

Abstract
One of the most applied reaction types to synthesize shape‐persistent organic cage compounds is the imine condensation reaction and it is assumed that the formed cages are thermodynamically controlled products due to the reversibility of the imine condensation. However, most of the synthesized imine cages reported are formed as precipitate from the reaction mixture and therefore rather may be kinetically controlled products. There are even examples in literature, where resulting cages are not soluble at all in common organic solvents to characterize or study their formation by NMR spectroscopy in solution. Here, a triptycene triamine containing three solubilizing n‐hexyloxy chains has been used to synthesize soluble congeners of prior insoluble cages. This allowed us to study the formation as well as the reversibility of cage formation in solution by investigating exchange of building blocks between the cages and deuterated derivatives thereof.
Keywords: dynamic covalent chemistry, imines, isotope labelling, shape-persistent organic cages, X-ray diffraction
Chains make the difference! By attaching three n‐hexyloxy chains to a triptycene triamine precursor, various soluble [4+4] and [4+6] imine cages were synthesized that were prior insoluble, revealing detailed insights in cage formation and exchange mechanisms by NMR spectroscopy in combination with gel permeation chromatography. The cubic [4+4] cage once formed is extraordinarily stable against exchange, although it is a clear thermodynamically controlled product.
Introduction
Shape‐persistent organic cages are fascinating synthetic targets, because their size, geometry, and the presence of functional groups in the exterior as well as the interior of their cavities can be precisely controlled.[1, 2, 3, 4, 5, 6, 7, 8, 9, 10] In addition to the shape‐persistency and thus the presence of an intrinsic pore or cavity, these molecular compounds can be seen as processable porous units, if soluble enough. This unique combination of porous cage properties has been used to decorate surfaces of quartz crystal microbalances,[11, 12] making cage‐containing membranes,[13, 14] coating columns with cages for gas chromatographic purposes,[15, 16] using cage‐films for sensing applications,[17] or generating porous liquids.[18, 19]
Nowadays, shape‐persistent organic cages are mainly synthesized by using reversible condensation reactions, such as by multiple imine bond formation or the generation of boronic esters from the corresponding diols and boronic acids.[20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60] These reactions used are often called dynamic covalent chemistry (DCC) reactions, allowing the system to reach thermodynamic equilibrium by self‐correction mechanisms due to the intrinsic reversibility.[5, 61, 62] However, not always is the formed cage also the favored product of thermodynamic equilibrium and the equilibrium needs to be shifted by either removing product by precipitation or, in case of formed water, by Dean‐Stark traps or other scavengers (e. g. molecular sieves).[39, 42, 57, 63] Especially when the formed cage is precipitating out of the reaction mixture, it is not always clear, if the cage is a thermodynamically or kinetically controlled product.[64, 65] In fact, for a large number of experimental protocols, pure cage is generated by precipitation, and it has to be questioned, if in these cases the cages are kinetically rather than thermodynamically controlled products, especially if calculated heat of formations do not fit the experimental observations.[42, 45, 46, 66]
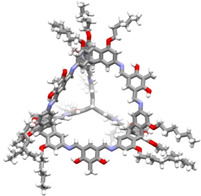
During our research on shape‐persistent imine cages,[8] there were a few examples, where the cages were completely insoluble in all common solvents, restricting the analysis to FT‐IR spectroscopy, mass spectrometry, elemental analysis and pore analysis by gas sorption.[41, 67, 68] These were the cubic [4+4] cage 1, which had high specific surface area of 1014 m2/g, high CO2 uptake (18.2 wt%) and a very good Henry selectivity or CO2 vs CH4 adsorption of 40;[41] the [4+6] exo‐functionalized cage 2, with similar specific surface area (1037 m2/g) and CO2 uptake (14.8 wt%);[67] and the [4+6] endo‐functionalized cage 3, with no alkyl substituent para to the phenolic OH of the used salicyldialdehyde (Figure 1).[38, 68]
Figure 1.
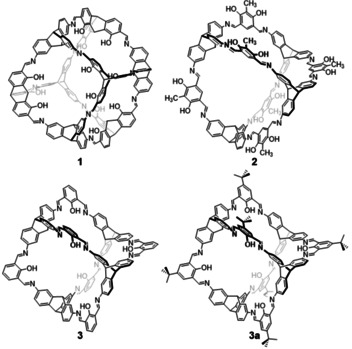
Molecular structures of insoluble imine cages (1–3)[41, 67, 68] and a derivative (3 a)[38] that is soluble in hot DMSO.
Although by monitoring the cage formation by time‐dependent mass spectrometry, polymer or oligomer formation as side reactions were excluded, suggesting that the products are pure cages and thus kinetically controlled products, final proof of structure by NMR spectroscopy or even single crystal X‐ray diffraction was lacking.
Here, we describe the synthesis and characterization of soluble congeners of cages 1–3 and investigations of the imine exchange reactions with their deuterated derivatives.
Results and Discussion
First, a triptycene triamine with solubilizung side chains needed to be synthesized (Scheme 1). Therefore, trihydroxytriptycene 4 [41] was transformed to the correspondig tris(hexyl ether) 5 in 92 % yield. As expected, the subsequent threefold Ir‐catalyzed borylation occured at the sterically least hindered positions to give 6 in 70 % yield. The trisboronic acid 6 was treated with hydroxylamine‐O‐sulfonic acid under basic conditions to transform the boronic ester units to amine groups.[69, 70] However, under no circumstances, we were able to get a full conversion to the trisamine 7 a (62 % yield) and always isolated some diamino monoboronic acid 7 b as side‐product in 9 % yield. The deuterated triamine 7 a–d39 was synthesized via the same route giving nearly the same yields in each step (Scheme 1). All compounds have been fully characterized (for details see Supporting Information) and for triptycenes 5 and 6 the solid state structures have been achieved by single‐crystal X‐ray diffraction (see Supporting Information).
