

Abstract
Objective
Osteoarthritis (OA) is initiated by pathogenic factors produced by multiple stimuli, including mechanical stress, metabolic stress, and/or inflammaging. This study was undertaken to identify novel low‐grade inflammation–associated pathogenic mediators of OA.
Methods
Candidate pathogenic molecules were screened using microarray data obtained from chondrocytes exposed to OA‐associated catabolic factors. In mice with OA generated by destabilization of the medial meniscus (DMM), low‐grade inflammation was induced by a high‐fat diet or endotoxemia. Functions of candidate molecules in OA pathogenesis were examined using primary‐culture chondrocytes from mice with DMM‐induced OA, following intraarticular injection of adenovirus expressing the candidate gene. Specific functions of candidate genes were evaluated using whole‐body gene‐knockout mice.
Results
Bioinformatics analysis identified multiple candidate pathogenic factors that were associated with low‐grade inflammation, including components of the Toll‐like receptor (TLR) signaling pathways (e.g., TLR‐2, TLR‐4, lipopolysaccharide binding protein [LBP], and CD14). Overexpression of the individual TLR signaling components in mouse joint tissue did not alter cartilage homeostasis. However, the low‐grade inflammation induced by a high‐fat diet or endotoxemia markedly enhanced posttraumatic OA cartilage destruction in mice, and this exacerbation of cartilage destruction was significantly abrogated in LBP−/− and CD14−/− mice. Additionally, LBP and CD14 were found to be necessary for the expression of matrix‐degrading enzymes in mouse chondrocytes treated with proinflammatory cytokines.
Conclusion
LBP and CD14, which are accessory molecules of TLRs, are necessary for the exacerbation of posttraumatic OA cartilage destruction resulting from low‐grade inflammation, such as that triggered by a high‐fat diet or endotoxemia.
INTRODUCTION
Osteoarthritis (OA) is a whole‐joint disease characterized by cartilage destruction, synovial inflammation, osteophyte formation, and subchondral bone remodeling (1). Among these OA manifestations, articular cartilage degradation is the central feature of OA. Although it is well understood that multiple cell types of joint tissue are involved in the disease process, the study of the pathogenesis of OA has largely focused on chondrocytes. OA cartilage destruction can be caused primarily by the up‐regulation of matrix‐degrading enzymes and/or the down‐regulation of cartilage extracellular matrix (ECM) molecules (2). Among the matrix‐degrading enzymes, matrix metalloproteinase 3 (MMP‐3), MMP‐13, and ADAMTS‐5 have been shown to play important roles in cartilage destruction in experimental OA (3, 4, 5). The expression levels of matrix‐degrading enzymes and cartilage ECM molecules are regulated by multiple pathogenic factors produced by chondrocytes and other cell types in joint tissue. These factors include proinflammatory cytokines such as interleukin‐1β (IL‐1β), IL‐6, and tumor necrosis factor (TNF) (6). We previously identified cellular catabolic mediators in chondrocytes, including the transcription factor hypoxia‐inducible factor 2α (HIF‐2α) (7), and the zinc importer ZIP8 (8). These cellular mediators exert catabolic functions in OA pathogenesis by up‐regulating matrix‐degrading enzymes and/or down‐regulating ECM molecules in articular chondrocytes (7, 8).
OA‐causing pathogenic factors can be produced by mechanical stress (9), metabolic stress (10), and/or inflammaging (11), which are all associated with low‐grade inflammation. Studies in humans and in animal models have demonstrated that low‐grade inflammation plays a crucial role in OA pathogenesis (12, 13). Innate immune pathways that involve pattern‐recognition receptors, such as the Toll‐like receptor (TLR) pathways, are crucial in OA inflammation (12, 13, 14, 15). Indeed, studies have shown that inflammation‐induced catabolic processes, including up‐regulation of matrix‐degrading enzymes and down‐regulation of cartilage ECM molecules, are tightly controlled by TLR‐dependent immune responses (16, 17). TLR signaling pathways are composed of multiple components, including the various TLRs; accessory molecules (cofactors), such as lipopolysaccharide binding protein (LBP) and CD14; upstream agonists, such as serum amyloid A (SAA) protein; and various downstream mediators, such as NF‐κB–related molecules (14, 15).
In preliminary experiments, we performed a bioinformatics analysis of various microarray data obtained from OA‐like chondrocytes to identify novel pathogenic factors involved in OA pathogenesis. We initially identified multiple components of TLR signaling pathways as being up‐regulated in chondrocytes exposed to OA‐associated catabolic factors (IL‐1β, HIF‐2α, and ZIP8). Accordingly, we selected subsets of TLR signaling components and characterized their potential functions in low‐grade inflammation–associated OA. We found that TLR‐2, TLR‐4, LBP, and CD14 have differential functions in OA pathogenesis associated with low‐grade inflammation in mice. In particular, we demonstrated that LBP and CD14 are necessary for the exacerbation of posttraumatic OA under conditions of low‐grade inflammation.
MATERIALS AND METHODS
Mice and experimental OA
All animal experiments were approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee. C57BL/6J‐background knockout (KO) mice (TLR2−/−, TLR4−/−, CD14−/−) were purchased from The Jackson Laboratory. BALB/c‐background LBP−/− mice were purchased from The Jackson Laboratory and backcrossed with C57BL/6J mice. Experimental OA was induced in 12‐week‐old mice by destabilization of the medial meniscus (DMM), and the mice were euthanized at 6, 8, or 10 weeks thereafter (18). We used a high‐fat diet model (19) and a metabolic endotoxemia model (20) to examine the impact of low‐grade inflammation. For the high‐fat diet model, 8‐week‐old mice were fed either a regular diet (10% of total calories from fat) or a high‐fat diet (60% of total calories from fat; Research Diets). DMM or sham operation was performed on mice at week 4 of exposure to a regular diet or a high‐fat diet, and the mice were euthanized at 6 weeks thereafter. For the endotoxemia model, DMM‐ or sham‐operated 12‐week‐old male mice were injected intraperitoneally with a subclinical low‐dose of lipopolysaccharide (LPS) (5 ng/kg body weight) every 3 days for 4 weeks (21). Mice were euthanized at 8 weeks after surgery for histologic analysis.
To examine the effects of overexpression of TLR signaling components in joint tissue, mice were injected intraarticularly (once weekly for 3 weeks) with 1 × 109 plaque‐forming units (PFU) (in a total volume of 10 μl) of the TLR signaling component–expressing adenoviruses Ad‐TLR‐2, Ad‐TLR‐4, Ad‐LBP, Ad‐CD14, and Ad‐SAA1. Mice were euthanized at 3 or 8 weeks after the first intraarticular injection. Intraarticular injections of Ad‐HIF‐2α and Ad‐ZIP8 were used as positive controls for OA pathogenesis (7, 8). All adenoviruses were purchased from Vector BioLabs.
