

Abstract
Fused in Sarcoma (FUS) is a ubiquitously expressed RNA/DNA-binding protein that plays different roles in the cell. FUS pathology has been reported in neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Mutations in FUS have also been linked to a subset of familial ALS. FUS is mainly localized in the nucleus although it shuttles between the nucleus and the cytoplasm. ALS-linked mutations cause the accumulation of the FUS protein in cytoplasm where it forms stress granule-like inclusions. The protein- and RNA-containing inclusions are reported to be positive of autophagosome markers and degraded by the autophagy pathway. However, the role of FUS in the autophagy pathway remains to be better understood. Using immunoblot and confocal imaging techniques in this study, we found that FUS knockout (KO) cells showed a decreased basal autophagy level. Rapamycin and bafilomycin A1 treatment showed that FUS KO cells were not able to initiate autophagy as efficiently as wild-type cells, suggesting that the autophagosome formation is affected in the absence of FUS. Moreover, using immunoblot and quantitative PCR techniques, we found that the mRNA and protein levels of the genes critical in the initial steps of the autophagy pathway (FIP200, ATG16L1 and ATG12) were significantly lower in FUS KO cells. Re-expressing FUS in the KO cells restored the expression of FIP200 and ATG16L1. Our findings demonstrate a novel role of FUS in the autophagy pathway, i.e. regulating the transcription of genes involved in early stages of autophagy such as the initiation and elongation of autophagosomes.
Keywords: Fused in Sarcoma (FUS), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), autophagy
Introduction
Fused in sarcoma (FUS) is a ubiquitously expressed RNA/DNA-binding protein that plays a role in different cellular processes such as DNA repair (Baechtold et al. 1999; Mastrocola et al. 2013; Wang et al. 2013), transcription (Wang et al. 2008; Tan et al. 2012; Schwartz et al. 2012; Bronisz et al. 2014; Dhar et al. 2014; Yang et al. 2014), RNA splicing (Yang et al. 1998; Dichmann & Harland 2012; Yang et al. 2014), nucleo-cytoplasmic RNA shuttling (Zinszner et al. 1997) and dendritic RNA transport (Fujii & Takumi 2005; Fujii et al. 2005; Sephton et al. 2014), protein translation and RNA decay (Kamelgarn et al. 2018; Lopez-Erauskin et al. 2018; Baron et al. 2019), and stress response (Gal et al. 2011; Sama et al. 2013; Andersson et al. 2008). FUS contains an N-terminal prion-like domain, a glycine rich region, an RNA recognition motif, a zinc finger domain flanked by two Arginine-Glycine-Glycine-rich domains, and a C-terminal nuclear localization sequence (NLS) (Sama et al. 2014). FUS is mainly localized in the nucleus, although it shuttles between the nucleus and the cytoplasm to transport RNAs (Fujii & Takumi 2005), and it is present in the cytoplasm of neuronal cells at lower levels (Andersson et al. 2008).
FUS pathology has been reported in neurodegenerative diseases amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Mutations in FUS have also been linked to a subset of familial ALS (Kwiatkowski et al. 2009). Many of the familial ALS-related FUS mutations are localized in the C-terminal NLS, causing mis-localization of FUS to the cytoplasm where it forms inclusions that are positive of stress granule markers (Bosco et al. 2010; Gal et al. 2011; Dormann et al. 2010; Arenas et al. 2020). Stress granules are dynamic cytoplasmic complexes of proteins, ribosomes, and RNAs that form under cellular stresses such as oxidative stress, hyperosmolar stress, or heat shock (Kedersha & Anderson 2002). Prolonged formation of SGs could become irreversible and form pathological inclusions, contributing to neurotoxicity and neurodegeneration.
There are two major protein degradation pathways: the ubiquitin-proteasome system (UPS) and the autophagy pathway. The UPS is responsible for the degradation of short-lived soluble proteins, while the autophagy pathway can degrade misfolded proteins as well as whole organelles (Ramesh & Pandey 2017). Macroautophagy (hereon referred to as ‘autophagy’) begins when the cells are under nutrient starvation or other stimuli. The initiation complex (ULK1-ATG13-FIP200) activates to form the phagophore (Hurley & Young 2017), which is subsequently elongated by two ubiquitin-like systems: ATG12:ATG5:ATG16L system and phosphatidylethanolamine (PE)-microtubule-associated protein 1A/1B-light chain 3 (LC3) system. The elongation of phagophore encloses misfolded proteins or damaged organelles, forming a double-membraned autophagosome (Mizushima 2007). The mature autophagosome fuses with the lysosome and its contents are degraded by acidic proteases in the auto-lysosome. Amino acids and other degradation products are recycled in the cytoplasm where they can be re-used for cell metabolism (Glick et al. 2010).
Neurons are post-mitotic cells that are very susceptible to protein aggregation, thus defects in protein homeostasis are highly detrimental to neurons. Defects in the autophagy pathway have been reported in neurodegenerative diseases such Alzheimer’s disease and ALS (Ryu et al. 2014; Boland et al. 2008; Kesidou et al. 2013). For instance, mutant FUS-positive SGs were co-localized with autophagosomes and autophagy deficient mouse embryonic fibroblasts (MEFs) showed an increase of SGs (Ryu et al. 2014). However, the specific role of FUS in the autophagy pathway remains to be better understood. In this study, we found that proteasome played a more prominent role in FUS turnover while autophagy had little impact on FUS in the radioimmunoprecipitation assay (RIPA) buffer -insoluble fraction. More importantly, we found that FUS knockout (KO) decreased the basal autophagy function. After the bafilomycin A1 (Baf-A1) treatment, FUS KO cells had lower levels of the autophagy flux. The rapamycin treatment failed to induce the autophagy levels in FUS KO cells, suggesting that FUS regulates initial stages of the autophagy pathway. Moreover, the mRNA and proteins levels of genes involved in autophagy initiation and phagophore elongation, such as Atg3, Atg16L1 and Fip200, were downregulated in FUS KO cells. FUS overexpression in the KO cells restored the mRNA and protein levels of Fip200 and Atg16L1. In summary, this study shows a new function of FUS in the cell, i.e. FUS affects the basal autophagy flux by regulating the transcription of genes required for autophagy initiation and phagophore elongation.