Scheme 1.
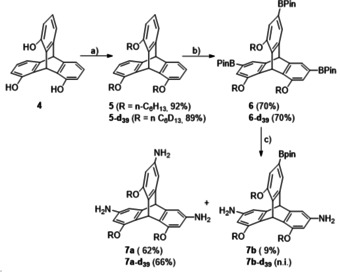
Synthesis of soluble 3,6,15‐triaminotriptycene 7 a. a) n‐hexylbromide (6 equiv.), K2CO3 (6 equiv.), DMF, 80 °C, 22 h. b) B2Pin2 (6 equiv.), 5 mol% [Ir(OMe)(COD)]2, 10 mol% 4,4’‐di‐tert‐butyl‐2,2’‐bipyridyl, THF, 80 °C, 42 h. c) H2N‐OSO3H (5.5 equiv.), NaOH(aq) (1 mol L−1, 15 equiv.), MeCN, r.t., 25 h. n.i.=not isolated.
Having now triamine 7 a at hand, this one was used for the condensation with dialdehydes 8, 9, 10 a and 10 b to give the cubic [4+4] cage 11, the [4+6] exo‐functionalized cage 12 and the [4+6] endo‐functionalized cages 13 and 14 (Scheme 2).
Scheme 2.
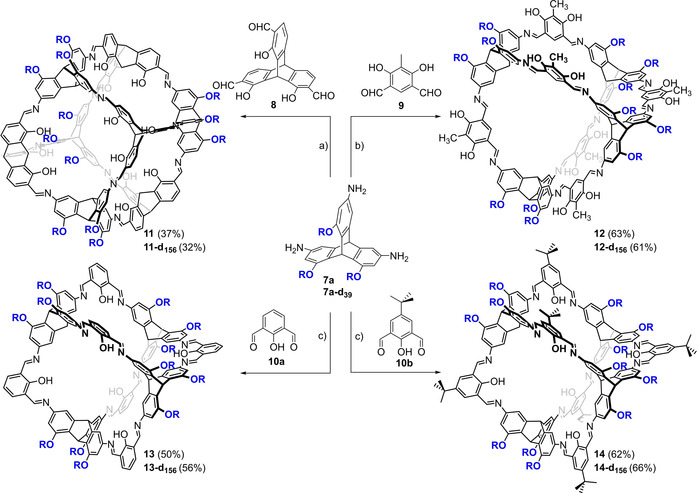
Synthesis of soluble imine cages. a) DMF, 5 mol% TFA (per amine 7 a), 150 °C, 3d; b) a) DMF, 5 mol% TFA (per amine 7 a), 120 °C, 3d; c) THF, 2 mol% TFA (per amine 7 a), room temperature, 4d. R=n‐hexyl.
We first investigated the formation of cubic cage 11 using the same conditions as described in literature for its unsubstituted congener 1.[41] Therefore, equivalent amounts of triamine 7 a and trialdehyde 8 were stirred for 3 days in DMF at 150 °C with 5 mol% TFA as catalyst, giving [4+4] cage in 37 % isolated yield. To achieve that, a combination of precipitation of oligomeric by‐products with a small amount of MeCN and further purification by recycling GPC with THF as eluent was necessary. It is worth mentioning that the best results were achieved with slightly higher concentration of reactands (c(7 a)=4.2 mol/L) than it was the case for the unsubstituted [4+4] cage 1 (here c(triamine) was 3.6 mol/L) and the temperature needed to be risen to 150 °C instead of 120 °C. Since the cage is well soluble, reactions were tested also in in other organic solvents such as DMSO‐d6, THF, DCM, toluene, CDCl3 or 1,4‐dioxane at various conditions (see the Supporting information), but yields never exceeded 10 %.
The reaction of amine 7 a and dialdehyde 9 to the exo‐functionalized [4+6] cage 12 was also performed in DMF at elevated temperature (120 °C, 3d) as it was the case for its unsubstituted congener 2.[67] In contrast to cubic cage 11, the reaction was much cleaner and by both 1H NMR (with the addition of an internal standard) and GPC of the reaction mixture it turned out that there are lesser by‐products or oligomeric intermedates formed (see discussion below). Thus, cage 12 was isolated in 63 % yield. It is also worth mentioning that in no case the reaction mixture became turbid and stayed all the time a clear solution, which excludes that the product formation is driven by precipitation and therefore is a kinetically controlled one.[64]
Different to the synthesis of cages 11 and 12, where high temperatures were needed, endo‐functionalized [4+6] cages 13 and 14 were formed in THF from salicyldialdehydes 10 a and 10 b at room temperature for four days and catalytic amounts of TFA (2 mol%) according to the original protocol for 3 b.[39] Here, cage formation was also very clean and the cages were isolated in 50 % (13) and 62 % (14) yield after purification by GPC. The deuterated analogues were synthesized the same way as the non‐deuterated ones, starting from triamine 7 a‐d156 , giving the cages, and in comparable yields of 32 % (11‐d156 ),61 % (12‐d156 ), 56 % (13‐d156 ) and 66 % (14‐d156 ).