Skeletal staining, histology, and immunohistochemistry
Skeletons from E18.5 whole‐mouse embryos were stained with Alcian blue and alizarin red (8, 22). Mouse knee joint samples were fixed with 4% paraformaldehyde, decalcified in 0.5M EDTA, embedded in paraffin, sectioned frontally at 5‐μm thickness, and stained with Safranin O (8, 22). Osteoarthritis Research Society International (OARSI) histologic OA severity grades (scale 0–6) (23) and synovitis scores (scale 0–3) were calculated as the average values obtained from 3 different sections selected at ~100‐μm intervals for each knee joint; each section was scored by 4 observers under blinded conditions (YW, JIY, SP, and JSC). OARSI grades were expressed as the maximum score observed among the medial femoral condyle, medial tibial plateau, lateral femoral condyle, and lateral tibial plateau (23). Synovitis was determined by scoring the extent of synovial inflammation using joint sections stained with Safranin O and hematoxylin (24), or by detecting synovial infiltration of monocyte chemotactic protein 1 (MCP‐1)–positive or F4/80–positive inflammatory cells (e.g., macrophages) (25). Osteophyte size was measured using an Aperio ImageScope (Leica) (8, 22). Subchondral bone sclerosis was indirectly examined by measuring the thickness of the subchondral bone plate from 3 joint sections in each mouse using an Aperio ImageScope (8, 22, 26). Alternatively, subchondral bone remodeling was examined by immunostaining with Osterix to identify osteoblast activation (27). The following antibodies were used for immunostaining: mouse anti‐SAA1 (Hycult Biotechnology), rabbit anti–TLR‐2 and mouse anti‐CD14 (Novus Biologicals), mouse anti–TLR‐4, rabbit anti–MCP‐1 and rabbit anti‐Osterix (Abcam), rabbit anti‐LBP (LifeSpan Biosciences), and rat anti‐F4/80 (Bio‐Rad).
Primary chondrocyte culture
Mouse articular chondrocytes were isolated from the femoral condyles and tibial plateaus of 5‐day‐old mice, and maintained as a monolayer in complete Dulbecco’s modified Eagle’s medium (DMEM) (28). On culture day 2, cells at passage 0 were treated with IL‐1β (GenScript) in complete medium or with IL‐6 (Merck Millipore) and LPS (Sigma‐Aldrich) under serum‐free conditions. For adenoviral overexpression of target genes, chondrocytes were cultured for 3 days, infected with adenovirus for 2 hours at the indicated multiplicity of infection (MOI), and cultured for an additional 36 hours prior to further analysis.
Enzyme‐linked immunosorbent assay (ELISA)
Mouse sera were collected from sham‐ or DMM‐operated mice fed with a regular diet or a high‐fat diet or administered phosphate buffered saline (PBS) or LPS, and serum aliquots were stored at −80°C until analysis. ELISA kits for IL‐1β, IL‐6, TNF, MCP‐1, CD14, and leptin were purchased from R&D Systems, and the ELISA kit for LBP was purchased from MyBioSource.
Reverse transcription–polymerase chain reaction (RT‐PCR), quantitative RT‐PCR, and Western blotting
Total RNA was extracted from primary‐culture chondrocytes and reverse transcribed, and the resulting complementary DNA was PCR amplified; primers and experimental conditions are summarized in Supplementary Table 1 (available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). For Western blotting, total cell lysates were prepared using radioimmunoprecipitation assay lysis buffer (8, 22). For detection of secreted proteins (SAA1, LBP, and CD14), 900 μl of serum‐free conditioned medium was subjected to trichloroacetic acid precipitation, and the proteins were fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and detected using anti‐SAA1, anti‐LBP, and anti‐CD14 antibodies.
Bioinformatics analysis of microarray data
Microarray data from chondrocytes stimulated with IL‐1β or overexpressing HIF‐2α or ZIP8 were previously deposited into Gene Expression Omnibus (GEO) under accession numbers GSE104794 (HIF‐2α), GSE104795 (ZIP8), and GSE104793 (IL‐1β). To identify dysregulated genes in the 3 GEO data sets, the full differential expression data sets were subjected to gene set enrichment analysis (GSEA) using GSEA software (Broad Institute). GSEA was performed using functional gene sets derived from the Molecular Signatures Database (MSigDB) of the canonical pathways, the KEGG pathway database, and gene ontology (GO) biologic process terms. Differentially expressed genes were selected using a |log2 fold change| cutoff of >1.5 and a significance level of P < 0.05. The genes identified as being significantly differentially expressed were subjected to pathway enrichment analysis using Enrichr (http://amp.pharm.mssm.edu/Enrichr/), with default parameters and annotations obtained from the Panther, Biocarta, or Reactome pathway databases. We used the ggplot2 R package to analyze the results of GO enrichment analysis and differentially expressed genes.
Statistical analysis
For statistical comparison of experimental groups, data were analyzed using the Shapiro‐Wilk normality test and Levene’s test for homogeneity of variance. Nonparametric data based on an ordinal grading system (OARSI and synovitis grades) were compared between 2 groups using the Mann‐Whitney U test, whereas the Kruskal‐Wallis test was used to compare nonparametric data from multiple groups. Direct comparisons between pairs of groups (in the multiple group comparisons of nonparametric data) were conducted using the Mann‐Whitney U test. Parametric data were compared between 2 independent experimental groups using the 2‐tailed t‐test. One‐way analysis of variance with the Bonferroni post hoc test was used to compare parametric data from ≥3 groups. P values less than 0.05 were considered significant. Parametric data are expressed as the mean ± SEM; 95% confidence intervals (95% CIs) are included for nonparametric data.
RESULTS
Up‐regulation of TLR signaling components in chondrocytes stimulated with OA‐associated catabolic factors
To identify novel factors in OA pathogenesis, we screened microarray data obtained from mouse articular chondrocytes treated with IL‐1β or overexpressing OA‐associated cellular catabolic mediators, such as HIF‐2α or ZIP8 (7, 8). GSEA of KEGG pathways and GO gene set analysis revealed that genes associated with the TLR signaling pathway exhibited the most significant enrichment in mouse chondrocytes stimulated with the examined catabolic factors (Figures 1A and B, and Supplementary Figures 1A and B, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). We further performed enrichment analysis to identify the functional categories of the differentially expressed genes, and we discovered that genes related to TLR signaling pathways were commonly enriched in chondrocytes stimulated with all 3 catabolic factors (Supplementary Figures 1C and D, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract).
Figure 1.
