

Abstract
The COVID-19 pandemic has resulted in 198 million reported infections and more than 4 million deaths as of July 2021 (covid19.who.int). Research to identify effective therapies for COVID-19 includes: (1) designing a vaccine as future protection; (2) de novo drug discovery; and (3) identifying existing drugs to repurpose them as effective and immediate treatments. To assist in drug repurposing and design, we determine two apo structures of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease at ambient temperature by serial femtosecond X-ray crystallography. We employ detailed molecular simulations of selected known main protease inhibitors with the structures and compare binding modes and energies. The combined structural and molecular modeling studies not only reveal the dynamics of small molecules targeting the main protease but also provide invaluable opportunities for drug repurposing and structure-based drug design strategies against SARS-CoV-2.
Keywords: SARS-CoV-2, SFX, ambient temperature, drug repurposing, main protease
Graphical abstract
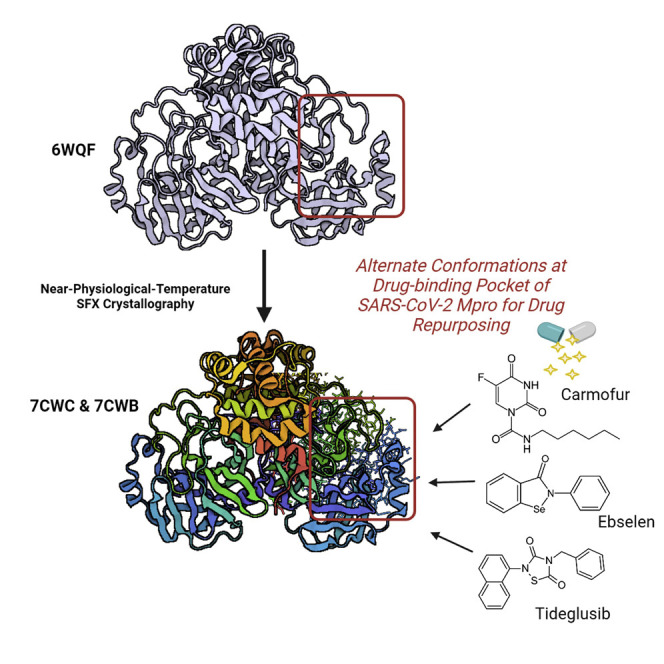
Durdağı et al. represent radiation damage-free high-resolution SARS-CoV-2 main protease SFX structures obtained at near-physiological temperature and performed MD simulation of apo-form proteins and three known main protease inhibitors. The structures reveal alternate conformation, while MD simulation indicates asymmetric behavior of the protein, which is invaluable information for immediate drug-repurposing studies.
Introduction
In late 2019, after the first patient was diagnosed with pneumonia of unknown etiology reported to the World Health Organization (WHO) from China, millions of cases followed in a short span of 4 months (WHO). On March 11, 2020, WHO declared the COVID-19 outbreak a pandemic, which originated from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. SARS-CoV-2 has a high spread rate (Ro) value, making the pandemic difficult to control (Petersen et al., 2020). Moreover, the lack of effective treatments to control the infection in high-comorbidity groups made this pandemic a major threat to global health (Ahn et al., 2020).
The first human coronaviruses causing a variety of human diseases, such as common cold, gastroenteritis, and respiratory tract diseases, were identified in the 1960s (Tyrrell and Bynoe, 1965). In 2002, a deadly version of coronavirus, SARS-CoV, was identified in China (Heymann and Rodier, 2004). SARS-CoV-2, the most recent member of the coronavirus family to be encountered, is a close relative of SARS-CoV and causes many systemic diseases (Andersen et al., 2020; Dutta and Sengupta, 2021; Braun et al., 2020). COVID-19 patients exhibit (1) high C-reactive protein and pro-inflammatory cytokine levels; (2) macrophage and monocyte infiltration to lung tissue; (3) atrophy of spleen and lymph nodes, which weakens the immune system; (4) lymphopenia; and (5) vasculitis (Zhang et al., 2020a, 2020b bib_Zhang_et_al_2020a; McGonagle et al., 2020). The release of a large amount of cytokines results in acute respiratory distress syndrome causing aggravation and widespread tissue injury, leading to multi-organ failure and death. Therefore, mortality in many severe cases of COVID-19 patients has been linked to the presence of the cytokine storm evoked by the virus (Ragab et al., 2020).
The SARS-CoV-2 genome encodes structural proteins including surface/spike glycoprotein (S), envelope (E), membrane (M), and nucleocapsid (N) proteins; and the main reading frames named ORF1a and ORF1b, which contain 16 non-structural proteins (NSP) (Gordon et al., 2020; Chen et al., 2020a, 2020b, 2020c). Among these, ORF1a/b encodes papain-like protease (PLpro), main protease (Mpro), a chymotrypsin-like cysteine protease, along with polyproteins named polyprotein1a (pp1a) and polyprotein1b (pp1b) (Astell et al., 2005). Encoded polyproteins are then proteolyzed to NSPs by precise Mpro and PLpro cleavages of the internal scissile bonds. NSPs are vital for viral replication, such as RNA-dependent RNA polymerase (RdRp) and Nsp13, which are used for the expression of structural proteins of the virus (Dai et al., 2020; Thiel et al., 2003; Ullrich and Nitsche, 2020; Ziebuhr et al., 2000). SARS-CoV Mpro has no homologous human protease that recognizes the same cleavage site (Pillaiyar et al., 2016). Therefore, drugs that target its active site are predicted to be less toxic and harmful to humans (Zhang et al., 2020a, 2020b). High sequence conservation of Mpro provides minimized mutation-caused drug resistance (Jin et al., 2020). Given its essential role in the viral life cycle (Dai et al., 2020; Thiel et al., 2003; Ullrich and Nitsche, 2020; Ziebuhr et al., 2000), the SARS-CoV-2 Mpro presents a major drug target requiring a detailed structural study.
Structure-based drug repurposing is a rapid method of identifying potential therapies that could be effective against COVID-19 compared with continuous investigative efforts (e.g., identification of new drugs and development of preventive vaccine therapies). The well-studied properties of drugs currently in clinical use mean that such compounds are better understood than their counterparts designed de novo. A putative drug candidate identified by drug-repurposing studies could make use of existing pharmaceutical supply chains for formulation and distribution, an advantage over developing new therapies (Chen et al., 2020a; Huang et al., 2020; Pushpakom et al., 2019; Jarada et al., 2020). In previous drug-repurposing studies, approved drug libraries were typically screened against the active site or an allosteric site of target protein structures, which were traditionally obtained by methods with limitations in revealing the enzyme structure, e.g., cryogenic temperature or radiation damage (Wang et al., 2020; Kneller et al., 2020). Such investigations generally only involve screening of drugs currently on the market (Zhou et al., 2020; Choudhary et al., 2020; Beck et al., 2020; Wang, 2020). Current structural biology-oriented studies that display a repurposing approach to SARS-CoV-2 research have focused on structure-based drug design, virtual screening, and a wide spectrum of inhibitors based on previously published tertiary protein structure alignments (Rathnayake et al., 2020; Chauhan and Kalra, 2020; Chen et al., 2020a, 2020b, 2020c; Muralidharan et al., 2021; Khan et al., 2021; Kumar et al., 2020; Joshi et al., 2020). Given the high-infectivity and persistence of SARS-CoV-2, any molecular candidates identified through drug-repurposing studies could be a potential treatment in the short term and should thus be investigated using empiric structural data.
In this work, we ascertain some conformational dynamics of Mpro via high-throughput serial crystallography using the macromolecular femtosecond crystallography (MFX) instrument from the Linac Coherent Light Source (LCLS), which provides ultrafast and ultrabright X-ray pulses allowing data collection without secondary radiation damage at ambient temperature (Sierra et al., 2019; Blaj et al., 2019; van Driel et al., 2020). Our structural data reveal the active site of SARS-CoV-2 Mpro with ambient-temperature radiation damage-free data and were used for subsequent in silico investigation of known Mpro inhibitors with the binding pocket residues.
Results
Ambient-temperature X-ray free-electron laser crystal structures of SARS-CoV-2 Mpro reveal alternate conformations of the drug-binding pocket
We determined two radiation damage-free serial femtosecond X-ray crystallography (SFX) crystal structures of SARS-CoV-2 Mpro in two crystal forms, monoclinic at 1.9 Å resolution and orthorhombic at 2.1 Å resolution (PDB: 7CWB and 7CWC, respectively) (Figures 1A and 1B) (Tables S1 and S2; Figure S1). We used an Mpro structure determined at ambient temperature using a rotating anode home X-ray source (Kneller et al., 2020) as our initial molecular replacement search model for structure determination (PDB: 6WQF). Two high-resolution SFX structures obtained in different space groups were superposed with an overall root-mean-square deviation (RMSD) of 1.0 Å (Figure S2). They reveal alternative active site residue conformations and dynamics at the atomic level, revealing several differences compared with the previously shown ambient-temperature structure of PDB: 6WQF (Kneller et al., 2020) (Figures 2A and 2B).
Figure 1.

Overview of Mpro crystal structures
(A) Cartoon representation of Mpro in space group C121. There is one molecule in the asymmetric unit cell colored in dark salmon (left). A biologically relevant dimeric form is generated by application of a symmetry operator which is colored in green.
(B) Cartoon representation of the Mpro in space group P212121 (right). Two chains of the dimer are colored in cyan and slate.
Figure 2.

Comparison of the SFX structure (PDB: 7CWB) with the ambient-temperature structure (PDB: 6WQF)
(A) The two structures align very well with an overall RMSD of 0.404 Å. The SFX structure is shown in gray, the ambient-temperature structure is shown in pink. The same coloring scheme is used in all panels. All the structural alignments are performed based on the superposition of Cα atoms throughout the manuscript.
(B) Superposition of the active site pocket reveals the significant conformational states. Residues with altered conformations were labeled and their positions are indicated.
(C) 2Fo − Fc electron density belonging to the active site residues is contoured at the 1σ level and colored in slate. H bonds and other interactions are indicated by dashed lines and distances are given in Å.
(D) Superposition of the PDB: 7CWB (SFX) and 6WQF active site reveals very similar states.
The unique N-terminal sequence affects the enzyme’s catalytic activity (Chang, 2010; Zhang et al., 2020a, 2020b). Besides the native monoclinic form of Mpro at 1.9 Å (Figure 1A), we also determined the structure of the Mpro with four additional N-terminal amino acids (generated by thrombin-specific N-terminal cleavage) at 2.1 Å resolution (Figure 1B). The structure obtained from this modified version of Mpro reveals that minor changes introduced at the N terminus can affect the three-dimensional structure of Mpro and promote orthorhombic crystal formation. The biologically relevant dimeric structure of native monomeric Mpro can be generated by adding the symmetry-related chain B (Figure 1A).
Each protomer of SARS-CoV-2 Mpro is formed by three major domains (Jin et al., 2020) (Figure S3). Domain I starts from the N terminus of the protein and includes an antiparallel β sheet structure. This β sheet forms a β barrel fold that ends at residue 100. Domain II of Mpro resides between residues 101 through 180 and mostly consists of antiparallel β sheets. The third domain of the Mpro is located between residues 181 through 306 and consists of mostly α helices and has a more globular tertiary structure (Figure S3). The intersection between domain I, domain II, and the loop region of domain III is the key region for enzyme activity (Figure S3), which forms the catalytic site and substrate binding pocket of the enzyme. The biologically active form of SARS-CoV-2 Mpro is a homodimer (Zhang et al., 2020a, 2020b). Previous biochemical studies of SARS-CoV Mpro suggested that there is a competition for the dimerization surface between domains I and III. In the absence of domain I, Mpro undergoes a new type of dimerization through domain III (Zhong et al., 2008). During our purifications, we repeatedly observed a combination of monomeric and dimeric forms of Mpro on size-exclusion chromatography steps that may be caused by this dynamic compositional and conformational equilibrium.