All new cages have been fully characterized (for details, see Supporting Information) and due to the good solubility, the cages 11–14 could in addition to MALDI‐TOF MS and IR be characterized in solution by both 1H and 13C NMR spectroscopy, which was not possible for the unsubstituted congeners 1, 2, and 3 and only at elevated temperatures for 3 a.[39] The 1H NMR spectra (all in THF‐d8) are depicted in Figure 2. For cages 11 and 12 all peaks were very sharp at room temperature, due to the intrinsic shape‐persistency and strong internal hydrogen bonds of the salicylimine units. Thus the OH protons are found as sharp peaks at 15.21 ppm for cubic cage 11 and 15.01 ppm for exo cage 12. In contrast, for cages 13 and 14 the ratio of imine units and OH groups is 2 : 1 and each OH group is shared by two imine units, resulting in a conformational flipping of the OHs. As a consequence, the peaks for the OH protons (13.84 ppm for 13 and 13.70 ppm for 14), as well as the imine protons (9.06 and 9.12 ppm) are broadened at room temperature. The same is observed for protons Hc (8.02 ppm) of cage 14. To get a further insight into the conformational dynamics of cage 14, 1H NMR spectra were recorded at several temperatures. A coalescence of the peak at Hc is observed at 265 K and for the OH protons (Ha) at 253 K, resulting in barrier energies for the conformational flipping of ΔG c ǂ=52 kJ/mol and 50 kJ/mol with frequencies of kc=322 s−1 and kc=300 s−1. By further decreasing the temperatures further splitting of the signals is observed. At 203 K the conformational movement seemed to be frozen and six sharp OH peaks are detected between 13.65 and 14.47 ppm (see Supporting Information).
Figure 2.
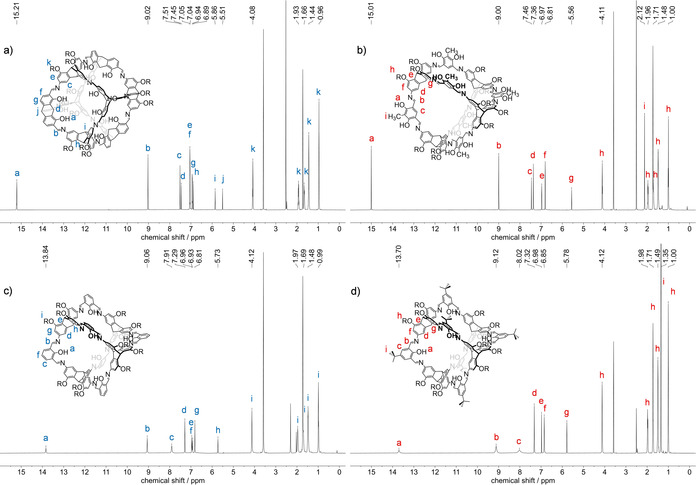
1H NMR spectra (600 MHz, THF‐d8, room temperature) of cage compounds 11 (a), 12 (b), 13 (c), and 14 (d). R=n‐hexyl.
By DOSY NMR spectrosopy the solvodynamic radii were determined to be r s=1.5 nm (D=3.1 ⋅ 10−10 m2 s−1) for cube 11, r s=1.4 nm (D=3.4 ⋅ 10−10 m2 s−1) for exo cage 12, r s=1.1 nm (D=4.2 ⋅ 10−10 m2 s−1) for endo cage 13, and r s=1.1 nm (D=4.2 ⋅ 10−10 m2 s−1) for endo cage 14 (see also Supporting Information) and correlate to the dimensions found by X‐ray diffraction.
For cages 11, 12, and 13 single crystals for X‐ray diffraction analysis have been obtained (Figure 3, for details, see Supporting Information). All compounds crystallize in the triclinic spacegroup P‐1. Single‐crystals of cubic cage 11 were grown by vapor diffusion of methanol into a saturated solution of 11 in ethyl acetate. Two molecules were found in the asymmetric unit and some of the hexyl chains could not be refined sufficiently. All imine groups are pointing into the same direction and the conformation is fixed by intramolecular hydrogen bonds to the adjacent OH groups. Therefore, the cubes are of a regular structure with two different triptycene units in the opposite corners. The diagonal distance of inner triptycene bridgehead carbons of the cubes is between 15.3 and 16.3 Å. The hexyl chains are all pointing more or less to the corners of a tetrahedron with an edge length of about 30 Å, which is in good agreement with the values found by DOSY NMR (see above). The cubic cages are relative loosely packed and mainly weak CH‐π interactions of hexyl chains with the aromatic triptycene backbones are found. Nevertheless, by running a probe sphere of 1.8 Å radius over the in silico desolvated crystal arrangement, a network of three‐dimensional pores can be found.
Figure 3.
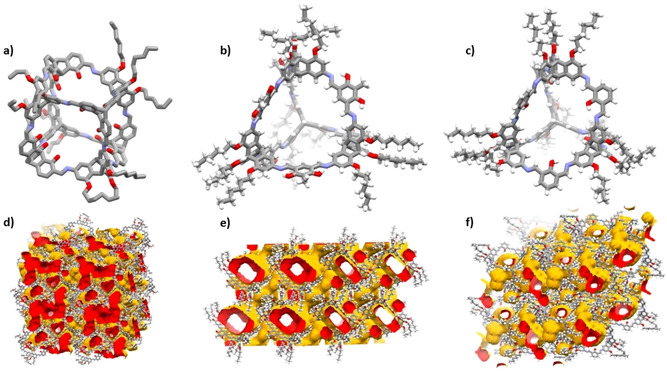
Single crystal X‐ray structures of cages 11 (a and d), 12 (b and e) and 13 (c and f). a–c) molecular structures as capped stick models. In a) hydrogens have been omitted for clarity and only one cage of two found in the asymmetric unit is depicted. Note: For cage 11 not all n‐hexyl chains could be satisfactorily refined. d–f) 2×2×2 unit cell with surface accessible voids for probe radius of 1.8 Å.
