division, but not vice versa [5]. However, ‘omics’ studies, biochemical studies, as well as single cell investigations have underscored the evidence that intimate connections between metabolism and growth with the cell cycle exist [7, 8]. While it is well described how metabolism affects cell division, this review will focus on the reverse, less well‐defined relationship how the cell cycle machinery controls metabolism and growth (Fig. 1).
Several studies in this past decade have challenged the view that there is a hierarchy of metabolism and growth over the cell division cycle [4]. Indeed, several core cell cycle regulators and components, notably cyclin D, CDKs, pRB and E2Fs, specifically target metabolic enzymes and pathways and thereby control metabolism and growth [9, 10]. The identification of specific metabolic targets of cell cycle regulators are increasingly appreciated in basic and clinical research [11]. These metabolic targets operate in different pathways, including glucose metabolism, oxidative metabolism [12] amino acid (AA) metabolism, nucleotide and lipid metabolism [10] and are involved in the delivery of metabolic precursors for cell growth. In the following chapter, we will discuss how cell cycle machinery directly regulates metabolic enzymes and pathways (Fig. 2).
Fig. 2.
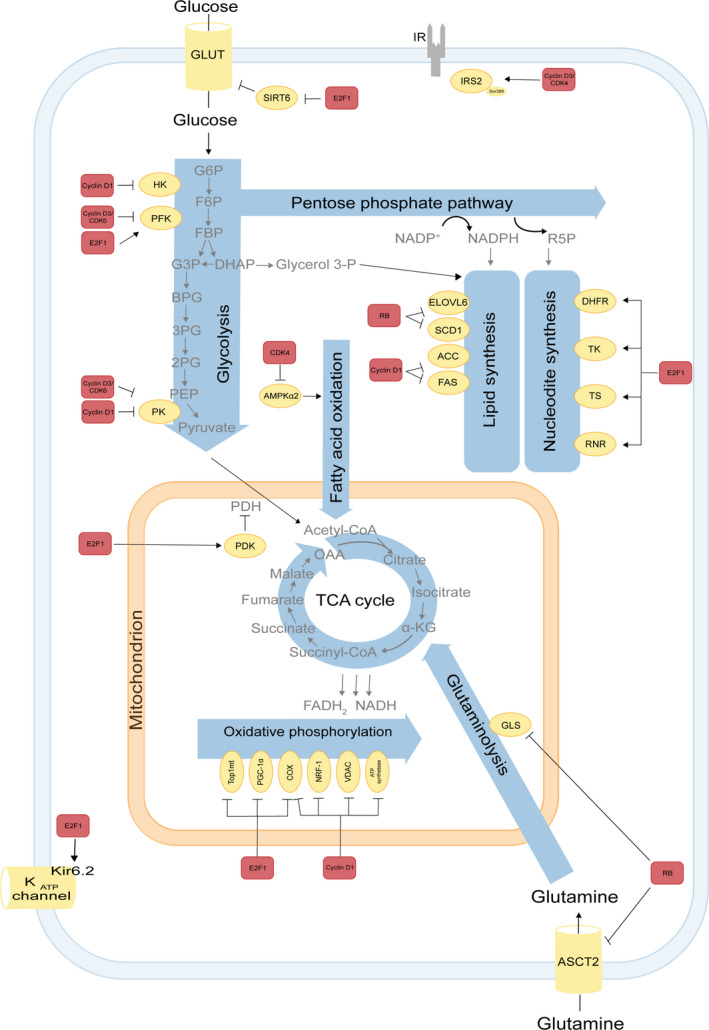
Participation of cell cycle regulators in metabolic pathways. The E2F‐pRB pathway is involved in several metabolic pathways. In glucose metabolism of pancreatic β‐cells E2F1 regulates Kir6.2, a key component of the KATP channel, controlling glucose‐induced insulin secretion [13]. Moreover, E2F1 also enhances glycolysis in bladder and prostate cancer cell lines through the SIRT6 that inhibits the transcription of several key glycolytic genes such as GLUT1 [22]. Furthermore, E2F1 stimulates glycolysis by upregulating the expression of the enzyme 6‐phophofructo‐2 kinase/fructose‐2,6‐bisphosphatase [20, 21]. E2F1 also induces expression of PDK1/4 in pancreatic cancer cells and PDK4 in the heart. PDKs inhibit PDH and thereby oxidative metabolism [23, 24]. In addition, associated to pRB E2F1 modulates the expression of key genes implicated in mitochondrial biogenesis or oxidative phosphorylation, such as mitochondrial topoisomerase 1 (Top1mt) [159], PGC‐1α [160], cytochrome c oxidase subunit 4 COX4, and cytochrome c oxidase subunit 7C [161, 162]. In nucleotide metabolism, E2F1 induces dihydrofolate reductase, thymidine kinase, TS and RNR [36, 38, 39]. Deletions of RB family members increase glutamine uptake and conversion to glutamate, mediated through elevated expression of the glutamine transporter ASCT2 and GLS1 activity in MEF [34] and upregulate enzymes involved in elongation, long chain fatty acid family member 6, and desaturation of fatty acids by stearoyl‐CoA desaturase 1 [163]. Cyclin D1 has an E2F1‐independent role in the control of oxidative metabolism. It directly modulates the activity of the transcription factor NRF1, the mitochondrial voltage‐dependent anion channel [122, 164], mitochondrial electron transport chain components COX and ATP synthase [165] thereby inhibiting oxidative phosphorylation. In addition, cyclin D1 inhibits HKII and pyruvate kinase (PK), which are involved in the glycolysis pathway as well as the lipogenic enzymes acetyl‐CoA carboxylase and fatty acid synthase levels [26]. In adipocytes, cyclin D3‐CDK4 complex phosphorylates IRS2 at serine 388, thereby creating a positive feedback loop that maintains adipocyte insulin signaling [17]. In addition, the CDK4 also represses FAO in an E2F1‐independent manner through direct phosphorylation and inhibition of AMPKα2. Cyclin D3–CDK6 kinase phosphorylates and inhibits the catalytic activity of two key enzymes in the glycolytic pathway, 6‐phosphofructokinase (isoform PFKP) and pyruvate kinase (isoform PKM2) in T‐cell acute lymphoblastic leukemia [27]. Interactions between the cell cycle machinery and metabolic enzymes indicated by arrows may be either direct or indirect. Color codes represent proteins belonging to the cell cycle machinery (red) or to metabolic pathways (yellow).
The cell cycle machinery directly regulates metabolic enzymes and metabolic pathways
Glucose metabolism and insulin signaling
In several organisms, glucose is a preferred carbon source. Indeed, glucose metabolism and cell cycle machinery are mutually regulated. On the one hand, glucose promotes elevated cyclin‐dependent kinase 4 (CDK4) activity, phosphorylation of pRB, which subsequently causes increased E2F1 transcriptional activity [13, 14]. Reciprocally, studies in genetically modified mice, in which the cell cycle regulators such as transcription factor E2F, CDK4, pRB, cyclin D, CDK2 and CDK5, are deficient, have emphasized that glucose homeostasis and insulin signaling can be regulated by the cell cycle machinery [10]. Our laboratory has previously shown that E2F1 directly maintains the expression of Kir6.2, a key component of the KATP channel involved in the regulation of glucose‐induced insulin secretion in non‐proliferating pancreatic β‐cells, both in vitro and in vivo. Consistently, Kir6.2 expression was decreased in the pancreas of E2f1−/− mice, leading to insulin secretion defects in these mice [13]. However, E2F1−/− mice are not diabetics. On the contrary, they have increased insulin sensitivity and show a decrease in white fat tissue. These phenomena are specific for E2F1, whereas β‐cell expansion can be compensated by E2F2. Consequently, E2f1/E2f2 double mutant mice develop insulin‐deficiency diabetes [15, 16].
Beside its important function in insulin‐producing β‐cells, cell cycle regulators such as CDK4 also have an impact on insulin target cells such as hepatocytes and adipocytes [17, 18]. In both cell types, CDK4 is a target and a regulator of insulin signaling. For instance, in liver, insulin activates cyclin D1‐CDK4, which in turn phosphorylates the histone acetyltransferase GCN5. GCN5 subsequently acetylates peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC‐1α), which leads to inhibition of the expression of gluconeogenic genes. This pathway, which involves cyclin D1‐CDK4, is responsible for controlling glucose metabolism by suppressing hepatic glucose production in mice [18]. In adipocytes, cyclin D3‐CDK4 complex controls insulin signaling by phosphorylation of the insulin receptor substrate 2 (IRS2) at serine 388, thereby creating a positive feedback loop that maintains adipocyte insulin signaling. Cdk4‐deficient mice showed impaired insulin signaling and were glucose intolerant. In contrast, mice with hyperactive CDK4, which carry a mutation in the Cdk4 gene (Cdk4R24C/R24C ) that abolishes the binding to members of the INK4 family of CDK inhibitors, were more glucose tolerant and showed increased insulin sensitivity. These results indicate that CDK4 activity is positively correlated with insulin sensitivity [17].
CDK2 is another cell cycle kinase, which is involved in the S‐phase entry and cell cycle progression in mammals. An important metabolic function of CDK2 was revealed by studying the phenotype of mice with pancreas‐specific loss of Cdk2, which are glucose intolerant primarily due to defects in glucose‐stimulated insulin secretion. This is accompanied by defects in β‐cell metabolism and a disturbed mitochondrial structure. Persistent defects in insulin secretion culminate in progressive deficits in β‐cell proliferation, reduced β‐cell mass and diabetes. These findings may be attributed directly to the deletion of Cdk2, which phosphorylates the transcription factor Foxo1 in a glucose‐dependent manner [19].
In addition to its role in cell cycle, E2F1 also regulates the expression of several genes that have established roles in glucose regulation. For example, E2F1 drives expression of the F‐type isoform of the glycolytic enzyme, 6‐phophofructo‐2‐kinase/fructose‐2,6‐bisphosphatase, which results in the synthesis of fructose‐2,6‐bisphosphate, a potent stimulator of glycolysis [20, 21]. Moreover, E2F1 also enhances glycolysis in bladder and prostate cancer cell lines through the suppression of the expression of sirtuin 6 (SIRT6), a NAD(+)‐dependent deacetylase that inhibits the transcription of several key glycolytic genes [22]. In parallel, E2F1 promotes glycolytic gene expression by blocking glucose oxidation in the mitochondria therefore enhancing the expression of the pyruvate dehydrogenase kinase (PDK) enzymes [23, 24]. PDKs are critical nutrient sensors and inhibitors of glucose oxidation, through phosphorylation of pyruvate dehydrogenase (PDH) [25]. While in the heart E2F1 regulates PDK4 [23], in pancreatic cancer cells E2F1 enhances the expression of PDK1 and PDK3 isoforms, which results in increased aerobic glycolysis and proliferation [24].
