

Abstract
Aim:
This study aimed to investigate the mechanism of reversal of multidrug resistance mediated by ABC transporters with tivozanib (AV-951 and KRN-951). Tivozanib is a potent inhibitor of VEGF-1, −2 and −3 receptors.
Materials & methods:
ABCB1- and ABCG2-overexpressing cell lines were treated with respective substrate antineoplastic agents in the presence or absence of tivozanib.
Results:
The results indicate that tivozanib can significantly reverse ABCB1-mediated resistance to paclitaxel, vinblastine and colchicine, as well as ABCG2-mediated resistance to mitoxantrone, SN-38 and doxorubicin. Drug efflux assays showed that tivozanib increased the intracellular accumulation of substrates by inhibiting the ABCB1 and ABCG2 efflux activity. Furthermore, at a higher concentration, tivozanib inhibited the ATPase activity of both ABCB1 and ABCG2 and inhibited the photolabeling of ABCB1 or ABCG2.
Conclusion:
We conclude that tivozanib at noncytotoxic concentrations has the previously unknown activity of reversing multidrug resistance mediated by ABCB1 and ABCG2 transporters.
Keywords: ABCB1, ABCG2, ABC transporter, multidrug resistance, tivozanib, tyrosine kinase inhibitor
A major obstacle to treating various cancers is their drug resistance to different chemotherapeutic drugs. ABC transporters play a major role in mediating multidrug resistance (MDR) in cancer cells [1,2]. ABC transporters increase the efflux of a broad class of cytotoxic drugs with energy derived from ATP hydrolysis [3]. Based on the sequence and organization of their nucleotide-binding folds, all 48 known human ABC transporter family members are divided into seven subfamilies, from A to G [4]. Even though there are conflicts regarding the function of ABC transporters in MDR, previous studies have shown that ABC transporters, such as ABCB1 (P-glycoprotein), ABCG2 (BCRP) and ABCC1 (MRP1), play major roles in the development of MDR [5–10]. ABCB1, with a molecular weight of 170 kDa, is widely expressed in the liver, kidney and GI tract, transporting toxins and xenobiotics out of cells [11,12]. ABCB1 is composed of two nucleotide-binding domains and two transmembrane domains [13]. ABCB1 has been extensively studied as it confers resistance to the widest variety of compounds, including vinca alkaloids, anthracyclines, epipodophyllotoxins and taxanes [3]. Another ABC family member, ABCG2, also known as BCRP, contains only one transmembrane domain and one nucleotide-binding domain and functions by either homodimerization or heterodimerization. ABCG2 is distributed in the placenta, the epithelium of the small intestine and colon, the liver canalicular and the brain microvessel endothelium [14,15] and is responsible for the efflux of chemotherapeutic drugs, such as mitoxantrone, the anthracyclines, methotrexate and indolocarbazole topoisomerase I inhibitors [16].
The extensively distributed ABC transporters and the many profiles of ABC transporter substrates lead to the ineffectiveness and failure of chemotherapy. Overcoming MDR and resensitizing tumorous tissues to anticancer drugs by either decreasing the expression of ABC transporter proteins or inhibiting the function of transporters could have a very positive impact on cancer chemotherapy [5,17,18]. The first-generation modulators of ABCB1, such as verapamil and cyclosporine A, were found to resensitize the effect of vincristine and vinblastine in ABCB1-overexpressing drug-resistant cell lines [19–21]. Since then, many other agents have been developed to overcome ABCB1- and ABCG2-mediated drug resistance, and some of these have been tested in clinical trials [22]. However, very few inhibitors have progressed into clinical trials because of their toxicity and low binding affinity [22,23]. Even for those inhibitors that have moved into clinical trials, there are constant debates regarding the clnical significance of ABC transporter inhibitors. For example, Phase I/II trials suggested that cyclosporine was useful for resistant acute myeloid leukemia patients [24]; however, a Phase II study using tariquidar showed limited clinical activity of cyclosporine for restoring sensitivity to anthracycline or taxane chemotherapy [25]. Clinical studies showed that a significant number of patients who overexpress MDR-linked ABC drug transporters were older patients with unfavorable cytogenetics, which are associated with relativly poor response to chemotherapy [26,27]. At the same time, because of the small amount of samples, it is hard to comment on the relative effectiveness of ABC transporter inhibitors. Consequently, researchers are still searching for more potent and less toxic inhibitors of ABC transporters. Tivozanib, a tyrosine kinase inhibitor, inhibits VEGF-1, −2 and −3 receptors [28]. Previous studies have shown that various tyrosine kinase inhibitors are able to inhibit ABC transporters [29,30]. Researchers have shown that tivozanib is effective in several xenograft tumors in animal models [28]. At present, there are ongoing clinical trials evaluating the effectiveness of tivozanib in breast, lung, colorectal and renal cancers [31]. Tivozanib combined with paclitaxel, which is an ABCB1 substrate, has recently undergone Phase I clinical trials [32]. For this reason, we decided to study the effect of tivozanib on ABC transporters when combined with their respective substrates.
Materials & methods
●. Chemicals
[3H]-paclitaxel (37.9 Ci/mmol) and [3H]-mitoxantrone (4 Ci/mmol) were purchased from Moravek Biochemicals (CA, USA). [125I]-iodoarylazidoprazosin (IAAP; 2200 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (MA, USA). The monoclonal antibodies BXP-34 (used against ABCG2) and C-219 (used against ABCB1) were purchased from Signet Laboratories (MA, USA). Santa Cruz Biotechnology (CA, USA) supplied the anti-β-actin monoclonal antibody (SC-8432). Selleck Chemicals (TX, USA) provided a free sample of tivozanib. The fumitremorgin C (FTC), a gift from SE Bates and RW Robey (NIH, MD, USA), was synthesized by the Thomas McCloud Developmental Therapeutics Program, Natural Products Extraction Laboratory, NCI, NIH. Mitoxantrone and cisplatin were purchased from Tocris Bioscience (MO, USA). Several other chemicals were purchased from Sigma Chemicals (MO, USA), including paclitaxel, doxorubicin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), vincristine, vinblastine and dimethyl sulfoxide (DMSO). DMEM, Roswell Park Memorial Institute 1640 medium, fetal bovine serum, penicillin/streptomycin and trypsin 0.25% were obtained from Hyclone Thermo Scientific (UT, USA).
●. Cell lines & cell cultures
The cell lines used in the following experiments include HEK293/pcDNA3.1, ABCG2-482-R2, ABCG2-482-G2, ABCG2-482-T7, HEK/ABCB1, KB-3-1 and KB-C2. HEK293/pcDNA3.1 was created by transfecting HEK293 cells with an empty pcDNA3.1 vector [33]. The wild-type ABCG2 trasnfectant cell line ABCG2-482-R2 and mutant ABCG2 transfectant cell lines ABCG2-482-G2 and ABCG2-482-T7 [33] were kindly provided by SE Bates and RW Robey. The ABCB1-overexpressing cell line HEK/ABCB1 was established by transfecting HEK/pcDNA3.1 cells with a pcDNA3.1 vector containing the full-length ABCB1 [34]. All transfected cell lines were cultured in medium with G418 at 2 mg/ml [33]. The ABCB1-overexpressing drug-resistant cell line KB-C2 was established by treating the parental human epidermoid carcinoma cell line KB-3-1 with increasing concentrations of colchicine [35]. All of the cell lines were grown as adherent monolayers at 37°C and 5% CO2 with DMEM culture medium (Hyclone Thermo Scientific), supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. These cell cultures were used throughout the following experiments.
●. Cytotoxicity by MTT assay
Cells were harvested with trypsin and counted in order to generate a suspension of 6 × 103 cells/well for all of the cell lines, except for KB-3–1, where 5 × 103 cells/well were seeded into 96-well plates. Tivozanib was dissovled in 1% DMSO in phosphate-buffered saline (PBS; v/v) and aliquots (20 μl) were tested over a series of concentrations. Control wells were treated with 1% aqueous DMSO. When the MTT assay was used for anticancer MDR reversal experiments, cells were preincubated with 2.5 or 5 μM of tivozanib, verapamil or FTC. At 1 h later, 20 μl/well at different concentrations of chemotherapeutic drugs were added into the appropriate wells. After incubating the chemotheraputic drugs for 72 h, 20 μl of MTT solution was added to each well at a rate of 4 mg/ml. In order to permit viable cells to convert the yellow-colored MTT into dark blue formazan crystals, the plates were incubated for another 4 h. After discarding the medium, we added 100 μl of DMSO into each well in order to dissolve the formazan crystals [36]. Using an Opsys™ Microplate Reader from DYNEX Technologies, Inc. (VA, USA), we measured absorbance at 570 nm. IC50 values were calculated from the cell killing assay as the concentrations of the anticancer drugs required for 50% inhibition of the cell growth. In addition, the degree of resistance was determined by dividing the IC50 for the MDR cells by the IC50 of the parental sensitive cells. The IC50 values were calculated using Bliss method [37].
●. Western blot analysis
The complete cell lysates were prepared by harvesting cells with a radioimmunoprecipitation assay buffer (PBS with 0.1% sodium dodecyl sulfate [SDS], 1% Nonidet™ P-40 [Sigma-Aldrich Chemical Co., MO, USA] 0.5% sodium deoxycholate and 100 mg/ml p-aminophenyl-methylsulfonyl fluoride) for 20 min on ice with further centrifugation (12,000 g at 4°C for 20 min). Each lane was loaded with identical complete cell lysates containing 20 μg of protein. Complete cell lysates were dissolved using 6–10% SDS-PAGE and then transferred to polyvinylidene fluoride membranes. After incubation in a blocking solution in a tris-buffered saline with Tween® (Amresco, OH, USA) buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl and 0.1% Tween 20) for 1 h at room temperature, the membranes were immunoblotted overnight with either C219 mouse monoclonal antibody against ABCB1 or monoclonal antibody BXP-34 against ABCG2 at 1:500 dilutions. β-actin at a 1:1000 dilution was used as an internal loading control. The polyvinylidene fluoride membrane was then incubated at room temperature with horseradish peroxide-conjugated secondary antibodies (1:1000 dilution) for another 2 h. The protein–antibody complex was detected by an enhanced chemiluminescence detection system (Amersham, NJ, USA) [38].