The two SARS-CoV-2 Mpro SFX crystal structures reveal a non-flexible core active site and the catalytic amino acid Cys145 (Figure S4). Temperature factor analysis revealed that the active site is surrounded by mobile regions (Figure S4). The presence of these mobile regions was observed in both SFX crystal structures, suggesting intrinsic plasticity rather than a structural artifact arising from the crystal lattice contacts (Figure S4; indicated with red circles). Furthermore, this intrinsic plasticity suggests that inhibitor molecules that interact via non-covalent bonds to the Mpro binding pocket would do so weakly (Gao et al., 2021). Our investigation of PDB structures with available electron densities identified that the majority of the Mpro inhibitors formed covalent bonds with the active site residue Cys145. In addition, few non-covalent inhibitors were identified among the available structures in September 2020, which have downloadable electron densities and exhibited weak electron densities (Figure S5.).
The cryogenic temperature structures of SARS-CoV-2 Mpro were summarized in an outstanding study and compared with an ambient-temperature PDB: 6WQF structure (Kneller et al., 2020). Thus, we compared our monoclinic ambient-temperature SFX structure with the previously published ambient-temperature X-ray structure PDB: 6WQF for observing minimization of secondary radiation damage effect on the SARS-CoV-2 Mpro structure by SFX technology. Both structures were largely similar with an RMSD value of 0.404 Å (Figure 2A). We observed significant conformational differences, especially in the side chains of Thr24, Ser46, Glu47, Leu50, Asn142, Cys145, Met165, and Gln189 residues (Figures 2B and S6). As suggested before, flexibility could conceivably help design by providing alternative binding sites for non-native molecules (Niu et al., 2008). Observed significant conformational differences are caused by intrinsic flexibility around the binding pocket in our SFX structure. This flexibility allows us to observe alternative side-chain conformations inaccessible to the ambient-temperature X-ray structure (PDB: 6WQF). The calculated bias-free composite omit map that covers the active region has been shown (Figures 3 B and S7). This structure offers insights into the active site of Mpro complementary to the PDB: 6WQF structure, which may be important for future ligand screening and in silico modeling studies. All three domains contribute to the formation of the active site of the protein. The intersection of domain I residue His41 and domain II residues Cys145 and His164 interact via a coordinated water molecule with Asp187 located at the N-terminal loop region of domain III to form the active site (Figures 2C and S8). The N-terminal loop of domain III is suggested to be involved in enzyme activity (Ma et al., 2020). The distance between Cys145 Sγ and His41 Nε2 is 3.7 Å, similar to the ambient-temperature structure PDB: 6WQF (Kneller et al., 2020). Oδ1-Oδ2 atoms of Asp187 and NH2-Nε atoms of Arg40 contribute to a salt bridge between these two residues and stabilize the positions of each other. The W5 water molecule (indicated with a red sphere at the figure) in the active site plays crucial roles for catalysis (Kneller et al., 2020). W5 forms triple H bonds with His41, His164, and Asp187 side chains with the distances of 2.8, 2.9, and 2.8 Å, respectively (Figures 2C and S8).
Figure 3.

Key crystal contacts of C121 crystal form
(A) The 2Fo − Fc electron density belonging to the N-terminal region of the SARS-CoV-2 main protease is contoured at the 1σ level and colored in gray. Regions of the symmetry-related molecule are colored in gray, 2Fo − Fc electron density is contoured at the 1σ level and colored in slate. H bonds are indicated by black dashed lines. Elimination of these H bonds was essential to obtain the second crystal form in the P212121 space group, with a more open inhibitor binding pocket for future soaking studies of the putative main protease inhibitors.
(B) Superposition of Cα atoms of our structure (PDB: 7CWB) was performed with the Mpro structure (PDB: 6WQF). Our structure is colored in salmon, while chain A of PDB: 6WQF is colored in gray. A composite omit map of the PDB: 7CWB active site is shown blue color.
When compared with the room temperature structure of Mpro PDB: 6WQF, our structure displays additional active site residue dynamics while it has an overall high similarity (Figures 2D, 4G, and 4H). Canonical chymotrypsin-like proteases contain a catalytic triad composed of Ser(Cys)-His-Asp(Glu) in their catalytic region; however, SARS-CoV-2 Mpro possesses a Cys145 and His41 catalytic dyad which distinguishes the SARS-CoV-2 Mpro from canonical chymotrypsin-like enzymes (Kneller et al., 2020; Gorbalenya and Snijder, 1996). During the catalysis, the thiol group of Cys145 is deprotonated by the imidazole of His41 and the resulting anionic sulfur nucleophilically attacks the carbonyl carbon of the substrate. After this initial attack, an N-terminal peptide product is released by abstracting the proton from the His41, resulting in the His41 becoming deprotonated again and a thioester is formed as a result. In the final step, the thioester is hydrolyzed, which results in a release of a carboxylic acid and the free enzyme, therefore restoring the catalytic dyad (Ullrich and Nitsche, 2020; Pillaiyar et al., 2016). Observed catalytic residue conformations of SFX structures are consistent with PDB: 6WQF and support the proposed Mpro catalytic mechanism (Figures 2C and 2D).
Figure 4.

Contributions of residues to the first three PCs
(A–F) (A–C) PDB: 7CWB and (D–F) 7CWC with chain A displayed on the right and chain B on the left side. The aligned trajectory frames were generated to interpolate between the most dissimilar structures in the distribution along specified PCs. The color scale from red to blue represents low to high displacements along specified PCs, with broadening of the tubes depicting the trajectory movements. The Cα atoms of catalytic residues His41 and Cys145 are displayed as spheres with cyan and lime colors, respectively.
(G and H) A dynamic cross-correlation matrix generated from the motions observed in PC space with values ranging from –1 (complete anti-correlation) to +1 (complete correlation) (G) for PDB: 7CWB and (H) for 7CWC. The boundary between chains A and B is denoted by dashed lines.
The crystal contact of the symmetry-related molecule with the N-terminal region of the Mpro is essential for the formation of the crystal lattice in the monoclinic C121 space group (Figures 3A and S9). There is an extensive network of hydrogen bonding interactions. The Ser1 amino group and carbonyl O atom engage in two hydrogen bonding interactions with carbonyl O and the backbone amide N of Phe140. The carbonyl group of Gly2 forms a hydrogen bond with the carbonyl Oγ atom of Ser139. The backbone amide group of Phe3 forms a second hydrogen bonding interaction with the carbonyl O atom of Gly138 in the C121 space group. The side-chain amine and amino group of Arg4 form a hydrogen bond with the carbonyl O atom of Lys137 (Figure S9). These interactions are crucial, and elimination of this hydrogen bond network by the addition of four N-terminal amino acids switches SARS-CoV-2 Mpro to the secondary crystal conformation in the orthorhombic P212121 space group (Figures S9 and S10). In the crystal form the active site pocket has a wider binding pocket than the cryogenic structure (Figure S11). This wider binding pocket may offer opportunities for obtaining more effective drug soaks for future structural studies of Mpro drug complexes guided by hybrid in silico methods.
All-atom molecular dynamics simulations of apo forms of Mpro reveals that the protomers display asymmetric behavior
To better understand the structural and dynamical properties of Mpro, molecular dynamics (MD) simulations were performed using our ambient-temperature SFX crystal structures (Figure 4). As the catalytically active form of Mpro is a homodimer, we have used the dimeric form of crystal structures. For the SFX structure obtained in space group C121 with a monomeric asymmetric unit, the relevant dimeric form is generated by symmetry operation. The MD simulations were run for 200 ns to elucidate the dynamic effects on the structures, specifically for the active site region. The evolutionary changes of atomic coordinates over time were monitored by calculating the RMSD for both chains, i.e., protomers (Figures S12 and S13 for PDB: 7CWB and 7CWC, respectively). The root-mean-square fluctuations (RMSF) were also computed based on Cα atoms for both protomers separately (Figures S14 and S15 for PDB: 7CWB and 7CWC, respectively) to determine the flexible regions. In simulations for both PDB: 7CWB and 7CWC, the protomers exhibit non-identical behavior as also observed by others in apo form of Mpro (Suárez and Díaz, 2020; Sheik Amamuddy et al., 2020), although, as expected, higher RMSF values were observed for the loop regions of protomers. In addition, the protomers of PDB: 7CWC display higher fluctuations compared with PDB: 7CWB around the loops covering the active site (such as loops containing residues 44–52 and 185–190) which could affect the accessibility of the active site by inhibitors (Figures S14 and S15).
Principal-component analysis correlates the inter-domain motions and its impact on the drug-binding pocket dynamics
The trajectory frames obtained from MD simulations were used to perform principal-component analysis (PCA) to determine the variations of conformers of protein structures, i.e., to observe the slowest motions during MD simulations. As PCA and PCA-based methods are useful to reveal intrinsically accessible movements, such as domain motions (Bahar et al., 2010), we have performed PCA for backbone atoms of dimeric units for the structures belonging to different space groups. We focused on the first three principal components (PCs), which show around 40% of the total variance in MD trajectories, to determine the regions of protein structures that display the highest variation (Figures S16 and S17 for PDB: 7CWB and 7CWC, respectively). The first three PCs were projected onto the protein structures to determine the contributions of each residue to specified PCs. The motions of specific regions with blue regions with higher thickness represent more mobile structural parts of the protein along the specified PCs. We also provide supplementary movies to illustrate the movements along the species PCs: in Videos S1, S2, and S3 the motions along PC1, PC2, and PC3 for PDB: 7CWB are displayed, respectively, while for PDB: 7CWC the movements along first three PCs are displayed in Videos S4, S5, and S6, respectively. In the movies, different colors represent the different domains of the enzyme such that domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B. In all of the movies presented, protomer A is shown on the right and protomer B is shown on the left. It can be seen that both in C121 and P212121 space group structures, again protomers A and B display asymmetric behavior and domain movements along all considered PCs (Figures 4A–4C for PDB: 7CWB and Figures 4D–4F for PDB: 7CWC) (Sheik Amamuddy et al., 2020). When the motions displayed by two dimeric forms are compared with each other, we observe that domain III is more mobile in PDB: 7CWB compared with PDB: 7CWC (Videos S1, S2, and S3 for PDB: 7CWB and M4-M6 for PDB: 7CWC). However, the loop region containing residues 45–53 around the catalytic site is more mobile for both protomers in PDB: 7CWC, while it is only mobile for chain B of PDB: 7CWB. Interestingly, protomer A of PDB: 7CWC is more mobile compared with protomer A of PDB: 7CWB, which could reflect the differences in dimeric interface due to the additional four N-terminal amino acids and missing interactions with chain B N-terminal residues in the dimer interface. However, the loop regions surrounding the binding pockets with residues 166–172 and 185–195 display higher flexibility in chain A of PDB: 7CWB, while its mobility is somewhat restricted in PDB: 7CWC.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
We have also performed cross-correlation analysis of residues along the PC space to understand the correlation between motions especially for different domains. The correlation between the motions along the first three PCs were plotted as dynamical cross-correlation maps, displayed in Figures 4G and 4H for PDB: 7CWB and 7CWC, respectively. Focusing on the correlation between motions within protomers initially, we see that motions of domains I and III as well as domains I and II are mostly anti-correlated, as shown by the purple regions in Figure 4G for PDB: 7CWB and Figure 4H for PDB: 7CWC. While we observed similar behavior for protomers A and B of PDB: 7CWC, the directions of domains I and III movements are different in protomers A and B of PDB: 7CWB, specifically for residues 189–192, which are at the linkage points of domains II and III and residues 45–50 of domain I bordering the catalytic site. While in protomer A, these loops move in the same directions, their movements are mostly anti-correlated in protomer B for the slowest motion along PC1 in PDB: 7CWB (Video S1, Figure 4G). This could cause differences in the accessibility of catalytic sites in a time-dependent manner in protomers of PDB: 7CWB. On the other hand, domains II and III within protomers have mixed correlations with each other in which motions of some residues are along the same directions, others are in opposite directions within both protomers A and B of PDB: 7CWB, while domain II has limited mobility as being more buried than other domains (Suárez and Díaz, 2020) except for the β strand segment of residues 166–172 of protomer A. Surprisingly, this segment also has some mobility, albeit limited, in protomer B of PDB: 7CWC instead of protomer A (Video S4; Figure 4H). Glu166 of this segment is actually an important residue that plays a role in stabilizing the substrate binding site S1 by interacting with Ser1 of the alternate protomer along with Phe140 (Ghahremanpour et al., 2020; Jin et al., 2020) and we observed that, along PC1, for protomer A of PDB: 7CWB and protomer B of PDB: 7CWC, the motions of these residues are correlated (Video S1 for PDB: 7CWB and M4 for PDB: 7CWC). This could have implications about information transfer from one protomer to the other, although other Mpro structures of SARS-CoV-2 need to be studied in atomistic detail. In addition, we observed that correlations between domains of protomers A and B for PDB: 7CWB and 7CWC are dissimilar. For instance, domains I of PDB: 7CWB have defined anti-correlated motions as can be seen from the purple areas in Figure 4H, while in PDB: 7CWC the movements of domain I are more ambiguous. There are also differences in the behavior of domain I of protomer A and domain II of protomer B in the dimer structures in which for PDB: 7CWB the motions of these domains are positively correlated while they are anti-correlated in PDB: 7CWC.
