

SUMMARY
Regulatory T (Treg) cell identity is defined by the lineage-specifying transcription factor (TF) Foxp3. Here we examined mechanisms of Foxp3 function by leveraging naturally occurring genetic variation in wild-derived inbred mice, which enables the identification of DNA sequence motifs driving epigenetic features. Chromatin accessibility, TF binding, and gene expression patterns in resting and activated subsets of Treg cells, conventional CD4 T cells, and cells expressing a Foxp3 reporter null allele revealed that the majority of Foxp3-dependent changes occurred at sites not bound by Foxp3. Chromatin accessibility of these indirect Foxp3 targets depended on the presence of DNA binding motifs for other TFs, including TCF1. Foxp3 expression correlated with decreased TCF1 and reduced accessibility of TCF1-bound chromatin regions. Deleting one copy of the Tcf7 gene recapitulated Foxp3-dependent negative regulation of chromatin accessibility. Thus, Foxp3 defines Treg cell identity in a largely indirect manner by fine-tuning the activity of other major chromatin remodeling TFs such as TCF1.
INTRODUCTION
Regulatory T (Treg) cells are a distinct subset of CD4 T cells required for suppression of lethal autoimmunity and dampening of inflammatory responses under a variety of physiological and pathophysiological conditions (Campbell and Rudensky, 2020; Josefowicz et al., 2012). Treg cells are defined by expression of the X-linked Forkhead family transcription factor (TF) Foxp3. Loss-of-function mutations in the Foxp3 gene in mice or humans cause impaired Treg cell development, leading to unrestrained activation of auto-reactive effector T cells and lethal multi-organ autoimmunity. Foxp3 expression can be induced in a subset of self-reactive CD4 single-positive thymocytes in response to T cell receptor (TCR) stimulation and interleukin-2 (IL-2) (Fontenot et al., 2005a; Fontenot et al., 2005b; Lio and Hsieh, 2008). How Foxp3 confers Treg cell identity to these cells is still unclear.
Unlike many other lineage-defining TFs acting at earlier stages of hematopoietic development, Foxp3 does not appear to drive widespread chromatin remodeling at its direct targets (Boller et al., 2016; Fasolino et al., 2020; Heinz et al., 2010; Li et al., 2018). Instead, Foxp3 has been found to bind predominantly to sites that have pre-established chromatin accessibility in conventional CD4 T cells (Samstein et al., 2012). Approximately 80% of these sites are constitutively accessible across many different immune cell types, with most of the remaining targets gaining accessibility after the double-positive thymocyte stage of T cell development, prior to Foxp3 induction (Yoshida et al., 2019). Although many regulatory elements across the genome undergo Treg cell-specific chromatin remodeling, very few of these elements are bound by Foxp3. These observations raise the possibility that most Foxp3 binding events do not affect gene activity and that Foxp3 functions in a largely indirect manner by controlling the expression of only a few key regulators with many downstream targets. However, it is also possible that more widespread direct Foxp3-dependent gene regulation has been overlooked or misinterpreted previously in comparisons between Treg and conventional CD4 T cells because the signals that precede and promote Foxp3 expression, including TCR and IL-2R activation, may profoundly alter the epigenetic landscape and obscure direct regulation by Foxp3. Indeed, our previous analysis of Treg “wannabe” cells expressing a Foxp3 reporter null allele (Foxp3GFPKO) found that Foxp3-dependent gene expression changes substantially intersect and interact with transcriptional changes induced immediately prior to Foxp3 expression (Gavin et al., 2007). Another model of Foxp3-dependent gene regulation holds that Foxp3 binding can act as a beacon for recruitment of stimulation-dependent cofactors. We reported such a mechanism in highly activated proliferating Treg cells responding to autoimmune inflammation, where Foxp3 recruits polycomb repressive complex 2 (PRC2) to mediate histone H3K27 tri-methylation and repression of nearby genes (Arvey et al., 2014). However, this model does not account for Foxp3-dependent lineage specification in newly differentiated, resting Treg cells.
The inability to distinguish direct Foxp3-dependent target gene regulation from indirect downstream effects highlights a more general limitation of genome-wide studies on TF function. Because many regulatory elements can be bound simultaneously by a multitude of TFs with interdependent expression patterns, it is often hard to disentangle direct gene regulation in cis from indirect regulation in trans. In this regard, analysis of TF binding site polymorphisms can provide important new insights. Naturally occurring genetic variation in wild-derived inbred mouse strains is a rich source of such polymorphisms, which can be leveraged to understand gene regulation (Fasolino et al., 2020; Heinz et al., 2013; Keane et al., 2011; Link et al., 2018; Soccio et al., 2015; Vierbuchen et al., 2017). Specifically, analysis of allelic bias in TF binding and chromatin accessibility in F1 offspring of laboratory mice and wild-derived inbred mice enables identification of DNA sequence motifs driving these epigenetic features (van der Veeken et al., 2019).
To gain new insights into the mechanisms underlying Foxp3-dependent gene regulation and Treg cell identity, we analyzed chromatin accessibility, TF binding and gene expression patterns in resting and activated subsets of Treg cells, conventional CD4 T cells, and cells expressing a Foxp3GFPKO reporter null allele and leveraged naturally occurring genetic variation in wild-derived inbred Cast/EiJ mice to dissect the cis-regulatory mechanisms driving these features. Interestingly, neither Foxp3 protein nor genetic variation in the Forkhead motif affected accessibility of Foxp3-bound chromatin regions in Treg cells, suggesting that only a small, statistically insignificant subset of these sites is directly regulated by Foxp3. In contrast, we found that TCF1 (encoded by the Tcf7 gene) and the corresponding Sox motif were critical determinants of Foxp3-dependent epigenetic changes. Foxp3 decreased the expression of TCF1, which acted as a major positive regulator of chromatin accessibility in conventional T cells and Foxp3GFPKO cells. Deleting one copy of Tcf7 in Foxp3-deficient Treg cells was sufficient to recapitulate a substantial portion of Foxp3-dependent negative regulation of chromatin accessibility. Our results suggest that Foxp3 defines Treg cell epigenetic identity in a largely indirect manner by fine-tuning the activity of other major TFs, such as TCF1.
RESULTS
Treg cells and Foxp3 prevent autoimmunity in (B6xCast) F1 mice
The wild-derived inbred mouse strain Cast/EiJ (Cast) has roughly 20 million annotated genetic variants relative to the C57BL/6 strain (Keane et al., 2011). This genetic variation can be leveraged to identify TF binding motif requirements for specific chromatin features by comparing the allele-specific effects of motif variation in heterozygous F1 mice (Figure S1) (van der Veeken et al., 2019). We thus sought to utilize the genetic variation in the (B6xCast) F1 genome to interrogate mechanisms underlying Treg cell lineage specification.
To confirm that Treg cell functions and their dependence on Foxp3 were preserved in F1 mice, we assessed the autoimmune sequelae of Foxp3 deficiency in male B6 and (B6xCast) F1 Foxp3GFPKO/Y mice (Figure 1A). Foxp3 deficiency caused lethal autoimmunity in B6 and F1 mice, characterized by splenomegaly and lymphadenopathy, massive T cell activation and expansion, and severe multi-organ inflammation (Figure 1B–G). Thus, the functions of Treg cells and Foxp3 are largely preserved on the F1 genetic background and these mice can be used to assess the genetic requirements for allele-specific Foxp3 binding and chromatin accessibility.
FIGURE 1: Foxp3-dependent suppression of autoimmune inflammation in B6/Cast F1 mice.
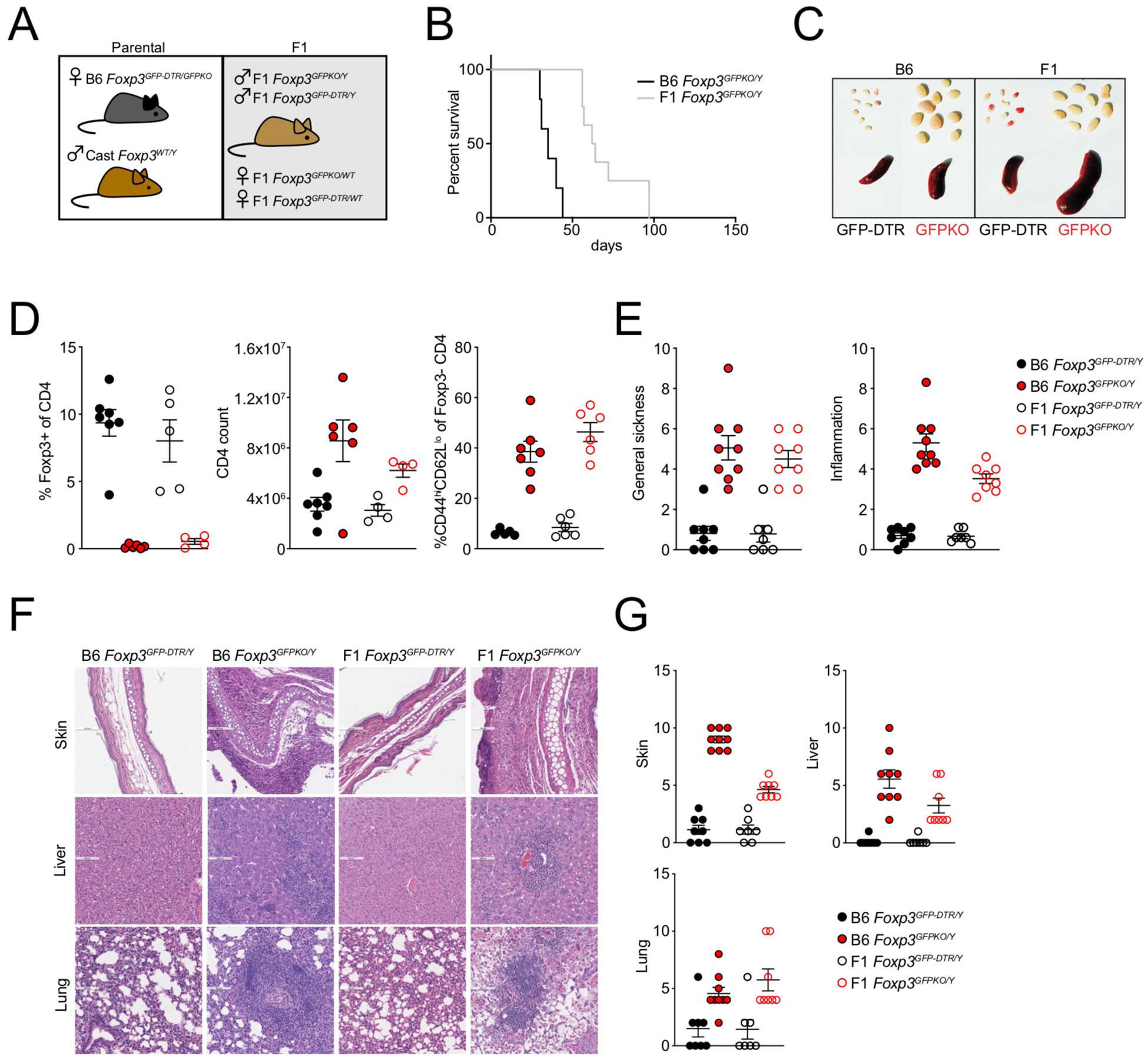
(A): Generation of experimental F1 mice. Female B6 Foxp3DTR-GFP/GFPKO mice were bred to male Cast/EiJ (Cast) mice.
(B): Survival of male Foxp3GFPKO/Y B6 (n=5) and B6/Cast F1 (n=8) mice.
(C): Spleens and lymph nodes of B6 and B6/Cast F1 Foxp3DTR-GFP and Foxp3GFPKO mice.