●. [3H]-paclitaxel & [3H]-mitoxantrone accumulation assays
For the [3H]-paclitaxel accumulation, equal amounts of HEK293/pcDNA3.1 or HEK/ABCB1 (5 × 106 cells) were preincubated at 37°C in DMEM (10% fetal bovine serum) with or without tivozanib (2.5 or 5 μM) for 2 h using verapamil (5 μM) as a positive control. For [3H]-mitoxantrone assays, 5 × 106 cells of HEK293/pcDNA3.1 or ABCG2-482-R2, ABCG2-482-G2 or ABCG2-482-T7 were treated with or without tivozanib (2.5 or 5 μM) or FTC (2.5 μM) for 2 h. Later, cells were added with 0.1 μM [3H]-paclitaxel or 0.1 μM [3H]-mitoxantrone and incubated for another 2 h. For the accumulation assay, the medium was removed after 2 h and the cells were rinsed three times with cold PBS. A 200-μl lysis buffer (pH 7.4, containing 1% Triton™ X-100 [Sigma-Aldrich Chemical Co.] and 0.2% SDS) was then added afterwards. Radioactivity was measured in a liquid scintillation counter [39].
●. [3H]-paclitaxel & [3H]-mitoxantrone efflux assays
For the efflux assay, the cells were prepared the same way as they were for the accumulation experiments. After being rinsed three times with cold PBS, the cells were incubated with or without reversal agents in a fresh medium at 37°C. At the beginning and after 30, 60 and 120 min, the same number of cells (1 × 106) was removed and washed three times with ice-cold PBS. The cells were then lysed and placed in the scintillation fluid in order to measure the radioactivity in a Packard TRI-CARB® 1900CA liquid scintillation analyzer (Packard Instrument Co., IL, USA) [40].
●. IAAP photoaffinity labeling
Crude membranes of High Five™ (Life Technologies, NY, USA) insect cells expressing ABCB1 (65 μg protein/100 μl) were incubated in 50 mM MES-Tris (pH 6.8) with increasing concentrations of tivozanib (0–20 μM) or 10 μM cyclosporine A at 37°C for 10 min, while crude membranes of MCF7-FLV cells expressing ABCG2 (25–30 μg protein/100 μl) were incubated in 50 mM Tris-HCl (pH 7.5) with tivozanib (0–20 μM) or 20 μM FTC at 37°C for 10 min. Samples were then transferred to a 4°C bath, and 4–5 nM IAAP was added under subdued light. The samples were photocrosslinked for 10 min under ultraviolet light (365 nm). ABCB1 samples were directly separated by electrophoresis and quantified, while ABCG2 samples were first immunoprecipitated with BXP-21 antibodies, as described previously [41], and then separated on 7% Tris-acetate gel and quantified [42]. The data were fitted with a one-phase decay equation using GraphPad® Prism 5.04 software (GraphPad software, Inc., CA, USA).
●. ATPase assay
Crude membranes (10 μg protein/100 μl) from High Five cells expressing ABCB1 or ABCG2 were incubated with increasing concentrations of tivozanib (0–20 μM) in the presence or absence of sodium orthovanadate (0.3 mM) in ATPase assay buffer (50 mM MES-Tris [pH 6.8], 50 mM KCl, 5 mM sodium azide, 1 mM ethylene glycol tetraacetic acid, 1 mM ouabain, 10 mM MgCl2 and 2 mM dithiothreitol). The reaction was initiated by the addition of 5 mM ATP, incubated for 20 min at 37°C and then terminated by the addition of 5% SDS solution. The amount of inorganic phosphate released was quantified with a colorimetric method, as described previously [43]. The data represent mean values from three independent experiments and error bars indicate standard deviation.
●. Statistical analysis
All experiments were repeated at least three times. Statistical differences were determined using the Student’s t-test at p < 0.05.
Results
●. Cytotoxicity of tivozanib on ABCB1-&ABCG2-overexpressing cell lines
In order to evaluate the cytotoxicity of tivozanib, a cell cytotoxicity assay was performed after treating different cell lines with increasing concentrations of tivozanib. It was shown that tivozanib was nontoxic (cell viability >85%) at a concentration less than 5 μM in the KB-3–1 and HEK293/pcDNA3.1 parental cells, as well as their respective ABCB1- and ABCG2-overexpressing cell lines (Figure 1). In the reversal study and the studies described below, we used 2.5 and 5 μM of tivozanib in order to study the effects of tivozanib on ABC transporters.
Figure 1. Cytotoxicity of tivozanib.
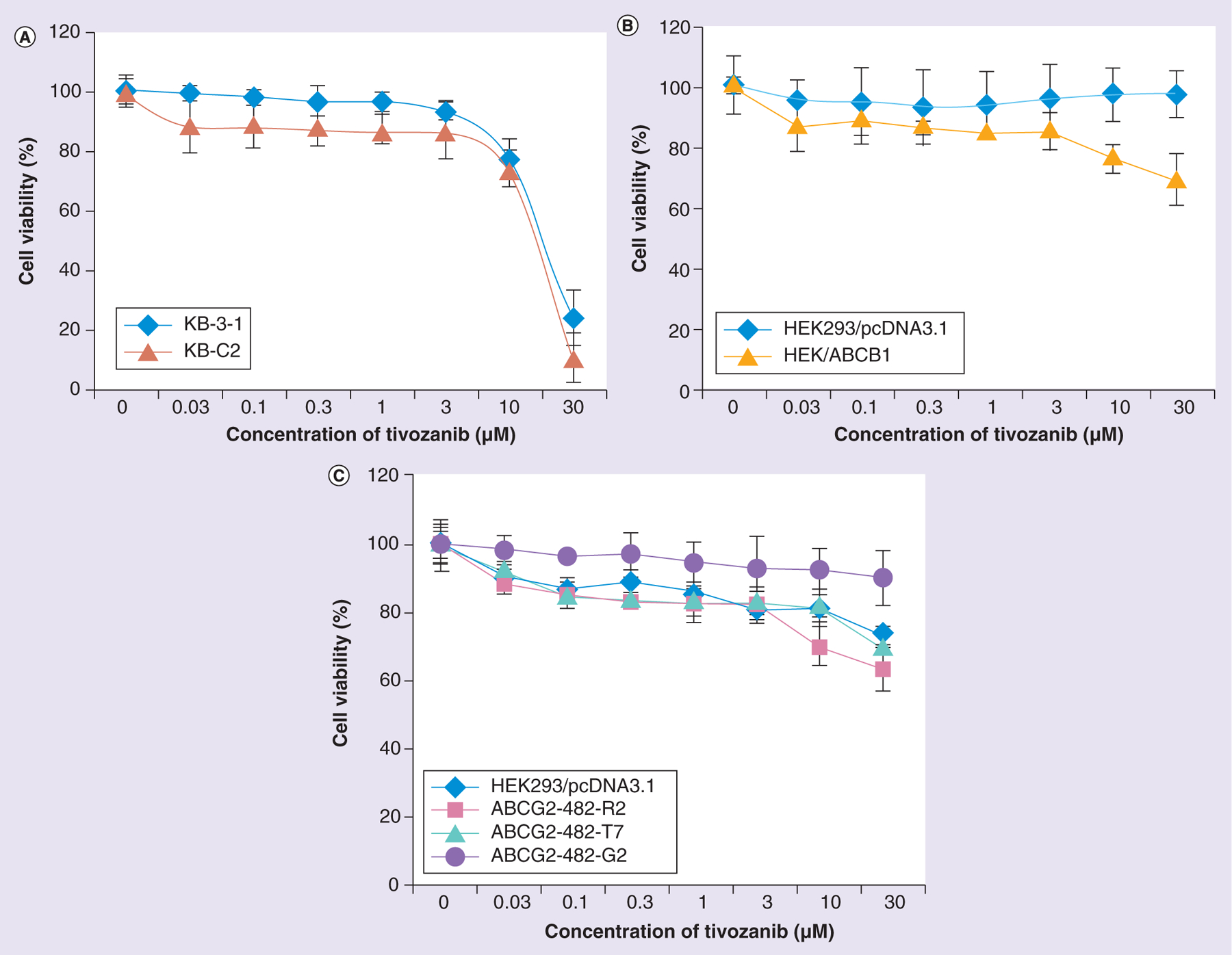
(A) Cytotoxicity of tivozanib in KB-3-1 and KB-C2 cell lines. (B) Cytotoxicity of tivozanib in HEK293/pcDNA3.1 and HEK/ABCB1 cell lines. (C) Cell cytotoxicity of tivozanib in HEK293/pcDNA3.1, ABCG2-482-R2, ABCG2-482-T7 and ABCG2-482-G2 cell lines. The data points represent the mean ± standard deviation of triplicate determinations.
●. Reversal effects of tivozanib on ABCB1-overexpressing cell lines
In order to determine the reversal effects of tivozanib on ABCB1-overexpressing cell lines, a cell cytotoxicity study was performed on the drug-resistant cell line KB-C2 and its parental cell line KB-3–1 using different chemotheraputic drugs. From Table 1, it is clear that after treating the parental cell line KB-3–1 with different concentrations of tivozanib and the positive control verapamil, there is no significantly change in the IC50 values in various substrates in KB-3–1, as it does not express the ABC transporter ABCB1. In the drug-resistant cell line KB-C2, the drug-resistant folds of KB-C2 when compared with the parental KB-3–1 cells were approximately 763.4-fold resistance towards paclitaxel, 162.2-fold resistance towards vinblastine and 495.0-fold resistance towards colchicine, with either the presence or absence of tivozanib, with cisplatin being a negative control, in which we found no change in the resistance folds either in the presence or absence of tivozanib. After adding tivozanib at 2.5 μM, the drug-resistance folds decreased to 56.1-fold towards paclitaxel, to 5.7-fold towards vinblastine and to 354.7-fold towards colchicine. After treating with tivozanib at 5 μM, tivozanib reversed drug resistance significantly, with 38.1-fold towards paclitaxel, 1.1-fold towards vinblastine and 22.2-fold towards colchicine. With the positive control verapamil at 5 μM, tivozanib decreased drug resistance to 1.4-fold towards paclitaxel, 1.1-fold towards vinblastine and 36.0-fold towards colchicine.
Table 1.