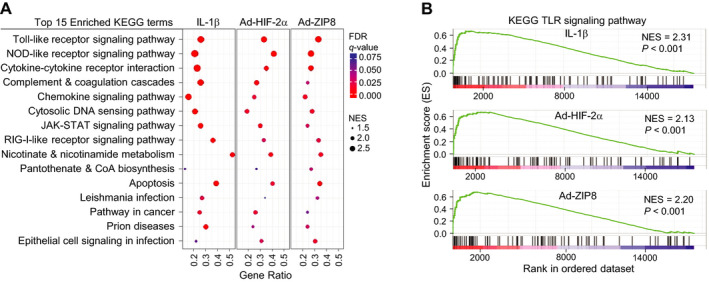
Transcriptional profiling of mouse chondrocytes treated with osteoarthritis‐associated pathogenic catabolic factors. Microarray data were obtained from primary‐culture mouse articular chondrocytes that had been treated for 36 hours with interleukin‐1β (IL‐1β) (1 ng/ml) or infected with 800 multiplicities of infection of control adenovirus, Ad‐HIF‐2α, or Ad‐ZIP8. A, Enrichment plot of the top 15 most significant KEGG terms with normalized enrichment scores (NES). Gene ratio is the number of core enriched genes divided by number of genes in a given KEGG gene set after filtering out the genes that were not found in the expression data set. B, Gene set enrichment analysis of microarray signals on KEGG pathways. NOD = nucleotide‐binding oligomerization domain; RIG‐1 = retinoic acid–inducible gene 1; CoA = coenzyme A; FDR = false discovery rate; TLR = Toll‐like receptor.
TLR signaling pathways regulate inflammation‐induced catabolic processes, such as the expression of matrix‐degrading enzymes, during OA pathogenesis (12, 13). Accordingly, we analyzed the microarray data in the context of TLR signaling components, including TLRs and their cofactors, upstream stimulators, and downstream targets (14, 15) (Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Exposure of chondrocytes to OA‐associated catabolic factors selectively up‐regulated certain TLR signaling components, including TLR‐2, TLR‐3, and TLR‐4; accessory molecules, such as LBP and CD14; upstream agonists, such as SAA protein; and downstream mediators, such as NF‐κB–related molecules (Figure 2A and Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). We validated these microarray data by qRT‐PCR analysis of chondrocytes treated with IL‐1β, Ad‐HIF‐2α, or Ad‐ZIP8. We also extended our analysis to include chondrocytes treated with IL‐6 or LPS. IL‐6 plays a key role in systemic inflammation and OA pathogenesis (29, 30), and LPS is thought to contribute to OA by stimulating TLR signaling (20). All of these catabolic stimulators caused significant up‐regulation of TLR‐2, LBP, CD14, and all of the SAA family members. In contrast, up‐regulation of TLR‐4, CD36, CD44, and LY96 was less marked or insignificant (Figures 2B and C, and Supplementary Figures 2A–E, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). These results collectively suggest that the significantly up‐regulated TLR signaling components may contribute to OA pathogenesis.
Figure 2.
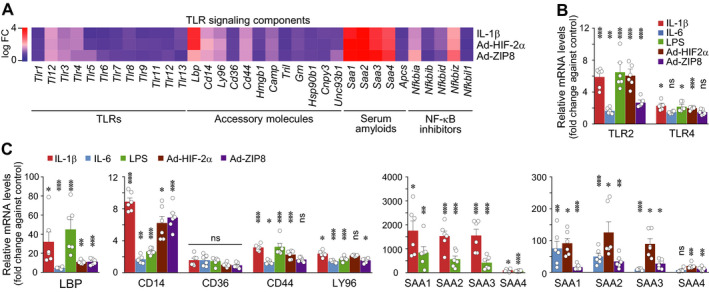
Osteoarthritis‐associated catabolic factors up‐regulate subsets of Toll‐like receptor (TLR) signaling components in chondrocytes. A, Chondrocytes were treated with interleukin‐1β (IL‐1β) (1 ng/ml) or infected with 800 multiplicities of infection of adenoviruses Ad‐HIF‐2α or Ad‐ZIP8. Vehicle and control adenovirus were used as controls. Heatmap shows expression of TLR signaling components in the chondrocytes after treatment. B and C, Primary‐culture chondrocytes were treated for 36 hours with IL‐1β (1 ng/ml), IL‐6 (100 ng/ml), lipopolysaccharide (LPS) (10 ng/ml), or 800 multiplicities of infection of control adenovirus, Ad‐HIF‐2α, or Ad‐ZIP8. Levels of mRNA for the TLR‐2 and TLR‐4 genes (B) as well as for other genes (C) were determined by quantitative reverse transcription–polymerase chain reaction analysis (n = 6 per group). Each symbol represents an individual mouse; bars show the mean ± SEM. * = P < 0.05; ** = P < 0.005; *** = P < 0.001, by one‐way analysis of variance with Bonferroni post hoc test. FC = fold change; LBP = lipopolysaccharide binding protein; NS = not significant.
Stimulation of TLR signaling pathways enhances posttraumatic OA in mice
Prior to characterizing the role of individual TLR signaling components in OA pathogenesis, we clarified whether conditions that stimulate TLR signaling pathways modulate OA pathogenesis in mice, in experiments using 2 models: high‐fat diet–induced low‐grade inflammation (19) and LPS‐mediated low‐grade inflammation (endotoxemia) (20). In mice, a high‐fat diet is known to cause low‐grade inflammation and can exacerbate the progression of OA (19). Consistently, we found that mice that were fed a high‐fat diet exhibited significant exacerbation of DMM‐induced OA manifestations, such as cartilage destruction, osteophyte formation, and synovial inflammation, without marked effects on subchondral bone plate thickness or the Osterix‐staining pattern indicative of osteoblast activation (Supplementary Figures 3A, B, and E, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Systemic low‐grade inflammation was determined by measuring circulating levels of inflammation markers, such as cytokines (IL‐1β, IL‐6, TNF), a chemokine (MCP‐1), an adipokine (leptin), and TLR components (LBP and CD14). A high‐fat diet under our experimental conditions had differential effects on the serum levels of these inflammation markers. We observed a decrease in IL‐6 levels, an increase in LBP levels, and no significant change in IL‐1β, TNF, MCP‐1, and CD14 levels (Supplementary Figure 4A, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Consistent with the synovitis score, a high‐fat diet in mice subjected to DMM markedly enhanced the synovial infiltration of MCP‐1– or F4/80–positive inflammatory cells (Supplementary Figures 4B and C, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract), indicating that a high‐fat diet enhanced inflammatory responses in joint tissue.