Materials and Methods
Cell culture and transfection
The study was not pre-registered. Institutional ethics approval was not required for your study. Custom-made materials will be shared upon reasonable request.
Neuro-2a (N2A), FUS KO N2A, N2A FLAG-FUS re-expression, and NSC34 cells were cultured in Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich, D5796) with 10% fetal bovine serum, penicillin-streptomycin, and amphotericin B at 37°C in 5% CO2/95% air with humidification. The pCMV10–3×FLAG-FUS plasmids, were generated as reported (Gal et al. 2011). Cells were treated with Baf-A1 (200 nM) for 6 hours, rapamycin (2 μM) for 4h, or MG-132 (5 μM) for 16 hours. FUS mutations were introduced using the QuickChange II Site-Directed Mutagenesis Kit (Agilent). N2A, FUS KO N2A and NSC34 cells were transfected with Lipofectamine 2000 (Thermo Fisher Scientific, 11668). siRNA-FUS (Santa Cruz, sc-40563) oligonucleotides were reversed-transfected into N2A and NSC34 cells at a final concentration of 1μM using Lipofectamine™ RNAiMAX (Thermo Fisher Scientific, 13778100). N2A cells were originally obtained from ATCC. NSC34 cells were originally obtained from Dr. Neil Cashman at University of British Columbia who generated NSC34 cells. The maximum number of passages used in this study was 20. Cell lines used in this study are not listed as a commonly misidentified cell line by the International Cell Line Authentication Committee.
Generation of FUS KO (ΔFUS) N2A cells and FLAG-FUS re-expression cells lines.
The FUS KO cells were generated by employing CRISPR technology. N2A cells were transfected with FUS double nickase CRISPR plasmid (Santa Cruz Biotechnology, sc-433326-NIC) following the manufacturer’s instructions. Clonal cell lines were isolated with serial dilution, and the FUS status of the clones was determined with immunoblotting.
FLAG-tagged FUS WT was transfected into FUS KO N2A cells using Lipofectamine 2000 as described above. Selection of stable cells with FLAG-FUS WT integration was performed with G-418 (Millipore Sigma G418-R). Clonal cells lines were isolated with serial dilution, and the FUS expression levels were compared to endogenous FUS in N2A cells by western blot, using anti-FUS and anti-FLAG antibodies. The KO and re-expression cell lines were authenticated with Western blot to confirm the FUS protein expression.
Immunofluorescence Microscopy
N2A and FUS KO cells were seeded on gelatin coated glass coverslips. Cells were rinsed with 1× PBS, fixed with 4% formaldehyde in 1× PBS, and permeabilized with 0.25 % (v/v) Triton-X100 in 1× PBS. The coverslips were blocked with 10% [w/v] bovine serum albumin in 1× PBS for 1h, followed by incubation with the primary antibody at 4°C overnight. Coverslips were rinsed with 1× PBS and incubated with secondary antibodies for 1 hour at 37°C. The primary antibody was mouse anti-FUS (Santa Cruz, sc-47711) and rabbit anti-LC3 (Millipore Sigma L8918). The secondary antibody was Alexa Fluor 488 donkey anti-mouse (Life Technologies, A-21202) and Alexa Fluor 568 donkey anti-rabbit (Life Technologies, A10042). All the samples were stained with 4′,6-Diamidino-2-phenylindole (DAPI) and mounted with Vectashield Mounting Medium (Vector Laboratories, H-1000–10). Confocal images were acquired using a Nikon A1 confocal microscope with a 40x objective. Quantification of cells containing LC3-positive autophagosomes was performed from multiple random fields with >150 cells.
Western blot analysis
Cells were lysed with 1x RIPA buffer (Millipore Sigma, 20–188) supplemented with protease inhibitor cocktail (Millipore Sigma, P8340, 1:500) and sodium orthovanadate (1 mM) Lysates were homogenized by sonication and centrifuged at 1,000g for 15 minutes at 4°C. Supernatant was collected and protein concentration was determined using a colorimetric Protein Assay Dye Reagent Concentrate (BIO-RAD, 5000006,). 6x SDS (sodium dodecyl sulphate) buffer sample was added to samples containing equal amounts of protein and heated for 5 min at 95°C. Proteins were resolved by 10% SDS-PAGE (sodium dodecyl sulphate–polyacrylamide gel electrophoresis) gel (with the exception of 15% gel for LC3 immunoblotting) and transferred to nitrocellulose membranes (Pall, 66485). The membranes were blocked with 5% non-fat dry milk in TBST (100 mM Tris-HCl, pH 7.5, 0.9% NaCl, 0.1% Tween-20) or bovine serum albumin for 1 hour.
