

Abstract
Venom-derived compounds are of broad interest in neuropharmacology and drug development. α-Conotoxins are small disulfide-containing peptides from Conus snails that target nicotinic acetylcholine receptors (nAChRs) and are in clinical development for non-opioid based treatment of intractable pain. Although refined by evolution for interaction with target prey receptors, enhancements of pharmacological properties are needed for use in mammalian systems. Therefore we synthesized analogs of α-conotoxin RgIA using a combination of selective penicillamine substitutions together with natural and non-natural amino acid replacements. This approach resulted in a peptide with 9000-fold increased potency on the human α9α10 nAChR and improved resistance to disulfide shuffling compared to the native peptide. The lead analog, RgIA-5474, potently blocked the α9α10 nAChR, but not opioid- or other pain-related targets. In addition, RgIA-5474 effectively reversed chemotherapy-induced neuropathic pain.
Keywords: nicotinic acetylcholine receptor, Conus, penicillamine, β3-homo tyrosine, analgesia
GRAPHICAL ABSTRACT
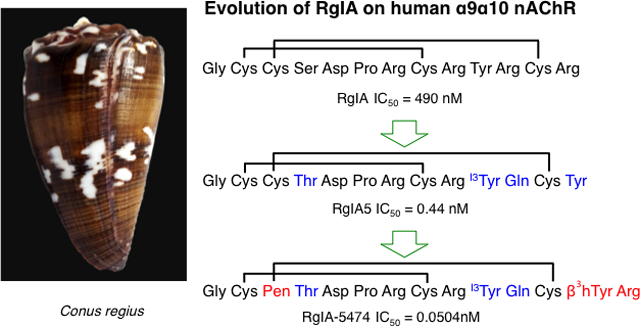
INTRODUCTION
Compounds produced by venomous predators are notable for their diverse structure, activity, and pharmacology. Minimal quantities of individual components may be sufficient to incapacitate prey or defend against a predator.1 A substantial fraction of venom components are small proteins and peptides; the latter are particularly amenable to solid-phase synthesis and potential drug development. Examples include angiotensin converting enzyme inhibitors for hypertension (e.g. captopril), antiplatelet drugs for heart attack (e.g. eptifibatide), and glucagon-like peptide 1 (GLP-1) agonists (e.g. exenatide) for treatment of type-II diabetes.2
Conus are predatory marine snails that hunt polychaete worms, mollusks and fish. There are over 830 known species of cone snails, each of which produces a unique cocktail of hundreds of distinct peptides targeted to a variety of receptors and ion channels.3 α-Conotoxins (α-Ctx), a subset of Conus peptides, are inhibitors of nicotinic acetylcholine receptors (nAChRs). nAChRs are pentameric ligand-gated ion channels with diverse receptor subtypes found in tissues, including neurons, immune cells, and skeletal muscle.4 Selective block of one particular subtype, the α9α10 nAChR, was shown to not only acutely alleviate neuropathic pain but also to accelerate recovery from nerve injury.5
Over 53 million people worldwide use opioids. With over 450 deaths per day from opioid overdoses,6 finding non-opioid drugs to treat severe to chronic pain is an urgent priority. Current clinically available medications treat pain but do not modify the underlying disease state. Previous studies have shown that the cone snail venom derived peptides α-Ctx Vc1.17, 8, 9 and RgIA10–12 and GeXIVA13, 14 produce analgesic activity mediated via α9α10 nAChRs. The analgesia elicited by the second-generation analog, RgIA4, lasted for several weeks after the treatment had been stopped.15 These findings indicate that the α9α10 nAChRs are promising targets for the development of novel medications for neuropathic pain and that Conus derived peptides have great potential for progression into potent analgesics.
Although promising in animal models, native α-conotoxins generally show low potency for human nAChRs. To address this, we designed a series of analogs, in which we introduced the cysteine surrogate, penicillamine, and other non-proteinogenic amino acids. A subset of the resulting analogs had picomolar affinities that were three orders of magnitude more potent than the parent analog. The highest affinity analog had extremely high selectivity, greatly improved serum stability, and potent analgesic activity.
RESULTS AND DISCUSSION
RgIA is a 13 amino acid, positively charged, disulfide-rich peptide expressed in the venom of the marine snail Conus regius. The bioactive form of the peptide has a globular disulfide bond connectivity (2Cys-8Cys, 3Cys-12Cys), potently blocks rat α9α10 nAChRs (IC50 = 1.49 nM),16 but shows a 300-fold decrease in potency on human α9α10 nAChRs. Previous studies with α-conotoxins have suggested the possible importance of the disulfide bridges not only for the overall structure stabilizing effect, but also for peptide interaction with the channel.17 A large decrease in activity was observed when either one of the RgIA cysteine bridges was replaced with a dicarba bridge.18 Trans- and cis-[3,12]-dicarba RgIA analogs exhibited over 600-fold loss of potency in blocking rat α9α10 nAChRs with IC50s of 1.15 μM and 1.47 μM respectively. No activity on α9α10 nAChRs was observed for both cis and trans analogs when such replacement was made for the [2,8]-disulfide bridge. A similar effect was observed for α-Ctx Vc1.1, a potent antagonist of α9α10 nAChRs and GABABRs.19, 20, 21
Our approach involved the use of penicillamine (Pen). We reasoned that this β,β-dimethyl substituted cysteine might introduce small, but favorable, local spatial constraints on the disulfide bridge as well as the overall peptide conformation. As a template for this study, we used RgIA4 a previously described analog of RgIA.22 Initially, each Cys residue in RgIA4 sequence was sequentially substituted with an L-Pen residue and tested on the human α9α10 nAChRs (Table 1).
Table 1.
L-Cys to L-Pen substitution in RgIA4 and their activity on human α9α10 nAChRs.
| Analog | Sequence | IC50 (nM)a | 95% CI (nM) |
|---|---|---|---|
| RgIA-5439 | G(Pen)CTDPRC(Cit) I3YQCY | >1200 | 702–2200 |
| RgIA-5408 | GC(Pen)TDPRC(Cit) I3YQCY | 1.3 | 1.03–1.6 |
| RgIA-5440 | GCCTDPR(Pen)(Cit) I3YQCY | >1000 | ND |
| RgIA-5409 | GCCTDPRC(Cit) I3YQ(Pen)Y | 24 | 11–50 |
| RgIA-5493 | G(Pen)CTDPR(Pen)(Cit) I3YQCY | > 1000 | NDb |
| RgIA-5446 | GC(Pen)TDPRC(Cit) I3YQ(Pen)Y | > 33 | ND |
| RgIA-5432c | GC(Pen)TDPRCR I3YQCY | 0.39 | 0.27–0.59 |
Data were collected by applying 100 μM ACh to Xenopus oocytes heterologously expressing the receptor. IC50 and 95% confidence intervals (CI) are expressed as the mean ± SEM from more than three separate oocytes. Ba2+ containing ND-96 buffer was utilized unless otherwise noted. Blue colored residues indicate change introduced into the sequence in this series of analogs. Amino acid abbreviations: Pen, L-penicillamine; Cit, L-citrulline, I3Y, 3-iodo-L-tyrosine. The C-terminus of each peptide depicted in this table is a free carboxyl group;
ND, not determined;
The position of the Pen residue had a very substantial effect on peptide potency. Substituting 2Cys or 8Cys, in the first disulfide bridge, with L-Pen resulted in over 800- or 600-fold drop in potency on the human α9α10 nAChR, respectively, compared to RgIA4 (IC50= 1.5 nM).22 The disulfide bridge between 2Cys and 8Cys has been proposed to be important for the interaction between α-conotoxins and neuronal nAChRs.17 The bulky β-methyls of Pen may disrupt that interaction. NMR studies may help determine what conformation effects are induced by the two methyl groups of the Pen residue. In contrast, substituting 12Cys in the second bridge only yielded an approximately 16-fold loss of potency. A comparable effect was observed when two L-Pen residues were introduced to replace the first and the second cysteine bridge, although 3Cys–12Cys tolerated such modification much better (IC50 >33 nM) than 2Cys–8Cys (IC50 >1000 nM). However, substitution with L-Pen in position 3 led to the RgIA-5408 analog that retained low-nanomolar affinity (IC50= 1.3 nM). The same trend was observed when 3Pen was introduced in another potent analog of RgIA, RgIA5.22 The resulting peptide, RgIA-5432, blocked the human α9α10 nAChRs equipotent to RgIA5 (IC50 = 0.39 nM and IC50= 0.44 nM, respectively).