Different non-covalent interactions stabilize the dimer interfaces in different space groups
The crystal structures that were obtained in this study were compared with four other Mpro structures crystallized recently. Selected Mpro structures were cryogenic apo form (Zhang et al., 2020a, 2020b) (PDB: 6Y2E and 7C2Y), holoprotein with non-covalent inhibitor, X77 (PDB: 6W63), and a room-temperature apo-form structure (Kneller et al., 2020) (PDB: 6WQF). Space groups of these structures are C121, P212121, P21212, and I121, respectively. The completeness of the structures was also evaluated: PDB: 6WQF, and 6Y2E crystallized in full-length sequence (1–306); however, PDB: 6W63 only lacks Gln306 at both C-terminal ends of its chains. Among compared structures, PDB: 7C2Y, which has the same space group as PDB: 7CWC, lacks 24 amino acid residues (chain A, 1–3, 141–142, 281–283, and 298; chain B, 1–2, 45–50, 139–142, and 277–279). In our structures, PDB: 7CWB has a full-length sequence; however, PDB: 7CWC lacks its last six amino acid residues from the C-terminal in chain A, and starts with Phe3 (lacks Ser1 and Gly2), and ends at Ser301, lacking the last five residues. When six structures (PDB: 6W63, 6Y2E, 6WQF, 7C2Y, 7CWB, and 7CWC) were compared based on hydrogen bonding interactions at the dimerization interfaces, more similarities were observed for the latter three (PDB: 7C2Y, 7CWB, and 7CWC). One of the main differences between our structures and the other four is the hydrogen bond between Ser139 (chain A) and Gln299 (chain B) (Figure S18). Although this hydrogen bond is not observed in other structures, the corresponding residues are close to each other; however, they are not within hydrogen bonding distance (Figure S19).
We also monitored all interface interactions throughout the MD simulations and compared PDB: 7CWB and 7CWC. Interestingly, the hydrogen bond between Ser139 (chain A) and Gln299 (chain B) was lost and the interaction was turned into a van der Waals interaction in PDB: 7CWB, and conserved by 64% of the simulation time. In the PDB: 7CWC structure, Ser139 (chain A) was not in the vicinity of Gln299 (chain B) (Figure S20). More interestingly, the same interaction but this time between Gln299 (chain A) and Ser139 (chain B), which is present at the PDB: 7CWB crystal structure, was only observed in 16% of the simulation time (Figure S21). In the PDB: 7CWC crystal structure, there was no hydrogen bonding interaction between Gln299 (chain A) and Ser139 (chain B). However, during simulation, this bond was formed and retained in 65% of the simulation time (Figure S21).
When static structures were compared based on hydrogen bond formation analysis, PDB: 7C2Y, 7CWB, and 7CWC clustered together, having the same hydrogen bonding network at the dimerization interface. The explanation for the hydrogen bond differences observed at the interface is the lack of amino acids at the N-terminal and C-terminal ends of the PDB: 7C2Y and 7CWC structures, compared with PDB: 7CWB.
Simulation trajectories of PDB: 7CWB and 7CWC were compared based on hydrogen bond occupancies. The major differences were Lys137 (chain A) and Arg4 (chain B), Asn142 (chain A) and Ser301 (chain B), and Arg298 (chain A) and Tyr118 (chain B). The first interaction was not observed in PDB: 7CWC; however, the latter two interactions were not present in PDB: 7CWB. The Lys137 (chain A) and Arg4 (chain B) interaction was conserved in 82% of the simulation times; Asn142 (chain A) and Ser301 (chain B), and Arg298 (chain A) and Tyr118 (chain B) interactions, retained 67% and 72% of the obtained trajectory frames, respectively (Figures S21 and S22). We also observed that, during MD simulations, in the case of PB: 7CWC, Gln299 (chain A) and Phe140 (chain B) come closer to each other, and this van der Waals contact was conserved in 64% of the simulation times and not observed in the case of PDB: 7CWB (Figure S22).
Non-covalent bound inhibitors form unstable complexes with Mpro
In their study, Jin et al. (2020) was screened in a library of around 10,000 compounds consisting of approved drugs, compounds in clinical trials, and natural products, using fluorescence resonance energy transfer assay. They identified six specific hit compounds, of which the half-maximal inhibitory concentration (IC50) values range from 0.67 to 21.4 μM. In this study, we used the top 3 hit compounds (i.e., Ebselen [IC50 = 0.67 μM], Tideglusib [IC50 = 1.55 μM], and Carmofur [IC50 = 1.90 μM]) for the investigation of ligand-target interactions. These compounds were docked to the PDB: 7CWB and 7CWC structures and all-atom MD simulations were performed for the top-docking poses. An induced fit docking (IFD) approach was conducted to better account for the flexibility of both of the ligands and the active site of the target structures. These three compounds were also docked to structures with PDB: 6W63 and 6Y2E (cryogenic) for comparison. MD simulations were performed using the same MD protocol as performed for apo-form structures. Results showed that, especially for apo-form dimer targets, Ebselen and Carmofur are quite flexible at the binding pocket throughout the simulations and, in most cases, they do not form a stable complex structure (Figure S23). Tideglusib has a more stable structure at the binding pocket of Mpro; however, its binding modes are different at the different targets considered in this study (Figure 5 ).
Figure 5.

Trajectory analyses of Tideglusib bound PDB: 7CWC and 6Y2E structures
(A) Conformational changes of Tideglusib at the binding pockets of PDB: 7CWC (left) and 6Y2E (right) throughout 200 ns MD simulations. In the color scale red-white-blue, red depicts simulation starting time, blue depicts simulation end time, in ns.
(B–D) (B) Representative binding poses of Tideglusib at PDB: 7CWC (left) and 6Y2E (right). Residues are colored with amino acid types: red, negatively charged; blue, positively charged; green, hydrophobic; cyan, hydrophilic. (C) Protein-ligand contacts throughout the MD simulations, PDB: 7CWC. The figure shows which residues interact with the ligand in each trajectory frame. Some residues make more than one specific contact with the ligand, which is represented by a darker shade of orange, according to the scale to the right of the plot. (D) 2D ligand interaction diagrams of Tideglusib, PDB: 7CWC (left) and 6Y2E (right).
Detailed MD simulations were performed for the stably bound SARS-CoV-2 Mpro inhibitor Tideglusib. Tideglusib initially docked to the binding pockets of dimeric Mpro structures PDB: 6Y2E and 7CWC using an IFD approach. Top-docking poses of these compounds were then used in all-atom MD simulations using the same MD protocols. While Tideglusib was structurally very stable at the binding pocket of PDB: 7CWC during the simulations, it was not so stable at the binding site of PDB: 6Y2E (Figure 5A). Representative trajectory frames (i.e., the frame that has the lowest RMSD to the average structures) were used in the comparison of binding modes (Figure 5B). Results showed that, while the binding mode of Tideglusib forms hydrogen bonds and π-π stacking interactions with Glu166, Gln189, and His172, respectively, at PDB: 7CWC its corresponding binding mode at PDB: 6Y2E only forms van der Waals type interactions with hydrophobic moieties. A timeline of protein-ligand contacts was visualized throughout the simulations (Figure 5C). Results showed that Thr25, Leu27, Met49, Glu166, and Gln189 form stable interactions with the ligand at PDB: 7CWC. However, corresponding interactions of the ligand at the active site of PDB: 6Y2E were not stable (Figure S24). Ligand interactions with protein residues that occur for more than 15% of the simulation time are also presented (Figure 5D). Comparison of binding pocket volumes of PDB: 7CWC and cryogenic Mpro structure (PDB; 6Y2E) shows that the latter has a bigger average binding pocket volume (Figure S25). Average binding pocket volumes were 174 and 142 Å3 for Tideglusib bound PDB: 6Y2E and 7CWC structures, respectively. Corresponding solvent-accessible surface area (SASA) values throughout the MD simulations also support this result. The SASA, which is the surface area of a molecule accessible by a water molecule of PDB: 7CWC is smaller than PDB: 6Y2E (Figure S25).
For comparison of monomer forms of Mpro, we applied an IFD protocol to predict the binding mode of Tideglusib at the binding pocket of PDB: 6WQF and 7CWB. Carbonyl oxygens of the thiadiazolidine ring of Tideglusib formed hydrogen bonds between Asn142, Gly143, and Glu166 from their backbone atoms. The naphthalene ring of the ligand formed a π-π stacking interaction with the His41 in the PDB: 7CWB structure. A similar binding mode of Tideglusib was observed when PDB: 6WQF was used. However, in this binding mode, His41 and Gly143 are found to be important. A backbone hydrogen bond was observed between one of the carbonyl oxygens of the thiadiazolidine ring and Gly143. His41 formed two π-π stacking interactions between the benzyl and the naphthalene rings of Tideglusib (Figures S26A and S26B). These two binding modes predicted by IFD were used in 200 ns classical all-atom MD simulations. Each Mpro-Tideglusib system is evaluated based on RMSD changes from the average structure and we obtained representative structures from corresponding trajectories. In the representative structure of the PDB: 7CWB-Tideglusib complex, we observed van der Waals interactions between surrounding residues: Thr25, His41, Cys44, Thr45, Ser46, Met49, Asn142, Cys145, His163, His164, Met165, Glu166, and Gln189. In the representative structure of the PDB: 6WQF-Tideglusib complex, in addition to the similar van der Waals interactions, Asn142 and Gln189 formed two hydrogen bonds with the carbonyl oxygens of the thiadiazolidine ring from their backbone atoms (Figures S26C and S26D). The alignment of the representative frames from PDB: 6WQF and 7CWB yielded an RMSD value of 0.97 Å (Figure S27).
Translational and rotational motions of Tideglusib were also examined by trajectory analyses. Conformational spaces that were explored by Tideglusib are given in Figure S28 . We also monitored the binding cavity volume throughout the MD simulations in both complexes and obtained average values of 154 and 127 Å3 for Tideglusib bound PDB: 7CWB and 6WQF structures, respectively ( Figure S29). In the IFD poses, corresponding binding cavity volumes were calculated as 251 and 238 Å3, respectively. Decreased binding cavity volume during the MD simulations may be correlated with the unstable conformations of Tideglusib in both structures. Overall, from the MD simulations of Tideglusib with the monomer forms of Mpro, we observed more stable conformations of Tideglusib at the binding pocket of PDB: 6WQF compared with PDB: 7CWB.
Discussion
SFX utilizes micro-focused, ultrabright, and ultrafast X-ray pulses to probe small crystals in a serial fashion. Structural information is obtained from individual snapshots, capturing Bragg diffraction of single crystals in random orientations (Martin-Garcia et al., 2016). The main advantages of SFX over its counterparts are the capability of working with micron- to nanometer-sized crystals, which does not necessitate lengthy and laborious crystal growth optimization steps and enables working with multiple crystal forms and space groups. It enables obtaining high-resolution structures of SARS-CoV-2 Mpro at physiologically meaningful temperature and has confirmed the dynamic regions of the active site. SFX offers great potential and provides critical information for future high-throughput structural drug screening and computational modeling studies with sensitivity to dynamics, resulting in production of detailed structural information.
