

Abstract
C-Glycosyl peptides/proteins are metabolically stable mimics of the native glycopeptides/proteins bearing O/N-glycosidic linkages, and are thus of great therapeutical potential. Herein, we disclose a protocol for the syntheses of vinyl C-glycosyl amino acids and peptides, employing a nickel-catalyzed reductive hydroglycosylation reaction of alkyne derivatives of amino acids and peptides with common glycosyl bromides. It accommodates a wide scope of the coupling partners, including complex oligosaccharide and peptide substrates. The resultant vinyl C-glycosyl amino acids and peptides, which bear common O/N-protecting groups, are amenable to further transformations, including elongation of the peptide and saccharide chains.
Subject terms: Peptides, Synthetic chemistry methodology, Carbohydrate chemistry
C-Glycosyl peptides/proteins are metabolically stable mimics of the native glycopeptides/proteins of great therapeutic potential, but their chemical synthesis is challenging. Here, the authors report a protocol for the synthesis of vinyl C-glycosyl amino acids and peptides, via a Ni-catalyzed reductive hydroglycosylation reaction of alkyne derivatives of amino acids and peptides with glycosyl bromides.
Introduction
Glycosylation of proteins, involving conjugation of saccharides onto the amino acid residues of proteins, represents a ubiquitous type of posttranslational modification. The added saccharides can then modulate the properties and functions of the proteins in various biological processes, such as in cell adhesion, signal transduction, and immune response1–5. In nature, more than 13 monosaccharides can join with eight amino acid residues to provide at least 41 distinct types of glycosidic linkages connecting the saccharides with the proteins6. These linkages are mostly O/N-glycosidic bonds with the hydroxyl and amido groups pending on serine, threonine, or asparagine residues7–9, with Man-Trp being the only C-glycosidic motif known to date10, 11 (Fig. 1a). The naturally occurring O/N-glycosidic linkages are metabolically vulnerable thus potentially hamper the therapeutical use of glycopeptide/proteins. Thus, the pursuit of hydrolytically stable linkages (e.g., C- or S-glycosidic bonds) in replace of the O/N glycosidic linkages has elicited great interest in the development of glycopeptide/protein drugs12–15.
Fig. 1. Ni-catalyzed syntheses of vinyl C-glycosyl amino acids and peptides.

a The N-linked core Man3GlcNAc2β1-Asn motif in glycoproteins, the O-linked tumor-associated carbohydrate antigen Sialyl Lewisx in glycolipids, and the native N/O/C-glycosidic linkages in glycoproteins. b Nickel-catalyzed reductive hydroglycosylation for access to vinyl C-glycopeptides and a plausible mechanism. c Structure of the glycosyl donors 1 and potential by-products 4 and 5. The glycosidic bonds are highlighted in red.
In comparison to the preparation of the native O/N- or artificial S/Se-glycosyl peptides16–21, the construction of C-glycosyl peptides is much more difficult and has lagged far behind22–25. Given the markedly lower nucleophilicity and higher pKa of C−H compared to the X−H (X = O, N, S) counterparts, the conventional glycosylation involving nucleophilic addition onto sugar oxocarbenium intermediates become frequently futile for C-glycosylation. Besides, the complex functionality of peptides are poorly tolerated with the glycosylation conditions. In recent years, transition metal-catalyzed C-glycosylation has gained great attention26–43 and a large variety of C-glycoside natural products as well as drug candidates have been successfully synthesized44–49. However, synthesis of complex C-glycosyl peptides, especially a convergent synthesis using oligosaccharides as donors still poses a formidable challenge, due to the following methodological limits: (i) scarcity of methods for construction of alkyl/alkenyl C-glycosidic bonds, in contrast to the well-studied aryl C-glycosylation; (ii) harsh reaction conditions, including high temperature, strong bases, stoichiometric amount of organometallic reagents, or metal additives that are poorly compatible with peptide substrates; (iii) use of large excess of sugar donors and/or insufficient anomeric selectivity, impeding convergent synthesis with expensive oligosaccharide donors; (iv) use of highly functionalized sugar donors, necessitating multistep transformations to procure the final glycopeptides. Recently, Chen50, Niu51, Ackermann52, 53, Liu54, Wang55, and co-workers have disclosed a series of methods for the synthesis of C-glycosyl amino acids via either C–H activation or radical addition strategies. Very recently, Wang et al. reported a stereodivergent synthesis of C-glycosamino acids using glycal donors via Pd/Cu dual catalysis56. Notwithstanding, straightforward and practical C-glycosylation methods are still in high demand to conquer the aforementioned limits.
Inspired by the recent breakthrough in the NiH-catalyzed hydrocarbonation of unsaturated bonds57–61, we envisioned the construction of vinyl C-glycosyl amino acids and peptides via a plausible reaction mechanism as depicted in Fig. 1b. Thus, the catalytic cycle started with a branch-selective insertion of NiH (B) to terminal alkyne 2 to form vinyl nickel species C, which was then oxidized by glycosyl bromide 1 via a bromine atom abstraction followed by anomeric radical trapping to form high-valent NiIII complex E. Subsequent reductive elimination delivered the desired C-glycoside 3 and catalyst A. The active NiH species B was regenerated by hydride transfer from the hydrosilane. It was expected that judicious choice of coupling partners and reaction conditions was required, in order to avoid the β-H/O/N elimination and anomeric reduction that would result in glycal 4 and tetrahydropyran 5 (Fig. 1c), to achieve useful anomeric α/β selectivity, and to secure wide compatibility of the functional groups and protecting groups on the saccharide and peptide substrates.
Here, we show that a wide variety of the easily accessible acetylenic amino acids/peptides and glycosyl bromides can be coupled regio- and stereoselectively under the catalysis of Ni to provide the metabolically stable vinyl C-glycosyl amino acids and peptides.