Cellular fractionation
Cells were sonicated and centrifuged at 100,000g for 15min at 4°C in order to obtain the RIPA soluble fraction in the supernatant. Consecutively, the pellet was collected, and urea buffer was added, followed by a centrifugation at 70,000g for 30min. The urea resolubilized fraction was collected in the supernatant and samples were analyzed through a western blot.
Antibodies
The antibodies used include: mouse anti-FUS (Santa Cruz, sc-47711), mouse monoclonal ANTI-FLAG® M2-Peroxidase (HRP) (Millipore Sigma, A8592), rabbit anti-β-actin (Cell Signaling 8457). Rabbit anti-LC3 (Millipore Sigma L8918), mouse anti-SQSTM1/p62 (H00008878-M01), autophagy induction (ULK1 Complex) antibody sampler kit (Cell Signaling 46486) containing ULK1 (D8H5) rabbit mAb (Cat #8054), Atg13 (D4P1K) rabbit mAb (Cat# 13273), FIP200 (D10D11) rabbit mAb (Cat#12436), Atg101 (E1Z4W) rabbit mAb (Cat#13492), phospho-ULK1 (Ser757) (D7O6U) rabbit mAb (Cat#14202), phospho-ULK1 (Ser555) (D1H4) rabbit mAb (Cat#5869). Autophagy antibody sampler kit (Cell Signaling, 4445) containing Beclin-1 (D40C5) rabbit mAb (Cat#3495), Atg5 (D5F5U) rabbit mAb (Cat#12994), Atg12 (D88H11) rabbit mAb (Cat#4180), Atg16L1 (D6D5) rabbit mAb (Cat#8089), Atg7 (D12B11) rabbit mAb (Cat#8558), Atg3 antibody (Cat#3415).
TaqMan qPCR Assay
RNA isolation was performed using Aurum Total RNA Mini Kit (BIO-RAD, 732–6820) following the manufacturer’s instructions. 1μg of isolated RNA was reverse transcribed using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, 18080). The resulting cDNA was subjected to Real-time PCR. Briefly, FAM-labeled Taqman probes for autophagy related genes and controls were obtained from the TaqMan Gene expression assay library from ThermoFisher Scientific. TaqMan® Fast Advanced Master Mix (ThermoFisher Scientific, 4444556, Thermo Fisher Scientific) was prepared following manufacturer’s instructions. The assay IDs for the FAM-labelled probes analyzed are: Mm00456545_m1 (Rbcc1, FIP200), Mm00509659_m1 (Atg101), Mm00512209_m1 (Atg7), Mm00471287_m1 (Atg3), Mm00503201_m1 (Atg12), Mm00513085_m1 (Atg16L1), Mm01612987_g1 (Rpl13a), Mm99999915_g1 (GAPDH), Mm00607939_s1 (Actin). The qPCR results were analyzed using the Δ Δ CT method.
Statistical analysis
Lane quantification for Western blot bands was performed using Image Lab software by BioRad. Statistical analysis was calculated with SigmaPlot 14.0 software. qPCR CT values and the Western blot band intensities were calculated and comparison between groups was performed using Anova with post hoc Tuckey HSD test. Student’s t test was used to determine statistical significance between two groups. A normality test was performed in ANOVA analysis. No outlines are identified. No statistical methods were employed to predetermine the sample size. Experiments were not blinded. All experiments were done in triplicates unless noted. According to the Journal policy, the Western blot and qPCR quantification data are presented as scatter plots with a bar representing the median. N numbers were specified where appropriate.
Results
Proteasome inhibition increased the accumulation of detergent insoluble FUS whereas autophagy modulation had little effect.
To evaluate the contribution of UPS and autophagy to FUS protein degradation, we performed a cellular fractionation to separate RIPA soluble and insoluble fractions (Figure 1A) in the presence of various UPS and autophagy inhibitors. NSC34 cells were transfected with GFP-tagged wild-type (WT), R521G, or R495X FUS followed by the treatment of the proteasome inhibitors MG132 (5 μM for 16 hours), the mechanistic target of rapamycin (mTORC) inhibitor rapamycin (2 μM for 4h), or the autophagy flux inhibitors Baf-A1 (200 nM for 6 hours). After the subcellular fractionation as depicted in Figure 1A, fractions were analyzed by Western blot. We found that FUS protein levels remained largely unchanged in the RIPA soluble fraction regardless WT or mutant FUS or any treatment. However, R495X showed a significantly higher level as compared to WT FUS in the urea re-solubilized (i.e. RIPA-insoluble) fractions in the non-treated cells (Figure 1B, lane 3 vs lane 1), suggesting that R495X mutant FUS formed insoluble protein inclusions as previously reported (Bosco et al. 2010; Gal et al. 2011; Dormann et al. 2010). In the samples treated with MG132, FUS protein levels in the RIPA soluble fraction did not change as compared to the non-treated cells. In contrast, the proteasome inhibitors increased the levels of WT, R521G and R495X FUS in the urea re-solubilized fraction as compared to non-treated cells (Figure 1B, lanes 4–6 vs lanes 1–3). Interestingly, the treatment of rapamycin (Figure 1B, lanes 7–9) or Baf-A1 (Figure 1B, lanes 10–12) did not change FUS protein levels in the urea re-solubilized fractions. These results suggest that the UPS pathway, but not autophagy, is the primary mechanism responsible for the turnover of WT and mutant FUS proteins to prevent the formation of FUS aggregates.
Figure 1. The proteasome pathway, but not autophagy, is the primary mechanism for FUS protein turnover.