SARS-CoV-2 Mpro catalyzes the precise cleavage events responsible for activation of viral replication and the expression of structural proteins (Jin et al., 2020). This has been the focus of several structural and biochemical studies, many of which have been performed at cryogenic temperatures, aiming to provide better understanding of the active site dynamics and reveal an inhibitor that affects the enzyme based on structural information. In our study, two crystal forms of Mpro, native and modified, were determined at ambient temperature with resolutions of 1.9 and 2.1 Å, respectively. The two produced crystal forms are optimal for co-crystallization and soaking, respectively. Co-crystallization experiments provide efficient interaction in the binding pocket as both drug and protein are stabilized before the formation of crystals. Due to the close crystal lattice contacts, co-crystallization should be the preferred method for larger molecules against the native form of Mpro in the space group C121. However, there are numerous successful soaking experiments with small non-covalent fragments (Douangamath et al., 2020). Residues at the N terminus of Mpro play a critical role in crystal packing. Elimination of the H-bonding network produced orthorhombic crystals in the P212121 space group yielding a wider binding pocket, increasing the probability of capturing an expanded number of protein-drug complexes by soaking. Obtaining two different crystal forms helped eliminate the artifacts introduced by specific lattice packing restraints of each crystal form and thereby increased the quality of the MD analysis. However, the presence of four additional residues at the N terminus could hinder soaking or co-crystallization efforts given that ∼200 ligand-bound structures of Mpro have been reported (Douangamath et al., 2020).
The high-resolution Mpro SFX structures presented here in two different crystal forms collectively revealed the intrinsic plasticity and dynamics around the enzyme's active site. Due to the anionic nature of Cys145, it seems challenging to design molecules that interact with the active site only through non-covalent bonds as it has a flexible region (Chang, 2010). These findings provide a structural basis for and are consistent with studies claiming that the majority of inhibitors form covalent bonds with the active site of Mpro through Cys145 (Figures S5.4–S5.37). In particular, unlike that suggested by some studies, Ebselen does not form a stable complex structure (Jin et al., 2020; Sies and Parnham, 2020; Menéndez et al., 2020; Zmudzinski et al., 2020; Weglarz-Tomczak et al., 2021), (Figure S30). The Cys145 residue is a key amino acid for catalytic activity, on the other hand the coordinated W5 molecule, regulating the catalytic reaction via triple hydrogen bonding interactions with His164 and Asp187, which stabilizes the positive charge of the His41 residue (Kneller et al., 2020). The active site residue conformations of SFX structures are consistent with previous ambient-temperature structures (PDB: 6WQF) (Figures 2C and 2D).
N-terminal loop residue Gln189 from domain III contributes to the stability of the potential inhibitors (Dai et al., 2020; Kneller et al., 2020) and, along with Asn142 and Ser46, is the active site residue forming the flank of the cavity (Figure 6 ). In a recent study, Asn142 and Gln189 have been indicated to interact with 11a (N-[(1S)-1-(cyclohexylmethyl)-2-[[(1S)-1-formyl-2-[(3S)-2-oxo-3- pyrrolidinyl]ethyl]amino]-2-oxoethyl]-1H-indole-2-carboxamide) and 11b (N-[(1S)-1-[(3-fluoro phenyl)methyl]-2-[[(1S)-1-formyl-2-[ (3S)-2-oxo-3-pyrrolidinyl]ethyl ]amino]-2-oxoethyl]-1H- indole-2-carboxamide) inhibitors that have proven in vitro effectiveness (Dai et al., 2020); along with these amino acids, Thr24 makes van der Waals interaction with the N3 inhibitor, and Ser46 undergoes conformational changes in the presence of this inhibitor (Jin et al., 2020) (PDB: 6LU7). N3 bound Mpro through Leu50 gives different side-chain conformations with SFX according to the PDB: 6WQF structure (Kneller et al., 2020) (Figure 2B). There were differences in the crystallization conditions between the SFX and PDB: 6WQF studies, the former making use of charge effects and the latter molecular crowding. Kneller et al. (2020) elegantly presented the major differences between cryogenic and ambient-temperature Mpro structures. The SFX study is eliminating potential artifacts to examine crucial amino acids at the atomic level, especially in terms of catalytic and inhibitor binding sites. An example of this is the decarboxylation of acidic residues, something frequently encountered at high X-ray doses (Figures S6–S8) (Martin-Garcia et al., 2016; Johansson et al., 2017). These two high-resolution SFX structures in different space groups reveal alternate active site residue conformations, intra- and inter-domain networks, and their dynamics at the atomic level. This information provides a better understanding of structural allosteric transitions of Mpro interacting with the inhibitors (Figures 5 and S23–S30). Therefore, considering the importance of the required sensitivity in drug design or the use of natural compounds in the studies, these active site residue conformations reveal the critical importance of our study more clearly.
Figure 6.

Different monoclinic crystal forms of the SARS-CoV-2 Mpro with catalytic site cavities
(A) C121 SFX, (B) PDB: 6WQF, and (C) 6Y2E, respectively. Water molecules in the catalytic cavity are shown with red spheres. The catalytic residue (Cys145) is highlighted in green and the flank cavity residues (Asn142, Ser46, Gln189) are highlighted in purple in the catalytic cavity of each crystal form C121 SFX (D), 6WQF (E), and 6Y2E (F), respectively. The distance between C (Asn142 Cγ-Ser46 Cβ-Gln189 Cδ) atoms of flank cavity residues is shown as a dashed line. All distances are given in Å.
There are many in silico docking studies performed based on cryogenic Mpro protein structures of SARS-CoV-2 (Dai et al., 2020; Pillaiyar et al., 2016; Jin et al., 2020; Ton et al., 2020; Durdagi et al., 2020). Although potent antiviral drug candidates have been identified and several vaccines are on the market, a desirable final cure has not yet been found. Having access to the alternative ambient-temperature structures of Mpro and observed conformational changes on active site residues will be a significant boon for the development of therapeutics by providing robust models for computational studies and a better understanding of ligand and inhibitor binding. At this point, our work has two original aspects. Firstly, we used a comprehensive platform, SFX, which helps to deeply understand the complexity of SARS-CoV-2 and the structural dynamics of the target protein Mpro at near-physiological temperatures. Secondly, we determined two high-resolution SFX structures of SARS-CoV-2 Mpro in two different space groups due to an upgraded high-throughput data collection setup offered by the MFX instrument of the LCLS-II.
A grave issue with antiviral drug research is the variability of the target proteins, as the mutations and modifications may render the found drugs ineffective (Dinesh et al., 2019). For this reason, working with a protein whose biochemical properties are conserved over time and carries significant importance among different strains. Evolutionarily, viruses bypass the host immune system by mimicking the proteins involved in the functioning of the host organism. For instance, the cytomegalovirus can mimic a common host protein to hijack normal cell growth machinery or the human immunodeficiency virus can mimic a high percentage of human T cell receptors (Root-Bernstein, 2017; Robertson, 2003). SARS-CoV-2 virus contains the PLpro enzyme, which is highly similar to the deubiquitinating enzymes as ubiquitin-specific proteases 7 and 14 (USP7, USP14) in human metabolism, which potentially pose the risk of interfering host metabolic pathways (Ratia et al., 2014). In addition, the spike protein, one of the other targeted proteins, has a higher mutation rate (Jia et al., 2020), and it includes a similar restriction site with epithelial channel protein (Anand et al., 2020). Besides, inhibitors that target the spike protein provide a more effective solution before the host cell is infected by the virus. As there is no Mpro homolog in the human genome, targeting this protease is therefore highly attractive due to reduced cross-reactivity and side effects, making it an ideal candidate for drug therapy (Goyal and Goyal, 2020) In contrast, covalent inhibitors may show cross-reactivity with human enzymes and are therefore less appealing as drug candidates.
Drug repurposing has been the preferred area of research in the insufficiency of time and resources in emergency cases, such as a novel pandemic; however, its impact on COVID-19 is debatable (e.g., remdesivir, hydroxychloroquine) (Beigel et al., 2020; Ip et al., 2021). Knowing the beneficial and detrimental effects of targeted drugs, along with well-established precautions, will help with time limitations. This procedure has emerged as a fundamental and very strategic approach, not only for prospective cohort design but also for many types of clinical trials. In particular, cross-sectional studies which require a large data pool, would benefit, as repurposed molecules have well-defined profiles and would not require prolonged pre-clinical studies. Hence, these molecules could be potential candidates to consider in the case of disease emergencies or outbreaks (Su and Sanger, 2017; Cavalla, 2013; Choo et al., 2019; Aguila and Cua, 2020). Considering all of this, adopting a drug-repurposing approach and using the known inhibitors Ebselen, Tideglusib, and Carmofur to carry out Mpro-based in silico molecular docking, MD simulations and post-MD analyses make our hybrid approach more specific for subsequent future studies. Our structural active site dynamics data further provide the nearest physiological template for future structure-based drug design and development of new Mpro inhibitors.
Limitations of the study
SARS-CoV-2 Mpro microcrystals transported to the LCLS for SFX data collection by non-temperature-controlled air cargo, which could negatively impact the diffraction quality of the crystals.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals and recombinant proteins | ||
| Tris | Sigma-Aldrich | Cat#93362 |
| Triton X-100 | Sigma-Aldrich | Cat#T9284 |
| Ni-NTA Agarose resin | QIAGEN | |
| PreScission | Genscript | Cat#Z02799 |
| Thrombin Protease | Cytiva | Cat#27-0846-01 |
| Terasaki Plate | Greiner Bio | Cat#654 102 |
| Pact Premier | Molecular Dimensions | Cat#MD1-29 |
| Pure Cotton | Ipek | N/A |
| Deposited data | ||
| Monoclinic structure of Mpro at ambient temperature | This Paper | PDB ID:7CWB |
| Orthorhombic structure of Mpro at ambient temperature | This Paper | PDB ID:7CWC |
| Structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77 | PDB Database | PDB ID:6W63 |
| Structural Plasticity of the SARS-CoV-2 3CL Mpro Active Site Cavity Revealed by Room Temperature X-ray Crystallography | Kneller et al., 2020 | PDB ID:6WQF |
| THE 1.28A CRYSTAL STRUCTURE OF 3CL MAINPRO OF SARS-COV-2 WITH OXIDIZED C145 (sulfinic acid cysteine) | PDB Database | PDB ID:6XKH |
| Crystal structure of the free enzyme of the SARS-CoV-2 (2019-nCoV) main protease | Zhang et al., 2020a, 2020b | PDB ID:6Y2E |
| The crystal structure of COVID-19 main protease in complex with an inhibitor N3 | Jin et al., 2020 | PDB ID:6LU7 |
| Experimental models: Cell lines | ||
| Rosetta 2(DE3) | Sigma-Aldrich | Cat#71400-M |
| E.coli BL21 (DE3) | Sigma-Aldrich | Cat#69450-M |
| Recombinant DNA | ||
| pET28a(+)_Construct-1 | Genscript | N/A |
| pET28a(+)_Construct-2 | Genscript | N/A |
| Software and algorithms | ||
| PSOCAKE | (Damiani et al., 2016) | https://github.com/lcls-psana/psocake |
| CHEETAH | (Barty et al., 2014) | https://github.com/biochem-fan/cheetah |
| CrystFEL | (White et al., 2012) | https://github.com/biochem-fan/CrystFEL |
| CCP4 | (Winn et al., 2011) | https://www.ccp4.ac.uk/ |
| Structure Refinement: PHENIX | (Adams et al., 2010) | http://www.phenix-online.org/ |
| Structure Modelling: COOT | (Emsley and Cowtan, 2004) | https://www.ccp4.ac.uk/ |
| PyMOL | Schrödinger, LLC | https://pymol.org/2/ |
| Maestro molecular modeling program | (Madhavi Sastry et al., 2013) | https://www.schrodinger.com/products/maestro |
| PROPKA | (Bas et al., 2008) | https://pypi.org/project/propka/ |
| R (Bio3D package) | (Grant et al., 2006; Yao and Grant, 2013) | https://www.r-project.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Hasan DeMirci (hdemirci@ku.edu.tr)
Materials availability
Any unique reagents/materials used in this study are available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
SARS-CoV-2 Main protease contains pET28a(+) transformed and cultured for protein expression in E.coli BL21 (DE3) cells. Growing of transformed cells was performed in LB with appropriate antibiotics at 37°C, overnight. OD600 was determined as 0.6 for induction of protein expression by addition of 0.4 mM IPTG and cells were cultured between 1-7 days at 18°C.