Penicillamine was previously used to increase conformational rigidity in oxytocin,23–26 a peptide hormone involved in different human physiological functions;27 while L-Pen substitution for 6Cys maintained agonistic activity,24 replacement of 1Cys resulted in a change from agonistic to antagonistic activity.23,25 Pen incorporation into somatostatin analogs resulted in substantial gain-of-function at the off-target μ-opioid receptor.28 In contrast, use of L- and D-Pen in encephalin provided an order of magnitude increased selectivity for δ-opioid receptors.29
In the present study, substitution with Pen resulted in widely varying effects depending on which of the four Cys residues was substituted. Three of the four positions caused large losses in activity. In contrast, substitution of 3Cys entirely preserved the activity. This indicates that selective Cys substitution can be essential for desired high affinity.
The spatial orientation of each cysteine’s side chain also plays an important role.30 As reported by Ren et al, individually substituting each L-Cys with D-Cys in the native RgIA led to inactive analogs on human α9α10 nAChRs when 2Cys or 12Cys were mutated (IC50 >10 μM), and analogs with drastic decreases in potency for 3Cys and 8Cys (IC50 = 6440 nM and 6950 nM respectively).31 We observed similar effects when each individual Cys in the sequence was replaced with D-Pen. The resulting analogs showed little or no activity on human α9α10 nAChRs at 10 μM. (data not shown).
Since the RgIA5 Pen-containing analog RgIA-5432 was the most potent out of the initially tested group of analogs we used it as a template to design the next set of peptides. The incorporation of Pen residues caused increased hydrophobicity of 3Pen RgIA-5432, and made it prone to precipitation. The addition of a positively charged L-Arg at the C-terminal position 14 improved the peptide’s solubility without affecting potency (Table 2). When both 13Tyr and 14Arg were substituted by D-Tyr and D-Arg, a minor ~4-fold drop in potency was observed for RgIA-5433, suggesting that spatial orientation of those C-terminal residues affects the peptide’s ability to bind to the receptor.
Table 2.
C-terminal substitutions and their impact on the potency of RgIA-5474 on blocking human α9α10 nAChRs.
| Analog | Sequence | IC50a (nM) | 95% CI (nM) |
|---|---|---|---|
| RgIA-5624b | GC(Pen)TDPRCRI3YQCYR | 0.51 | 0.34–0.75 |
| RgIA-5433 | GC(Pen)TDPRCRI3YQCyr | 2.1 | 1.4–3.2 |
| RgIA-5474 | GC(Pen)TDPRCRI3YQC(β3hY)R | 0.0504 | 0.037–0.069 |
| RgIA-5477 | GC(Pen)TDPRCRI3YQC(β3hY)r | 0.36 | 0.28–0.46 |
Data were collected by applying 100 μM ACh to Xenopus oocytes heterologously expressing the receptor. IC50 and 95% confidence intervals (CI) are expressed as the mean ± SEM from 3–5 separate oocytes. Ba2+ containing ND-96 buffer was utilized. Blue colored residues indicate changes introduced into the sequence in this series of analogs. Amino acid abbreviations: Pen, L-penicillamine; I3Y, 3-iodo-L-tyrosine; y, D-Tyr; r, D-Arg; β3hY, β3-homo tyrosine. The C-terminus of each peptide depicted in this table is a free carboxyl group;
A previous RgIA structure-activity relationship (SAR) study indicated that residues in the first loop: 5Asp, 6Pro, and 7Arg, were responsible for orienting the peptide for binding to the nAChR and 9Arg, was important for selectivity towards the α9α10 subtype of nAChR.32 Previous SAR data also revealed that an Arg13Tyr mutation can enhance RgIA potency.22, 33 We sought to potentially capitalize on these findings. β-amino acids are homologs of natural α-amino acids, with an extra methylene group immediately before the carboxylic group in the backbone of the amino acids. The side chain can be positioned either on the α- or β-carbon (β2- or β3-analog, respectively), or can be present in both positions. These amino acids have been used to synthesize peptidomimetics (in combination with or without α-amino acids) with retained biological activity and improved resistance to proteolysis.34,35,36 Therefore in order to maintain the side-chain position of 13Tyr, we substituted this residue with β3-homo tyrosine (β3hY), yielding the analog RgIA-5474 with an IC50 = 0.0504 nM. Together the changes present in the RgIA-5474 analog account for over 9000-fold increased potency over the native RgIA on the human α9α10 nAChR.
At saturating concentrations (10 nM) RgIA-5474 was characterized by very slow off-rate kinetics compared to RgIA, with 3% (±1.7) recovery after 5 min peptide washout compared to > 99% recovery for RgIA at 10 μM (data not shown). These results suggest that the 14Arg residue confers high affinity, in part, via modulation of the koff. When D-Arg was incorporated in position 14 the potency dropped 7-fold (IC50 = 0.36 nM) (Table 2) compared to the all-L analog, further implicating the importance of the optimal positioning of 14Arg side chain within the receptor.
In order to investigate the role of C-terminal Arg, we prepared a series of RgIA-5474 analogs where 14Arg was substituted by other positively charged, neutral, or negatively charged amino acids (Table 3). Substitution with 14Lys and 14Orn (ornithine, a non-proteinogenic amino acid with a side chain one methylene group shorter than Lys that terminates with a primary amine) resulted in analogs with a 7- and 5-fold drop in potency, respectively. A neutral 14Cit (citrulline, an uncharged arginine analog, terminating in a carbamoyl amino group) substitution led to an 8-fold potency drop. Thus, the addition of 14Arg may enable formation of one or more hydrogen-bonds via its guanidinium group with receptor residues at or adjacent to the core binding site. Substitution with a negatively charged Glu in position 14 caused a ~28-fold drop in potency, with an IC50 of 1.4 nM for RgIA-5687.
Table 3.