(A) Schematic representing the protein fractionation process. (B) NSC34 cells were transfected with GFP-FUS WT, R521G, or R495X and treated with 200 nM Baf-A1 for 6 hours, or 5 μM MG132 for 16 hours, or 2 μM rapamycin for 4 hours. Immunoblotting was performed using the indicated antibodies.
Loss of FUS reduced the basal level of autophagy.
Although the insoluble FUS level was not affected by the autophagy modulation, we asked whether the absence of FUS would affect the autophagy pathway. We used siRNA to knockdown FUS and examined the levels of autophagy markers LC3-I and LC3-II. The LC3-II/LC3-I ratio was reduced by approximately 70% (p < 0.005) in the FUS knockdown cells as compared to WT N2A cells (Figure 2A). Similarly, the LC3-II/LC3-I ratio decreased by approximately 75% (p < 0.05) in FUS KO N2A cells generated using the CRISPR technology (Figure 2B). In addition, confocal imaging analysis showed that the formation of LC3-positive autophagosomes decreased in FUS KO cells as compared to WT N2A cells (Figure 2C-D). Together, these data suggest that FUS might play an important role in the basal autophagy pathway. We next examined whether the ALS-linked FUS mutations would affect the levels of basal autophagy. FUS KO N2A cells were transfected with FLAG-tagged WT, R521G, R495X or P515L FUS, along with WT N2A cells and non-transfected FUS KO cells as controls. Consistent with Figure 2B, FUS KO cells showed a decreased LC3-II/LC3-I ratio. Moreover, introducing WT FUS back in the KO cells restored the LC3-II/LC3-I ratio (Figure 2E-F), confirming that FUS is important in the basal autophagy pathway. However, neither the LC3-II/LC3-I ratio nor the p62 level changed between any ALS mutant FUS and WT FUS (Figure 2E-G), indicating that mutant FUS did not affect the autophagy flux under the conditions in this study. The expression level of WT or mutant FUS was comparable to the endogenous FUS level in WT N2A cells (Figure 2E).
Figure 2. Loss of FUS reduced the basal level of autophagy.

(A) N2A cells were transfected with FUS siRNA using Lipofectamine™ RNAiMAX. 48h after transfection cells were harvested and immunoblotting was performed using the indicated antibodies. (B) N2A and FUS KO cells were cultured and immunoblotting was performed using the indicated antibodies. (C) Confocal imaging of FUS and LC3 in N2A and FUS KO cells. Scale bars are 10 μm. (D) Quantification of the percentage of cells containing LC3-positive autophagosomes from multiple random viewfields (N > 150 cells). **, p ≤0.01. (E) FUS KO cells were transfected with FLAG-tagged WT FUS, FUS-R495X, R521G, P525L. WT N2A cells and FUS KO cells with no transfection were included as positive and negative controls. 48h after transfections cells were collected and immunoblotting was performed with the indicated antibodies. (F-G) Quantification of (E) from three independent experiments. Results are shown in scatter plots with the bar representing the median. Student’s t-test was performed for individual comparisons against FLAG-FUS WT. **, p ≤0.01. N.S., no significance.
FUS interfered with the initiation of the autophagy pathway.
To test whether FUS plays a role in the autophagy initiation, phagophore elongation, or autophagy flux stage, we treated N2A and N2A FUS KO cells with Baf-A1 or rapamycin, followed by Western blots of autophagy markers. Baf-A1 disrupts the late stages of the autophagy flux by inhibiting the autophagosome-lysosome fusion and preventing autolysosome acidification (Mauvezin et al. 2015). Consistent with earlier results, the ratio of LC3-II/LC3-I was significantly lower in FUS KO cells as compared to WT cells without Baf-A1 (Figure 3A-B). After the Baf-A1 treatment (200 nM, 6 hrs), the LC3-II/LC3-I ratio increased significantly in WT N2A cells (Figure 3A, lane 2 vs lane 1) as well as FUS KO cells (Figure 3A, lane 4 vs lane 3). However, even in the presence of Baf-A1, the LC3-II/LC3-I ratio was still significantly lower in FUS KO cells compared to WT cells (Figure 3A, lane 4 vs lane 2). These results suggest that FUS KO did not affect the late stages of autophagosome-lysosome fusion or autophagy flux, but rather affected the early stages of the autophagy pathway.
Figure 3. FUS knockout impaired the initiation of the autophagy pathway.

(A) N2A and FUS KO cells were treated with 200 nM Baf-A1 for 6h. Cells were harvested, and immunoblotting was performed with the indicated antibodies. (B) Quantification of (A) from four independent experiments. One Way Anova was performed to determine statistical significance. *p ≤0.05; **p≤0.01 (C) N2A and FUS KO cells were treated with 2 μM rapamycin for 4h. Cells were harvested, and immunoblotting was performed with the indicated antibodies. (D) Quantification of (C) from three independent experiments. One Way Anova was performed to determine statistical significance. *p ≤0.05; **p≤0.01; N.S., not significant. All quantification results are shown in scatter plots with the bar representing the median.
Rapamycin is a well-stablished mTOR inhibitor that induces the initiation of autophagy (Sarkar et al. 2009). After the rapamycin treatment (2 μM, 4 hrs), the LC3-II/LC3-I ratio significantly increased in WT N2A cells as compared to non-treatment (Figure 3C-D, lane 2 vs lane 1). In contrast, the LC3-II/LC3-I ratio did not change in FUS KO cells in the presence of rapamycin (Figure 3C, lane 4 vs lane 3), suggesting that FUS KO cells were not able to respond to the rapamycin-mediated initiation of autophagy.
FUS affected the levels of proteins critical in early phases of autophagy.
