

Graphical abstract
Keywords: Mutual exclusivity, Synthetic lethality, Cancer genomics, Computational genomics
Abstract
Mutual Exclusivity analysis of genomic aberrations contributes to the exploration of potential synthetic lethal (SL) relationships thus guiding the nomination of specific cancer cells vulnerabilities. When multiple classes of genomic aberrations and large cohorts of patients are interrogated, exhaustive genome-wide analyses are not computationally feasible with commonly used approaches. Here we present Fast Mutual Exclusivity (FaME), an algorithm based on matrix multiplication that employs a logarithm-based implementation of the Fisher’s exact test to achieve fast computation of genome-wide mutual exclusivity tests; we show that brute force testing for mutual exclusivity of hundreds of millions of aberrations combinations can be performed in few minutes. We applied FaME to allele-specific data from whole exome experiments of 27 TCGA studies cohorts, detecting both mutual exclusivity of point mutations, as well as allele-specific copy number signals that span sets of contiguous cytobands. We next focused on a case study involving the loss of tumor suppressors and druggable genes while exploiting an integrated analysis of both public cell lines loss of function screens data and patients’ transcriptomic profiles. FaME algorithm implementation as well as allele-specific analysis output are publicly available at https://github.com/demichelislab/FaME.
1. Introduction
Synthetic lethality (SL) is a type of genetic interaction in which simultaneous perturbation of two genes results in cell death [1]. Initial evidence of this process was found in Drosophila [2], and further studied in yeast models [3]. Importantly, SL plays a key role in cancer biology as it translates into ad hoc search for tumor cell vulnerabilities [1], [4] that can ultimately lead to novel cancer cell specific therapies [5]. Remarkable examples are represented by the use of PARP inhibitors for BRCA2 mutant ovarian and breast cancers [6], [7] and by PRMT5 inhibitors in the context of MTAP deletion [8], [9]. Over the past few years, additional SL relationships were nominated and validated in vitro and/or in vivo [10], [11], [12]. Genetic rearrangements of ERG were demonstrated to induce synthetic sickness upon point mutations (or inhibition) in SPOP in prostate cancer cell line models as well as in patients derived xenografts [13]. WRN showed to be selectively essential in multiple micro satellite instability cancer models [14]. In the light of the ever-increasing availability of large-scale genomic data of human tumors and the importance of SL interactions towards the ultimate goal of personalizing cancer patient’s treatment, extensive and comprehensive genomic searches are becoming mandatory. In fact, tumor data from large collections of genomic profiles can be agnostically explored to search for potential SL interactions through the identification of mutual exclusive (ME) aberrations (i.e. aberrations that co-occur significantly less often than expected); the underlying principle is that cancer cells that have lost both genes of an SL pair do not survive, thus SL interactions can be identified by analyzing somatic copy number alterations (SCNA) and somatic mutation data that co-occur significantly less than expected (ME events) [15]. Methods for detecting ME from large genomic datasets have been combined with transcriptomics approaches [16], [17], pathway and network analysis [15], [18], metabolic models [19], [20], and validated through cell-lines screenings and clinical outcome data [16], [17]. Although some methods with improved performance and statistical framework for ME detection have been proposed in the past years [21], [22], the hurdle of comprehensively testing for mutual exclusivity (or co-occurrence) of aberrations across the whole genome has not been addressed in a computational efficient manner. Some approaches leverage on protein–protein interaction networks [23] or mutations impact and molecular annotation [24] to reduce the computational burden. Optimization of computational performance becomes especially relevant when multiple types of aberrations are considered, therefore increasing the number of combinations to be tested. For instance, a gene can be perturbed by loss-of-function single nucleotide variants (SNV), by DNA loss of both alleles (homozygous deletion), or, in case of haploinsufficiency, by the loss of a single allele (hemizygous deletion); similarly, gain of function mutations and genomic gain of one or multiple copies of DNA can associate with oncogenic gene perturbation. Further, allele-specific copy number alterations can add complexity to this scenario as in the case of copy number neutral loss of heterozygosity, CN-LOH [25], recently proved as relevant in the context of cancer vulnerabilities [26]. A brute force approach for the agnostic detection of ME instances (or co-occurrence) while considering a spectrum of mutational states and their combinations would lead to a combinatorial explosion that affects its feasibility. Here we present FaME (Fast Mutual Exclusivity), a tool that addresses such efficiency problems. FaME, as opposed to other methods for ME detection [23], [24], can be applied to any binarized data and does not need any additional prior information. We demonstrate its performance by testing for ME across allele-specific copy number states from the TCGA collection, provide a catalog of ME pairs from 27 tumor type studies, and finally corroborate our approach towards SL interactions nomination through the integration of matched transcriptomics and cell line-based loss-of-function data.