Cage 12 was crystallized by slow solvent evaporation of a concentrated solution of 12 in dichloromethane. The compound shows also a very symmetric and shape‐persistent cage scaffold, due to the hydrogen‐bond stabilized conformationally fixed imine units. The triptycenes form a regular tetrahedron as well with a distance of the inner bridgehead carbons between 12.6 and 13.0 Å. Again, the hexyl chains are pointing into the corner of a tetrahedron, with the largest distance of the peripheral CH3 units of the hexyl chains of 34 Å. The cages 12 are similar loose packed as cages 11 and the hexyl chains of one triptycene corner of a cage molecule are pointing into the cavity of another blocking the access of those. However, this tight arrangement cannot be observed for the three other triptycene units, which results in large three‐dimensional pores (Figure 3e).
Single‐crystals of cage 13 were gained by slow diffusion of acetonitrile into a DMF solution of the cage. Similar as found for 12, hexyl chains of one of the triptycene units is pointing into the cavity of an adjacent cage, but the others not. The cage scaffold shows the same type of imine bond arrangement as previously found for other endo‐functionalized [4+6] salicylimine cages.[39, 71] Namely, one is hydrogen‐bonded to the inner OH groups, the other one not, with the nitrogen lone pair pointing outside of the cavity. Similar as found for 12, the largest distance of peripheral CH3 groups is also 34 Å. As for the other two cages, three‐dimensional pores have been found despite some of the hexyl‐chains are sticking into the cavities of adjacent molecules.
Although pores can be found for all three cages by analyzing their single crystal structures, it has to be kept in mind that disordered solvate molecules have been omitted for solving the structures in all three cases and the solvate molecules most likely contribute in stabilizing the structure. Furthermore, no “stronger” aggregation motifs are found, than loose CH‐π interactions. Therefore, it is expected that by desolvating the crystals, the ordered structure will collapse, resulting in a low porosity.[48, 72] Thus, no gas sorption experiments have been performed.
As mentioned in the introduction, we were interested to get further insight into the thermodynamic or kinetic stability of the cages under reaction conditions. To exclude any precipitation effects, with both soluble type of cages, non‐deuterated and deuterated in hand with a difference of 39 mass units per incorporated triptycene amine, possible exchange reactions were studied to get insight into the reversibility of cage formation (Scheme 3,Table 1 and Figure 4).[64] First, we applied the exact reaction conditions (with catalytic amount of TFA) to all the cage mixtures without any alterations (Table 1, Entries 1, 6 and 11) and found no scrambling for all three cage pairs. With the addition of a few amount of water (Table 1, Entries 2, 7, and 12) no exchange of amine units was observed for cubic cages 11/11‐d156 (Table 1, Entry 2) and some scrambling for cage 14/14‐d156 was observed after 3 days and after 7 days the intensities of MS signals of the scrambled cages 14‐d39; 14‐d78 and 14‐d117 did not increased much in comparison to the three days, suggesting that the exchange is very slow. For 12/12‐d156 some scrambling was observed after 3 days and a clear dynamic exchange of molecular building blocks was detected after 7 days. It is worth mentioning that increasing the water content for 11/11‐d156 *to 12 equivalents (one per imine bond) still no scrambling was observed, whereas cage 14/14‐d156 scrambled faster.
Scheme 3.

Scrambling experiments of deuterated and non‐deuterated cage pairs under various conditions. For additives and results see Table 1.
Table 1.
Scrambling of cages and deuterated cages according to Scheme 3.
|
Entry |
Cage Pair |
Additives |
Scrambling[d] observed after |
|
|---|---|---|---|---|
|
3 days |
7 days |
|||
|
1 |
11/11‐d156 |
TFA[a] |
× |
× |
|
2 |
11/11‐d156 |
TFA,[a] wate,r[a] |
× [b] |
× |
|
3 |
11/11‐d156 |
p‐toluidine[a] |
× |
× |
|
4 |
11/11‐d156 |
p‐toluidine[a], water[a] |
× |
× |
|
5 |
11/11‐d156 |
TFA[a], p‐toluidine[a] |
× |
× |
|
6 |
12/12‐d156 |
TFA[a] |
× |
× |
|
7 |
12/12‐d156 |
TFA[a], water[a] |
(✔) |
✔ |
|
8 |
12/12‐d156 |
p‐toluidine[a] |
× |
× |
|
9 |
12/12‐d156 |
p‐toluidine[a], water[a] |
(✔) |
✔ |
|
10 |
12/12‐d156 |
TFA[a], p‐toluidine[a] |
(✔) |
✔ |
|
11 |
14/14‐d156 |
TFA[c] |
× |
× |
|
12 |
14/14‐d156 |
TFA[c], water [c], |
(✔) [b] |
(✔) |
|
13 |
14/14‐d156 |
p‐toluidine[b] |
× |
× |
|
14 |
14/14‐d156 |
p‐toluidine[b], water[b] |
× |
× |
|
15 |
14/14‐d156 |
TFA[b], p‐toluidine[b] |
✔ |
✔ |
[a] 5 mol% related to the overall amount of substance (mol). [b] 2 mol% related to the overall amount of substance (mol). [c] 24 equivalents of water does not changed the outcome. [d] detected by MALDI‐TOF from an aliquot of the reaction mixture.
