

ABSTRACT
Glioblastoma (GBM) is the most common and aggressive brain malignancy in adults and is currently incurable with conventional therapies. The use of chimeric antigen receptor (CAR) modified T cells has been successful in clinical treatment of blood cancers, except solid tumors such as GBM. This study generated two third-generation CARs targeting different epitopes of ephrin type-A receptor 2 (EphA2) and examined their anti-GBM efficacy in vitro and in tumor-bearing mice. We observed that these two types of T cells expressing CAR (CAR-T) targeting EphA2 could be activated and expanded by EphA2 positive tumor cells in vitro. The survival of tumor-bearing mice after EphA2 CAR-T cell treatment was significantly improved. T cells transduced with one of the two EphA2 CARs exhibited better anti-tumor activity, which is related to the upregulation of CXCR-1/2 and appropriate interferon-γ (IFN-γ) production. CAR-T cells expressed excessively high level of IFN-γ exhibited poor anti-tumor activity resulting from inducing the upregulation of PD-L1 in GBM cells. The combination of CAR-T cells with poor anti-tumor activity and PD1 blockade improved the efficacy in tumor-bearing mice. In conclusion, both types of EphA2 CAR-T cells eliminated 20%-50% of GBM in xenograft mouse models. The appropriate combination of IFN-γ and CXCR-1/2 levels is a key factor for evaluating the antitumor efficiency of CAR-T cells.
KEYWORDS: Glioblastoma, Chimeric antigen receptor, EphA2, IFN-γ, PD-l1
Introduction
Glioblastoma (GBM) is the most common and aggressive primary brain malignancy in adults.1 Due to its high morbidity, high mortality and low cure rates, GBM has caused a severe social and medical burden worldwide. It is still incurable with conventional therapies.2 Therefore, the development of novel therapies for GBM is necessary to improve the outcome and prognosis of patients with GBM.
T lymphocytes have been found in the cerebrospinal fluid (CSF) of healthy individuals, which are important for immune surveillance.3 However, in patients with GBM, most T cells are exhausted.4–6 This is due to the ability of GBM cells to induce local and systemic immunosuppression. Immune checkpoints, particularly programmed cell death-1 (PD-1) and its ligand (PD-L1), can suppress T lymphocytes activity. The binding of PD-1 to PD-L1 can induce apoptosis and the exhaustion of activated immune cells, limiting the efficacy of adaptive immunotherapy, and poses a significant challenge to the development of new therapies.
Chimeric antigen receptor (CAR) modified T cell therapy is an adoptive T cell therapy emerging as a promising strategy to treat cancer. CARs are synthetic molecules that redirect T cells to eradicate tumors through specific recognition of surface proteins expressed on tumor cells, consisting of an extracellular tumor antigen-binding domain linked to hinge, transmembrane, and intracellular signaling domains.7–9 T cells expressing CAR (CAR-T cells) can directly identify tumor-associated antigens through single-chain variable fragment (scfv) of the extracellular domain, then are activated by intracellular signal transduction, releasing multiple cytokines, such as perforin, granzyme, interferon-γ (IFN-γ), and tumor necrosis factor (TNF), and induce tumor cell apoptosis.10,11 Therefore, CAR-T cells act in an major histocompatibility complex (MHC)-independent manner and ingeniously combine the abilities of antibody-specific recognition of antigens and cytotoxicity of T lymphocytes.
CAR-T cell therapy is an adoptive T-cell therapy, that targets tumor cells directly. Emerging evidence indicates that CAR-T cells also acts in harmony with the endogenous immune system within the tumor microenvironment (TME). In the TME, a group of tumor-infiltrating immune cells including T regulatory cells (Tregs), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) play inhibitory roles in the antitumor efficacy of adoptively transferred tumor-targeted effector T cells.12–14 Furthermore intratumoral CAR-T cells are associated with non-CAR immune cell activation within the TME, with beneficial and pathological effects.15
CAR-T cell therapy was first applied to treat hematologic B-cell malignancies, and it provided effective and encouraging results.16,17 Two new drugs (Kymrial and Yescata) are available to treat hematologic malignancies in the United States and Europe.18,19 However, CAR-T cell treatment for solid tumors has shown limited antitumor activity and is still in the experimental stage.
Several GBM-CAR-T therapy candidate targets have been examined for evaluating their feasibility and reliability, such as IL-13 receptor subunit alpha 2 (IL13Ra2), human epidermal growth factor receptor 2 (HER2), and epidermal growth factor receptor variant III (EGFRvIII), which have exhibited encouraging anti-tumor effects.12,20,21 Ephrin type-A receptor 2 (EphA2) is a member of the ephrin receptor family and plays important roles in a diverse array of developmental, physiological, and disease processes. Recent studies have indicated that EphA2 overexpression is associated with a poor prognosis in multiple types of tumors, such as esophageal, ovarian, and lung cancers.22,23 EphA2 is overexpressed in GBM, although poorly expressed in the normal brain, making it an attractive target for the development of novel therapeutic strategies.24–26 At present, at least two independent groups have developed different EphA2 specific CARs to treat GBM and obtained encouraging pre-clinical results.27–29
This study generated two third-generation CARs targeting different EphA2 epitopes. T cells transduced with different CARs exhibited inhibitory effects on GBM. High-throughput RNA sequencing was used to reveal the molecular mechanisms, and a list of gene expression patterns was obtained for CAR-T cell efficiency prediction.
Materials and methods
Cell lines
The human GBM cell lines U87, U373, U251, and the retrovirus packaging cell line PG13 were purchased from the American Type Culture Collection (ATCC). U251 and U373 cells expressing eGFP and firefly luciferase were generated by retroviral infection. All these cells were maintained in Dulbecco’ s modified Eagle’ s medium (Lonza), containing 10% fetal bovine serum (Biosera) and 10,000 IU/mL penicillin/10,000 μg/mL streptomycin (EallBio Life Sciences).
Generation of retroviral vectors encoding EphA2-specific CARs
Two different EphA2-scFvs (D2-1A7 and D2-1B1) coding sequences were synthesized by GeneArt (Invitrogen) and then subcloned into the SFG retroviral vector. All cloning of the CARs were verified by sequencing. Retroviral particles were generated by PG13 cells after transient transfection.
Generation of CAR T cells
Human peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by Lymphoprep (MP Biomedicals) gradient centrifugation. To generate EphA2-CAR T cells, T cells in PBMCs were stimulated with anti-CD3 and anti-CD28 beads, and then infected with retrovirus. On day 7, the T cells were subjected to CAR expression detection and then expanded in X–VIVOTM15 serum-free medium (Lonza) which containing 5% GemCellTM Human Serum AB (Gemini Bio) and IL-2 (SL PHARM). This research was approved by the Beijing Shijitan Hospital Institutional Review Board and informed consent was obtained from all the participants.