Effects of tivozanib on ABCB1-mediated drug resistance to paclitaxel, vinblastine and colchicine in KB-3-1 and KB-C2 cell lines.
| IC50 ± SD†; nM (resistance fold) | ||
|---|---|---|
| Paclitaxel | 6.5 ± 1.3 (1.0)‡ | 4931.4 ± 244.5 (763.4) |
| Paclitaxel +tivozanib 2.5 μM | 5.4 ± 2.6 (0.8) | 362.4 ± 83.2 (56.1)** |
| Paclitaxel + tivozanib 5 μM | 8.9 ± 1.7 (1.4) | 246.2 ± 77.9 (38.1)** |
| Paclitaxel + verapamil 5 μM | 6.7 ± 1.6 (1.0) | 8.7 ± 3.2 (1.4)** |
| Vinblastine | 0.2 ± 0.01 (1.0)‡ | 34.4 ± 1.6 (162.2) |
| Vinblastine + tivozanib 2.5 μM | 0.2 ± 0.03 (1.1) | 1.2 ± 0.4 (5.7)* |
| Vinblastine + tivozanib 5 μM | 0.2 ± 0.05 (1.2) | 0.2 ± 0.04 (1.1)* |
| Vinblastine + verapamil 5 μM | 0.2 ± 0.01 (0.8) | 0.2 ± 0.06 (1.1)* |
| Colchicine | 19.0 ± 3.4 (1.0)‡ | 8544.6 ± 603.0 (495.0) |
| Colchicine + tivozanib 2.5 μM | 20.8 ± 1.2 (1.1) | 6739.7 ± 211.7 (354.7)* |
| Colchicine + tivozanib 5 μM | 20.4 ± 1.7 (1.1) | 421.2 ± 25.0 (22.2)** |
| Colchicine + verapamil 5 μM | 19.1 ± 1.1 (1.0) | 684.2 ± 12.2 (36.0)** |
| Cisplatin | 2893.7 ± 223.1 (1.0)‡ | 3158.8 ± 433.2 (1.1) |
| Cisplatin + tivozanib 2.5 μM | 2727.8 ± 291.4 (1.0) | 2916.5 ± 354.4 (1.0) |
| Cisplatin + tivozanib 5 μM | 2614.7 ± 206.1 (1.0) | 3091.8 ± 514.3 (1.1) |
| Cisplatin + verapamil 5 μM | 2765.1 ± 141.4 (1.0) | 2661.0 ± 244.2 (0.9) |
Values represent the mean ± SD of at least three independent experiments, each performed in triplicate.
Resistance fold was calculated by dividing the IC50 values of paclitaxel, vinblastine, colchicine or cisplatin of KB-C2 cells in the presence or absence of reversal agents by the IC50 values of substrates of the KB-3–1 cell line.
p < 0.05;
p < 0.01.
SD: Standard deviation.
As various factors are known to contribute to drug resistance in drug-selected cell lines the use of ABCB1 gene-transfected cells (without exposure to any anticancer drug) allows us to assess the effect of tivozanib more effectively on the function of this transporter. From Table 2, we found that, in ABCB1-overexpressing HEK/ABCB1 cells, the drug resistance folds to different ABCB1 transporter substrates were 296.2-fold towards paclitaxel, 344.6-fold towards vincristine and 27.1-fold towards colchicine. Cisplatin was used as a negative control drug since it is not a substrate of the ABCB1 transporter. After adding tivozanib at 2.5 or 5 μM, tivozanib significantly decreased the drug resistance to paclitaxel, vincristine and colchicine. These results are consistent with the previous results from the drug-resistant cancer cell line KB-C2 and its parental cell line KB-3–1.
Table 2.
Effects of tivozanib on drug resistance to paclitaxel, vincristine and colchicine in HEK293/pcDNA3.1 and HEK/ABCB1 cell lines.
| IC50 ± SD†; nM (resistance fold) | ||
|---|---|---|
| Paclitaxel | 28.3 ± 4.3 (1.0)‡ | 8483.1 ± 726.4 (296.2) |
| Paclitaxel + tivozanib 2.5 μM | 21.9 ± 4.1 (0.8) | 1296.8 ± 383.4 (59.2)* |
| Paclitaxel + tivozanib 5 μM | 24.9 ± 3.7 (0.9) | 277.2 ± 44.7 (9.8)** |
| Paclitaxel + verapamil 5 μM | 20.0 ± 4.0 (0.7) | 26.8 ± 3.4 (0.9)** |
| Vincristine | 4.1 ± 0.9 (1.0)‡ | 1413.9 ± 159.7 (344.6) |
| Vincristine + tivozanib 2.5 μM | 3.6 ± 0.9 (0.9) | 751.1 ± 31.9 (183.2)* |
| Vincristine + tivozanib 5 μM | 3.8 ± 0.7 (0.9) | 44.8 ± 9.1 (10.9)** |
| Vincristine + verapamil 5 μM | 3.6 ± 0.6 (0.9) | 4.3 ± 2.8 (1.0)** |
| Colchicine | 20.4 ± 3.1 (1.0)‡ | 552.4 ± 66.2 (27.1) |
| Colchicine + tivozanib 2.5 μM | 24.8 ± 4.2 (1.2) | 381.6 ± 59.1 (18.7)* |
| Colchicine + tivozanib 5 μM | 23.5 ± 1.3 (1.2) | 125.1 ± 26.1 (6.1)* |
| Colchicine + verapamil 5 μM | 20.0 ± 1.5 (1.0) | 128.9 ± 16.2 (6.3)* |
| Cisplatin | 2914.2 ± 235.6 (1.0)‡ | 2251.6 ± 172.0 (0.8) |
| Cisplatin + tivozanib 2.5 μM | 3037.1 ± 273.2 (1.0) | 2233.0 ± 189.3 (0.8) |
| Cisplatin + tivozanib 5 μM | 2873.7 ± 352.7 (1.0) | 2211.1 ± 160.8 (0.8) |
| Cisplatin + verapamil 5 μM | 2671.1 ± 161.5 (1.0) | 2649.4 ± 293.6 (0.9) |
Values represent the mean ± SD of at least three independent experiments, each performed in triplicate.
Resistance fold was calculated by dividing the IC50 values of paclitaxel, vincristine, colchicine or cisplatin of HEK/ABCB1 cells in the presence or absence of reversal agents by the IC50 values of substrates of the HEK293/pcDNA3.1 cell line.
p < 0.05;
p < 0.01.
SD: Standard deviation.
●. Reversal effects of tivozanib on ABCG2-overexpressing cell lines
Previous research showed that, after transfecting HEK293 with a pcDNA3.1 vector containing full-length ABCG2 mutated at amino acid 482, coding either arginine (R), glycine (G) or threonine (T), different resistance folds to ABCG2 substrates were observed [22]. In order to look into the reversal of drug resistance in ABCG2-overexpressing cell lines, tivozanib was added to the ABCG2-overexpressing cell lines ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7. As shown in Table 3, before treating with reversal agents, the ABCG2-overexpressing cell lines show drug resistance to the ABCG2 transporter substrates mitoxantrone, SN-38 and doxorubicin. After being treated with tivozanib at 2.5 or 5 μM, tivozanib enhanced the sensitivity of ABCG2-overexpressing cell lines to the conventional chemotherapeutic drugs mitoxantrone, SN-38 and doxorubicin without changing the IC50 values of the parental cell line HEK293/pcDNA3.1.
Table 3.
Effects of tivozanib on drug resistance in ABCG2-overexpressing cell lines.
| IC50 ± SD†; nM (resistance fold) | ||||
|---|---|---|---|---|
| Mitoxantrone | 35.7 ± 8.6 (1.0)‡ | 166.7 ± 14.1 (4.7) | 179.2 ± 15.4 (5.0) | 189.9 ± 11.1 (5.3) |
| Mitoxantrone + tivozanib 2.5 μM | 31.1 ± 9.8 (0.9) | 99.0 ± 5.6 (2.8)* | 38.0 ± 9.3 (1.1)* | 74.9 ± 14.5 (2.1)* |
| Mitoxantrone + tivozanib 5 μM | 25.6 ± 5.0 (0.7) | 77.4 ± 6.2 (2.2)* | 26.9 ± 2.3 (0.8)* | 24.4 ± 4.0 (0.7)* |
| Mitoxantrone + FTC 2.5 μM | 37.5 ± 6.7 (1.0) | 28.7 ± 2.6 (0.8)* | 29.9 ± 2.5 (0.7)* | 26.9 ± 4.6 (0.8)* |
| SN-38 | 2.1 ± 1.3 (1.0)‡ | 71.6 ± 9.9 (34.1) | 152.1 ± 11.9 (72.4) | 162.1 ± 18.4 (81.0) |
| SN-38 + tivozanib 2.5 μM | 2.3 ± 1.4 (1.0) | 20.7 ± 5.4 (9.9)* | 22.3 ± 5.7 (10.6)* | 62.1 ± 8.9 (29.6)* |
| SN-38 + tivozanib 5 μM | 2.1 ± 0.8 (1.0) | 13.3 ± 3.2 (6.3)* | 5.4 ± 1.1 (2.6)* | 5.6 ± 1.4 (2.7)* |
| SN-38 + FTC 2.5 μM | 2.0 ± 1.2 (1.0) | 2.4 ± 1.0 (1.1)* | 1.7 ± 1.3 (0.8)* | 2.4 ± 1.2 (1.1)* |
| Doxorubicin | 16.6 ± 5.2 (1.0)‡ | 62.1 ± 2.5 (3.8) | 167.5 ± 14.3 (10.1) | 133.2 ± 30.3 (8.0) |
| Doxorubicin + tivozanib 2.5 μM | 18.4 ± 2.2 (1.1) | 42.2 ± 7.2 (2.5)* | 20.8 ± 4.5 (1.3)* | 29.0 ± 5.7 (1.8)* |
| Doxorubicin + tivozanib 5 μM | 19.9 ± 5.5 (1.2) | 30.0 ± 5.0 (1.8)* | 17.1 ± 3.3 (1.1)* | 21.0 ± 6.4 (1.3)* |
| Doxorubicin + FTC 2.5 μM | 17.0 ± 7.0 (1.0) | 21.7 ± 11.1 (1.3)* | 18.0 ± 1.9 (1.0)* | 17.0 ± 9.5 (1.0)* |
| Cisplatin | 2296.5 ± 253.4 (1.0) | 1588.7 ± 247.5 (0.7) | 1764.0 ± 208.6 (0.7) | 2128.0 ± 224.1 (0.9) |
| Cisplatin + tivozanib 2.5 μM | 2611.0 ± 243.6 (1.1) | 1851.6 ± 385.1 (0.8) | 1863.4 ± 177.7 (0.8) | 2610.8 ± 343.7 (1.1) |
| Cisplatin + tivozanib 5 μM | 2468.9 ± 191.7 (1.1) | 1626.0 ± 242.9 (0.7) | 2106.4 ± 166.1 (1.3) | 2533.0 ± 203.5 (1.1) |
| Cisplatin + FTC 2.5 μM | 2531.2 ± 343.9 (1.1) | 1998.3 ± 163.9 (0.9) | 2543.5 ± 159.1 (1.1) | 2525.6 ± 253.0 (1.1) |
Values represent the mean ± SD of at least three independent experiments, each performed in triplicate.