Metabolic endotoxemia, which is a low‐level elevation of gut‐derived endotoxin (LPS) in blood, also causes low‐grade inflammation and activates TLR signaling pathways (20). However, no direct evidence has been published to date regarding the in vivo role of circulating LPS in OA pathogenesis. We therefore examined the effects of intraperitoneal injection of a subclinical low dose of LPS (21) in DMM‐operated mice and found that this treatment significantly enhanced DMM‐induced OA cartilage destruction (Supplementary Figures 3C and D, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). However, other OA manifestations, such as osteophyte formation and subchondral bone sclerosis (i.e., subchondral bone plate thickness and Osterix staining for osteoblast activation), were not modulated by LPS administration (Supplementary Figures 3C–E, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Although synovitis scores were similar in mice administered PBS or LPS, the synovial infiltration of MCP‐1– or F4/80–positive cells was markedly increased in mice that were administered LPS (Supplementary Figures 4E and F, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract), indicating that local inflammatory responses occurred in the joint tissue of these mice. Unexpectedly, with a high‐fat diet, LPS administration caused significant increases in the circulating levels of inflammation markers such as IL‐1β, IL‐6, TNF, MCP‐1, LBP, and CD14 (Supplementary Figure 4D, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Collectively, our results suggest that conditions that stimulate low‐grade inflammation and TLR signaling pathways can enhance posttraumatic OA cartilage destruction in mice, but may not affect the other OA manifestations examined.
Overexpression of individual TLR signaling components in joint tissue is not sufficient to cause OA in mice
To examine the possible functions of the up‐regulated TLR signaling components in OA pathogenesis, we used adenovirus‐mediated overexpression in mouse joint tissue. Our group previously described adenoviruses that could effectively deliver transgenes to joint tissue (7, 8, 22). Here, we confirmed that intraarticular injection of individual adenoviruses caused marked overexpression of SAA1, TLR‐2, TLR‐4, LBP, or CD14 in articular cartilage (Supplementary Figure 5A, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). While overexpression of the previously characterized catabolic factors (HIF‐2α or ZIP8) (7, 8) caused cartilage erosion, osteophyte formation, and subchondral bone sclerosis, we found that overexpression of SAA1, TLR‐2, TLR‐4, LBP, or CD14 did not cause any OA‐like change in the joint tissue (Figure 3A). This suggests that overexpression of these individual TLR signaling components is not sufficient to cause OA development in mice.
Figure 3.
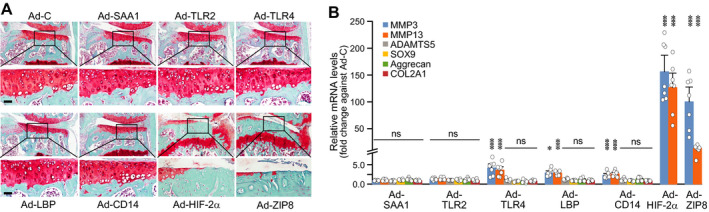
Overexpression of individual Toll‐like receptor (TLR) signaling components in joint tissue does not cause osteoarthritis‐like changes in mice. A, Wild‐type mice were intraarticularly injected (once weekly for 3 weeks) with 1 × 109 plaque‐forming units of control adenovirus (Ad‐C) or adenovirus expressing TLR signaling components (Ad‐SAA1, Ad‐TLR‐2, Ad‐TLR‐4, Ad‐LBP, or Ad‐CD14). Ad‐HIF‐2α and Ad‐ZIP8 were used as positive controls for OA pathogenesis. Mice were euthanized at 8 weeks after the first intraarticular injection. Representative images of joint sections are shown. The boxed areas in upper rows are shown at higher magnification in lower rows. Bars = 50 μm. B, Primary‐culture mouse articular chondrocytes were infected with 800 multiplicities of infection of Ad‐C, Ad‐SAA1, Ad‐TLR‐2, Ad‐TL‐R4, Ad‐LBP, Ad‐CD14, Ad‐HIF‐2α, or Ad‐ZIP8 for 36 hours. Relative levels of mRNA for the indicated molecules were determined by quantitative reverse transcription–polymerase chain reaction analysis (n = 7 per group). Each symbol represents an individual mouse; bars show the mean ± SEM. * = P < 0.05; *** = P < 0.001, by one‐way analysis of variance with Bonferroni post hoc test. NS = not significant.
We also examined the impact of up‐regulated TLR signaling components on the expression of matrix‐degrading enzymes and cartilage ECM molecules, as OA cartilage destruction can be regulated by the balance of these catabolic and anabolic factors (2). Unexpectedly, we found that overexpression of TLR‐4, LBP, or CD14 caused significant up‐regulation of MMP‐3 and MMP‐13 mRNA, but did not modulate the expression levels of SOX9, type II collagen, and aggrecan mRNA (Figure 3B and Supplementary Figure 5C, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Because MMP‐3 and MMP‐13 are known to play crucial roles in OA cartilage destruction (3, 4), it might seem contradictory that intraarticular injection of Ad‐TLR‐4, Ad‐LBP, or Ad‐CD14 did not cause cartilage degeneration. However, MMP‐3 and MMP‐13 were up‐regulated to much smaller degrees by the overexpression of TLR‐4, LBP, or CD14 compared to the overexpression of OA‐associated HIF‐2α or ZIP8 (Figure 3B, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). This could possibly explain why overexpression of TLR‐4, LBP, or CD14 in the mouse joint tissue was not associated with OA pathogenesis.
Differential regulation of posttraumatic OA cartilage destruction by TLR‐2, LBP, and CD14 under conditions of low‐grade inflammation
Next, we investigated the role of the individual TLR signaling components in DMM‐induced OA. To this end, we used TLR2−/−, TLR4−/−, LBP−/−, and CD14−/− mice, which are viable and exhibit normal development (31, 32, 33, 34). We confirmed the normal skeletal development of these mice in E18.5 embryos and verified the deletion of target genes in primary‐culture chondrocytes (Supplementary Figures 6A and B, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). In both wild‐type (WT) and KO mice, all examined OA manifestations (cartilage destruction, osteophyte formation, and subchondral bone plate thickening) were observed at 6 weeks after DMM and exacerbated at 8 and 10 weeks after surgery (Supplementary Figure 7A, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Among the KO mice, LBP−/− and CD14−/− mice exhibited significant amelioration of cartilage destruction at 10 weeks, but not at 6 or 8 weeks, post‐DMM (Supplementary Figure 7B). However, osteophyte formation and subchondral bone sclerosis were not modulated in these mice at the examined time points after DMM (Supplementary Figure 7B).
We further examined the effects of the various gene deletions on DMM‐induced OA under conditions with or without low‐grade inflammation (i.e., regular diet versus high‐fat diet, and PBS versus LPS). A high‐fat diet caused marked increases in body weight in both WT mice and KO mice (Supplementary Figures 8A and B, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Although individual KO mice exhibited differential gains of body weight under a high‐fat diet, the leptin levels in sera were not significantly different among the KO mice (Supplementary Figure 8C). Thus, the observed body weight differences did not appear to be due to differences in serum leptin levels. Additionally, serum levels of IL‐6 and TNF were similar between WT and KO mice (CD14−/− and LBP−/−) under both regular diet and high‐fat diet conditions. However, IL‐1β levels were significantly elevated in LBP−/− mice, but not in CD14−/− mice under regular diet and high‐fat diet conditions (Supplementary Figure 8D).