Another example of elevated glycolytic enzyme expression regulated by a cell cycle regulator is the hexokinase II (HKII). HKII catalyzing the first steps of glycolysis and elevated RNA and protein expression levels of HKII were found in mammary glands of mice expressing antisense cyclin D1 [26].
Moreover, a recent study identified interaction of cyclin D3‐CDK6 with nine glycolytic enzymes in human cancer cells [27]. Two of these interaction partners, 6‐phosphofructokinase (PFKP) and pyruvate kinase M2 (PKM2) catalyze irreversible and rate‐limiting steps in glycolysis and were further characterized by the authors. While PFKP catalyze the reaction of glucose‐6‐phosphate to fructose‐bis‐phosphate, PKM2 converts phospho‐enol‐pyruvate to pyruvate. Cyclin D3‐CDK6 kinase phosphorylates and inhibits the catalytic activity of PFKP and PKM2 resulting in redirection of glycolytic intermediates into the pentose phosphate and serine pathways in T‐cell acute lymphoblastic leukemia [27].
Cell cycle regulators also target glucose metabolism in plants and yeast, indicating that regulation of central carbon metabolism by CDKs is conserved between organisms. In Arabidopsis thaliana, several enzymes of glycolysis and mitochondrial metabolism have been identified as direct CDK‐A (the CDK that drives G1/S/G2 in plants) substrates [28]. Furthermore, two labs independently found that yeast carbohydrate metabolism is regulated by CDK1 (the only cell cycle CDK in yeast) [29, 30]. The enzymes neutral trehalase and glycogen phosphorylase are activated by CDK to liquidate the carbohydrate storage molecules trehalose and glycogen, thereby generating glucose. This regulation may optimize the decision to undertake a final cell division in nutrient‐limited environments, when cells are faced with sudden nutrient depletion [29] or are approaching the stationary phase [30]. Thus, CDK directly regulate the increase of glycolytic flux at the G1/S transition to ensure sufficient carbon and energy supply and coordinate cell cycle, metabolism and growth [9].
Amino acids and nucleotide metabolism
Glutamine is the most abundant AA in human sera and is essential for cell growth and proliferation. While cells have the ability to synthesize glutamine, this AA is considered a ‘conditionally’ essential AA. Under the demands of cell growth, the need for glutamine exceeds the cell’s synthetic capacity and cells depend on enhanced glutamine uptake [31]. Glutamine fulfills a variety of biosynthetic functions as it serves as a source of carbon and nitrogen for AA, protein, lipids, and nucleotide biosynthesis. One of the best understood roles of glutamine is a ready supply of carbon for TCA anaplerosis and other cellular pathways. Glutamine also plays an important role in maintaining redox homeostasis and facilitation of certain enzymatic reactions. In particular, glutamine‐derived glutamate serves as the major carbon source for glutathione biosynthesis in multiple cell systems [32]. Glutamine catabolism is a regulated process, and the signaling mechanisms that control glutamine metabolism are being studied intensively. Two independent studies discovered metabolic changes in the utilization of glutamine after loss of pRB in mammalian cells or the fly ortholog RBF1 in Drosophila [33, 34]. Deletions of the three RB family members (pRB, p107, and p130) in immortalized mouse embryonic fibroblasts (MEF) increases glutamine uptake and conversion to glutamate, mediated through elevated expression of the glutamine transporter ASCT2 and glutaminase 1 (GLS1) activity, respectively [34]. In addition, RBF1/pRB deficient cells exhibited increased glutamine incorporation into glutathione [33, 34]. Glutamine deprivation significantly reduced glutathione levels within Rb‐inactivated MEFs and led to increased generation of reactive oxygen species (ROS) [34], suggesting that these cells are more susceptible to oxidative stress. Furthermore, data from RBF1‐depleted larvae indicated that supplementation of the antioxidant N‐acetyl‐cysteine rescued fasting due to reduction of oxidative stress and suppression of glutathione levels, thereby increasing the availability of glutamine carbon for nucleotide production [33]. The catabolism of glutamine is not only important for ATP production but is also required to synthesize metabolic precursors that are required by the dividing cell, such as deoxynucleotide triphosphates. Loss of all three RB family proteins led to increased incorporation of glutamine carbon into aspartate, a phenomenon also observed in RBF1‐repressed Drosophila larvae and in pRB suppressed tumor cells [33].
The TCA metabolite malate is the primary source for aspartate, which provides the carbon backbone for pyrimidine synthesis, and contributes an amino group to purine bases. The observation that RB1 loss alters the metabolic flux into nucleotide synthesis is consistent with previous findings [35]. Among the first described E2F1‐regulated genes are dihydrofolate reductase, thymidylate synthase (TS), ribonucleotide reductase (RNR) and thymidine kinase, which are involved in nucleotide synthesis and are known to be induced by E2F1 [36, 37, 38, 39]. Indeed, the transcription of each of these genes increases in the absence of pRB [40]. These studies strongly emphasize that cells harboring inactivated pRB, such as some cancer cells, could be selectively targeted by anti‐glutamine strategies such as glutaminase inhibitors [35]. A recent study in indicated that CB‐839 administration significantly reduced tumor growth in both KP‐6634 and KPH2‐7215 allografts and these tumors showed increased glutamine levels with concurrent decreased glutamate and aspartate levels, confirming that CB‐839 effectively blocks glutamine breakdown and production of downstream metabolites [41].
Recently, a study in a model of esophageal squamous cell carcinoma (ESCC) demonstrated the contribution of F‐box protein 4 (FBXO4)‐cyclin D1 axes in regulating glutamine addiction through its role as a suppressor of RB function and an activator of mammalian target of rapamycin complex 1 or mechanistic target of rapamycin complex 1 (mTORC1). Deregulated FBXO4‐cyclin D1 lead to compromised energy production from oxidative phosphorylation due to mitochondrial dysfunction and reprogramming of cellular metabolism. This paradox of energy production/consumption makes these cells vulnerable, which offers new therapeutic opportunities. A combined treatment with GLS1 inhibitor telaglenastat (CB‐839) and targeting mitochondrial respiration effectively induced apoptosis and suppressed cell proliferation in vitro and in vivo, providing a promising strategy to treat human ESCC and to overcome CDK4/6 inhibitor (palbociclib) resistance [42]. In contrast, Tarrado‐Castellarnau et al. [43] characterized the metabolic reprogramming that follows CDK4/6 depletion in HCT116 colorectal cancer cells. They demonstrated that CDK4/6 inhibition increases mitochondrial metabolism through elevated utilization of glutamine and enhanced mitochondrial respiratory capacity, which is in agreement with the results from Franco et al. [8] in a pancreatic cancer cell model. Additional experiments showed that CDK4/6 depletion increased glutathione, NADPH, and ROS levels while it impaired fatty acid synthesis in HCT116 cells, all of which are processes where glutamine is or can be involved.
Beside increased glutaminolysis, CDK4/6 depletion leads to mTORC1 activation and compromised adaptation to hypoxia in a MYC‐dependent manner. These dependencies render cells highly sensitive to therapeutic combinations of CDK4/6 inhibitors with inhibitors of glutaminase or mTOR [43]. The co‐treatment with CDK4/6 inhibitor palbociclib and the GLS1 inhibitor telaglenastat (CB‐839) is currently in phase 1 and 2 clinical trials in patients with KRAS‐mutated colorectal cancer and KRAS‐mutated non‐small‐cell lung cancer (NCT03965845).
Collectively, the tight coordination between cell cycle and metabolism is becoming evident across different proliferating cell types, differentiated tissues as well as on organismal level. Cell cycle regulator play an important role in coordinating the cell division cycle and metabolism and ensuring that the energy demands of cells are met.
This chapter highlights the fact that studies of cell cycle regulator have uncovered a series of direct links to enzymes involved in metabolism. These connections are especially interesting given that mutations that drive tumorigenesis often reprogram central carbon metabolism. Metabolic reprogramming is considered to enhance the ability of cancer cells to sustain the intermediates needed for synthesis of proteins, lipids, and DNA. Therefore, mechanistic studies will be required to completely understand species and tissue dependent effects, because different cell types may have different strategies to supply the appropriate amount of energy and anabolic precursors. Further uncovering the role of cell cycle regulators in health and disease may open new strategies for treatments.
Cell cycle regulators control cellular growth through mTORC1 signaling
mTORC1 is a protein complex that senses nutrient, energy and redox status to regulate protein synthesis in diverse biological processes such as cell growth, proliferation, motility and autophagy. mTORC1 activity and lysosomes are mutually regulated. The lysosomes are able to store metabolites and sense AAs and lipids which lead to activate mTORC1. Reciprocally, when mTORC1 is active, inhibitory phosphorylation of transcription factor EB (TFEB) prevents the transcription of genes that regulate lysosomal biogenesis. However, in starvation conditions, when mTORC1 is inactive, TFEB accumulates in the nucleus leading to the transcriptional and translational responses that enhance lysosomal biogenesis and activity [44]. Thus, mTORC1 plays a key role in regulating lysosomal adaptation and expansion in response to metabolic challenges [45]. Moreover, mTORC1 also controls lysosomal membrane dynamics by regulating the localized calcium signals which are important to generate the fusion of endolysosomal membranes [46, 47]. mTORC1 controls the activity of some lysosomal calcium channels including mucolipin 1 (MCOLN1), which is upregulated during starvation, to mediate the increase in lysosomal degradative capacity [48]. Interestingly, calcium signaling resulting from MCOLN1 channel, also leads to mTORC1 activation [49] and thus it can trigger clathrin‐dependent lysosomal tubulation and fission as a mechanism to restore free lysosomes once autophagy is finished [50]. These data indicate that the complex regulation of nutrient sensing and the role of mTORC1 governing calcium signaling and lysosomal dynamics have a great impact in cellular nutrient balance.
The connection between cell cycle regulators and the mTOR pathway was evidenced in several studies. For instance, a study by Ladu et al. investigated the molecular mechanisms underlying oncogenic cooperation between c‐Myc and E2F1 in relationship to hepatocellular carcinoma. Coexpression of c‐Myc and E2F1 in c‐Myc/E2F1 transgenic mice (Alb/c‐Myc/Alb/E2F1) suggests that E2F1 functions as an important anti‐apoptotic factor in human and rodent liver cancer that counteract c‐Myc‐driven apoptosis via activation of PIK3CA/Akt/mTOR and c‐Myb/COX‐2 pathways [51]. Consistent with this observation, E2F1 regulates cell growth through the activation of mTORC1 independently of the canonical PI3K/Akt/TSC pathway. Mechanistically, E2F1 links lysosomal trafficking and mTORC1 activation by transcriptional regulation of the v‐ATPase subunit, ATP6V0B [52, 53].