Method details
Gene construct design and cloning
Construct-1 with Native N- & C-terminals
The published Severe Acute Respiratory Syndrome CoronaVirus-2 main protease (SARS-CoV-2 Mpro) crystal structure revealed that the N-terminal serine residue is involved in the critical C121 space group crystal lattice contact (5)(PDB ID: 6LU7). We designed the native Mpro construct with the following amino acid sequence to obtain the native enzyme with no modifications at the N- & C-terminal ends. MSAVLQ(native_Mpro_cleavage_site)SGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTSEDMLNPNYEDLLIRKSNHNFLVQAGNVQLRVIGHSMQNCVLKLKVDTANPKTPKYKFVRIQPGQTFSVLACYNGSPSGVYQCAMRPNFTIKGSFLNGSCGSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGNFYGPFVDRQTAQAAGTDTTITVNVLAWLYAAVINGDRWFLNRFTTTLNDFNLVAMKYNYEPLTQDHVDILGPLSAQTGIAVLDMCASLKELLQNGMNGRTILGSALLEDEFTPFDVVRQCSGVTFQ(PreScission_protease_cleavage_site)GPHHHHHH∗ (∗ is stop). The corresponding gene was synthesized by Genscript, USA and cloned into pET28a(+) bacterial vector by using NdeI and BamHI restriction cleavage sites at 5’ and 3’ ends respectively. N-terminal canonical SARS-CoV-2 Mpro autocleavage cut site is indicated by green and purple which generated the native N-terminus. C-terminus had the PreScision™ restriction site shown in red which is used to generate the native C-terminus after Ni-NTA hexa-histidine affinity purification chromatography. In-frame hexa-histidine tag and stop codon were shown in blue color.
Construct-2 with modified N-terminus and native C-terminus
To eliminate the critical N-terminus crystal contact to obtain a new apo crystal form we designed the modified Mpro construct with the following amino acid sequence inserted in th E. coli vector pET28a(+) MGSSHHHHHHSSGLVPR(thrombin_cleavage_site)GSHMSGFRKMAFPSGKVEGCMVQVTCGTTTLNGLWLDDVVYCPRHVICTSEDMLNPNYEDLLIRKSNHNFLVQAGNVQLRVIGHSMQNCVLKLKVDTANPKTPKYKFVRIQPGQTFSVLACYNGSPSGVYQCAMRPNFTIKGSFLNGSCGSVGFNIDYDCVSFCYMHHMELPTGVHAGTDLEGNFYGPFVDRQTAQAAGTDTTITVNVLAWLYAAVINGDRWFLNRFTTTLNDFNLVAMKYNYEPLTQDHVDILGPLSAQTGIAVLDMCASLKELLQNGMNGRTILGSALLEDEFTPFDVVRQCSGVTFQ∗ (∗ is stop). The gene was synthesized by Genscript, USA and cloned into pET28a(+) bacterial overexpression vector by using NdeI and BamHI restriction sites at 5’ and 3’ ends respectively. N-terminal hexa-histidine tag (labeled in purple) and modified SARS-CoV-2 Mpro thrombin cleavage site which is part of the pET28a(+) vector were indicated blue sequence which generated the modified N-terminus with four extra residues as follows (GSHM) shown in blue and underlined. C-terminus had the in-frame stop codon that was used to generate the native C-terminus by ribosome during bacterial overexpression shown in green asterisk.
Protein expression
Both constructs were transformed into E. coli BL21 Rosetta-2 strain. 12 liters of six independent bacterial cell cultures containing target Mpro protein genes were grown in either regular LB-Miller media or Terrific Broth (TB) supplemented with 35 μg/ml chloramphenicol and 50 μl/ml kanamycin at 37°C. Cultures were incubated by using New Brunswick Innova 4430R shaker at 110 rpm until they reached OD600 about 0.8-1.2 for each culture. Recombinant protein expression was induced by Isopropyl β-D-1-thiogalactopyranoside (IPTG) with a final concentration of 0.4 mM. Incubation for protein production was performed at 18°C for minimum of 24 hours and maximum of 7 days. Cells were harvested at 4°C by using Beckman Allegra 15R desktop centrifuge at 3500 rpm for 20 minutes. Protein expression was confirmed by precast TGX-mini protean gradient SDS-PAGE from BioRad.
Protein purification
Standard chromatography purification methods were applied to both constructs with slight modifications as described below. Soluble Mpro proteins were purified by first dissolving the bacterial cells in the lysis buffer containing 50 mM Tris pH 7.5, 300 mM NaCl, 5% v/v Glycerol supplemented with 0.01% Triton X-100 followed by sonication (Branson W250 sonifier, USA). After sonication step, cell lysate was centrifuged by using The Beckman Optima™ L-80 XP Ultracentrifuge at 40000 rpm for 30 minutes at 4°C by using Ti45 rotor (Beckman, USA). After ultracentrifugation, the pellet which contained membranes and insoluble debris was discarded and clear supernatant applied to nickel affinity chromatography by using a Ni-NTA agarose resin (QIAGEN, USA). To purify the Mpro protein, first the chromatography column was equilibrated by flowing 3 column volume of the loading buffer containing 20 mM Tris-HAc pH 7.5, 5 mM Imidazole, 150 mM NaCl. After equilibration, the supernatant containing the overexpressed Mpro protein was loaded into the Ni-NTA agarose column at 2 ml/minute flow rate. Unbound proteins were removed by washing with 5 column volumes of the loading buffer to clear the non-specific binding. After washing, hexa-histidine tagged Mpro proteins were eluted from the column with the elution buffer containing 20 mM Tris-HAc pH 7.5, 150 mM NaCl, 250 mM Imidazole in 35 ml of total volume. After elution, purified protein was placed in a 3 kDa cut off dialysis membrane and dialyzed against the buffer containing 20 mM Tris-HAc pH 7.5, 150 mM NaCl overnight to get rid of the excess Imidazole. After the dialysis step, we applied 1:100 stoichiometric molar ratio 3C protease (PreScission protease, GenScript, USA) to cleave the C-terminal hexa-histidine tag of construct-1 with native N- & C-terminals. For construct-2 with modified N-terminus we used thrombin protease (Sigma, USA) to get rid of the N-terminal hexa-histidine tag. Both PreCision and thrombin cleavage have been performed overnight at 4°C. In the final purification step, to remove the cleaved hexa-histidine tag and other non-specific binding proteins we applied the solution to reverse Ni-NTA chromatography and collected the unbound fractions containing the untagged Mpro protein. The pure Mpro was concentrated by ultrafiltration columns from Millipore to a final concentration of 25 mg/ml and added 1 mM final concentration of DTT and stored at -80°C until crystallization trials.
Crystallization of Mpro protein for SFX crystallography at XFEL
For initial crystallization screening, we employed sitting-drop microbatch under oil screening method by using 72 well Terasaki crystallization plates (Greiner-Bio, Germany). Purified Mpro protein at 25 mg/ml mixed with 1:1 volumetric ratio with ∼3500 commercially available sparse matrix crystallization screening conditions. The sitting drop solutions were then covered with 20 μl of 100% paraffin oil (Tekkim Kimya, Turkey). All the crystallization experiments were performed at ambient-temperature. For our native construct-1 we were able to obtain multiple hit conditions and among them the best crystals were obtained at Pact Premier™ crystallization screen 1 condition #39 from Molecular Dimensions, UK. The best crystallization condition has contained 100 mM MMT buffer pH 6.0 and 25 % w/v PEG 1500 [MMT buffer; DL-Malic acid, 4-Morpholine Ethane Sulfonic acid (MES) monohydrate, 2-Amino-2-(hydroxymethyl)-1,3- propanediol (TRIS)-HCl]. For the modified construct only one crystallization condition yields the macrocrystals. After multiple optimization of the seeding protocol by using crystals obtained by microbatch under oil, we scaled up the batch crystallization volume to a total of 14 ml for native construct-1 and total volume of 50 ml for modified construct-2. Microcrystals 1-5 × 5-10 × 10-20 μm3 in size were passed through 100 micron plastic mesh filters (Millipore, USA) in the same mother liquor composition to eliminate the large single crystals and other impurities before the data collection. Crystal concentration was approximated to be 1010-1011 particles per ml based on light microscopy. Due to COVID19 travel restrictions none of the initial crystals or the batched crystalline slurry were able to be pretested for their diffraction quality before the scheduled XFEL beamtime.
Transport of Mpro microcrystal for SFX studies at MFX instrument at the LCLS
1.9 ml total volume of crystal slurry was transferred to 2 ml screw top cryovial (Wuxi NEST biotechnology, China cat#607001). To absorb the mechanical shocks during transport from Istanbul to Menlo Park, CA these vials were wrapped loosely by Kimwipes (Kimberly-Clark, USA) and placed in 20 ml screw top glass vials and tightly closed to provide insulation during transport via air. The vials were wrapped with excess amounts of cotton (Ipek, Turkey) and placed in a Ziploc™ bag (SC Johnson, USA) to provide both an added layer of insulation and mechanical shock absorption. The Ziploc™ bags were placed in a styrofoam box that was padded with ∼1 kg of cotton to provide more insulation and mechanical shock absorption during the transport. The styrofoam box was sealed and wrapped with an additional layer of 1 cm thick loose cotton layer and duck taped all around to further insulate the delicate Mpro crystals during ambient-temperature transport. All these packing materials and techniques provided us with crystals diffracting to 1.9 Å - 2.1 Å resolution as described below.
MESH sample injection for Mpro crystals
The 1.6 ml sample reservoir was loaded with Mpro crystal slurry in their unaltered mother liquor as described above. We used standard Microfluidic Electrokinetic Sample Holder (MESH) (Sierra et al., 2012, 2016) injector for our sample injection. The sample capillary was a 200 μm ID × 360 μm OD × 1.0 m long fused silica capillary. The applied voltage on the sample liquid was typically 2500-3000 V, and the counter electrode was grounded. The sample flow rate was typically between 2.5 and 8 μl/min.
Data collection and analysis for SFX studies at LCLS
The SFX experiments with native Mpro microcrystals were carried out at the LCLS beamtime ID: mfx17318 at the SLAC National Accelerator Laboratory (Menlo Park, CA) at ambient temperature. The LCLS X-ray beam with a vertically polarized pulse with duration of 30 fs was focused using compound refractive beryllium lenses to a beam size of ∼6 × 6 μm full width at half maximum (FWHM) at a pulse energy of 0.8 mJ, a photon energy of 9.8 keV (1.25 Å) and a repetition rate of 120 Hz. OM monitor (Mariani et al., 2016) and PSOCAKE (Damiani et al., 2016; Thayer et al., 2017) were used to monitor crystal hit rates, analyze the gain switching modes and determine the initial diffraction geometry of the new ePix10k2M detector (van Driel et al., 2020). A total of 1,163,413 detector frames were collected in 2h47m7s continuously with the new from native (construct-1) Mpro microcrystals. A total of 686,808 detector frames were collected in 1h36m20s continuously with the new ePix10k2M Pixel Array Detector from modified (construct-2) Mpro microcrystals. The total beamtime needed for native (construct-1) and modified (construct-2) datasets were 2h59m17s and 1h50m59s respectively. The MFX beamline equipped with the new ePix10k 2M detector that was operated in dynamic gain mode and MESH injector system has no blockages during the data collection. Individual diffraction pattern hits were defined as frames containing more than 30 Bragg peaks with a minimum signal-to-noise ratio larger than 4.5, which were a total of 208,839 and 214,355 images for native and modified respectively. The detector distance was set to 118 mm, with an achievable resolution of 2.1 Å at the edge of the detector (1.64 Å in the corner). An example diffraction pattern is shown in Figure S1.