WT and KO mice that were fed a regular diet exhibited similar degrees of DMM‐induced OA manifestations at 6 weeks post‐DMM. However, a high‐fat diet caused more severe cartilage destruction in DMM‐operated WT mice, and this enhanced OA manifestation was significantly eliminated in TLR2−/−, LBP−/−, and CD14−/− mice (Figures 4A and B, and Supplementary Figure 7, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). Osteophyte formation was also enhanced in high‐fat diet−fed WT mice and DMM‐operated WT mice, but this was eliminated only in TLR2−/− mice (Figures 4A and B). The increases in subchondral bone plate thickness and Osterix staining for activated osteoblasts were not modulated by a high‐fat diet, and no differential change in these parameters were seen among the various KO mice (Figures 4A and B, and Supplementary Figure 9A, http://onlinelibrary.wiley.com/doi/10.1002/art.41679/abstract). These results suggest that TLR signaling components differentially regulate the high‐fat diet–induced exacerbation of posttraumatic OA under our examined experimental conditions.
Figure 4.
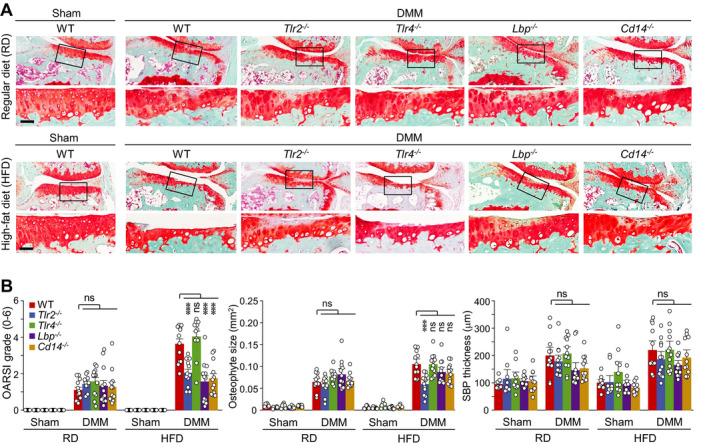
Deficiency of Tlr2, Lbp, or Cd14 abrogates the high‐fat diet (HFD)–induced acceleration of posttraumatic osteoarthritis cartilage destruction in mice. Wild‐type (WT) mice and TLR2−/−, TLR4−/−, LBP−/−, or CD14−/− mice subjected to sham surgery or destabilization of the medial meniscus (DMM) were fed a regular diet (RD) or high‐fat diet, and then euthanized 6 weeks after the surgery. A, Representative immunostained images of joint sections. The boxed areas in upper rows are shown at higher magnification in lower rows. Bars = 50 μm. B, Scoring of Osteoarthritis Research Society International (OARSI) histologic severity grade, osteophyte size, and subchondral bone plate (SBP) thickness (n = 10 mice per sham group and 12 mice per DMM group). Each symbol represents an individual mouse; bars show the mean and 95% confidence interval (for OARSI grade) or the mean ± SEM (for osteophyte size and SBP thickness). *** = P < 0.001, by Mann‐Whitney U test for OARSI grade, and by one‐way analysis of variance with Bonferroni post hoc test for osteophyte size and SBP thickness. NS = not significant.
Intraperitoneal injection of LPS also exacerbated DMM‐induced cartilage destruction without significantly affecting other manifestations of OA, such as osteophyte formation, synovitis, and subchondral bone sclerosis (Supplementary Figures 3C–E). Similar to our findings in high‐fat diet–fed mice, LPS administration in WT mice enhanced DMM‐induced OA cartilage destruction, and this was significantly eliminated in LBP−/− and CD14−/− mice (Figures 5A and B). Unlike our findings in high‐fat diet–fed mice, however, TLR2−/− and TLR4−/− mice exhibited no significant modulation of the LPS‐induced exacerbation of OA cartilage destruction (Figures 5A and B). Additionally, none of the KO mice showed any marked change in osteophyte formation or subchondral bone sclerosis (i.e., subchondral bone plate thickness and Osterix staining for activated osteoblasts) (Figures 5A and B, and Supplementary Figure 9B). Thus, our results collectively suggest that LBP and CD14 are necessary for the enhanced posttraumatic OA cartilage destruction seen under the low‐grade inflammation caused by a high‐fat diet or LPS administration.
Figure 5.
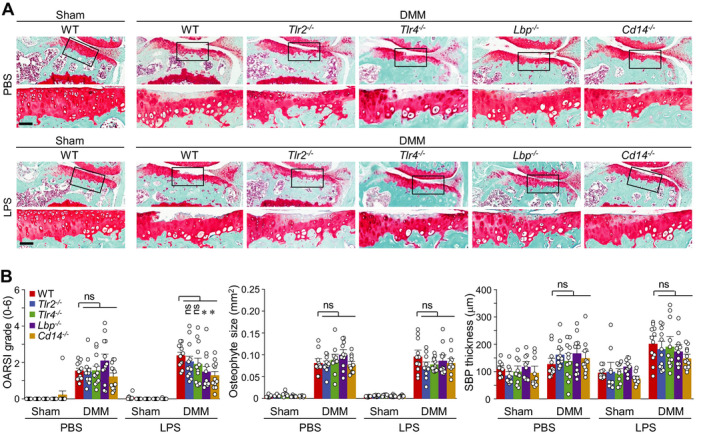
Deficiency of Lbp or Cd14 abrogates lipopolysaccharide (LPS)–induced acceleration of posttraumatic osteoarthritis cartilage destruction in mice. Mice subjected to sham surgery or destabilization of the medial meniscus (DMM) (wild‐type [WT], TLR2−/−, TLR4−/−, LBP−/−, or CD14−/−) were intraperitoneally injected with phosphate buffered saline (PBS) or a subclinical low dose of LPS for 4 weeks, and euthanized 8 weeks after the surgery. A, Representative immunostained images of joint sections. The boxed areas in upper rows are shown at higher magnification in lower rows. Bars = 50 μm. B, Scoring of Osteoarthritis Research Society International (OARSI) grade, osteophyte size, and subchondral bone plate (SBP) thickness (n = 10 mice per sham group and 12 mice per DMM group). Each symbol represents an individual mouse; bars show the mean and 95% confidence interval (for OARSI grade) or the mean ± SEM (for osteophyte size and SBP thickness). * = P < 0.05, by Mann‐Whitney U test for OARSI grade and by one‐way analysis of variance with post hoc Bonferroni test for osteophyte size and SBP thickness. NS = not significant.