We next analyzed the expression of proteins involved in the ULK1 initiation complex that is composed of ULK1, Atg13, Atg101 and FIP200. This complex promotes or suppresses the autophagy process depending on the nutrient status of the cell (Wang et al. 2015). We tested the expression of Atg13, Atg101, FIP200, total ULK1 and phosphorylated ULK1 in WT and FUS KO N2A cells (Figure 4A-B). Atg13, Atg101, total ULK1 expression did not change between WT and FUS KO cells. The phosphorylation on UKL1 Ser757 or Ser555 did not change in FUS KO cells either (Figure 4C). Interestingly, FIP200 expression was significantly reduced in FUS KO cells as compared to WT cells (Figure 4A-B).
Figure 4. FUS regulated the protein and mRNA levels of proteins involved in early phases of the autophagy pathway.

N2A and FUS KO cells were cultured and immunoblotting was performed using the indicated antibodies. (B-C) Quantification of (A) from three independent t-test was performed for individual comparisons against N2A WT cells. *, p ≤0.05; N.S., not significant. (D) N2A and FUS KO cells were cultured and immunoblotting was performed using the indicated antibodies. (E) Quantification of (D) from three independent t-test was performed for individual comparisons against WT N2A WT cells. *, p ≤0.05; N.S., not significant. (F-G). N2A and FUS KO cells were cultured and harvested for RNA isolation, followed by reverse transcription and quantitative PCR using TaqMan probes for the indicated genes. Actin probe was used to normalize gene expression. Results from three (F) or four (G) independent experiments are shown. Student’s t-test was performed for individual comparisons against N2A WT cells. *, p ≤0.05; N.S., not significant. All quantification results are shown in scatter plots with the bar representing the median.
We also evaluated the proteins involved in the phagohore elongation (Xie & Klionsky 2007): Atg3, Atg5, Atg12, Atg7 and Ath16L1 (Figure 4D-E). Atg3, Atg16L1, and the Atg5-Atg12 complex showed a significant decrease in FUS KO cells as compared to WT cells. However, the protein level of Atg5 alone did not change between FUS KO and WT cells, indicating that only Atg12 protein was affected in the absence of FUS. Similarly, Atg7 did not change in FUS KO cells. Taken together, these results suggest that FUS likely plays an important role in the autophagy initiation and phagophore elongation stages by regulating the expression of FIP200, Atg 3, Atg 12 and Atg16L1.
FUS regulated the transcription of genes involved in autophagy initiation and phagophore elongation.
Since FUS regulated the protein levels, we examined whether FUS might regulated the transcription of corresponding genes involved in the initial phases of autophagy using quantitative RT-PCR. For genes involved in the autophagy initiation, the mRNA levels of FIP200 showed a significant decrease (~57%) in FUS KO cells (Figure 4F) while Atg101 and Atg7 mRNA levels did not significantly change between the two groups. For genes involved in the phagophore elongation, the mRNA levels of Atg3 (~ 48%), ATG16L1 (~54%), and Atg12 (~48%) decreased significantly in FUS KO cells (Figure 4G). Atg7 was used as a negative control and did not change significantly. These results suggest that FUS likely plays a role in regulating the gene expression of those critical to the autophagy initiation and phagophore elongation.
FUS re-expression restored expression of autophagy related genes.
To further solidify our conclusion, we examined whether re-expression of FUS in the FUS KO cells can restore the expression of the affected autophagy related genes. To this end, we generated a cell line that stably expresses FLAG-tagged WT FUS in N2A-FUS KO cells. The FUS re-expression level was comparable to the endogenous FUS level in WT N2A cells. FUS re-expression largely restored the protein levels of Atg16L1 (100%) and FIP200 (~80%) as compared to FUS KO cells (Figure 5A-B). Similarly, FUS re-expression also increased the mRNA levels of Atg16L1 and FIP200 as compared to the FUS KO cells (Figure 5C). The mRNA levels of Atg16L1 and FIP200 were restored to ~60% in WT N2A cells. The results support that FUS plays a role in regulating the transcription of the genes involved in the autophagy initiation and phagphore elongation.
Figure 5. FUS expression restored the affected autophagy genes.

(A) N2A, FUS KO, and FUS re-expression (KO + WT) cells were cultured and immunoblotting was performed using the indicated antibodies. (B) Quantification of (A) from three independent experiments. One Way Anova was performed to determine statistical significance. *p ≤0.05; **p≤0.01; ***p≤0.005; N.S., not significant. (C) N2A, FUS KO, and FUS re-expression cells were cultured and harvested for RNA isolation, followed by reverse transcription and quantitative PCR using TaqMan probes for the indicated genes. Actin probe was used to normalize gene expression. Results from three independent experiments are shown. Student’s t-test was performed for individual comparisons against N2A WT cells. *, p ≤0.05; N.S., not significant. All quantification results are shown in scatter plots with the bar representing the median.
DISCUSSION
FUS is a DNA/RNA binding protein that has been linked to familial cases of ALS, and its pathology has been reported in FTD patients (Mackenzie et al. 2010). Research has found that FUS plays multiple roles in various pathways inside the cell. We hereby report a previously unknown function of FUS, i.e. FUS regulates the initiation of autophagy by mediating the transcription of genes that are critical to autophagosome formation.