2. Material and methods
The description of the core algorithm proposed in this work is reported in section 3 Calculation.
2.1. TCGA studies inclusion criteria and data preprocessing
Whole Exome Sequencing (WES) BAM files of the TCGA collection available on January 2018 were downloaded (N. BAM = 22,196) through the Genomic Data Commons (GDC) [27]). The study inclusion criteria [25] led to the selection of a total number of 8,183 normal-tumor pairs across 27 tumor types (see Table S1, S2). Briefly, samples were excluded if one of the following applied: kit annotation was ambiguous, gender information was not available, MuTect2 SNV calls from GDC were not available, the genetic distance check [28] for the verification of the correct annotation of paired samples failed, or no primary tumor data was available. If more than one pair of samples was available for the same patient, we retained the one with the highest tumor purity. Last, we excluded studies with < 60 patients, prior to tumor ploidy and purity correction [29]. Quality filters on tumor purity identified 4,950 pairs as adequate for downstream mutual exclusivity investigations exploiting allele specific profiles and stratification for allele specific ploidy (asP) [25]. The in house SPICE allele specific pipeline was applied to generate a comprehensive analysis of matched normal and tumor WES aligned data (https://github.com/demichelislab/SPICE-pipeline-CWL) and autosomal data were retained for downstream analyses. Briefly, leveraging the allele specific gene classification by CLONETv2 [29], we had extended the conventional five-level classification of copy number status (Amp, Gain, WT, Hemi del, Homo del) to a ten-level allele-specific copy number classification (Fig. S1, introducing CN-LOH (2,0), Gain-LOH (3,0 and 4,0), Amp-LOH (5+,0), Gain-Unb (3,1), and Amp-Unb (4,1; 4,2; 5,1; 5,2; 5,3;...). We label as unbalanced (Unb) all the events involving autosomal chromosomes in which the difference between the number of copies of the two alleles is greater or equal to two and each allele copy number is greater or equal to one. Expression data for each TCGA study were obtained from recount2 project [30] and scaled counts were used. Genomic data used in this work can be downloaded from https://github.com/demichelislab/SPICE-pipeline#pre-computed-data.
2.2. Tumor suppressor genes, oncogenes and druggable gene lists
Lists of tumor suppressors (TS) and oncogenes (OG) were obtained from Futreal et al and Zhao et al [31], [32]. For TS, only genes present in both lists were kept (Table S3). Druggable genes list is based to Pharmgkb database (https://www.pharmgkb.org/) [33] and Dgi database (http://www.dgidb.org/) [34] (Table S4). The utilized gene model is reported in Table S5.
2.3. Analysis of mutually exclusive pairs of genomic events
2.3.1. Analysis of experimentally validated SL pairs selected from the literature
First, we selected SL events from the literature by strictly considering manuscripts with in vitro and/or in vivo experimental validation (see Table S6). Although likely not exhaustive, this list provides SL events for a first benchmark of FaME. The FaME algorithm was applied to the set of 30 pairs and to random pairs of genes with similar aberration frequencies as comparison. We proceeded as follows: one thousand sets of pairs of random genes were generated from the SPICE processed data; genes on the same chromosome as the corresponding literature gene were excluded at each iteration. To control for the aberration frequency distributions, a similarity was calculated using the mean absolute error as distance and selecting the closest 10 genes, each one from a distinct cytoband. The number of significant results from the literature pairs was compared to the distribution of the percentage of significant pairs from the random sets using a T test statistics (p < 0.005). The allele-specific states Amp-LOH, Amp-Unb, Gain-LOH were excluded from the computations due to low incidence. Genes with low allele specific copy number (asCN) quality signal (corresponding to the lower 5th percentile of asCN available calls per genes) were excluded from the computations.