(D): CD4 T cell composition in lymph nodes of B6 and B6/Cast F1 Foxp3DTR-GFP and Foxp3GFPKO mice determined by flow cytometry. For Figures C-D, mice were analyzed at 3 weeks of age; data points were accumulated over multiple experiments.
(E): Sickness and inflammation scores based on combined histological assessment of skin, liver, and lung from B6 Foxp3DTR-GFP (n=8), B6/Cast F1 Foxp3DTR-GFP (n=7), B6 Foxp3GFPKO (n=9) and B6/Cast F1 Foxp3GFPKO (n=7) mice.
(F): H&E staining of skin, liver, and lung from B6 and B6/Cast F1 Foxp3DTR-GFP and Foxp3GFPKO mice.
(G): Pathology scores of individual tissues.
For E-G, data are derived from accumulated formaldehyde-preserved tissues from multiple experiments with 1–4 mice per group.
DNA sequence motif requirements for Treg cell-specific chromatin accessibility patterns
To better understand the mechanisms defining Treg cell-specific transcriptional and epigenetic identity, we performed ATAC-seq and RNA sequencing (RNA-seq) analysis of resting CD44loCD62Lhi and activated CD44hiCD62Llo GFP+ Treg and GFP− conventional CD4 T (Tcon) cells isolated from (B6xCast) F1 mice (Figure 2A). We identified ~65,000 accessible chromatin regions, including ~28,000 regions undergoing accessibility changes (p < 0.05) in one or more cell state comparisons. RNA-seq analysis revealed ~12,500 differentially expressed genes (p < 0.05) across multiple comparisons (Figure 2B,C).
FIGURE 2: TF binding motif requirements for Treg cell-specific chromatin accessibility.
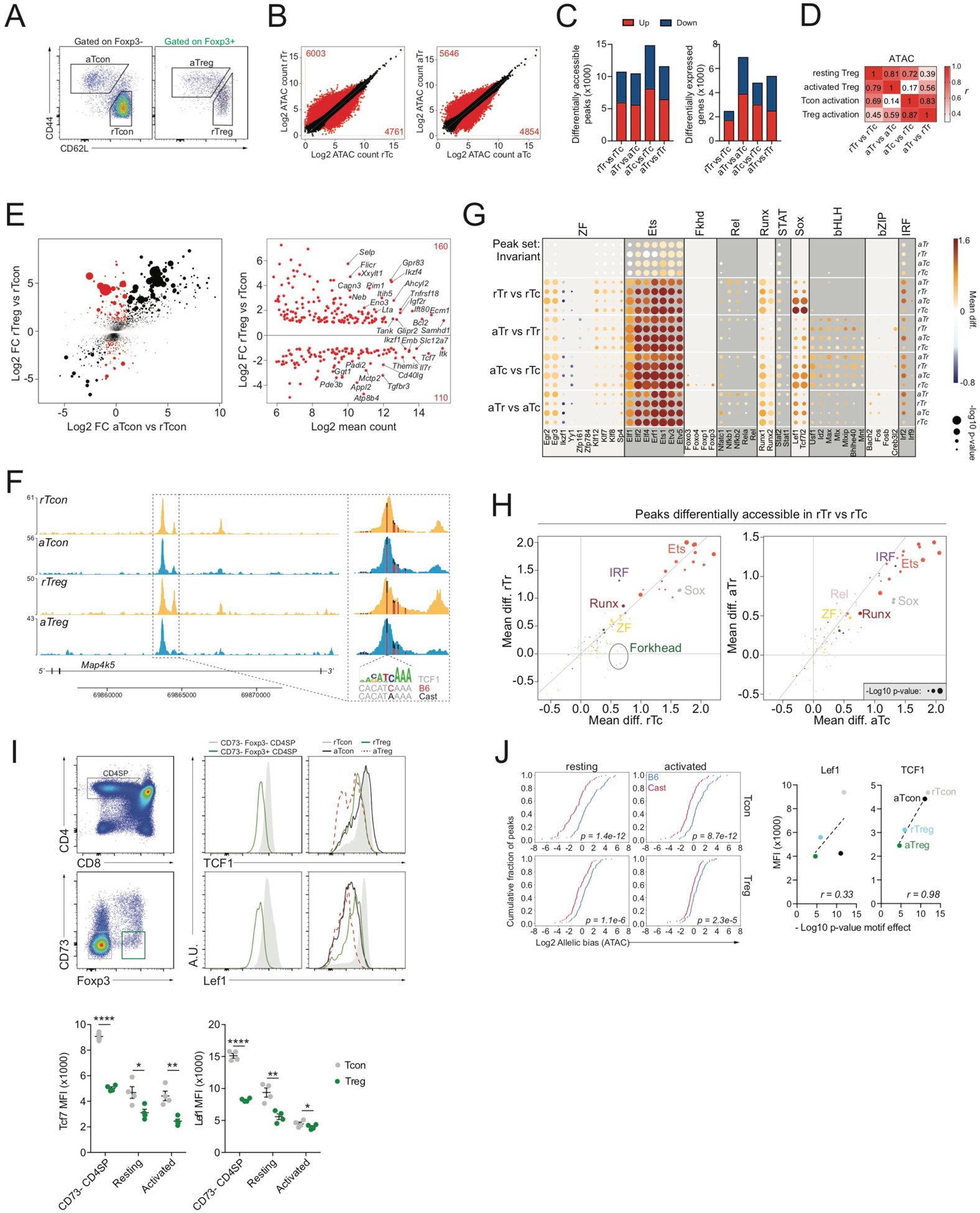
(A): Flow cytometric isolation of resting and activated CD4+ Tcon (Foxp3−) and Treg (Foxp3+) from pooled spleen and peripheral lymph nodes.
(B): ATAC-seq analysis of resting Tcon (rTc), resting Treg (rTr), activated Tcon (aTc), and activated Treg (aTr) cells. Differentially accessible (DA) chromatin regions (p<0.05) are highlighted in red.
(C): DA chromatin regions and gene expression changes across multiple comparisons.
(D): Pearson correlation (r) between chromatin accessibility changes identified across multiple comparisons. Peak sets were identified as follows: resting Treg (DA in rTr vs rTc), activated Treg (DA in aTr vs aTc), Tcon activation (DA in aTc vs rTc), Treg activation (DA in aTr vs rTr).
(E): Gene expression changes in rTreg vs rTcon were compared to gene expression changes in aTcon vs rTcon. Genes undergoing strong and very significant gene expression changes (p<10e-5, log2 FC>1) in rTreg vs rTcon were selected, while excluding genes that underwent similar changes in aTcon vs rTcon (log2 FC<1). The resulting Treg-specific gene signature (270 genes) is highlighted in red. Dots are scaled according to the −log10 p-value in rTreg vs rTcon. The right panel shows the gene expression levels and changes for these signature genes in rTreg vs rTcon.
(F): ATAC-seq tracks from indicated populations. Vertical bars mark the position of genetic variants, with color indicating the ratio of sequencing reads derived from each allele. A TCF7 motif variant is highlighted.
(G): Effect of genetic variation in select TF binding motifs on allelic bias in chromatin accessibility across cell types and peak sets. Mean diff. indicates the difference in mean log2 allelic bias (B6/Cast) for peaks with stronger motif matches on the B6 allele versus peaks with stronger motifs on the Cast allele. Positive and negative values are indicative of a positive or negative effect of the intact motif on chromatin accessibility. Data points are scaled according to the −log10 p-value of a t-test comparing mean log2 allelic bias of peaks with stronger motif matches on the B6 allele to peaks with stronger motifs on the Cast allele (see also Figure S1).
(H): Effect of TF binding motif variation on accessibility of peaks DA in rTr vs rTc (resting Treg signature). Data points are colored according to motif family.
(I): Flow cytometry plots of Lef1 and TCF1 levels in different subpopulations of thymic and peripheral CD4 T cells.
(J): Effect of the TCF7/Lef1 motif on chromatin accessibility of resting Treg signature peaks in different cell types. Correlation between Lef1 and TCF1 protein levels measured by intracellular flow cytometry and p-value of motif effect determined as shown in Figure S1.
Analysis of differentially expressed genes and differentially accessible chromatin regions across four cell state comparisons identified resting Treg (rTreg) (differential in rTreg versus rTcon cells) and activated Treg (aTreg) programs (differential in aTreg versus aTcon), a Tcon activation program (differential in aTcon versus rTcon cells), and a Treg activation program (differential in aTreg versus rTreg cells) (Figure 2B,C). These programs were non-mutually exclusive and correlated across multiple cell state comparisons (Figure 2D, S2A). For example, many genes that were differentially expressed in rTreg versus rTcon cells, such as Ctla4, Il2ra, and Ikzf2, were also differentially expressed in aTcon versus rTcon cells. Only a small subset of genes underwent highly Treg cell-specific transcriptional changes that were not induced in aTcon versus rTcon, including Samhd1, Ikzf4, Gpr83, Tcf7, Il7r, Themis, Cd40lg, and Pde3b (Figure 2E, S2B). Thus, the transcriptome and epigenome of Treg cells consist of a combination of relatively rare Treg cell-specific features and a variation of a common T cell activation program.
To better understand the regulation of Treg cell epigenetic identity, we utilized pervasive natural DNA sequence variation in the F1 genome to find out whether polymorphisms in particular TF binding motifs affected chromatin accessibility patterns. Specifically, we subdivided peaks with sequence variant-containing TF binding motifs into those with stronger motif matches on the B6 (reference) allele and those with stronger motif matches on the Cast (alternative) allele and measured the allelic ratio of ATAC-seq reads for these peaks (Figure 2F). If a particular motif exerts a positive effect on chromatin accessibility by recruiting an activator, then the allele with the stronger motif match should, on average, be more accessible, whereas for a repressor-recruiting motif, the allele with the stronger motif match should be less accessible (Figure S1). A global analysis of the effects of known TF binding motifs on chromatin accessibility across cell states and peak groups revealed that the vast majority of motifs influencing local chromatin states acted as positive regulators (Figure 2G). Motifs with a negative effect on accessibility were rare and included motifs for specific zinc-finger TFs such as Ikzf1 and YY1. The strongest positive regulators of chromatin accessibility were Ets motifs, which were invariably active across all cell types, activation states, and peak groups. In contrast, other motif families, such as Runx, Sox, and basic helix-loop-helix (bHLH), showed cell state- or peak group-specific effects. Importantly, we observed no significant effect of Forkhead motif variation on Treg cell-specific accessible chromatin regions (Figure 2G, H). In contrast, we found that Sox motifs acted as strong positive regulators of rTreg cell chromatin features and were preferentially effective in Tcon cells compared with Treg cells (Figure 2G, H). Of note, the effect of Sox motif variation on Treg cell-specific accessible chromatin regions was strongly correlated with protein levels of one particular Sox motif-binding TF, namely TCF1 (Figure 2I, J). Although decreased expression of TCF1 was largely Treg cell-specific, expression of a closely related TF, Lef1, was also reduced in aTcon versus rTcon cells (Figure 2I). TCF1 and Lef1 expression was decreased early during Treg cell development in CD73− CD4 SP thymocytes (Figure 2I), which have recently undergone TCR rearrangement and still express RAG recombinase (Owen et al., 2019). These observations suggest that Sox motif-binding TFs, and TCF1 in particular, play a major role in shaping Treg cell-specific chromatin accessibility.
Forkhead motif variation affects Foxp3 binding, but not chromatin accessibility
To gain insights into the contribution of Foxp3 to Treg cell epigenetic identity, we analyzed the effects of genetic variation on Foxp3 binding and accessibility of Foxp3-bound chromatin regions. We quantified Foxp3 binding patterns by chromatin immunoprecipitation (ChIP) sequencing and by CUT&RUN in bulk GFP+ Treg cells sorted from Foxp3DTR-GFP/Y (B6xCast) F1 mice (Skene and Henikoff, 2017) (Figure 3A). Approximately 75% of Foxp3-bound sites identified by CUT&RUN were also identified by ChIP-seq, and binding intensity at these overlapping sites was positively correlated across methods (Figure 3B–D). Foxp3-bound sites only identified by one method showed reduced binding intensity compared with sites identified by both methods. Thus, ChIP-seq and CUT&RUN identified a similar set of Foxp3-bound sites.