Flow cytometry
Flow cytometry was performed on a FACSCanto Plus instrument (BD Biosciences) and FlowJo v.10 (FlowJo, LLC) was used for data analysis. Genetically modified T cells were detected after staining with APC-labeled mouse anti-human CD3 antibody (BD Biosciences), PE-labeled mouse anti-human CD8 antibody (BD Biosciences), BV421 labeled mouse anti-human CD4 antibody (BD Biosciences), and goat anti-mouse IgG (Fab specific) F(ab′)2 fragment-FITC antibody (Sigma). GBM cells was stained with Alexa fluor 700 labeled mouse anti-human EphA2 (R&D System) and then subjected to flow cytometry to examine the cell surface EphA2 expression.
RNA interference
GBM cells were transfected with small interfering RNAs (siRNAs) using Lipofectamine 3000 (Thermo Fisher Scientific) for 48 hours. The cells were then collected for RNA and protein extraction.
Sequences for siRNAs:
Si-Control, ACGUGACACGUUCGGAGAA; si-IFNGR1, UUAUACUGGAUCUCACUUC; si-IFNGR2, UUCGUAGCAAGAUAUGUUG; si-PD-L1, UCUCUCUUGGAAUUGGUGG.
Cell binding assay
CAR-T cells were washed twice by sterile PBS and then stained with 0.5 μM CSFE (Thermo Fisher Scientific) solution. After blocking and wash, the CAR-T cells were incubated with pre-plated GBM cells for 5 min at 37°C. The cells were then washed by pre-warmed PBS three times and then fixed by 4% paraformaldehyde. Images were captured before and after wash. Experiment were repeat for 3 times and cells numbers were counted in at least 3 independent field.
Immunoblotting
Cells were washed with PBS three times and then the protein was extracted using RIPA buffer. The protein samples were quantified using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), and then denatured in sodium dodecyl sulfate (SDS)/β-mercaptoethanol sample buffer. Samples (10 μg) were separated on a 15% SDS-polyacrylamide gel and blotted onto polyvinylidene fluoride membranes (Millipore) by electrophoretic transfer. The membrane was incubated with mouse anti-human CD247 (BD Biosciences) overnight at 4°C, and then the specific protein-antibody complex was detected using HRP conjugated goat anti-mouse secondary antibody (Santa Cruz Biotechnology). Detection of the chemiluminescence reaction was carried out using an ECL kit (Thermo Fisher Scientific). The experiment was repeated at least three times.
Real-time RT-PCR
Total RNA was extracted from cells using TRIzol Reagent (Invitrogen) following the manufacturer’s instructions. The quantity and purity of RNA were measured using a Nanodrop One spectrophotometer (Thermo Fisher Scientific). Only samples with the appropriate absorbance measurements (A260/A280 of ~2.0, and A260/A230 of 1.9–2.2) were considered for inclusion in this study. cDNA was synthesized using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) and then amplified using SYBR Green PCR Master Mix (Thermo Fisher Scientific) with gene-specific primers. GAPDH was used as an internal control. Relative gene expression was calculated using the 2− ΔΔCt method.
T cell proliferation assay
T cells were labeled with 5 μM CFSE (Thermo Fisher Scientific) following the manufacturer’s protocol. A co-culture of 1 × 106 CFSE-labeled T cells and 5 × 105 target cells was done for 72 h, and T cell proliferation was analyzed by flow cytometry to confirm the existence of discrete peaks.
In vitro killing assay
Mock or anti-EphA2 CAR T cells were incubated with U87, U373 or U251 cells at 1:1, 2.5:1, 5:1 or 10:1 ratio (Effectors: Targets, E: T) in X–VIVOTM15 medium for 24 h. Supernatants were removed and collected for measurement of IFNγ by ELISA. The luciferases activities were monitored using the IVIS imaging system (IVIS, Xenogen, Alameda, CA, USA).
Analysis of cytokine production
The CAR-T cells were co-cultured with GBM cells at an E: T ratio of 10:1 for 24 hours. The supernatants were collected and subjected to IFN-γ detection. IFN-γ levels were measured using the Human IFN-gamma DuoSet ELISA kit (R&D system) according to the manufacturer’s instructions.
Impedance-based tumor cell killing assay
Using the xCELLigence impedance-based system, continuous tumor cell death was evaluated over 24 h. U373 and U251 tumor cells were plated in a 96-well, resistor-bottomed plate at 10,000 cells per well. After 24 h, 100,000 T cells (10:1 seeding ratio) were added in triplicate, and NT cells served as controls, at which point cell index values correlating to U373 or U251 adherence were normalized. Impedance-based measurements of the normalized cell index were collected every 15 min, which were determined by measuring of the impedance of current across the transistor plate caused by tumor cell adherence.
Xenograft mouse model with subcutaneous glioma cells injection
Six- to eight-week-old NOD-SCID mice were purchased from Charles River Laboratories. The right flank of female NOD-SCID mice were injected with 5 × 106 U251-eGFP-Luc cells to construct the xenograft mouse model. Ten days after tumor cell injection, 3 × 107 CAR T cells were injected directly into the tumor. Tumor development was monitored using IVIS (IVIS, Xenogen, Alameda, CA, USA), and the mice were sacrificed when the diameter of the tumor reached 20 mm.
For the PD1 antibody co-treatment experiment, NOD-SCID mice were injected subcutaneously with 1 × 107 U251-eGFP-Luc cells to construct the xenograft mouse model. Five days post-tumor xenograft, mice were treated with a total of 3 × 107 CAR T cells or non-transduced control T cells, followed by peritumoral (p.t.) administration of 200 μg PD1 antibody (Bio X Cell). On the day of 13, the mice were administrated a second dose of PD1antibody (Bio X Cell) and then the tumor growth was monitored using IVIS (IVIS, Xenogen, Alameda, CA, USA).
All experiments including mice were approved by the Beijing Shijitan Hospital Institutional Review Board.
Xenograft mouse model with glioma cells intracranial implantation
Six to eight week-old NOD-SCID mice were anesthetized with a ketamine/xylazine cocktail solution. Animals were secured in a stereotaxic head frame, a 1 cm midline scalp incision was made, and 2 × 105 U373-eGFP-luc cells in 5 μL PBS were injected into the left striatum (coordinates: 2.5 mm lateral and 0.5 mm posterior to the bregma) through a burr hole in the skull using a 10 μL BD syringe to deliver tumor cells to a 3.5 – mm intraparenchymal depth. The burr hole in the skull was sealed with bone wax, and the incision was closed using medical adhesive glue (COMPONT). Two weeks after the tumor cells injection, 3 × 107 CAR T cells were injected through tail vein and tumor growth was monitored using the IVIS in vivo imaging system (IVIS, Xenogen, Alameda, CA, USA). All experiments with mice were approved by the Beijing Shijitan Hospital Institutional Review Board.
Immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) sections of tumor specimens were deparaffinized and then incubated with rabbit anti-PD-L1 or rabbit anti-Ki67 primary antibody (Cell Signaling Technology) at 4°C overnight. After incubation with HRP – conjugated goat anti-rabbit secondary antibody, the signal was detected using DAB Substrate kit (Abcam) following the manufacture’s instructions. Images were obtained using a microscopy (Nikon). Three different random images were captured for each sample at 400× magnification and the relative density of PD-L1 signal was quantified by Image J v1.49.
RNA high-throughput sequencing
Samples containing 2 µg total RNA each were used as input material for generating the sequencing libraries using the NEBNext®Ultra™ RNA Library Prep Kit (#E7530L, NEB) following the manufacturer’s. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEB Next First Strand Synthesis Reaction Buffer (5X). First-strand cDNA was synthesized using random hexamer primers and RNase H. Second strand cDNA synthesis was subsequently performed using buffer, dNTPs, DNA polymerase I, and RNase H. The library fragments were purified with QiaQuick PCR kits, eluted with EB buffer, and then terminal repair, A-tailing, and adapters were implemented. To complete the library, the products were retrieved and PCR was performed. The clustering of index-coded samples was performed on a cBot Cluster Generation system with TruSeq SR Cluster kit, v3-cBot-HS (Illumina Inc.), according to the manufacturer’s protocol. Subsequently, the library was subjected to an Illumina NovaSeq 6000 System platform (Illumina Inc.) for sequencing. Raw data were first processed using custom Perl scripts. Clean data (clean reads) were read by removing reads containing poly-N with 5′- adapter contaminants, without 3′- adapter or the insert tag, or containing poly-A, -T, -G or -C from raw data, as well as low-quality reads.
The differentially expressed genes between two CAR-T cells were subjected to gene ontology (GO), KEGG pathway, and subcellular localization analysis using an online bioinformatics tool: DAVID Bioinformatics Resources 6.8. Data visualization and analysis were processed by custom Rstudio scripts following the packages (ggplot2 and Treemap). Fisher’s exact test was used for the gene-enrichment analysis.
Statistical analysis
All experiments were performed at least in triplicate and GraphPad Prism version 8.0.2 (GraphPad Software) was used for statistical analysis. Data are presented as mean ± standard deviation. The differences between means were tested using appropriate tests. Overall survival of mice with GBM xenografts was measured using the Kaplan–Meier method, with Cox proportional hazard regression analysis for group comparison. Statistical significance was set at p < .05.
Results
Overexpression of EphA2 in glioblastoma cell lines
To evaluate the clinical treatment potential of EphA2 specific CAR-T cells, the level of EphA2 on the cell surface of three GBM cell lines (U251, U87, and U373) was first detected by flow cytometry. As shown in (Figure S1), EphA2 was expressed on the cell surface of 93.45% U87 cells, 97.75% U251 cells, and 93.40% U373 cells, indicating that EphA2 can be used as a target for CAR design.
Generation of EphA2 CAR-T cells
To generate EphA2 specific CAR-T cells, we developed retroviral vectors encoding third-generation (CD28.4–1BBζ) CAR based on two different EphA2-specific mAbs. These two different CARs contained the same N-terminal leader sequence, one of the EphA2-scFvs (D2-1A7 and D2-1B1), the CD28 transmembrane domain, and signaling domains derived from CD28.4–1BB and CD3.ζ (Figure 1a).
Figure 1.
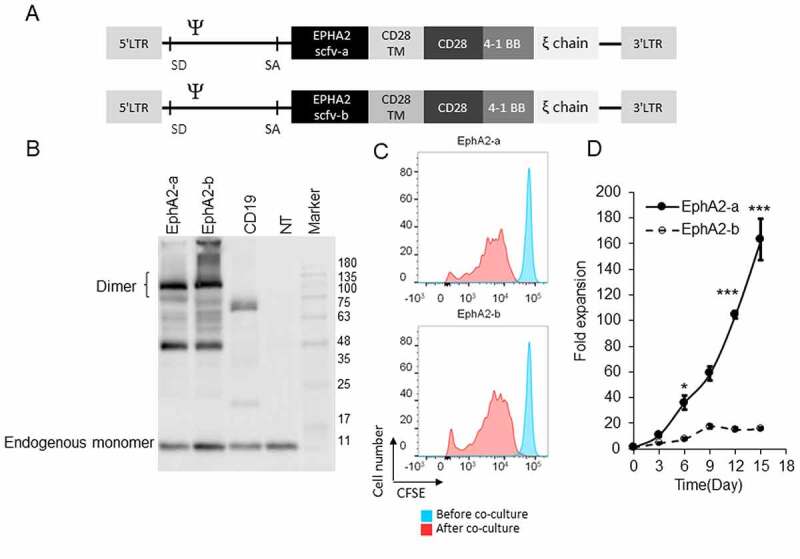
Developing EphA2-Specific CAR-T Cells (a) Schematic diagram of two different chimeric antigen receptors (CARs) targeting EphA2. It consists of EphA2 scFv, the hinge, transmembrane (TM) region of CD28, CD28 and 4–1BB signaling domain, and human CD3ζ chain. (b) CAR-T cells were subjected to immunoblotting to detect the expression of full-length EphA2-CARs by using CD3ζ antibody. (c) Different EphA2-CAR-T cells were labeled with CFSE and then co-cultured with U251 cells for 72 h. The extent of T cell proliferation is reflected by the loss of incorporated CFSE. (d) CAR-T cells were co-cultured with U251 cells for 15 days at an E:T ratio of 2:1. T cells were stimulated every three days with fresh U215 cells, and T cells were counted before the addition of U251 cells. Results were analyzed by student’s t-test, and a p < .05 was considered significant. *p < .05, ***p < .001. SD, splice donor; SA, splice acceptor
Primary T cells were isolated from healthy donor peripheral blood mononuclear cells and stimulated with anti-CD3 and anti-CD28 beads. The T cells were infected with one of the retroviruses and then subjected to immunoblotting 7 days after transduction to examine their efficiency. As shown in (Figure S2a), approximately 30% of the cells were CAR positive, and there were no significant differences between these two groups (Figure 1b). Cell proliferation was detected by flow cytometry 3 days after CFSE staining and direct cell counting. The CAR-T cells exhibited good viability and proliferation capacity (Figure 1c and Figure 1d), and the CD4/CD8 ratios were similar between the two groups (Figure S2B).