Resistance fold was calculated by dividing the IC50 values of mitoxantrone, SN-38, doxorubicin or cisplatin of ABCG2-overexpressing cell lines in the presence or absence of reversal agents by the IC50 values of substrates of the HEK293/pcDNA3.1 cell line.
p < 0.05.
FTC: Fumitremorgin C; SD: Standard deviation.
●. Effect of tivozanib on ABCB1 & ABCG2 protein expression levels
The reversal of ABCB1- and ABCG2-mediated MDR can be achieved either by downregulating the protein levels of ABCB1 and/or ABCG2 or by inhibiting their functions. In order to detect whether there are any changes in the protein expression of ABCB1 and ABCG2 in the presence of tivozanib, western blotting was performed. After treating the ABCB1-overexpressing drug-resistant cell line KB-C2 with 5 μM tivozanib at 0, 24, 48 and 72 h, the expression levels of ABCB1 were determined.
Figure 2A shows that after treating KB-C2 with tivozanib at the aforementioned time intervals, the protein levels of ABCB1 were not changed in the drug-resistant cell line KB-C2. Similar results were found in the ABCB1-transfected cell line HEK/ABCB1, as shown in Figure 2B. After incubating with 5 μM tivozanib for 0, 24, 48 and 72 h, western blotting was performed in the ABCG2-transfected cell line HEK/ABGC2/R2 and the mutant cell lines ABCG2-482-G2 and ABCG2-482-T7. The results of this are shown in Figure 2C. These results show that tivozanib does not change the protein expression of the ABC transporters ABCB1 and ABCG2.
Figure 2. Expression of ABCB1 and ABCG2 is not affected by treatment with tivozanib.
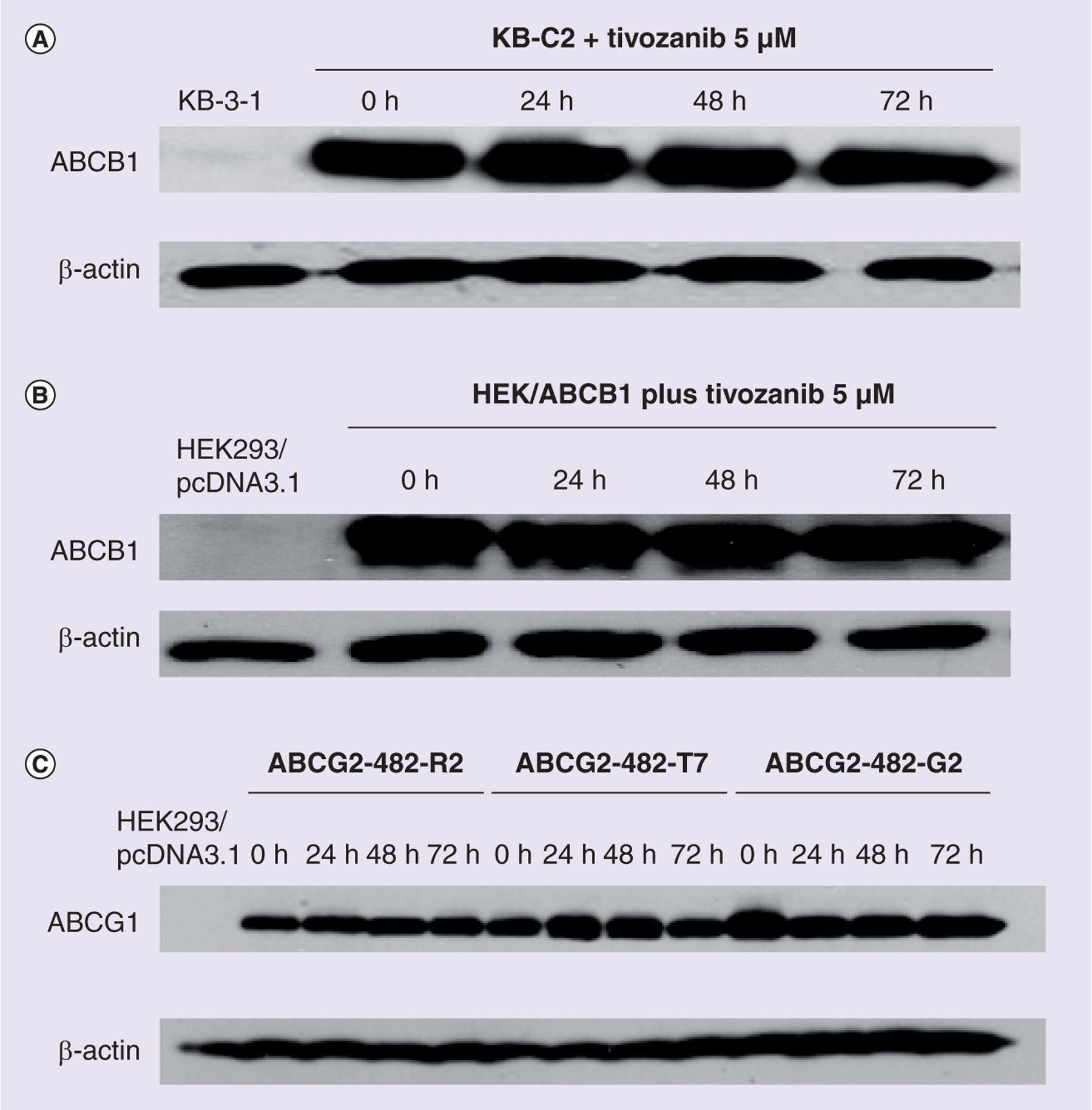
Western blot analysis showing that tivozanib (5 μM) does not alter the expression of ABCB1 in (A) the KB-C2 and (B) the ABCB1-transfected cell line HEK/ABCB1, as well as (C) the expression of ABCG2 protein in ABCG2-transfected cell lines. After pretreating with 5 μM tivozanib for 0, 24, 48 and 72 h in ABCB1- and ABCG2-overexpressing cell lines, the cells were lysed and proteins were extracted and analyzed. Equal amount of proteins (25 μg) were loaded into each well and immunoblot analysis was performed using ABCB1 and ABCG2 monoantibodies. β-actin was used for an internal control for loading. Representative results are shown, and similar results were obtained in two other trials.
●. Tivozanib increases the intracellular accumulation of [3H]-paclitaxel & [3H]-mitoxantrone
In order to understand whether tivozanib can inhibit the function of ABCB1 and/or ABCG2, a drug accumulation assay was performed. Figure 3A shows that in the ABCB1-transfected cell line HEK/ABCB1, the intracellular accumulation of [3H]-paclitaxel is much lower when compared with its accumulation in the parental cell line HEK293/pcDNA3.1. Here, HEK/ABCB1 without adding tivozanib or verapamil acts as the control group. After being treated with tivozanib at 2.5 or 5 μM for 4 h in HEK293/pcDNA3.1 and HEK/ABCB1, the intracellular accumulation of [3H]-paclitaxel increased. This is comparable to what happened with the positive control inhibitor of ABCB1. A similar experiment was performed using the wild-type ABCG2- (ABCG2-482-R2) and mutant ABCG2- (ABCG2-482-G2 and ABCG2-482-T7) transfected cell lines in the presence and absence of tivozanib or FTC. In this experiment, the intracellular accumulation of [3H]-mitoxantrone, which is a substrate of the ABCG2 transporter, was tested. Figure 3B shows that incubating with tivozanib 2.5 or 5 μM causes the intracellular accumulation of [3H]-mitoxantrone to increase greatly. In this case, FTC 2.5 μM was used as the positive control inhibitor of ABCG2, which increases the intracellular accumulation of [3H]-mitoxantrone in ABCG2-overexpressing cell lines.
Figure 3. Effects of tivozanib on the accumulation of [3H]-paclitaxel and [3H]-mitoxantrone.
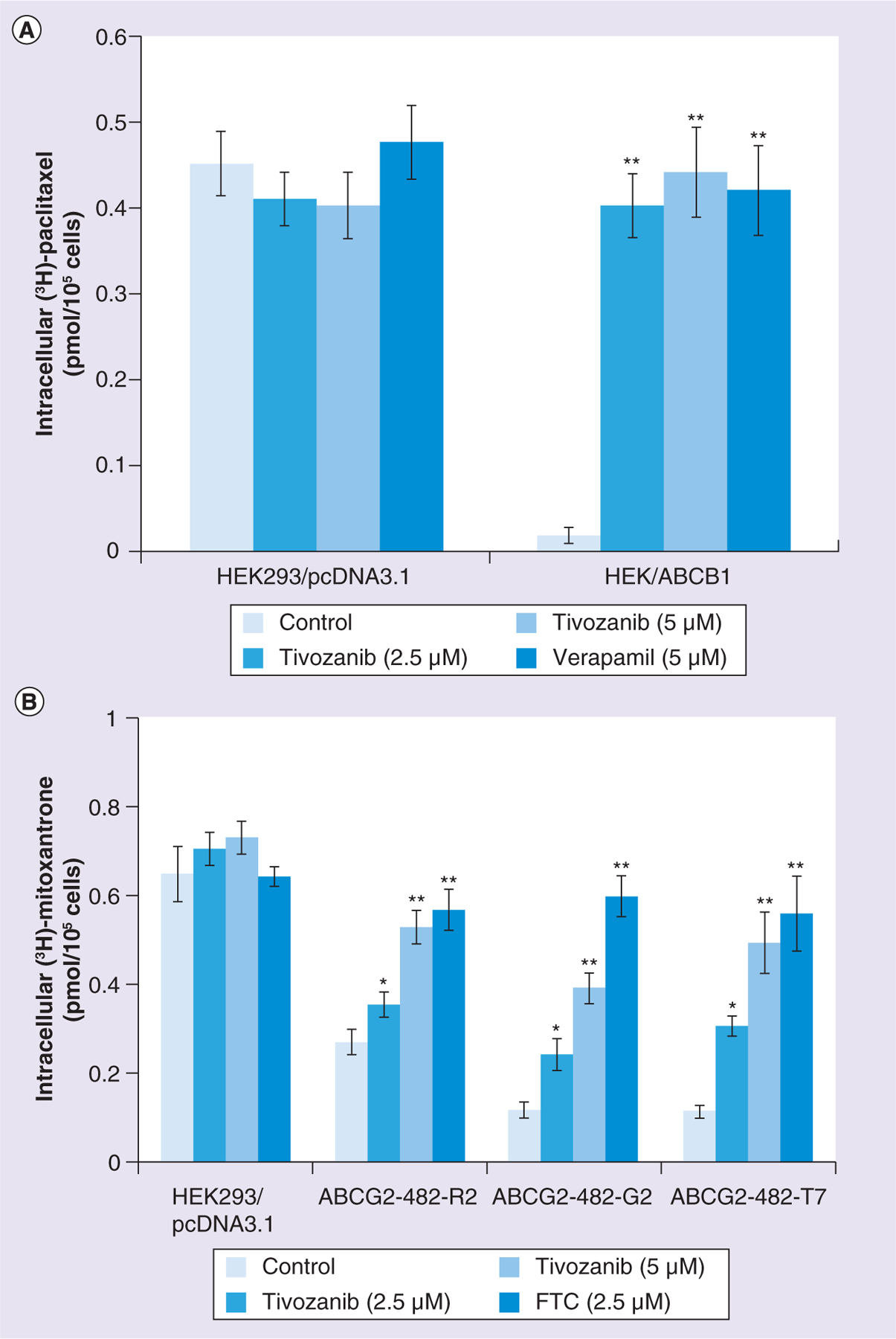
(A) Accumulation of [3H]-paclitaxel in HEK293/pcDNA3.1 cells and the ABCB1-transfected cell line HEK/ABCB1. (B) Accumulation of tivozanib in HEK293/pcDNA3.1 cells and the ABCG2-transfected cell lines ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7. Cells were pretreated with or without 2.5 or 5 μM tivozanib for 2 h, then incubated with 0.1 μM [3H]-paclitaxel or 0.1 μM [3H]-mitoxantrone for another 2 h at 37°C. The bars represent the mean ± standard deviation of triplicate determinations. Experiments were performed at least three times independently.*p < 0.05; **p < 0.01, for values versus those in the control group.