Data processing; hit finding, indexing and scaling
The diffraction patterns were collected at the MFX instrument at the LCLS using the ePix10k2M detector (van Driel et al., 2020). The raw data images were subjected to detector corrections with CHEETAH (Barty et al., 2014), as well as for hit finding based on Bragg reflections. The hit finding parameters for all datasets classifying a hit were as follows (using peakfinder8): a minimum pixel count of 2 above an adc-threshold of 500 with a minimum signal to noise ratio of 7 was considered a peak, and an image containing at least 20 peaks was classified as a crystal hit. The crystal hits were then indexed using the software package CrystFEL (White et al., 2012; White, 2019) version 9.0 (10White et al., 2020) using the peaks found by CHEETAH. Indexing was attempted using the indexing algorithms from XGANDALF (Gevorkov et al., 2019), DIRAX (Duisenberg, 1992), MOSFLM (Powell et al., 2013) and XDS (Kabsch, 2010) in this order. After an approximate cell was found, the data was indexed using cell axis tolerances of 5 Å and angle tolerances of 5° (--tolerance option in CrystFEL). The integration radii were set to 2, 3, 5 and the “multi” option was switched on to enable indexing of multiple crystal lattices in a single image. The indexed reflections were subsequently integrated and merged using PARTIALATOR (White et al., 2016) applying the unity model over 3 iterations and the max-ADU set to 7500. The complete reflection intensity list from CrystFEL was then scaled and cut using the TRUNCATE program from the CCP4 suite (Winn et al., 2011) prior to further processing.
For the native Mpro protein crystals the final set of indexed patterns, containing 168,655 frames (80.7% indexing rate), was merged into a final dataset (Overall CC∗ = 0.999; 1.8 Å cutoff) for further analysis (C121, unit cell: a = 114.0 Å, b = 53.5 Å, c = 45.0 Å; α = 90°, β = 102°, γ = 90°). The final resolution cutoff was estimated to be 1.9 Å using a combination of CC∗ (Karplus and Diederichs, 2012) and other refinement parameters. The final dataset had overall Rsplit = 6.31%, and CC∗ = 0.865 in the highest resolution shell. For the N-terminally modified Mpro protein crystals the final set of indexed patterns, containing 157,976 frames (73.6% indexing rate), was merged into a final dataset (Overall CC∗ = 0.999; 2.1 Å cutoff) for further analysis (P212121, unit cell: a = 69.2 Å, b = 104.3 Å, c = 105.6 Å; α = β = γ = 90°). The final resolution cutoff was estimated to be 2.1 Å using a combination of CC∗ and other refinement parameters. The final dataset had overall Rsplit = 5.91%, and CC∗ = 0.678 in the highest resolution shell.
Structure determination and refinement of Apo Mpro structures
We determined two ambient-temperature Mpro structures by using two crystal forms in space group C121 and P212121 structures using the automated molecular replacement program PHASER (McCoy et al., 2007) implemented in PHENIX (Adams et al., 2010) with the previously published ambient-temperature structure as a search model (Kneller et al., 2020) (PDB ID: 6WQF). This choice of starting search model minimized experimental temperature variations between the two structures. Coordinates of the 6WQF were used for initial rigid body refinement with the PHENIX software package. After simulated-annealing refinement, individual coordinates and TLS parameters were refine(d. We also performed composite omit map refinement implemented in PHENIX to identify potential positions of altered side chains and water molecules were checked in program COOT (Emsley and Cowtan, 2004), and positions with strong difference density were retained. Water molecules located outside of significant electron density were manually removed. The Ramachandran statistics for native monoclinic Mpro structure (PDB ID: 7CWB) (most favored / additionally allowed / disallowed) were 96.7 / 3.0 / 0.3 % respectively. Ramachandran statistics for orthorhombic Mpro structure (PDB ID: 7CWC) (most favored / additionally allowed / disallowed) were 96.5 / 2.4 / 0.1 % respectively. The structure refinement statistics are summarized in Table S1. Structure alignments were performed using the alignment algorithm of PyMOL (www.schrodinger.com/pymol) with the default 2σ rejection criterion and five iterative alignment cycles. All X-ray crystal structure figures were generated with PyMOL.
Temperature factor analysis and generation of ellipsoids
The two ambient-temperature Mpro in space group C121 and P212121 were examined to generate ellipsoid structures based on b-factor with PyMOL and these two structures were compared with the Mpro structures at 100 K (PDB ID: 6XKH) to provide better understanding on the flexibility of atoms, side chains and domains. The all ellipsoid structures were colored with rainbow selection on PyMOL.
Molecular modeling studies
We have used different crystal structures of Mpro available in literature as well as the obtained crystal structures in this study (PDB IDs: 7CWB and 7CWC) as target structures for molecular docking and MD simulations. The biologically-relevant dimeric form of 7CWB was generated by application of a symmetry operator. As the crystal structures in this study were obtained at ambient-temperature, for comparison another ambient-temperature structure of Mpro (PDB ID: 6WQF) in apo form was also selected as target structure. Another apo form structure of Mpro (PDB ID: 6Y2E) in dimeric form was also chosen for comparison. Additionally, Mpro structure bound to a non-covalent inhibitor (PDB ID: 6W63) in both monomeric and dimeric forms was utilized as target structure. For ligands, we have considered three compounds that have shown promising inhibitory activity based on the high-throughput screening of around 10.000 compounds by Jin et al., namely; Ebselen (IC50 = 0.67 ± 0.09 μM), Tideglusib (IC50 = 1.55 ± 0.30 μM) and Carmofur (IC50 = 1.82 ± 0.06 μM) (Jin et al., 2020).
All the target structures considered in this study were firstly prepared using Protein Preparation module of Maestro molecular modeling program in which missing atoms were added, water molecules not in the vicinity of co-crystallized ligands were removed and bond orders were assigned ( Madhavi Sastry et al., 2013). The protonation states of amino acids at physiological pH were adjusted using PROPKA (Bas et al., 2008) to optimize the hydrogen binding and charge interactions. As a final step of preparation, a restrained minimization was performed with OPLS3e force field parameters (Harder et al., 2016).
The structures of three compounds were taken from PubChem; Ebselen (PubChem ID, 3194), Tideglusib (PubChem ID: 11313622) and Carmofur (PubChem ID: 2577). The compounds also needed preparation hence, LigPrep module (Schrödinger Release 2018-4, 2018) of Maestro molecular modeling package was employed with OPLS3e force field parameters (Harder et al., 2016). The ionization states of the molecules were predicted by Epik module (Shelley et al., 2007) at physiological pH of 7.4.
The prepared target protein and ligand structures were used for molecular docking studies. We have employed a grid-based docking method, Induced Fit Docking (IFD) protocol of Maestro (Sherman et al., 2006) which uses Glide (Friesner et al., 2004, 2006; Halgren et al., 2004) and Prime (Jacobson et al., 2004) to induce adjustments in receptor structures with flexible ligand sampling options. For target structures in apo form (structures with PDB IDs: 7CWB, 7CWC, 6WQF and 6Y2E), binding sites for docking studies were defined by centering grids at the centroid of a set of residues, namely His41, Cys145 and Glu166. On the other hand, for target structures in holo form (structures with PDB ID: 6W63 both monomeric and dimeric form) binding sites were defined by centering the grids at the centroid of the co-crystallized ligand molecule. In the dimeric form of Mpro, only one chain was considered for docking studies. IFD protocol involves subsequent phases, including (i) initial docking of the compounds with the rigid receptor in which 20 poses per ligand are retained; (ii) refining the residues (within 5 Å of the ligand) in complex using Prime module (Jacobson et al., 2004), and (iii) redocking of each protein/ligand complex structure within 30 kcal/mol of the lowest-energy structure and within the top 20 structures overall. During Glide redocking, standard precision (SP) option was chosen. The docking poses were scored and ranked based on GlideScore and poses with the lowest scores, i.e. top-docking poses were selected for further studies at each target protein.
The selected docking poses at each considered target structure of Mpro with the three compounds were subjected to MD studies. The apo form structures obtained in this study were also subjected to MD simulations. For comparison reasons, MD simulations were also performed for the holo form structure (PDB ID: 6W63) with its co-crystallized ligand, X77. The target protein-ligand complexes were placed in simulation boxes with orthorhombic shape in which box sizes were calculated based on buffer distance of 10.0 Å along all three dimensions and solvated with explicit water molecules of SPC (Berendsen et al., 1987) model. The simulation systems were neutralized by the addition of counter ions (Na+ or Cl− depending on the charge of the systems) and 0.15 M NaCl solution was added to adjust concentration of the solvent systems. All atom MD simulations package Desmond (Bowers et al., 2006) was employed. Proceeding the production MD simulations, the systems were equilibrated using relaxation protocols of Desmond package in which a series of minimizations and short MD simulations which were performed with small time-steps at lower temperature and restrains on the non-hydrogen solute atoms in the initial stages and slowly time-steps were increased as well as simulation temperature and restrains on solute atoms were released. The production simulations were performed under constant pressure and temperature conditions, i.e. NPT ensemble. Temperature was set as 310 K while being controlled by Nose–Hoover thermostat (Nosé, 1984; Hoover, 1985). The pressure was set as atmospheric pressure of 1.01325 bar with isotropic pressure coupling and controlled by Martyna–Tobias–Klein barostat (Martyna et al., 1994). Smooth particle mesh Ewald method (Essmann et al., 1995) was utilized to calculate long range electrostatic interactions with periodic boundary conditions (PBC). For short range electrostatics and Lennard-Jones interactions, the cut-off distance was set as 9.0 Å. The multi-step integrator RESPA was employed in which the time steps were varied for interaction types as followed in fs for: bonded, 2.0; near 2.0 and far 6.0.
Principal components analysis (PCA), a statistical data processing method, were performed to reduce the large-dimensional data by extracting large amplitude motions onto collective sets. A covariance matrix were generated from MD trajectory data for backbone atoms of protein structures as follow.
Here, i and j represent the backbone atom number, i.e. residue numbers of proteins while N is the number of backbone atoms considered in analysis. The Cartesian coordinates of atoms were denoted by and for ith and jth atom, respectively with and representing the time-averaged values over MD simulations. By diagonalization of covariance matrix, a collection of eigenvectors and corresponding eigenvalues were obtained. The eigenvectors of the diagonalized matrix were referred to as principal components (PCs) and constitute a linear basis set that matched the distribution of observed structures. The corresponding eigenvalues of the diagonalized matrix display the variance of the distribution along each PCs. In this study, we have utilized the Bio3D package (Grant et al., 2006; Yao and Grant, 2013), a platform independent R package to perform PCA for considered simulation systems. The trajectories obtained from independent MD simulations were concatenated and frames were aligned with respect to the initial (reference) frame before PCA.
The correlation of atomic displacements was evaluated by cross-correlation analysis to appreciate the coupling of motions. The magnitudes of all pairwise cross-correlation coefficients were investigated to assess the extent of atomic displacement correlations for each simulation system in principal component space. The normalized covariance matrix of atomic fluctuations was calculated as
where and are the displacements of residues i and j, i.e., mean square atomic fluctuations. The values of varies between -1 to 1 with representing completely correlated motions (same period and same phase), representing completely anticorrelated motions (same period and opposite phase) while 0 value indicates motions are uncorrelated (Ichiye and Karplus, 1991; McCammon and Harvey, 1987) Bio3D package (Grant et al., 2006; Yao and Grant, 2013) in R environment was employed to generate atom-wise cross-correlations of motions observed in PCs 1 to 3 and dynamical cross-correlation map, or DCCM were generated and displayed as a graphical representation of cross-correlation coefficients.