Two EphA2 CAR-T cells exhibited different anti-tumor activity in vitro
Subsequently, EphA2-CAR-T cells were co-cultured with GBM cells (U87, U251, and U373) at E:T ratios from 1:1 to 10:1 to examine their anti-tumor activity. As shown in (Figure 2a),EphA2-a-CAR-T cells lysed more than 76.05% of the tumor cells at a low E:T ratio (1:1) and more than 85% of the tumor cells at a high E:T ratio (10:1) after 24 h of co-culture, which had a significantly higher tumor cell killing rate than NT and EphA2-b-CAR-T cells (Figure 2a). We observed an extremely high IFN-γ level in the medium from co-cultured EphA2-b-CAR-T cell, compared to that from the EphA2-a group (Figure 2b). Results from the real-time cell growth monitoring (RTCA) system indicated that EphA2-a and EphA2-b-CAR-T cells could repress the growth of tumor cells when compared with control T cells (Figure 2c). EphA2-a-CAR-T cells exhibited the highest tumor repression capacity in vitro (Figure 2c).
Figure 2.
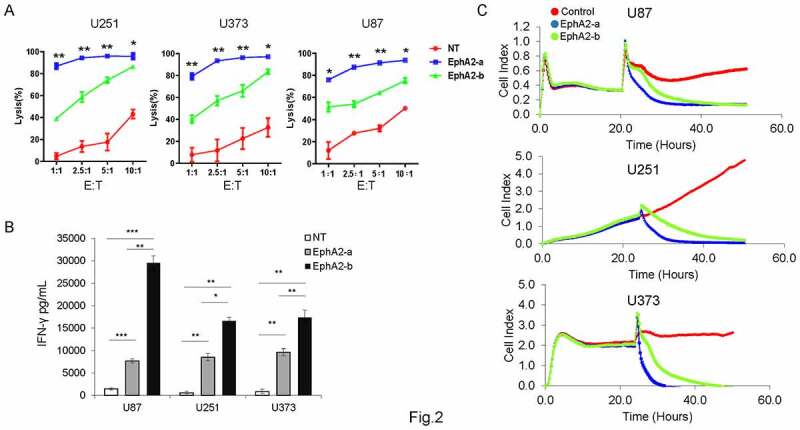
Comparison of antitumor effects of different EphA2 CAR-T cells In vitro. NT or EphA2 CAR-T cells were co-cultured with different target cells at different E:T ratios (1:1, 2.5:1, 5:1, and 10:1) for 24 h. (a) The cell lysis rate was quantified by examining the luciferase activity. (b) The supernatants were collected to evaluate IFN-γ levels by ELISA (E:T = 10:1). (c) Continuous graphical output of cell index values up to the 50 h time point was monitored using the xCELLigence impedance system. Results were analyzed by one-way ANOVA, and statistical significance was set at *p < .05, **P < .01, ***p < .001
Two EphA2 CAR-T cells exhibited different anti-tumor activity in vivo
To further examine the anti-GBM activity of EphA2-CAR-T cells in vivo, U251-eGFP-Luc cells were injected subcutaneously into NOD-SCID mice to generate a GBM xenograft mouse model. On day 10, T cells were injected directly into the tumor with non-transduced T cells (NT) as the negative control, and tumor growth was monitored for 70 days. As shown in (Figure 3a), mice treated with EphA2-a-CAR-T cells showed significantly prolonged survival compared to control mice. EphA2-b-CAR-T cell treated mice had prolonged survival compared to control mice; however, the difference was not significant. Similar results were observed in an intracranial glioma xenograft mouse model. As shown in (Figure 3b), both types of CAR-T cells repressed tumor growth, whereas EphA2-a -CAR-T cells exhibited better anti-GBM activity than the other.
Figure 3.
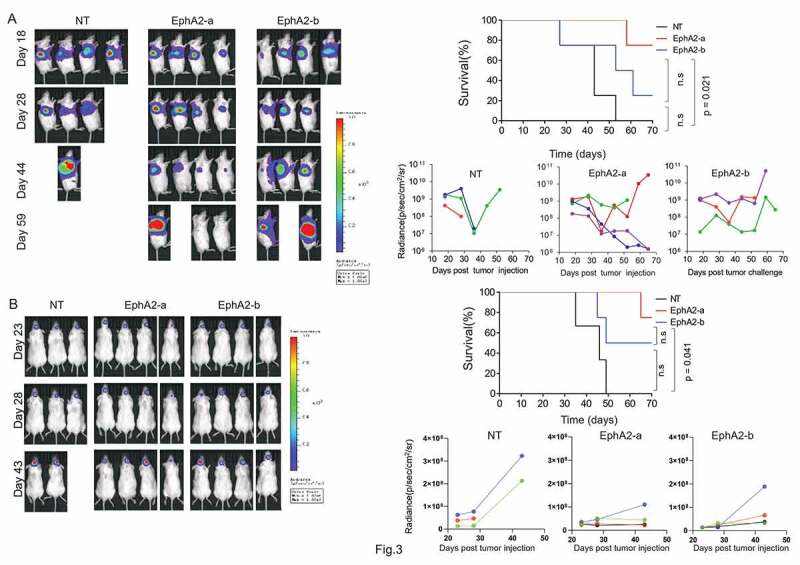
Comparison of antitumor effects of different EphA2 CAR-T cells in xenograft mouse models. (a) 5 × 106 eGFP-Luc-U251 cells were injected subcutaneously into the left flank of 6–8 week-old female NOD-SCID mice. Ten days after injection, the tumor bearing mice were treated with 3 × 107 EphA2 CAR T cells through direct injection into the tumor, with un-transduced T cells (NT) as control. The tumor growth was monitored using the IVIS system with a tumor diameter of 2 cm as the endpoint. Quantitative bioluminescence (radiance = photons/cm2/sr) imaging data for all mice are shown. Overall survival of mice with GBM xenografts was measured using the Kaplan–Meier method, with Cox proportional hazard regression analysis for group comparison. A p-value less than 0.05 was considered significant. (b) Six to eight week-old NOD-SCID mice were anesthetized and then 2 × 105 cells were injected into the left striatum through a burr hole in the skull. Two weeks after tumor cells injection, 3 × 107 CAR-T cells were injected through the tail vein. Thereafter, the tumor growth was monitored using in vivo imaging system IVIS. Quantitative bioluminescence (radiance = photons/cm2/sr) imaging data for all mice are shown. Overall survival of mice with GBM xenografts were measured using the Kaplan–Meier method, with Cox proportional hazard regression analysis for group comparison. A p-value less than 0.05 was considered significant
Analysis of the differentially expressed genes using high throughput RNA sequencing
To explore why EphA2-a-CAR-T cells have better tumor repression activity than EphA2-b-CAR-T cells, high throughput RNA sequencing was used to examine the differentially expressed genes between EphA2-a and EphA2-b-CAR-T cells. Raw data were submitted to the Gene Expression Omnibus database (accession code GSE163833). We found 1090 upregulated and 1228 downregulated genes in the EphA2-a-CAR-T cells compared with EphA2-b-CAR-T cells after co-culture with GMB cells (Figure S3). The top 400 differentially expressed genes were subjected to gene ontology analysis, and we found that genes regulating adaptive immune response, potassium ion transport, and transmembrane receptor protein tyrosine kinase signaling pathway were upregulated in EphA2-a-CAR-T cells. The expression of genes in IFN-γ mediated signaling pathway, JAK-STAT cascade, inflammation response and type I interferon signaling pathway were relatively low in EphA2-a-CAR-T cells (Figure 4). In addition, both the up-regulated and downregulated genes in EphA2-a-CAR-T cells were mostly enriched in the extracellular region and cell membrane. After protein-protein interaction (PPI) analysis (Figure S4 and S5), we found that two clusters including 12 genes 42 edges, and 10 genes 43 edges were at the center of the downregulated PPI network (Figure S5).