FTC: Fumitremorgin C.
●. Tivozanib decreases the intracellular efflux of [3H]-paclitaxel & [3H]-mitoxantrone
In order to clarify whether the increased intracellular accumulation of [3H]-paclitaxel and [3H]-mitoxantrone occurs due to inhibiting the efflux of these chemotherapeutic drugs, an efflux assay was run, as described in the ‘Materials & methods’ section. Figure 4A shows that, as time passed, the amount of [3H]-paclitaxel that was expelled out into the extracellular medium was much greater in HEK/ABCB1 than in HEK293/pcDNA3.1 cells. Incubating with tivozanib as the reversal agent significantly inhibited the extrusion of [3H]-paclitaxel. Verapamil at 5 μM was used as the positive control inhibitor of ABCB1 and also decreased the efflux of [3H]-paclitaxel. Meanwhile, in the parental cell line HEK293/pcDNA3.1, tivozanib and verapamil did not alter the rate of efflux of [3H]-paclitaxel. This is probably because HEK293/pcDNA3.1 cells do not express the protein ABCB1. Similar results were found in the ABCG2-transfected cell line ABCG2-482-R2, as shown in Figure 4B. Tivozanib significantly decreased [3H]-mitoxantrone efflux by inhibiting the ABCG2 transporter in ABCG2-482-R2 cells, which is comparable to what happened when the positive control inhibitor of ABCG2 FTC was used.
Figure 4. Effects of tivozanib on the efflux of [3H]-paclitaxel and [3H]-mitoxantrone.
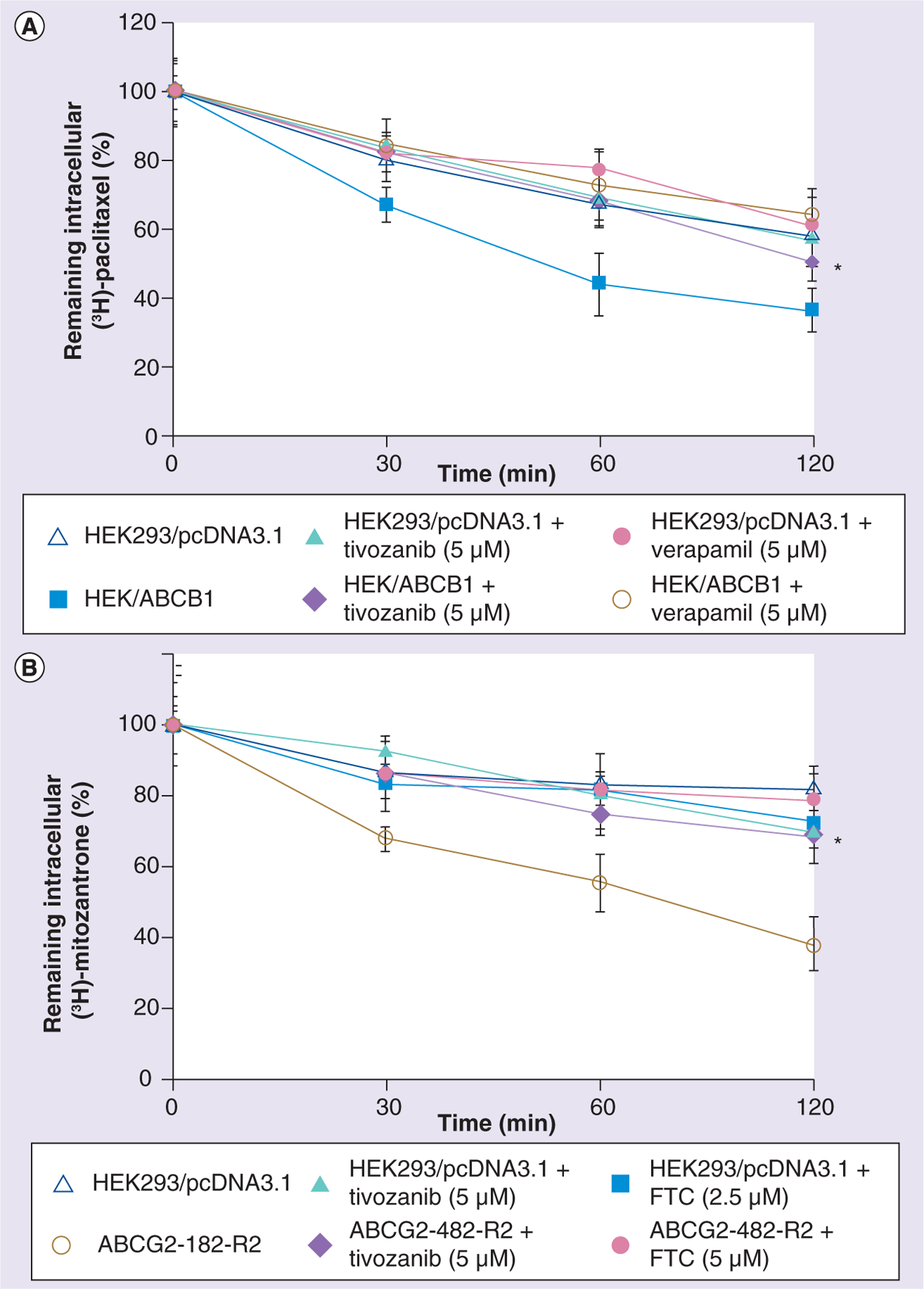
(A) Tivozanib decreases the efflux of [3H]-paclitaxel in HEK293/pcDNA3.1 cells and the transfected cell line HEK/ABCB1. (B) Tivozanib decreases the efflux of [3H]-mitoxantrone in HEK293/pcDNA3.1 cells and the transfected cell line ABCG2-482-R2. After pretreating cells with or without tivozanib 5 μM for 2 h, cells were incubated with 0.1 μM [3H]-paclitaxel or 0.1 μM [3H]-mitoxantrone for another 2 h at 37°C. After being rinsed three times with cold phosphate-buffered saline, cells were incubated with or without reversal agents in a fresh medium at 37°C. After 0, 30, 60 or 120 min, the same numbers of cells (1 × 106) were removed and washed three times with ice-cold phosphate-buffered saline. The cells were then lysed and placed in scintillation fluid in order to measure their radioactivity. The data points represent the mean ± standard deviation of triplicate determinations. Experiments were performed at least three times independently. *p < 0.05 for values versus those in the control group for (A) HEK/ABCB1 plus tivozanib (5 μM) and (B) ABCG2-482-R2 plus tivozanib (5 μM).
FTC: Fumitremorgin C.
●. Tivozanib inhibits the IAAP photoaffinity labeling of ABCB1 & ABCG2
Figure 5A shows that tivozanib inhibits the photoaffinity labeling of ABCB1 with IAAP in a concentration-dependent manner, reaching a maximum of 80% inhibition at concentrations higher than 10 μM (IC50: 2.9 μM). These results demonstrate that tivozanib can bind to the same binding sites of IAAP in the transmembrane domains of ABCB1. In Figure 5B, tivozanib is shown to inhibit the photoaffinity labeling of ABCG2 with IAAP in a concentration-dependent manner, reaching a maximum of 60–65% inhibition at concentrations higher than 1 μM. The IC50 was calculated using the values corrected for the inhibition produced by 20 μM FTC (i.e., FTC-insensitive (32%) IAAP incorporation was subtracted from all values). Thus, the calculated IC50 is 0.07 μM. These results show that tivozanib can bind to the binding sites of IAAP in ABCG2, with significantly higher binding affinity than that for ABCB1.
Figure 5. Tivozanib inhibits the photoaffinity labeling of ABCG2 and ABCB1 with [125I]-iodoarylazidoprazosin.