The role of charge in position 14 of RgIA-5474 on blocking human α9α10 nAChRs.
| Analog | Sequence | IC50 (nM)a | 95% CI (nM) |
|---|---|---|---|
| RgIA-5474 | GC(Pen)TDPRCRI3YQC(β3hY)R | 0.051 | 0.027–0.095 |
| RgIA-5702 | GC(Pen)TDPRCRI3YQC(β3hY)K | 0.34 | 0.24–0.47 |
| RgIA-5672 | GC(Pen)TDPRCRI3YQC(β3hY)Orn | 0.26 | 0.15–0.45 |
| RgIA-5686 | GC(Pen)TDPRCRI3YQC(β3hY)Cit | 0.40 | 0.27–0.601 |
| RgIA-5687 | GC(Pen)TDPRCRI3YQC(β3hY)E | 1.4 | 0.57–3.5 |
Data were collected by applying 100 μM ACh to Xenopus oocytes heterologously expressing the receptor. IC50 and 95% confidence intervals (CI) are expressed as the mean ± SEM from 3–5 separate oocytes. Ca2+ containing ND-96 buffer was utilized. Blue colored residues indicate change introduced into the sequence in this series of analogs. Amino acid abbreviations: Pen, L-penicillamine; I3Y, 3-iodo-L-tyrosine; β3hY, β3-homo tyrosine; Orn, L-ornithine; Cit, L-citrulline. The C-terminus of each peptide depicted in this table is a free carboxyl group. Concentration-response curves and the Hill slope values are shown in Figure S8 and Table S2.
The combined data suggest that there are several components contributing to the high potency of RgIA-5474: the original mutations introduced into the first (Ser4Thr) and second (Tyr10I3Tyr, Arg11Gln) loop of the peptide, combined with the C-terminal modification (Arg13Tyr). In the new set of analogs, β3-homo tyrosine positioned the side chain of 14Arg by one methylene group farther from the peptide’s core, potentially allowing for more optimal interaction between the guanidinium group and the channel. The observed increase in potency of RgIA-5474 also appeared to be a result of the presence of Pen in the sequence. To assess this, we synthesized an analog of RgIA-5474 in which the 3Pen residue was replaced by 3Cys. That substitution led to 17-fold drop in potency consistent with the importance of 3Pen (RgIA-5711, IC50 of 0.85 nM, 95% CI = 0.51–1.4 nM; the concentration-response curve and the Hill slope value are shown in Figure S9 and Table 2 respectively).
We also examined the stability of RgIA-5474 in human serum and compared it to RgIA4. Twenty-five percent serum concentration was utilized to assure that the reaction speed was linearly dependent on the serum concentration and that the peptide concentration was not rate limiting.37 As illustrated in Figure 1, RgIA-5474 retained its integrity, with 94% (±0.7) globular form and only 6% (±0.7) ribbon form present at 24 h, whereas substantial disulfide reshuffling of RgIA4 occurred with 64% (±0.5) of the globular form and 36% (±0.5) of the ribbon form present (calculated based on the area under the HPLC peak, n= 6). These data indicate a protective effect of the penicillamine on the disulfide bridge, potentially by the β,β-dimethyl groups providing steric hindrance and slowing down the reshuffling process.
Figure 1.
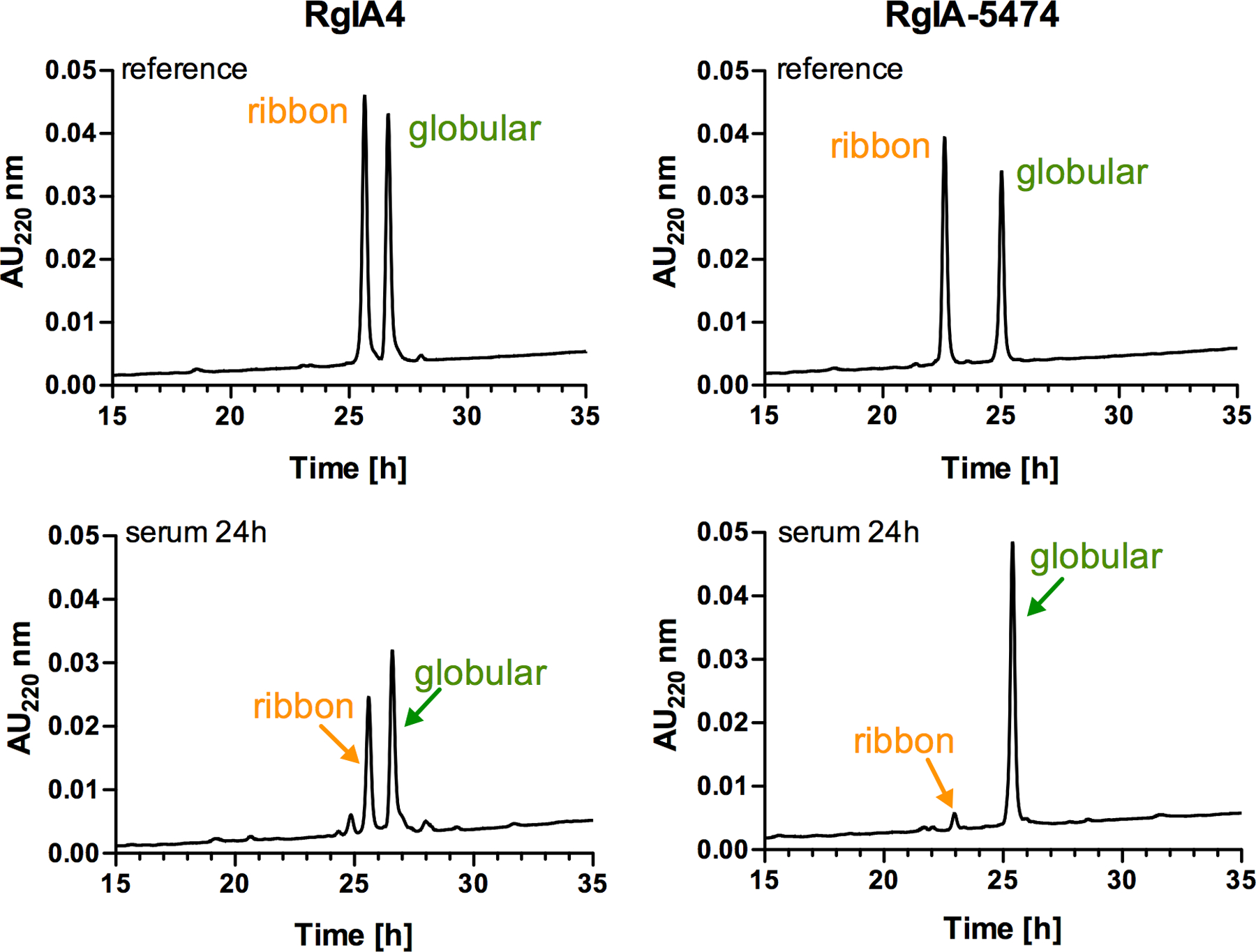
Stability of RgIA4 and RgIA-5474 in 25% human serum. Top panel: HPLC traces of a mixture (~1:1) of the globular and ribbon forms of the reference peptides RgIA4 and RgIA-5474. Bottom panel: serum stability at 24h (n=6). The reaction was monitored by RP-HPLC, using a C18 column and the gradient: ranging from 10% to 50% of buffer B in 40 min, with a flow rate of 1mL/min. Serum stability data for RgIA5 is shown in Figure S10.