Figure 4.
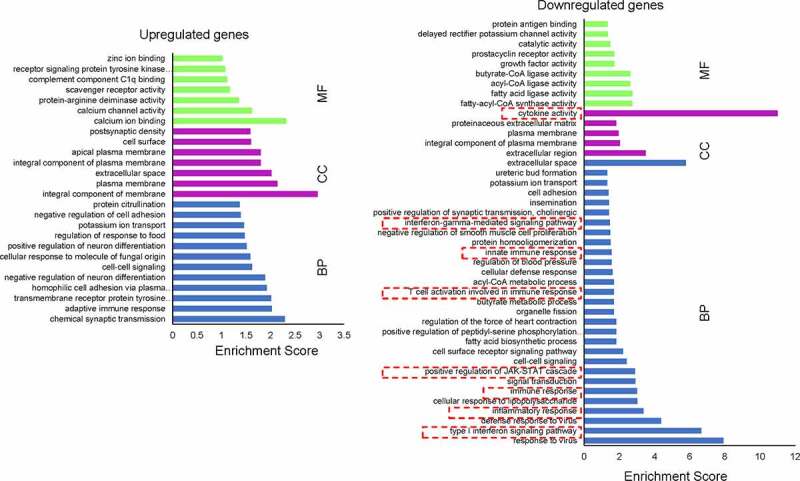
Gene ontology enrichment analysis of differentially expressed genes after co-culture. The differentially expressed genes between the two CAR-T cells were subjected to GO analysis using the online bioinformatics tool: DAVID Bioinformatics Resources 6.8. Fisher’s exact test was used for the gene enrichment analysis. BP, biological process; CC, cellular component; MF, molecular function
To confirm the RNA-seq results, we collected the CAR-T cells 4 h after being co-cultured with U87, U251, or U373 cells, and then detected 3 candidate genes by RT-qPCR. We found that both EphA2-CAR-T cells had significantly high expression of the IFNG genes before and after co-culture with GBM cells (Figure 5a). Compared with EphA2-a-CAR-T cells, EphA2-b-CAR-T cells had high expression of IFNG before GBM cells co-culture, and 4 h after co-culturing with U251 and U87 cells. Similarly, EphA2-b-CAR-T cells expressed higher levels of CXCL8 than EphA2-a-CAR-T cells before and after tumor cell co-culture. EphA2-b-CAR-T cells expressed 11.87 fold higher IL-21 than EphA2-a-CAR-T cells, however, no significant difference was found after tumor cell co-culture.
Figure 5.
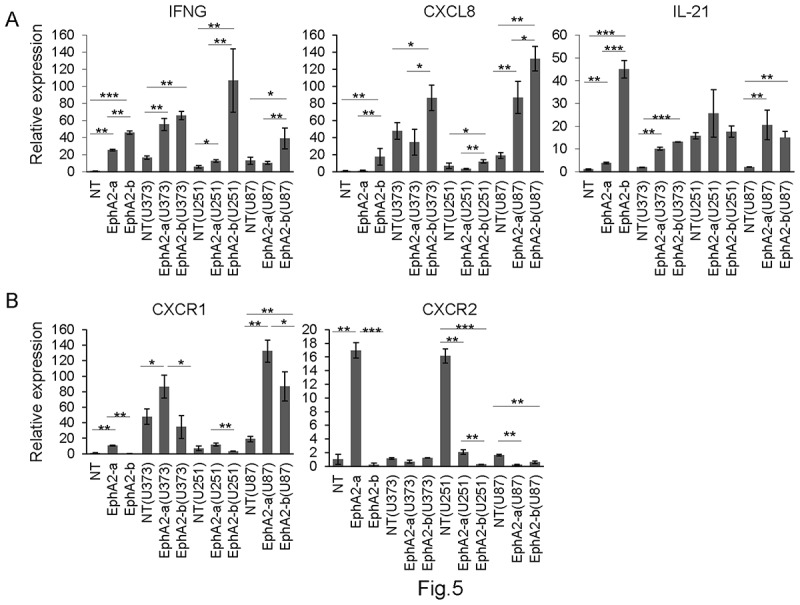
Low IFN-γ and CXCL8 levels relate to better anti-tumor activity in EphA2-CAR-T cells. EphA2 CAR-T cells before or after co-culture with GBM cells were subjected to RT-qPCR to examine the expression of IFN-γ, CXCL8, IL-21, CXCR1, and CXCR2. Results were analyzed by one-way ANOVA and statistical significance was set at p < .05. *p < .05, **P < .01
Different scFvs may have variable affinity to antigens, and furthermore influence the cell-cell interactions between CAR-T cells and target cells. To reveal the mechanism of differentially expressed genes in these two EphA2-CAR-T cells, we examined the interactions between T and tumor cells with a simple cell binding assay. As shown in (Figure S6), EphA2-a-CAR-T cells are more easily to recognize and bind with GMB cells, which should be pivotal factor to induce the different gene expression profile.
It has been reported that T cells are suppressed by various mechanisms at the tumor site, among which PD-1/PD-L1 axis-mediated functional inhibition plays a key role.30–32 It is known that overexpressed IFN-γ in the TME can bind to its receptor, subsequently activating the JAK/STAT signaling pathway, which further induces PD-L1 expression in the tumor cells.33,34 To understand whether the extremely high IFN-γ level contributed to the worse anti-tumor activity of EphA2-b-CAR-T cells via inducing PD-L1 in GBM cells, we examined the mRNA level of PD-L1 in GBM cells co-cultured with CAR-T cells at two different time points. As shown in (Figure 6a), the GBM cells cultured with EphA2-b-CAR T cells expressed significantly increased PD-L1 compared with EphA2-a-CAR T cells at 30 min after co-culture with tumor cells. When the co-culture time was increased to 4 h, the co-cultured GBM cells expressed more than 8-fold higher PD-L1 expression than wild-type GBM cells. However, PD-L1 levels in GBM cells co-cultured with two different EphA2-CAR T cells were similar.