Regulation of matrix‐degrading enzyme expression in chondrocytes by LBP and CD14
Finally, given our finding that LBP and CD14 are required for the exacerbation of posttraumatic OA cartilage destruction under conditions of low‐grade inflammation, we sought to elucidate some potential mechanisms that could be related to this enhanced cartilage destruction. We examined the impact of LBP and CD14 on the expression levels of the matrix‐degrading enzymes MMP‐3, MMP‐13, and ADAMTS‐5, which are crucial for OA cartilage destruction in animal models (3, 4, 5). Treatment of WT chondrocytes with IL‐1β, IL‐6, or LPS caused marked up‐regulation of MMP‐3, MMP‐13, and ADAMTS‐5, and these effects were significantly inhibited in LBP−/− mouse chondrocytes (Figure 6A). Deficiency of CD14 in chondrocytes also eliminated the up‐regulation of these matrix‐degrading enzymes caused by IL‐6 or LPS, but not IL‐1β (Figure 6B). Based on the established catabolic function of IL‐6 (29, 30) and the potential role of LPS (20) in OA pathogenesis, these results suggest that the expression modulation of these matrix‐degrading enzymes by LBP and CD14 may contribute to the ability of the latter factors to exacerbate OA cartilage destruction.
Figure 6.
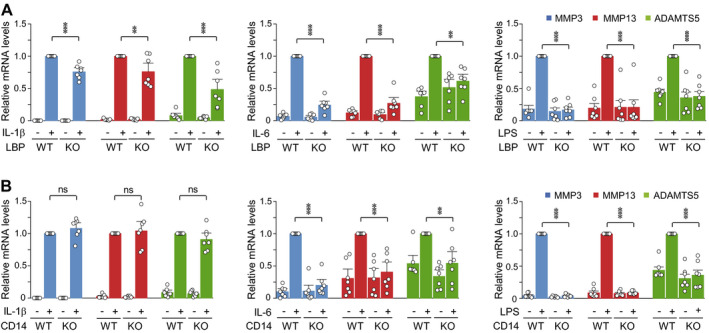
Deficiency of Lbp or Cd14 abrogates the up‐regulation of matrix‐degrading enzymes following treatment of mouse chondrocytes with osteoarthritis‐associated catabolic factors. Wild‐type (WT) and LBP−/− mouse chondrocytes (A) and CD14−/− mouse chondrocytes (B) were isolated, cultured, and treated for 36 hours with interleukin‐1β (IL‐1β) (1 ng/ml), IL‐6 (100 ng/ml), or lipopolysaccharide (LPS) (10 ng/ml). Levels of mRNA for matrix‐degrading enzymes (matrix metalloproteinase 3 [MMP‐3] MMP‐13, and ADAMTS‐5) were determined by quantitative reverse transcription–polymerase chain reaction analysis. Each symbol represents an individual mouse; bars show the mean ± SEM (n = 7 per group). ** = P < 0.005; *** = P < 0.001, by one‐way analysis of variance with Bonferroni post hoc test. KO = knockout; NS = not significant.
DISCUSSION
In this study, we have demonstrated that multiple components of the TLR signaling pathways, such as TLR‐2, TLR‐4, LBP, CD14, and SAAs, are up‐regulated in chondrocytes treated with OA‐associated catabolic factors. Overexpression of a specific TLR signaling component alone in joint tissue did not affect DMM‐induced posttraumatic OA manifestations under our experimental conditions. Moreover, knockout of Tlr2 or Tlr4 does not modulate posttraumatic OA. This is consistent with previous reports that deficiency of Tlr2 and/or Tlr4 does not affect DMM‐induced cartilage damage (35, 36). In contrast, we found that LBP−/− and CD14−/− mice exhibited slight but significant amelioration of posttraumatic OA cartilage destruction at 10 weeks, but not at 6 or 8 weeks, post‐DMM. Our findings in CD14−/− mice were similar to the results of a previous study by Sambamurthy et al (37), in which the authors found that CD14 deficiency was associated with less severe cartilage destruction at a later stage of OA (19 weeks post‐DMM), but not at 6 weeks post‐DMM.
Our key finding is that LBP and CD14 exacerbate posttraumatic cartilage destruction under conditions of low‐grade inflammation. We verified systemic low‐grade inflammation by detecting circulating levels of inflammation markers in the high‐fat diet model (i.e., leptin and LBP) and the metabolic endotoxemia model (i.e., IL‐1β, IL‐6, TNF, MCP‐1, LBP, and CD14). Circulating levels of inflammation markers have been reported to vary depending on the type of diet, duration of feeding, time point at which feeding is initiated, etc. For instance, some studies showed that a high‐fat diet increased levels of IL‐6, TNF, MCP‐1, and leptin levels (38), whereas other studies showed insignificant changes or no changes of these molecules (39). LPS‐induced endotoxemia is also known to increase IL‐6, TNF, and MCP‐1 blood levels (21).
We confirmed the presence of local synovial inflammation in both models by detecting infiltrating MCP‐1– or F4/80–positive inflammatory cells in the inflamed synovial tissue. Low‐grade inflammation stimulates OA pathogenesis by producing inflammatory mediators, including cytokines, microbial factors (LPS), and/or lipid metabolites (10). Innate immune pathways, such as TLR signaling pathways, are known to play pivotal roles in these processes (12, 13, 14, 15). TLRs are activated by damage‐associated molecular patterns to trigger proinflammatory mediator secretion and joint inflammation (12, 15). The expression of TLRs is also increased in the lesional areas of cartilage in OA patients (16). We observed up‐regulation of various TLR signaling components in chondrocytes treated with OA‐associated catabolic factors. The most prominent up‐regulations were seen in LBP and CD14, which are accessory proteins for multiple TLRs and interact with various signaling molecules, including LPS (14, 15). LBP binds with high affinity to LPS and presents LPS to CD14 to enable TLR‐4 to respond to LPS (14, 15). Therefore, it is logical to consider that LBP and CD14 would play essential roles in exacerbation of the posttraumatic cartilage destruction resulting from intraperitoneal injection of LPS.
In the high‐fat diet model, a high‐fat diet causes low‐grade inflammation, in which TLR signaling pathways play pivotal roles (12, 19). Therefore, it is likely that the LBP‐ and CD14‐mediated regulations of TLR signaling pathways are responsible for the high‐fat diet–induced acceleration of cartilage destruction. Given that mice fed high‐fat diets exhibit an elevation in plasma LPS levels (20, 40), we speculate that LBP and CD14 can relay LPS signaling in these mice to regulate the exacerbation of cartilage destruction. However, the role of CD14 and LBP in low‐grade inflammation–induced exacerbation of posttraumatic OA could be more complicated in OA pathogenesis associated with metabolic disorders. Indeed, CD14−/− mice exhibit impaired glucose intolerance under a high‐fat diet (41). LBP−/− mice exhibit no significant alterations in blood glucose levels, but develop glucose intolerance and insulin resistance (42).