Analog RgIA-5474 was the most active peptide from this series of analogs and was highly selective for the α9α10 nAChR. We tested the lead compound on several different subtypes of the human and rat nAChR (Table 4) and found it to be inactive (IC50 >10,000 nM) on all subtypes tested, with the exception of the human α7 nAChR, for which it had 2000-fold lower potency compared to the human α9α10 nAChR. RgIA-5474 retained its potency for rat α9α10 nAChR with an IC50 of 0.39 nM, and a significant decrease in potency was observed for rat α7. RgIA-5474 was additionally tested against μ-, δ- and κ-opioid receptor subtypes, N- and L-type Ca2+ channels, serotonin-, and norepinephrine-transporters, and a wide panel of relevant receptors and ion channels and showed low or no activity at micromolar levels (Table S3–S7, Supporting Information). Thus, RgIA-5474 is a highly selective peptide.
Table 4.
Subtype selectivity of RgIA-5474.
| Subtypes of nAChR | humana IC50 [nM] (95%CI) | rata IC50 [nM] (95%CI) |
|---|---|---|
| α9α10 | 0.051 (0.027–0.095) | 0.39 (0.33–0.48) |
| α2β2 | >10,000 | >10,000 |
| α2β4 | >10,000 | >10,000 |
| α3β2 | >10,000 | >10,000 |
| α3β4 | >10,000 | >10,000 |
| α4β2 | >10,000 | >10,000 |
| α4β4 | >10,000 | >10,000 |
| α6/α3β4 | >10,000 | >10,000 |
| α6/α3β2β3 | >10,000 | >10,000 |
| α7 | 115 (102–130) | >333 |
| α1β1δε | >10,000 | NAb |
Data was collected in Ca 2+ containing ND-96 buffer and 100 μM ACh application. IC50 and 95% confidence intervals (CI) are expressed as the mean ± SEM from 3–5 separate oocytes;
peptide was not tested on this subtype. Concentration-response curves and the Hill slope values are shown in Figure S11–12 and Table S2.
Finally, we examined the peptide for analgesic activity (Figure 2). Chemotherapy-induced peripheral neuropathy is a major side effect of platinum-containing drugs used to treat various cancers. The consequent pain can be disabling and is associated with a significant increase in opioid use. In most cases, the pain is only partially reversible; symptoms may last for years, and neuronal damage may be permanent.38 At present, there are no approved drugs to prevent or treat neuropathy, often forcing premature cessation of chemotherapy treatment. We tested RgIA-5474, using a well-established mouse oxaliplatin-induced peripheral neuropathy model.22, 39 RgIA-5474 successfully reversed chemotherapy-induced cold-allodynia or painful cold sensitization.
Figure 2.
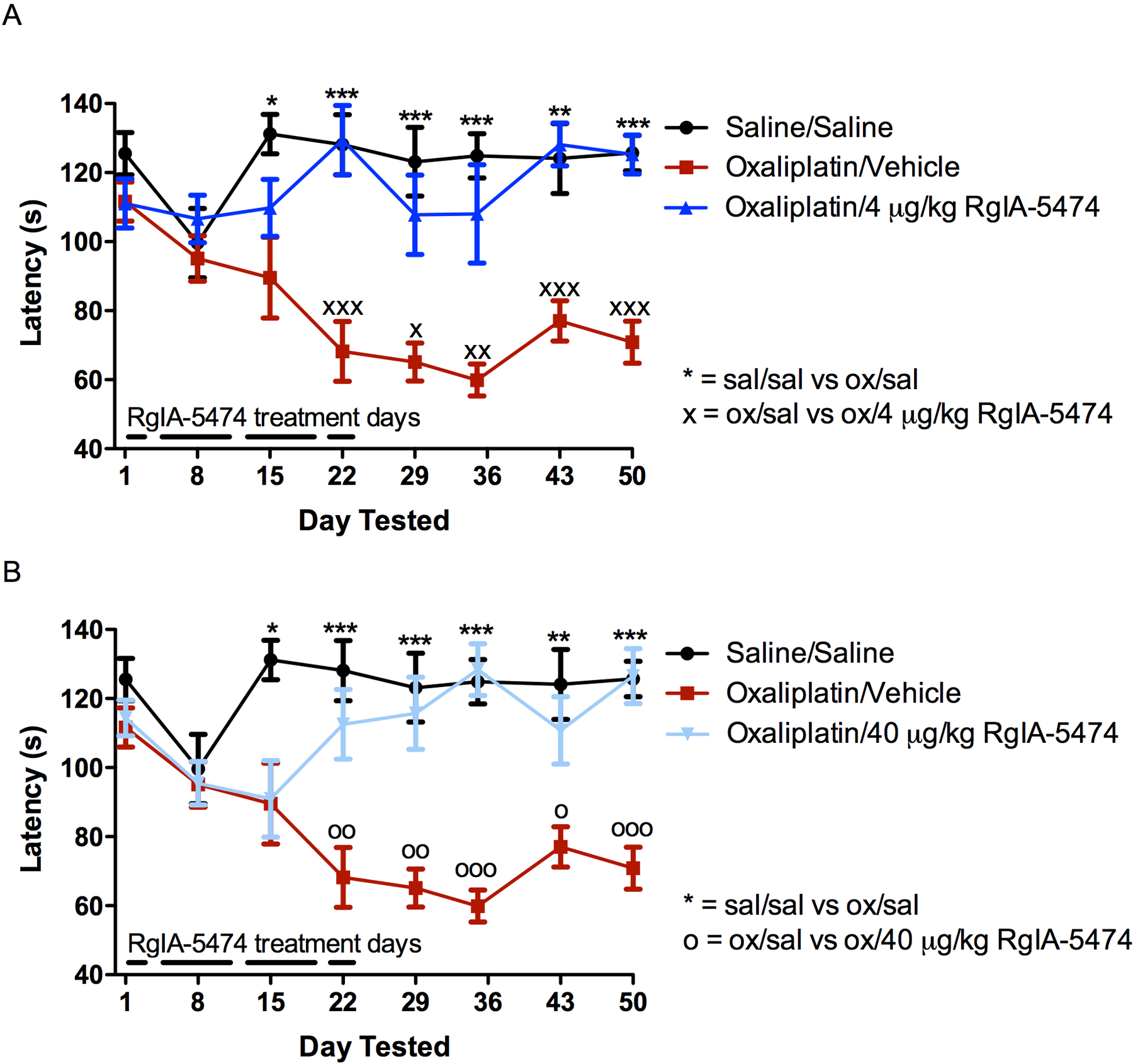
Effect of RgIA-5474 in oxaliplatin-induced peripheral neuropathy in mice. Mice were injected i.p. with oxaliplatin (ox; 3.5 mg/kg) daily as described in Materials and Methods. Control animals were treated with vehicle. RgIA-5474 was dissolved in sterile saline (sal) and injected s.c. daily. Withdrawal latency was used as a measure of cold allodynia. The cold allodynia test was performed on days 8, 15, and 22 at 24 h after RgIA-5474 administration at 4 μg/kg (panel A) and 40 μg/kg (panel B). Values are expressed as the mean ± SEM from eight mice for each experimental determination. ***P < 0.001, **P < 0.01, *P < 0.05 significantly different from vehicle.