Interface analysis
Interface analysis of crystal structures and MD trajectories were carried out with the GetContacts PYTHON scripts (https://getcontacts.github.io/). Two different approaches were followed, in the first one only hydrogen bonds at the dimerization interface were taken into account, and in the second approach all possible interactions, namely; salt bridges, pi-cation, pi-pi stacking, T-stacking, van der Waals (vdW), and hydrogen bonds were calculated. If the distance between the acceptor and the donor atoms is <3.5 Å and the angle <70° hydrogen bond is defined between atom groups. Salt bridges were defined between atoms of negatively charged [ASP (OD1, OD2) and GLU (OE1, OE2)] and positively charged [LYS (NZ) and ARG (NH1, NH2)], where distances were <4.0 Å. T-stacking, pi-cation, and pi-stacking distance criteria were 5.0, 6.0, and 7.0 Å, respectively. Hydrophobic and vdW interactions were calculated based on atom R (radii), if the distance between atoms is less than the sum of R of atom A, R of atom B, and 0.5 Å.
One frame and trajectory-based calculations were performed. One frame calculations were applied for the crystal structures, and “1” means the corresponding residues are in contact, and “0” means no interaction. However, in trajectory-based calculations, we used all available 2000 trajectory frames obtained throughout the performed MD simulations. For the interaction frequencies, we applied a 0.6 threshold to only take into account the contacts that occurred at least 60% of the simulations.
Quantification and statistical analysis
Data collection and refinement statistics for X-ray crystallography shown in Table S1 and the section of ‘‘method details’’. The atom distances of the protein model and RMSDs were measured by pyMOL.
Acknowledgments
Authors would like to dedicate this manuscript to the memory of John C. H. Spence. H.D. acknowledges support from National Science Foundation (NSF) Science and Technology Centers grant NSF-1231306 (Biology with X-ray Lasers, BioXFEL) and The Scientific and Technological Research Council of Turkey (TUBITAK) grant (118C270). H.D. would like to thank Michelle Young, Ritu Khurana, Lori Anne Love, and Tracy Chou for their invaluable support and discussions. The authors would like to thank James Fraser and Galen Correy for their critical reading of our manuscript and their comments. Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-76SF00515. The HERA system for in helium experiments at MFX was developed by Bruce Doak and funded by the Max-Planck Institute for Medical Research. Research was supported by the DOE Office of Science through the National Virtual Biotechnology Laboratory, a consortium of DOE national laboratories focused on response to COVID-19, with funding provided by the Coronavirus CARES Act. The numerical calculations reported in this paper were partially performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources). S.D. and I.E. acknowledge TRUBA for the computational resources. S.D. acknowledges support from Bahcesehir University (BAU) Scientific Research Projects (BAP) grant (BAU2020-0101).
Author contributions
H.D. and S.D. designed and coordinated the project. H.D., C.D., M.Y., C.B., F.B.E., G.Y., O.G., S.O.B., A.S., F.A., and G.T. prepared the samples. B.H., M.L., M.H.S., M.S.H., A.B., V.M., Z.S., F.P., C.H.Y., C.K., and R.G.S. performed the sample delivery and on-site data collection. H.D., C.D., B.D., M.Y., C.B., I.E., F.B.E., S.C., G.Y., M.D.O., O.G., E.D., S.O.B., A.S., E.A., B.Y., A.B.P., O.G., A.D.Y., O.C., S.O., and E.H.D. performed the remote data collection. S.B., O.M.Y., A.B., A.T., G.K.K., Z.S., F.P., and C.H.Y. executed the data processing and reduction. S.D., B.D., T.A., I.E., S.C., M.D.O., B.A., K.S., L.O., I.T., A.O., and E.E. performed the in silico analysis. Structures were refined by H.D. Data were analyzed by H.D., E.S., S.D., C.D., E.D., B.Y., I.E., B.D., and T.A. The manuscript was prepared by H.D. and S.D. with input from all the coauthors.
Declaration of interests
The authors declare no competing interests.
Published: December 2, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.07.007.
Supplemental information
Data and code availability
The 3D electron density map of SARS-CoV-2 Main protease has been deposited in the ProteinDataBank under accession numbers 7CWB, 7CWC. This paper does not report original code. Any additional information required to reanalyze the data reported is available from the lead contact upon request.
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguila E.J.T., Cua I.H.Y. Repurposed GI drugs in the treatment of COVID-19. Dig. Dis. Sci. 2020;65:2452–2453. doi: 10.1007/s10620-020-06430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn D.G., Shin H.J., Kim M.H., Lee S., Kim H.S., Myoung J., Kim B.T., Kim S.J. Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19) J. Microbiol. Biotechnol. 2020;30:313–324. doi: 10.4014/jmb.2003.03011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P., Puranik A., Aravamudan M., Venkatakrishnan A.J., Soundararajan V. SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. eLife. 2020;9:1–7. doi: 10.7554/eLife.58603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020;26:450–452.. doi: 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astell C.R., Holt R.A., Jones S.J.M., Marra M.A. Coronaviruses with Special Emphasis on First Insights Concerning SARS. Birkhäuser Basel; 2005. Genome organization and structural aspects of the SARS-related virus; pp. 101–128. [DOI] [Google Scholar]
- Bahar I., Lezon T.R., Bakan A., Shrivastava I.H. Normal mode analysis of biomolecular structures: functional mechanisms of membrane proteins. Chem. Rev. 2010;110:1463–1497. doi: 10.1021/cr900095e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barty A., Kirian R.A., Maia F.R.N.C., Hantke M., Yoon C.H., White T.A., Chapman H. Cheetah: software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 2014;47:1118–1131. doi: 10.1107/S1600576714007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins: Struct. Funct. Bioinformatics. 2008 doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- Beck B.R., Shin B., Choi Y., Park S., Kang K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020;18:784–790. doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., et al. Remdesivir for the treatment of Covid-19—final report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., Grigera J.R., Straatsma T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987;91:6269–6271. [Google Scholar]
- Blaj G., Dragone A., Kenney C.J., Abu-Nimeh F., Caragiulo P., Doering D., Kwiatkowski M., Markovic B., Pines J., Weaver M., et al. AIP Conference Proceedings. 60062nd. Vol. 2054. AIP; 2019. Performance of ePix10K, a high dynamic range, gain auto-ranging pixel detector for FELs; pp. 1–9. [Google Scholar]
- Bowers K.J., Sacerdoti F.D., Salmon J.K., Shan Y., Shaw D.E., Chow E., Xu H., Dror R.O., Eastwood M.P., Gregersen B.A., et al. Proceedings of the 2006 ACM/IEEE Conference on Supercomputing - SC ’06 (New York, New York, USA: ACM Press) ,; 2006. Molecular dynamics—scalable algorithms for molecular dynamics simulations on commodity clusters; p. 43. Chemistry. [Google Scholar]
- Braun F., Lütgehetmann M., Pfefferle S., Wong M.N., Carsten A., Lindenmeyer M.T., Nörz D., Heinrich F., Meißner K., Wichmann D., et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet. 2020;396:597–598. doi: 10.1016/S0140-6736(20)31759-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalla D. Predictive methods in drug repurposing: gold mine or just a bigger haystack? Drug Discov. Today. 2013;18:523–532. doi: 10.1016/j.drudis.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Chang G.G. Molecular Biology of the SARS-Coronavirus. Springer; 2010. Quaternary structure of the SARS coronavirus main protease; pp. 115–128. [DOI] [Google Scholar]
- Chauhan A., Kalra S. Identification of potent COVID-19 main protease (MPRO) inhibitors from flavonoids. Preprints. 2020 [Google Scholar]
- Chen H., Cheng F., Li J. IDrug: integration of drug repositioning and drug-target prediction via cross-network embedding. PLoS Comput. Biol. 2020 doi: 10.1371/journal.pcbi.1008040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.W., Yiu C.P.B., Wong K.Y. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CLpro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res. 2020 doi: 10.12688/f1000research.22457.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo O.S., Yoon D., Choi Y., Jo S., Jung H.M., An J.Y., Choung Y.H. Drugs for hyperlipidaemia may slow down the progression of hearing loss in the elderly: a drug repurposing study. Basic Clin. Pharmacol. Toxicol. 2019;124:423–430. doi: 10.1111/bcpt.13150. [DOI] [PubMed] [Google Scholar]
- Choudhary S., Malik Y.S., Tomar S. Identification of SARS-CoV-2 cell entry inhibitors by drug repurposing using in silico structure-based virtual screening approach. Front. Immunol. 2020;11:1664. doi: 10.3389/fimmu.2020.01664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani D., Dubrovin M., Gaponenko I., Kroeger W., Lane T.J., Mitra A., O’Grady C.P., Salnikov A., Sanchez-Gonzalez A., Schneider D., et al. Linac Coherent Light Source data analysis using psana. J. Appl. Crystallogr. 2016;49:672–679. [Google Scholar]
- Dinesh D.C., Tamilarasan S., Rajaram K., Bouřa E. Antiviral drug targets of single-stranded RNA viruses causing chronic human diseases. Curr. Drug Targets. 2019;21:105–124. doi: 10.2174/1389450119666190920153247. [DOI] [PubMed] [Google Scholar]
- Douangamath A., Fearon D., Gehrtz P., Krojer T., Lukacik P., Owen C.D., Resnick E., Strain-Damerell C., Aimon A., Ábrányi-Balogh P., et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020 doi: 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Driel T.B., Nelson S., Armenta R., Blaj G., Boo S., Boutet S., Doering D., Dragone A., Hart P., Haller G., et al. The ePix10k 2-megapixel hard X-ray detector at LCLS. J. Synchrotron Radiat. 2020;27:608–615. doi: 10.1107/S1600577520004257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duisenberg A.J.M. Indexing in single-crystal diffractometry with an obstinate list of reflections. J. Appl. Crystallogr. 1992;25:92–96. [Google Scholar]
- Durdagi S., Aksoydan B., Dogan B., Sahin K., Shahraki A. Screening of clinically approved and investigation drugs as potential inhibitors of COVID-19 main protease: a virtual drug repurposing study. ChemRxiv. 2020 [Google Scholar]
- Dutta S., Sengupta P. SARS-CoV-2 and male infertility: possible multifaceted pathology. Reprod. Sci. 2021;28:23–26. doi: 10.1007/s43032-020-00261-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Section D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- Friesner R.A., Banks J.L., Murphy R.B., Halgren T.A., Klicic J.J., Mainz D.T., Repasky M.P., Knoll E.H., Shelley M., Perry J.K., et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision Glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Gao X., Qin B., Chen P., Zhu K., Hou P., Wojdyla J.A., Wang M., Cui S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11:237–245.. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevorkov Y., Yefanov O., Barty A., White T.A., Mariani V., Brehm W., Tolstikova A., Grigat R.R., Chapman H.N. XGANDALF—extended gradient descent algorithm for lattice finding. Acta Crystallogr. Sect. A Found. Adv. 2019;75:694–704. doi: 10.1107/S2053273319010593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghahremanpour M.M., Tirado-Rives J., Deshmukh M., Ippolito J.A., Zhang C.H., Cabeza De Vaca I., Liosi M.E., Anderson K.S., Jorgensen W.L. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020;11:2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Snijder E.J. Viral cysteine proteinases. Perspect. Drug Discov. Des. 1996;6:64–86. doi: 10.1007/BF02174046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal B., Goyal D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020;22:297–305. doi: 10.1021/acscombsci.0c00058. [DOI] [PubMed] [Google Scholar]
- Grant B.J., Rodrigues A.P.C., ElSawy K.M., McCammon J.A., Caves L.S.D. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., Banks J.L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Harder E., Damm W., Maple J., Wu C., Reboul M., Xiang J.Y., Wang L., Lupyan D., Dahlgren M.K., Knight J.L., et al. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theor. Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Heymann D.L., Rodier G. Global surveillance, national surveillance, and SARS. Emerg. Infect. Dis. 2004 doi: 10.3201/eid1002.031038. 10/2/03-1038_article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695. doi: 10.1103/PhysRevA.31.1695. [DOI] [PubMed] [Google Scholar]