FIGURE 3: The forkhead motif regulates Foxp3 binding but not accessibility of Foxp3 targets.
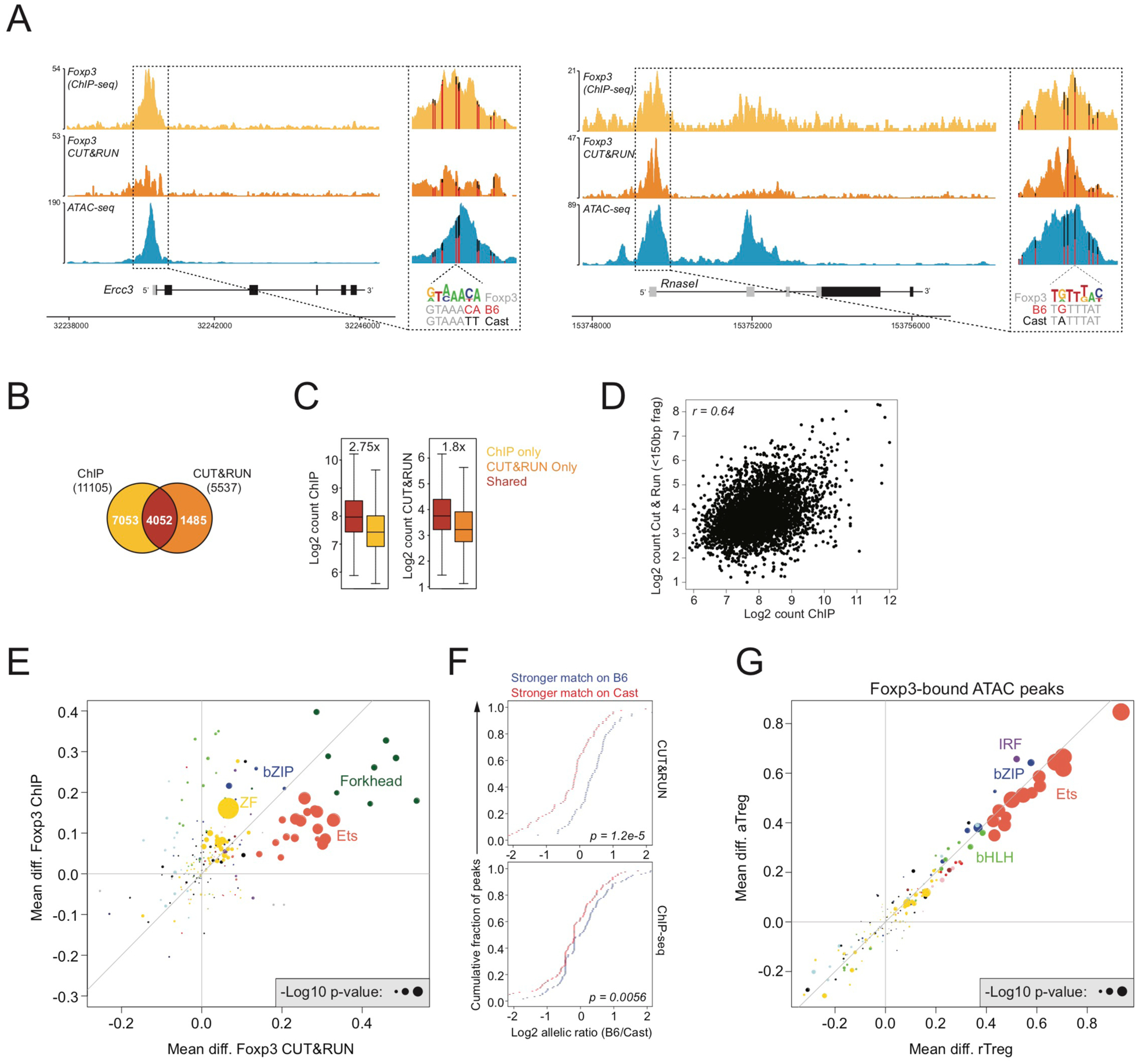
(A): Foxp3 ChIP-seq, Foxp3 CUT&RUN, and ATAC-seq (rTreg) tracks. Vertical bars mark the position of genetic variants, with color indicating the ratio of sequencing reads derived from each allele. A Foxp3 motif variant is highlighted.
(B): Overlap between Foxp3 binding sites identified by ChIP or CUT&RUN in bulk Treg cells from pooled spleen and lymph nodes of Foxp3DTR-GFP B6/Cast F1 mice.
(C): Read counts at Foxp3 binding sites identified by ChIP-seq, CUT&RUN, or by both techniques.
(D): Pearson correlation between Foxp3 binding intensity measured by ChIP and CUT&RUN for sites detected by both techniques.
(E): Effect of genetic variation in TF binding motifs on allelic bias in Foxp3 binding, measured by ChIP or CUT&RUN. See Figure S1.
(F): Effect of Foxp3 motif variation on allelic bias in Foxp3 binding by ChIP (bottom) or CUT&RUN (top).
(G): Effect of genetic variation in TF binding motifs on allelic bias in chromatin accessibility (ATAC-seq) of Foxp3-bound sites.
To characterize the requirements for Foxp3 recruitment, we next analyzed the effect of TF binding motif variation on allelic bias in Foxp3 binding measured by ChIP-seq or CUT&RUN. We identified Forkhead and Ets motifs as the main determinants of Foxp3 binding, with Forkhead motif variation showing the strongest effect (Figure 3E, F). Interestingly, although Ets, IRF, and bZIP motif variation had a strong effect on the accessibility of Foxp3-bound chromatin, Forkhead motif variation did not (Figure 3G). These observations suggest that Ets, IRF, and bZIP TFs create accessible chromatin sites to which Foxp3 can bind. Thus, although the Forkhead motif contributes to recruitment of Foxp3, it does not typically affect chromatin accessibility of Foxp3-bound sites.
Foxp3-dependent epigenetic changes are largely driven by trans-acting intermediates
To further dissect a potential role of Foxp3 in driving gene expression and chromatin accessibility changes, we compared Treg cells and Foxp3GFPKO cells from healthy heterozygous Foxp3DTR-GFP/WT and Foxp3GFPKO/WT (B6xCast) F1 females using ATAC-seq and RNA-seq analysis. To account for potential effects of cellular activation, we isolated “resting” and “activated” cell subsets based on cell surface expression of CD44 and CD62L (Figure 4A). ATAC-seq analysis revealed approximately 3,000 peaks with a more than 2-fold change in chromatin accessibility in resting wild-type (WT) Treg and Foxp3-deficient Treg “wannabe” cells (Figure 4B). In activated cells, Foxp3 deficiency had a much more dramatic effect, with approximately 10,000 peaks showing differential accessibility. Similarly, Foxp3-dependent changes in gene expression were more numerous and more dramatic in the activated than in the resting state (Figure 4C). Interestingly, we found that only a subset of the highly Treg cell-specific transcriptional changes (Figure 2E, S2B) were Foxp3 dependent, including decreased expression of Pde3b, Abhd15, Id2, Tcf7, Themis, and Cd40lg and increased expression of Phlpp1, P2ry10, and Samhd1 (Figure 2E, S2B). Thus, Foxp3 expression induced major changes in the transcriptome and epigenome of F1 Treg cells, including a small number of highly Treg cell-specific changes and many other changes that were less Treg cell specific.
FIGURE 4: Foxp3-dependent chromatin accessibility changes are largely driven by trans-acting intermediates.
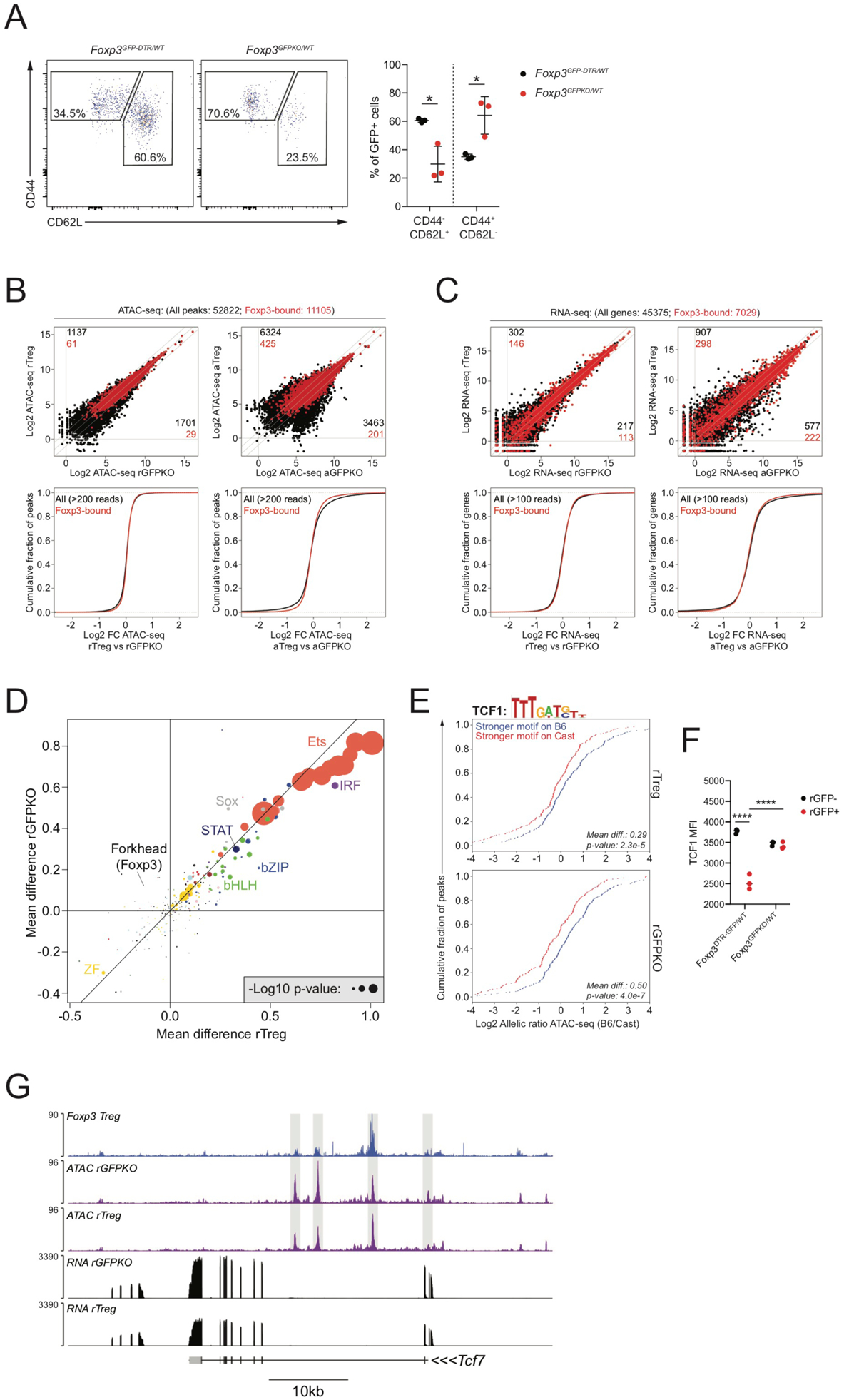
(A): CD44 and CD62L expression on GFP+ cells isolated from Foxp3DTR-GFP/WT and Foxp3GFPKO/WT B6/Cast F1 females.