A principle problem with the opioid class of medications is the development of tolerance. Chronic use of opioids results in decreased therapeutic effect and the need for a higher dose to produce the initial analgesia. This phenomenon frequently leads to accidental overdose when an opioid is stopped and is subsequently re-initiated at the higher dose. It is interesting to note that tolerance was not observed with a constant dose of RgIA-5474. Indeed, the therapeutic effect was not apparent until after 15 doses of drug were given over a period of three weeks. Furthermore, the therapeutic effect of RgIA-5474 persisted for four-weeks after cessation of treatment. This remarkable enduring analgesia is consistent with a disease-modifying effect that has been observed for other compounds that target the α9α10 nAChR.40
CONCLUSIONS
Utilizing selective incorporation of penicillamine and β3-homo tyrosine we dramatically improved the activity of the naturally occurring peptide, making it more potent, selective and resistant to disulfide reshuffling. We also validated the important role of the disulfides on the activity of this class of peptides and indicated additional residues contributing to the potency on the α9α10 nAChRs. Thus, RgIA-5474 represents a novel template for the development of high potency, selective, non-opioid analgesics.
Experimental Section
All experiments referred to in the manuscript are described in detail in the supporting information. Experiments involving the use of animals were approved and performed in compliance with the Institutional Animal Care and Use Committee (IACUC) of the University of Utah that conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The IACUC protocols were as follows: #20-07003 and #20-07005.
Solid Phase Peptide Synthesis.
All peptides described herein were synthesized on 0.05 mmol scale using an Apex 396 automated peptide synthesizer (AAPPTec; Louisville, KY), applying standard solid-phase Fmoc (9-fluorenylmethyloxycarbonyl) protocols using Fmoc-Tyr(tBu) Wang resin (0.49 mmol/g load; Peptides International, Louisville, KY), Fmoc-Arg(Pbf)-Wang resin (0.3 mmol/g load; Peptides International), Fmoc-Lys(Boc)-Wang resin (0.3 mmol/g load; Peptides International), Fmoc-Orn(Boc)-Wang resin (0.73 mmol/g; Bachem, Torrance, CA), Fmoc-Cit-Wang resin (0.42 mmol/g; Santa Cruz Biotechnology, Dallas, TX), Fmoc-Glu(OtBu)-Wang resin LL (Millipore Sigma, Burlington, MA) and Fmoc-Asp(OtBu)-Wang resin (0.51 mmol/g, Peptides International). All standard amino acids, Fmoc-Pen(Trt)-OH (Pen), Fmoc-Cit-OH and Fmoc-beta-HTyr(tBu)-OH were purchased from AAPPTec except for Fmoc-3-iodo-L-Tyr-OH (Peptides International)(I3Y). Side-chain protection for the following amino acids was as follows: Arg, 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl (Pbf); Thr, Tyr, beta-HTyr, tert-butyl (tBu); Lys, tert-butyloxycarbonyl (Boc); Gln, trityl (Trt). To promote the correct folding of the disulfide bridge a trityl protected Pen residue was paired either with another Pen (Trt) or with trityl protected Cys residue, while the other pair of Cys residues were acetamidomethyl (Acm) protected. Coupling activation was achieved with 1 eq of 0.4 M benzotriazol-1-yloxytripyrrolidino-phosphonium hexafluorophosphate (ChemImpex; Wood Dale, IL) and 2 eq of 2 M N,N-diisopropylethyl amine (Millipore Sigma, St. Louis, MO) in N-methyl-2-pyrrolidone (Fisher Scientific; Waltham, MA) as the solvent. For each coupling reaction, a 10-fold excess of the standard amino acid and 5-fold excess of the special amino acid was used, and the reaction was carried out for 60 and 90 min respectively. Fmoc deprotection was performed for 20 min with 20% (v/v) piperidine (Alfa Aesar; Tewksbury, MA) in N,N-dimethylformamide (Fisher Scientific). We note that the use of non-natural amino acids increases the cost of materials for synthesis of linear peptide. For a 0.05 mmol scale synthesis, the approximate cost per residue, based on the price of the amino acids and resins, is $1.2 for RgIA, $2.9 for RgIA4 and RgIA5 and $9 for RgIA-5474.
Peptide Cleavage, Purification and Oxidative Folding.
The protocols described for RgIA-5474 cleavage from the resin, purification and two-step oxidative folding were used for all newly synthesized peptides describe herein.
RgIA-5474 Cleavage and Purification.
The peptides were cleaved from the resin using Reagent K consisting of trifluoroacetic acid (TFA)/phenol/ethanedithiol/thioanisol/H2O (9:0.75:0.25: 0.5:0.5 by volume)(Fisher Scientific, Millipore Sigma, Millipore Sigma and Acros Organics respectively). Next, the cleavage mixture was filtered and precipitated with 150 mL of cold methyl-tert-butyl ether (MTBE; Fisher Scientific). The crude peptide was then precipitated by centrifugation at 7,000 × g for 7 min and washed twice with 150 mL cold MTBE. The crude peptide was diluted with 50 mL of the HPLC buffer B and purified by reverse-phase (RP) HPLC using a preparative C18 Vydac column (218TP101522, 250×22 mm, 5-μm particle size) eluted with a linear gradient ranging from 10% to 50% buffer B in 40 min at a flow rate 20 mL/min. The HPLC buffers were 0.1% (vol/vol) TFA in water (buffer A) and 0.092% TFA (vol/vol) in 60% aqueous acetonitrile (Fisher Scientific) (vol/vol) (buffer B). The eluent was monitored by measuring absorbance at 220/280 nm. Purity of the peptide was assessed by analytical C18 Vydac RP-HPLC (218TP54, 250×4.6 mm, 5-μm particle size) using the same gradient as described above with a flow rate 1 mL/min. From ~200 mg resin cleaved 4,318 nmols of linear RgIA-5474 was prepared. The HPLC trace of the linear RgIA-5474 is provided in Figure S1, Supporting Information.
RgIA-5474 First Disulfide Bond Formation (3Pen-12Cys).
In a solution consisting of 20 mM potassium ferricyanide (0.659 g; 2 mmol; Millipore Sigma) and 0.1M Tris base (1.21 g; 10 mmol; Millipore Sigma), dissolved in 100 mL of nano-pure water, 4,318 nmols of linear RgIA-5474, diluted with buffer A to total volume of 150 mL, was added drop-wise (peptide final concentration was approximately 20 μM and the pH was 7.5). The reaction was carried out for 45 min at room temperature, then terminated by diluting it with 250 mL buffer A to drop the pH. The reaction mixture was then passed through a disposable C18 cartridge (Thermo Fisher, Hypersep Spe C18 1000 mg/8 mL), and the peptide was eluted using buffer B. The efficiency of the reaction as well as the purity of the peptide was analyzed by analytical RP-HPLC as described for the linear peptide. From 4,318 nmols of the linear peptide, of 2,387 nmols of monocyclic RgIA-5474 was obtained 55 % yield, 85 % purity). The HPLC trace of the monocyclic RgIA-5474 is provided in Figure S2, Supporting Information.
RgIA-5474 Second Disulfide Bond Formation (2Cys-8Cys).