Figure 6.
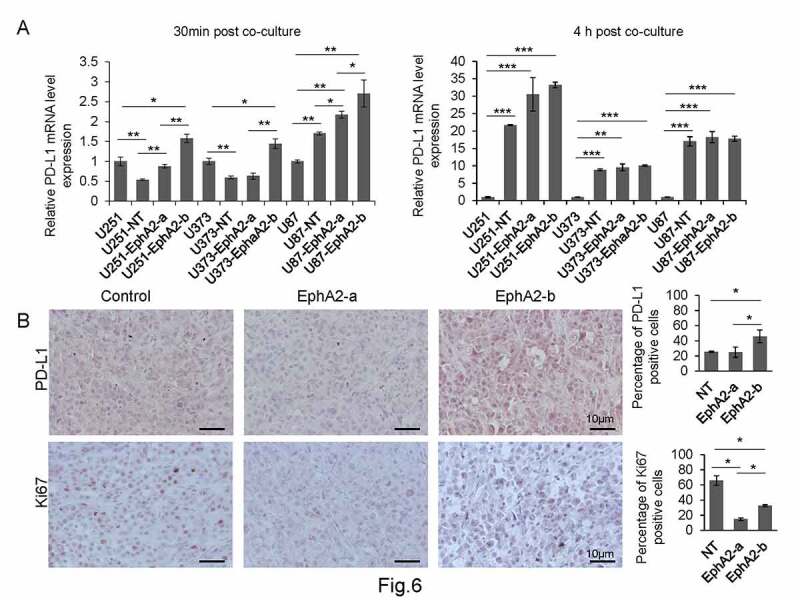
EphA2-b-CAR-T cells induced upregulated PD-L1 expression in GBM cells. (a) GBM cells were co-cultured with CAR-T cells at an E:T ratio of 2:1 for 30 min and 4 h. The GBM cells were then subjected to RT-qPCR to determine the PD-L1 level. (b) The PD-L1 and Ki-67 levels in the tumors from CAR-T cells treated mice were detected by immunohistochemistry. The positive signal was quantified by Image J. Results were analyzed One-way ANOVA and statistical significance was set at p < .05. *p < .05, **P < .01, ***p < .001
We detected PD-L1 levels in tumor samples from CAR-T cell treated xenograft mouse models by immunohistochemistry. As shown in (Figures 6B), 45.89% of tumor cells were PD-L1 positive in the EphA2-b-CAR-T cells treated group, significantly higher than the percentage in the EphA2-a-CAR-T cell-treated group. In contrast, the percentage of Ki67-positive tumor cells in the EphA2-b-CAR-T group was 32.46%, significantly higher than that in the EphA2-a-CAR-T group.
To verify whether EphA2-b-CAR-T cells induced increased PD-L1 expression in GBM cells through the IFN-γ-IFN receptor pathway, we knocked down IFNGR1, IFNGR2, or PG-L1 by siRNAs separately (Figure 7aandFigure 7b), and then performed an in vitro killing assay. As shown in Figure 7c, increased anti-GBM activity was observed in both EphA2-a and EphA2-b-CAR-T cells, and EphA2-b-CAR-T cells exhibited similar tumor cell killing activity as EphA2-a-CAR-T cells.
Figure 7.
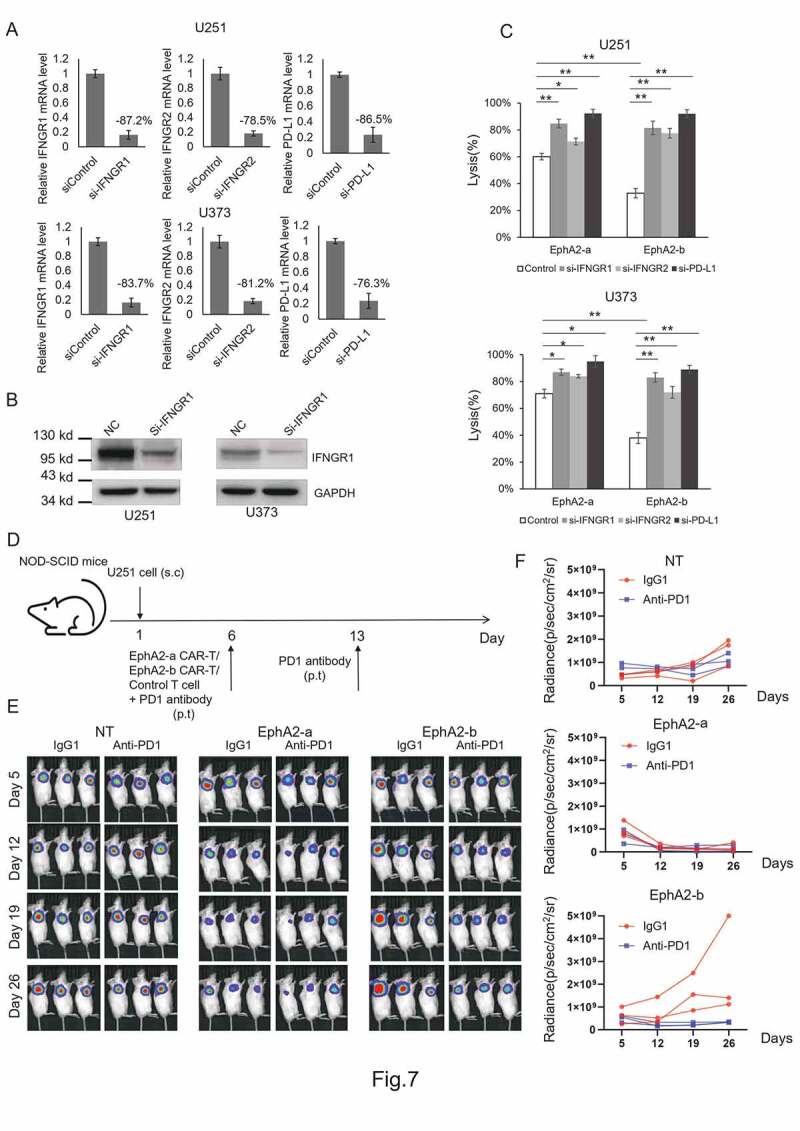
EphA2-b-CAR-T treatment combined with PD1 blockade exhibited enhanced antitumor activity. U251 and U373 cells were transiently transfected with one of the siRNAs to knock down the expression of IFNGR1, IFNGR2 or PG-L1 separately (a and b), and then the cells were subjected to an in vitro killing assay at an E:T = 1:1. The cell lysis rates were determined by detecting luciferase activity (c). NOD-SCID mice were injected subcutaneously with 1 × 107 U251.eGFP.Luc cells to construct a xenograft mouse model. Five days post-tumor xenograft, mice were treated with a total of 3 × 107 CAR-T cells or non-transduced control T cells peritumorally, followed by peritumoral (p.t.) administration of 200 μg PD1 antibody. On day 13, the mice were administered a second dose of PD1antibody, and tumor growth was monitored using IVIS (D, E, and F)
Finally, we treated tumor-bearing mice with CAR-T cells combined with PD1 antibody (Figure 7d-f). In the NT group, the PD1 antibody co-treatment did not inhibit tumor growth. EphA2-a-CAR-T cell treatment dramatically repressed the tumor growth regardless of the PD1 antibody treatment. The combination of EphA2-b-CAR-T cell with PD1 blockade could boost CAR-T antitumor activity indicating that PD1-PD-L1 interaction plays an important role in repressing the antitumor activity of the EphA2-b-CAR-T cells. Based on the results established herein, we proposed a model for the IFN-γ-PD-L1 axis regulating EphA2-CAR-T cell therapy against GBM (Figure 8).