2.3.2. Tumor suppressors and druggable gene pairs genomic-transcriptomic integrated analysis
For the analysis of TS and druggable genes we proceeded as follows: 1. performed study-specific FaME analysis on diploid samples using hemizygous deletion and CN-LOH status; 2. significantly mutually exclusive pairs at the genomic level were then tested for corresponding transcripts. We therefore quantified for each transcript the 3rd quartile of the expression range by using aberrant samples only; those values were then used to subdivide the expression space, this time considering all the samples, in 4 quadrants (g1_low-g2_low, g1_low-g2_high, g1_high-g2_low, and g1_high-g2_high). 3. we performed a co-independence test and use Pearson’s residuals to define gene pairs for which the g1_low-g2_low expression quadrant is depleted.
2.3.3. Validation of genomic-transcriptomic nominated pairs on cell lines screen data
Synthetic lethal candidate pairs were analyzed using data from Project Achilles (https://doi.org/10.1101/720243); briefly, for each pair we stratified diploid cell-lines (defined using Picnic score [35]) by the status of the tumor-suppressor gene and extracted the CERES scores for the druggable gene [36]. CERES is a computational method to estimate gene-dependency levels from essentiality screens while accounting for the copy number–specific effect. For each tumor suppressor gene of a nominated pair, we then calculated the odds-ratio (OR) and the enrichment significance for the number of homo- and hemizygous deletions in cell-lines with CERES scores lower than the first quartile of the distribution of scores (i.e. the cells for which the presence of the gene is more essential) for the corresponding druggable gene with respect to all the other cell-lines.
2.3.4. Correction for multiple hypothesis
As FaME performs multiple tests, FDR correction is applied by default considering all tests in each run. In this study, for the explorative genome-wide exhaustive analyses, we applied FDR per tumor-type and aberration combination (Table S7) on all results with OR < 1 (i.e. putative ME pairs).
2.4. Benchmarks
2.4.1. FaME performance assessment
To compare FaME against a baseline runtime, we created a naive implementation of the core elements of the method (computation of contingency tables and Fisher’s exact tests). This naive implementation computes the contingency tables and iteratively tests one pair at a time without leveraging either one of matrix multiplication, fast Fisher’s test function or parallelism (it still uses R vectorized operations to compute the counts of the single tests). A single randomly generated 1000 × 1000 (“genes” x “samples”) matrix was used as input, so that about 30% of the cells are set to 1 while the rest are 0. For the comparative analysis against the naive implementation, the core of the FaME method (computation of contingency tables and Fisher’s exact tests) was run on the same input and without any parallelism.
As both the Fisher’s exact test and the Matrix multiplication are influenced by sample size (due to the Fisher’s exact test performing a number of iterations that depends on the values specified in the contingency table), we calculated the runtimes for the main phases of FaME while varying the number of samples. For each run we randomly generated a matrix of binary values (with probability to select 1 at 0.3 i.e. ~ 30% of the entries in the matrix are expected to be set to 1) of 1000 × 100000 “genes” × “samples” and used it as input for both the Matrix multiplication and the Fisher’s exact test. We subset the columns in order to change the number of samples. Both phases were run in parallel using 40 cores; each phase was run 10 times for each different setting.
We then ran an exhaustive evaluation of FaME performance while varying the number of samples and of genes. We created two input matrices sampled from pan-cancer TCGA data for both homozygous deletions and amplifications. The matrices were created to have size equal to the maximum combination that we tested (30000 × 10000 – genes × samples) and were then subset to simulate smaller datasets. We tested all the possible combinations of the following sizes genes: {10, 100, 1000, 10000, 15,000 20000, 25000, 30000} and samples: {1000, 2500, 5000, 10000}. For each combination, we ran the computation 10 times and we collected the duration of the computation. This test was run by using a linux HPC machine with 40 cores and 256 Gb of memory (we tested the runtime using 10 and 20 cores as well). The complete results of the performance characterization are available in Table S17 where we report all the runtimes for each combination of parameter and each replicate.