(B): ATAC-seq analysis of CD44loCD62Lhi Treg (rTreg), CD44hiCD62Llo Treg (aTreg) and reporter-null expressing counterparts (rGFPKO and aGFPKO). Black dots: all ATAC-seq peaks, red dots: Foxp3-bound peaks. Number of all peaks (black) or Foxp3-bound peaks (red) undergoing >2-fold change in chromatin accessibility between cell types is shown. CDF plots (bottom) show quantification of accessibility changes.
(C): Similar to panel B, showing gene expression changes at all genes (black) and Foxp3-bound genes (red).
(D): Effect of TF binding motif variation on allelic bias in chromatin accessibility in rTreg and rGFPKO cells. Dots are colored according to TF family. See Figure S1.
(E): Effect of TCF1 motif variation on chromatin accessibility in rTreg (top) and rGFPKO (bottom) cells. Blue lines indicate peaks with stronger motif matches on the B6 allele, red lines indicate peaks with stronger motif matches on the Cast allele, as described in Figure S1.
(F): TCF1 protein levels in Treg and Foxp3GFPKO cells measured by flow cytometry. (****: p<0.0001; by two-way ANOVA).
(G): Track examples showing Foxp3 binding, chromatin accessibility, and gene expression at the Tcf7 locus.
We next sought to identify the earliest Foxp3-dependent transcriptional changes through analysis of developing thymic Treg cells, reasoning that these early changes may precipitate some of the downstream indirect effects of Foxp3 observed in mature Treg cells. To distinguish recently generated Treg and Foxp3GFPKO cells from their recirculating mature counterparts, we isolated GFP+ cells that were negative for CD73 staining. The effect of Foxp3 deficiency on the transcriptome of thymic CD73−Foxp3+ cells was less pronounced than in mature rTreg cells from peripheral lymphoid organs, with only a handful of genes showing more than 2-fold differential expression, including Pde3b, Tdrp, Prr5l, and Fgl2 (Figure S3C–D). Thus, Foxp3-dependent transcriptional and epigenetic changes accumulate gradually during Treg cell development and maturation.
We next looked specifically at the regulation of Foxp3-bound genes and chromatin regions. Surprisingly, we found that gene expression and chromatin accessibility of the vast majority of Foxp3-bound sites were unaffected by Foxp3 in rTreg or aTreg cells (Figure 4B, C). Accordingly, Foxp3-bound genes as a whole showed no enrichment for gene expression or accessibility changes. Thus, although Foxp3 has a strong, widespread effect on chromatin accessibility and gene expression, these changes occur preferentially at sites that are not bound by Foxp3. Our observations suggest that only a small subset of Foxp3-bound genes is subject to Foxp3-dependent regulation, whereas the majority is unaffected. Thus, the pervasive Foxp3-dependent chromatin accessibility and gene expression changes at non-bound sites must be mediated indirectly through the activity of one or more Foxp3-dependent trans-regulatory factors.
To identify these putative Foxp3-dependent “trans regulators,” we first performed motif enrichment analysis on the chromatin regions that were differentially accessible in Treg cells versus Foxp3GFPKO cells after correcting for their activation status. Chromatin regions that were more accessible in rTreg versus rFoxp3GFPKOcells were enriched for multiple TF-binding motifs, including nuclear factor κB (NF-κB) and bZIP motifs, whereas sites that were less accessible in rTreg versus rFoxp3GFPKO cells were most strongly enriched for Sox family motifs (Figure S4A). Chromatin regions differentially accessible in aTreg versus aFoxp3GFPKO cells were characterized by strong nuclear receptor (NR) motif enrichment at sites with higher accessibility in aFoxp3GFPKO cells, consistent with the increased expression of Rorc and other Th17-associated transcripts in these cells (Figure S4B, C).
We next searched for TF binding motifs whose effect on allele-specific chromatin accessibility was modulated in a Foxp3-dependent manner. Analysis of the effects of TF binding motif variation on chromatin accessibility in rTreg and rFoxp3GFPKOcells indicated that Ets, IRF, STAT, Sox, bZIP, and bHLH motifs acted as positive regulators of chromatin accessibility (Figure 4D). Motifs that differentially affected chromatin accessibility in Treg cells and their Foxp3-deficient counterparts included a Sox motif that showed preferential activity in Foxp3GFPKO cells (Figure 4D, E). Accordingly, we found that Foxp3 bound to the Tcf7 locus and that its expression was associated with a reduction in TCF1 levels (Figure 4F,G, S3A). Thus, our results raise the possibility that TCF1 acts as a trans-acting intermediary contributing to Foxp3-dependent establishment of Treg cell epigenetic identity.
Foxp3-dependent reduction in accessibility of TCF1-bound chromatin
To confirm a potential role of TCF1 in driving Foxp3-dependent chromatin accessibility changes in Treg cells, we next analyzed genome-wide TCF1 binding patterns in resting CD44loCD62Lhi Treg and Foxp3GFPKO cells isolated from healthy heterozygous Foxp3DTR-GFP/WT and Foxp3GFPKO/WT (B6xCast) F1 females, respectively. Using TCF1 CUT&RUN, we identified 7,774 TCF1-bound accessible chromatin regions (Figure 5A, B). TCF1 binding patterns were similar between cell types, with 181 sites showing higher (>2 fold) TCF1 binding patterns were similar between cell types, with 181 sites showing higher (>2 fold) TCF1 binding in Treg cells and 309 sites showing higher binding in Foxp3GFPKO cells. Genetic variation in the TCF1 (Sox) motif had a strong effect on allelic bias in TCF1 binding, suggesting that the presence of intact TCF1 motifs is an essential determinant of TCF1 binding (Figure 5C).
FIGURE 5: Foxp3-dependent repression of chromatin accessibility at TCF1 targets.
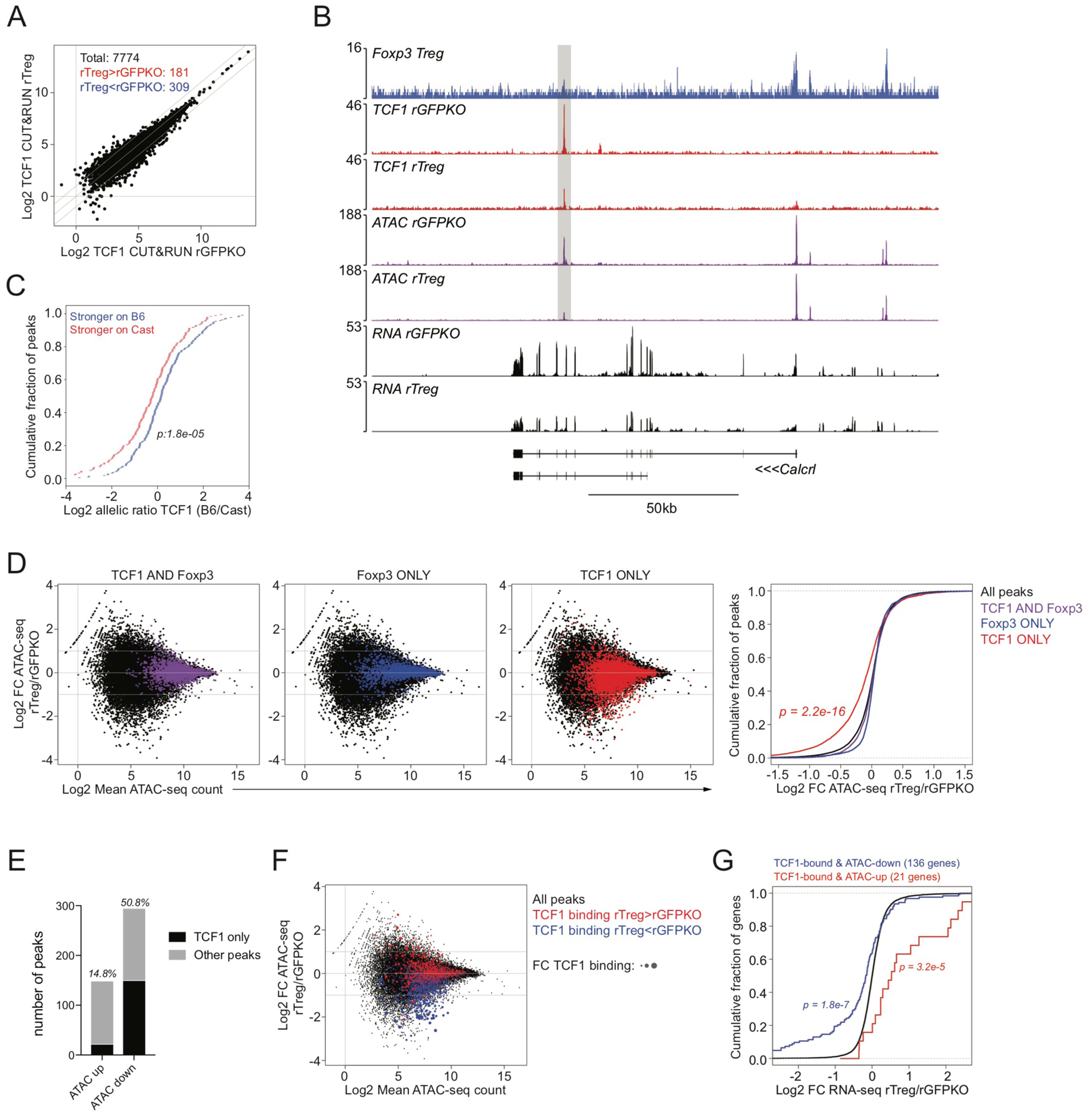
(A): Normalized TCF1 CUT&RUN counts at genomic regions overlapping ATAC-seq peaks in resting CD44loCD62Lhi Treg and resting CD44loCD62Lhi GFPKO cells isolated from Foxp3DTR-GFP/WT or Foxp3GFPKO/WT B6/Cast F1 females, respectively. Total number of TCF1-bound ATAC-seq peaks is shown. Diagonal lines indicate 2-fold difference in binding between cell types.
(B): Track examples of Foxp3 and TCF1 CUT&RUN, ATAC-seq, and RNA-seq at the Calcrl locus. A Foxp3-unbound site with reduced accessibility and reduced TCF1 binding in resting Treg vs GFPKO cells is shown.
(C): Effect of genetic variation in the TCF1 motif on allelic bias in TCF1 CUT&RUN in resting Treg cells. See Figure S1.
(D): Scatter plots showing mean ATAC-seq count and log2 fold change in ATAC-seq counts in rTreg vs rGFPKO cells. Black dots represent all ATAC-seq peaks. Colored dots represent peaks bound by both TCF1 and Foxp3 (left, purple), Foxp3 only (center, blue), or TCF1 only (right, red). Accessibility changes at these peak sets are summarized in the CDF plot (far right). Sites bound by TCF1, but not Foxp3, are significantly less accessible in rTreg cells vs rGFPKO cells (one-sided Kolmogorov-Smirnov test comparing red and black distributions).
(E): TCF1 binding to ATAC-seq peaks with higher (ATAC up) or lower (ATAC down) accessibility (>2 fold) in rTreg vs rGFPKO cells. ATAC-seq peaks with at least 100 reads in one cell type are included in the analysis. TCF1 only denotes peaks with TCF1 binding, but no Foxp3 (red dots in panel D).
(F): Scatter plots showing mean ATAC-seq count and log2 fold change in ATAC-seq counts in resting Treg vs GFPKO cells, similar to (D). Red dots have stronger TCF1 binding in rTreg cells vs rGFPKO cells, blue dots have stronger TCF1 binding in rGFPKO cells vs rTreg cells. Dots are scaled according to the absolute fold change in TCF1 binding. Sites with relatively stronger TCF1 binding in GFPKO cells show preferential Foxp3-dependent loss of chromatin accessibility.
(G): CDF plot showing expression changes for all genes (black) and TCF1-bound genes that lost (blue) or gained (red) chromatin accessibility in rGFPKO vs rTreg cells.