Cyclin D‐CDK4 and CDK6 also couple the cell cycle machinery to cell growth via activation of mTORC1. In a mouse model for resistance to anti‐HER2 targeted therapy, tumor cells were characterized by the loss of CDK inhibitors such as p16INK4a and overexpression of cyclin D1, resulting in increased CDK4/6 activities [54]. The authors revealed that the downregulation of CDK4 leads to reduced tuberous sclerosis complex 2 (TSC2) phosphorylation, which activates TSC2, inhibiting mTOR and its substrate p70‐S6K. The negative feedback loop induced on kinases of the epidermal growth factor receptor (EGFR) family can be suppressed by CDK4/6 inhibitors, resulting in increased phosphorylation of EGFR or HER2 and AKT, but without simultaneous activation of mTOR or p70‐S6K due to the effect of TSC2 dephosphorylation [54]. Despite the direct interaction between cyclin D1 and TSC2 [55], the first to show a direct phosphorylation of TSC2 by CDK4 or CDK6 were Romero‐Pozuelo et al. in D rosophila [56]. Recently, they also found that cyclin D‐CDK4/6 activates mTORC1 by binding and phosphorylation of TSC2 in multiple human and mouse cell lines and pharmacological inhibition of CDK4/6 leads to a rapid, TSC2‐dependent reduction of mTORC1 activity [57]. In addition to these findings, another connection between CDK4 and mTORC1 activation was revealed. On the one hand, directly through the phosphorylation of FLCN, which regulates the recruitment of mTORC1 to the lysosomal surface in response to AAs, and indirectly, through the regulation of lysosomal function [58].
Altogether, these studies indicate that cyclin D‐CDK4/6 activates mTORC1, thereby promoting cell proliferation both directly via phosphorylation of RB and indirectly by promoting cell growth [57]. The link between TSC2‐mTOR or FLCN‐mTOR and cyclin D1‐CDK4/6 may provide new biomarkers and strategies in clinical trials. Additionally, synergistic treatments of mTOR and CDK4/6 already demonstrated therapeutic potential in a wide variety of human cancers [59].
Cell cycle regulators and metabolism in the endolysosomal pathways
The endosomal pathways in proliferating cells
Proliferating cells are in constant need of nutrients to synthesize proteins, lipids and DNA and to successfully remodel the plasma membrane when mitosis occurs. The endocytic pathway mediates nutrient uptake, the composition and internalization of plasma membrane receptors and signal transduction. These signals trigger biological functions that under endocytic trafficking regulation connect the cytoplasmic cellular processes with its environment. Importantly, internalization and trafficking of membrane‐bound receptors may affect the localization of specialized signaling cascades [60].
In 1975, the discovery and characterization of clathrin [61] marked the starting point to describe the several regulators of endocytosis which control the clathrin‐dependent and independent pathways for membrane internalization. Endocytosis is a ubiquitous cellular mechanism that mediates the transport of extracellular macromolecules across the plasma membrane and it is mediated through the binding of transmembrane carriers. The newly formed vesicles detach from the plasma membrane and traffic toward their intracellular destination [60]. Although clathrin‐mediated endocytosis (CME) is the best characterized endocytic pathway and the principal uptake mechanism to support essential functions in the cell, including the insulin receptor internalization [62], other clathrin‐independent pathways can mediate the uptake of extracellular lipids and proteins and the elimination of activated receptors from the plasma membrane [63]. Adaptor proteins like AP‐2 complex or epsins bind clathrin or phospholipids of the coated pits to initiate endocytosis and facilitate vesicle formation and budding. After endocytosis, clathrin‐coated‐vesicles are fused within the early endosome, which is a control point for sorting receptors, allowing them to be directed to recycling endosomes and thus back to the cell surface, or instead directed to the multivesicular endosomes and subsequently toward the late endosome and finally to the lysosome for degradation [64].
It is widely accepted that endocytosis occurs in homogenous rates during G1, S and G2 phases of the cell cycle and that CME is more constitutively active at any given time than clathrin‐independent endocytosis (CIE) which varies more due to cell migration, membrane receptors activation and membrane tension [65]. However, some evidence points toward differences in the uptake of cargos depending on the cellular environment. For instance, endocytosis and membrane lipid composition are influenced by cellular adaptability to the population context. Studying the CME activity in different human cell lines, it was found that in vitro cultures with a high local cell density maintain high CME activity and keep in place mechanisms to successfully control this activity [66].
Epidermal growth factor receptor and its ligand EGF have been well studied in driving cells through G1 and under sustained signals engaging cell cycle progression by the induction of cyclin D‐ CDK4/6 complex activation. Consequently, EGFR internalization by endocytosis has been a topic of great interest. When EGFR is activated, it can be rapidly internalized by CME and CIE mechanisms. EGFR sorting to early endosomes, late endosomes and recycling endosomes determine the fate of EGFR to lysosomal degradation and thus signal attenuation, or recycling to the plasma membrane and thus altering proliferation, cellular homeostasis and cell death [67]. In a model of chemotherapy‐induced senescence, it was found that CDK4 induced the upregulation of enhancer of zeste homolog 2 (EZH2) methylase and that EZH2 depletion inhibited cell emergence from senescence. Among the proteins involved in receptor endocytosis, proteomic analysis revealed that clathrin‐associated/assembly/adaptor protein medium 1 (AP2M1) plays an important role in transmitting secreted signals by senescent cells, suggesting that the CDK4–EZH2–AP2M1 pathway may regulate specific receptors controlling senescence escape [68].
During mitosis, endocytosis of specific receptors contributes to their equal or asymmetrical distribution between the daughter cells. However, studies investigating the dynamics of endocytosis during mitosis have been controversial. Some studies in HeLa and BSC1 cells have shown that CME normally occurs through all the phases of cell division, while recycling of internalized membrane decreased sharply during metaphase suggesting that its decrease accounts for the reduction in the surface area observed when an expanded interphase cell becomes a rounded mitotic cell [69]. These findings were confirmed later in a model using mouse keratinocytes undergoing natural cell division [70]. On the contrary, other studies reported that CME stops during mitosis when cells were arrested by addition of nocodazole [71] or synchronized by washout of RO‐3306 a CDK1 inhibitor [72]. In a HeLa cell model using nocodazole, CDK1 was reported to phosphorylate and inactivate epsin in a mechanism involving Ral (Ras‐like) signaling, leading to suppression of endocytosis [73]. More recently, it was shown that compounds used to produce mitotic arrest or mitotic synchrony affect CME. The use of nocodazole eliminated coated pits at the plasma membrane, and RO‐3306 washout increased coated pit lifetimes during mitosis of synchronized cells thus affecting the dynamics of the formation of clathrin‐coated pits indicating that in chemical arrested mitotic cells CME stops but it does not during unperturbed mitosis [74]. In line with this, it was previously described in budding yeast that CDK1 is not required for endocytosis but is necessary for proper polarized growth, highlighting the role of CDK1 in the regulation of membrane dynamics during growth and cell cycle progression [75].
Collectively, the above examples highlight the importance of studying endocytosis through the cell cycle by approaches that are based on natural cell division models to avoid some in vitro culture bias when cell cycle arrest compounds are used.
The autophagy‐lysosomal pathway during the cell cycle
Autophagy mediates the catabolism of cytoplasmic material by its sequestration and targeting into the lysosome for degradation. Under specific signals for targeting cytoplasmic material for degradation, cargoes are membrane isolated in a double‐membrane autophagic vesicle, the autophagosome. After fusion with lysosomes (autophagolysosome), the cargoes incorporated in the autophagosome are degraded and the digestive products are released to contribute to cellular metabolism (reviewed in Ref. [76]). In colon carcinoma cells, the activation of autophagy using different inducers was observed preferentially in G1 and S phases determined by concomitant measurement of cell cycle markers and the cytoplasmic aggregation of the widely used marker of autophagososmes GFP‐microtubule‐associated proteins 1A/1B light chain 3B (LC3) in autophagic vesicles [77]. More recently, in a model using lung and colon cancer cell lines for flow cytometry detection of LC3‐II, it was reported that basal but also starvation and rapamycin‐induced autophagy, lead to accumulation of LC3‐II in all cell cycle phases [78]. However, using leukemia cell lines it was observed that the levels of LC3‐II increased by 30–40% during S phase compared to G1 phase and increase again when cells are in G2 phase, under different starvation conditions [79]. By quantification of autophagic vesicles identified by electron microscopy in normal rat kidney cells, it was observed a significant reduction in volume of autophagosomes during prometaphase and anaphase [80]. More recently, it was demonstrated in different human cell lines that CDK1 phosphorylates vacuolar protein sorting 34 (VPS34) which is required for autophagy. Enhanced CDK1 phosphorylation of VPS34 (T159) during mitosis negatively regulates the lipid kinase activity of VPS34 and impairs the interaction with an essential autophagy regulatory protein, beclin‐1. Furthermore, in vivo analysis in mouse brains showed that CDK5 can also phosphorylate VPS34 on T159 and T668 thus contributing to the downregulation of autophagy [49]. However, decrease autophagic vesicles could also be a result of the fast clearance of autophagosomes due to increase autophagic flux. Therefore, by shortly inhibiting autophagic flux using chloroquine, it was shown that the number of LC3‐II signals in both interphase and unperturbed mitotic cells, was increased in different human cell lines. Additionally, this study reported that only during late but not during early mitosis an increase in LC3‐II signal is observed [81]. Altogether, these studies suggest that different experimental approaches, nutrient deprivation and pharmacological inhibitors have to be carefully selected to avoid biased conclusions and that further studies supported by different experimental approaches are required to complement the understanding of the autophagic status at specific phases of the cell cycle.
Cell cycle regulators influence the intertwine between endolysosomal pathways and metabolism
Nutrient uptake and the sensing of extracellular changes are mediated by plasma membrane proteins. Responding to these environmental cues is critical for cell survival and adaptations that will determine cell fate or the coordination of cellular responses in multicellular organisms. The controlled endocytosis of signaling receptors, nutrient transporters and carriers determine the activity of these membrane bounded regulators of cellular metabolism. Consequently, endocytic membrane trafficking directly influences the signaling pathways mediated by the intracellular master regulators of metabolism.
AMP‐activated protein kinase (AMPK) is a highly conserved protein sensor of the relative value of Adenosine monophosphate, ADP and ATP molecules within the cell. Upon activation, AMPK inhibits anabolic processes and promotes catabolic reactions to generate ATP [82]. AMPK has among its targets some proteins that either undergo endocytosis or control it. For instance, in order to replenish ATP levels by enhancing glucose uptake, AMPK can control the endocytic traffic of glucose transporters, including GLUT1 in a variety of tissues and GLUT4 in muscle [83]. Upon metabolic insufficiency, AMPK directly phosphorylates thioredoxin interacting protein, TXNIP (at S308), which is an adaptor for clathrin. TXNIP phosphorylation inhibits its binding to GLUTs thus impairing their endocytosis from the plasma membrane and ultimately enhancing glucose uptake [84]. In L6 myoblasts, AMPK contributes to regulating GLUT4 endocytosis by inhibiting clathrin‐ and caveolae‐independent endocytosis indicating that GLUT4 internalization also depends on cholesterol‐ and dynamin‐dependent routes [85].