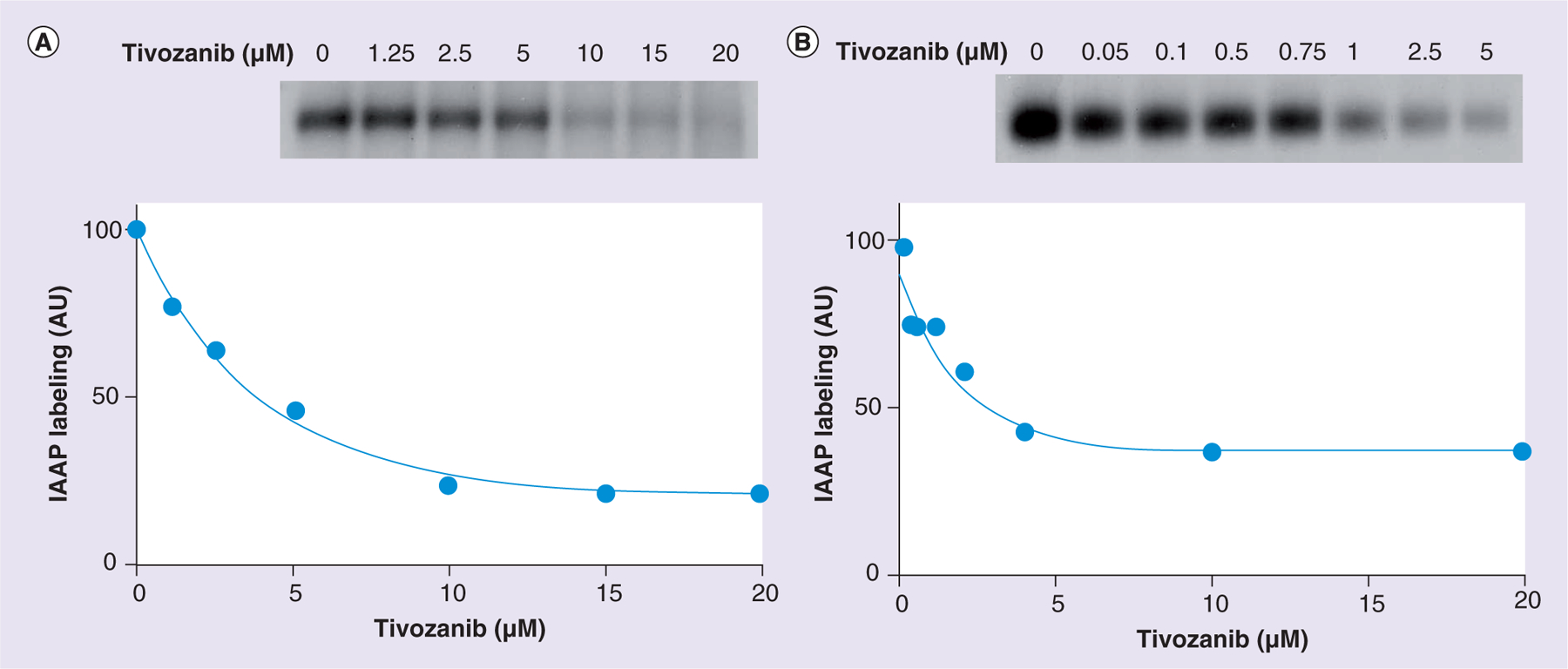
(A) Crude membranes of High Five™ (Life Technologies, NY, USA) insect cells expressing ABCB1 (65 μg protein/100 μl) were incubated in 50 mM MES-Tris(pH 6.8) with increasing concentrations of tivozanib (0–20 μM) or 10 μM cyclosporine A at 37°C for 10 min. Samples were then transferred to a 4°C bath, and 4–5 nM IAAP was added under subdued light. The samples were photocrosslinked for 10 min under ultraviolet light (365 nm) followed by electrophoresis and quantification, as described previously. A representative autoradiogram from one of two independent experiments is shown. In the graph, the data points represent the means of two independent experiments. The data were fitted with a one-phase decay equation using GraphPad® Prism 5.04 software (GraphPad software, Inc., CA, USA). (B) Crude membranes of MCF7-FLV cells expressing ABCG2 (25–30 μg protein/100 μl) were incubated in 50 mM Tris-HCl (pH 7.5) with increasing concentrations of tivozanib (0–20 μM) or 20 μM fumitremorgin C at 37°C for 10 min. Samples were then transferred to a 4°C bath, and 4–5 nM IAAP was added under subdued light. Samples were photocrosslinked for 10 min under ultraviolet light (365 nm) followed by immunoprecipitation with BXP-21 antibodies, as described previously. Finally, samples were separated by electrophoresis and quantified, as described previously. A representative autoradiogram from one of two independent experiments is shown. In the graph, the data points represent the means of two independent experiments. The data were fitted with a one-phase decay equation using GraphPad Prism 5.04 software.
IAAP: [125I]-iodoarylazidoprazosin.
●. Modulation of the basal ATPase activity of ABCB1 & ABCG2 by tivozanib
The effect of tivozanib on the basal ATPase activity of ABCB1 was studied in crude membranes expressing ABCB1, as shown in Figure 6A. Tivozanib slightly stimulates (10–15%) basal ATP hydrolysis by ABCB1 at lower concentrations, with the maximum effect observed at 0.25–1.0 μM. At concentrations higher than 1.0 μM, a decrease in ATPase activity is observed, reaching a plateau of 60% of the basal ATPase activity of ABCB1. However, the stimulatory effect is very marginal compared with its inhibitory effect at higher concentrations. The effect of tivozanib on the basal ATPase activity of ABCG2 was studied in crude membranes expressing ABCG2, and the results of this are shown in Figure 6B. As was observed for ABCB1, tivozanib stimulated the basal ATPase activity of the transporter at lower concentrations, and at higher concentrations (5 μM), an approximately 15–20% decrease in activity of ABCG2 was observed. This stimulatory effect is maximal at 0.5–1.0 μM and more significant than that observed for ABCB1; tivozanib increases the activity by 60% versus the 10–15% observed for ABCB1.
Figure 6. Modulation of the basal ATPase activity of ABCB1 and ABCG2 by tivozanib.
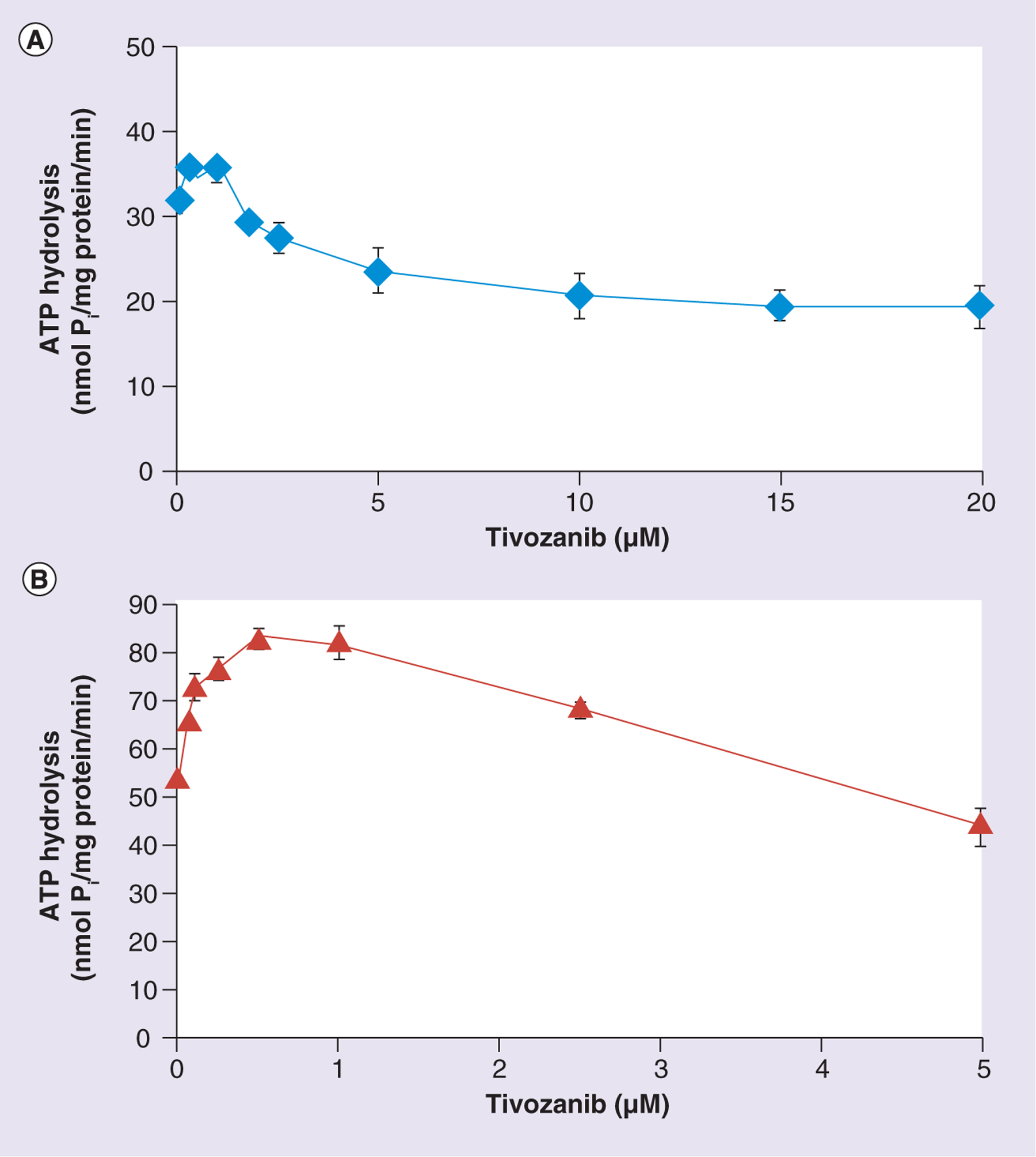
Crude membranes (10 μg protein/100 μl) from High Five™ (Life Technologies, NY, USA) insect cells expressing (A) ABCB1 or (B) ABCG2 were incubated with increasing concentrations of tivozanib (0–20 μM) in the presence or absence of sodium orthovanadate (Vi) (0.3 mM) in an ATPase assay buffer, as described in the ‘Materials & methods’ section. The reaction was initiated by the addition of 5 mM ATP, incubated for 20 min at 37°C and then terminated by the addition of 5% sodium dodecyl sulfate solution. The amount of Pi released was quantified using a colorimetric reaction. The data points represent the means from at least three independent experiments, while the bars represent the standard deviation values.
Pi: Inorganic phosphate.