We next analyzed the relationship between TCF1 binding and Foxp3-dependent chromatin accessibility changes. Our earlier findings suggested a model in which Foxp3-dependent inhibition of Sox TF activity causes Treg cell-specific loss of chromatin accessibility. To avoid potentially confounding effects of TCF1 and Foxp3 co-occupancy, we first separated ATAC-seq peaks into those bound by TCF1 and Foxp3 together (TCF1 and Foxp3) or alone (TCF1 only and Foxp3 only, respectively). We found that ATAC-seq peaks bound by TCF1 in the absence of Foxp3 showed reduced accessibility in rTreg cells versus rFoxp3GFPKO cells (Figure 5D). We identified TCF1 binding in the absence of Foxp3 at approximately 50% of sites with lower chromatin accessibility in rTreg versus rFoxp3GFPKO cells, whereas only 15% of sites with higher accessibility showed similar TF occupancy (Figure 5E). Moreover, we found that sites with strongly reduced TCF1 binding in rTreg cells versus rFoxp3GFPKO cells also showed the strongest reduction in chromatin accessibility (Figure 5F). Thus, a moderate Foxp3-dependent reduction in TCF1 protein levels in Treg cells may lead to selectively diminished TCF1 binding at a subset of sites and the associated decrease in chromatin accessibility. Finally, genes closest to these TCF1-bound sites were expressed at lower levels in rTreg cells versus rFoxp3GFPKO cells (Figure 5G). Thus, our observations suggest that Foxp3 reduces the expression of TCF1, a major positive regulator of chromatin accessibility, to drive Treg cell-specific repression of chromatin accessibility and gene expression. This mechanism may account for up to half of the genomic regions undergoing Foxp3-dependent reductions in chromatin accessibility in Treg cells.
Reducing TCF1 levels recapitulates Foxp3-dependent negative regulation of chromatin accessibility in Foxp3GFPKO cells
We sought to provide further experimental support for the above model by testing whether the observed Foxp3-dependent reduction in chromatin accessibility of TCF1-bound sites could be recapitulated simply by lowering TCF1 protein levels in Treg “wannabe” cells lacking Foxp3. For these experiments, we made use of a Foxp3ΔEGFPiCre allele in which a GFP-Cre cassette is inserted into the Foxp3 3′ UTR and the Foxp3 coding sequence is disrupted through introduction of a premature stop codon (Charbonnier et al., 2019). Foxp3ΔEGFPiCre mice thus enable Cre-mediated recombination specifically in Foxp3 reporter null-expressing cells. To rescue Foxp3-dependent repression of TCF1 levels, we generated female Foxp3ΔEGFPiCre/WTTcf7FL/WT mice. Foxp3 reporter null-expressing cells from these animals indeed showed reduced TCF1 protein levels relative to Foxp3ΔEGFPiCre/wtTcf7WT/WT controls (Figure 6A). Importantly, deleting one copy of Tcf7 was not sufficient to rescue the autoimmune phenotype of hemizygous Foxp3ΔEGFPiCre/Y reporter null male mice, arguing that down-modulation of TCF1 is not the sole requirement for Treg cell function (data not shown). We also found that Tcf7FL/WT and Tcf7WT/WT reporter null-expressing cells did not exhibit strong differential expression of Foxp3-dependent genes (Figure S5A–C). However, certain Treg cell-specific transcriptional features, such as reduced expression of Themis and Cd40lg and increased expression of Dusp4 and Axl, could be recapitulated by loss of a single copy of the Tcf7 gene.
FIGURE 6: Downregulation of TCF1 rescues Foxp3-dependent repression of chromatin accessibility in the absence of Foxp3.
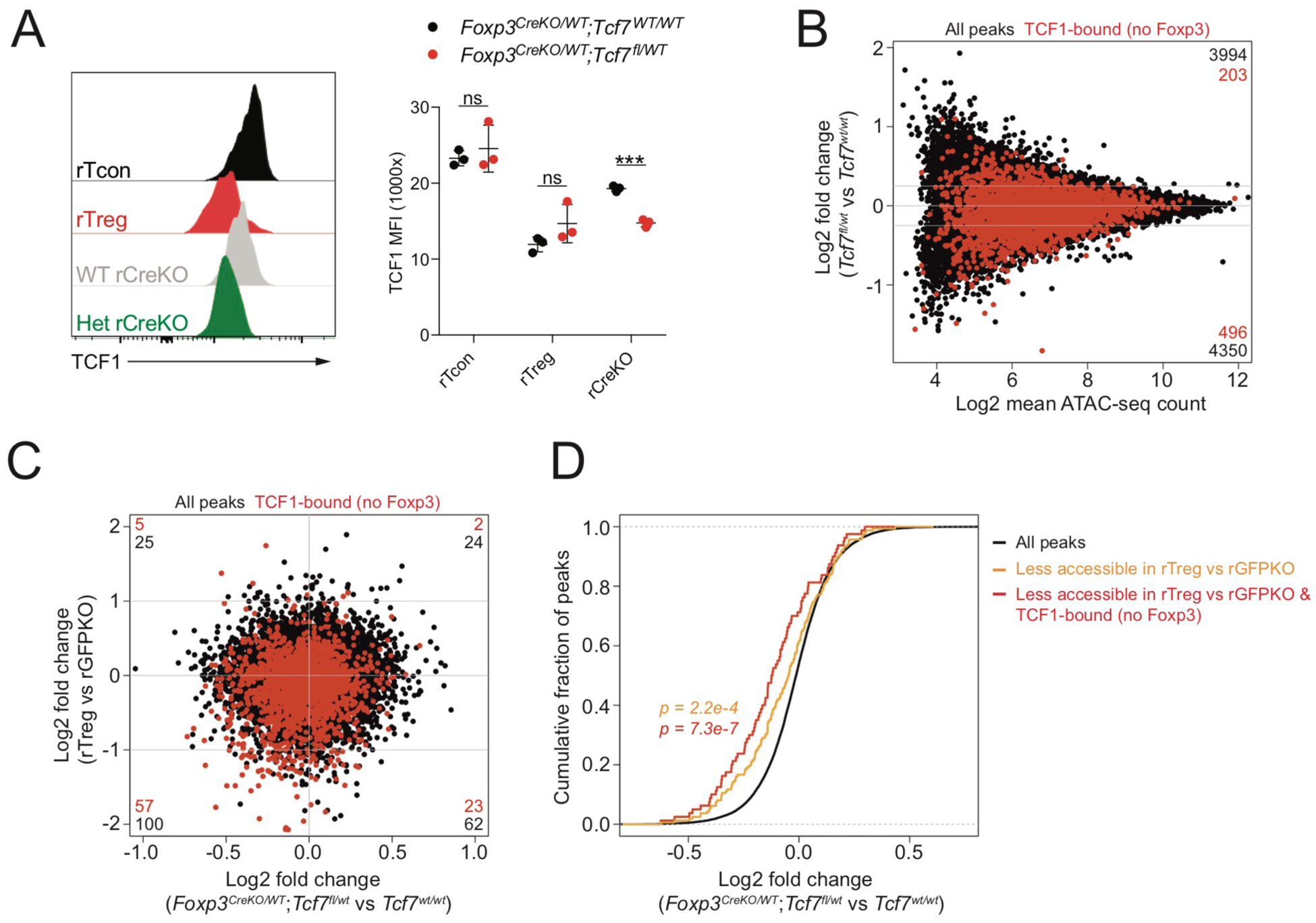
(A): TCF1 protein levels in indicated populations of CD44loCD62Lhi CD4 T cells from pooled spleen and lymph nodes of female Foxp3ΔEGFPiCre/WTTcf7WT/WT or Foxp3ΔEGFPiCre/WTTcf7fl/WT mice. P-values from multiple t-tests (***: p<0.001).
(B): ATAC-seq analysis of WT and Tcf7 heterozygous rGFPKO cells. Peaks bound by TCF1, but not by Foxp3, are highlighted in red. Number of peaks in each section of the plot is indicated.
(C): Fold change – fold change plot showing chromatin accessibility in rTreg vs rGFPKO cells compared to WT vs Tcf7 heterozygous rGFPKO cells.
(D): TCF1-dependent changes in chromatin accessibility at all peaks (black), all peaks less accessible in WT rTreg vs rGFPKO cells, and TCF1-bound peaks less accessible in WT rTreg vs rGFPKO cells. P-value from one-sided Kolmogorov-Smirnov test comparing red or orange distributions to the black distribution.
To determine the effect of reduced TCF1 protein levels on chromatin accessibility, we performed ATAC-seq analysis on WT and Tcf7 heterozygous resting CD44loCD62Lhi Foxp3ΔEGFPiCre-expressing cells. We found that reduced TCF1 protein levels were associated with a moderate reduction in chromatin accessibility of previously identified TCF1-bound sites (Figure 6B). Importantly, we found that chromatin regions that underwent Foxp3-dependent negative regulation in WT Treg cells versus Foxp3GFPKO reporter null cells were also less accessible in Tcf7heterozygous versus Tcf7 WT Foxp3ΔEGFPiCre cells and that this effect was particularly evident at sites that were bound by TCF1 but not by Foxp3 (Figure 6C,D). This observation argues that reduced TCF1 protein levels can, to a substantial degree, account for Foxp3-dependent negative regulation of chromatin accessibility in Treg cells.
Integration of positive and negative Foxp3-dependent trans-regulatory modules
The observation that Tcf7 heterozygosity could cause Treg cell-specific chromatin accessibility changes but was not sufficient to rescue Treg cell function or most of the Treg cell-specific gene expression program suggested that the activity of additional TFs may intersect with TCF1-dependent regulation. We first considered the possibility that TCF1 and the closely related TF Lef1 acted in a partially redundant manner to control the accessibility of shared targets. Functional redundancy between these two TFs is well documented (Okamura et al., 1998). Consistently, CUT&RUN analysis revealed that ~90% of TCF1 targets were also bound by Lef1 (Figure S6A, B). Moreover, we found that expression of Lef1 was also reduced in rTreg cells relative to rTcon or rFoxp3GFPKO cells (Figure 2I, S3A), and although reducing TCF1 levels in Foxp3ΔEGFPiCre/WTTcf7FL/WT mice was sufficient to mimic Foxp3-dependent chromatin accessibility changes at TCF1-bound sites, the magnitude of these changes was typically lower in Tcf7FL/WT versus Tcf7WT/WT than in rTreg versus rFoxp3GFPKO cells (Figure 6C). These observations are consistent with the notion that TCF1 and Lef1 may act in a partially redundant or dose-dependent manner.
Additionally, it was possible that lower TCF1 and Lef1 activity in Treg cells could, in some cases, be compensated for by increased activity of other TFs. Indeed, we found that bZIP and AP1-IRF composite (Batf) motifs were enriched in chromatin regions with higher accessibility in rTreg versus rFoxp3GFPKO cells (Figure S3A) and that genetic variation in bZIP and IRF family motifs had a substantial effect on chromatin accessibility in rTreg cells (Figure 4D). To explore a potential interaction between binding of bZIP and IRF family TFs and TCF1, we performed CUT&RUN for the bZIP and IRF family members c-Jun and IRF4 in rTreg cells (Figure S6A). We identified ~4,500 c-Jun-bound sites and a relatively small number (~800) of IRF4-bound sites. Although sites bound by TCF1 and Lef1 had lower chromatin accessibility in rTreg versus rFoxp3GFPKO cells, this association was only evident at regions not bound by c-Jun or IRF4 (Figure S6C–E). Conversely, sites bound by c-Jun and IRF4, but not TCF1 or Lef1, were more accessible in rTreg versus rFoxp3GFPKO cells whereas those co-bound by TCF1 and Lef1 were not. Thus, non-overlapping TCF1/Lef1- and c-Jun/IRF4-bound sites exhibited Foxp3-dependent decreases and increases in chromatin accessibility, respectively, whereas sites co-occupied by TCF1/Lef1 and c-Jun/IRF4 underwent minimal net changes.