Small GTPases called Rab localize to specific intracellular membranes and function as regulators of different steps in the intracellular traffic pathways [86]. AMPK also contributes to exocytosis by inhibiting the GAP activity of AS160 [87] and promoting the activity of specific Rab proteins (including Rab 8A, 10 and 14), thus leading to GLUT4 delivery to the plasma membrane of adipocytes [88, 89]. Another membrane‐bound transporter regulated by AMPK is the Na/K‐ATPase which is essential for the establishment of gradients of these ions to generate the negative membrane potential of cells in an ATP‐consuming process. In hypoxic alveolar cells, Ca2+/calmodulin‐dependent protein kinase kinase‐β‐dependent AMPK activation leads to Na/K‐ATPase endocytosis [90]. CD36 traffic from recycling endosomes to the cell membrane is important in fatty acid uptake and utilization. For instance, in CD36‐deficient mice, upon cardiomyocyte contraction signals, the gain in fatty acid uptake dependent on AMPK is abolished. This finding highlights the importance of the regulation of CD36 traffic by AMPK to modulate the cell surface CD36 levels contributing to the balance in the utilization of external versus external fatty acids [91].
AMPK also phosphorylates proteins involved in the control of CME including, clathrin heavy chain, AP2 β2 subunit, dynamin 1, some other traffic coat proteins such as PTRF/cavin or Sec23/24, and several Rab proteins (reviewed in Ref. [92]).
Interestingly, AMPK has been described as a target of cell cycle regulators. CDK4 has been reported to directly phosphorylate AMPKα2 subunit (Ser345, Ser377, Thr485 and Ser529) inhibiting its kinase activity in a mechanism that highlights the role of CDK4 in promoting anabolism by blocking catabolic processes such as fatty acid oxidation (FAO) in MEFs and murine muscle, and the blockage of whole‐body oxidative metabolism [93]. Interestingly in an in vitro and in vivo model of hepatocellular carcinoma, palbociclib, a CDK4/6 inhibitor, induced autophagy and apoptosis by a mechanism activating AMPKα (T172). However, the autophagic and apoptotic effect was shown to be independent of CDK4/6 activity, but instead palbociclib inhibited protein phosphatase 5 leading to upregulation of phospho‐AMPK [94]. Additionally, CDK1 also phosphorylates both catalytic AMPK α1 and AMPKα2 (Ser377 and Thr485) subunits and the β1 regulatory subunit (Thr19 and Ser40) in HeLa cells mitotically arrested [95]. In the same study using U2OS cells CDK1 mitotic phosphorylation of AMPK catalytic subunits is, at least in part, essential for proper early mitotic progression.
The above‐mentioned studies exemplify how challenging is to investigate metabolic reprogramming in highly proliferative cells due to the complexity and the intertwine of different signaling pathways. However, understanding this complex network will shed light on the molecular players by which metabolic signaling regulates endocytic trafficking during cell cycle progression. A computational modeling approach might contribute to solve these questions. The study of molecular oscillations during different rhythms of the cell cycle shows that it is controlled by a dynamic system caused by a cluster of coupled oscillators, where the CDK oscillator is coupled with mitochondrial metabolic and transcriptional oscillators [96]. These findings highlight the strong correlation between cell cycle regulators and metabolic alterations and encourage for further investigation in in vivo models.
Cell cycle regulators target endolysosomal pathway compartments
Cumulative evidence demonstrates that lysosomes are key effectors for the degradation of intracellular and extracellular proteins derived from the secretory, endocytic and autophagic pathways (reviewed in Ref. [76]).
In human mammary gland tissues, the downregulation of cyclin D1 (cyclin D1KE/KE model) or pharmacological inhibition of CDK4/6 exhibited increased autophagic activity as the number of autophagosomes and LC3‐II levels were increased [97]. In line with this, endogenous cyclin D1 was shown to inhibit autophagic flux in transformed human breast cancer cells [98]. A specialized autophagic process, the selective autophagy of lipid stores, called lipophagy, has been linked to cyclin D1 regulation. Using a hepatocellular regeneration model it was observed that cyclin D1 diminished autophagosomes formation in AML12 cells, thus inhibiting lipid droplets (LD) catabolism via lipophagy, shown by the decreased colocalization of LC3B with LD upon cyclin D1 knockdown [99].
Additionally, in a xenograft model of breast cancer, pharmacological inhibition and genetic inactivation of CDK4 increases lysosomal mass but impairs autophagic flux as electron microscopy revealed higher density of autophagosomes and lysosomes, where lysosomes showed accumulation non‐digested material leading to induced cell senescence. In vitro, CDK4 depleted MDA‐MB‐231 cells downregulated the expression of cathepsins B and D. The tumors of mice treated with the CDK4 inhibitor abemaciclib increased the expression of the lysosomal marker LAMP1 and the genes regulated by TFEB, indicating that CDK4 plays a critical role in the regulation of lysosomal biogenesis in vivo [58].
To study the role of lysosomal storage in cell damage, a model of lysosomal accumulation was established in human fibroblasts. Lysosomal impairment causes accumulation of sphingolipids, which are structural components within the plasma membrane but are also important in the control of signal transduction pathways to maintain cellular homeostasis. The sucrose‐induced lysosomal accumulation impaired autophagic flux and consequently reduced the lysosomal sphingolipid catabolism that induce changes in lipid composition in these cells. Interestingly, lysosome‐plasma membrane fusion was increased leading to ectopic sphingolipid catabolism at the plasma membrane, which was associated with the onset of the cell cycle arrest since ~ 30% of downregulated genes encode for proteins involved in cell cycle progression, including CDK1 [100].
Neuronal cytoskeleton structure is critical for brain development and growth and endocytosis of synaptic vesicles is pivotal for synaptic transmission. Interestingly, CDK5 plays a key role as the kinase that re‐phosphorylates dynamin I [101] and amphiphysin I [102] upon the termination of synaptic vesicle endocytosis, in nerve terminals. Brains of Cdk5 −/− mice lack cortical laminar structure and cerebellar foliation which is associated with perinatal mortality [103]. CDK5 also participates in the recycling endosomal trafficking. AATYK1A, a major isoform of the kinase Apoptosis‐associated tyrosine kinase 1 (AATYK1) expressed in neurons, localizes at principally in the pericentrosomal endocytic recycling compartments (ERC) in CHO‐K1 cells. CDK5 phosphorylation of AATYK1A (Ser34) abolishes the function of AATYK1A in the formation of the pericentrosomal ERC. Since the pericentrosomal ERC increases in confluent cell culture conditions [104], the phosphorylation of AATYK1A by CDK5 controls cell‐confluence‐dependent recycling endosomes localization, and as a consequence AATYK1A‐phospho‐mimic mutant, impairs the traffic of recycling endosomes demonstrated by decreased transport of internalized transferrin back to the surface membrane [105].
An unbiased approach to study the lysosomal protease activity of single living cells used fluorescence signal from a probe that monitors lysosomal biogenesis and the trafficking pathway from the endoplasmic reticulum to the lysosomes. By performing a compound screening that included several CDK inhibitors in HeLa cells such as, kenpaullone, purvalanol A, alsterpaullone, CDK2/9 inhibitor, and CDK4 inhibitor, it was suggested that CDK activity maintains lysosomal biogenesis. Further analysis depleting individual CDKs and rescuing their expression, confirmed that CDK5 is involved in lysosomal activity. Depletion of CDK5 increased the size of lysosomes, upregulated the mRNA levels of cathepsin D and tended to increase the levels of lysosomal acid phosphatase activity in a manner independent of TFEB and cell cycle arrest. These findings suggest that other transcription factors might be phosphorylated by CDK5 to suppress lysosomal activity [106].
There is still much to be uncovered regarding the molecular mechanisms used by cell cycle regulators to alter the fate of the endolysosomal vesicles and the metabolites that are target for degradation or recycling. However, the control of metabolic signals exerted on cellular trafficking during cell cycle progression is an growing field to better understand cell physiology and pathology.
Cell cycle regulators in mitochondrial dynamics
Mitochondria are generally renowned for being the ‘powerhouses’ of the cell due to their fundamental role in ATP production via oxidative phosphorylation. They are ubiquitous endosymbiotic double‐membrane organelles thought to have been originated from an ancient integration of an alpha‐proteobacterium into a nucleated cell [107, 108]. Throughout numerous years of co‐evolution, most of the bacterial original traits were loaned to the host cell. In return, the organelle has gained primordial functions in orchestrating intermediate cell metabolism, calcium homeostasis, stress management, free radicals’ production, innate immunity, and apoptotic cell death through the release of cytochrome c [109, 110, 111, 112, 113, 114]. Moreover, mitochondria have safeguarded a fraction of their own small circular polycistronic genome, which encodes for thirteen proteins belonging to the mitochondrial electron transport chain [115].
Considering the multifaceted physiologic functions that mitochondria perform and emphasizing the importance of their dynamic behavior, it is intuitive to hypothesize that cell cycle regulators might directly influence them. In that way, a potential bidirectional regulation would ensure the provision of sufficient fuel and metabolites for cell growth and cell proliferation.
Mitochondrial biogenesis can be broadly defined as the symmetrical division and subsequent growth of pre‐existing organelles since they cannot be engendered de novo [116, 117]. This process requires communication between the organelle and the nucleus [118] and is coordinated at the transcriptional level by PPARγ‐coactivators (PGC‐1α and PGC‐1β) and by nuclear respiratory factors 1 and 2 (NRF1 and NRF2) [119, 120, 121].
Over the past years, cell cycle regulators have been implicated in mitochondrial biogenesis. Hepatocytes lacking cyclin D were found to display increased mitochondrial size and function as a result of higher NRF1 expression levels. Conversely, the same authors reported that overexpression of cyclin D lead to a two‐fold decrease in mitochondrial activity; which was dependent on CDK activity [122]. Besides, the atypical CDK‐activating kinase CDK7 has been also claimed to regulate mitochondrial biogenesis. When it is active forming a trimeric complex with cyclin H and ménage‐à‐trois 1 (MAT1), CDK7 acts as a transcriptional regulator of PGC‐1 members. Certainly, Sano et al. [123] documented that MAT1 deletion in mouse hearts caused a decrease in CDK7 activity, which resulted in a general defect of transcriptional activation mediated by PGC‐1 members.
Nevertheless, the core of mitochondrial dynamism goes way beyond via extremely vigorous cycles of balanced fusion and fission, shaping the organelle’s meshwork to meet metabolic demands [124]. Fusion, the joining of two neighbor mitochondria into a larger and tubular one, and fission, the division of the organelle, have been proven to be essential for life [125, 126, 127].