Removal of the acetamidomethyl groups and the second disulfide bridge formation was accomplished by iodine oxidation. 76 mg of iodine (2 mmols; Acros Organics) was added to 15 mL of acetonitrile and stirred until completely dissolved. Then, 45 mL of nanopure water was added followed by 1.8 mL of TFA. The monocyclic RgIA-5474 solution of 2,387 nmols diluted with 90 mL buffer A, was dripped into 60 mL of the 10 mM iodine solution (prepared as described above) and allowed to react for 10 min at room temperature. Peptide final concentration was kept close to 20 μM. The reaction was quenched by adding 5–10 drops of 1 M freshly prepared ascorbic acid (0.176 g, 1 mmol; Research Products International, Mount Prospect, IL) solution in water (1 mL) until the reaction mixture became transparent. The reaction was then diluted 5-fold with buffer A and subsequently purified by RP-HPLC using a preparative C18-column as described for the linear peptide to obtain 1,568 nmols of the fully folded RgIA-5474. Purity and final yield of RgIA-5474 was determined by RP-HPLC using analytical C18 column using the gradients described earlier for linear peptides and was determined to be 96% and 36% (based on the starting amount of the linear peptide) respectively. The calculated mass of RgIA-5474 [MH]+1= 1887.75 was verified to be [M+H]+1= 1887.59 by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry or electrospray ionization (ESI) at the Mass Spectrometry and Proteomics Core Facility at the University of Utah. The HPLC trace of the fully folded RgIA-5474 is provided in SI Figure S3. The purity for all for all fully folded peptides was ≥95% and was determined using analytical RP-HPLC. Mass Spec results, purity and retention times (RT) for all fully folded peptides are provided in Table S1, Supporting Information.
Two-Electrode Voltage-Clamp Recording.
Xenopus laevis (Xenopus1, Dexter, MI) oocytes were used to heterologously express cloned rat or human nAChR subtypes. Recordings were made 1–5 d post-injection. Oocytes were voltage-clamped at −70 mV at room temperature and pulsed for 1 second, every 60 seconds, with a bolus of acetylcholine, RgIA analogs were flowed on as described previously in detail.41, 42 Where noted, Ba2+ was substituted for Ca2+ to reduce current run-up following antagonist block. Concentration-response curves for the inhibition of α9α10 and α7 nAChR with the Hill slope values are shown in Supporting Information, Figures S6–S11 and Table S2 respectively.
Serum-Stability Assay.
A modified protocol reported by Zheng et al was followed.43 150 nmols of RgIA4 and RgIA-5474 were dissolved in 250 μL water and this solution was added into 750 μL of human serum solution (from human male AB plasma, sterile filtered and defrosted only once and pre-centrifuged 15 min to remove lipids, 250 μL of supernatant was added to 500 μL water; Millipore Sigma) making a total solution of 25% human serum. Twenty-five percent serum concentration was utilized to assure that the reaction speed was linearly dependent on the serum concentration and that the peptide concentration was not rate limiting.37 Final concentration of the peptides in the solution was 150 μM. The peptide-serum solution was incubated at 37°C and an individual 100 μL sample of the solution was removed at 24 h, treated with 50 μL ice cold acetonitrile (HPLC grade) and cooled on ice for 15 mins. The suspension was then centrifuged at 13,000 rpm for 5 mins at room temperature. Next 10 μL of the supernatant was removed and diluted in 90 μL of buffer A (0.1% TFA in water) to make the HPLC sample. The samples were analyzed by RP-HPLC with a linear gradient of 10–50% buffer B over 40 mins. Peptide peak areas were integrated at 280 nm and the % of peptide left, compared to the initial was graphed. The serum stability experiments were repeated 2 times in triplicates for each peptide. The synthesis of the reference peptide RgIA4 was described previously22 and the ribbon RgIA4 was synthesized following the protocol described for RgIA4, and was obtained with 99% purity (RT and MS data are provided in SI Table S1). The ribbon RgIA-5474 was prepared synthetically, following the protocol described for RgIA-5474, with the disulfide bridge formed in the following order: 3Pen-8Cys first and 2Cys-12Cys second. The ribbon RgIA-5474 was obtained with 99% purity (RT and MS data are provided in SI Table S1).
Receptor Pharmacology.
Synthetic RgIA-5474 was tested on non-nAChRs targets in the CYP Inhibition assays by Eurofins Cerep, Pharma Discovery Service, Celle l’Evescault, France. Automated patch-clamp electrophysiology was used for assessing the human ether-a-go-go–related gene (hERG) function. RgIA-5474 was also tested at the Dept. of Veterans Affairs Medical Center Research Service (R&D-22) in Portland, OR on serotonin, dopamine, and opioid receptors, as well as biogenic amine transporters. Detailed description of the assays and the data are presented in the SI Tables S3–S7.
Oxaliplatin-Induced Peripheral Neuropathy in Mice.
Neuropathy in mice was induced as described by Cavaletti et al.44 Oxaliplatin (MedChem Express, Monmouth Junction, NJ) was dissolved at 0.875 μg/μL in 0.9% NaCl, sterile filtered. RgIA-5474 was dissolved at 0.01 and 0.001 μg/μL in 0.9% NaCl, sterile filtered. Mice were divided into four equally sized groups (n = 8 animals) and injected with saline (i.p.) + saline (s.c.), oxaliplatin (3.5 mg/kg i.p.) + saline (s.c.), oxaliplatin (3.5 mg/kg, i.p.) + RgIA-5474 (40 μg/kg, s.c.), and oxaliplatin (3.5 mg/kg, i.p.) + RgIA-5474 (4 μg/kg, s.c.). Mice were injected once per day on Wednesday, Thursday, and Friday, and again on Monday and Tuesday. Mice were injected 5 times per week rather than 7 times per week, for convenience, based on prior data with a related compounds.22 We did not assess if the analgesic effect was superior with a 7 times vs. 5 times per week dosing. Mice were tested on Wednesdays before daily injection occurred (24-h post last injection). This injection pattern was repeated for an additional 2 weeks until the 22nd day; and then mice were tested weekly on Wednesdays to track the effects. Testing was conducted using a cold-plate test chamber (IITC, Inc. Life Science). Animals were allowed to acclimate in the chamber at room temperature (23 °C) for 2–5 min. Temperature was then lowered at a rate of 10 °C per minute. The testing was stopped when the animal lifted both forepaws and shaking or licking occurred. Alternating lifting of forepaws was not scored. Throughout the study period, experimenters were blinded as to the identity of the injected compounds. Data was analyzed with one-way ANOVA using Dunnett’s multiple comparison test (GraphPad Prism, GraphPad Software, San Diego, CA).
Supplementary Material
Figure S1-S3: HPLC traces of the linear, monocyclic and fully-folded RgIA-5474 (JPEG)
Table S1: Purity, retention time, MS data for all fully folded peptide analogs
Figure S4-S5: HPLC traces of the fully folded peptide analogues (JPEG)
Figure S6-S8: Concentration-response curves for the inhibition of human α9α10 nAChR (JPEG)
Figure S9: Concentration-response curves for the inhibition of human α9α10 nAChR by RgIA-5711 (JPEG)
Figure S10: Stability of RgIA5 in 25% serum (JPEG)
Figure S11: Concentration-response curves for the inhibition of human α7 nAChR by RgIA-5474 (JPEG)
Figure S12: Concentration-response curves for the inhibition of rat α9α10 nAChR by RgIA-5474 (JPEG)
Table S2: Hill slope values for blockade of human and rat α9α10 and human α7 nAChRs.