Together, our results suggest that Foxp3 shapes epigenetic features of Treg cells by modulating the activity of multiple trans-regulatory factors with partially overlapping targets.
DISCUSSION
Despite numerous previous studies, the mechanisms through which Foxp3 confers Treg cell epigenetic identity have remained obscure. Here we characterized the effect of naturally occurring cis-regulatory variation in mice on TF binding, chromatin accessibility, and gene expression in Treg cells, Tcon cells, and Foxp3GFPKO-expressing cells to uncover novel mechanisms of Foxp3-dependent epigenetic regulation.
Allelic biases in chromatin accessibility and gene expression are very common in (B6xCast) F1 mice, affecting thousands of genes and regulatory elements (van der Veeken et al., 2019). Most genetic variants are associated with only modest allelic biases rather than complete mono-allelic activity. It has been reported previously that “suboptimization,” or mutational fine-tuning of suboptimal TF recognition motifs may be required to ensure an appropriate balance between enhancer strength and tissue specificity (Farley et al., 2015a; Farley et al., 2015b). Thus, the subtle changes in TF binding strength observed between mouse strains may be highly consequential and evolutionarily relevant.
By analyzing the effects of TF binding site polymorphisms on allele-specific Foxp3 occupancy patterns in (B6xCast) F1 animals, we identified critical sequence requirements for Foxp3 recruitment. Foxp3 binding was strongly modulated by the presence of intact Forkhead motifs, consistent with the observation that mutations disrupting the Forkhead domain of Foxp3 cause autoimmunity (Bennett et al., 2001; Brunkow et al., 2001; Chatila et al., 2000). In contrast, chromatin accessibility of Foxp3-bound sites was largely independent of the Forkhead motif and, instead, was strongly dependent on the presence of Ets and bZIP motifs, which also contributed to Foxp3 binding. Thus, Ets and bZIP family TFs seem to drive the accessibility of most of the Forkhead motif-containing regulatory elements to which Foxp3 binds. Although Foxp3 has been shown to interact with a large number of sequence-specific TFs, including NFAT, Runx1, GATA3, RelA, Ikzf1–Ikzf4, and many others (Kwon et al., 2017; Rudra et al., 2012), DNA binding motifs for these factors did not typically affect Foxp3 localization or the accessibility of Foxp3-bound sites. It is possible that these factors contribute to Foxp3 function at only a small number of sites or contribute in a manner that is independent of their direct DNA-binding activity. It is also conceivable that sequence motif requirements for the binding of these factors may be less stringent because of cooperative effects of other complex components.
Strikingly, we found that the vast majority of Foxp3-bound sites were unaffected by Foxp3 in terms of chromatin accessibility and gene expression. Although Foxp3 expression caused widespread transcriptional and epigenetic changes, these mostly occurred at sites that were not directly bound by Foxp3. These observations suggest that only a small fraction of Foxp3-bound sites is actually affected by its presence and that these few direct Foxp3 targets drive significant downstream changes. What exactly distinguishes the few genes that are affected by Foxp3 binding from the many that are not is still unclear. Because Foxp3 forms large and heterogeneous multi-protein complexes, it is possible that the presence of a unique and complex combination of cofactors enables Foxp3 to act only in rare instances (Kwon et al., 2017; Rudra et al., 2012). Alternatively, it may be that the absence of other TFs, whose activity may be dominant, enables Foxp3 to act preferentially at low-complexity cis-regulatory elements. Given the limited number of sites subject to direct regulation, this question cannot easily be addressed statistically through motif enrichment analysis and will require a case-by-case investigation of candidates.
It is important to note that, in addition to a small number of bona fide Treg cell-specific features, many of the gene expression changes typically associated with Treg cell identity and suppressive function, such as increased expression of CD25, CTLA4, and IL-10 and reduced expression of IL-2 can also be induced in a Foxp3-independent manner in Tcon cells (Arvey et al., 2014). Thus, although increased or more stable expression of these molecules is associated with Treg cells, they do not define the lineage per se. This observation suggests that much of the Treg cell epigenetic program can, in principle, be driven by common TFs that are present in all T cells. We propose that modulation of the amount or activity of such factors by Foxp3 is a major mechanism of Treg cell lineage specification. Our work identifies the TF TCF1 as one of the key trans-acting intermediaries driving Foxp3-dependent epigenetic programming. TCF1 has a critical role in early T cell development and drives acquisition of T cell-specific accessible chromatin regions (Johnson et al., 2018; Verbeek et al., 1995; Weber et al., 2011). We found that Foxp3 bound to the Tcf7locus and that Foxp3 expression was associated with decreased TCF1 mRNA and protein levels. Using allele-specific analysis of chromatin accessibility in (B6xCast) F1 mice, we found that intact Sox motifs acted as major positive regulators of chromatin accessibility. The effect of Sox motif variants was reduced markedly in Treg cells relative to Tcon or Foxp3GFPKO cells, consistent with a pronounced Foxp3-dependent reduction in the accessibility of TCF1-bound chromatin regions. In the absence of Foxp3, lowering TCF1 levels by genetic deletion of one Tcf7 allele in Foxp3 reporter null cells reduced chromatin accessibility in a pattern similar to the Foxp3-induced accessibility changes observed in WT Treg cells. Thus, we propose that Foxp3 represses TCF1 expression to drive a reduction of chromatin accessibility in Treg cells. Importantly, reduced expression of TCF1 and the related TF Lef1 are observed in Treg cells in mice and humans (Arvey et al., 2015).
Several recent studies aimed to characterize the role of TCF1 and Lef1 as well as upstream components of the Wnt signaling pathway in Treg cells. They found that Treg cell-specific deletion of Tcf7 and Lef1 causes spontaneous autoimmunity (Xing et al., 2019; Yang et al., 2019). Conversely, increasing TCF1 transcriptional activity by stabilizing β-catenin in Treg cells also causes inflammation and impairs suppressive function (Keerthivasan et al., 2014; van Loosdregt et al., 2013). Thus, it is likely that TCF1 and Lef1 activity must be maintained in a certain range to allow proper Treg cell functionality.
Our study first and foremost supports the notion that Foxp3 regulates Treg cell transcriptional and epigenetic identity in a largely indirect manner. Although we characterized a key role of TCF1 in this process, it is likely that additional intermediary factors contribute to Foxp3-dependent regulation. Indeed, although reducing TCF1 levels did partially restore a Foxp3-dependent epigenetic program in Foxp3 reporter null-expressing cells, this was not sufficient to rescue the disease and massive immune cell activation resulting from Foxp3 deficiency in hemizygous Foxp3ΔEGFPiCre/Y mice. Thus, it is likely that TCF1 and other Foxp3-regulated genes jointly establish Treg cell identity and functionality. Our allele-specific analysis identified motifs for several TF families, including bZIP and IRF, that were associated with a Foxp3-dependent increase in chromatin accessibility. We also found that c-Jun- and IRF4-bound sites not overlapping with TCF1, Lef1, or Foxp3 binding were more accessible in Treg cells versus Foxp3GFPKO cells. Because Treg cells express numerous bZIP and IRF family TFs as well as upstream signaling molecules that converge on these pathways, the exact mechanisms driving Foxp3-dependent regulation of bZIP and IRF targets still remain to be determined. It is likely that these and other Foxp3-dependent gene-regulatory modules operate together with the TCF1-dependent module to control distinct aspects of Treg cell biology. Addressing this hypothesis will require combinatorial tuning of multiple Foxp3-dependent genes in immediate precursor cells lacking Foxp3 expression. We propose that the early Foxp3-dependent genes identified in our RNA-seq analysis of thymic CD73− Treg and reporter null cells are most likely to have a major role in driving establishment of Treg cell identity.
Our work demonstrates that Foxp3 imparts pervasive epigenomic and transcriptional changes in Treg cells in a largely indirect manner, using TCF1 as one of several critical intermediaries.
Limitations of study:
Although the Foxp3-dependent reduction in TCF1 protein and transcript levels occurred early after Foxp3 induction and coincided with binding of Foxp3 to regulatory elements within the Tcf7 locus, our study does not formally demonstrate that the former is a direct consequence of the latter. Such proof would require mutating multiple putative Foxp3 binding motifs within several regulatory elements surrounding the Tcf7 locus in Treg cells and Foxp3GFPKO cells. Although direct repression of Tcf7 by Foxp3 would explain our results, it is equally possible that Tcf7 is repressed indirectly by another Foxp3-dependent factor. The distinguishing features of the few genes subject to direct Foxp3-dependent regulation remain unknown.
Although deletion of one copy of Tcf7 in cells was sufficient to recapitulate a considerable fraction of Foxp3-dependent chromatin accessibility changes observed in Treg versus Foxp3-reporter null cells, this was not sufficient to rescue most Foxp3-dependent gene expression changes or suppressive function. Our results suggest that generation of a Treg cell mimic in the absence of Foxp3 would likely require modulation of Tcf7 and Lef1, which may act in a partially redundant manner, together with other early Foxp3-dependent intermediaries identified in our study.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alexander Rudensky (rudenska@mskcc.org)
Materials Availability
This study did not generate new unique reagents
Data and code availability
All next generation sequencing data generated in this paper were deposited in Gene Expression Omnibus (GEO) under SuperSeries accession number GSE154680. Code is available from the authors on request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Animals were housed at the Memorial Sloan Kettering Cancer Center (MSKCC) animal facility under specific pathogen free (SPF) conditions on a 12-hour light/dark cycle under ambient conditions with free access to food and water. All studies were performed under protocol 08-10-023 and approved by the MSKCC Institutional Animal Care and Use Committee. Mice used in this study had no previous history of experimentation or exposure to drugs. Male Cast/Eij mice were purchased from Jackson Laboratory and bred to female Foxp3DTR-GFP/GFPKO mice to generate experimental F1 mice. Adult mice (>6 weeks old) were used for experiments unless otherwise indicated. Tcf7-floxed mice were generated by breeding Tcf7GFP-flox mice (Jackson Laboratory: 030909) to a Flp-recombinase expressing strain. Generation of Foxp3DTR-GFP, Foxp3GFPKO, and Foxp3ΔEGFPiCre mice was previously described (Charbonnier et al., 2019; Gavin et al., 2007; Kim et al., 2007).
METHOD DETAILS
Cell staining and flow cytometry
Single cell suspensions of spleens and pooled peripheral lymph nodes were prepared in ice-cold cell isolation buffer (PBS with 2mM EDTA and 1% FCS) and subject to red blood cell lysis using ACK buffer (150mM NH4Cl, 10mM KHCO3, 0.1mM Na2EDTA, pH7.3). Cells were washed in PBS and stained with GhostDye stain to mark dead cells.
Cell surface antigens were stained at 4 degrees for 15 minutes using a mixture of fluorophore-conjugated antibodies. For transcription factor staining, cells were fixed and permeabilized using the eBioscience Foxp3/Transcription Factor Staining Buffer Set and stained according to manufacturer protocol. Cells were washed and passed through a 100μm nylon mesh before analysis on a BD LSR II flow cytometer. For cell sorting experiments, single cell suspensions were enriched for CD4 T cells using the Dynabeads FlowComp mouse CD4 kit and stained as described above. Cell populations were sorted on a BD Aria II flow cytometer. Post-sort purity was routinely analyzed and typically higher than 95%.
Histology and pathology scoring
Ear skin, liver, pancreas, stomach, kidney, and lung samples from B6 Foxp3DTR-GFP (n=8), (B6xCast) F1 Foxp3DTR-GFP (n=7), B6 Foxp3GFPKO (n=9) and (B6xCast) F1 Foxp3GFPKO (n=7) mice were isolated and preserved in 10% neutral buffered formalin. Histology was performed by HistoWiz Inc. (histowiz.com) using a Standard Operating Procedure and fully automated workflow. Samples were processed, embedded in paraffin and sectioned at 4μm. Hematoxylin and Eosin staining were performed on Tissue-Tek automated slide stainer (Sakura) using HistoWiz’s standard protocol. After staining, sections were dehydrated and film coverslipped using a TissueTek-Prisma and Coverslipper (Sakura). Whole slide scanning (40x) was performed on an Aperio AT2 (Leica Biosystems).