Fusion of the outer mitochondrial membrane (OMM) is controlled by the dynamin family of GTPases, namely mitofusins 1 and 2 (MFN1/2), and supported by cardiolipin [128]. Regarding the inner mitochondrial membrane fusion, the effector unit is the optic atrophy 1 protein together with MFN1 [129]. On the other side, OMM fission has widely been assumed to be essentially mediated by the large cytosolic GTPase dynamin‐related protein 1 (DRP1) in association with its receptor proteins [130, 131, 132]. DRP1 activity is strongly regulated by post‐translational modifications, comprising phosphorylation, acetylation, ubiquitination, S‐nitrosylation, and SUMOylation. By far, the most studied among them is phosphorylation, which can occur at various residues with antagonistic effects. Acquired phosphorylation at human Ser616 residue has been documented to stimulate DRP1‐mediated fission. Oppositely, modifications over human Ser637 halt fission and curb DRP1 action tilting the balance toward fusion [133]. Several proteins (such as CDK1 and CDK5) have been documented to modify DRP1 by direct phosphorylation [134], but the extent to which other elements, including other cell cycle regulators, might be controlling DRP1 activity remains enigmatic.
Albeit fission is primordial for adequate mitochondrial fitness, cell metabolism, and proliferation, excessive fragmentation has been disclosed as an early event promoting cell apoptosis and resulting in human diseases [135, 136, 137]. In this context, the use of mdivi‐1, a DRP1 small‐molecule inhibitor, was a promising approach to better elucidate the fission process and its physiological role during cell division [138]. However, this molecule was recently rejected to be specific since it also inhibits the mitochondrial complex I [139]. Complementary procedures for the upcoming years will be needed to separately pinpoint all DRP1 actions.
During the last decades, breakthrough experiments have revealed that mitochondria undergo stereotyped changes during the cell cycle, thus modulating entry, progression through, exit, and either quiescence or senescence states. They basically include an interconnected mitochondrial network during G1, which evolves to a single giant hyperfused tubule at the G1‐to‐S transition, and an extensive breakdown just prior to mitosis [134, 140, 141, 142]. Increased fusion at G1 phase has been largely correlated with more cristae, oxidative metabolism boosting, and higher performance of sustained ATP production, which forces cells to enter the cell cycle and permits cell growth and subsequent DNA synthesis [124, 143]. Moreover, it mixes components, promotes homogeneity, and it has been postulated as a rescue mechanism by diluting mitochondrial damage [127]. The underlying mechanism provoking fusion lies on the E3‐ubiquitin ligase anaphase‐promoting complex/cyclosome (APC/C). During the course of its well‐known role in regulating mitosis, APC/C together with Cdc20 cofactors orchestrates sister chromatid separation and final cell cycle exit [144]. Then, Cdh1 substitutes Cdc20 ensuring low cyclin levels during G1 phase and, finally, mitogenic signals inhibit APC/C‐Cdh1 allowing the formation of CDK‐cyclin modules to overcome the restriction point [145]. Moreover, APC/C‐Cdh1 is recognized to target DRP1, which gets directed to ubiquitination and proteasomal cleavage, hence sparing mitochondrial fission. Indeed, Horn et al. illustrated, using HeLa cells, that DRP1 levels were minimum at G1 phase in the presence of APC/C‐Cdh1 [146].
Furthermore, in early G1 phase CDK4/6‐cyclin D complexes phosphorylate the retinoblastoma protein family to release E2F transcription factors, which will regulate the G1‐to‐S phase transition. In parallel, this axis induces the expression of NRF1 [147] and links proliferation to mitochondrial metabolism [148]. Taken together, these results show that high‐performing hyperfused organelles due to DRP1 repression and E2F1 activity are a prerequisite for cyclin E build‐up, the molecular effector of S‐phase input [140, 149]. Certainly, the induction of mitochondrial fusion was found sufficient to accumulate cyclin E [140], whereas improper cyclin E activity leads to genomic instability [150].
In onward with the cell cycle progression, augmented levels of typical S‐phase CDK‐cyclins are known to phosphorylate and inhibit APC/C‐Cdh1, thus urging a peak in DRP1 protein levels [145]. In addition, Wang et al. demonstrated that CDK1, along with cyclin B1, extensively phosphorylates the mitochondrial complex I to provide suitable ATP for a rapid G2 cell growth [151]. Right before entering in mitosis, CDK1/5‐cyclin B modules largely phosphorylate DRP1 at S616 residue bringing about massive mitochondrial fission. In order to help in promoting this event, Park and Cho [152] observed that the profusion MFN1 protein is degraded in a CDK1‐dependent manner (allegedly by a coordinated action with the E3 ubiquitin ligase MARCH5), and later on the same authors and others also emphasized the importance of DRP1 receptors for the cell cycle progression [141].
Already in the XX century, Christiansen determined that the partitioning of mitochondria in symmetric cell division is equitable between daughter cells [153]. In this sense, mitochondrial fragmentation during G2‐to‐M transition is envisioned to be a mechanism ensuring the unbiased organelles’ heritage [134]. At last, the upstream activation of CDK1/5‐DRP1 activity is brought about the mitotic kinase Aurora A. This protein has been recognized to phosphorylate Ras‐like proto‐oncogene A (RALA), which accumulates in the OMM accompanied by its effector RALA‐binding protein 1 (RALBP1). In the end, RALA/RALBP1 recruit CDK1/5‐cyclin B allowing its function since loss of either RALA or RALBP1 hinders cell proliferation [154].
Multiple lines of evidence have led to the consensus that DRP1‐mediated mitochondrial fission before mitosis is an essential phenomenon. Indeed, extended tubular structures cause defects at the G2‐to‐M transition and can even trigger senescence or apoptotic cell death [134, 155, 156]. Despite the fact that it is true that cells lacking DRP1 are capable of completing cell division, filamentous organelles disturb their equal partition to daughter cells as seen in different cell lines [155, 156]. After the mitotic cycle is achieved, daughter cells rebuild a partially intertwined and fused mitochondrial network caused, in part, by the reactivation of APC/C‐Cdh1 complexes.
Peculiarly, very limited evidence relates DRP1 S637 phosphorylation or profusion proteins’ regulation to the normal cell cycle progression. Only a few insights emerged some years ago in a yeast model [157], yet it remains to be assessed whether putative modifications in these proteins might also be regulating mitochondrial dynamics during division. What is clear is that cumulative evidence links decreased levels of DRP1 S637 with cancer cells. For example, Rehman et al. showed that reversion of DRP1 phosphorylation imbalances between Ser616 and Ser637 residues in vitro could reduce cancer cell proliferation and activate apoptosis instead [158]. Nonetheless, an important limitation of all these studies is the use of drugs or chemicals to induce cell cycle arrest because they generate artifactual states that could be far off physiological conditions. Therefore, it becomes essential the use of more translational approaches and in vivo techniques, although it becomes a challenging endeavor.
Collectively, the basic concept put forward by several investigations is that mitochondria follow a specific dynamic patterning to properly develop a plethora of cell functions, including cell growth and proliferation. Furthermore, cell cycle regulators vastly influence mitochondrial function and dynamics in an integrated manner during cell division. Reasonably, mitochondrial dynamics, cell cycle, and overall metabolism reciprocally influence each other in vivo. However, the comprehension of these processes is not straightforward since relevant differences exist across cell types and we cannot rule out a spectrum of still unknown tissue‐specific regulatory mechanisms. For this reason, and even though we have tried to provide a comprehensive overview in this topic, it is unfeasible to integrate all observations in a single model.
Concluding remarks and Future directions
The participation of cell cycle regulators in the control of cell proliferation, survival, cell signaling, and cancer is well established. This canonical pathway comprises the CDKs, retinoblastoma protein pRB, and the E2F transcription factors that regulate the expression of genes involved in the cell cycle progression. However, under unfavorable energetic conditions, cell division, which requires a high biosynthetic activity, cannot take place. We discussed here the concept that (some) cell cycle regulators play, in addition to the control of the cell cycle, crucial roles in the control of several metabolic processes. The distinct functions of cell cycle regulators in the different cellular compartments, including endosomes and lysosomes, or mitochondria have been described (Fig. 3). The overall control of the cellular function depends, indeed, on the compartmentalization of diverse metabolic processes in specific cellular structures, each of them having a specific function that contributes to coordinating a metabolic response. The specific role of cell cycle regulators in the coordination of such response with cell proliferation remains, however, not fully understood. Moreover, it would be of interest to expand the concept of cell proliferation beyond the cell cycle, including the described metabolic processes, which are of most importance for the cell to grow and divide. This new concept could switch the conception of CDKs from cell cycle regulators to considering them as a cellular hub for energy homeostasis, in particular in proliferating cells.
Fig. 3.
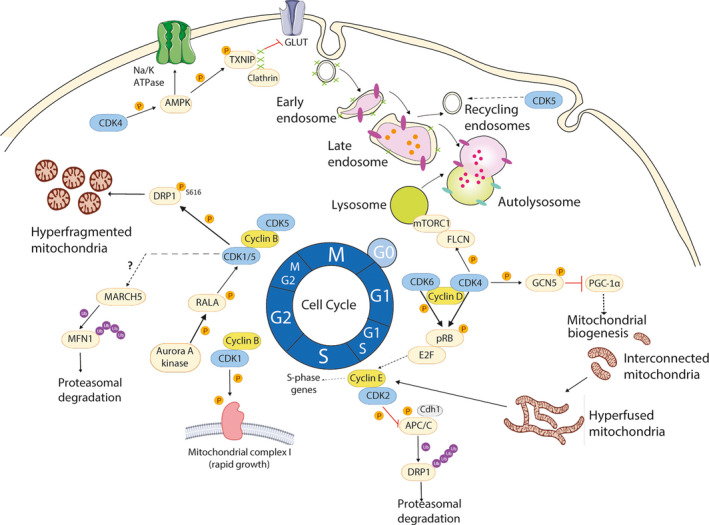
Overview on cell cycle regulators' modulation of mitochondrial dynamics and the endolysosomal pathway during the cell cycle. At the G1 phase, mitochondria are interconnected, and then evolve to a giant hyperfused network when reaching the G1‐to‐S phase transition. This profusion activity is accomplished by APC/C, causing DRP1’s proteasomal degradation. However, the canonical CDK4/6‐cyclin D axis allows the build‐up of cyclin E. Then, it binds CDK2 inhibiting APC/C and restoring the fusion/fission balance. In parallel, CDK4 also targets GCN5 to inhibit mitochondrial biogenesis via PGC‐1α during G1 phase. The mitotic Aurora A kinase activates CDK1/5‐cyclin B, which, in turn, cause massive mitochondrial fission thanks to DRP1 function. Additionally, CDK5 also regulates the recycling endosomal trafficking (see text). Cell cycle regulators, such as CDK4, also participate in the activities of master metabolic regulators, including mTORC1 through FLCN phosphorylation which contributes to lysosomal functionality. Moreover, CDK4 can regulate AMPK activation. Active AMPK can promote the endocytosis of the Na/K‐ATPase or promote glucose uptake by inhibiting the endocytosis of GLUTs mediated by clathrin and its adaptor protein TXNIP.