We found that neither activating autophagy by rapamycin nor inhibiting autophagy by Baf-A1 had any significant effect on the level of insoluble FUS in cells (Figure 1). In contrast, inhibiting the ubiquitin-proteasome system significantly increased the level of insoluble FUS in the RIPA-insoluble fraction. Moreover, the expression of mutant FUS had little effect on the levels of autophagy markers (Figure 2F-G). The role of autophagy in FUS mediated ALS has been debatable. One study reported that impairment of the proteasome pathway replicated ALS phenotypes in mice with TAR DNA-binding protein 43 (TDP-43) and FUS proteinopathy, but impairment of the autophagy pathway by motor neuron-specific knock-out of Atg7 did not show ALS phenotype (Tashiro et al. 2012). This is consistent with our findings that UPS, but not autophagy, is primarily responsible for the turnover of WT and mutant FUS to prevent the accumulation of FUS in the RIPA-insoluble fraction (Figure 1B). However, another study showed that torkinib, an autophagy-enhancing compound that functions as mTOR inhibitor, reduced FUS cytoplasmic aggregates and rescued motor function in P525L induced pluripotent stem cells (iPSC)-derived motor neurons (Marrone et al. 2019). Thus, it remains possible that both UPS and autophagy pathways contribute to the degradation of toxic protein aggregates in various experimental systems in neurodegenerative diseases (Limanaqi et al. 2020).
Our results showed that re-expression of mutant FUS in FUS KO cells had little affect on autophagy as compared to cells re-expressed WT FUS (Figure 2E-G). This is consistent with a previous study showing that the expression of FUS mutant R521C did not impair autophagy (Ryu et al. 2014). However, another study reported that overexpressing mutant FUS affects autophagy by impairing early stages of Rab-1 dependent autophagy pathway (Soo et al. 2015). A recent study elegantly showed that the over-expression of WT and mutant FUS by impairing the FUS autoregulation produced progressive ALS-like phenotypes. Moreover, FUS overexpression altered the autophagy-lysosome pathway and protein homeostasis (Ling et al. 2019). It is noted that the level of WT or mutant FUS re-expression in FUS KO cells was comparable to the endogenous FUS level in N2A cells in this study, which is different from other studies in which FUS was over-expressed at high levels on top of the endogenous mouse FUS. It is conceivable that mutant FUS affected the autophagy pathway differently in different experimental systems.
More interestingly, FUS knockdown or KO cells showed significantly decreased levels of autophagy flux marker (Figure 2A-B). The formation of LC3-positive autophagosomes was also reduced in FUS KO cells (Figure 2C-D). Moreover, the FUS KO cells did not respond to rapamycin treatment as WT cells did (Figure 3C-D). Consistently, the level of LC3-II increased in the presence of Baf-A1, but the level in FUS KO cells was significantly lower than that in WT cells (Figure 3A-B). The results suggest that FUS is involved in the LC3-II lipidation, phagophore elongation and autophagosome formation. It is well documented that the ALS-linked mutations cause cytoplasmic accumulation of FUS protein (Bosco et al. 2010; Gal et al. 2011; Dormann et al. 2010), resulting in a reduced level of FUS in the nucleus. The results regarding the effect of FUS knockdown and KO on autophagy in this study provide an additional mechanism how mutant FUS would disrupt the autophagy pathway.
Furthermore, we demonstrated that the mRNA and protein levels of FIP200, Atg3, Atg16L1 and Atg12 decreased in FUS KO cells (Figure 4). Moreover, the mRNA and proteins levels of the affected genes were restored by expression of FUS (Figure 5). Consistent with gene and protein levels changes, the autophagosome marker LC3-II/LC3-I ratio was also increased when WT FUS was expressed in FUS KO cells (Figure 2E). ULK1, FIP200 and Atg13 form a complex that is required for ULK1 localization to the isolation membrane and subsequent phagophore formation (Jung et al. 2009; Hosokawa et al. 2009a; Ganley et al. 2009). Atg101 is associated with the ULK1 complex by interacting with Atg13 (Hosokawa et al. 2009b). Among the four genes/proteins examined, only FIP200 was affected by FUS KO. FIP200 has been reported to be an essential regulator of autophagy in Drosophila melanogaster (Kim et al. 2013) and mammalian cells (Hara et al. 2008). In both studies, when the FIP200 was silenced, the induction of autophagy activity was severely impacted. Thus, we conclude that the regulation of FIP200 expression by FUS is a critical mechanism in autophagy initiation.
Atg (AuTophaGy-related) genes function as a ubiquitin-like conjugation system to facilitate LC3-II lipidation and phagophore elongation (Xie & Klionsky 2007). Among five Atg genes examined in this study, the levels of Atg3, Atg16L1 and Atg5-Atg12 complex decreased consistently. The Atg5-Atg12 complex conjugates with Atg16L1 (Mizushima et al. 2003) and facilitate the attachment of phosphatidylethanolamine (PE) to the C-terminal glycine of Atg8 (LC3) (Nakatogawa 2013), which promotes phagophore elongation (Xie & Klionsky 2007; Fujita et al. 2008; Saitoh et al. 2008). These studies support our model (Figure 6) that FUS regulates the expression of genes critical to LC3 lipidation, phagophore elongation, and autophagosome formation.
Figure 6. Proposed model of the role of FUS in the autophagy pathway.