The limitations of this study include the use of single–time point evaluations of OA manifestations in relatively young mice (12 weeks old) under conditions of low‐grade inflammation (6 weeks after exposure to a high‐fat diet and 8 weeks after LPS administration). In our experimental setting, knockout of Tlr2, but not Tlr4, ameliorated the high‐fat diet–induced exacerbation of posttraumatic cartilage destruction at 6 weeks post‐DMM. Tlr2‐deficient mice are known to exhibit protection from insulin resistance induced by a high‐fat diet (43). Insulin resistance is associated with metabolic OA (10), which may explain the observed TLR‐2–specific contributions in mice fed a high‐fat diet. However, in contrast to our observations, a previous study showed that TLR‐4 is necessary for high‐fat diet–induced cartilage catabolism in middle‐aged female mice (13–15 months old) (44). Both TLR‐2 and TLR‐4 were not required for the LPS‐induced stimulation of cartilage destruction under our current experimental conditions. However, LPS appears to regulate OA pathogenesis by stimulating LBP, CD14, and TLR‐4 signaling pathways (15, 20, 45). Therefore, the function of TLR‐4 should be further elucidated by analysis of mice of different ages at various time points post‐DMM. Given that both LBP and CD14 are accessory proteins for multiple TLRs (14), it may be that deficiency of a single TLR is not sufficient to inhibit the LPS‐induced stimulation of cartilage degeneration. Indeed, a recent study revealed that TLR‐2 and TLR‐4 have redundant functions (46). Finally, although we focused our efforts on elucidating the pathogenic mechanisms of OA cartilage destruction, analysis of symptomatic outcomes, such as OA pain‐related behaviors, might provide further insight into the functions of LBP and CD14 in OA pathogenesis and facilitate the development of new therapeutic approaches to the disease.
In conclusion, we identified various TLR signaling pathway–related molecules as potential pathogenic factors in OA. Among them, we demonstrated that the TLR accessory molecules LBP and CD14 are essential to the low‐grade inflammation–induced exacerbation of posttraumatic OA cartilage destruction. Thus, our results suggest that LBP and CD14 may regulate the processes of meta‐inflammation and/or inflammaging in the pathogenesis of OA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Chun had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Won, Yang, Chun.
Acquisition of data
Won, Yang, Park.
Analysis and interpretation of data
Won, Yang, Chun.
Supporting information
Supplementary Material
Supported by the National Research Foundation of Korea (grants 2016R1A3B1906090, 2016R1A5A1007318, and 2019M3A9A8065758) and the Korea Health Industry Development Institute (grant H114C3484).
No potential conflicts of interest relevant to this article werereported.
References
- 1.Martel‐Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis [review]. Nat Rev Dis Primers 2016;2:16072. [DOI] [PubMed] [Google Scholar]
- 2.Mehana EE, Khafaga AF, El‐Blehi SS. The role of matrix metalloproteinases in osteoarthritis pathogenesis: an updated review. Life Sci 2019;234:116786. [DOI] [PubMed] [Google Scholar]
- 3.Blom AB, van Lent PL, Libregts S, Holthuysen AE, van der Kraan PM, van Rooijen N, et al. Crucial role of macrophages in matrix metalloproteinase–mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheum 2007;56:147–57. [DOI] [PubMed] [Google Scholar]
- 4.Little CB, Barai A, Burkhardt D, Smith SM, Fosang AJ, Werb Z, et al. Matrix metalloproteinase 13–deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum 2009;60:3723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005;434:644–7. [DOI] [PubMed] [Google Scholar]
- 6.Wojdasiewicz P, Poniatowski LA, Szukiewicz D. The role of inflammatory and anti‐inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm 2014;2014:561459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang S, Kim J, Ryu JH, Oh H, Chun CH, Kim BJ, et al. Hypoxia‐inducible factor‐2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat Med 2010;16:687–93. [DOI] [PubMed] [Google Scholar]
- 8.Kim JH, Jeon J, Shin M, Won Y, Lee M, Kwak JS, et al. Regulation of the catabolic cascade in osteoarthritis by the zinc‐ZIP8‐MTF1 axis. Cell 2014;156:730–43. [DOI] [PubMed] [Google Scholar]
- 9.Punzi L, Galozzi P, Luisetto R, Favero M, Ramonda R, Oliviero F. Post‐traumatic osteoarthritis: overview on pathogenic mechanisms and role of inflammation. RMD Open 2016;2:e000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berenbaum F, Griffin TM, Liu‐Bryan R. Metabolic regulation of inflammation in osteoarthritis [review]. Arthritis Rheumatol 2017;69:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis [review]. Nat Rev Rheumatol 2016;12:412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson WH, Lepus CM, Wang Q, Raghu H, Mao R, Lindstrom TM, et al. Low‐grade inflammation as a key mediator of the pathogenesis of osteoarthritis [review]. Nat Rev Rheumatol 2016;12:580–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van den Bosch MH, van Lent PL, van der Kraan PM. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthritis Cartilage 2020;28:532–43. [DOI] [PubMed] [Google Scholar]
- 14.Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll‐like receptors and their function [review]. Nat Rev Immunol 2012;12:168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez R, Villalvilla A, Largo R, Gualillo O, Herrero‐Beaumont G. TLR4 signalling in osteoarthritis‐finding targets for candidate DMOADs [review]. Nat Rev Rheumatol 2014;11:159–70. [DOI] [PubMed] [Google Scholar]
- 16.Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY, Oh HJ, et al. The catabolic pathway mediated by Toll‐like receptors in human osteoarthritic chondrocytes. Arthritis Rheum 2006;54:2152–63. [DOI] [PubMed] [Google Scholar]
- 17.Abdollahi‐Roodsaz S, Joosten LA, Koenders MI, van den Brand BT, van de Loo FA, van den Berg WB. Local interleukin‐1‐driven joint pathology is dependent on Toll‐like receptor 4 activation. Am J Pathol 2009;175:2004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 2007;15:1061–9. [DOI] [PubMed] [Google Scholar]