Of particular interest is the complex relation between cell cycle regulators and mitochondrial biology. In overall, it can be concluded that an active involvement exists between mitochondrial dynamics in its broadest sense and cell cycle regulators, exerting a pivotal role in controlling cell growth and proliferation beyond their canonical functions. Nevertheless, there are some fundamental questions and mechanistic details of widespread interest that remain to be answered. For example, how cell cycle regulators and mitochondrial dynamics interact in stem cells as their organelles’ partitioning is not symmetrical? Once all the interconnections are better understood, we could determine whether pathological mitochondrial remodeling is a cause or a disease consequence and act in accordance. Nowadays, there is a powerful rationale for targeting mitochondria, yet the available molecules are still raw tools with no ability to discern between healthy and altered cells. For that, subcellular selective addressing becomes of paramount concern, gaining therapeutic benefit likewise minimizing side effects or drug resistance. Thus, deciphering the interactions between mitochondrial dynamics and cell cycle regulators is, and surely will be, a stimulating field of research.
To conclude, new studies should provide novel and original insights on the mechanisms leading the cell cycle pathways to regulate a dual proliferative and the adapted metabolic response.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
KH, LF, and LCLE conceptualized the scope. KH AMA, LF, and LCLE wrote the manuscript. KH, AMA, and LCLE prepared the figures. KH, LF, and LCLE revised and corrected the manuscript.
Acknowledgements
We acknowledge the Swiss National Science Foundation grant (31003A_143369).
[Correction added on 15 December 2020 , after first online publication: Peer review history is not available for this article, so the peer review history statement has been removed.]
References
- 1.Malumbres M & Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153–166. [DOI] [PubMed] [Google Scholar]
- 2.van den Heuvel S & Dyson NJ (2008) Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 9, 713–724. [DOI] [PubMed] [Google Scholar]
- 3.Murray AW (2004) Recycling the cell cycle: cyclins revisited. Cell 116, 221–234. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Winkler S, Schlottmann FP, Kohlbacher O, Elias JE, Skotheim JM & Ewald JC (2019) Multiple layers of phospho‐regulation coordinate metabolism and the cell cycle in budding yeast. Front Cell Dev Biol 7, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ewald JC (2018) How yeast coordinates metabolism, growth and division. Curr Opin Microbiol 45, 1–7. [DOI] [PubMed] [Google Scholar]
- 6.Goranov AI & Amon A (2010) Growth and division–not a one‐way road. Curr Opin Cell Biol 22, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai L & Tu BP (2012) Driving the cell cycle through metabolism. Annu Rev Cell Dev Biol 28, 59–87. [DOI] [PubMed] [Google Scholar]
- 8.Franco J, Balaji U, Freinkman E, Witkiewicz AK & Knudsen ES (2016) Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep 14, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solaki M & Ewald JC (2018) Fueling the cycle: CDKs in carbon and energy metabolism. Front Cell Dev Biol 6, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fajas L (2013) Re‐thinking cell cycle regulators: the cross‐talk with metabolism. Front Oncol 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galluzzi L, Kepp O, Vander Heiden MG & Kroemer G (2013) Metabolic targets for cancer therapy. Nat Rev Drug Discov 12, 829–846. [DOI] [PubMed] [Google Scholar]
- 12.Lopez‐Mejia IC & Fajas L (2015) Cell cycle regulation of mitochondrial function. Curr Opin Cell Biol 33, 19–25. [DOI] [PubMed] [Google Scholar]
- 13.Annicotte JS, Blanchet E, Chavey C, Iankova I, Costes S, Assou S, Teyssier J, Dalle S, Sardet C & Fajas L (2009) The CDK4‐pRB‐E2F1 pathway controls insulin secretion. Nat Cell Biol 11, 1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feliers D, Frank MA & Riley DJ (2002) Activation of cyclin D1‐Cdk4 and Cdk4‐directed phosphorylation of RB protein in diabetic mesangial hypertrophy. Diabetes 51, 3290–3299. [DOI] [PubMed] [Google Scholar]
- 15.Iglesias A, Murga M, Laresgoiti U, Skoudy A, Bernales I, Fullaondo A, Moreno B, Lloreta J, Field SJ, Real FXet al. (2004) Diabetes and exocrine pancreatic insufficiency in E2F1/E2F2 double‐mutant mice. J Clin Invest 113, 1398–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li FX, Zhu JW, Tessem JS, Beilke J, Varella‐Garcia M, Jensen J, Hogan CJ & DeGregori J (2003) The development of diabetes in E2f1/E2f2 mutant mice reveals important roles for bone marrow‐derived cells in preventing islet cell loss. Proc Natl Acad Sci USA 100, 12935–12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagarrigue S, Lopez‐Mejia IC, Denechaud PD, Escote X, Castillo‐Armengol J, Jimenez V, Chavey C, Giralt A, Lai Q, Zhang Let al. (2016) CDK4 is an essential insulin effector in adipocytes. J Clin Invest 126, 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, Chim H, Lim JH, Ruan HB, Yang Xet al. (2014) Cyclin D1‐Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 510, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SY, Lee JH, Merrins MJ, Gavrilova O, Bisteau X, Kaldis P, Satin LS & Rane SG (2017) Loss of cyclin‐dependent kinase 2 in the pancreas links primary beta‐cell dysfunction to progressive depletion of beta‐cell mass and diabetes. J Biol Chem 292, 3841–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darville MI, Antoine IV, Mertens‐Strijthagen JR, Dupriez VJ & Rousseau GG (1995) An E2F‐dependent late‐serum‐response promoter in a gene that controls glycolysis. Oncogene 11, 1509–1517. [PubMed] [Google Scholar]
- 21.Fernandez de Mattos S, Lam EW & Tauler A (2002) An E2F‐binding site mediates the activation of the proliferative isoform of 6‐phosphofructo‐2‐kinase/fructose‐2,6‐bisphosphatase by phosphatidylinositol 3‐kinase. Biochem J 368, 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu M, Seto E & Zhang J (2015) E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget 6, 11252–11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsieh MC, Das D, Sambandam N, Zhang MQ & Nahle Z (2008) Regulation of the PDK4 isozyme by the Rb‐E2F1 complex. J Biol Chem 283, 27410–27417. [DOI] [PubMed] [Google Scholar]
- 24.Wang LY, Hung CL, Chen YR, Yang JC, Wang J, Campbell M, Izumiya Y, Chen HW, Wang WC, Ann DKet al. (2016) KDM4A coactivates E2F1 to regulate the PDK‐dependent metabolic switch between mitochondrial oxidation and glycolysis. Cell Rep 16, 3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugden MC & Holness MJ (2006) Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem 112, 139–149. [DOI] [PubMed] [Google Scholar]
- 26.Sakamaki T, Casimiro MC, Ju X, Quong AA, Katiyar S, Liu M, Jiao X, Li A, Zhang X, Lu Yet al. (2006) Cyclin D1 determines mitochondrial function in vivo . Mol Cell Biol 26, 5449–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, Gao H, Yang G, Williams JA, Suski JMet al. (2017) The metabolic function of cyclin D3‐CDK6 kinase in cancer cell survival. Nature 546, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harashima H, Dissmeyer N, Hammann P, Nomura Y, Kramer K, Nakagami H & Schnittger A (2016) Modulation of plant growth in vivo and identification of kinase substrates using an analog‐sensitive variant of CYCLIN‐DEPENDENT KINASE A;1. BMC Plant Biol 16, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ewald JC, Kuehne A, Zamboni N & Skotheim JM (2016) The yeast cyclin‐dependent kinase routes carbon fluxes to fuel cell cycle progression. Mol Cell 62, 532–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao G, Chen Y, Carey L & Futcher B (2016) Cyclin‐dependent kinase co‐ordinates carbohydrate metabolism and cell cycle in S. cerevisiae . Mol Cell 62, 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Pavlova NN & Thompson CB (2017) Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J 36, 1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altman BJ, Stine ZE & Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16, 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicolay BN, Gameiro PA, Tschöp K, Korenjak M, Heilmann AM, Asara JM, Stephanopoulos G, Iliopoulos O & Dyson NJ (2013) Loss of RBF1 changes glutamine catabolism. Genes Dev 27, 182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, Dean DC & Clem BF (2014) Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicolay BN & Dyson NJ (2013) The multiple connections between pRB and cell metabolism. Curr Opin Cell Biol 25, 735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeGregori J, Kowalik T & Nevins JR (1995) Cellular targets for activation by the E2F1 transcription factor include DNA synthesis‐ and G1/S‐regulatory genes. Mol Cell Biol 15, 4215–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dou QP, Zhao S, Levin AH, Wang J, Helin K & Pardee AB (1994) G1/S‐regulated E2F‐containing protein complexes bind to the mouse thymidine kinase gene promoter. J Biol Chem 269, 1306–1313. [PubMed] [Google Scholar]
- 38.Good L, Chen J & Chen KY (1995) Analysis of sequence‐specific binding activity of cis‐elements in human thymidine kinase gene promoter during G1/S phase transition. J Cell Physiol 163, 636–644. [DOI] [PubMed] [Google Scholar]
- 39.Good L, Dimri GP, Campisi J & Chen KY (1996) Regulation of dihydrofolate reductase gene expression and E2F components in human diploid fibroblasts during growth and senescence. J Cell Physiol 168, 580–588. [DOI] [PubMed] [Google Scholar]
- 40.Angus SP, Wheeler LJ, Ranmal SA, Zhang X, Markey MP, Mathews CK & Knudsen ES (2002) Retinoblastoma tumor suppressor targets dNTP metabolism to regulate DNA replication. J Biol Chem 277, 44376–44384. [DOI] [PubMed] [Google Scholar]
- 41.Lee P, Malik D, Perkons N, Huangyang P, Khare S, Rhoades S, Gong YY, Burrows M, Finan JM, Nissim Iet al. (2020) Targeting glutamine metabolism slows soft tissue sarcoma growth. Nat Commun 11, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qie S, Yoshida A, Parnham S, Oleinik N, Beeson GC, Beeson CC, Ogretmen B, Bass AJ, Wong KK, Rustgi AKet al. (2019) Targeting glutamine‐addiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat Commun 10, 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tarrado‐Castellarnau M, de Atauri P, Tarragó‐Celada J, Perarnau J, Yuneva M, Thomson TM & Cascante M (2017) De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol Syst Biol 13, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, Monfregola J, Medina DL, Lippincott‐Schwartz J & Ballabio A (2018) mTOR‐dependent phosphorylation controls TFEB nuclear export. Nat Commun 9, 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Settembre C & Ballabio A (2014) Cell metabolism: autophagy transcribed. Nature 516, 40–41. [DOI] [PubMed] [Google Scholar]