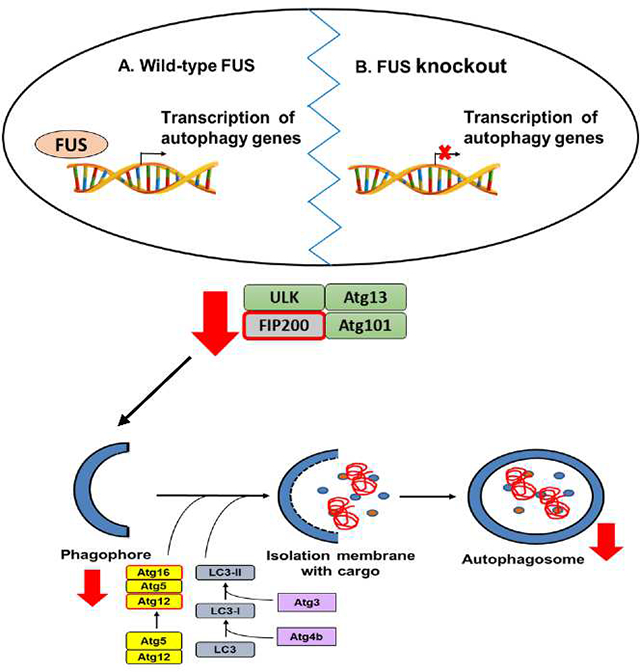
We propose that FUS is involved in transcription regulation of genes involved in autophagy initiation and phagophore elongation. Lack of FUS in the cells causes downregulation at the gene expression and protein levels, causing a decrease in autophagosome formation, and thus the reduction in basal autophagy flux.
It is noted that it is yet to be determined in future studies how FUS regulates the expression of FIP200, Atg3, Atg16L1 and Atg12 genes. FUS was reported to be associated with active chromatin (Yang et al. 2014), to interact with nuclear receptors (Powers et al. 1998), transcription factors (Hallier et al. 1998), and RNA polymerase II and the TFIID complex (Schwartz et al. 2012), suggesting that FUS is a part of the transcription machinery. However, it is unclear how FUS specifically regulate these genes while it does not change other Atg genes. It is conceivable that there are specific elements in the regulatory region of these genes that specifically interact with FUS. Future studies are needed to determine such FUS-interacting elements for those genes. Alternatively, FUS can regulate splicing and mutant FUS was reported to cause aberrant splicing alterations (Qiu et al. 2014; Zhou et al. 2013). Thus, it is possible that FUS KO can also cause abnormal splicing such as intron retention and change mRNA homeostasis, leading to alterations in mRNAs of autophagy genes. This alternative mechanism remains to be tested in future studies.
Our study discovered a novel function of FUS in protein homeostasis. Specifically, FUS regulates the early stages of autophagy by modulating the transcription of genes that play a role in autophagy initiation (FIP200) and phagophore elongation (Atg12 and Atg16L1). The lack of FUS causes the downregulation of the gene expression and protein levels, leading to decreased phagophore formation, and thus the reduction in basal autophagy flux (Figure 6). The function of FUS in autophagy in vivo remains to be further examined in future studies. The significance of FUS regulation of autophagy genes in the context of neurodegenerative diseases such as ALS and FTD is also to be better understood.
Acknowledgements
This study was supported, in part, by National Institute of Neurological Disorder and Stroke Grant R01NS077284 and R01NS115507, Muscular Dystrophy Association grant MDA352743, and Department of Veteran Affairs Merit Review Award I01 BX002149 (to H.Z.). A.A was supported by National Institute of Environmental Health Sciences Training Grant T32ES007266.
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- ATG
Autophagy-related
- Baf-A1
bafilomycin A1
- FTD
frontotemporal dementia
- FUS
Fused in Sarcoma
- GFP
green fluorescent protein
- iPSCs
induced pluripotent stem cells
- KO
knockout
- LC3
microtubule-associated protein 1A/1B-light chain 3
- mTORC
mechanistic target of rapamycin
- NLS
nuclear localization sequence
- PE
phosphatidylethanolamine
- RIPA
radioimmunoprecipitation assay buffer
- SDS-PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- SG
stress granule
- siRNA
small interference RNA UPS: ubiquitin-proteasome system
- WT
wild-type
Footnotes
Open Science Badges
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. More information about the Open Science badges can be found at https://cos.io/our-services/open-science-badges/
Conflict of interest disclosure
No conflict of interest.