Figure 8.
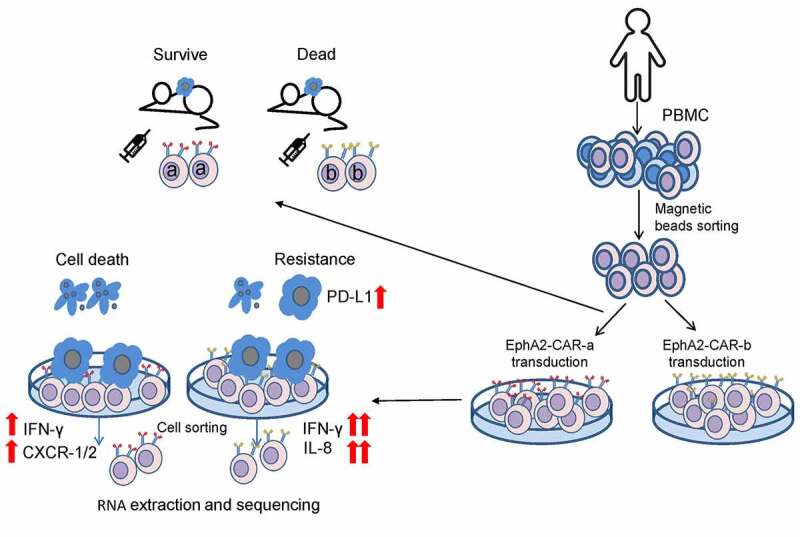
Experiment workflow and proposed model of better anti-tumor efficacy after EphA2-a-CAR-T cells treatment
Discussion
GBM is one of the most common and aggressive primary brain malignancies in humans, and has high morbidity, mortality, and recurrence rate. The use of CAR-T cells has been successful in treating blood cancers clinically, except solid tumors such as GBM. EphA2 is overexpressed in GBM cells, and at least two independent groups have tried EphA2 specific CAR-T cells to treat GBM.27–29 This study successfully generated two third-generation CARs targeting EphA2 and evaluated their tumor repression capacity in vitro and in vivo. T cells transduced with any one of these two CARs exhibited anti-tumor activity both in vitro and in xenograft mouse models, which is consistent with the findings of other researchers and provides a powerful candidate tool for clinical GBM treatment.
Despite the significant breakthrough of CAR-T therapy in terms of its clinical curative effects, the CAR-T cells generated from different laboratories or by different methods exhibit inconsistent anti-tumor efficiency. Several factors affect the anti-tumor efficiency of CAR-T cells, such as target antigen affinity, terminal differentiation, and off-target toxicity. However, there is still no technical standard to evaluate CAR-T cell efficiency.35 This study compared the gene expression profiles of T cells transduced with two different CARs targeting different epitopes of the same protein. Several candidate genes regulating the Wnt signaling pathway, cell viability, and apoptosis have the potential to become gene markers for CAR-T cell evaluation.
Interferon gamma is a dimerized soluble cytokine critical for innate and adaptive immunity and is mainly secreted by activated T cells. IFN-γ plays a key role in the activation of cellular immunity and, subsequently, in the stimulation of the antitumor immune response. However, excessive IFN-γ production is one of the main sources of CAR-T cell toxicity.36,37 In addition, high IFN-γ levels in the TME can induce the overexpression of PD-L1 in tumor cells, which further binds to PD-1 in T cells and induces T cell exhaustion. This study observed that CAR-T cells secreted an extremely high level of IFN-γ had weak anti-tumor activity against GBM in vitro and in vivo. These findings indicate that excessively high IFN-γ production can be used as a negative evaluation criterion for CAR-T cells. Furthermore, PD-1 knockout combined with CAR-T therapy is a good strategy to overcome the IFNγ-PD-L1-PD-1 obstacle.
CXCL-8, also known as IL-8, is a chemotactic factor that attracts neutrophils, basophils, and T-cells during the inflammatory process. In addition, CXCL-8 acts as an important multifunctional cytokine that modulates tumor proliferation, invasion, and migration in an autocrine or paracrine manner.38 In the TME, tumor cells can overexpress CXCL-8 naturally, and ionizing radiation has been found to significantly increase the production of CXCL-8 in tumors.39 The CXCL-8 receptors CXCR1 or CXCR2 modified CAR-T cells exhibit increased migration and persistence when treating solid tumors,40 indicating that CXCL-8 can be leveraged for CAR T cell therapy. In this study, we found that EphA2-a-CAR T cells expressed less CXCL-8 and more CXCR1 and CXCR2. The upregulation of CXCR1 and CXCR2 may sensitize T cells to CXCL-8 in the TME contributing to better anti-tumor activity.
As one of the most important parameters of scFv, affinity has been modulated to improve the specificity of CARs, and reduce “on-target, off-tumor” side effects. The altered affinity of scFvs can impact CAR signaling and other effector functions, such as cytokine production, cell proliferation, and in vivo persistence. In this study, the two CARs were developed by targeting two different epitopes of EphA2, and they should have different affinities for EphA2. The difference in affinity is related to altered avidity, which directly determine the anti-tumor efficiency of CAR-T cells. Therefore, better anti-GBM activity of EphA2-a-CAR-T cells, and differentially expressed genes should mostly result from different T-tumor cell interactions, which are determined by their different affinity and avidity.
In conclusion, we generated two third-generation EphA2 CARs which can eliminate 20% – 50% GBM in xenograft mouse models and provide a candidate gene list to evaluate the efficiency of CAR T cells.
Supplementary Material
Funding Statement
Beijing Municipal Science and Technology Commission, Brain Science Research Fund (Z161100000216136);
Disclosure statement
No competing interest.
Consent for publication
Written informed consent was obtained from all the participants.