- 46.Cao Q, Zhong XZ, Zou Y, Murrell‐Lagnado R, Zhu MX & Dong X‐P (2015) Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J Cell Biol 209, 879–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cang C, Zhou Y, Navarro B, Seo Y‐J, Aranda K, Shi L, Battaglia‐Hsu S, Nissim I, Clapham DE & Ren D (2013) mTOR regulates lysosomal ATP‐sensitive two‐pore Na(+) channels to adapt to metabolic state. Cell 152, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao Q, Yang Y, Zhong XZ & Dong XP (2017) The lysosomal Ca(2+) release channel TRPML1 regulates lysosome size by activating calmodulin. J Biol Chem 292, 8424–8435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T, Shen Y, Rameh L, Yankner B, Tsai LHet al. (2010) Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell 38, 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y & Yu L (2017) Recent progress in autophagic lysosome reformation. Traffic 18, 358–361. [DOI] [PubMed] [Google Scholar]
- 51.Ladu S, Calvisi DF, Conner EA, Farina M, Factor VM & Thorgeirsson SS (2008) E2F1 inhibits c‐Myc‐driven apoptosis via PIK3CA/Akt/mTOR and COX‐2 in a mouse model of human liver cancer. Gastroenterology 135, 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meo‐Evoli N, Almacellas E, Massucci FA, Gentilella A, Ambrosio S, Kozma SC, Thomas G & Tauler A (2015) V‐ATPase: a master effector of E2F1‐mediated lysosomal trafficking, mTORC1 activation and autophagy. Oncotarget 6, 28057–28070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Real S, Meo‐Evoli N, Espada L & Tauler A (2011) E2F1 regulates cellular growth by mTORC1 signaling. PLoS One 6, e16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, Ramm S, Palmer AC, Yuzugullu H, Varadan Vet al. (2016) Overcoming therapeutic resistance in HER2‐positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29, 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zacharek SJ, Xiong Y & Shumway SD (2005) Negative regulation of TSC1‐TSC2 by mammalian D‐type cyclins. Cancer Res 65, 11354–11360. [DOI] [PubMed] [Google Scholar]
- 56.Romero‐Pozuelo J, Demetriades C, Schroeder P & Teleman AA (2017) CycD/Cdk4 and discontinuities in Dpp signaling activate TORC1 in the drosophila wing disc. Dev Cell 42, 376–387.e5. [DOI] [PubMed] [Google Scholar]
- 57.Romero‐Pozuelo J, Figlia G, Kaya O, Martin‐Villalba A & Teleman AA (2020) Cdk4 and Cdk6 couple the cell‐cycle machinery to cell growth via mTORC1. Cell Rep 31, 107504. [DOI] [PubMed] [Google Scholar]
- 58.Martinez‐Carreres L, Puyal J, Leal‐Esteban LC, Orpinell M, Castillo‐Armengol J, Giralt A, Dergai O, Moret C, Barquissau V, Nasrallah Aet al. (2019) CDK4 Regulates lysosomal function and mTORC1 activation to promote cancer cell survival. Cancer Res 79, 5245–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olmez I, Brenneman B, Xiao A, Serbulea V, Benamar M, Zhang Y, Manigat L, Abbas T, Lee J, Nakano Iet al. (2017) Combined CDK4/6 and mTOR inhibition is synergistic against glioblastoma via multiple mechanisms. Clin Cancer Res 23, 6958–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Disanza A, Frittoli E, Palamidessi A & Scita G (2009) Endocytosis and spatial restriction of cell signaling. Mol Oncol 3, 280–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pearse BM (1976) Clathrin: a unique protein associated with intracellular transfer of membrane by coated vesicles. Proc Natl Acad Sci USA 73, 1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hall C, Yu H & Choi E (2020) Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp Mol Med 52, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ferreira APA & Boucrot E (2018) Mechanisms of carrier formation during clathrin‐independent endocytosis. Trends Cell Biol 28, 188–200. [DOI] [PubMed] [Google Scholar]
- 64.Le Roy C & Wrana JL (2005) Clathrin‐ and non‐clathrin‐mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol 6, 112–126. [DOI] [PubMed] [Google Scholar]
- 65.Bitsikas V, Correa IR Jr & Nichols BJ (2014) Clathrin‐independent pathways do not contribute significantly to endocytic flux. Elife 3, e03970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Snijder B, Sacher R, Ramo P, Damm EM, Liberali P & Pelkmans L (2009) Population context determines cell‐to‐cell variability in endocytosis and virus infection. Nature 461, 520–523. [DOI] [PubMed] [Google Scholar]
- 67.Tomas A, Futter CE & Eden ER (2014) EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol 24, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le Duff M, Gouju J, Jonchere B, Guillon J, Toutain B, Boissard A, Henry C, Guette C, Lelievre E & Coqueret O (2018) Regulation of senescence escape by the cdk4‐EZH2‐AP2M1 pathway in response to chemotherapy. Cell Death Dis 9, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boucrot E & Kirchhausen T (2007) Endosomal recycling controls plasma membrane area during mitosis. Proc Natl Acad Sci USA 104, 7939–7944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Devenport D, Oristian D, Heller E & Fuchs E (2011) Mitotic internalization of planar cell polarity proteins preserves tissue polarity. Nat Cell Biol 13, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fielding AB, Willox AK, Okeke E & Royle SJ (2012) Clathrin‐mediated endocytosis is inhibited during mitosis. Proc Natl Acad Sci USA 109, 6572–6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC & Chen L (2006) Selective small‐molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci USA 103, 10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosse C, L'Hoste S, Offner N, Picard A & Camonis J (2003) RLIP, an effector of the Ral GTPases, is a platform for Cdk1 to phosphorylate epsin during the switch off of endocytosis in mitosis. J Biol Chem 278, 30597–30604. [DOI] [PubMed] [Google Scholar]
- 74.Tacheva‐Grigorova SK, Santos AJ, Boucrot E & Kirchhausen T (2013) Clathrin‐mediated endocytosis persists during unperturbed mitosis. Cell Rep 4, 659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCusker D, Royou A, Velours C & Kellogg D (2012) Cdk1‐dependent control of membrane‐trafficking dynamics. Mol Biol Cell 23, 3336–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lawrence RE & Zoncu R (2019) The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol 21, 133–142. [DOI] [PubMed] [Google Scholar]
- 77.Tasdemir E, Maiuri MC, Tajeddine N, Vitale I, Criollo A, Vicencio JM, Hickman JA, Geneste O & Kroemer G (2007) Cell cycle‐dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle 6, 2263–2267. [DOI] [PubMed] [Google Scholar]
- 78.Kaminskyy V, Abdi A & Zhivotovsky B (2011) A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy 7, 83–90. [DOI] [PubMed] [Google Scholar]
- 79.Warnes G (2015) Flow cytometric assays for the study of autophagy. Methods 82, 21–28. [DOI] [PubMed] [Google Scholar]
- 80.Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X, Backer JM & Lucocq JM (2002) Inhibition of autophagy in mitotic animal cells. Traffic 3, 878–893. [DOI] [PubMed] [Google Scholar]
- 81.Li Z, Ji X, Wang D, Liu J & Zhang X (2016) Autophagic flux is highly active in early mitosis and differentially regulated throughout the cell cycle. Oncotarget 7, 39705–39718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garcia D & Shaw RJ (2017) AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fazakerley DJ, Holman GD, Marley A, James DE, Stockli J & Coster AC (2010) Kinetic evidence for unique regulation of GLUT4 trafficking by insulin and AMP‐activated protein kinase activators in L6 myotubes. J Biol Chem 285, 1653–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TEet al. (2013) AMPK‐dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 49, 1167–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Antonescu CN, Diaz M, Femia G, Planas JV & Klip A (2008) Clathrin‐dependent and independent endocytosis of glucose transporter 4 (GLUT4) in myoblasts: regulation by mitochondrial uncoupling. Traffic 9, 1173–1190. [DOI] [PubMed] [Google Scholar]
- 86.Stenmark H & Olkkonen VM (2001) The Rab GTPase family. Genome Biol 2, REVIEWS3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sakamoto K & Holman GD (2008) Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am J Physiol Endocrinol Metab 295, E29–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus Met al. (2005) Characterization of the role of the Rab GTPase‐activating protein AS160 in insulin‐regulated GLUT4 trafficking. J Biol Chem 280, 37803–37813. [DOI] [PubMed] [Google Scholar]
- 89.Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS & Lienhard GE (2005) AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase‐activating protein domain. Biochem J 391, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M & Sznajder JI (2011) Hypoxia leads to Na, K‐ATPase downregulation via Ca(2+) release‐activated Ca(2+) channels and AMPK activation. Mol Cell Biol 31, 3546–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Habets DD, Coumans WA, El Hasnaoui M, Zarrinpashneh E, Bertrand L, Viollet B, Kiens B, Jensen TE, Richter EA, Bonen Aet al. (2009) Crucial role for LKB1 to AMPKalpha2 axis in the regulation of CD36‐mediated long‐chain fatty acid uptake into cardiomyocytes. Biochim Biophys Acta 1791, 212–219. [DOI] [PubMed] [Google Scholar]
- 92.Rahmani S, Defferrari MS, Wakarchuk WW & Antonescu CN (2019) Energetic adaptations: metabolic control of endocytic membrane traffic. Traffic 20, 912–931. [DOI] [PubMed] [Google Scholar]
- 93.Lopez‐Mejia IC, Lagarrigue S, Giralt A, Martinez‐Carreres L, Zanou N, Denechaud PD, Castillo‐Armengol J, Chavey C, Orpinell M, Delacuisine Bet al. (2017) CDK4 phosphorylates AMPKalpha2 to inhibit its activity and repress fatty acid oxidation. Mol Cell 68, 336–349. e6. [DOI] [PubMed] [Google Scholar]
- 94.Hsieh FS, Hung MH, Wang CY, Chen YL, Hsiao YJ, Tsai MH, Li JR, Chen LJ, Shih CT, Chao TIet al. (2017) Inhibition of protein phosphatase 5 suppresses non‐small cell lung cancer through AMP‐activated kinase activation. Lung Cancer 112, 81–89. [DOI] [PubMed] [Google Scholar]
- 95.Stauffer S, Zeng Y, Santos M, Zhou J, Chen Y & Dong J (2019) Cyclin‐dependent kinase 1‐mediated AMPK phosphorylation regulates chromosome alignment and mitotic progression. J Cell Sci 132, jcs236000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arata Y & Takagi H (2019) Quantitative studies for cell‐division cycle control. Front Physiol 10, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brown NE, Jeselsohn R, Bihani T, Hu MG, Foltopoulou P, Kuperwasser C & Hinds PW (2012) Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res 72, 6477–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Casimiro MC, Di Sante G, Di Rocco A, Loro E, Pupo C, Pestell TG, Bisetto S, Velasco‐Velazquez MA, Jiao X, Li Zet al. (2017) Cyclin D1 restrains oncogene‐induced autophagy by regulating the AMPK‐LKB1 signaling axis. Cancer Res 77, 3391–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu H, Ploeger JM, Kamarajugadda S, Mashek DG, Mashek MT, Manivel JC, Shekels LL, Lapiro JL & Albrecht JH (2019) Evidence for a novel regulatory interaction involving Cyclin D1, lipid droplets, lipolysis, and cell cycle progression in hepatocytes. Hepatol Commun 3, 406–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Samarani M, Loberto N, Solda G, Straniero L, Asselta R, Duga S, Lunghi G, Zucca FA, Mauri L, Ciampa MGet al. (2018) A lysosome‐plasma membrane‐sphingolipid axis linking lysosomal storage to cell growth arrest. FASEB J 32, 5685–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, Hansra G, McClure SJ, Sarcevic B, Boadle RAet al. (2003) Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol 5, 701–710. [DOI] [PubMed] [Google Scholar]