Figure 4.
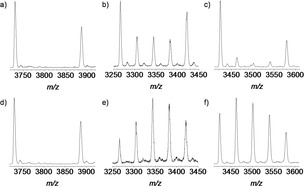
Examples of MALDI‐TOF mass spectra (linear mode) of scrambling experiments for cage pairs 11/11‐d156 (a and d), 12/12‐d156 (b and e) and 14/14‐d156 (c and f) after 7 days according to conditions of Table 1. (a: Entry 2; b: Entry 7; c: Entry 12; d: Entry 5; e: Entry 10 and f: Entry 15)
We also tested the addition of p‐toluidine that acts as a nucleophile to open imine bonds and thus the cage to foster scrambling. Whereas toluidine alone does not lead to any exchange of units for all three systems (Table 1, Entries 3, 8, and 13), in combination with water, the [4+6] exo cages 12/12‐d156 is the only one that is showing a clear scrambling after 7 days (Table 1, Entry 9). Finally, we examined the combination of TFA and toluidine and found that both [4+6] cages 12 and 14 scramble (Table 1, Entries 10 and 15) while the cubic [4+4] cage 11 does not (Table 1, Entry 5).
By these observations cage scrambling (if occurring) is not taken place via any imine metathesis, but rather by an opening up of imine bonds[73] or even disassembly of cages to fragments triggered by a nucleophile (water or amine) which is accelerated by the addition of a Brønsted acid that attacks the imine nitrogen and thus activating the imine carbon for the addition of the nucleophile. The most important result of the scrambling experiments is that the cubic [4+4] cage 11 does not undergo any scrambling at all, suggesting that it is energetically locked, which may be simply explained by the fact that in comparison to the two other systems, three imine bonds need to be broken to “rip off” one molecular building block unit, instead of two for cages 12, 13 and 14.
Although by the scrambling experiments insight into reversibility of cage formation was gained, it does not answer the question, if these soluble cages are thermodynamically or kinetically controlled products. The good solubility of the cages and their intermediates allowed us to study their formation more detailed by monitoring the reactions in DMF‐d7 (for cages 11 and 12) or THF‐d8 (cages 13 and 14) by 1H NMR spectroscopy in combination with GPC. Therefore, after certain time periods, the 1H NMR spectrum of the reaction mixture that contained a defined amount of 1,3,5‐trimethoxybenzene as internal standard was recorded and an aliquot of each reaction investigated by GPC.
Again, most interestingly is the formation of [4+4] cubic cage 11. In the beginning of the reaction the 1H NMR spectrum shows a lot of broad overlapping peaks, that could originate from mixtures of oligomeric and polymeric condensation products larger than the cages or from smaller cage intermediates. The latter can most likely be excluded by the corresponding GPC trace (Figure 5a), because no peaks with retention times larger than the cubic cage (56 min, 2nd cycle) could be found, but very large peaks at lower retention time (47 min, 2nd cycle), with a broad and less intense shoulder were found between these two peaks. Therefore, it is concluded that in the beginning of the reaction indeed species larger in size than the cage 11 are formed. By time, this first peak decreases and the peak of cage 11 increases until after 3 days no more of the first one is observed. The corresponding NMR spectra show relative clean signals for cage 11 after 4 days. In one experiment, the first peak was isolated by r‐GPC and investigated by 1H NMR spectroscopy, showing only very broad and undefined peaks in the aromatic region, which is expected for a mixture of larger oligomers and polymers and not for smaller fragments of the cage 11. From these experiments the formation of cage 11 is indeed a DCC reaction with the product being the thermodynamically controlled one.
Figure 5.
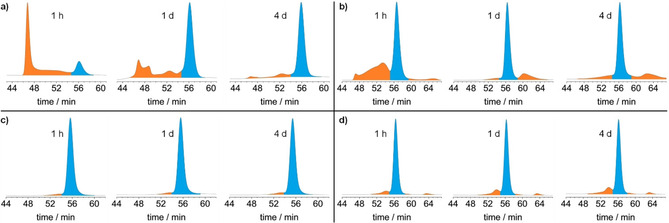
GPC traces of aliquots taken from reaction mixtures of cage synthesis in deuterated solvents (DMF‐d7 for a and b; THF‐d8 for c and d) under typical reaction conditions (see Scheme 2). a) [4+4] cube 11; b) [4+6] exo cage 12; c) [4+6] endo cage 14 and d) [4+6] endo cage 13. Depicted is always the second cycle of a recycling‐GPC for better peak separation. The blue colored peaks are assigned to the respective cage compound.
A similar, although much cleaner, reaction was observed for exo‐functionalized [4+6] cage 12. Here, already the first 1H NMR spectrum after one hour mainly showed cage signals and the peaks in the GPC trace of cage 12 vs. larger species were already clearly shifted towards cage fraction (Figure 5 b). This implies that cage 12 is also a thermodynamically controlled product, but is formed much faster, which is in agreement with a faster “self‐correction” mechanism of the system due to the above described fact that only two imine units need to be cleaved here, rather than three for the system leading to cage 11. For cages 13 and 14 by both 1H NMR as well as GPC almost no side‐product formation was observed even after 2 hours at room temperature, assuming that the reaction is very fast, so the cages seem to be both kinetically and thermodynamically formed products (Figure 5 c and d). When decreasing the amount of added TFA to 0.5 mol% or even to 0.0 mol%, the reaction was slowed down allowing us to monitor the cage formation process by GPC and 1H NMR spectroscopy. In both cases broad peaks with smaller retention times (corresponding to oligomeric intermediates larger than the cage) are found by GPC that diminish by time, revealing that the [4+6] cages 13 and 14 are also thermodynamically favored products, but the reaction is just much faster than found for cage 11.