Discussion
MDR is developed when cancer cells become resistant to chemotheraputic drugs. The decreased intracellular concentration of anticancer drugs due to reduced influx and/or increased efflux is one of the main mechanisms that contributes to the phenomenon of MDR. Decreasing uptake or increasing efflux of chemotheraputic drugs has gained extensive attention, and attempts are being made to find a way to conquer MDR and resensitize cancer cells to cytotoxic drugs [44]. Members of the ABC transporter family, which is one of the superfamilies of transporters, are expressed in many kinds of cancer cells. They play a significant role in the efflux of xenobiotics or chemotherapeutics from cancer cells [45]. For decades, efforts have been underway in order to try to reverse anticancer drug resistance by inhibiting the function of ABC transporters. Tyrosine kinases, including PDGF receptors, VEGF receptors, the stem cell factor receptor c-KIT, FLT3 and the glial cell-line derived neurotrophic factor receptor (RET) are transmembrane proteins that are located at the surface of the cell membrane [46]. After a ligand binds to a receptor, dimerization of the receptor causes autophosphorylation of the cytoplasmic domains, thus activating the tyrosine kinase [47]. Tyrosine kinase activites are involved in embryogenesis, angiogenesis, cell proliferation and antiapoptotic signaling [48]. Recently, researchers have shown that several tyrosine kinase inhibitors that compete with ATP for the catalytic site of kinases inhibit ABC transporters by either being a substrate or a modulator [49–51]. ABC transporters, especially ABCB1 and ABCG2, may interact with tyrosine kinase inhibitors [52]. In the current study, we looked into the effects of one tyrosine kinase inhibitor, tivozanib, on ABC transporters. Tivozanib is a selective VEGF inhibitor. It is effective in the treatment of various solid cancers, such as breast, lung, prostate, colon, liver and renal cell carcinoma, without significant adverse side effects [53–57].
Tivozanib was first tested in a cytotoxicity study with an MTT assay. From the results obtained with this assay, we chose the noncytotoxic concentrations of 2.5 and 5 μM as the concentrations for investigation in the following experiments. A reversal study, the results of which are presented in Table 1, showed that tivozanib at 5 μM is able to reverse the MDR in the ABCB1-overexpressing, drug-resistant cell line KB-C2 towards the ABCB1 substrates colchicine, paclitaxel and vinblastine. It must be noted that tivozanib has no effect on cisplatin, since cisplatin is not the substrate of ABCB1. In order to minimize any other effects that might contribute to drug-selected resistance in cancer cell lines, a reversal study in the ABCB1-transfected cell line HEK/ABCB1 was performed. The MTT assay results in Table 2 show that tivozanib also sensitized HEK/ABCB1 cells to the ABCB1 substrates colchicine, paclitaxel and vincristine. These results suggest that tivozanib significantly decreased ABCB1-mediated MDR in ABCB1-overexpressing cell lines. A reversal study in HEK293/pcDNA3.1 cells and ABCG2-overexpressing cell lines was also performed. According to the results in Table 3, tivozanib also reversed ABCG2-mediated drug resistance towards the ABCG2 substrates SN-38, mitoxantrone and doxorubicin. The scope of the current study was widened to include ABCC1- and ABCC10-mediated MDR [58]. Our results on these two pumps showed no change in the resistance folds in the transfected cell lines HEK/ABCC1 and HEK/ABCC10 (data not shown). This suggests that tivozanib selectively inhibits ABCB1- and ABCG2-mediated MDR. In order to determine whether tivozanib down-regulates the protein levels of ABC transporters, an immunoblotting analysis was performed. The immunoblotting results shown in Figure 2A & B indicate that tivozanib does not alter the protein expression level of the ABCB1 transporter in both the drug-resistant cancer cell line KB-C2 and ABCB1 gene-transfected cell line HEK/ABCB1. Similar results can be found in the ABCG2-overexpressing cell lines, as shown in Figure 2C. In that case, we found that tivozanib reverses drug resistance without altering the expression levels of ABC transporters. Another possibility is that tivozanib inhibits the function of ABC transporters, thus reversing MDR. In order to look into this possibility further, [3H]-paclitaxel accumulation assays in HEK293/pcDNA3.1 cells and the ABCB1-overexpressing cell line HEK/ABCB1 were performed. The results in Figure 3 show that tivozanib at 5 μM significantly increases the intracellular accumulation of [3H]-paclitaxel in HEK/ABCB1 cells, being similar to the level in the parental cell line HEK293/pcDNA3.1. The efflux assay results in Figure 4 show that tivozanib reduces the efflux of [3H]-paclitaxel in HEK/ABCB1 cells at different times. Thus, we arrive at the conclusion that tivozanib reverses ABCB1-mediated drug resistance by inhibiting the function of the ABCB1 pump instead of downregulating the protein expression level of ABCB1. When similar studies were performed in the ABCG2-overexpressing cell line ABCG2-482-R2 with [3H]-mitoxantrone, tivozanib significantly increased the intracellular accumulation of [3H]-mitoxantrone in ABCG2-482-R2 cells. This is the same as inhibiting the efflux of the ABCG2 pump instead of increasing the influx of the substrate. Furthermore, photoaffinity labeling studies (the results of which are shown in Figure 5) demonstrated that tivozanib inhibited the photoaffinity labeling of ABCB1 and ABCG2 with IAAP in a concentration-dependent manner. These results demonstrate that tivozanib binds to the drug-binding pocket in the transmembrane domains of ABCB1 and ABCG2, strongly suggesting that tivozanib inhibits the function of ABCB1 and ABCG2 by direct interation with these transporters. Tivozanib could possibly act as a substrate of both ABCB1 and ABCG2 transporters, competing for the binding sites with other substrates of these two transporters; this could possibly explain the fact that tivozanib selectively inhibits the function of ABCB1 and ABCG2 due to the binding specificity. ATPase assays were performed in order to determine the modulation of the basal ATPase activity of ABC drug transporters by tivozanib. According to Ambudkar et al., there are three classes of inhibitors [59]: class I inhibitors increase ATPase activity at low concentrations, while inhibiting ATPase activity at high concentration; class II inhibitors increase the activity of ATPase in a concentration-dependent manner; and class III inhibitors inhibit ATPase activity without any stimulation. Tivozanib could be a class I inhibitor. Tivozanib slightly stimulates the basal ATP hydrolysis by ABCB1 at lower concentrations (0.25–1.0 μM), indicating that it probably interacts with higher affinity with this transporter. At tivozanib concentrations higher than 1.0 μM, a decrease in the ATPase activity is observed, reaching a plateau of 60% of the basal ATPase activity of ABCB1. Thus, in the concentrations we used in the present study (2.5 and 5 μM), tivozanib could possibly act as an inhibitor of the ATPase activity of ABCB1 transporters. The decrease in the ATPase activity is not as obvious for the ABCG2 transporter at high concentrations of tivozanib; only a marginal (15–20%) decrease in basal activity was observed at 5 μM.
The anticancer activity of tivozanib is currently being widely investigated in preclinical research and clinical trials [60]. In a Phase II trial, tivozanib showed a statistically significant improvement in median progression-free survival for more than 12 weeks compared with placebo (49 vs 21%; p = 0.001) in the subpopulation of patients with renal cell carcinoma [61]. Tivozanib was well tolerated in patients with advanced renal cell carcinoma, and the most common adverse events were hypertension, diarrhea and hand–foot skin reactions [61]. However, no reports have compared the toxicity profile of tivozanib with other ABC transporter inhibitors, such as cyclosporine A, valspodar or tariquidar. Further study is needed regarding the toxicity profile of tivozanib. A Phase III trial recently showed that subjects on tivozanib had a longer progression-free survival period than those on sorafenib (another VEGF receptor kinase inhibitor) in the overall population (median: 11.9 vs 9.1 months; p = 0.042) [62]. Tivozanib is also administered in combination with chemotherapy drugs. Paclitaxel, a typical chemotherapeutic drug for breast cancer, is the substrate of the ABCB1 transporter. Recently, a Phase I trial of tivozanib with paclitaxel was performed in metastic breast cancer [32]. Even though there have been no clinical studies investigating tivozanib being used as an ABC transporter inhibitory agent, tivozanib might act as an ABCB1 inhibitor that is beneficial in combination regimens, which could be clinically significant.
Conclusion & future perspective
We conclude that tivozanib at noncytotoxic concentrations has a previously unknown activity in reversing MDR mediated by ABCB1 and ABCG2 transporters by directly blocking the drug efflux function of ABCB1 and ABCG2 without altering protein expression. Thus, tivozanib may become a new adjuvant agent to improve the effectiveness of the conventional chemotherapeutic drugs in the future.
EXECUTIVE SUMMARY.
Increased efflux of structurally and mechanistically unrelated antineoplastic agents by ABC transporters is the hallmark of multidrug resistance (MDR). The human ABCB1, ABCC1 and ABCG2 transporters have been established as potential mediators of MDR.
Various small-molecule tyrosine kinase inhibitors and compounds from marine sources have been tested for their ability to reverse MDR mediated by ABC transporters. However, they fail to reach clinical significance due to their toxic effects.
Tivozanib, a potent inhibitor of VEGF-1, −2 and −3 receptors, significantly decreases MDR mediated by ABCB1 and ABCG2.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays showed that tivozanib was able to resensitize ABCB1- and ABCG2-overexpressing cell lines to the substrates of the ABCB1 and ABCG2 transporters, respectively.
Tivozanib does not alter ABCB1 and/or ABCG2 protein expression levels.
Western blotting showed that tivozanib does not change the expression of ABCB1 in KB-C2 cells and theABCB1-transfected cell line HEK/ABCB1, nor the expression of the ABCG2 protein in ABCG2-transfected cell lines.
Tivozanib dependently inhibits the efflux function and increases the accumulation of ABCB1 and ABCG2 substrate antineoplastic drugs in ABCB1- and ABCG2-overexpressing cells, respectively.
Tivozanib increases the intracellular accumulation of [3H]-paclitaxel and [3H]-mitoxantrone.
Tivozanib decreases the intracellular efflux of [3H]-paclitaxel and [3H]-mitoxantrone.
[125I]-iodoarylazidoprazosin photolabeling studies demonstrate that tivozanib can bind to the same binding sites as[125I]-iodoarylazidoprazosin in the transmembrane domains of ABCB1 and ABCG2.
Tivozanib at concentrations used in the reversal studies (2.5 and 5 μM) decreases the ATPase activity of ABCB1 and ABCG2 transporters.
Thus, tivozanib at nontoxic concentrations, in combination with antineoplastic agents, can reverse the MDR mediated by ABCB1 and ABCG2.
Acknowledgements
The authors thank S Akiyama (Kagoshima University, Japan) for the KB-3-1 and KB-C2 cell lines and SE Bates and RW Robey (NIH, MD, USA) for the fumitremorgin C and ABCG2-transfected cell lines. The authors are thankful to Selleck Chemicals for kindly providing tivozanib as a free sample.