Results
Reaction design and optimization
To implement the hypothesis, α-mannosyl bromides were initially selected as glycosyl donors and n-hexyne as a model alkyne acceptor. Conditions optimization was proven tedious, and competing by-products from β-H/O elimination (i.e., 4) or anomeric reduction (i.e., 5) were obtained concomitantly in many of the cases (see Supplementary Figs. 26–35). Fortunately, extensive surveys of various parameters, including the protecting groups on the sugar bromides, Ni catalyst, bipyridine ligand, phosphine additive, base, silane, reaction atmosphere, and solvent, led to optimal Conditions I and Conditions II for the conjugation of mannose and glucosamine type saccharides (1a and 1b) with N-Boc-L-Pra-OMe (2a) (Pra = propargylglycine), yielding vinyl glycosyl amino acids 3aa and 3ba in 77 and 85% yield, respectively (Fig. 2). The 1,1-disubstituted alkene moiety in the product is well diagnostic in the 1H NMR spectra by two singlet signals at high field (e.g., 5.74 and 5.51 ppm for 3aa; 5.10 and 4.94 ppm for 3ba)57. Besides, the anomeric H of α-glycoside 3aa presents as a singlet at 4.75 ppm, while the anomeric H of β-glycoside 3ba is a doublet at 5.06 ppm (d, J = 10.6 Hz). Some key findings with GlcNPhth bromide 1b as the donor are listed in Fig. 2. Thus, an inert atmosphere was essential for the successful transformation (entries 2 and 3). The absence of dtbbpy ligand completely shut down the reaction (entry 5). Ni(0) could also be used as a catalyst albeit leading to lower yields (entries 6 and 7). The phosphine additive (R)-Tol-BINAP or Ph3P was found to be fully oxidized into Tol-BINAP(O)2 or Ph3P(O) after the reaction, and its absence only slightly diminished the coupling yield (entry 4). Besides, the chirality of Tol-BINAP did not affect the β-selectivity of the glycosylation (see Supplementary Figs. 30, 31, and 35). Therefore, the phosphine additive was not involved in the catalytic cycle, whereas it might facilitate the dissolution of NiCl2(DME) and formation of NiCl2(dtbbpy) as the actual catalyst, in addition, a role as a residue O2 scavenger was also possible62, 63.
Fig. 2. Optimized reaction conditions I and II and the control experiments for Conditions II.

a Reaction conditions: 1b (0.1 mmol), 2a (2.0 equiv.), NiCl2(DME) (10 mol%), dtbbpy (12 mol%), (R)-Tol-BINAP (10 mol%), PMHS (2.5 equiv.), Na2CO3 (2.5 equiv.), THF (1 mL), 30 °C, Ar, 36 h. The yields were determined by 1H NMR using CH2Br2 as an internal standard. b Isolated yield. DME dimethoxyethane, DEMS diethoxymethylsilane, DMAc N,N-dimethylacetamide, dtbbpy 4,4′-di-tert-butyl-2,2′-bipyridine, PMHS poly(methylhydrosiloxane), Tol-BINAP 2,2′-bis(di-p-tolylphosphino)-1,1′-binaphthyl, w/o without. In red are the formed C-glycosidic bonds.
It is worth noting that no epimerization of the amino acids was observed in the reaction, as determined by careful HPLC analysis (see Supplementary Figs. 40 and 41), testifying the mild reaction conditions using weak bases (Na2CO3) and mild temperature (<35 °C) for the present C-glycosylation.
Substrate scope
With the optimal conditions in hand, we explored the scope of the present method. Firstly, a variety of acetylenic amino acid derivatives, which were easily prepared (see Supplementary Figs. 13–25), were examined to couple with mannosyl bromide 1a (Fig. 3). Gratifyingly, the frequently used amino protecting groups for peptide synthesis, such as Boc (3aa), Cbz (3ab), Fmoc (3ac) were well tolerated, and so did the carboxylic acid protecting groups, such as Bn (3ad) and tBu (3ae). Expectedly, N-
Fig. 3. C-Mannosylation of alkyne derivatives of amino acids and peptides.

1a (0.1 mmol), 2 (2.0 equiv.), NiCl2(DME) (10 mol%), dtbbpy (12 mol%), PPh3 (20 mol%), DEMS (2.5 equiv.), Na2CO3 (2.5 equiv.), DME/DMAc (10/1, v/v, 1 mL), 25–35 °C, Ar, 36 h. Isolated yields are reported. In red are the formed C-glycosidic bonds.
Phth-L-Pra-OMe reacted smoothly to afford 3af in 73% yield; alkynes easily derived from natural amino acids via ether or amide linkages, such as propargyl Ser and Tyr ethers (3ag and 3ah), hept-6-ynoyl Lys amide (3ai), and propargylamino Asn (3aj) were also shown to be suitable substrates. Significantly, the current nickel-catalyzed coupling reaction was highly compatible with the peptide bonds, and thus could be readily applied to the C-glycosylation of dipeptides and tripeptides. Indeed, a panel of the vinyl C-glycosyl dipeptides, including N-Boc-(Man-vinyl)-Ala-Phe-OMe (3ak), N-Boc-(Man-vinyl)-Ala-Leu-OMe (3al), N-Boc-(Man-vinyl)-Ala-Ile-OMe (3am), N-Boc-(Man-vinyl)-Ala-Pro-OMe (3an), N-Boc-Trp-(Man-vinyl)-Ala-OMe (3ao), O-Bn-N-Boc-Thr-(Man-vinyl)-Ala-OMe (3ap), N-Boc-Phe-(Man-vinyl)-Ala-OMe (3aq), and tripeptides, including N-Boc-(Man-vinyl)-Ala-Ala-Val-OMe (3ar), N-Boc-(Man-vinyl)-Ala-Tle-Ala-OMe (3as), and N-Boc-(Man-vinyl)-Ala-Ala-O-tBu-Thr-OMe (3at) were successfully prepared in 52–80% yields. It was noted that no epimerization of the amino acid residues was observed.
GlcNAcβ-Asn represents the most common glycosyl amino acid motif on nuclear and cytoplasmic glycoproteins6, bringing the synthesis of the glucosamine-based C-glycopeptides an important subject. As shown in Fig. 4, the optimal GlcNPhth bromide donor 1b could be installed not only to amino acid (3ba), but also to dipeptides and tripeptides derivatives with the Pra moiety located either at the terminal (3bl, 3bt, and 3bp) or at an interior position (3bu) in satisfactory yields and exclusive β-selectivity. Notably, the peptide sequence of 3bt simulates the consensus sequence of Asn-X-Thr/Ser (X can be any amino acid except Pro) in the native N-glycan where GlcNAc can be attached.
Fig. 4. Scope of C-glycosylation with glucosamine donor 1b.

1b (0.1 mmol), 2 (2.0 equiv.), NiCl2(DME) (10 mol%), dtbbpy (12 mol%), (R)-Tol-BINAP (10 mol%), PMHS (2.5 equiv.), Na2CO3 (2.5 equiv.), THF (1 mL), 30 °C or as noted, Ar, 36 h. Isolated yields are reported. In red are the formed C-glycosidic bonds.
Next, we turned to test with other types of pyranosides (Fig. 5). N-Phth-galactosamine bromide was smoothly coupled with Pra 2a, delivering 3ca with complete β-selectivity in 79% yield. For xylose, 4-(trifluoromethyl)benzoyl group was used as protecting groups to facilitate separation of the coupling products via silica-gel chromatography, thus (Xyl-vinyl)-
Fig. 5. C-Glycosylation with various mono- and disaccharide bromides.

See SI for detailed conditions, which might vary slightly from Conditions I and II, and isolated yields are reported. In red are the formed C-glycosidic bonds.
Ala (3da) was obtained as β/α anomers with a ratio of 3:1 in 65% yield, in that the β anomer adopted 4C1 conformation (J1,2 = 9.7 Hz) and the α anomer adopted 1C4 conformation (H1 showed a singlet signal). Mannosamine and rhamnose bromides also reacted smoothly with 2a, providing 3ea and 3fa in 72 and 93% yield, respectively. In addition, the orthogonally protected C-GlcN amino acids 3ga and 3ha were obtained in 72 and 58% yield from the corresponding glucosamine donors bearing 6-O-TBDPS and 4-O-Bn groups, respectively. Glucosyl bromide was also tested, and the desired (Glc-vinyl)-Ala (3ia) was obtained in 52% yield, albeit without β/α selectivity (β/α = 1:1). Moreover, the scope could be expanded to disaccharide bromide donors, with the fully protected N-Boc-(Galβ(1,3)GlcNβ-vinyl)-Ala-OMe (3ja),
N-Boc-(GlcNβ(1,4)GlcNβ-vinyl)-Ala-OMe (3ka), N-Boc-(Rhaα(1,2)Rhaα-vinyl)-Ala-OMe (3la), and N-Boc-(Fucα(1,3)GlcNβ-vinyl)-Ala-OMe (3ma) being prepared in synthetically useful yields (54–68%).
Intriguingly, this method could also be extended to internal acetylenic amino acids. As exemplified in Fig. 6, when unsymmetrically substituted alkyne 2v was used as the coupling partner, cis-hydroglycosylation with Man bromide 1a and GlcN bromide 1b occurred smoothly, leading to the corresponding regio-isomeric C-glycosides (Man 3av1 and 3av2 and GlcN 3bv1 and 3bv2, respectively) in moderate yields (52 and 47%) and varied regioselectivity (r.r. = 3:1 and 8:1).
Fig. 6. C-Glycosylation with an internal acetylenic amino acid.
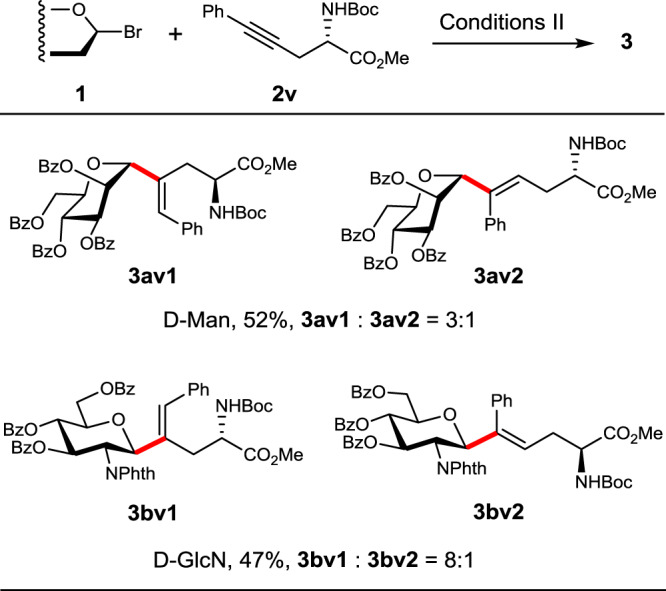
In red are the formed C-glycosidic bonds.
The attained stereoselectivity of the C-glycosylation could be attributed to the predominant conformation of the glycosyl radical intermediate, which is stabilized by the interaction of SOMO of the anomeric unpaired electron with lone pair of the ring oxygen and the σ* of the adjacent C2–O/C2–N bond64–67. Thus, a mannose-derived radical adopts preferentially a 4C1 conformation, leading to the 1,2-trans (α-selectivity) product in the C-glycosylation. A glucose-derived radical adopts a flexible B2,5 conformation, thus the stereoselectivity of C-glycosylation can be shifted from 1,2-cis (α-selectivity) to 1,2-trans (β-selectivity) by using a bulkier protecting group on C2-OH; and for a glucosamine-derived radical bearing the bulky NPhth group at C2, exclusive 1,2-trans (β-selectivity) product can be attained. Due to the lack of C5 substituent, a xylose-derived radical can adopt both the B2,5 conformation and 1C4 conformation, thus resulting in a 1,2-trans (β-selectivity) dominated C-glycosylation.
To probe the occurrence of the glycosyl radical (species D in Fig. 1b), we conducted a radical clock experiment (Fig. 7)31,35. Thus, δ-olefinic 1-bromo glucoside 6 and alkyne 2a were subjected to the standard Conditions II; the desired ring-closure product 7 was isolated in 33% yield with mild diastereoselectivity (d.r. = 3:2). Though not conclusive, this result supports the intermediacy of an anomeric radical species.
Fig. 7. A radical clock experiment.
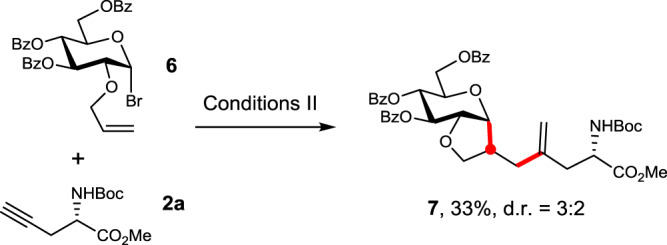
In red are the formed bonds.
Synthetic utilities
To demonstrate the potential utilities of the current method, we also examined a scale-up reaction and further transformations of the resulting vinyl C-glycosyl amino acid. Thus, compound 3ba (1.96 g) was obtained in a 65% yield at a 3.6 mmol scale reaction (Fig. 8a, A; see Supplementary Fig. 37). The transformation of the N-Phth to the native NHAc residue is critical for the synthesis of GlcNAcβ-Asn mimics, fortunately, this was realized selectively via sequential treatment with 80% N2H4·H2O, HOAc, and Ac2O, leading to the desired C-GlcNAc amino acid 8 in 60% yield and >99% de value (see Supplementary Figs. 42 and 43). The orthogonally protected 3ba and 3ga allow subsequent elongation of the peptide and saccharide chains. Indeed, the subjection of 3ga to desilylation followed by fucosylation under the mild Au(I)-catalyzed glycosylation conditions68 afforded disaccharide 9 in 68% yield (see Supplementary Fig. 44). Alternatively, the subjection of 3ba to the cleavage of the N-Boc group followed by peptide synthesis led to C-glycosyl dipeptide 10 in 93% yield (see Supplementary Fig. 45). These transformations showcased the potential of the current protocol for the synthesis of complex and biologically relevant glycopeptides. In addition, three examples of deprotection under strong basic conditions (with LiOH) were conducted, leading to glycosyl amino acids and peptides 11–13 in excellent yields (Fig. 8b).
Fig. 8. Scale-up reaction, subsequent transformation, and C-glycosylation with complex saccharides.

a A gram-scale synthesis of C-glycosyl amino acid and subsequent transformations. Conditions and reagents: A. NiCl2(DME) (10 mol%), dtbbpy (15 mol%), (R)-Tol-BINAP (6.0 mol%), PMHS (2.5 equiv.), Na2CO3 (2.5 equiv.), THF (0.1 M), 25–28 °C, Ar, 48 h, 65%. B. i) 80% N2H4·H2O, MeOH, 0 °C, 9 h; ii) HOAc/MeOH (1/4, v/v), 70 °C, 1.5 h; iii) Ac2O, Et3N, CH2Cl2, 6 h, 60% over three steps. C. i) HF·pyridine, pyridine, 0 °C→rt, 2 h, 85%; ii) Au(PPh3)NTf2 (10 mol%), 4 Å MS, CH2Cl2, 0 °C→rt, 0.5 h, 81%. D. i) CH2Cl2/TFA (2/1, v/v), 0 °C→rt, 1.5 h; ii) N-Cbz-O-tBu-L-serine (1.5 equiv.), HOBt (1.5 equiv.), DIPEA (4.0 equiv.), EDCI (1.5 equiv.), DMF, -10 °C→rt, 6 h, 93% over two steps. b Deprotection of acyl groups. Conditions: LiOH (7.5 equiv), MeOH/H2O (4/1, v/v, 0.01 M), rt, 10 h. c Convergent C-glycosylation of complex oligosaccharides with amino acid, see SI for details. In red are the formed C-glycosidic bonds.
Finally, we further assessed the feasibility of convergent assembly of C-glycosyl peptides using biologically intriguing oligosaccharides (Fig. 8c). Using a branched pentasaccharide bromide as a donor and Boc-l-Pra-OMe 2a as acceptor, the desired C-glycosyl amino acid 14 was successfully obtained in ~39% yield (see Supplementary Fig. 36), with the saccharide being relevant to the O-antigen of the lipopolysaccharides of Pseudomonas syringae69. Using a trisaccharide bromide as a donor, the coupled 15 was obtained in a satisfactory 58% yield, which bears the tumor-associated Lewisx antigen70.
Discussion
We have developed a nickel-catalyzed hydroglycosylation reaction for the straightforward synthesis of vinyl C-glycosyl amino acids and peptides. A variety of glycosyl bromides can be used as limiting reagents, and excellent 1,2-trans diastereoselectivity is attained for C2-axially substituted pyranosides (e.g., Man, ManN, and Rha) or C2-equatorially substituted 2-aminopyranosides (e.g., GlcN and GalN). A wide substrate scope has been proven and also a gram-scale reaction has been demonstrated. The resultant C-glycosyl amino acids and peptides, which bear common N- and O-protecting groups, could be readily transformed into various mimics of the native O/N-glycosyl peptides. The late-stage C-glycosylation with complex oligosaccharide bromides has also been successful. Additionally, the nascent vinyl group in the products would provide a special handle for further derivatization. All these features render the present protocol a promising method for the preparation of C-glycosyl peptides of biological and therapeutical significance.
Methods
General procedure A (Conditions I) for the NiH-catalyzed reductive hydroglycosylation of acetylenic amino acid and peptides
To an oven-dried 10 mL Schlenk tube (Titan, TF891910) containing a Teflon coated magnetic stirring bar were added glycosyl bromide 1 (0.1 mmol), NiCl2(DME) (2.2 mg, 10 mol%), dtbbpy (3.2 mg, 12 mol%), PPh3 (5.2 mg, 20 mol%), and Na2CO3 (25 mg, 2.5 equiv.). The tube was sealed with a rubber cap and parafilm, and evacuated then refilled with Ar for at least five cycles. The acetylenic amino acid or peptide derivative 2 was dissolved in solvent (DME/DMAc = 1:1, 1.0 mL) and the solution was injected into the reaction tube (this substrate could be added directly with glycosyl bromide if it was solid). When stirring, (EtO)2MeSiH (40 µL, 2.5 equiv.) was injected via a microliter syringe. Otherwise noted, the tube was moved to an oil bath preheated to 33–35 °C and kept stirring for 36 h. The reaction mixture was diluted with CH2Cl2 (20 mL) and filtered. After concentration, the residue was purified by column chromatography on silica gel or preparative TLC to afford the desired product 3.
General procedure B (Conditions II) for the NiH-catalyzed reductive hydroglycosylation of acetylenic amino acid and peptides
To an oven-dried 10 mL Schlenk tube (Titan, TF891910) containing a Teflon coated magnetic stirring bar were added glycosyl bromide 1 (0.1 mmol), NiCl2(DME) (2.2 mg, 10 mol%), dtbbpy (3.2 mg, 12 mol%), (R)-Tol-BINAP (6.7 mg, 10 mol%), and Na2CO3 (25 mg, 2.5 equiv.). The tube was sealed with a rubber cap and parafilm, and evacuated then refilled with Ar for at least five cycles. The acetylenic amino acid or peptide derivative 2 was dissolved in THF (1.0 mL), and the solution was injected into the reaction tube (this substrate could be added directly with glycosyl bromide if it was solid). When stirring, PMHS (32 µL, 2.5 equiv.) was injected via a microliter syringe. Otherwise noted, the tube was kept stirring under an indicated temperature of 30 °C for 36 h. The reaction mixture was diluted with CH2Cl2 (20 mL) and filtered. After concentration, the residue was purified by flash column chromatography on silica gel or preparative TLC to afford the desired product 3.
Supplementary information
Acknowledgements
Financial support from the National Key Research & Development Program of China (2018YFA0507602), National Natural Science Foundation of China (22031011 and 21621002), Key Research Program of Frontier Sciences of CAS (ZDBS-LY-SLH030), Strategic Priority Research Program of CAS (XDB20020000), Youth Innovation Promotion Association of CAS (2020258), Shanghai Municipal Science and Technology Major Project, and Shanghai Committee of Science and Technology (17JC1405300) are acknowledged.
Author contributions
B.Y., Y.-H.L., and D.Z. conceived the project. Y.-H.L., Y.-N.X., T.G., B.W., H.L., and Z.H. conducted the synthetic work. Y.-H.L., P.X., and D.Z. conducted the data analysis. B.Y., Y.-H.L., and P.X. wrote the manuscript. All authors discussed the results and commented on the paper.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information file, including experimental details, characterization data, and 1H and 13C NMR spectra of new compounds. All data are available from the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review informationNature Communications thanks Xiang-Guo Hu and the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Peng Xu, Email: peterxu@sioc.ac.cn.
Biao Yu, Email: byu@sioc.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-25127-z.
References
- 1.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 2.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 3.Varki A. Biological roles of glycans. Glycobiology. 2017;27:3–49. doi: 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buskas T, Ingale S, Boons GJ. Glycopeptides as versatile tools for glycobiology. Glycobiology. 2006;16:113R–136R. doi: 10.1093/glycob/cwj125. [DOI] [PubMed] [Google Scholar]
- 5.Schjoldager KT, Narimatsu Y, Joshi HJ, Clausen H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020;21:729–749. doi: 10.1038/s41580-020-00294-x. [DOI] [PubMed] [Google Scholar]
- 6.Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43R–56R. doi: 10.1093/glycob/12.4.43R. [DOI] [PubMed] [Google Scholar]
- 7.Schmaltz RM, Hanson SR, Wong C-H. Enzymes in the synthesis of glycoconjugates. Chem. Rev. 2011;111:4259–4307. doi: 10.1021/cr200113w. [DOI] [PubMed] [Google Scholar]
- 8.Herzner H, Reipen T, Schultz M, Kunz H. Synthesis of glycopeptides containing carbohydrate and peptide recognition motifs. Chem. Rev. 2000;100:4495–4537. doi: 10.1021/cr990308c. [DOI] [PubMed] [Google Scholar]
- 9.Zechel DL, Withers SG. Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc. Chem. Res. 2000;33:11–18. doi: 10.1021/ar970172. [DOI] [PubMed] [Google Scholar]
- 10.Hofsteenge J, et al. New type of linkage between a carbohydrate and a protein: C-glycosylation of a specific tryptophan residue in human RNase Us. Biochemistry. 1994;33:13524–13530. doi: 10.1021/bi00250a003. [DOI] [PubMed] [Google Scholar]
- 11.de Beer T, Vliegenthart JF, Löffler A, Hofsteenge J. The hexopyranosyl residue that is C-glycosidically linked to the side chain of tryptophan-7 in human RNase Us is alpha-mannopyranose. Biochemistry. 1995;34:11785–11789. doi: 10.1021/bi00037a016. [DOI] [PubMed] [Google Scholar]
- 12.Tamburrini A, Colombo C, Bernardi A. Design and synthesis of glycomimetics: recent advances. Med. Res. Rev. 2020;40:495–531. doi: 10.1002/med.21625. [DOI] [PubMed] [Google Scholar]
- 13.Apostola CR, Hayb M, Polt R. Glycopeptide drugs: a pharmacological dimension between “small molecules” and “biologics”. Peptides. 2020;131:170369. doi: 10.1016/j.peptides.2020.170369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hevey R. Strategies for the development of glycomimetic drug candidates. Pharmaceuticals. 2019;12:55. doi: 10.3390/ph12020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moradi SV, Hussein WM, Varamini P, Simerska P, Toth I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem. Sci. 2016;7:2492–2500. doi: 10.1039/C5SC04392A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Companon I, et al. Structure-based design of potent tumor-associated antigens: modulation of peptide presentation by single-atom O/S or O/Se substitutions at the glycosidic linkage. J. Am. Chem. Soc. 2019;141:4063–4072. doi: 10.1021/jacs.8b13503. [DOI] [PubMed] [Google Scholar]
- 17.Zhu S, Samala G, Sletten ET, Stockdill JL, Nguyen HM. Facile triflic acid-catalyzed α-1,2-cis-thio glycosylations: scope and application to the synthesis of S-linked oligosaccharides, glycolipids, sublancin glycopeptides, and TN/TF antigens. Chem. Sci. 2019;10:10475–10480. doi: 10.1039/C9SC04079J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji P, Zhang Y, Gao F, Bi F, Wang W. Direct, stereoselective thioglycosylation enabled by an organophotoredox radical strategy. Chem. Sci. 2020;11:13079–1308. doi: 10.1039/D0SC04136J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tegl G, et al. Facile formation of β-thioGlcNAc linkages to thiol-containing sugars, peptides, and proteins using a mutant GH20 hexosaminidase. Angew. Chem. Int. Ed. 2019;58:1632–1637. doi: 10.1002/anie.201809928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floyd N, Vijayakrishnan B, Koeppe JR, Davis BG. Thiyl glycosylation of olefinic proteins: S-linked glycoconjugate synthesis. Angew. Chem. Int. Ed. 2009;48:7798–7802. doi: 10.1002/anie.200903135. [DOI] [PubMed] [Google Scholar]
- 21.Pachamuthu K, Schmidt RR. Synthetic routes to thiooligosaccharides and thioglycopeptides. Chem. Rev. 2006;106:160–187. doi: 10.1021/cr040660c. [DOI] [PubMed] [Google Scholar]
- 22.Aldhoun M, Massi A, Dondoni A. Click azide−nitrile cycloaddition as a new ligation tool for the synthesis of tetrazole-tethered C-glycosyl α-amino acids. J. Org. Chem. 2008;73:9565–9575. doi: 10.1021/jo801670k. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Ben RN. C-Linked galactosyl serine AFGP analogues as potent recrystallization inhibitors. Org. Lett. 2005;7:2385–2388. doi: 10.1021/ol050677x. [DOI] [PubMed] [Google Scholar]
- 24.Palomo C, et al. Design and synthesis of a novel class of sugar-peptide hybrids: C-linked glyco β-amino scids through a stereoselective “acetate” mannich reaction as the key strategic element. J. Am. Chem. Soc. 2002;124:8637–8643. doi: 10.1021/ja026250s. [DOI] [PubMed] [Google Scholar]
- 25.Dondoni A, Marra A. Methods for anomeric carbon-linked and fused sugar amino acid synthesis: the gateway to artificial glycopeptides. Chem. Rev. 2000;100:4395–4421. doi: 10.1021/cr9903003. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, Yu B. Recent advances in the chemical synthesis of C-glycosides. Chem. Rev. 2017;117:12281–12356. doi: 10.1021/acs.chemrev.7b00234. [DOI] [PubMed] [Google Scholar]
- 27.Bokor É, et al. C-Glycopyranosyl arenes and hetarenes: synthetic methods and bioactivity focused on antidiabetic potential. Chem. Rev. 2017;117:1687–1764. doi: 10.1021/acs.chemrev.6b00475. [DOI] [PubMed] [Google Scholar]
- 28.Ghouilem J, de Robichon M, Le Bideau F, Ferry A, Messaoudi S. Emerging organometallic methods for the synthesis of C-branched (hetero)aryl, alkenyl, and alkyl glycosides: C−H functionalization and dual photoredox approaches. Chem. Eur. J. 2021;27:491–511. doi: 10.1002/chem.202003267. [DOI] [PubMed] [Google Scholar]
- 29.Gong H, Sinisi R, Gagné MR. A room temperature Negishi cross-coupling approach to C-alkyl glycosides. J. Am. Chem. Soc. 2007;129:1908–1909. doi: 10.1021/ja068950t. [DOI] [PubMed] [Google Scholar]
- 30.Gong H, Gagné MR. Diastereoselective Ni-catalyzed Negishi cross-coupling approach to saturated, fully oxygenated C-alkyl and C-aryl glycosides. J. Am. Chem. Soc. 2008;130:12177–12183. doi: 10.1021/ja8041564. [DOI] [PubMed] [Google Scholar]
- 31.Nicolas L, et al. Diastereoselective metal-catalyzed synthesis of C-aryl and C-vinyl glycosides. Angew. Chem. Int. Ed. 2012;51:11101–11104. doi: 10.1002/anie.201204786. [DOI] [PubMed] [Google Scholar]
- 32.Koester DC, Kriemen E, Werz DB. Flexible synthesis of 2-deoxy-C-glycosides and (1→2)-, (1→3)-, and (1→4)-linked C-glycosides. Angew. Chem. Int. Ed. 2013;52:2985–2989. doi: 10.1002/anie.201209697. [DOI] [PubMed] [Google Scholar]
- 33.Zhao C, Jia X, Wang X, Gong H. Ni-Catalyzed reductive coupling of alkyl acids with unactivated tertiary alkyl and glycosyl halides. J. Am. Chem. Soc. 2014;136:17645–17651. doi: 10.1021/ja510653n. [DOI] [PubMed] [Google Scholar]
- 34.Zhu F, Rourke MJ, Yang T, Rodriguez J, Walczak MA. Highly stereospecific cross-coupling reactions of anomeric stannanes for the synthesis of C-aryl glycosides. J. Am. Chem. Soc. 2016;138:12049–12052. doi: 10.1021/jacs.6b07891. [DOI] [PubMed] [Google Scholar]
- 35.Adak L, et al. Synthesis of aryl C-glycosides via iron-catalyzed cross coupling of halosugars: stereoselective anomeric arylation of glycosyl radicals. J. Am. Chem. Soc. 2017;139:10693–10701. doi: 10.1021/jacs.7b03867. [DOI] [PubMed] [Google Scholar]
- 36.Zhu F, Rodriguez J, O’Neill S, Walczak MA. Acyl glycosides through stereospecific glycosyl cross-coupling: rapid access to C(sp3)-linked glycomimetics. ACS Cent. Sci. 2018;4:1652–1662. doi: 10.1021/acscentsci.8b00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakamoto K, Nagai M, Ebe Y, Yorimitsu H, Nishimura T. Iridium-catalyzed direct hydroarylation of glycals via C–H activation: ligand-controlled stereoselective synthesis of α- and β-C-glycosyl arenes. ACS Catal. 2019;9:1347–1352. doi: 10.1021/acscatal.8b04686. [DOI] [Google Scholar]
- 38.de Robichon M, et al. Access to C-aryl/alkenylglycosides by directed Pd-catalyzed C–H functionalisation of the anomeric position in glycal-type substrates. Chem. Commun. 2019;55:11806–11808. doi: 10.1039/C9CC05993H. [DOI] [PubMed] [Google Scholar]
- 39.Wang Q, et al. Palladium-catalysed C−H glycosylation for synthesis of C-aryl glycosides. Nat. Catal. 2019;2:793–800. doi: 10.1038/s41929-019-0324-5. [DOI] [Google Scholar]
- 40.Li M, et al. Visible-light-induced Pd-catalyzed radical strategy for constructing C-vinyl glycosides. Org. Lett. 2020;22:6288–6293. doi: 10.1021/acs.orglett.0c02053. [DOI] [PubMed] [Google Scholar]
- 41.Lv W, Chen Y, Wen S, Ba D, Cheng G. Modular and stereoselective synthesis of C-aryl glycosides via Catellani reaction. J. Am. Chem. Soc. 2020;142:14864–14870. doi: 10.1021/jacs.0c07634. [DOI] [PubMed] [Google Scholar]
- 42.Ma Y, et al. Highly stereoselective synthesis of aryl/heteroaryl-C-nucleosides via the merger of photoredox and nickel catalysis. Chem. Commun. 2019;55:14657–14660. doi: 10.1039/C9CC07184A. [DOI] [PubMed] [Google Scholar]
- 43.Wei Y, Ben-zvi B, Diao T. Diastereoselective synthesis of aryl C-glycosides from glycosyl esters via C-O bond homolysis. Angew. Chem. Int. Ed. 2021;60:9433–9438. doi: 10.1002/anie.202014991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitamura K, Ando Y, Matsumoto T, Suzuki K. Total synthesis of aryl C-glycoside natural products: strategies and tactics. Chem. Rev. 2018;118:1495–1598. doi: 10.1021/acs.chemrev.7b00380. [DOI] [PubMed] [Google Scholar]
- 45.Yang G, Schmieg J, Tsuji M, Franck RW. The C-glycoside analogue of the immunostimulant α-galactosylceramide (KRN7000): synthesis and striking enhancement of activity. Angew. Chem. Int. Ed. 2004;43:3818–3822. doi: 10.1002/anie.200454215. [DOI] [PubMed] [Google Scholar]
- 46.Goodwin NC, et al. Discovery of LX2761, a sodium-dependent glucose cotransporter 1 (SGLT1) inhibitor restricted to the intestinal lumen, for the treatment of diabetes. J. Med. Chem. 2017;60:710–721. doi: 10.1021/acs.jmedchem.6b01541. [DOI] [PubMed] [Google Scholar]
- 47.Mydock-McGrane L, et al. Antivirulence C-mannosides as antibiotic-sparing, oral therapeutics for urinary tract infections. J. Med. Chem. 2016;59:9390–9408. doi: 10.1021/acs.jmedchem.6b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boehlich GJ, Schützenmeister N. β-Selective C-glycosylation and its application in the synthesis of scleropentaside A. Angew. Chem. Int. Ed. 2019;58:5110–5113. doi: 10.1002/anie.201900995. [DOI] [PubMed] [Google Scholar]
- 49.Zou L-J, et al. Cyanide-free synthesis of glycosyl carboxylic acids and application for the synthesis of scleropentaside A. Org. Lett. 2020;22:8302–8306. doi: 10.1021/acs.orglett.0c02949. [DOI] [PubMed] [Google Scholar]
- 50.Wang Q, et al. Total synthesis of C-α-mannosyl tryptophan via palladium-catalyzed C–H glycosylation. CCS Chem. 2020;2:1729–1736. [Google Scholar]
- 51.Shang W, et al. Generation of glycosyl radicals from glycosyl sulfoxides and its use in the synthesis of C-linked glycoconjugates. Angew. Chem. Int. Ed. 2021;60:385–390. doi: 10.1002/anie.202009828. [DOI] [PubMed] [Google Scholar]
- 52.Wu J, Kaplaneris N, Ni S, Kaltenhäuser F, Ackermann L. Late-stage C(sp2)–H and C(sp3)–H glycosylation of C-aryl/alkyl glycopeptides: mechanistic insights and fluorescence labeling. Chem. Sci. 2020;11:6521–6526. doi: 10.1039/D0SC01260B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, Subramanian P, Martinazzoli O, Wu J, Ackermann L. Glycopeptides by linchpin C−H activations for peptide-carbohydrate conjugation by manganese(I)-catalysis. Chem. Eur. J. 2019;25:10585–10589. doi: 10.1002/chem.201902788. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y, et al. Palladium-catalysed C(sp3)−H glycosylation for the synthesis of C-alkyl glycoamino acids. Angew. Chem. Int. Ed. 2020;59:3491–3494. doi: 10.1002/anie.201914184. [DOI] [PubMed] [Google Scholar]
- 55.Ji P, et al. Visible-light-mediated, chemo- and stereoselective radical process for the synthesis of C-glycoamino acids. Org. Lett. 2019;21:3086–3092. doi: 10.1021/acs.orglett.9b00724. [DOI] [PubMed] [Google Scholar]
- 56.Yan X, et al. Stereodivergent synthesis of C-glycosamino acids via Pd/Cu dual catalysis. Sci. China Chem. 2021;64:552–557. doi: 10.1007/s11426-020-9930-7. [DOI] [Google Scholar]
- 57.Lu XY, et al. 1,1-Disubstituted olefin synthesis via Ni-catalyzed Markovnikov hydroalkylation of alkynes with alkyl halides. Chem. Commun. 2016;52:5324–5327. doi: 10.1039/C6CC00176A. [DOI] [PubMed] [Google Scholar]
- 58.Lu X, et al. Practical carbon–carbon bond formation from olefins through nickel-catalyzed reductive olefin hydrocarbonation. Nat. Commun. 2016;7:11129. doi: 10.1038/ncomms11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He Y, Cai Y, Zhu S. Mild and regioselective benzylic C–H functionalization: Ni-catalyzed reductive arylation of remote and proximal olefins. J. Am. Chem. Soc. 2017;139:1061–1064. doi: 10.1021/jacs.6b11962. [DOI] [PubMed] [Google Scholar]
- 60.Wang Z, Yin H, Fu GC. Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins. Nature. 2018;563:379. doi: 10.1038/s41586-018-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bera S, Mao R, Hu X. Enantioselective C(sp3)–C(sp3) cross-coupling of non-activated alkyl electrophiles via nickel hydride catalysis. Nat. Chem. 2021;13:270–277. doi: 10.1038/s41557-020-00576-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lipshutz BH, Frieman BA. CuH in a bottle: a convenient reagent for asymmetric hydrosilylations. Angew. Chem. Int. Ed. 2005;44:6345–6348. doi: 10.1002/anie.200500800. [DOI] [PubMed] [Google Scholar]
- 63.Zhu S, Buchwald SL. Enantioselective CuH-catalyzed anti-Markovnikov hydroamination of 1,1-disubstituted alkenes. J. Am. Chem. Soc. 2014;136:15913–15916. doi: 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giese B. The stereoselectivity of intermolecular free radical reactions. Angew. Chem. Int. Ed. 1989;28:969–980. doi: 10.1002/anie.198909693. [DOI] [Google Scholar]
- 65.Roe BA, Boojamra CG, Griggs JL, Bertozzi CR. Synthesis of β-C-glycosides of N-acetylglucosamine via Keck allylation directed by neighboring phthalimide groups. J. Org. Chem. 1996;61:6442–6445. doi: 10.1021/jo960819o. [DOI] [PubMed] [Google Scholar]
- 66.Togo, H., Wei, H., Waki, Y. & Yokoyama, M. C-Glycosidation technology with free radical reactions. Synlett 700–717 (1998).
- 67.Abe H, Shuto S, Matsuda A. Highly α- and β-selective radical C-glycosylation reactions using a controlling anomeric effect based on the conformational restriction strategy. A study on the conformation–anomeric effect–stereoselectivity relationship in anomeric radical reactions. J. Am. Chem. Soc. 2001;123:11870–11882. doi: 10.1021/ja011321t. [DOI] [PubMed] [Google Scholar]
- 68.Yu B. Gold(I)-catalyzed glycosylation with glycosyl o-alkynylbenzoates as donors. Acc. Chem. Res. 2018;51:507–516. doi: 10.1021/acs.accounts.7b00573. [DOI] [PubMed] [Google Scholar]
- 69.Chao Z, Zhang J, Kong F. A concise synthesis of two isomeric pentasaccharides, the O repeat units from the lipopolysaccharides of P. syringae pv. porri NCPPB 3364T and NCPPB 3365. Carbohydr. Res. 2004;339:43–49. doi: 10.1016/j.carres.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 70.Kobayashi M, Morita T. Significant expression patterns of Lewis X-related antigens as a prognostic predictor of low-stage renal cell carcinomas. Anticancer Res. 2010;30:593–600. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information file, including experimental details, characterization data, and 1H and 13C NMR spectra of new compounds. All data are available from the corresponding authors upon reasonable request.