Tables S3-S7: Binding activity of RgIA-5474 on ion channels, receptors and transporters
Acknowledgments
We would like to acknowledge the support of Dr. David White and NIDA’s Addiction Treatment Discovery Program for data provided for use in this manuscript.
Funding Sources
This work was supported by: National Institutes of Health R35 GM136430 and Office of the Assistant Secretary of Defense for Health Affairs under Award No. W81XWH-17-1-0413.
ABBREVIATIONS
- nAChRs
nicotinic acetylcholine receptors
- α-Ctx
α-conotoxin
- Pen
L-penicillamine
- GABABRs
γ-Aminobutyric acid type B receptors
- β3hY
β3-homo tyrosine
- I3Y
3-iodo-L-tyrosine
- Cit
L-citrulline
- Orn
L-ornithine
- RP-HPLC
reverse-phase high-performance liquid chromatography
- RT
retention time
- hERG
human ether-a-go-go–related gene
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
The University of Utah has filed patent applications on conopeptides including those listed in the present manuscript and on which JG and JMM are listed as inventors.
REFERENCES
- 1.Dutertre S; Jin A-H; Vetter I; Hamilton B; Sunagar K; Lavergne V; Dutertre V; Fry BG; Antunes A; Venter DJ; Alewood PF; Lewis RJ Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nature Communications 2014, 5, 3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pennington MW; Czerwinski A; Norton RS Peptide therapeutics from venom: Current status and potential. Bioorganic & Medicinal Chemistry 2018, 26, 2738–2758. [DOI] [PubMed] [Google Scholar]
- 3.MolluscaBase eds. (2021). MolluscaBase. Accessed at http://www.molluscabase.org on2021-03-17. doi: 10.14284/448 [DOI]
- 4.Albuquerque EX; Pereira EFR; Alkondon M; Rogers SW Mammalian nicotinic acetylcholine receptors: from structure to function. Physiological Reviews 2009, 89, 73–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Livett BG; Sandall DW; Keays D; Down J; Gayler KR; Satkunanathan N; Khalil Z Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon 2006, 48, 810–829. [DOI] [PubMed] [Google Scholar]
- 6.publication, U. N. World Drug Report 2019. In Sales No. E.19.XI.8, p www.unodc.org/wdr2019.
- 7.Sandall DW; Satkunanathan N; Keays DA; Polidano MA; Liping X; Pham V; Down JG; Khalil Z; Livett BG; Gayler KR A novel α-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry 2003, 42, 6904–6911. [DOI] [PubMed] [Google Scholar]
- 8.Satkunanathan N; Livett B; Gayler K; Sandall D; Down J; Khalil Z Alpha-conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Research 2005, 1059, 149–158. [DOI] [PubMed] [Google Scholar]
- 9.Yu R; Kompella SN; Adams DJ; Craik DJ; Kaas Q Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J Med Chem 2013, 56, 3557–3567. [DOI] [PubMed] [Google Scholar]
- 10.Vincler M; Wittenauer S; Parker R; Ellison M; Olivera BM; McIntosh JM Molecular mechanism for analgesia involving specific antagonism of α9α10 nicotinic acetylcholine receptors. Proceedings of the National Academy of Sciences 2006, 103, 17880–17884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Cesare Mannelli L; Cinci L; Micheli L; Zanardelli M; Pacini A; McIntosh MJ; Ghelardini C α-Conotoxin RgIA protects against the development of nerve injury-induced chronic pain and prevents both neuronal and glial derangement. PAIN 2014, 155, 1986–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pacini A; Micheli L; Maresca M; Branca JJV; McIntosh JM; Ghelardini C; Di Cesare Mannelli L The α9α10 nicotinic receptor antagonist α-conotoxin RgIA prevents neuropathic pain induced by oxaliplatin treatment. Experimental Neurology 2016, 282, 37–48. [DOI] [PubMed] [Google Scholar]
- 13.Luo S; Zhangsun D; Harvey PJ; Kaas Q; Wu Y; Zhu X; Hu Y; Li X; Tsetlin VI; Christensen S; Romero HK; McIntyre M; Dowell C; Baxter JC; Elmslie KS; Craik DJ; McIntosh JM Cloning, synthesis, and characterization of αO-conotoxin GeXIVA, a potent α9α10 nicotinic acetylcholine receptor antagonist. Proceedings of the National Academy of Sciences 2015, 112, E4026–E4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang H; Li X; Zhangsun D; Yu G; Su R; Luo S The α9α10 nicotinic acetylcholine receptor antagonist αO-conotoxin GeXIVA[1,2] alleviates and reverses chemotherapy-induced neuropathic pain. Marine Drugs 2019, 17, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christensen SB; Hone AJ; Roux I; Kniazeff J; Pin J-P; Upert G; Servent D; Glowatzki E; McIntosh JM RgIA4 potently blocks mouse α9α10 nAChRs and provides long lasting protection against oxaliplatin-induced cold allodynia. Frontiers in Cellular Neuroscience 2017, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azam L; McIntosh JM Molecular basis for the differential sensitivity of rat and human α9α10 nAChRs to α-conotoxin RgIA. Journal of Neurochemistry 2012, 122, 1137–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tabassum N; Tae H-S; Jia X; Kaas Q; Jiang T; Adams DJ; Yu R Role of CysI–CysIII disulfide bond on the structure and activity of α-conotoxins at human neuronal nicotinic acetylcholine receptors. ACS Omega 2017, 2, 4621–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chhabra S; Belgi A; Bartels P; van Lierop BJ; Robinson SD; Kompella SN; Hung A; Callaghan BP; Adams DJ; Robinson AJ; Norton RS Dicarba analogues of α-conotoxin RgIA. Structure, stability, and activity at potential pain Targets. Journal of Medicinal Chemistry 2014, 57, 9933–9944. [DOI] [PubMed] [Google Scholar]
- 19.van Lierop BJ; Robinson SD; Kompella SN; Belgi A; McArthur JR; Hung A; MacRaild CA; Adams DJ; Norton RS; Robinson AJ Dicarba α-conotoxin Vc1.1 analogues with differential selectivity for nicotinic acetylcholine and GABAB receptors. ACS Chemical Biology 2013, 8, 1815–1821. [DOI] [PubMed] [Google Scholar]
- 20.Sadeghi M; Carstens BB; Callaghan BP; Daniel JT; Tae H-S; O’Donnell T; Castro J; Brierley SM; Adams DJ; Craik DJ; Clark RJ Structure–activity studies reveal the molecular basis for GABAB-receptor mediated inhibition of high voltage-activated calcium channels by α-conotoxin Vc1.1. ACS Chemical Biology 2018, 13, 1577–1587. [DOI] [PubMed] [Google Scholar]
- 21.Klimis H; Adams DJ; Callaghan B; Nevin S; Alewood PF; Vaughan CW; Mozar CA; Christie MJ A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain 2011, 152, 259–266. [DOI] [PubMed] [Google Scholar]