References
- Andersson MK, Stahlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, Nilsson O and Aman P (2008) The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC cell biology 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas A, Chen J, Kuang L, Barnett KR, Kasarskis EJ, Gal J and Zhu H (2020) Lysine acetylation regulates the RNA binding, subcellular localization and inclusion formation of FUS. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baechtold H, Kuroda M, Sok J, Ron D, Lopez BS and Akhmedov AT (1999) Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem 274, 34337–34342. [DOI] [PubMed] [Google Scholar]
- Baron DM, Matheny T, Lin YC et al. (2019) Quantitative proteomics identifies proteins that resist translational repression and become dysregulated in ALS-FUS. Hum Mol Genet 28, 2143–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH and Nixon RA (2008) Autophagy Induction and Autophagosome Clearance in Neurons: Relationship to Autophagic Pathology in Alzheimer’s Disease. The Journal of Neuroscience 28, 6926–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco DA, Lemay N, Ko HK et al. (2010) Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet 19, 4160–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronisz A, Carey HA, Godlewski J, Sif S, Ostrowski MC and Sharma SM (2014) The multifunctional protein fused in sarcoma (FUS) is a coactivator of microphthalmia-associated transcription factor (MITF). J Biol Chem 289, 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SK, Zhang J, Gal J, Xu Y, Miao L, Lynn BC, Zhu H, Kasarskis EJ and St Clair DK (2014) FUsed in sarcoma is a novel regulator of manganese superoxide dismutase gene transcription. Antioxidants & redox signaling 20, 1550–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichmann DS and Harland RM (2012) fus/TLS orchestrates splicing of developmental regulators during gastrulation. Genes Dev 26, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D, Rodde R, Edbauer D et al. (2010) ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO journal 29, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG and Takumi T (2005) The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Current biology : CB 15, 587–593. [DOI] [PubMed] [Google Scholar]
- Fujii R and Takumi T (2005) TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 118, 5755–5765. [DOI] [PubMed] [Google Scholar]
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T and Yoshimori T (2008) The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 19, 2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J and Zhu H (2011) Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol Aging 32, 2323 e2327–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Lam DH, Wang J, Ding X, Chen S and Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. The Journal of biological chemistry 284, 12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D, Barth S and Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallier M, Lerga A, Barnache S, Tavitian A and Moreau-Gachelin F (1998) The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J Biol Chem 273, 4838–4842. [DOI] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL and Mizushima N (2008) FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. The Journal of cell biology 181, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T et al. (2009a) Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular biology of the cell 20, 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T and Mizushima N (2009b) Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5, 973–979. [DOI] [PubMed] [Google Scholar]
- Hurley JH and Young LN (2017) Mechanisms of Autophagy Initiation. Annu Rev Biochem 86, 225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Jun CB, Ro S-H, Kim Y-M, Otto NM, Cao J, Kundu M and Kim D-H (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular biology of the cell 20, 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamelgarn M, Chen J, Kuang L, Jin H, Kasarskis EJ and Zhu H (2018) ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proceedings of the National Academy of Sciences 115, E11904–E11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N and Anderson P (2002) Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochemical Society transactions 30, 963–969. [DOI] [PubMed] [Google Scholar]
- Kesidou E, Lagoudaki R, Touloumi O, Poulatsidou KN and Simeonidou C (2013) Autophagy and neurodegenerative disorders. Neural regeneration research 8, 2275–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Park HL, Park H-W, Ro S-H, Nam SG, Reed JM, Guan J-L and Lee JH (2013) Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy 9, 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ Jr., Bosco DA, Leclerc AL et al. (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, N.Y.) 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- Limanaqi F, Biagioni F, Gambardella S, Familiari P, Frati A and Fornai F (2020) Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. International journal of molecular sciences 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Dastidar SG, Tokunaga S et al. (2019) Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Erauskin J, Tadokoro T, Baughn MW et al. (2018) ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 100, 816–830 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IRA, Neumann M, Bigio EH et al. (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta neuropathologica 119, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone L, Drexler HCA, Wang J et al. (2019) FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol 138, 67–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA and Tibbetts RS (2013) The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem 288, 24731–24741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Nagy P, Juhász G and Neufeld TP (2015) Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nature communications 6, 7007–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2007) Autophagy: process and function. Genes Dev 21, 2861–2873. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y and Yoshimori T (2003) Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 116, 1679–1688. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H (2013) Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays in biochemistry 55, 39–50. [DOI] [PubMed] [Google Scholar]
- Powers CA, Mathur M, Raaka BM, Ron D and Samuels HH (1998) TLS (translocated-in-liposarcoma) is a high-affinity interactor for steroid, thyroid hormone, and retinoid receptors. Mol Endocrinol 12, 4–18. [DOI] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y et al. (2014) ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 124, 981–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh N and Pandey UB (2017) Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Frontiers in molecular neuroscience 10, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HH, Jun MH, Min KJ, Jang DJ, Lee YS, Kim HK and Lee JA (2014) Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol Aging 35, 2822–2831. [DOI] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH et al. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268. [DOI] [PubMed] [Google Scholar]
- Sama RR, Ward CL, Kaushansky LJ, Lemay N, Ishigaki S, Urano F and Bosco DA (2013) FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. Journal of cellular physiology 228, 2222–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sama RRK, Ward CL and Bosco DA (2014) Functions of FUS/TLS From DNA Repair to Stress Response: Implications for ALS. ASN NEURO 6, 1759091414544472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA and Rubinsztein DC (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death & Differentiation 16, 46–56. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ and Cech TR (2012) FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev 26, 2690–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Tang AA, Kulkarni A et al. (2014) Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proc Natl Acad Sci U S A 111, E4769–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soo KY, Sultana J, King AE, Atkinson RAK, Warraich ST, Sundaramoorthy V, Blair I, Farg MA and Atkin JD (2015) ALS-associated mutant FUS inhibits macroautophagy which is restored by overexpression of Rab1. Cell Death Discovery 1, 15030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Riley TR, Coady T, Bussemaker HJ and Manley JL (2012) TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc Natl Acad Sci U S A 109, 6030–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro Y, Urushitani M, Inoue H et al. (2012) Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J Biol Chem 287, 42984–42994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DW, Peng ZJ, Ren GF and Wang GX (2015) The different roles of selective autophagic protein degradation in mammalian cells. Oncotarget 6, 37098–37116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ and Tsai LH (2013) Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16, 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Arai S, Song X et al. (2008) Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z and Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nature Cell Biology 9, 1102–1109. [DOI] [PubMed] [Google Scholar]
- Yang L, Embree LJ, Tsai S and Hickstein DD (1998) Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 273, 27761–27764. [DOI] [PubMed] [Google Scholar]
- Yang L, Gal J, Chen J and Zhu H (2014) Self-assembled FUS binds active chromatin and regulates gene transcription. Proc Natl Acad Sci U S A 111, 17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liu S, Liu G, Oztürk A and Hicks GG (2013) ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet 9, e1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Sok J, Immanuel D, Yin Y and Ron D (1997) TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110 ( Pt 15), 1741–1750. [DOI] [PubMed] [Google Scholar]