- Huang F., Zhang C., Liu Q., Zhao Y., Zhang Y., Qin Y., Li X., Li C., Zhou C., Jin N., et al. Identification of amitriptyline HCl, flavin adenine dinucleotide, azacitidine and calcitriol as repurposing drugs for influenza A H5N1 virus-induced lung injury. Plos Pathog. 2020;16:1–16. doi: 10.1371/journal.ppat.1008341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiye T., Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins: Struct. Funct. Genet. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- Ip A., Ahn J., Zhou Y., Goy A.H., Hansen E., Pecora A.L., Sinclaire B.A., Bednarz U., Marafelias M., Sawczuk I.S., et al. Hydroxychloroquine in the treatment of outpatients with mildly symptomatic COVID-19: a multi-center observational study. BMC Infect. Dis. 2021 doi: 10.1186/s12879-021-05773-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M.P., Pincus D.L., Rapp C.S., Day T.J.F., Honig B., Shaw D.E., Friesner R.A. A hierarchical approach to all-atom protein loop prediction. Proteins: Struct. Funct. Bioinformatics. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Jarada T.N., Rokne J.G., Alhajj R. A review of computational drug repositioning: strategies, approaches, opportunities, challenges, and directions. J. Cheminform. 2020 doi: 10.1186/s13321-020-00450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y., Shen G., Zhang Y., Huang K.-S., Ho H.-Y., Hor W.-S., Yang C.-H., Li C., Wang W.-L. Analysis of the mutation dynamics of SARS-CoV-2 reveals the spread history and emergence of RBD mutant with lower ACE2 binding affinity. BioRxiv. 2020 doi: 10.1101/2020.04.09.034942. 2020.04.09.034942. [DOI] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Johansson L.C., Stauch B., Ishchenko A., Cherezov V. A bright future for serial femtosecond crystallography with XFELs. Trends Biochem. Sci. 2017;42:749–762. doi: 10.1016/j.tibs.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi T., Joshi T., Sharma P., Mathpal S., Pundir H., Bhatt V., Chandra S. In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking. Eur. Rev. Med. Pharmacol. Sci. 2020;24:4529–4536. doi: 10.26355/eurrev_202004_21036. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R.J., Jha R.K., Amera G.M., Jain M., Singh E., Pathak A., Singh R.P., Muthukumaran J., Singh A.K. Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2021;39:2679–2692. doi: 10.1080/07391102.2020.1753577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D.W., Phillips G., O’Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020 doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Y., Singh H., Patel C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health. 2020;13:1210–1223. doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Mariani V., Morgan A., Yoon C.H., Lane T.J., White T.A., O’Grady C., Kuhn M., Aplin S., Koglin J., Barty A., et al. OnDA: online data analysis and feedback for serial X-ray imaging. J. Appl. Crystallogr. 2016;49:1073–1080. doi: 10.1107/S1600576716007469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Garcia J.M., Conrad C.E., Coe J., Roy-Chowdhury S., Fromme P. Serial femtosecond crystallography: a revolution in structural biology. Arch. Biochem. Biophys. 2016;602:32–47. doi: 10.1016/j.abb.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCammon J.A., Harvey S.C. Cambridge University Press; 1987. Dynamics of Proteins and Nucleic Acids. [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGonagle D., O’Donnell J.S., Sharif K., Emery P., Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020;2:437–445. doi: 10.1016/S2665-9913(20)30121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menéndez C.A., Byléhn F., Perez-Lemus G.R., Alvarado W., de Pablo J.J. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Sci. Adv. 2020 doi: 10.1126/sciadv.abd0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan N., Sakthivel R., Velmurugan D., Gromiha M.M. Computational studies of drug repurposing and synergism of lopinavir, oseltamivir and ritonavir binding with SARS-CoV-2 protease against COVID-19. J. Biomol. Struct. Dyn. 2021;39:2673–2678. doi: 10.1080/07391102.2020.1752802. [DOI] [PubMed] [Google Scholar]
- Niu C., Yin J., Zhang J., Vederas J.C., James M.N.G. Molecular docking identifies the binding of 3-chloropyridine moieties specifically to the S1 pocket of SARS-CoV Mpro. Bioorganic Med. Chem. 2008;16:293–302. doi: 10.1016/j.bmc.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984;81:511–519. doi: 10.1063/1.447334. [DOI] [Google Scholar]
- Petersen E., Koopmans M., Go U., Hamer D.H., Petrosillo N., Castelli F., Storgaard M., Al Khalili S., Simonsen L. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020 doi: 10.1016/S1473-3099(20)30484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.-H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell H.R., Johnson O., Leslie A.G.W. Autoindexing diffraction images with iMosflm. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013;69:1195–1203. doi: 10.1107/S0907444912048524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushpakom S., Iorio F., Eyers P.A., Escott K.J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., et al. Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019;18:41–58. doi: 10.1038/nrd.2018.168. [DOI] [PubMed] [Google Scholar]
- Ragab D., Salah Eldin H., Taeimah M., Khattab R., Salem R. The COVID-19 cytokine storm; what we know so far. Front. Immunol. 2020;11:1446. doi: 10.3389/fimmu.2020.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathnayake A.D., Zheng J., Kim Y., Perera K.D., Mackin S., Meyerholz D.K., Kashipathy M.M., Battaile K.P., Lovell S., Perlman S., et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Translational Med. 2020;12:1–11. doi: 10.1126/scitranslmed.abc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Kilianski A., Baez-Santos Y.M., Baker S.C., Mesecar A. Structural basis for the ubiquitin-linkage specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014;10:e1004113. doi: 10.1371/journal.ppat.1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D. US FDA approves new class of HIV therapeutics. Nat. Biotechnol. 2003;21:470–471. doi: 10.1038/nbt0503-470. [DOI] [PubMed] [Google Scholar]
- Root-Bernstein R. Human immunodeficiency virus proteins mimic human T cell receptors inducing cross-reactive antibodies. Int. J. Mol. Sci. 2017;18:2091. doi: 10.3390/ijms18102091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheik Amamuddy O., Verkhivker G., Tastan Bishop O. Impact of emerging mutations on the dynamic properties the SARS-CoV-2 main protease: an in silico investigation. J. Chem. Inf. Model. 2020 doi: 10.1021/acs.jcim.0c00634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelley J.C., Cholleti A., Frye L.L., Greenwood J.R., Timlin M.R., Uchimaya M. Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Computer-Aided Mol. Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Sherman W., Day T., Jacobson M.P., Friesner R.A., Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Sierra R.G., Laksmono H., Kern J., Tran R., Hattne J., Alonso-Mori R., Lassalle-Kaiser B., Glöckner C., Hellmich J., Schafer D.W., et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr. Section D Biol. Crystallogr. 2012;68:1584–1587. doi: 10.1107/S0907444912038152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra R.G., Gati C., Laksmono H., Dao E.H., Gul S., Fuller F., Kern J., Chatterjee R., Ibrahim M., Brewster A.S., et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods. 2016;13:59–62. doi: 10.1038/nmeth.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra R.G., Batyuk A., Sun Z., Aquila A., Hunter M.S., Lane T.J., Liang M., Yoon C.H., Alonso-Mori R., Armenta R., et al. The macromolecular femtosecond crystallography instrument at the linac coherent light source. J. Synchrotron Radiat. 2019;26:346–357. doi: 10.1107/S1600577519001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H., Parnham M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free Radic. Biol. Med. 2020;156:107–112. doi: 10.1016/j.freeradbiomed.2020.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su E.W., Sanger T.M. Systematic drug repositioning through mining adverse event data in ClinicalTrials.gov. PeerJ. 2017;5:3154. doi: 10.7717/peerj.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez D., Díaz N. SARS-CoV-2 main protease: a molecular dynamics study. J. Chem. Inf. Model. 2020;60:5815–5831. doi: 10.1021/acs.jcim.0c00575. [DOI] [PubMed] [Google Scholar]
- Thayer J., Damiani D., Ford C., Dubrovin M., Gaponenko I., O’Grady C.P., Kroeger W., Pines J., Lane T.J., Salnikov A., et al. Data systems for the Linac coherent light source. Adv. Struct. Chem. Imaging. 2017;3:3. doi: 10.1186/s40679-016-0037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V., Ivanov K.A., Putics Á., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E.J., Rabenau H., Doerr H.W., et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Ton A., Gentile F., Hsing M., Ban F., Cherkasov A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inform. 2020;39:1–8. doi: 10.1002/minf.202000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell D.A.J., Bynoe M.L. Cultivation of a novel type of common-cold virus in organ cultures. BMJ. 1965 doi: 10.1136/bmj.1.5448.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020;30:127377. doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020;60:3277–3286. doi: 10.1021/acs.jcim.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., He S., Deng W., Zhang Y., Li G., Sun J., Zhao W., Guo Y., Yin Z., Li D., et al. Comprehensive insights into the catalytic mechanism of Middle East respiratory syndrome 3C-like protease and severe acute respiratory syndrome 3C-like protease. ACS Catal. 2020;10:5871–5890. doi: 10.1021/acscatal.0c00110. [DOI] [PubMed] [Google Scholar]
- Weglarz-Tomczak E., Tomczak J.M., Talma M., Burda-Grabowska M., Giurg M., Brul S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021 doi: 10.1038/s41598-021-83229-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.A. Processing serial crystallography data with crystFEL: a step-by-step guide. Acta Crystallogr. Sect. D Struct. Biol. 2019;75:219–233. doi: 10.1107/S205979831801238X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.A., Kirian R.A., Martin A.V., Aquila A., Nass K., Barty A., Chapman H.N. CrystFEL: a software suite for snapshot serial crystallography. J. Appl. Cryst. 2012;45:335–341. [Google Scholar]
- White T.A., Mariani V., Brehm W., Yefanov O., Barty A., Beyerlein K.R., Chervinskii F., Galli L., Gati C., Nakane T., et al. Recent developments in CrystFEL. J. Appl. Crystallogr. 2016;49:688–689. doi: 10.1107/S1600576716004751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White T.A., Kirian R.A., Beyerlein K.R., Aquila A., Martin A.V., Galli L., Yoon C.H., Nass K., Zatsepin N.A., Barty A., et al. 2020. CrystFEL - Release Notes for Version 0.9.0. [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X.-Q., Grant B.J. Domain-opening and dynamic coupling in the α-subunit of heterotrimeric G proteins. Biophysical J. 2013;105:L08–L10. doi: 10.1016/j.bpj.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Zhao Y., Zhang F., Wang Q., Li T., Liu Z., Wang J., Qin Y., Zhang X., Yan X., et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clin. Immunol. 2020;214 doi: 10.1016/j.clim.2020.108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong N., Zhang S., Zou P., Chen J., Kang X., Li Z., Liang C., Jin C., Xia B. Without its N-finger, the main protease of severe acute respiratory syndrome coronavirus can form a novel dimer through its C-terminal domain. J. Virol. 2008;82:4227–4234. doi: 10.1128/JVI.02612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Hou Y., Shen J., Huang Y., Martin W., Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- Zmudzinski M., Rut W., Olech K., Granda J., Giurg M., Burda-Grabowska M., Zhang L., Sun X., Lv Z., Nayak D., et al. Ebselen derivatives are very potent dual inhibitors of SARS-CoV-2 proteases—PLpro and Mpro in in vitro studies. BioRxiv. 2020 doi: 10.1101/2020.08.30.273979. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Protomer A is displayed on the right while protomer B is displayed on the left. Domain I is colored in light blue, domain II is colored in cyan, and domain III is colored in dark salmon for both protomers A and B.
Data Availability Statement
The 3D electron density map of SARS-CoV-2 Main protease has been deposited in the ProteinDataBank under accession numbers 7CWB, 7CWC. This paper does not report original code. Any additional information required to reanalyze the data reported is available from the lead contact upon request.