- 22.Romero HK; Christensen SB; Di Cesare Mannelli L; Gajewiak J; Ramachandra R; Elmslie KS; Vetter DE; Ghelardini C; Iadonato SP; Mercado JL; Olivera BM; McIntosh JM Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proceedings of the National Academy of Sciences 2017, 114, E1825–E1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz H; Du Vigneaud V Synthesis of 1-L-penicillamine-oxytocin, 1-D-penicillamine-oxytocin, and 1-deaminopenicillamine-oxytocin, potent inhibitors of the oxytocic response of oxytocin1. Journal of Medicinal Chemistry 1966, 9, 647–650. [DOI] [PubMed] [Google Scholar]
- 24.Schulz H; Du Vigneaud V Synthesis and some pharmacological properties of 6-L-penicillamine-oxytocin. Journal of Medicinal Chemistry 1967, 10, 1037–1039. [DOI] [PubMed] [Google Scholar]
- 25.Bélec L; Lubell WD; Maletinska L; Slaninová J The influence of steric interactions on the conformation and biology of oxytocin. Synthesis and analysis of penicillamine6-oxytocin and penicillamine6-5-tert-butylproline7-oxytocin analogs. The Journal of Peptide Research 2001, 58, 263–273. [DOI] [PubMed] [Google Scholar]
- 26.Meraldi JP; Hruby VJ; Brewster AI Relative conformational rigidity in oxytocin and (1-penicillamine)-oxytocin: a proposal for the relationship of conformational flexibility to peptide hormone agonism and antagonism. Proceedings of the National Academy of Sciences 1977, 74, 1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter CS; Kenkel WM; MacLean EL; Wilson SR; Perkeybile AM; Yee JR; Ferris CF; Nazarloo HP; Porges SW; Davis JM; Connelly JJ; Kingsbury MA Is oxytocin “nature’s medicine”? Pharmacological Reviews 2020, 72, 829–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelton JT; Gulya K; Hruby VJ; Duckles SP; Yamamura HI Conformationally restricted analogs of somatostatin with high mu-opiate receptor specificity. Proceedings of the National Academy of Sciences 1985, 82, 236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosberg HI; Hurst R; Hruby VJ; Gee K; Yamamura HI; Galligan JJ; Burks TF Bis-penicillamine enkephalins possess highly improved specificity toward delta opioid receptors. Proceedings of the National Academy of Sciences 1983, 80, 5871–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zouridakis M; Papakyriakou A; Ivanov IA; Kasheverov IE; Tsetlin V; Tzartos S; Giastas P Crystal structure of the monomeric extracellular domain of α9 nicotinic receptor subunit in complex with α-conotoxin RgIA: molecular dynamics insights Into RgIA binding to α9α10 nicotinic receptors. Frontiers in Pharmacology 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren J; Zhu X; Xu P; Li R; Fu Y; Dong S; Zhangsun D; Wu Y; Luo S d-Amino acid substitution of α-conotoxin RgIA identifies its critical residues and improves the enzymatic stability. Marine Drugs 2019, 17, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ellison M; Feng Z-P; Park AJ; Zhang X; Olivera BM; McIntosh JM; Norton RS α-RgIA, a novel conotoxin that blocks the α9α10 nAChR: structure and identification of key receptor-binding residues. Journal of Molecular Biology 2008, 377, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huynh PN; Harvey PJ; Gajewiak J; Craik DJ; Michael McIntosh J Critical residue properties for potency and selectivity of α-Conotoxin RgIA towards α9α10 nicotinic acetylcholine receptors. Biochem Pharmacol 2020, 181, 114124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ondetti MA; Engel SL Bradykinin analogs containing .beta.-homoamino acids. Journal of Medicinal Chemistry 1975, 18, 761–763. [DOI] [PubMed] [Google Scholar]
- 35.Steer D; Lew R; Perlmutter P; Smith AI; Aguilar M-I Inhibitors of metalloendopeptidase EC 3.4.24.15 and EC 3.4.24.16 Stabilized against proteolysis by the incorporation of β-amino acids. Biochemistry 2002, 41, 10819–10826. [DOI] [PubMed] [Google Scholar]
- 36.Gopi HN; Ravindra G; Pal PP; Pattanaik P; Balaram H; Balaram P Proteolytic stability of β-peptide bonds probed using quenched fluorescent substrates incorporating a hemoglobin cleavage site. FEBS Letters 2003, 535, 175–178. [DOI] [PubMed] [Google Scholar]
- 37.Jenssen H; Aspmo SI Serum stability of peptides. In Peptide-based drug design. Methods in molecular biology™, Otvos L, Ed. Humana Press: 2008; Vol. 494. [DOI] [PubMed] [Google Scholar]
- 38.Fallon MT Neuropathic pain in cancer. Br J Anaesth 2013, 111, 105–111. [DOI] [PubMed] [Google Scholar]
- 39.Di Cesare Mannelli L; Micheli L; Farina C; Scherz M; Ghelardini C Effects of dimiracetam on oxaliplatin-induced hyperalgesia and allodynia in the rat. Journal of Clinical Oncology 2015, 33, e20650–e20650. [Google Scholar]
- 40.Hone AJ; Servent D; McIntosh JM α9-containing nicotinic acetylcholine receptors and the modulation of pain. Br J Pharmacol 2018, 175, 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McIntosh JM; Plazas PV; Watkins M; Gomez-Casati ME; Olivera BM; Elgoyhen AB A Novel α-conotoxin, PeIA, cloned from conus pergrandis, discriminates between rat α9α10 and α7 nicotinic cholinergic receptors. Journal of Biological Chemistry 2005, 280, 30107–30112. [DOI] [PubMed] [Google Scholar]
- 42.Hone AJ; Kaas Q; Kearns I; Hararah F; Gajewiak J; Christensen S; Craik DJ; McIntosh JM Computational and functional mapping of human and rat α6β4 nicotinic acetylcholine receptors reveals species-specific ligand-binding motifs. Journal of Medicinal Chemistry 2021, 64, 1685–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng N; Christensen SB; Blakely A; Dowell C; Purushottam L; McIntosh JM; Chou DH-C Development of conformationally constrained α-RgIA analogues as stable peptide antagonists of human α9α10 nicotinic acetylcholine receptors. Journal of Medicinal Chemistry 2020, 63, 8380–8387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cavaletti G; Alberti P; Frigeni B; Piatti M; Susani E Chemotherapy-induced neuropathy. Current Treatment Options in Neurology 2011, 13, 180–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1-S3: HPLC traces of the linear, monocyclic and fully-folded RgIA-5474 (JPEG)
Table S1: Purity, retention time, MS data for all fully folded peptide analogs
Figure S4-S5: HPLC traces of the fully folded peptide analogues (JPEG)
Figure S6-S8: Concentration-response curves for the inhibition of human α9α10 nAChR (JPEG)
Figure S9: Concentration-response curves for the inhibition of human α9α10 nAChR by RgIA-5711 (JPEG)
Figure S10: Stability of RgIA5 in 25% serum (JPEG)
Figure S11: Concentration-response curves for the inhibition of human α7 nAChR by RgIA-5474 (JPEG)
Figure S12: Concentration-response curves for the inhibition of rat α9α10 nAChR by RgIA-5474 (JPEG)
Table S2: Hill slope values for blockade of human and rat α9α10 and human α7 nAChRs.
Tables S3-S7: Binding activity of RgIA-5474 on ion channels, receptors and transporters