Financial & competing interests disclosure
This work was supported by funds from RayBiotech, the National Natural Science Foundation of China no. 81071290 (X Chen) and the National Natural Science Foundation for Distinguished Young Scholars of China no. 81225013 (X Chen). EE Chufan and SV Ambudkar were supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Footnotes
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Gillet JP, Efferth T, Remacle J.Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim. Biophys. Acta 1775(2), 237–262 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv 1(1), 27–42 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov 5(3), 219–234 (2006). [DOI] [PubMed] [Google Scholar]; • Demonstrates that multidrug resistance in cancer cells heavily relies on active efflux by ABC transporters.
- 4.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11(7), 1156–1166 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin. J. Cancer 31(2), 58–72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun YL, Patel A, Kumar P, Chen ZS. Role of ABC transporters in cancer chemotherapy. Chin. J. Cancer 31(2), 51–57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen ZS, Tiwari AK. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 278(18), 3226–3245 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murray S, Briasoulis E, Linardou H, Bafaloukos D, Papadimitriou C. Taxane resistance in breast cancer: mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev 38(7), 890–903 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Seymour L, Bezwoda WR, Dansey RD. P-glycoprotein immunostaining correlates with ER and with high Ki67 expression but fails to predict anthracycline resistance in patients with advanced breast cancer. Breast Cancer Res. Treat 36(1), 61–69 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Kim H, Kim K, No JH, Jeon YT, Jeon HW, Kim YB. Prognostic value of biomarkers related to drug resistance in patients with advanced epithelial ovarian cancer. Anticancer Res. 32(2), 589–594 (2012). [PubMed] [Google Scholar]
- 11.Cordon-Cardo C, O’Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J. Histochem. Cytochem 38(9), 1277–1287 (1990). [DOI] [PubMed] [Google Scholar]
- 12.Kimura Y, Morita SY, Matsuo M, Ueda K. Mechanism of multidrug recognition by MDR1/ABCB1. Cancer Sci. 98(9), 1303–1310 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sauna ZE, Kim IW, Ambudkar SV. Genomics and the mechanism of P-glycoprotein (ABCB1). J. Bioenerg. Biomembr 39(5–6), 481–487 (2007). [DOI] [PubMed] [Google Scholar]; • Demonstrates that prehydrolysis transition-like state formation takes place before the energy required for translocation of a substrate across the cell membrane is acquired during ATP hydrolysis in ABCB1. In this study, tivozanib is shown to inhibit ATPase activity.
- 14.Mo W, Zhang JT. Human ABCG2: structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol 3(1), 1–27 (2012). [PMC free article] [PubMed] [Google Scholar]
- 15.Maliepaard M, Scheffer GL, Faneyte IF et al. Subcellular localization and distribution of the breast cancer resistance protein transporter in normal human tissues. Cancer Res. 61(8), 3458–3464 (2001). [PubMed] [Google Scholar]
- 16.Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22(47), 7340–7358 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Shi Z, Liang YJ, Chen ZS et al. Overexpression of Survivin and XIAP in MDR cancer cells unrelated to P-glycoprotein. Oncol. Rep 17(4), 969–976 (2007). [PubMed] [Google Scholar]
- 18.Tiwari AK, Sodani K, Dai CL, Ashby CR Jr, Chen ZS. Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr. Pharm. Biotechnol 12(4), 570–594 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Myers CJ, Griffiths AJ, Kraus SR, Martin RR. Double stranded RNA in natural isolates of Neurospora. Curr. Genet 13(6), 495–501 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Goldberg H, Ling V, Wong PY, Skorecki K. Reduced cyclosporin accumulation in multidrug-resistant cells. Biochem. Biophys. Res. Commun 152(2), 552–558 (1988). [DOI] [PubMed] [Google Scholar]
- 21.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 41(5), 1967–1972 (1981). [PubMed] [Google Scholar]
- 22.Ahmed-Belkacem A, Pozza A, Macalou S, Perez-Victoria JM, Boumendjel A, Di Pietro A. Inhibitors of cancer cell multidrug resistance mediated by breast cancer resistance protein (BCRP/ABCG2). Anticancer Drugs 17(3), 239–243 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist 8(5), 411–424 (2003). [DOI] [PubMed] [Google Scholar]; •• Reports that three randomized trials have shown statistically significant benefits of the use of an ABCB1 inhibitor in combination with chemotherapy.
- 24.Gruber A, Bjorkholm M, Brinch L et al. A Phase I/II study of the MDR modulator valspodar (PSC 833) combined with daunorubicin and cytarabine in patients with relapsed and primary refractory acute myeloid leukemia. Leuk. Res 27(4), 323–328 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Pusztai L, Wagner P, Ibrahim N et al. Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 104(4), 682–691 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Greenberg PL, Lee SJ, Advani R et al. Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: a Phase III trial (E2995). J. Clin. Oncol 22(6), 1078–1086 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slovak ML, Kopecky KJ, Cassileth PA et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group study. Blood 96(13), 4075–4083 (2000). [PubMed] [Google Scholar]
- 28.Pal SK, Bergerot PG, Figlin RA. Tivozanib: current status and future directions in the treatment of solid tumors. Expert Opin. Investig. Drugs 21(12), 1851–1859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi Z, Tiwari AK, Shukla S et al. Inhibiting the function of ABCB1 and ABCG2 by the EGFR tyrosine kinase inhibitor AG1478. Biochem. Pharmacol 77(5), 781–793 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi Z, Peng XX, Kim IW et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 67(22), 11012–11020 (2007). [DOI] [PubMed] [Google Scholar]
- 31.De Luca A, Normanno N. Tivozanib, a pan-VEGFR tyrosine kinase inhibitor for the potential treatment of solid tumors. IDrugs 13(9), 636–645 (2010). [PubMed] [Google Scholar]
- 32.Mayer EL, Scheulen ME, Beckman J et al. A Phase I dose-escalation study of the VEGFR inhibitor tivozanib hydrochloride with weekly paclitaxel in metastatic breast cancer. Breast Cancer Res. Treat. 140(2), 331–339 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Robey RW, Honjo Y, Morisaki K et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br. J. Cancer 89(10), 1971–1978 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Amino acid mutations at the 482 position in the ABCG2 gene can affect substrate and antagonist specificity. In the current study, tivozanib reverses wild-type as well as mutant ABCG2-mediated multidrug resistance.
- 34.Robey RW, Shukla S, Finley EM et al. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1®. Biochem. Pharmacol 75(6), 1302–1312 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akiyama S, Fojo A, Hanover JA, Pastan I, Gottesman MM. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somat. Cell Mol. Genet 11(2), 117–126 (1985). [DOI] [PubMed] [Google Scholar]
- 36.Carmichael J, Degraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 47(4), 936–942 (1987). [PubMed] [Google Scholar]
- 37.Shi Z, Liang YJ, Chen ZS et al. Reversal of MDR1/P-glycoprotein-mediated multidrug resistance by vector-based RNA interference in vitro and in vivo. Cancer Biol. Ther 5(1), 39–47 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Deng W, Dai CL, Chen JJ et al. Tandutinib (MLN518) reverses multidrug resistance by inhibiting the efflux activity of the multidrug resistance protein 7 (ABCC10). Oncol. Rep 29(6), 2479–2485 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sodani K, Tiwari AK, Singh S et al. GW583340 and GW2974, human EGFR and HER-2 inhibitors, reverse ABCG2- and ABCB1-mediated drug resistance. Biochem. Pharmacol 83(12), 1613–1622 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun YL, Kathawala RJ, Singh S et al. Zafirlukast antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Anticancer Drugs 23(8), 865–873 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Rai V, Gaur M, Shukla S, Ambudkar SV, Komath SS, Prasad R. Conserved Asp327 of walker B motif in the N-terminal nucleotide binding domain (NBD-1) of Cdr1p of Candida albicans has acquired a new role in ATP hydrolysis. Biochemistry 45(49), 14726–14739 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauna ZE, Ambudkar SV. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J. Biol. Chem 276(15), 11653–11661 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 292, 504–514 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Shukla S, Ohnuma S, Ambudkar SV. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr. Drug Targets 12(5), 621–630 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gottesman MM. Mechanisms of cancer drug resistance. Annu. Rev. Med 53, 615–627 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J. Clin. Oncol 25(7), 884–896 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Pawson T Regulation and targets of receptor tyrosine kinases. Eur. J. Cancer 38(Suppl. 5), S3–S10 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Pytel D, Sliwinski T, Poplawski T, Ferriola D, Majsterek I. Tyrosine kinase blockers: new hope for successful cancer therapy. Anticancer Agents Med. Chem 9(1), 66–76 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab. Dispos 37(2), 359–365 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dai CL, Tiwari AK, Wu CP et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 68(19), 7905–7914 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shukla S, Chen ZS, Ambudkar SV. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist. Updat 15(1–2), 70–80 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozvegy-Laczka C, Cserepes J, Elkind NB, Sarkadi B. Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist. Updat 8(1–2), 15–26 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Bukowski RM. Third generation tyrosine kinase inhibitors and their development in advanced renal cell carcinoma. Front. Oncol 2, 13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakamura K, Taguchi E, Miura T et al. KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res. 66(18), 9134–9142 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Taguchi E, Nakamura K, Miura T, Shibuya M, Isoe T. Anti-tumor activity and tumor vessel normalization by the vascular endothelial growth factor receptor tyrosine kinase inhibitor KRN951 in a rat peritoneal disseminated tumor model. Cancer Sci. 99(3), 623–630 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berge EM, Bowles DW, Flaig TW, Lam ET, Jimeno A. Tivozanib: practical implications for renal cell carcinoma and other solid tumors. Drugs Today (Barc.) 49(5), 303–315 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Heng DY, Kollmannsberger C, Chi KN. Targeted therapy for metastatic renal cell carcinoma: current treatment and future directions. Ther. Adv. Med. Oncol 2(1), 39–49 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kathawala RJ, Wang YJ, Ashby CR Jr, Chen ZS. Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin. J. Cancer doi: 10.5732/cjc.013.10122 (2013) (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol 39, 361–398 (1999). [DOI] [PubMed] [Google Scholar]
- 60.Pal SK, Bergerot PG, Figlin RA. Tivozanib: current status and future directions in the treatment of solid tumors. Expert Opin. Investig. Drugs 21(12), 1851–1859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nosov DA, Esteves B, Lipatov ON et al. Antitumor activity and safety of tivozanib (AV-951) in a Phase II randomized discontinuation trial in patients with renal cell carcinoma. J. Clin. Oncol 30(14), 1678–1685 (2012). [DOI] [PubMed] [Google Scholar]
- 62.Motzer RJ, Nosov D, Eisen T et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: results from a Phase III trial. J. Clin. Oncol 31(30), 3791–3799 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]