- 19.Sansone V, Appledield RC, De Luca P, Pecoraro V, Gianola S, Pascale W, et al. Does a high‐fat diet affect the development and progression of osteoarthritis in mice? A systemic review. Bone Joint Res 2019;8:582–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang Z, Kraus VB. Does lipopolysaccharide‐mediated inflammation have a role in OA? [review]. Nat Rev Rheumatol 2016;12:123–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geng S, Chen K, Yuan R, Peng L, Maitra U, Diao N, et al. The persistence of low‐grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun 2016;7:13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi WS, Lee G, Song W, Koh JT, Yang J, Kwak JS, et al. The CH25H‐CYP7B1‐RORα axis of cholesterol metabolism regulates osteoarthritis [letter]. Nature 2019;566:254–8. [DOI] [PubMed] [Google Scholar]
- 23.Glasson SS, Chambers MG, van den Berg WB, Little CB. The OARSI histopathology initiative: recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010;18:S17–23. [DOI] [PubMed] [Google Scholar]
- 24.Krenn V, Morawietz L, Burmester GR, Kinne RW, Mueller‐Ladner U, Muller B, et al. Synovitis score: discrimination between chronic low‐grade and high‐grade synovitis. Histopathology 2006;49:358–64. [DOI] [PubMed] [Google Scholar]
- 25.Mendez ME, Murugesh DK, Sebastian A, Hum NR, McCloy SA, Kuhn EA, et al. Antibiotic treatment prior to injury improves post‐traumatic osteoarthritis outcomes in mice. Int J Mol Sci 2020;21:6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Leong DJ, Xu L, He Z, Wang A, Navati M, et al. Curcumin slows osteoarthritis progression and relieves osteoarthritis‐associated pain symptoms in a post‐traumatic osteoarthritis mouse model. Arthritis Res Ther 2016;18:128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 27.Lin C, Liu L, Zeng C, Cui ZK, Chen Y, Lai P, et al. Activation of mTORC1 in subchondral bone preosteoblasts promotes osteoarthritis by stimulating bone sclerosis and secretion of CXCL12. Bone Res 2019;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc 2008;3:1253–60. [DOI] [PubMed] [Google Scholar]
- 29.Jones SA, Jenkins BJ. Recent insights into targeting the IL‐6 cytokine family in inflammatory diseases and cancer [review]. Nat Rev Immunol 2018;18:773–89. [DOI] [PubMed] [Google Scholar]
- 30.Ryu JH, Yang S, Shin Y, Rhee J, Chun CH, Chun JS. Interleukin‐6 plays an essential role in hypoxia‐inducible factor 2α–induced experimental osteoarthritic cartilage destruction in mice. Arthritis Rheum 2011;63:2732–43. [DOI] [PubMed] [Google Scholar]
- 31.Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, et al. Toll‐like receptor 2 is required for innate, but not acquired, host defense to Borrelia burgdorferi. J Immunol 2002;168:348–55. [DOI] [PubMed] [Google Scholar]
- 32.Vogel SN, Hansen CT, Rosenstreich DL. Characterization of a congenitally LPS‐resistant, athymic mouse strain. J Immunol 1979;122:619–22. [PubMed] [Google Scholar]
- 33.Jack RS, Fan X, Bernheiden M, Rune G, Ehlers M, Weber A, et al. Lipopolysaccharide‐binding protein is required to combat a murine gram‐negative bacterial infection [letter]. Nature 1997;389:742–5. [DOI] [PubMed] [Google Scholar]
- 34.Moore KJ, Andersson LP, Ingalls RR, Monks BG, Li R, Arnaout MA, et al. Divergent response to LPS and bacteria in CD14‐deficient murine macrophages. J Immunol 2000;165:4272–80. [DOI] [PubMed] [Google Scholar]
- 35.Nasi S, Ea HK, Chobaz V, van Lent P, Liote F, So A, et al. Dispensable role of myeloid differentiation primary response gene 88 (MyD88) and MyD88‐dependent Toll‐like receptors (TLRs) in a murine model of osteoarthritis. Joint Bone Spine 2014;81:320–4. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Zhao X, Liu‐Bryan R. Role of TLR2 and TLR4 in regulation of articular chondrocyte homeostasis. Osteoarthritis Cartilage 2020;28:669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambamurthy N, Zhou C, Nguyen V, Smalley R, Hankenson KD, Dodge GR, et al. Deficiency of the pattern‐recognition receptor CD14 protects against joint pathology and functional decline in a murine model of osteoarthritis. PLoS One 2018;13:e0206217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avtanski D, Pavlov V, Tracey KJ, Poretsky L. Characterization of inflammation and insulin resistance in high‐fat diet‐induced male C57BL/6J mouse model of obesity. Animal Model Exp Med 2019;2:252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van der Heijden RA, Bijzet J, Meijers WC, Yakala GK, Kleemann R, Nguyen TQ, et al. Obesity‐induced chronic inflammation in high fat diet challenged C57BL/6J mice is associated with acceleration of age‐dependent renal amyloidosis. Sci Rep 2015;5:16474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Ding W, Wang HL, Dai LL, Zong WH, Wang YZ, et al. Gut microbiota and obesity‐associated osteoarthritis. Osteoarthritis Cartilage 2019;27:1257–65. [DOI] [PubMed] [Google Scholar]
- 41.Young JL, Mora A, Cerny A, Czech MP, Woda B, Kurt‐Jones EA, et al. CD14 deficiency impacts glucose homeostasis in mice through altered adrenal tone. PLoS One 2012;7:e29688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gavaldà‐Navarro A, Moreno‐Navarrete JM, Quesada‐López T, Cairó M, Giralt M, Fernández‐Real JM, et al. Lipopolysaccharide‐binding protein is a negative regulator of adipose tissue browning in mice and humans. Diabetologia 2016;59:2208–18. [DOI] [PubMed] [Google Scholar]
- 43.Ehses JA, Meier DT, Wueest S, Rytke J, Boller S, Wielinga PY, et al. Toll‐like receptor 2‐deficient mice are protected from insulin resistance and β cell dysfunction induced by a high‐fat diet. Diabetologia 2010;53:1795–806. [DOI] [PubMed] [Google Scholar]
- 44.Kalaitzoglou E, Lopes EB, Fu Y, Herron JC, Flaming JM, Donovan EL, et al. TLR4 promotes and DAP12 limits obesity‐induced osteoarthritis in aged female mice. JBMR Plus 2018;3:e10079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bobacz K, Sunk IG, Hofstaetter JG, Amoyo L, Toma CD, Akira S, et al. Toll‐like receptors and chondrocytes: the lipopolysaccharide‐induced decrease in cartilage matrix synthesis is dependent on the presence of Toll‐like receptor 4 and antagonized by bone morphogenetic protein 7. Arthritis Rheum 2007;56:1880–93. [DOI] [PubMed] [Google Scholar]
- 46.Ji Y, Sun S, Shrestha N, Darragh LB, Shirakawa J, Xing Y, et al. Toll‐like receptors TLR2 and TLR4 block the replication of pancreatic β cells in diet‐induced obesity. Nat Immunol 2019;20:677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material