Conclusion
To summarize, three congeners of prior insoluble larger imine cages have been synthesized and characterized by attaching solubilizing sidechains to the triptycene triamine precursor used in the condensation reaction with several aldehydes. In addition, the deuterated derivatives of all these cages have been generated, too, allowing to study exchange of the triamine building blocks of cages under various conditions. Whereas the cubic [4+4] cage and its deuterated derivative do not undergo any exchange at all, the [4+6] exo‐functionalized cage scrambles as soon as a catalytic amount of TFA and some nucleophile (water or p‐toluidine) is present. TFA or p‐toluidine (as basic catalyst) alone do not trigger the exchange reaction, but p‐toluidine plus water does. The [4+6] endo cages show only a significant exchange with p‐toluidine and TFA, but surprisingly in the presence of TFA and water it seems that this reaction is relatively slow.
The good solubility of the cages and intermediates also allowed us to study the formation of cages by time. Whereas, the [4+6] endo cages are formed very quickly (after 1 h the reaction is already almost complete), the [4+4] cage formation was slow, even at 150 °C. Most important, before the [4+4] cage is formed much larger species (oligomers and polymers) are generated (detected by GPC), that are consumed by time, clearly indicating that the [4+4] cage is a thermodynamically controlled product, although once formed it is very stable against the investigated exchange of building blocks. This is in contrast to the observations made before for the unsubstituted cubic cage 1, which is not soluble at all and there, precipitation most likely is the kinetic driving force.
In conclusion, this study once more demonstrates, how large the difference in cage formation mechanisms can be, even for cages that only slightly vary in structure, such as the [4+6] endo and exo‐cages. The good solubility of the here presented cages, will further allow us to chemically transform these to more stable compounds, such as amide or carbamate cages[74, 75] or study their conversion to transition metal or boroquinol complexes.[76]
Experimental Section
For Experimental Details, see the Supporting Information.
Deposition Number(s) 2060523 (5), 2060524 (6), 2060525 (12), 2060526 (11), and 2060527 (13) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the European Research Council ERC in the frame of the consolidators grant CaTs n DOCs (grant no. 725765) and Deutsche Forschungsgemeinschaft DFG under Germany's Excellence Strategy Cluster 3D Matter Made to Order (EXC‐2082/1 – 390761711) for funding this project. Open access funding enabled and organized by Projekt DEAL.
M. Holsten, S. Feierabend, S. M. Elbert, F. Rominger, T. Oeser, M. Mastalerz, Chem. Eur. J. 2021, 27, 9383.
References
- 1.Beuerle F., Gole B., Angew. Chem. Int. Ed. 2018, 57, 4850–487; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4942–4972. [Google Scholar]
- 2.Hasell T., Cooper A. I., Nat. Rev. Mater. 2016, 1, 16053. [Google Scholar]
- 3.Little M. A., Cooper A. I., Adv. Funct. Mater. 2020, 30, 1909842. [Google Scholar]
- 4.Acharyya K., Mukherjee P. S., Angew. Chem. Int. Ed. 2019, 58, 8640–8653; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8732–8745. [Google Scholar]
- 5.Mastalerz M., Angew. Chem. Int. Ed. 2010, 49, 5042–5053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5165–5175. [Google Scholar]
- 6.Mastalerz M., Synlett 2013, 24, 781–786. [Google Scholar]
- 7.Zhang G., Mastalerz M., Chem. Soc. Rev. 2014, 43, 1934–1947. [DOI] [PubMed] [Google Scholar]
- 8.Mastalerz M., Acc. Chem. Res. 2018, 51, 2411–2422. [DOI] [PubMed] [Google Scholar]
- 9.Rue N. M., Sun J., Warmuth R., Isr. J. Chem. 2011, 51, 743–768. [Google Scholar]
- 10.Skowronek P., Warzajtis B., Rychlewska U., Gawronski J., Chem. Commun. 2013, 49, 2524–2526. [DOI] [PubMed] [Google Scholar]
- 11.Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Adv. Mater. 2012, 24, 6049–6052. [DOI] [PubMed] [Google Scholar]
- 12.Brutschy M., Schneider M. W., Mastalerz M., Waldvogel S. R., Chem. Commun. 2013, 49, 8398–8400. [DOI] [PubMed] [Google Scholar]
- 13.Hasell T., Zhang H., Cooper A. I., Adv. Mater. 2012, 24, 5732–5737. [DOI] [PubMed] [Google Scholar]
- 14.Bushell A. F., Budd P. M., Attfield M. P., Jones J. T. A., Hasell T., Cooper A. I., Bernardo P., Bazzarelli F., Clarizia G., Jansen J. C., Angew. Chem. Int. Ed. 2013, 52, 1253–1256; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1291–1294. [Google Scholar]