Overall Inflammation and severity of sickness was evaluated by a board-certified pathologist. The pathologist was blinded to mouse strain(s) and experimental conditions. A 6-point scale was established with possible scores of 0, 2, 4, 6, 8, and 10. Overall inflammatory score was calculated as the following:
Overall sickness score was calculated as the following:
RNA-seq library preparation and sequencing
Cell populations were double sorted straight into 1ml Trizol reagent (Thermo Fisher: 15596018). After addition of 200 μL chloroform, RNA was precipitated from the aqueous phase by addition of isopropanol and linear acrylamide. RNA was washed with 75% ethanol and resuspended in RNase-free water. RNA extraction from low-input samples was performed using the miRNAeasy Micro Kit (Qiagen: 217084) on the QIAcube Connect (Qiagen) according to manufacturer’s protocol. After RiboGreen quantification and quality control by Agilent BioAnalyzer, 0.4–2.0ng total RNA was amplified using the SMART-Seq v4 Ultra Low Input RNA Kit (Clontech: 63488), with 12 cycles of amplification. Subsequently, 1.5–15ng of amplified cDNA was used to prepare libraries with the KAPA Hyper Prep Kit (Kapa Biosystems KK8504), using 8 cycles of PCR. Samples were barcoded and run on a HiSeq 4000 or HiSeq 2500 in either a 50bp/50bp end run, using the HiSeq 3000/4000 SBS Kit (HiSeq 4000) or HiSeq Rapid SBS Kit v2 (HiSseq 2500) (Illumina).
ATAC-seq library preparation and sequencing
ATAC-seq libraries were prepared as described (Buenrostro et al., 2013). 5×104 cells were washed in ice cold PBS and lysed using ATAC-seq lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). Following lysis, cells were incubated in 1x transposition buffer (Nextera TD buffer) containing 2.5 μL Nextera Tn5 transposase for 30 min at 37 degrees. Transposed DNA fragments were isolated using QIAGEN MinElute Reaction Cleanup kit and amplified using barcoded primers with Illumina adaptor sequences. The number of cycles used for library amplification was determined using a quantitative PCR side-reaction. Amplified libraries were purified and size selected using Ampure XP beads using consecutive purifications with bead-to-sample ratios of 0.5 and 1.8. After PicoGreen quantification and quality control by Agilent BioAnalyzer, libraries were run on a HiSeq 4000 in a 50bp/50bp or 100bp/100bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina). The loading concentration was 2nM and a 5% spike-in of PhiX was added to the run to increase diversity and for quality control purposes.
ChIP-seq library preparation and sequencing
For Foxp3 ChIP, Treg and naïve conventional CD4 T cells were isolated from pooled spleen and lymph nodes of male Foxp3DTR-GFP (B6xCast) F1 mice. Cells were fixed in 1% paraformaldehyde for 5 min, fixation was quenched by addition of glycine and cell pellets were stored at −80 degrees. Chromatin was prepared using the Covaris TruChIP chromatin shearing kit and sheared for 4 minutes on a Covaris E220 focused ultrasonicator. Chromatin was incubated overnight at 4 degrees with a polyclonal rabbit antibody against Foxp3 (prepared in house). On the following day, immune complexes were precipitated using protein A Dynabeads for 1.5 hours at 4 degrees. Beads were washed twice in nuclear lysis buffer containing 0.1% SDS, twice in high salt nuclear lysis buffer containing 0.1% SDS and 500mM NaCl, twice in ChIP wash buffer (20mM Tris-HCl pH 8, 140 mM NaCl, 1mM EDTA, 250mM LiCl, 0.5% Na-deoxycholate, 0.5% NP40), and twice in 1X TE buffer. Enriched chromatin fragments were eluted by incubation in elution buffer (0.1M NaHCO3, 1% SDS) for 15 min at 37 degrees in two sequential elution steps. 50μL decrosslinking solution (0.2M NaCl, 0.1M EDTA, 0.4M Tris-HCl pH7.5) and 10μL of proteinase K (10mg/mL) were added and samples were decrosslinked overnight at 65 degrees. DNA was isolated using QIAGEN PCR purification Kit. Immunoprecipitated DNA was quantified by PicoGreen and the size was evaluated on a High Sensitivity BioAnalyzer chip. When possible, fragments between 100 and 600 bp were size selected using AMPure XP beads. Illumina libraries were prepared using the KAPA HTP Library preparation kit (Kapa Biosystems KK8234) according to the manufacturer’s instructions with 8 or 12 cycles of PCR. Barcoded libraries were run on the Hiseq 2500 in high output mode or HiSeq 4000 in a 50bp/50bp paired end run, using the TruSeq SBS Kit v4 (HiSeq 2500) or HiSeq 3000/4000 SBS Kit (HiSeq 4000) (Illumina).
Preparation of protein A/G-MNase fusion protein
Plasmid expressing pAG-MNase was a gift from Steven Henikoff (Addgene plasmid #123461; http://n2t.net/addgene:123461 ; RRID:Addgene_123461) (Meers et al., 2019). Plasmid was transformed into BL21-CodonPlus (DE3)-RIPL competent cells (Agilent: #230280). A single antibiotic resistant colony was cultured in LB broth supplemented with 50 μg/ml Kanamycin at 37°C. Protein expression was induced by addition of 0.5 mM Isopropyl β-D-1-thiogalactopyranoside at O.D. 0.6 followed by culturing the cells at 30°C for an additional 16 hours. Cells were pelleted and resuspended in Lysis Buffer containing 10 mM Tris-HCl pH 7.5, 300 mM NaCl, 10 mM Imidazole, 5 mM beta-mercaptoethanol, and EDTA-free protease inhibitor (SigmaAldrich #11836170001), and lysed by sonication with a Branson S250D sonifier, output level 3, 5 s intervals for ~30 rounds until no longer viscous. The lysate was cleared by centrifugation at 12,000 RPM for 20 minutes and the supernatant was incubated with HisPur Ni-NTA Resin (Thermo Fisher: #88221) at 4°C for 1 hour. Protein-bound resin was washed twice with five bed volumes of 10 mM Tris-HCl pH 7.5, 300 mM NaCl, 20 mM Imidazole, 0.03% ZWITTERGENT 3–10 Detergent (EMD Millipore cat. #693021) supplemented with EDTA-free protease inhibitor. Protein was eluted twice with 10 mM Tris-HCl pH 7.5, 300 mM NaCl, 250 mM Imidazole supplemented with EDTA-free protease inhibitor, and imidazole was removed by concentrating the eluate using a 10 kDa centrifugal filter unit (Millipore) followed by 10-fold dilution with PBS for three times. Glycerol was then added to a final concentration of 50% for storage.
CUT&RUN library preparation and sequencing
CUT&RUN libraries were prepared as described by Skene et al with the modifications described below (Skene and Henikoff, 2017). Because Concanavalin-A (ConA) is a well-known T cell mitogen, we avoided the use of ConA-coated beads for cell isolation and handling. 100–500k cells per replicate were collected in a V-bottom 96 well plate by centrifugation and washed in antibody buffer (buffer 1 (1x permeabilization buffer from eBioscience Foxp3/Transcription Factor Staining Buffer Set diluted in nuclease free water, 1X EDTA-free protease inhibitors, 0.5mM spermidine) containing 2mM EDTA). Cells were incubated with antibodies (1:200 dilution) for 1h on ice. After 2 washes in buffer 1, cells were incubated with pA/G-MNase at 1:4000 dilution in buffer 1 for 1h at 4 degrees. Cells were washed twice in buffer 2 (0.05% (w/v) saponin, 1X EDTA-free protease inhibitors, 0.5mM spermidine in PBS) and resuspended in calcium buffer (buffer 2 containing 2mM CaCl2) to activate MNase. Following a 30 minute incubation on ice, 2x stop solution (20mM EDTA, 4mM EGTA in buffer 2) was added and cells were incubated for 10 minutes in a 37 degree incubator to release cleaved chromatin fragments. Supernatants were collected by centrifugation and DNA was extracted using a Qiagen MinElute kit.
CUT&RUN libraries were prepared using the Kapa Hyper Prep Kit (Kapa Biosystems KK8504) and Kapa UDI Adapter Kit (Kapa Biosystems KK8727) according to manufacturers protocol with the modifications described below. A-tailing temperature was reduced to 50 degrees to avoid melting of short DNA fragments and reaction time was increased to 1h to compensate for reduced enzyme activity as described by Liu et al (Liu et al., 2018). Following the adapter ligation step, 3 consecutive rounds of Ampure purification were performed using a 1.4x bead to sample ratio to remove excess unligated adapters while retaining short adapter-ligated fragments. Libraries were amplified for an average of 15 cycles using a 10 second 60 °C annealing/extension step to enrich for shorter library fragments. Following amplification, libraries were purified using 3 consecutive rounds of Ampure purification with a 1.2x bead to sample ratio to remove amplified primer dimers while retaining short library fragments. A 0.5x Ampure purification step was included to remove large fragments prior to sequencing. After PicoGreen quantification and quality control by Agilent BioAnalyzer, libraries were run on a HiSeq 4000 in a 50bp/50bp or 100bp/100bp paired end run, using the HiSeq 3000/4000 SBS Kit (Illumina). The loading concentration was 2nM and a 5% spike-in of PhiX was added to the run to increase diversity and for quality control purposes.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mapping of allele-specific reads
Allele-specific alignment of sequencing reads was performed as previously described (van der Veeken et al., 2019). To ensure unbiased mapping of sequencing reads from F1 mice, we applied a diploid genome alignment strategy (Chen et al., 2016; Crowley et al., 2015; Huang et al., 2014). Briefly, the genetic variants of Cast mice were obtained from the mouse genome project (Keane et al., 2011). A pseudo-Cast genome was constructed by modifying the B6 reference genome with SNPs, insertions and deletions from the wild-derived inbred strain. The sequencing reads were aligned to the B6 and pseudo Cast genome in parallel. After converting the genomic coordinates of pseudo-genome-aligned reads back to the corresponding B6 coordinates, the allelic origins of the reads were determined by comparing the mapping scores of the two alignments. For each read, the alignment with the higher score was retained. In cases where the diploid genome alignment produced identical scores for both genomes, one of the alignments was selected randomly. The final BAM files produced using this strategy contain both variant-containing reads that were aligned in an allele-specific manner, and non-variant-containing reads that aligned equally well to both genomes. Reads aligned to multiple loci were removed from further analysis.
RNA-seq analysis
Paired-end RNA-seq reads were mapped to the diploid genome using STAR (Dobin et al., 2013) with the following parameter settings: $STAR–runMode alignReads–readFilesCommand zcat–outSAMtype BAM–outBAMcompression 6–outFilterMultimapNmax 1–outFilterMatchNmin 30–alignIntronMin 20–alignIntronMax 20000–alignEndsType Local. Allele-specific reads were counted as described above. Gene annotations were downloaded from Ensembl release 83, which is based on mouse genome assembly GRCm38. DESeq2 (Love et al., 2014) was used to identify differentially expressed genes.