Ethics approval and consent to participate
This research was approved by the Beijing Shijitan Hospital Institutional Review Board.
Availability of data and material
The high throughput sequencing data has been submitted to the Gene Expression Omnibus database (accession code GSE163833)
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Abbreviations
| GBM | Glioblastoma |
| CAR | Chimeric antigen receptor |
| EphA2 | Ephrin type A receptor 2 |
| RTCA | Real-time cell analysis |
| TNF | Tumor necrosis factor |
| IFN-γ | Interferon-γ |
| IL13Ra2 | IL-13 receptor subunit alpha 2 |
| HER2 | Human epidermal growth factor receptor 2 |
| EGFRvIII | Epidermal growth factor receptor variant III |
| PBMC | Peripheral blood mononuclear cell |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| PPI | Protein-protein interaction |
References
- 1.Omuro A, DeAngelis LM.. Glioblastoma and other malignant gliomas: a clinical review. JAMA J Am Med Assoc. 2013;310(17):1842–11. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 3.Kivisakk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, Wu L, Baekkevold ES, Lassmann H, Staugaitis SM, et al. Human cerebrospinal fluid central memory CD4+T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A. 2003;100(14):8389–8394. doi: 10.1073/pnas.1433000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, Zimmer J, Chekenya M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264(1–2):71–83. doi: 10.1016/j.jneuroim.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Raychaudhuri B, Rayman P, Huang P, Grabowski M, Hambardzumyan D, Finke JH, Vogelbaum MA. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol. 2015;122(2):293–301. doi: 10.1007/s11060-015-1720-6. [DOI] [PubMed] [Google Scholar]
- 6.Elliott LH, Brooks WH, Roszman TL. Cytokinetic basis for the impaired activation of lymphocytes from patients with primary intracranial tumors. J Immunol. 1984;132:1208–1215. [PubMed] [Google Scholar]
- 7.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16(9):566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gacerez AT, Arellano B, Sentman CL. How chimeric antigen receptor design affects adoptive T cell therapy. J Cell Physiol. 2016;231(12):2590–2598. doi: 10.1002/jcp.25419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez-Lostao L, Anel A, Pardo J. How do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res off J Am Assoc Cancer Res. 2015;21(22):5047–5056. doi: 10.1158/1078-0432.CCR-15-0685. [DOI] [PubMed] [Google Scholar]
- 11.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, Bercovici N, Guérin M, Biton J, Ouakrim H, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A. 2018;115(17):E4041–E50. doi: 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen PH, Lipschitz M, Weirather JL, Jacobson C, Armand P, Wright K, Hodi FS, Roberts ZJ, Sievers SA, Rossi J, et al. Activation of CAR and non-CAR T cells within the tumor microenvironment following CAR T cell therapy. JCI Insight. 2020;5(12). doi: 10.1172/jci.insight.134612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DAN, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116(20):4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheridan C. First approval in sight for Novartis’ CAR-T therapy after panel vote. Nat Biotechnol. 2017;35(8):691–693. doi: 10.1038/nbt0817-691. [DOI] [PubMed] [Google Scholar]
- 19.Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nat Rev Clin Oncol. 2018;15(1):31–46. doi: 10.1038/nrclinonc.2017.128. [DOI] [PubMed] [Google Scholar]
- 20.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. Regression of glioblastoma after chimeric antigen receptor T-Cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA oncol. 2017;3(8):1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wykosky J, Debinski W. The EphA2 receptor and ephrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res MCR. 2008;6(12):1795–1806. doi: 10.1158/1541-7786.MCR-08-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishigaki H, Minami T, Morimura O, Kitai H, Horio D, Koda Y, Fujimoto E, Negi Y, Nakajima Y, Niki M, et al. EphA2 inhibition suppresses proliferation of small-cell lung cancer cells through inducing cell cycle arrest. Biochem Biophys Res Commun. 2019;519(4):846–853. doi: 10.1016/j.bbrc.2019.09.076. [DOI] [PubMed] [Google Scholar]
- 24.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16(1):9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res MCR. 2005;3(10):541–551. doi: 10.1158/1541-7786.MCR-05-0056. [DOI] [PubMed] [Google Scholar]
- 26.Wykosky J, Gibo DM, Debinski W. A novel, potent, and specific ephrinA1-based cytotoxin against EphA2 receptor expressing tumor cells. Mol Cancer Ther. 2007;6(12):3208–3218. doi: 10.1158/1535-7163.MCT-07-0200. [DOI] [PubMed] [Google Scholar]
- 27.Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, Rainusso N, Wu M-F, Liu H, Kew Y, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther J Am Soc Gene Ther. 2013;21(3):629–637. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2-CAR T Cells for the adoptive immunotherapy of Glioma. Mol Ther Methods Clin Dev. 2018;9:70–80. doi: 10.1016/j.omtm.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, Wu M-F, Orange JS, Sumazin P, Man T-K, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-oncology. 2018;20(4):506–518. doi: 10.1093/neuonc/nox182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183(6):2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Z, Fourcade J, Pagliano O, Chauvin JM, Sander C, Kirkwood JM, Zarour HM. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8 +T cells. Cancer Res. 2015;75(8):1635–1644. doi: 10.1158/0008-5472.CAN-14-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19(6):1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian J, Wang C, Wang B, Yang J, Wang Y, Luo F, Xu J, Zhao C, Liu R, Chu Y, et al. The IFN-gamma/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy. J Neuroinflammation. 2018;15(1):290. doi: 10.1186/s12974-018-1330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang E, Cesano A, Butterfield LH, Marincola F. Improving the therapeutic index in adoptive cell therapy: key factors that impact efficacy. J Immunother Cancer. 2020;8(2):e001619. doi: 10.1136/jitc-2020-001619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine 2011;365:725––33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthys P, Dillen C, Proost P, Heremans H, Van Damme J, Billiau A. Modification of the anti-CD3-induced cytokine release syndrome by anti-interferon-gamma or anti-interleukin-6 antibody treatment: protective effects and biphasic changes in blood cytokine levels. Eur J Immunol. 1993;23(9):2209–2216. doi: 10.1002/eji.1830230924. [DOI] [PubMed] [Google Scholar]
- 38.Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, Chen Y, Han X, Wu K. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. 2016;31:61–71. doi: 10.1016/j.cytogfr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh RK, Gutman M, Reich R, Bar-Eli M. Ultraviolet B irradiation promotes tumorigenic and metastatic properties in primary cutaneous melanoma via induction of interleukin 8. Cancer Res. 1995;55:3669–3674. [PubMed] [Google Scholar]
- 40.Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, Dyson KA, Grippin AJ, Deleyrolle LP, Zhang W, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. doi: 10.1038/s41467-019-11869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The high throughput sequencing data has been submitted to the Gene Expression Omnibus database (accession code GSE163833)