- 15.Kewley A., Stephenson A., Chen L., Briggs M. E., Hasell T., Cooper A. I., Chem. Mater. 2015, 27, 3207–3210. [Google Scholar]
- 16.Zhang J.-H., Xie S.-M., Wang B.-J., He P.-G., Yuan L.-M., J. Chromatogr. A 2015, 1426, 174–182. [DOI] [PubMed] [Google Scholar]
- 17.Alexandre P.-E., Zhang W.-S., Rominger F., Elbert S. M., Schröder R. R., Mastalerz M., Angew. Chem. Int. Ed. 2020, 59, 19675–19679; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 19843–19847. [Google Scholar]
- 18.Giri N., Del Pópolo M. G., Melaugh G., Greenaway R. L., Rätzke K., Koschine T., Pison L., Gomes M. F. C., Cooper A. I., James S. L., Nature 2015, 527, 216. [DOI] [PubMed] [Google Scholar]
- 19.Mastalerz M., Nature 2015, 527, 174. [DOI] [PubMed] [Google Scholar]
- 20.MacDowell D., Nelson J., Tetrahedron Lett. 1988, 29, 385–386. [Google Scholar]
- 21.Jazwinski J., Lehn J.-M., Lilienbaum D., Ziessel R., Guilhem J., Pascard C., J. Chem. Soc. Chem. Commun. 1987, 1691–1694. [Google Scholar]
- 22.Drew M. G. B., McDowell D., Nelson J., Polyhedron 1988, 7, 2229–2232. [Google Scholar]
- 23.Skowronek P., Gawronski J., Org. Lett. 2008, 10, 4755–4758. [DOI] [PubMed] [Google Scholar]
- 24.Liu X., Liu Y., Li G., Warmuth R., Angew. Chem. Int. Ed. 2006, 45, 901–904; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 915–918. [Google Scholar]
- 25.Hong S., Rohman M. R., Jia J., Kim Y., Moon D., Kim Y., Ko Y. H., Lee E., Kim K., Angew. Chem. Int. Ed. 2015, 54, 13241–13244 Angew. Chem. 2015, 127, 13439–13442. [DOI] [PubMed] [Google Scholar]
- 26.Mukhopadhyay R. D., Kim Y., Koo J., Kim K., Acc. Chem. Res. 2018, 51, 2730–2738. [DOI] [PubMed] [Google Scholar]
- 27.Acharyya K., Mukherjee S., Mukherjee P. S., J. Am. Chem. Soc. 2013, 135, 554–557. [DOI] [PubMed] [Google Scholar]
- 28.Acharyya K., Mukherjee P. S., Chem. Eur. J. 2014, 20, 1646–1657. [DOI] [PubMed] [Google Scholar]
- 29.Acharyya K., Mukherjee P. S., Chem. Commun. 2015, 51, 4241–4244. [DOI] [PubMed] [Google Scholar]
- 30.Wang X., Wang Y., Yang H., Fang H., Chen R., Sun Y., Zheng N., Tan K., Lu X., Tian Z., Cao X., Nat. Commun. 2016, 7, 12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qu H., Wang Y., Li Z., Wang X., Fang H., Tian Z., Cao X., J. Am. Chem. Soc. 2017, 139, 18142–18145. [DOI] [PubMed] [Google Scholar]
- 32.Jin Y., Voss B. A., Noble R. D., Zhang W., Angew. Chem. Int. Ed. 2010, 49, 6348–6351; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 6492–6495. [Google Scholar]
- 33.Jiao T., Chen L., Yang D., Li X., Wu G., Zeng P., Zhou A., Yin Q., Pan Y., Wu B., Hong X., Kong X., Lynch V. M., Sessler J. L., Li H., Angew. Chem. Int. Ed. 2017, 56, 14545–14550; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14737–14742. [DOI] [PubMed] [Google Scholar]
- 34.Cao N., Wang Y., Zheng X., Jiao T., Li H., Org. Lett. 2018, 20, 7447–7450. [DOI] [PubMed] [Google Scholar]
- 35.Jiao T., Wu G., Chen L., Wang C.-Y., Li H., J. Org. Chem. 2018, 83, 12404–12410. [DOI] [PubMed] [Google Scholar]
- 36.Zheng X., Zhu W., Zhang C., Zhang Y., Zhong C., Li H., Xie G., Wang X., Yang C., J. Am. Chem. Soc. 2019, 141, 4704–4710. [DOI] [PubMed] [Google Scholar]
- 37.Mateus P., Delgado R., Brandão P., Carvalho S., Félix V., Org. Biol. Chem. 2009, 7, 4661–4673. [DOI] [PubMed] [Google Scholar]
- 38.Mastalerz M., Chem. Commun. 2008, 4756–4758. [DOI] [PubMed] [Google Scholar]
- 39.Schneider M. W., Oppel I. M., Ott H., Lechner L. G., Hauswald H.-J. S., Stoll R., Mastalerz M., Chem. Eur. J. 2012, 18, 836–847. [DOI] [PubMed] [Google Scholar]
- 40.Schneider M. W., Oppel I. M., Mastalerz M., Chem. Eur. J. 2012, 18, 4156–4160. [DOI] [PubMed] [Google Scholar]
- 41.Elbert S. M., Rominger F., Mastalerz M., Chem. Eur. J. 2014, 20, 16707–16720. [DOI] [PubMed] [Google Scholar]
- 42.Lauer J. C., Zhang W.-S., Rominger F., Schröder R. R., Mastalerz M., Chem. Eur. J. 2018, 24, 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beaudoin D., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2017, 56, 1244–1248; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1264–1268. [Google Scholar]
- 44.Schick T. H. G., Lauer J. C., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2019, 58, 1768–1773; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1782–1787. [Google Scholar]
- 45.Greenaway R. L., Santolini V., Bennison M. J., Alston B. M., Pugh C. J., Little M. A., Miklitz M., Eden-Rump E. G. B., Clowes R., Shakil A., Cuthbertson H. J., Armstrong H., Briggs M. E., Jelfs K. E., Cooper A. I., Nat. Commun. 2018, 9, 2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pugh C. J., Santolini V., Greenaway R. L., Little M. A., Briggs M. E., Jelfs K. E., Cooper A. I., Cryt. Growth Des. 2018, 18, 2759–2764. [Google Scholar]