ATAC-seq analysis
Paired-end ATAC-seq reads were aligned to the diploid genome using STAR (Dobin et al., 2013) with the splicing alignment feature switched off, using the following parameters: $STAR–runMode alignReads–readFilesCommand zcat–outSAMtype BAM–outBAMcompression 6–outFilterMultimapNmax 1–outFilterMatchNmin 30–alignIntronMax 1–alignEndsType Local. A chromatin accessibility atlas was created using all replicates from all cell types. Briefly, ATAC-seq peaks for individual replicates were called using MACS2 (Zhang et al., 2008). Reproducibility of the peaks among replicates of a cell type was evaluated as previously described (Li et al., 2011). After removing the irreproducible peaks (IDR < 0.05), the chromatin accessibility atlas was constructed by aggregating all the reproducible peaks from the three cell types. Next, ATAC-seq reads were counted for peaks, and DESeq2 (Love et al., 2014) was used to identify the differentially accessible regions.
ChIP-seq analysis
Paired-end ChIP-seq reads were mapped to the diploid genome using STAR (Dobin et al., 2013) with the splicing alignment feature switched off, using the following parameters: $STAR–runMode alignReads–readFilesCommand zcat–outSAMtype BAM–outBAMcompression 6–outFilterMultimapNmax 1–outFilterMatchNmin 70–outFilterMatchNminOverLread 0.85–alignIntronMax 1–alignEndsType Local. Peaks were called using MACS2 (Zhang et al., 2008). Reproducibility of the peaks was evaluated as described (Li et al., 2011) and a peak atlas was obtained by retaining the reproducible peaks (IDR < 0.1)
CUT&RUN Analysis
Similar to ChIP-seq analysis, paired-end CUT&RUN reads were mapped to the diploid genome using STAR (Dobin et al., 2013) with the splicing alignment feature switched off. We set the parameters as following to run alignment: $STAR --runMode alignReads --genomeLoad NoSharedMemory --readFilesCommand zcat --outSAMtype BAM SortedByCoordinate --outBAMcompression 6 --outFilterMultimapNmax 1 --outFilterMatchNmin 40 --outFilterMatchNminOverLread 0.4 --seedSearchStartLmax 15 --alignIntronMax 1 --alignEndsType Local. The aligned read pairs were retained if their fragment length was between 50 to 500 bp. Peaks were called using MACS2 with its nomodel setting (Zhang et al., 2008). Reproducibility of the peaks was evaluated as described (Li et al., 2011) and a peak atlas was obtained by retaining the reproducible peaks (IDR < 0.2). CUT&RUN peaks that do not overlap ATAC-seq peaks were further removed from the atlas.
Transcription factor binding motif analysis
Alternative-allele sequences for B6 ATAC-seq peak atlas regions were generated by modifying the B6 sequences with known genetic variants from the Cast genome (Keane et al., 2011). We scanned both the reference and alternative allele peak sequences with FIMO (Grant et al., 2011) to identify peaks containing (Pc < c10−4) transcription factor binding motifs (TFBM) from the Cis-BP database (Weirauch et al., 2014). We used all directly determined Mus musculus motifs in the Cis-BP database as well as motifs that were inferred from closely related similar TFs. The genomic coordinates of Cast motifs were converted back to the B6 coordinate system in order to merge the allele-specific motif scanning results. When significant motif matches were found on both the reference and alternative alleles, the strongest match was determined according to the FIMO hits with a more significant FIMO p value indicating a better match to the position weight matrix (PWM). Using this approach, variant-containing TFBMs could be categorized as stronger on the reference or stronger on the alternative allele. These allelically-biased motif matches were further associated with the allelic imbalance of the ATAC-seq peaks in which the motif resides. A t-test was used to compare the mean allelic ratio of ATAC-seq reads from peaks containing stronger motif matches on the reference allele to those containing stronger motif matches on the alternative allele.
For motif enrichment analysis, motif scan, as described above, was performed on different peaks subsets as well as a background group consisting of randomly selected peaks from the accessibility atlas. Peaks containing a specific motif with one or multiple occurrences was counted once for both foreground and background groups. Fisher’s exact test was used to determine the significance of motif enrichment, followed by Benjamini-Hochberg procedure to obtain the false discovery rate.
Supplementary Material
Table S1: Overview of sequencing data generated in this study (Related to STAR METHODS)
ACKNOWLEDGEMENTS
We thank all members of the Rudensky lab for discussions and technical assistance. We thank Stephen Henikoff for providing protein A/G-MNase plasmid. This study was supported by NIH grants R01AI034206 and U54 CA209975; NIH/NCI Cancer Center Support Grant P30 CA008748; The Ludwig Center at Memorial Sloan Kettering Cancer Center; and the Parker Institute for Cancer Immunotherapy. A.G. was supported by an Irvington fellowship of the Cancer Research Institute. R.B.P. was supported by a “la Caixa” Fellowship from “la Caixa” Foundation (LCF/BQ/AA17/11610016). A.Y. Rudensky is an investigator with the Howard Hughes Medical Institute. We acknowledge the use of the MSKCC Integrated Genomics Operation Core, funded by NIH/NCI Cancer Center Support Grant P30 CA008748, Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
Footnotes
DECLARATION OF INTERESTS
A.Y.R. is an SAB member and has equity in Sonoma Biotherapeutics and Vedanta Biosciences and is a co-inventor or has IP licensed to Takeda, which is unrelated to the content of this study.
REFERENCES
- Arvey A, van der Veeken J, Plitas G, Rich SS, Concannon P, and Rudensky AY (2015). Genetic and epigenetic variation in the lineage specification of regulatory T cells. Elife 4, e07571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, and Rudensky AY (2014). Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol 15, 580–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, and Ochs HD (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 27, 20–21. [DOI] [PubMed] [Google Scholar]
- Boller S, Ramamoorthy S, Akbas D, Nechanitzky R, Burger L, Murr R, Schübeler D, and Grosschedl R (2016). Pioneering Activity of the C-Terminal Domain of EBF1 Shapes the Chromatin Landscape for B Cell Programming. Immunity 44, 527–541. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, and Ramsdell F (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 27, 68–73. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, and Rudensky A (2020). Roles of Regulatory T Cells in Tissue Pathophysiology and Metabolism. Cell Metab 31, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonnier LM, Cui Y, Stephen-Victor E, Harb H, Lopez D, Bleesing JJ, Garcia-Lloret MI, Chen K, Ozen A, Carmeliet P, et al. (2019). Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat Immunol 20, 1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, and Bowcock AM (2000). JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest 106, R75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Rozowsky J, Galeev TR, Harmanci A, Kitchen R, Bedford J, Abyzov A, Kong Y, Regan L, and Gerstein M (2016). A uniform survey of allele-specific binding and expression over 1000-Genomes-Project individuals. Nat Commun 7, 11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Zhabotynsky V, Sun W, Huang S, Pakatci IK, Kim Y, Wang JR, Morgan AP, Calaway JD, Aylor DL, et al. (2015). Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nat Genet 47, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, and Levine MS (2015a). Regulatory Principles Governing Tissue Specificity of Developmental Enhancers. Cold Spring Harb Symp Quant Biol 80, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, and Levine MS (2015b). Suboptimization of developmental enhancers. Science 350, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasolino M, Goldman N, Wang W, Cattau B, Zhou Y, Petrovic J, Link VM, Cote A, Chandra A, Silverman M, et al. (2020). Genetic Variation in Type 1 Diabetes Reconfigures the 3D Chromatin Organization of T Cells and Alters Gene Expression. Immunity 52, 257–274.e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Dooley JL, Farr AG, and Rudensky AY (2005a). Developmental regulation of Foxp3 expression during ontogeny. J Exp Med 202, 901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, and Rudensky AY (2005b). A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 6, 1142–1151. [DOI] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, and Rudensky AY (2007). Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Allison KA, Kaikkonen MU, Orozco LD, and Glass CK (2013). Effect of natural genetic variation on enhancer selection and function. Nature 503, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Holt J, Kao CY, McMillan L, and Wang W (2014). A novel multi-alignment pipeline for high-throughput sequencing data. Database (Oxford) 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Georgakilas G, Petrovic J, Kurachi M, Cai S, Harly C, Pear WS, Bhandoola A, Wherry EJ, and Vahedi G (2018). Lineage-Determining Transcription Factor TCF-1 Initiates the Epigenetic Identity of T Cells. Immunity 48, 243–257.e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, and Rudensky AY (2012). Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30, 531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, Weber C, Emmanuel AO, Sun T, Bentrem DJ, et al. (2014). β-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci Transl Med 6, 225ra228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8, 191–197. [DOI] [PubMed] [Google Scholar]
- Kwon HK, Chen HM, Mathis D, and Benoist C (2017). Different molecular complexes that mediate transcriptional induction and repression by FoxP3. Nat Immunol 18, 1238–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann Appl Stat 5, 1752–1779. [Google Scholar]
- Li R, Cauchy P, Ramamoorthy S, Boller S, Chavez L, and Grosschedl R (2018). Dynamic EBF1 occupancy directs sequential epigenetic and transcriptional events in B-cell programming. Genes Dev 32, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, et al. (2018). Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell 173, 1796–1809.e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio CW, and Hsieh CS (2008). A two-step process for thymic regulatory T cell development. Immunity 28, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, et al. (2018). Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442.e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers MP, Bryson TD, Henikoff JG, and Henikoff S (2019). Improved CUT&RUN chromatin profiling tools. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura RM, Sigvardsson M, Galceran J, Verbeek S, Clevers H, and Grosschedl R (1998). Redundant regulation of T cell differentiation and TCRalpha gene expression by the transcription factors LEF-1 and TCF-1. Immunity 8, 11–20. [DOI] [PubMed] [Google Scholar]
- Owen DL, Mahmud SA, Sjaastad LE, Williams JB, Spanier JA, Simeonov DR, Ruscher R, Huang W, Proekt I, Miller CN, et al. (2019). Thymic regulatory T cells arise via two distinct developmental programs. Nat Immunol 20, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, Leslie C, Shaffer SA, Goodlett DR, and Rudensky AY (2012). Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 13, 1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. (2012). Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, Emmett MJ, Briggs ER, Fang B, Everett LJ, et al. (2015). Genetic Variation Determines PPARγ Function and Anti-diabetic Drug Response In Vivo. Cell 162, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veeken J, Zhong Y, Sharma R, Mazutis L, Dao P, Pe’er D, Leslie CS, and Rudensky AY (2019). Natural Genetic Variation Reveals Key Features of Epigenetic and Transcriptional Memory in Virus-Specific CD8 T Cells. Immunity 50, 1202–1217.e1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loosdregt J, Fleskens V, Tiemessen MM, Mokry M, van Boxtel R, Meerding J, Pals CE, Kurek D, Baert MR, Delemarre EM, et al. (2013). Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity 39, 298–310. [DOI] [PubMed] [Google Scholar]
- Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, Oosterwegel M, Wilson A, MacDonald HR, and Clevers H (1995). An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature 374, 70–74. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol Cell 68, 1067–1082.e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber BN, Chi AW, Chavez A, Yashiro-Ohtani Y, Yang Q, Shestova O, and Bhandoola A (2011). A critical role for TCF-1 in T-lineage specification and differentiation. Nature 476, 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S, Gai K, Li X, Shao P, Zeng Z, Zhao X, Chen X, Paradee WJ, Meyerholz DK, Peng W, et al. (2019). Tcf1 and Lef1 are required for the immunosuppressive function of regulatory T cells. J Exp Med 216, 847–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang BH, Wang K, Wan S, Liang Y, Yuan X, Dong Y, Cho S, Xu W, Jepsen K, Feng GS, et al. (2019). TCF1 and LEF1 Control Treg Competitive Survival and Tfr Development to Prevent Autoimmune Diseases. Cell Rep 27, 3629–3645.e3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Lareau CA, Ramirez RN, Rose SA, Maier B, Wroblewska A, Desland F, Chudnovskiy A, Mortha A, Dominguez C, et al. (2019). The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912.e820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Overview of sequencing data generated in this study (Related to STAR METHODS)
Data Availability Statement
All next generation sequencing data generated in this paper were deposited in Gene Expression Omnibus (GEO) under SuperSeries accession number GSE154680. Code is available from the authors on request.