- 102.Tomizawa K, Sunada S, Lu YF, Oda Y, Kinuta M, Ohshima T, Saito T, Wei FY, Matsushita M, Li STet al. (2003) Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin‐mediated endocytosis of synaptic vesicles. J Cell Biol 163, 813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna PHC, Brady RO, Martin LJ & Kulkarni AB (1996) Targeted disruption of the cyclin‐dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 93, 11173–11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takahashi M, Murate M, Fukuda M, Sato SB, Ohta A & Kobayashi T (2007) Cholesterol controls lipid endocytosis through Rab11. Mol Bio Cell 18, 2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Takano T, Tsutsumi K, Saito T, Asada A, Tomomura M, Fukuda M & Hisanaga S‐I (2010) AATYK1A phosphorylation by Cdk5 regulates the recycling endosome pathway. Genes Cells 15, 783–797. [DOI] [PubMed] [Google Scholar]
- 106.Ishii S, Matsuura A & Itakura E (2019) Identification of a factor controlling lysosomal homeostasis using a novel lysosomal trafficking probe. Sci Rep 9, 11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Margulis L (1975) Symbiotic theory of the origin of eukaryotic organelles; criteria for proof. Symp Soc Exp Biol. 29, 21–38. [PubMed] [Google Scholar]
- 108.Lane N & Martin W (2010) The energetics of genome complexity. Nature 467, 929–934. [DOI] [PubMed] [Google Scholar]
- 109.Tait SW & Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11, 621–632. [DOI] [PubMed] [Google Scholar]
- 110.Antico Arciuch VG, Elguero ME, Poderoso JJ & Carreras MC (2012) Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal 16, 1150–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Galluzzi L, Kepp O, Trojel‐Hansen C & Kroemer G (2012) Mitochondrial control of cellular life, stress, and death. Circ Res 111, 1198–1207. [DOI] [PubMed] [Google Scholar]
- 112.Rizzuto R, De Stefani D, Raffaello A & Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13, 566–578. [DOI] [PubMed] [Google Scholar]
- 113.Vafai SB & Mootha VK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. [DOI] [PubMed] [Google Scholar]
- 114.Schieber M & Chandel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24, R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kurland CG & Andersson SG (2000) Origin and evolution of the mitochondrial proteome. Microbiol Mol Biol Rev 64, 786–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jornayvaz FR & Shulman GI (2010) Regulation of mitochondrial biogenesis. Essays Biochem 47, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11, 872–884. [DOI] [PubMed] [Google Scholar]
- 118.Kelly DP & Scarpulla RC (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18, 357–368. [DOI] [PubMed] [Google Scholar]
- 119.Puigserver P, Wu Z, Park CW, Graves R, Wright M & Spiegelman BM (1998) A cold‐inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839. [DOI] [PubMed] [Google Scholar]
- 120.Scarpulla RC (2002) Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 286, 81–89. [DOI] [PubMed] [Google Scholar]
- 121.Diaz F & Moraes CT (2008) Mitochondrial biogenesis and turnover. Cell Calcium 44, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PMet al. (2006) Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci USA 103, 11567–11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sano M, Izumi Y, Helenius K, Asakura M, Rossi DJ, Xie M, Taffet G, Hu L, Pautler RG, Wilson CRet al. (2007) Menage‐a‐trois 1 is critical for the transcriptional function of PPARgamma coactivator 1. Cell Metab 5, 129–142. [DOI] [PubMed] [Google Scholar]
- 124.Liesa M & Shirihai OS (2013) Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mishra P & Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wai T & Langer T (2016) Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27, 105–117. [DOI] [PubMed] [Google Scholar]
- 127.Spurlock B, Tullet J, Hartman Jl & Mitra K (2020) Interplay of mitochondrial fission‐fusion with cell cycle regulation: possible impacts on stem cell and organismal aging. Exp Gerontol 135, 110919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE & Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liesa M, Palacin M & Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89, 799–845. [DOI] [PubMed] [Google Scholar]
- 130.Kageyama Y, Zhang Z & Sesaki H (2011) Mitochondrial division: molecular machinery and physiological functions. Curr Opin Cell Biol 23, 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Otera H, Ishihara N & Mihara K (2013) New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta 1833, 1256–1268. [DOI] [PubMed] [Google Scholar]
- 132.Kalia R, Wang RY, Yusuf A, Thomas PV, Agard DA, Shaw JM & Frost A (2018) Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.van der Bliek AM, Shen Q & Kawajiri S (2013) Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5, a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Taguchi N, Ishihara N, Jofuku A, Oka T & Mihara K (2007) Mitotic phosphorylation of dynamin‐related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282, 11521–11529. [DOI] [PubMed] [Google Scholar]
- 135.Frank S, Gaume B, Bergmann‐Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL & Youle RJ (2001) The role of dynamin‐related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1, 515–525. [DOI] [PubMed] [Google Scholar]
- 136.Wasiak S, Zunino R & McBride HM (2007) Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177, 439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Suen DF, Norris KL & Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22, 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Qi X, Qvit N, Su YC & Mochly‐Rosen D (2013) A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 126, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam‐Vizi V, Cherok E, Khalil A, Yadava N, Ge SXet al. (2017) The Putative Drp1 Inhibitor mdivi‐1 Is a reversible mitochondrial complex i inhibitor that modulates reactive oxygen species. Dev Cell 40, 583–594. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mitra K, Wunder C, Roysam B, Lin G & Lippincott‐Schwartz J (2009) A hyperfused mitochondrial state achieved at G1‐S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA 106, 11960–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lee S, Park YY, Kim SH, Nguyen OT, Yoo YS, Chan GK, Sun X & Cho H (2014) Human mitochondrial Fis1 links to cell cycle regulators at G2/M transition. Cell Mol Life Sci 71, 711–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen H & Chan DC (2017) Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab 26, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Horbay R & Bilyy R (2016) Mitochondrial dynamics during cell cycling. Apoptosis 21, 1327–1335. [DOI] [PubMed] [Google Scholar]
- 144.Peters JM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7, 644–656. [DOI] [PubMed] [Google Scholar]
- 145.Salazar‐Roa M & Malumbres M (2017) Fueling the cell division cycle. Trends Cell Biol 27, 69–81. [DOI] [PubMed] [Google Scholar]
- 146.Horn SR, Thomenius MJ, Johnson ES, Freel CD, Wu JQ, Coloff JL, Yang CS, Tang W, An J, Ilkayeva ORet al. (2011) Regulation of mitochondrial morphology by APC/CCdh1‐mediated control of Drp1 stability. Mol Biol Cell 22, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Benevolenskaya EV & Frolov MV (2015) Emerging links between E2F control and mitochondrial function. Cancer Res 75, 619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mori K, Uchida T, Fukumura M, Tamiya S, Higurashi M, Sakai H, Ishikawa F & Shibanuma M (2016) Linkage of E2F1 transcriptional network and cell proliferation with respiratory chain activity in breast cancer cells. Cancer Sci 107, 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Parker DJ, Iyer A, Shah S, Moran A, Hjelmeland AB, Basu MK, Liu R & Mitra K (2015) A new mitochondrial pool of cyclin E, regulated by Drp1, is linked to cell‐density‐dependent cell proliferation. J Cell Sci 128, 4171–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Siu KT, Rosner MR & Minella AC (2012) An integrated view of cyclin E function and regulation. Cell Cycle 11, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann‐Hogiu S, Ahmed KM, Chromy BA, Nantajit Det al. (2014) Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell‐cycle G2/M progression. Dev Cell 29, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Park YY & Cho H (2012) Mitofusin 1 is degraded at G2/M phase through ubiquitylation by MARCH5. Cell Div 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Christiansen EG (1949) Orientation of the mitochondria during mitosis. Nature 163, 361. [DOI] [PubMed] [Google Scholar]
- 154.Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD & Counter CM (2011) RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat Cell Biol 13, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Yet al. (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11, 958–966. [DOI] [PubMed] [Google Scholar]
- 156.Pena‐Blanco A, Haschka MD, Jenner A, Zuleger T, Proikas‐Cezanne T, Villunger A & Garcia‐Saez AJ (2020) Drp1 modulates mitochondrial stress responses to mitotic arrest. Cell Death Differ 27, 2620–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Neutzner A & Youle RJ (2005) Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae . J Biol Chem 280, 18598–18603. [DOI] [PubMed] [Google Scholar]
- 158.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C & Archer SL (2012) Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J 26, 2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Goto Y, Hayashi R, Kang D & Yoshida K (2006) Acute loss of transcription factor E2F1 induces mitochondrial biogenesis in HeLa cells. J Cell Physiol 209, 923–934. [DOI] [PubMed] [Google Scholar]
- 160.Scimè A, Grenier G, Huh MS, Gillespie MA, Bevilacqua L, Harper ME & Rudnicki MA (2005) Rb and p107 regulate preadipocyte differentiation into white versus brown fat through repression of PGC‐1alpha. Cell Metab 2, 283–295. [DOI] [PubMed] [Google Scholar]
- 161.Bieda M, Xu X, Singer MA, Green R & Farnham PJ (2006) Unbiased location analysis of E2F1‐binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 16, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Polager S, Kalma Y, Berkovich E & Ginsberg D (2002) E2Fs up‐regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 21, 437–446. [DOI] [PubMed] [Google Scholar]
- 163.Muranaka H, Hayashi A, Minami K, Kitajima S, Kohno S, Nishimoto Y, Nagatani N, Suzuki M, Kulathunga LAN, Sasaki Net al. (2017) A distinct function of the retinoblastoma protein in the control of lipid composition identified by lipidomic profiling. Oncogenesis 6, e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tchakarska G, Roussel M, Troussard X & Sola B (2011) Cyclin D1 inhibits mitochondrial activity in B cells. Cancer Res 71, 1690–1699. [DOI] [PubMed] [Google Scholar]
- 165.Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima Jet al. (2010) Transcriptional role of cyclin D1 in development revealed by a genetic‐proteomic screen. Nature 463, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]