- 47.Jiang S., Bacsa J., Wu X., Jones J. T. A., Dawson R., Trewin A., Adams D. J., Cooper A. I., Chem. Commun. 2011, 47, 8919–8921. [DOI] [PubMed] [Google Scholar]
- 48.Jelfs K. E., Wu X., Schmidtmann M., Jones J. T. A., Warren J. E., Adams D. J., Cooper A. I., Angew. Chem. Int. Ed. 2011, 50, 10653–10656; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10841–10844. [Google Scholar]
- 49.Kunde T., Nieland E., Schröder H. V., Schalley C. A., Schmidt B. M., Chem. Commun. 2020, 56, 4761–4764. [DOI] [PubMed] [Google Scholar]
- 50.Mastalerz M., Wagner P., Rominger F., Zhang W.-S., Gross J. H., Elbert S. M., Schröder R. R., Angew. Chem. Int. Ed. 2021, 60, 8896–8904; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 8978–8986. [Google Scholar]
- 51.Christinat N., Scopelliti R., Severin K., Angew. Chem. Int. Ed. 2008, 47, 1848–1852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1874–1878. [Google Scholar]
- 52.Hutin M., Bernardinelli G., Nitschke J. R., Chem. Eur. J. 2008, 14, 4585–4593. [DOI] [PubMed] [Google Scholar]
- 53.Nishimura N., Kobayashi K., Angew. Chem. Int. Ed. 2008, 47, 6255–6258; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6351–6354. [Google Scholar]
- 54.Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 1516–1520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1542–1546. [Google Scholar]
- 55.Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 5126–5130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5226–5230. [Google Scholar]
- 56.Elbert S. M., Regenauer N. I., Schindler D., Zhang W.-S., Rominger F., Schröder R. R., Mastalerz M., Chem. Eur. J. 2018, 24, 11438–11443. [DOI] [PubMed] [Google Scholar]
- 57.Klotzbach S., Scherpf T., Beuerle F., Chem. Commun. 2014, 50, 12454–12457. [DOI] [PubMed] [Google Scholar]
- 58.Klotzbach S., Beuerle F., Angew. Chem. Int. Ed. 2015, 54, 10356–10360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10497–10502. [Google Scholar]
- 59.Beuerle F., Klotzbach S., Dhara A., Synlett 2016, 27, 1133–1138. [Google Scholar]
- 60.Schäfer N., Bühler M., Heyer L., Röhr M. I. S., Beuerle F., Chem. Eur. J. 2021, 27, 6077–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lehn J.-M., Chem. Eur. J. 1999, 5, 2455–2463. [Google Scholar]
- 62.Rowan S. J., Cantrill S. J., Cousins G. R. L., Sanders J. K. M., Stoddart J. F., Angew. Chem. Int. Ed. 2002, 41, 898–952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 938–993. [Google Scholar]
- 63.Schneider M. W., Lechner L. G., Mastalerz M., J. Mater. Chem. 2012, 22, 7113–7116. [Google Scholar]
- 64.Schick T. H. G., Rominger F., Mastalerz M., J. Org. Chem. 2020, 85, 13757–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Rycke N., Marrot J., Couty F., David O. R. P., Tetrahedron Lett. 2010, 51, 6521–6525. [Google Scholar]
- 66.Greenaway R. L., Santolini V., Pulido A., Little M. A., Alston B. M., Briggs M. E., Day G. M., Cooper A. I., Jelfs K. E., Angew. Chem. Int. Ed. 2019, 58, 16275–16281; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 16421–16427. [Google Scholar]
- 67.Schneider M. W., Hauswald H.-J. Siegfried, Stoll R., Mastalerz M., Chem. Commun. 2012, 48, 9861–9863. [DOI] [PubMed] [Google Scholar]
- 68.M. Holsten, M. W. Schneider, I. M. Oppel, F. Rominger, M. Mastalerz, unpublished results.
- 69.Wagner P., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2018, 57, 11321–11324; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11491–11494. [Google Scholar]
- 70.Voth S., Hollett J. W., McCubbin J. A., J. Org. Chem. 2015, 80, 2545–2553. [DOI] [PubMed] [Google Scholar]
- 71.Mastalerz M., Schneider M. W., Oppel I. M., Presly O., Angew. Chem. Int. Ed. 2011, 50, 1046–1051; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1078–1083. [Google Scholar]
- 72.Kohl B., Rominger F., Mastalerz M., Chem. Eur. J. 2015, 21, 17308–17313. [DOI] [PubMed] [Google Scholar]
- 73.Ro S., Rowan S. J., Pease A. R., Cram D. J., Stoddart J. F., Org. Lett. 2000, 2, 2411–2414. [DOI] [PubMed] [Google Scholar]
- 74.Bhat A. S., Elbert S. M., Zhang W.-S., Rominger F., Dieckmann M., Schröder R. R., Mastalerz M., Angew. Chem. Int. Ed. 2019, 58, 8819–8823; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8911–8915. [Google Scholar]
- 75.Hu X.-Y., Zhang W.-S., Rominger F., Wacker I., Schröder R. R., Mastalerz M., Chem. Commun. 2017, 53, 8616–8619. [DOI] [PubMed] [Google Scholar]
- 76.Elbert S. M., Wagner P., Kanagasundaram T., Rominger F., Mastalerz M., Chem. Eur. J. 2017, 23, 935–945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary