

Abstract
Propylamycin (4’-deoxy-4’-propylparomomycin) is a next generation aminoglycoside antibiotic that displays increased antibacterial potency over the parent, coupled with reduced susceptibility to resistance determinants and reduced ototoxicity in the guinea pig model. Propylamycin nevertheless is inactivated by APH(3’)-Ia, a specific aminoglycoside phosphotransferase isozyme that acts on the primary hydroxy group of the ribofuranosyl moiety (at the 5”-position). To overcome this problem we have prepared and studied the antibacterial and antiribosomal activity of various propylamycin derivatives carrying amino or substituted amino groups at the 5”-position in place of the vulnerable hydroxy group. We find that the introduction of an additional basic amino group at this position, while overcoming the action of the aminoglycoside phosphoryltransferase isozymes acting at the 5”-position as anticipated, results in a significant drop in selectivity for the bacterial over the eukaryotic ribosomes that is predictive of increased ototoxicity. In contrast, 5”-deoxy-5”-formamidopropylamycin retains the excellent across-the-board levels of antibacterial activity of propylamycin itself, while circumventing the action of the offending aminoglycoside phosphotransferase isozymes, and affording even greater selectivity for the bacterial over the eukaryotic ribosomes. Other modifications to address the susceptibility of propylamycin to the APH(3’)-Ia isozyme including deoxygenation at the 3’-position and incorporation of a 6’,5”-bis(hydroxyethylamino) modification offer no particular advantage.
Keywords: Aminoglycoside modifying enzymes, ribosomal methyltransferases, antibacterial, antiribosomal, ototoxicity
Graphical Abstract
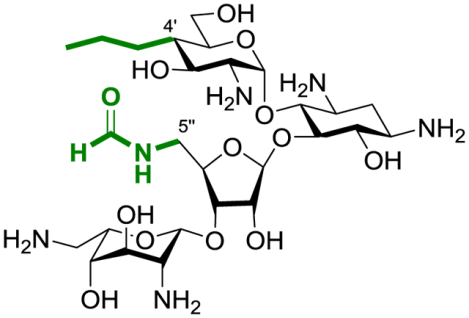
Synopsis: The TOC graphic shows the structure of the optimal compound with the two key features colored green for emphasis.
We recently described propylamycin 1 (Figure 1),1 a next generation 2-deoxystreptamine-type (DOS) aminoglycoside antibiotic (AGA)2–5 derived from the parent paromomycin 2 by substitution of the 4’-hydroxy group in ring I by a propyl group. Propylamycin displays superior in vitro antibacterial activity to the parent against a broad selection of ESKAPE pathogens and improved in vivo activity against E. coli in neutropenic mouse thigh and septicemia models. Propylamycin retains full activity against clinical isolates carrying various aminoglycoside modifying enzymes (AMEs)6–11 that impart resistance against the parent, including the AAC(3), and AAC(6’) aminoglycoside acetyltransferases, the aminoglycoside nucleotidyltransferases ANT(4’,4”), and aminoglycoside phosphotransferases from the APH(3’) family. Additionally, propylamycin retains full activity in the presence of the G1405 ribosomal methyltransferases (RMTs) that completely abrogates the activity of all DOS-type AGAs in current clinical use.12–15 Finally, at the target level, propylamycin shows enhanced selectivity for inhibition of protein synthesis by the bacterial ribosome over hybrid bacterial ribosomes carrying the complete decoding A site of the human mitochondrial, A1555G mutant mitochondrial, and cytoplasmic ribosomes resulting in reduced ototoxicity in a guinea pig model. In spite of these multiple attributes, we have subsequently found that, like 3’-deoxyparomomycin or lividomycin B 3,16 propylamycin is susceptible to inactivation by a subclass of APH(3’)s acting at both the 3’- and the 5”-hydroxy groups, namely the APH(3’,5”)s.17
Figure 1.
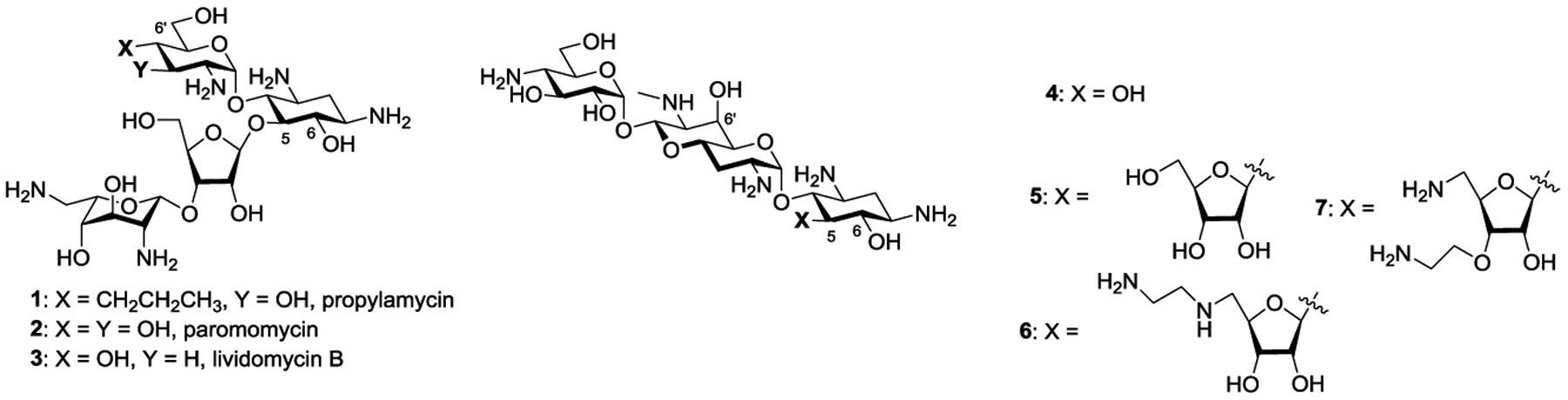
Propylamycin, Paromomycin, Lividomycin B and the Apralogs.
In seeking to overcome the susceptibility of propylamycin toward the APH(3’,5”)s, we were guided by our efforts on the development of the apralogs, namely 5-O-furanosylated analogs of the atypical DOS-type AGA apramycin 4.18, 19 Thus, while the simple 5-O-β-D-ribofuranosyl apralog 5, described originally by Japanese scientists and subsequently by Fridman and coworkers,20, 21 was susceptible to APH(3’,5”)s, derivatives lacking the primary hydroxy group in the furanosyl ring were protected from the action of these AMEs. In particular, the aminodeoxy derivatives 6 and 7 (Figure 1) were found to exhibit improved in vitro and in vivo antibacterial activity over apramycin itself in the presence of multiple relevant AMEs, including the APH(3’,5”)s, and RMTs. In addition to displaying improved activity 6 and 7 also showed increased selectivity, as determined by a cell-free ribosomal translation assay and as borne out by reduced cochleotoxicity in ex vivo mouse cochlear ex-plant studies.
Combining the attributes of propylamycin 1 and apralogs 6 and 7, we designed and report here on the synthesis and evaluation of the antiribosomal and antibacterial activities of the 5”-aminodeoxy analogs 8–13 of propylamycin (Figure 2). In order to help distinguish between the 3’ and 5”-prongs of the APH(3’,5”)s, we also report the synthesis and assessment of 3’-deoxypropylamycin 14. Finally, exploring an alternative avenue to the introduction of amino functionality at the 5”-position, we describe a 6’,5”-bis(hydroxyethylamino) analog 15 of propylamycin (Figure 2).
Figure 2.
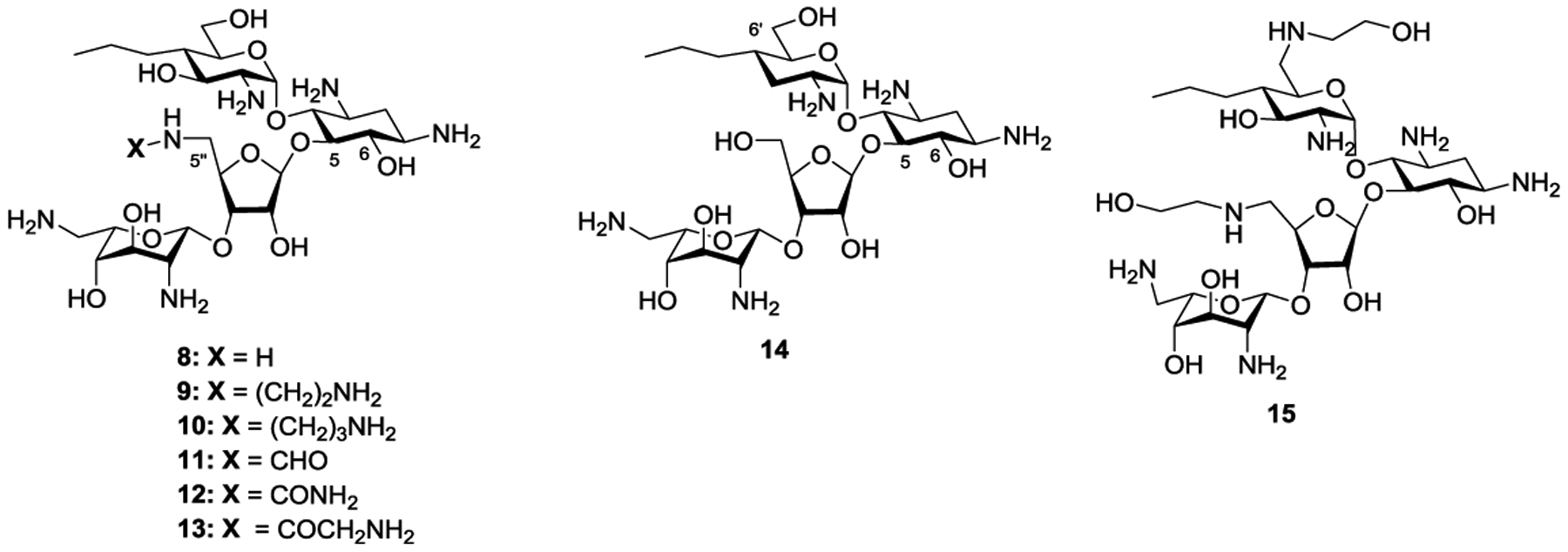
Target Propylamycin Derivatives.
Results and Discussion
Synthesis.
The synthesis of the 5”-aminodeoxy analogs 8–13 of propylamycin 1 was based on that developed for propylamycin itself, with suitable modification for functionalization of the primary hydroxy group in the ribofuranoside side chain. Briefly, the paromomycin 4’,6’-O-benzylidene pentatrifluoroacetamide 16, obtained from paromomycin as described previously,1 was converted to the 5”-O-silyl ether 17 by standard means in 95% yield. Perbenzoylation with benzoic anhydride in the presence of 4-dimethylaminopyridine in pyridine to give 18 in 92% yield, was followed by desilylation using tetrabutylammonium fluoride under standard conditions to give the mono-ol 19 suitable for introduction of the requisite amino group in 99% yield. Triflation with triflic anhydride in the presence of 2,6-lutidine was followed by displacement with sodium azide giving the azide 20 in 77% yield overall. Staudinger reaction with trimethylphosphine to give amine 21 in 40% yield was followed by conversion to the benzyl carbamate 22 in 87% yield under standard conditions, hydrolysis of the benzylidene acetal with aqueous acetic acid to give diol 23 in 86% yield, and regioselective monobenzoylation with benzoyl cyanide to give 24 in 91% yield, primed for introduction of the propyl chain. To this end triflation of mono-ol 24 was followed by displacement with potassium iodide in acetone affording the galacto-configured axial iodide 25 in 83% yield. Treatment of iodide 25 with allyl phenyl sulfone in 1,2-dichlorobenzene with initiation by triethylborane and oxygen at 0 °C1 then gave the 4’-deoxy-4’-C-propyl derivative 26 in 40% yield (Scheme 1).
Scheme 1.
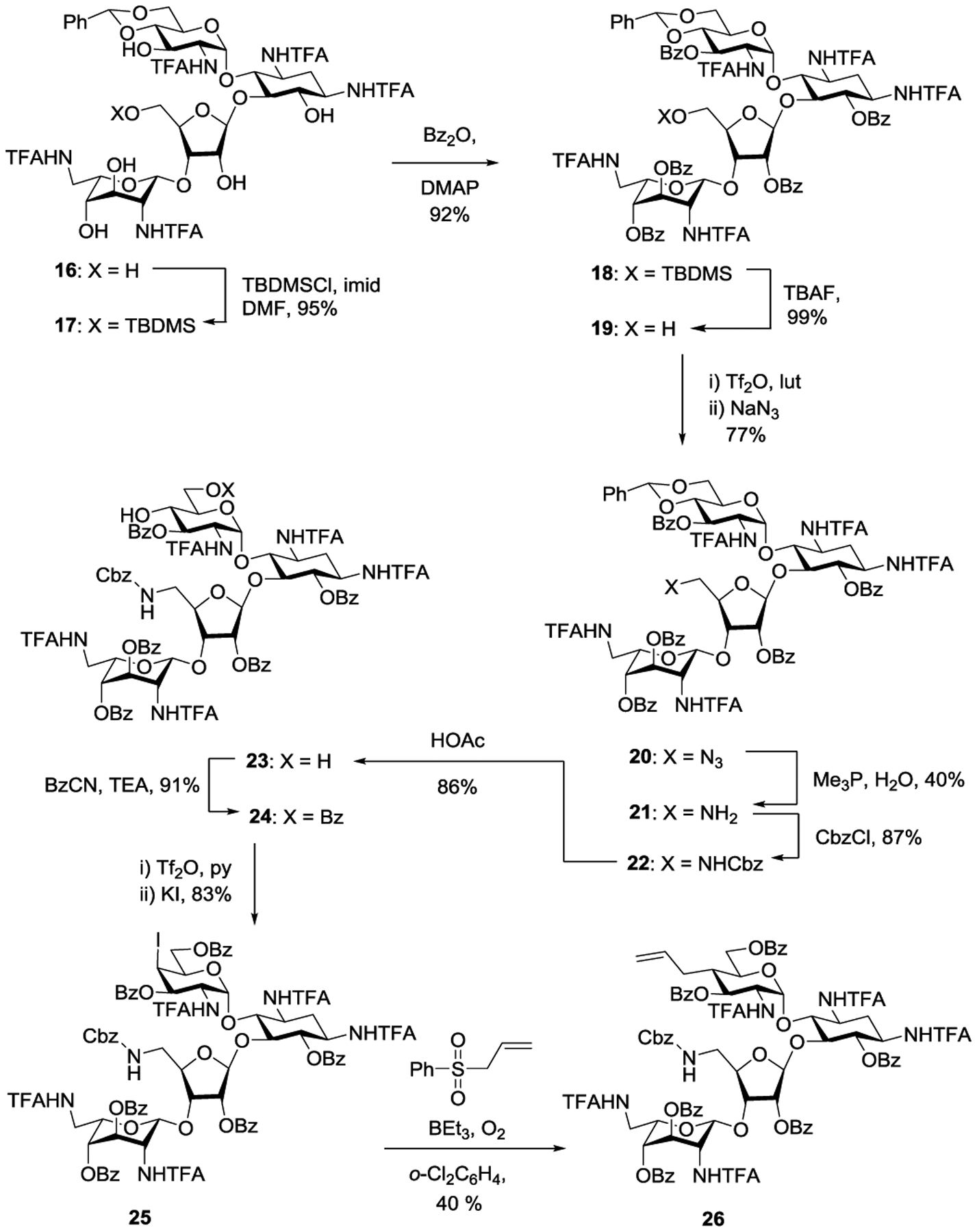
Synthesis of Key Intermediate 26 en route to propylamycin derivatives 8–13
In addition to the allylation of 25 by the action of allyl phenyl sulfone with initiation by triethylborane and oxygen, which mirrored the earlier synthesis of propylamycin itself (Scheme 1), we chose to briefly explore the use of photocatalytic initiation for allylation. A variety of transition metal-based photocatalytic systems have been employed to promote the coupling of alkyl iodides with allyl phenyl sulfone and its congeners, including palladium and manganese-based catalysts22, 23 and iridium-based systems24 typically in the presence of a tertiary amine as sacrificial electron source, and indeed we have employed one such Ir-based system in our laboratories in the course of a synthesis of bradyrhizose.25 Here, we elected to examine the metal-free 1,3,4,5-tetra(N-carbazolyl)-2,6-dicyanobenzene (4-CzIPN) photocatalyst in combination with triethylamine as overall reductant26, 27 as applied recently to allylation of alkyl iodides with allyl phenyl sulfone as trap.28 Working with N-tert-butyloxycarbonyl-4-iodopiperidine we were able to reproduce, the literature photocatalytic allylation reaction,28 but were frustrated to find that application of the same conditions to iodide 25 resulted only in the formation of the elimination product 27, which ultimately was traced to competing elimination of the axial iodide by the triethylamine (Scheme 2).
Scheme 2.
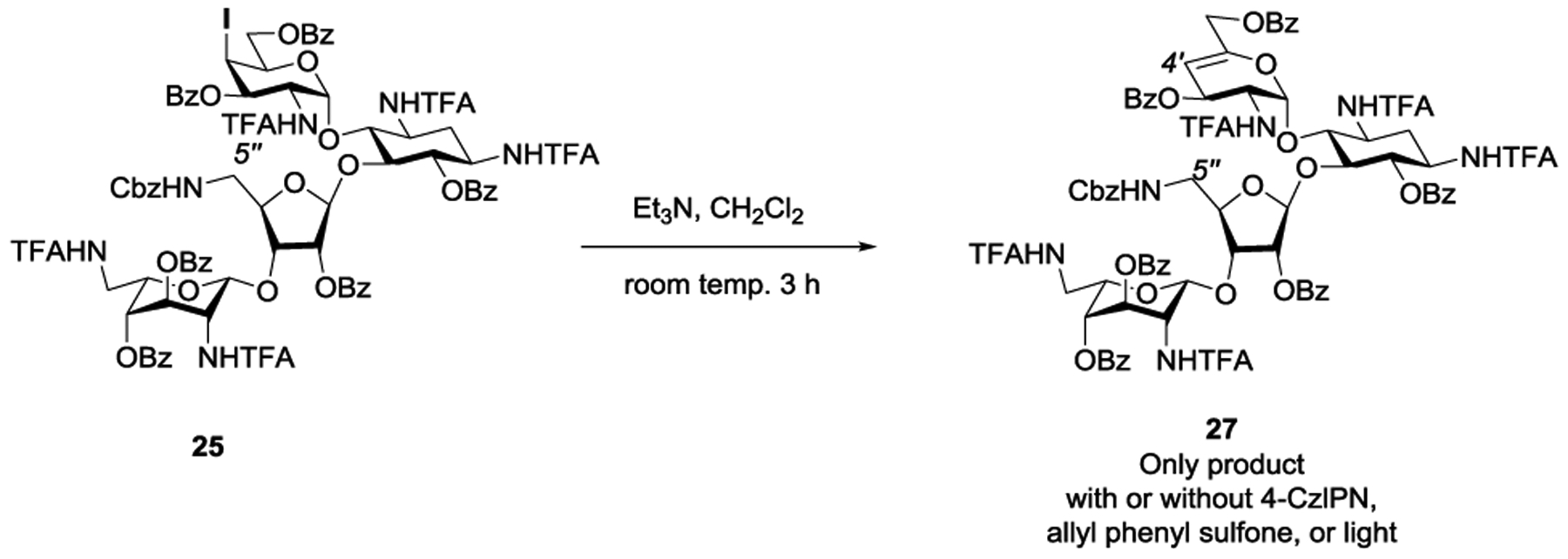
Competing Elimination of Attempted Photocatalytic Allylation of 25
We therefore prepared the corresponding equatorial iodide 29 by Lattrel-Dax epimerization of the gluco-alcohol 24 to its galacto-epimer 28,29–31 followed by triflation and displacement with potassium iodide (Scheme 3). White light irradiation of equatorial iodide 29 in the presence of 5 mol % CzIPN and triethylamine in DMSO afforded a 1:1.2 mixture of the desired allylation product 26 and the reduction product 30-H, but none of the elimination product 27, thereby clearly demonstrating the configuration sensitive nature of the elimination reaction. A slight improvement of the 26:30-H ratio to 1:1 was achieved by using an excess (5 equiv) of allyl phenyl sulfone. The hydrogen atom required for the formation of reduction product 30-H is not derived from the solvent DMSO as was revealed by the use of d6-DMSO. However, replacement of triethylamine by d15-triethylamine resulted in the formation of 3:1 mixture of 26:30-H with approximately 50% incorporation of deuterium (1:1 30-H:30-D), suggesting that triethylamine is the ultimate hydrogen atom source for the formation of 30-H, and that reduction can be at least in part suppressed leading to improved yields of allylation by means of the kinetic isotope effect, as is known for other radical C-C bond forming processes.32 Overall, while these brief studies were ultimately unsuccessful in providing a practical route to 26, they do serve to outline some limitations in the application of photocatalytic processes to the complex substrates particularly when tertiary amines are required as overall reductant.
Scheme 3.
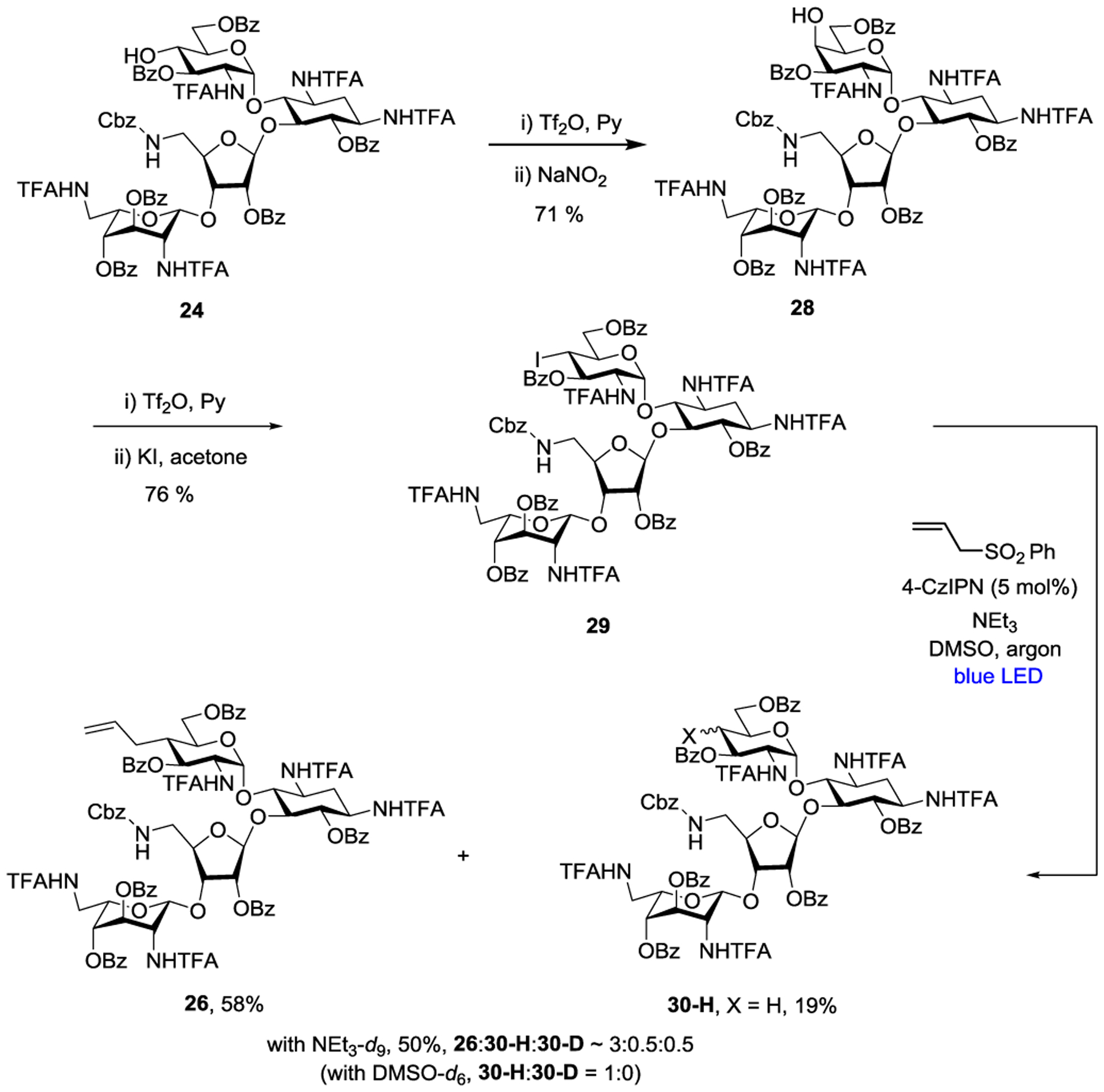
Competing Photocatalytic Allylation and Reduction of Equatorial Iodide 29
Returning to the synthesis of the target aminodeoxy propylamycin analogs 8–13, hydrogenation of 26 over palladium on charcoal in 80% acetic acid afforded the pivotal amine 31 in quantitative yield. Reductive amination of 31 with a pair of trifluoracetamide substituted aldehydes 32 and 33, prepared as described in the supporting information, afforded the corresponding amines 34 and 35, which were immediately protected as the corresponding trifluoracetamides 36 and 37 for ease of purification in moderate overall yield for the two steps. Acylation of amine 31 with benzyloxycarbamoyl chloride, formic acetic anhydride, benzylisocyanate and N-trifluoroacetylglycine, the latter with the aid of HATU, afforded the corresponding 5”-amido derivatives 38–41 in good to excellent yield. Finally, deprotection of 36–41, yielding 8–13 was achieved by a two step protocol, designed to prevent O→N benzoyl group migration,1 involving initial removal of the benzoate esters with magnesium methoxide in dry methanol followed by heating with aqueous barium hydroxide in moderate to good overall yield following HPLC purification (Scheme 4).
Scheme 4.
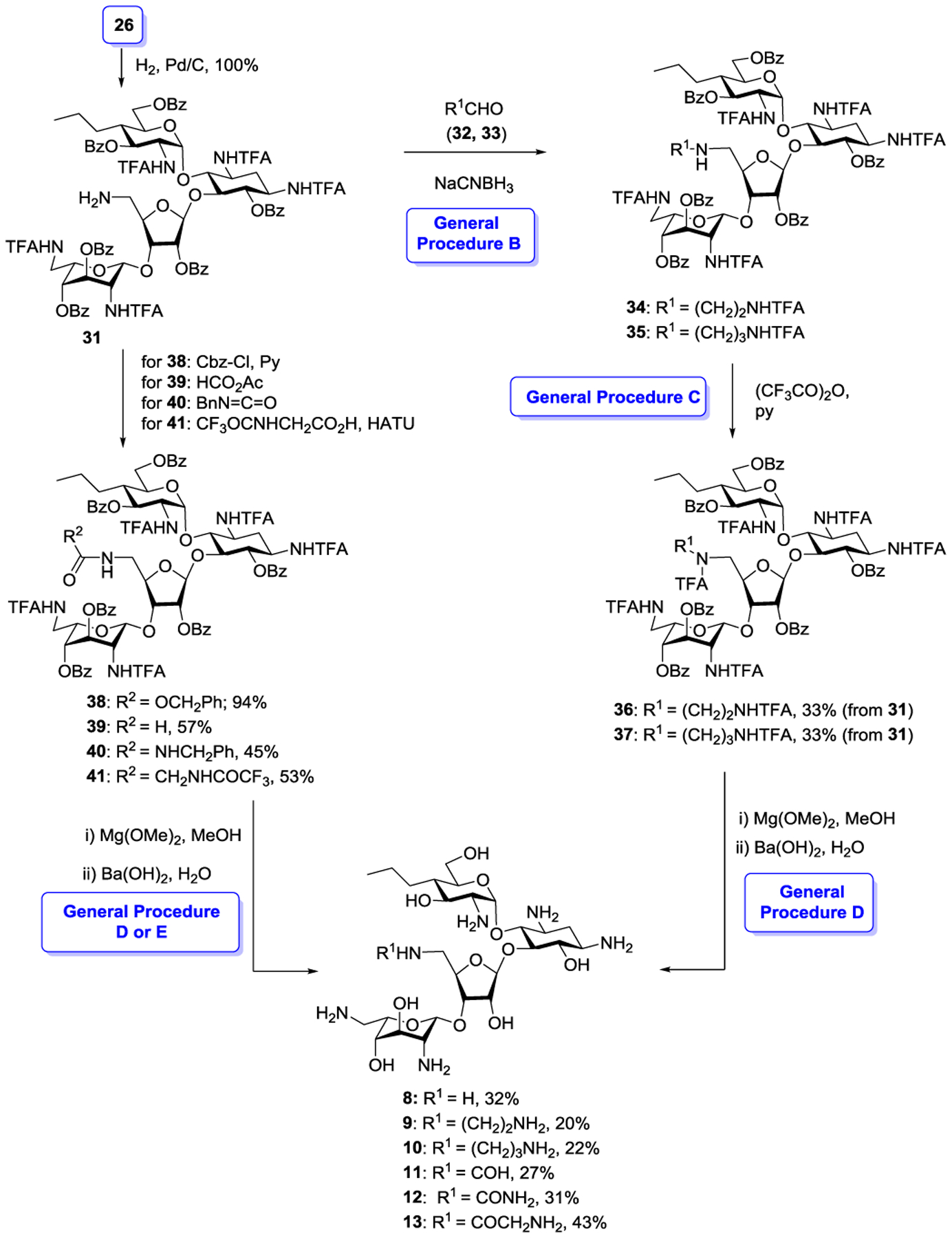
Completion of 5”-N-Functionalized Propylamycin Derivatives 8–13
For the synthesis of 3’-deoxypropylamycin 14 we developed a route based on cleavage of ring I from a suitable protected paromomycin derivative followed by reinstallation of the modified ring I. A suitable glycosyl donor for this purpose 45 was obtained from the 1,6-anhydro pyranose derivative 42, itself available from 1,6-anhydroglucose via the Cerny epoxide as described previously,33 by ring opening with S-trimethylsilyl thiophenol in the presence of zinc iodide34 followed by treatment with aqueous methanolic potassium carbonate to give the thioglycoside 43 in 87% yield as a 2.8:1 α:β mixture of anomers. Acetylation of 43 under standard conditions gave 44 in 97% yield and was followed by oxidation with Selectfluor35 giving the glycosyl sulfoxide 45 in 98% yield as a mixture of isomers. Sulfoxide 45 was coupled to acceptor 46, prepared as previously described,36 under the aegis of triflic anhydride and in the presence of the hindered base 2,4,6-tri-tert-butylpyrimidine affording 47 as a 12:1 α:β mixture in 48% yield. Hydrogenolysis of 47 over Pearlman’s catalyst followed by heating with aqueous barium hydroxide and finally chromatography over Sephadex C25 gave the target compound 14 in 41% yield (Scheme 5).
Scheme 5.
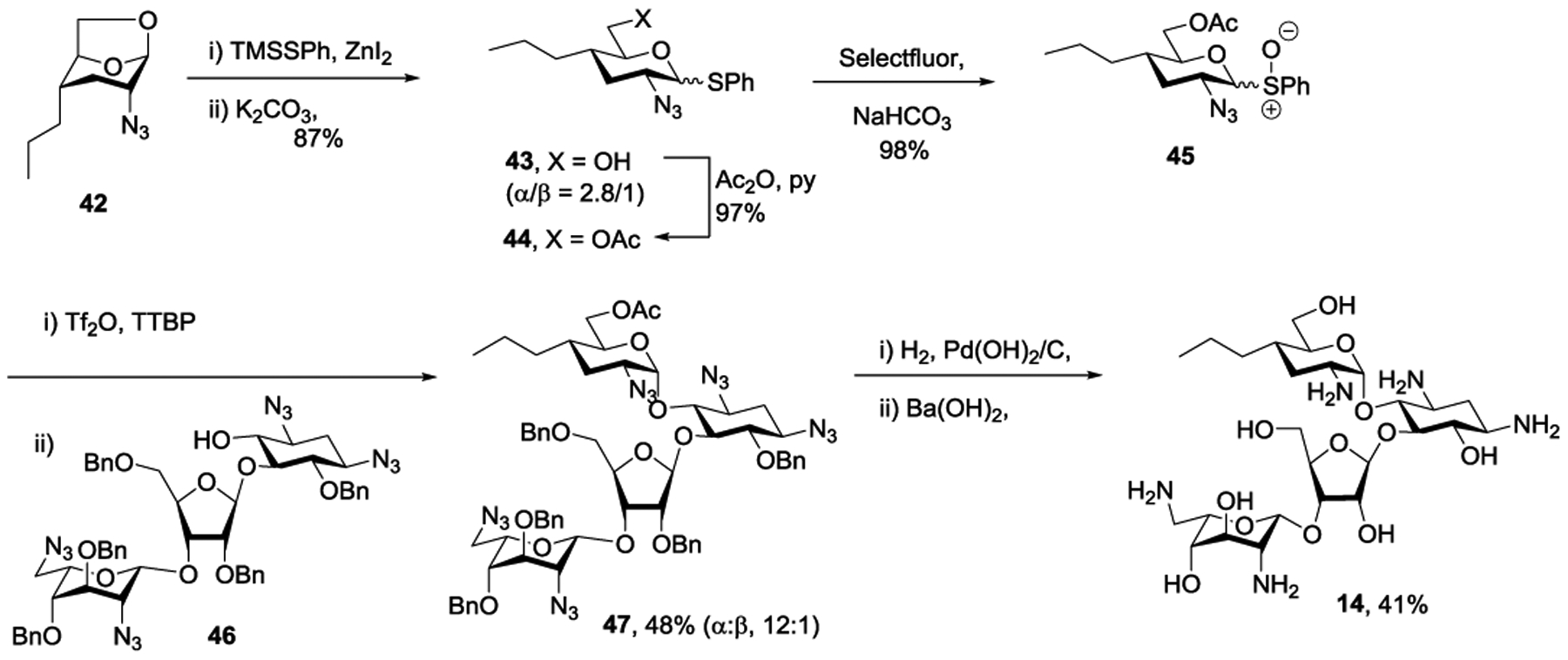
Synthesis of 3’-Deoxypropylamycin 14
Finally, exploring an alternative avenue to the introduction of an amino group at the 5”-position, propylamycin 1 was converted in 86% yield to the penta-N-benzyloxycarbamoyl derivative 48. This was followed by selective sulfonylation of the two primary hydroxy groups with 2,4,6-triisopropylbenzene sulfonyl chloride in pyridine giving 49 in 45% yield. Heating to 100 °C with ethanolamine then afforded the doubly substituted derivative 50 in 40% yield, and was followed by hydrogenolysis and chromatography over Sephadex to give 15 in 21% yield (Scheme 6).
Scheme 6.
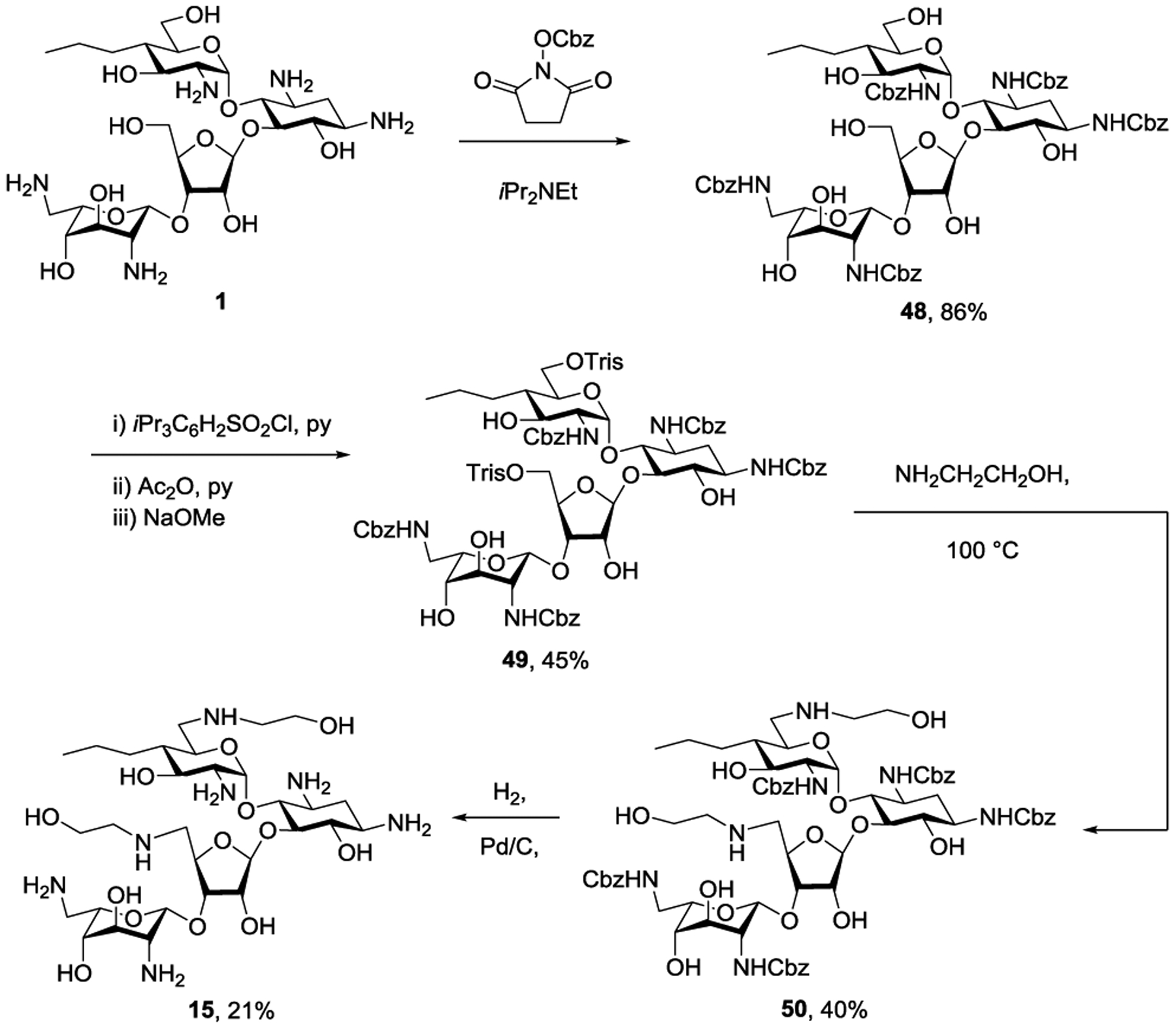
Synthesis of 6’,5”-Dideoxy-6’,5”-bis(ethanolamino)propylamycin 15
Antibacterial Activity Against Wild-type Bacterial Strains
All newly prepared compounds and the comparators propylamycin 1, paromomycin 2, and lividomycin B 3 were tested for activity against a panel of ESKAPE pathogens sourced from the diagnostic department of the Institute of Medical Microbiology, at the University of Zurich. This panel was made up of a Gram-positive methicillin-resistant Staphylococcus aureus (MRSA) strain, and a panel of wild-type Gram negative pathogens (Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Acinetobacter baumannii, Pseudomonas aeruginosa) (Table 1). Although we have previously demonstrated the parent propylamycin to show excellent activity against Mycobacteria,1 we excluded them from this study because of their low incidence of APH(3’)s. The replacement of the hydroxy group at the 5”-position of propylamycin 1 by a primary amine as in 8 resulted in an approximately two-fold reduction in activity across the range of ESKAPE pathogens assayed, with the exception of the P. aeruginosa strain for which a two-fold improvement in activity was seen. No advantage was gained in terms of activity by the elaboration of the 5”-amine in 3 into two diamino alkyl chains as in 9 and 10, rather a gradual drop-off in activity was observed as the chain was lengthened and a further amino group incorporated. Notably, the protection of the 5”-amino group in 3 in the form of the formamide 11 restored activity across the board to levels seen in propylamycin itself, and comparable to those of the clinical AGAs gentamicin and plazomicin. The corresponding urea and glycinamide modification at the 5”-position, 12 and 13, were not as effective as the formamide but nevertheless retained good activity. 3’-Deoxypropylamycin 14 was marginally less active than propylamycin itself, but was notably more active than 3’-deoxyparomomycin (lividomycin B) 3 thereby clearly highlighting the general increase in activity conferred by the 4’-deoxy-4’-propyl modification. Finally, the 6’,5”-bis(hydroxyethylamino) derivative 15 was notably less active than propylamycin, consistent with the effects of the analogous modification on paromomycin reported earlier.37
Table 1.
Antibacterial Activities Against Wild-Type E. coli and ESKAPE Pathogens (MIC, μg/mL)a
| Species | MRSA | E. coli | K. pneu. | Enterob. | A. baum. | P. aerug. b |
|---|---|---|---|---|---|---|
| Strain | AG038 | AG001 | AG215 | AG290 | AG225 | AG220 |
| Propylamycin 1 | 1–2 | 1 | 0.25–0.5 | 0.5 | 1–2 | 8–16 |
| Paromomycin 2 | 2 | 2–4 | 1 | 2 | 2–4 | >128 |
| Lividomycin B 3 | 4 | 4–8 | 2 | 2 | 4 | 4 |
| 8 | 2 | 2 | 1 | 1 | 2 | 4 |
| 9 | 1–2 | 2–4 | 1 | 4 | 4 | 4 |
| 10 | 2 | 2–4 | 2 | 8 | 8 | 16 |
| 11 | 1 | 1 | 1 | 1 | 2 | 8 |
| 12 | 2 | 4 | 2–4 | 2–4 | 2–4 | 128 |
| 13 | 2 | 2–4 | 2 | 2 | 2 | 4 |
| 14 | 2 | 1–2 | 1 | 1 | 2 | 4 |
| 15 | 4 | 4 | 2 | 2 | 32 | >64 |
| Gentamicin | 1–2 | 0.5–1 | 0.25 | 0.25 | 0.5–1 | 1 |
| Plazomicin | 2 | 1 | 0.5 | 0.5 | 2 | 2–4 |
All values were determined in duplicate using twofold dilution series.
P. aeruginosa carries a chromosomal APH(3’) gene, which principally affects the 3’-hydroxy group.
Antibacterial Activity Against Resistant Bacterial Strains
To determine the ability of the various modifications to overcome resistance due to the presence of AMEs, particularly the APHs acting at the 3’- and 5”-positions, we turned to a panel of engineered E. coli with each member carrying a specific resistance determinant (Table 2). In addition to surveying four APH isoforms, we also investigated the AAC(3)-IV AME known to be problematic in the apramycin series,18, 19 and two RMTs acting on G1405, which seriously abrogate the activity of all DOS-type AGAs in current clinical use (Table 2). All four APHs included in this study seriously curtailed the activity of paromomycin 2, indicating them to act at either the 3’, or the 5”-positions, or both. In contrast, the strains carrying APH(3’)-IIa, APH(3’)-IIb, and APH(3’)-VI were all susceptible to 3’-deoxyparomomycin (lividomycin B) 3, while only the APH(3’)-Ia carrying strain was not. APH(3’) isoforms IIa, IIb and VI therefore act only at the 3’-position, while isoform Ia acts at both the 3’ and 5”-positions. Thus, it is clear that 4’-deoxy-4’-propyl modification that characterizes propylamycin is effective in blocking the action of APHs at the adjacent 3’-position, but does not hinder the action of isoforms acting at the 5”-position. Modification of propylamycin at the 5”-position to remove the hydroxy group is effective at overcoming the action of APH(3’)-Ia and affords a series of compounds that retain excellent activity in the presence of each of the four APH(3’) isoforms investigated. Consistent with the activity levels against the ESKAPE pathogens (Table 1) the optimal compound in this series was the 5”-deoxy-5”-formamide 11. The profile of 3’-deoxypropylamycin 14 in the face of the various APH(3’) isoforms was essentially unchanged with respect to propylamycin itself indicating the high level of protection afforded by the 4’-deoxy-4’-propyl modification against those isoforms acting solely at the 3’-position. Interestingly, the 6’,5”-(bishydroxyethylamino) derivative 15 was susceptible to inactivation by APH(3’)-Ia suggesting that this isoform is sufficiently accommodating to accept the hydroxy group of the 5”-(hydroxyethylamino)-substituent as substrate. Most compounds retained their full activity in the presence of the AAC(3)-IV, ArmA and RmtB resistance determinants suggesting that the various modifications introduced at the 5”-position little affect resilience toward the AAC(3)-IV itself or to target modification by ArmA or RmtB.
Table 2.
Activities Against E. coli in the Presence of Specific Resistance Determinants (MIC, μg/mL)a
| Resistance det | WT-parental | APH(3’)-Ia | APH(3’)-IIa | APH(3’)-IIb | APH(3’)-VI | AAC(3)-IV | ArmA | RmtB |
|---|---|---|---|---|---|---|---|---|
| Strain | EC026 | EC189 | EC191 | EC125 | EC141 | EC118 | EC102 | EC103 |
| Propylamycin 1 | 0.25–0.5 | 64–128 | 0.5–1 | 0.5–1 | 0.5 | 1–2 | 0.5 | 0.5 |
| Paromomycin 2 | 0.5–1 | 128 | 64 | 64–128 | 64 | 1–2 | 0.5–1 | 0.5–1 |
| Lividomycin B 3 | 1–2 | >128 | 1–2 | 0.5 | 1 | 2 | 1 | 1–2 |
| 8 | 0.5–1 | 2–4 | 1–2 | 0.5–1 | 0.5–1 | 2–4 | 2–4 | 2–4 |
| 9 | 0.5–1 | 4 | 1–2 | 0.5–1 | 0.5 | 2 | 1–2 | 2 |
| 10 | 1–2 | 8 | 4 | 4 | 1–2 | 8 | 4 | 4–8 |
| 11 | 0.5–1 | 2 | 0.5–1 | 0.5 | 0.25 | 2 | 2 | 4 |
| 12 | 1 | 8–16 | 2–4 | 2–4 | 0.5–1 | 16 | 8 | 4 |
| 13 | 0.5–1 | 2–4 | 1–2 | 0.5–1 | 0.5 | 4 | 4 | 2–4 |
| 14 | 1 | >128 | 1–2 | 0.5–1 | 1 | 2–4 | 1 | 2 |
| 15 | 2 | >32 | 4 | 4 | 1–2 | 16 | 4 | 4–8 |
All values were determined in duplicate using twofold dilution series.
To further assess the susceptibility of the compounds prepared to AMEs we screened against a series of clinical isolates with acquired resistance determinants (Table 3). With respect to the isolates carrying the APH(3’) and AAC(3) determinants, the results are consistent with those reported in Table 2 for the engineered strains. Notably, strain AG163 carrying the APH(3’)-I resistance determinant inactivated those compounds with a hydroxy group at the 5”-position and additionally the 5”-(hydroxyethylamino) derivative 15 fully consistent with the observations from the genetically engineered strains. No compounds were found to display significant susceptibility to the presence of AAC(6’).
Table 3.
Activities Against E. coli in the Presence of Acquired Resistance Determinants (MIC, μg/mL)a
| Resistance det | WT-parental | AAC(3)-II | AAC(3’)-IV | APH(3’)-I | APH(3’)-II | AAC(6’) |
|---|---|---|---|---|---|---|
| Strain | AG001 | AG003 | AG173 | AG163 | AG166 | AG175 |
| Propylamycin 1 | 1 | 2 | 2 | >128 | 1–2 | 2–4 |
| Paromomycin 2 | 2–4 | 4 | 8–16 | >128 | >128 | 4 |
| Lividomycin B 3 | 4–8 | 8 | 8–16 | >128 | 8 | 8 |
| 8 | 2 | 4–8 | 4 | 16 | 2–4 | 4–8 |
| 9 | 2–4 | 2–4 | 4 | 4–8 | 4 | 4 |
| 10 | 2–4 | 4–8 | 8–16 | 16–32 | 4 | 4–8 |
| 11 | 1 | 2 | 4–8 | 4–8 | 2 | 2–4 |
| 12 | 4 | 4–8 | 32 | 32 | 16–32 | 4–8 |
| 13 | 2–4 | 4–8 | 8 | 8–16 | 4 | 4–8 |
| 14 | 1–2 | 4 | 4–8 | >128 | 4 | 4 |
| 15 | 4 | 8 | 4 | >32 | 16 | 8–16 |
All values were determined in duplicate using twofold dilution series.
Activity and Selectivity at the Drug Target
Turning to the investigation of activity at the target level, the ribosomal decoding A site is the well-established target for aminoglycosides in general2, 11 and for propylamycin 1 in particular.1 We employed cell-free translation assays to test the ability of the various compounds to inhibit protein synthesis by wild-type bacterial ribosomes as previously described (Table 4).38 Consistent with expectation, and the mode of action of the DOS type AGAs by binding to the ribosomal decoding A site,11, 39–43 the pattern of IC50 values for inhibition of protein synthesis by the bacterial ribosome largely followed that for antibacterial activity against the wild-type ESKAPE pathogens (Table 1), with the most active compounds being propylamycin 1, and 5”-deoxy-5”-formamidopropylamycin 11.
Table 4.
Antiribosomal Activities and Selectivities (IC50, μM).
| Antiribosomal Activity | Selectivity | ||||||
|---|---|---|---|---|---|---|---|
| wt | Mit13 | A1555G | Cyt14 | Mit13 | A1555G | Cyt14 | |
| Propylamycin 1 | 0.022±0.005 | 150±51 | 56±17 | 61±14 | 6818 | 2545 | 2773 |
| Paromomycin 2 | 0.033±0.006 | 128±67 | 12±3 | 38±6 | 3879 | 364 | 1152 |
| Lividomycin B, 3 | 0.084±0.030 | 182±35 | 24±1 | 98±12 | 2167 | 286 | 1140 |
| 8 | 0.14±0.04 | 62±24 | 38±9 | 9.0±1.6 | 443 | 271 | 64 |
| 9 | 0.091±0.008 | 55±10 | 36±8 | 31±2 | 604 | 396 | 341 |
| 10 | 0.46±0.09 | 41±12 | 25±4 | 24±2 | 89 | 54 | 52 |
| 11 | 0.034±0.014 | 505±208 | 118±12 | 175±14 | 14991 | 3471 | 5147 |
| 12 | 0.078±0.007 | 318±69 | 189±66 | 194±15 | 4077 | 2423 | 2487 |
| 13 | 0.090±0.018 | 40±16 | 10±4 | 6.1±0.4 | 444 | 111 | 68 |
| 14 | 0.064±0.005 | 204±25 | 93±31 | 305±58 | 3188 | 1453 | 4766 |
| 15 | 0.40±0.04 | 89±6 | 54±6 | 68±3 | 222 | 135 | 170 |
| Plazomicin | 0.065±0.020 | 107±33 | 5.9±2.1 | 299±82 | 1633 | 90 | 4569 |
| Gentamicin | 0.029±0.009 | 15±4 | 0.80±1.5 | 74±41 | 517 | 28 | 2552 |
As AGA binding to the cognate decoding A sites of the human mitochondrial and especially the A1555G mutant mitochondrial ribosomes in the cochlea is considered to be one of the main causes of AGA-induced ototoxicity,11, 44–50 we also screened for inhibition of protein synthesis by a set of humanized bacterial ribosomes in which the complete bacterial decoding A site has been replaced by that of the human mitochondrial (Mit13) or A1555G mutant mitochondrial ribosome (A1555G) (Figure 3).51 Finally, to assess the possibility of broader systemic toxicity, we screened for inhibition of protein synthesis by similarly engineered bacterial ribosomes carrying the human cytosolic decoding A site (Cyt14) (Figure 3). Consistent with our earlier observations and as borne out by in-vivo studies using the guinea pig model for ototoxicity,1 propylamycin 1 exhibits increased selectivity over the parent paromomycin 2 for the A site of the bacterial over the human mitochondrial and especially the mutant mitochondrial ribosome; it also exhibits consistently high selectivity for the A site of the bacterial over the human cytosolic ribosome (Table 1). The selectivity profile of 3’-deoxyparomomycin 3 on the other hand is close to that of paromomycin itself. In contrast, the 5”-deoxy-5”-amino derivatives of propylamycin 8, 9; and 10 show much reduced selectivity for each of the mitochondrial, mutant mitochondrial and cytosolic ribosomes suggestive of a reduced therapeutic index for these compounds. Compound 13, which also carries a basic amino group at the 5”-position albeit with an acetyl spacer, also suffers from this phenomenon. These observations are intriguing as we have previously demonstrated that apralogs carrying basic amino groups at the 5-position of the appended ribofuranosyl ring, as exemplified by 6, show reduced affinity for each of the three humanized ribosomes that is reflected in increased selectivity and reduced outer hair cell loss in the ex-vivo mouse cochlear explant system.18, 19 In contrast, Baasov and coworkers have demonstrated that 5”-amino-5”-deoxyribostamycin derivatives show increased affinity for the cytosolic ribosome which, coupled with reduced antibacterial activity, renders them attractive for the treatment of genetic diseases due to the mutation of an amino acid codon to a premature stop codon, and is reflected in the ongoing clinical trials of ELX-02 (Figure 4).52–54
Figure 3.
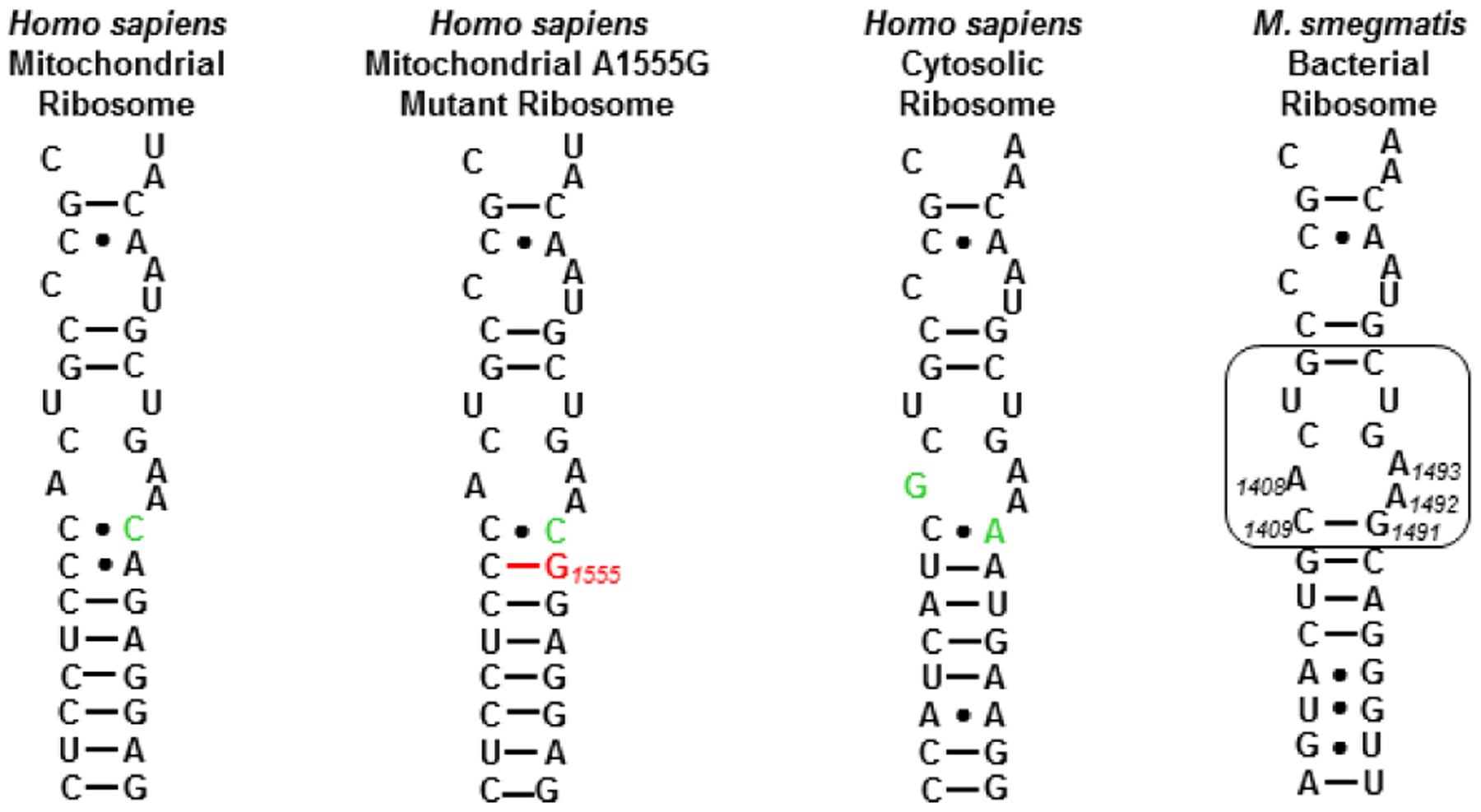
Decoding A sites of prokaryotic and eukaryotic ribosomes. The bacterial AGA binding pocket is boxed. The bacterial numbering scheme is illustrated for the AGA binding pocket. Changes from the bacterial ribosome binding pocket are coloured green. The A1555G mutant conferring hypersusceptibility to AGA ototoxicity is coloured red.
Figure 4.
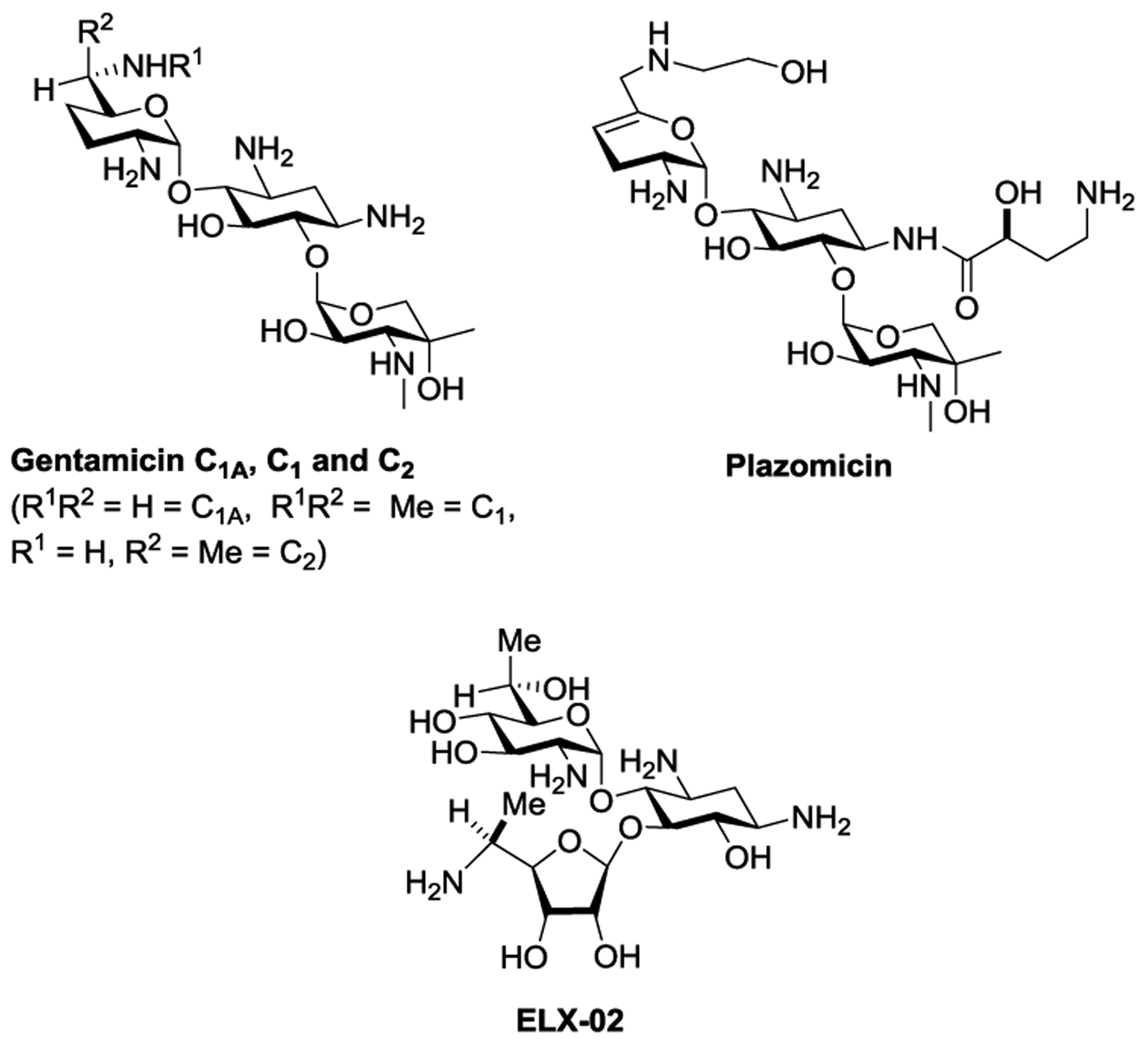
Structures of the Clinical Drugs Gentamicin and Plazomicin and of the Experimental Drug ELX-02
In contrast to the 5”-amines, the 5”-formamido derivative 11 showed excellent selectivity for the bacterial ribosome over the three humanized ribosomes surpassing that of propylamycin itself, while the ureido derivative 12 showed across the board selectivity comparable to propylamycin. 3-Deoxypropylamycin 14 was similar to propylamycin itself and showed good across the board selectivity. Finally, the activity of the formamido derivative 11 at the target level is comparable to that of the clinical AGAs gentamicin and plazomicin, while its selectivity is vastly superior.
Conclusions
Replacement of the 5”-hydroxy group in propylamycin by either an amino group or a derivatized amino group overcomes the susceptibility of this AGAs to inactivation by APH(3’) isozymes capable of acting at the 5”-position. Unfortunately, and in contrast to the apralog series, this type of substitution conveys a small reduction in inhibition of the bacterial ribosome and an increase in inhibition of the eukaryotic ribosomes resulting overall in significantly reduced selectivity. In contrast, the 5”-formamido derivative of propylamycin combines propylamycin-like levels of antibacterial activity with increased selectivity for the bacterial over the eukaryotic ribosomes, all while overcoming the APH(3’)-resistance determinants that act at the 5”-position. The 3’-deoxypropylamycin derivative shows no advantage over propylamycin itself, with the presence of the propyl group at the 4’-position being sufficient to retard the action of the APH(3’) AMEs on the 3’-hydroxy group.
Methods.
Cell-free luciferase translation assays.
Cell-free in-vitro translation inhibition assays were performed using luciferase mRNA and bacterial S30 extracts containing either wild-type bacterial or human hybrid ribosomes.55 In brief, firefly luciferase mRNA was transcribed in vitro using T7 RNA polymerase (Thermo) using a plasmid as template in which the mammalian promoter in pGL4.14 (Promega) has been replaced by theT7 bacteriophage promoter. Test articles in aqueous solution containing 0.3% Tween20 were dispensed into white 96-well plates (Eppendorf) using the TECAN D300e digital dispenser. The test article dispension volume was balanced to a total of 1.5 μL by 0.3% Tween20 in water. The reaction volume was brought to 15 μL by addition of 13.5 μL Translation Master Mix comprised of bacterial S30 extract, 0.2 mM amino acid mix, 6 μg tRNA (Sigma), 0.4 μg hFluc mRNA, 0.3 μL protease inhibitor (cOmplete, EDTA-free, Roche), 12 U RNAse inhibitor (Ribolock, Thermo Scientific), and 6 μL S30 premix without amino acids (Promega). Dispension and mixing of reagents was performed on ice prior to incubating the sealed plates at 37 °C. After 30 minutes of incubation, the reaction was stopped on ice and 75 μL of luciferase assay reagent (Promega) was added to each well. Luminescence was recorded with a plate reader (BIO-TEK FLx800, Witec AG, Littau, Switzerland).
Antibacterial inhibition assays.
The minimal inhibitory concentrations (MIC) of synthesized compounds were determined by broth microdilution assays according to CLSI reference methodology M0756 as described previously.57 A summary of bacterial strains used in this study is provided in Table S1. Clinical bacterial isolates were obtained from the diagnostic laboratories of the Institute of Medical Microbiology, University of Zurich. Whole genome sequencing of the bacterial isolates and bioinformatic annotation of resistance genes was done as described previously.57
Supplementary Material
Acknowledgments.
We thank the NIH (AI123352) for support of the work in the USA and Switzerland. Work in Switzerland was additionally supported by the Swiss National Science Foundation under the framework of JPIAMR RIBOTARGET. Research in Latvia leading to some of these results was conducted as part of the ND4BB European Gram-Negative Antibacterial Engine (ENABLE) Consortium (www.nd4bb-enable.eu) and has received funding from the Innovative Medicines Initiative Joint Undertaking under grant agreement n_115583, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and The European Federation of Pharmaceutical Industries and Associations (EFPIA) companies in kind contribution. The ENABLE project is also financially supported by contributions from Academic and Small and medium-sized enterprise (SME) partners. RZ thanks the Latvian Institute of Organic Synthesis for support.
Footnotes
Conflict of Interest. SNH, AV, ECB and DC are cofounders of and equity holders in Juvabis AG, a biotech start-up working in the area of aminoglycoside development.
References
- 1.Matsushita T; Sati GC; Kondasinghe N; Pirrone MG; Kato T; Waduge P; Kumar HS; Cortes Sanchon A; Dobosz-Bartoszek M; Shcherbakov D; Juhas M; Hobbie SN; Schrepfer T; Chow CS; Polikanov YS; Schacht J; Vasella A; Böttger EC; Crich D, Design, Multigram Synthesis, and in Vitro and in Vivo Evaluation of Propylamycin: A Semisynthetic 4,5-Deoxystreptamine Class Aminoglycoside for the Treatment of Drug-Resistant Enterobacteriaceae and Other Gram-Negative Pathogens. J. Am. Chem. Soc 2019, 141, 5051–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Böttger EC; Crich D, Aminoglycosides: Time for Resurrection of a Neglected Class of Antibacterials? ACS Infect. Dis 2020, 6, 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jospe-Kaufman M; Siomin L; Fridman M, The Relationship Between the Structure and Toxicity of Aminoglycoside Antibiotics Bioorg. Med. Chem. Lett 2020, 30, 127218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan-Krohn T; Manetsch R; O’Doherty GA; Kirby JE, New Strategies and Structural Considerations in Development of Therapeutics for Carbapenem-Resistant Enterobacteriaceae. Translational Res. 2020, 2020, 14–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi Y; Igarashi M, Destination of Aminoglycoside Antibiotics in the ‘Post-Antibiotic Era’. J. Antibiotics 2018, 71, 4–14. [DOI] [PubMed] [Google Scholar]
- 6.Bacot-Davis VR; Bassenden AV; Berghuis AM, Drug-Target Networks in Aminoglycoside Resistance: Hierarchy of Priority in Structural Drug Design. Med. Chem. Commun 2016, 7, 103–113. [Google Scholar]
- 7.Magnet S; Blanchard JS, Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev 2005, 105, 477–497. [DOI] [PubMed] [Google Scholar]
- 8.Yang L; Ye XS, Development of Aminoglycoside Antibiotics Effective Against Resistant Bacterial Strains. Curr. Top. Med. Chem 2010, 10, 1898–1926. [DOI] [PubMed] [Google Scholar]
- 9.Garneau-Tsodikova S; Labby KJ, Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. Med. Chem. Commun 2016, 7, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zárate SG; De la Cruz Claure ML; Benito-Arenas R; Revuelta R; Santana AG; Bastida A, Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules 2018, 23, 284, doi: 10.3390/molecules23020284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armstrong ES; Kostrub CF; Cass RT; Moser HE; Serio AW; Miller GH, Aminoglycosides. Antibiotic Discovery and Development, Dougherty TJ; Pucci MJ, Springer Science+Business Media: New York, 2012; 229–269. [Google Scholar]
- 12.Livermore DM; Mushtaq S; Warner M; Zhang J-C; Maharjan S; Doumith M; Woodford N, Activity of Aminoglycosides, Including ACHN-490, Against Carbapenem-Resistant Enterobacteriaceae Isolates. J. Antimicrob. Chemother 2011, 66, 48–53. [DOI] [PubMed] [Google Scholar]
- 13.Cox G; Ejim L; Stogios PJ; Koteva K; Bordeleau E; Evdokimova E; Sieron AO; Savchenko A; Serio AW; Krause KM; Wright GD, Plazomicin Retains Antibiotic Activity against Most Aminoglycoside Modifying Enzymes. ACS Infect. Dis 2018, 4, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonousi A; Sarpe VA; Brilkova M; Schacht J; Vasella A; Böttger EC; Crich D, Effects of the 1-N-(4-Amino-2S-hydroxybutyryl) and 6’-N-(2-Hydroxyethyl) Substituents on Ribosomal Selectivity, Cochleotoxicity and Antibacterial Activity in the Sisomicin Class of Aminoglycoside Antibiotics. ACS Infect. Dis 2018, 4, 1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doi Y; Wachino JI; Arakawa Y, Aminoglycoside Resistance: The Emergence of Acquired 16S Ribosomal RNA Methyltransferases. Infect. Dis. Clin. North Am 2016, 30, 523–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Courvalin P; Davies J, Plasmid-Mediated Aminoglycoside Phosphotransferase of Broad Substrate Range That Phosphorylates Amikacin. Antimicrob. Agents Chemother 1977, 11, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright GD; Thompson PR, Aminoglycoside Phosphotransferases: Proteins, Structure, and Mechanism. Front. Biosci 1999, 4, d9–21. [DOI] [PubMed] [Google Scholar]
- 18.Quirke JCK; Rajasekaran P; Sarpe VA; Sonousi A; Osinnii I; Gysin M; Haldimann K; Fang Q-J; Shcherbakov D; Hobbie SN; Sha S-H; Schacht J; Vasella A; Böttger EC; Crich D, Apralogs: Apramycin 5-O-Glycosides and Ethers with Improved Antibacterial Activity and Ribosomal Selectivity and Reduced Susceptibility to the Aminoacyltranserferase (3)-IV Resistance Determinant. J. Am. Chem. Soc 2020, 142, 530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonousi A; Quirke JCK; Waduge P; Janusic T; Gysin M; Haldimann K; Xu S; Hobbie SN; Sha S-H; Schacht J; Chow CS; Vasella A; Böttger EC; Crich D, An Advanced Apralog with Increased in-vitro and in-vivo Activity toward Gram-negative Pathogens and Reduced ex-vivo Cochleotoxicity. Chem. Med. Chem 2021, 16, 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abe Y; Nakagawa S; Naito T; Kawaguchi H, Synthesis and Activity of 6-O-(3-Amino-3-deoxy-α-D-glucopyranosyl)- and 5-O-(β-D-ribofuranosyl)apramycins. J. Antibiotics 1981, 34, 1434–1446. [DOI] [PubMed] [Google Scholar]
- 21.Zada SL; Baruch BB; Simhaev L; Engel H; Fridman M, Chemical Modifications Reduce Auditory Cell Damage Induced by Aminoglycoside Antibiotics. J. Am. Chem. Soc 2020, 142, 3077–3087. [DOI] [PubMed] [Google Scholar]
- 22.Sumino S; Uno M; Huang H-J; Wu Y-K; Ryu I, Palladium/Light Induced Radical Alkenylation and Allylation of Alkyl Iodides Using Alkenyl and Allylic Sulfones Org. Lett 2018, 20, 1078–1081. [DOI] [PubMed] [Google Scholar]
- 23.Wang X; Dong J; Li Y; Liu Y; Wang Q, Visible-Light-Mediated Manganese-Catalyzed Allylation Reactions of Unactivated Alkyl Iodides J. Org. Chem 2020, 85, 7459–7467. [DOI] [PubMed] [Google Scholar]
- 24.Goddard J-P; Ollivier C; Fensterbank L, Photoredox Catalysis for the Generation of Carbon Centered Radicals. Acc. Chem. Res 2016, 49, 1924–1936. [DOI] [PubMed] [Google Scholar]
- 25.Ngoje P; Crich D, Synthesis of Bradyrhizose from d-Glucose. Org. Lett 2020, 22, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo J; Zhang J, Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)−C(sp2) Cross Coupling. ACS Catal. 2016, 6, 873–877. [Google Scholar]
- 27.Uoyama H; Goushi K; Shizu K; Nomura H; Adachi C, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–238. [DOI] [PubMed] [Google Scholar]
- 28.Constantin T; Zanini M; Regni A; Sheikh NS; Julía F; Leonori D, Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367, 1021–1026. [DOI] [PubMed] [Google Scholar]
- 29.Lattrell R; Lohaus G, Attempted Total Synthesis of Cephalosporin Derivatives. II. Substitution Reactions with trans-3-(Sulfonyloxy)-2-azetidinones. Synthesis of cis-3-(Acylamino)-4-(alkylthio)-2-azetidinones. Liebigs Ann. Chem 1974, 901–920. [Google Scholar]
- 30.Albert R; Dax K; Link RW; Stuetz AE, Carbohydrate Triflates: Reaction with Nitrite, Leading Directly to epi-Hydroxy Compounds. Carbohydr. Res 1983, 118, C5–C6. [Google Scholar]
- 31.Dong H; Pei Z; Ramström O, Stereospecific Ester Activation in Nitrite-Mediated Carbohydrate Epimerization. J. Org. Chem 2006, 71, 3306–3309. [DOI] [PubMed] [Google Scholar]
- 32.Clive DLJ; Cantin M; Khodabocus A; Kong X; Tao Y, Protecting Group Improvement by Isotopic Substitution: Synthesis of the Quinone System of Fredericamycin A. Tetrahedron 1993, 49, 7917–7930. [Google Scholar]
- 33.Pirrone MG; Matsushita T; Vasella A; Crich D, Stereospecific Synthesis of Methyl 2-Amino-2,4-dideoxy-6S-deuterio-α-D-xylopyranoside and Methyl 2-Amino-2,4-dideoxy-6S-deuterio-4-propyl-α-D-glucopyranoside: Side Chain Conformation of the Novel Aminoglycoside Antibiotic Propylamycin. Carbohydr. Res 2020, 491, 107984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L-X; Sakairi N; Kuzuhara H, 1,6-Anhydro-β-D-glucopyranose Derivatives as Glycosyl Donors for Thioglycosidation Reactions. J. Chem. Soc, Perkin Trans 11990, 1677–1682. [Google Scholar]
- 35.Vincent SP; Burkart MD; Tsai C-Y; Zhang Z; Wong C-H, Electrophilic Fluorination-Nucleophilic Addition Reaction Mediated by Selectfluor: Mechanistic Studies and New Applications. J. Org. Chem 1999, 64, 5264–5279. [DOI] [PubMed] [Google Scholar]
- 36.Sonousi A; Vasella A; Crich D, Synthesis of a Pseudodisaccharide Suitable for Synthesis of Ring I Modified 4,5–2-Deoxystreptamine Type Aminoglycoside Antibiotics. J. Org. Chem 2020, 85, 7583–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sati GC; Shcherbakov D; Hobbie S; Vasella A; Böttger EC; Crich D, N6′, N6′′′, and O4′-Modifications to Neomycin Affect Ribosomal Selectivity Without Compromising Antibacterial Activity. ACS Infect. Dis 2017, 3, 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duscha S; Boukari H; Shcherbakov D; Salian S; Silva S; Kendall A; Kato T; Akbergenov R; Perez-Fernandez D; Bernet B; Vaddi S; Thommes P; Schacht J; Crich D; Vasella A; Böttger EC, Identification and Evaluation of Improved 4’-O-(Alkyl) 4,5-Disubstituted 2-Deoxystreptamines as Next Generation Aminoglycoside Antibiotics. mBio 2014, 5, doi: 10.1128/mBio.01827-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brodersen DE; Clemons WM; Carter AP; Morgan-Warren R; Wimberly BT; Ramakrishnan V, The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit Cell 2000, 103, 1143–1154. [DOI] [PubMed] [Google Scholar]
- 40.Moazed D; Noller HF, Interaction of Antibiotics with Functional Sites in 16S Ribosomal RNA. Nature 1987, 327, 389–394. [DOI] [PubMed] [Google Scholar]
- 41.François B; Russell RJM; Murray JB; Aboul-ela F; Masquid B; Vicens Q; Westhof E, Crystal Structures of Complexes Between Aminoglycosides and Decoding A Site Oligonucleotides: Role of the Number of rings and Positive Charges in the Specific Binding Leading to Miscoding. Nucleic Acids Res. 2005, 33, 5677–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fourmy D; Recht MI; Blanchard SC; Puglisi JD, Structure of the A Site of Escherichia coli 16S Ribosomal RNA Complexed with an Aminoglycoside Antibiotic. Science 1996, 274, 1367–1371. [DOI] [PubMed] [Google Scholar]
- 43.Lin J; Zhou D; Steitz TA; Polikanov YS; Gagnon MG, Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Ann. Rev. Biochem 2018, 87, 451–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang M; Karasawa T; Steyger PS, Aminoglycoside-Induced Cochleotoxicity: A Review. Front. Cell. Neurosci 2017, 11, 308; Doi: 10.3389/fncel.2017.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Böttger EC; Schacht J, The Mitochondrion: A Perpetrator of Acquired Hearing Loss. Hearing Res 2013, 303, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prezant TR; Agapian JV; Bohlman MC; Bu X; Öztas S; Qiu W-Q; Arnos KS; Cortopassi GA; Jaber L; Rotter JI; Shohat M; Fischel-Ghodsian N, Mitochondrial Ribosomal RNA Mutation Associated with Both Antibiotic-Induced and Non-Syndromic Deafness. Nat. Genetics 1993, 4, 289–294. [DOI] [PubMed] [Google Scholar]
- 47.Huth ME; Ricci AJ; Cheng AG, Mechanisms of Aminoglycoside Ototoxicity and Targets of Hair Cell Protection. Int. J. Otolaryngol 2011, 937861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hobbie SN; Akshay S; Kalapala SK; Bruell C; Shcherbakov D; Böttger EC, Genetic Analysis of Interactions with Eukaryotic rRNA Identify the Mitoribosome as Target in Aminoglycoside Ototoxicity. Proc. Natl. Acad. Sci., USA 2008, 105, 20888–20893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hobbie SN; Bruell CM; Akshay S; Kalapala SK; Shcherbakov D; Böttger EC, Mitochondrial Deafness Alleles Confer Misreading of the Genetic Code. Proc. Natl. Acad. Sci., USA 2008, 105, 3244–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qian Y; Guan M-X, Interaction of Aminoglycosides with Human Mitochondrial 12S rRNA Carrying the Deafness-Associated Mutation. Antimicrob. Agent. Chemother 2009, 53, 4612–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hobbie SN; Kalapala SK; Akshay S; Bruell C; Schmidt S; Dabow S; Vasella A; Sander P; Böttger EC, Engineering the rRNA Decoding Site of Eukaryotic Cytosolic Ribosomes in Bacteria. Nucl. Acids Res 2007, 35, 6086–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sabbavarapu NM; Pienko T; Zalman B-H; Trylska J; Baasov T, Exploring Eukaryotic versus Prokaryotic Ribosomal RNA Recognition with Aminoglycoside Derivatives. Med. Chem. Commun 2018, 9, 503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crawford DK; Alroy I; Sharpe N; Goddeeris MM; Williams G, ELX-02 Generates Protein via Premature Stop Codon Read-Through without Inducing Native Stop Codon Read-Through Proteins. J. Pharmacol. Expt. Therap 2020, 374, 264–272. [DOI] [PubMed] [Google Scholar]
- 54.Shalev M; Baasov T, When Proteins Start to Make Sense: Fine-Tuning of Aminoglycosides for PTC Suppression Therapy. Med. Chem. Commun 2014, 5, 1092–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matt T; Ng CL; Lang K; Sha S-H; Akbergenov R; Shcherbakov D; Meyer M; Duscha S; Xie J; Dubbaka SR; Perez-Fernandez D; Vasella A; Ramakrishnan V; Schacht J; Böttger EC, Dissociation of Antibacterial Activity and Aminoglycoside Ototoxicity in the 4-Monosubstituted 2-Deoxystreptamine Apramycin. Proc. Natl. Acad. Sci., USA 2012, 109, 10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clinical Laboratory Standards Institute (2015). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically-Tenth Edition: Approved Standard M07-A10. CLSI, Wayne, PA, USA. [Google Scholar]
- 57.Juhas M; Widlake E; Teo J; Huseby DL; Tyrrell JM; Polikanov Y; Ercan O; Petersson A; Cao S; Aboklaish AF; Rominski A; Crich D; Böttger EC; Walsh TR; Hughes DE; Hobbie SN, In-vitro Activity of Apramycin Against Multidrug-, Carbapenem-, and Aminoglycoside-Resistant Enterobacteriaceae and Acinetobacter baumannii. J. Antimicrob. Chemother 2019, 74, 944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.