2.4.2. Comparison with other tools and databases
For the ME detection performance comparative analyses, we focused on de-novo approaches that do not require a-priori information such as protein interaction networks [37], in line with FaME. Specifically, we opted for WesME [38] and WeXt [39]. The former introduces a heuristic approach for the computation of ME between gene pairs, accounting for mutation frequencies. The latter implements a permutation-based method, aimed at the identification of modules of mutually exclusive genes. We tested FaME ability to identify mutually exclusive pairs (compared to WesME) and mutually exclusive triplets (compared to WeXt). To do that, we followed the work of Liu, Sisheng, et al. [24] and constructed in-silico binary matrices, with genes in rows and patients in columns, mimicking the presence or absence of mutations. At each of 100 iterations, we introduced a perfect mutually exclusive pair or triplet of genes aberrations and applied each of the three tools, ranking the output accordingly to each tool-specific ME measure. In the case of FaME, we considered (1- p_value)*sign(log(OR)) as ranking procedure to also distinguish between mutual exclusive or co-occurring observations. Tests' p-values were used for WesME and WeXt. We then explored the tools' ability to rank the in silico mutual exclusive event in the top ranked results, upon different coverage (i.e. fraction of patients with a mutation on either one of the mutually exclusive genes) settings and overall mutations probabilities. Every other mutation was added by sampling each gene mutation probability from a uniform distribution with a priori defined mean. For each setting, we generated matrices of 200 samples and 100 (for pairs) or 50 (for triplets) genes. As FaME is implemented to inspect gene pairs, we measured triplets ME as the mean OR of all pairs involved in each possible triplet, aggregating their p-values according to Fisher's method (https://doi.org/10.2307/2681650). We finally benchmarked time performance at each iteration (we included FaME triplet computation in the triplets benchmarking) and for different matrix sizes.
To inspect FaME's ability to detect ME events corresponding to known SL pairs, we downloaded collections of synthetic lethal and non-synthetic lethal pairs from the SynLethDb database [40] (at 14/07/2021). Considering the broad collection of pairs, we selected the top 100 synthetic-lethal and non-synthetic lethal pairs, based on the SynLethDb statistical score, to use as positive and negative controls. A pair was considered as identified if with p-value < 0.05 and OR < 1 in at least one tumor type. We evaluated FaME performance considering the combinations of the following aberration states: SNV, Homo-del, Hemi-del, CN-LOH, Gain-LOH, Amp-LOH.
2.5. A web app gene pairs visualizer
A web application based on the shiny R library was created to allow users to explore the processed genomic data and pair-wise mutual exclusivity between groups of selected genes within one or multiple tumor types. The app uses the R bindings for the python Altair library (a library to create interactive web plots based on the Vega plotting standard) to show an interactive visualization of the allele specific status of one or more selected genes. The user can select specific sets of samples using the lineup.js data interactive visualization. The app will show a representation of the chosen set of samples and aberrations for the selected genes. The plot shows the data with samples sorted to highlight potential relationships between the selected genes (either mutual exclusivity or co-occurrence). The web application computes the p-values of the Fisher’s exact tests between all the possible pairs of selected genes. Each sample is considered aberrant based on its status with respect to the aberration type(s) specified by the user and non-aberrant otherwise. Libraries include: Shiny: https://shiny.rstudio.com/, Altair for R: https://vegawidget.github.io/altair/; Altair for python: https://altair-viz.github.io/; Vega: http://vega.github.io/vega-lite/; Lineup js: https://lineup.js.org/. The web app gene pairs visualizer is available at the address http://apps.demichelislab.eu/fame. Fig. S8 shows an example of analysis performed using the web app.
3. Calculation
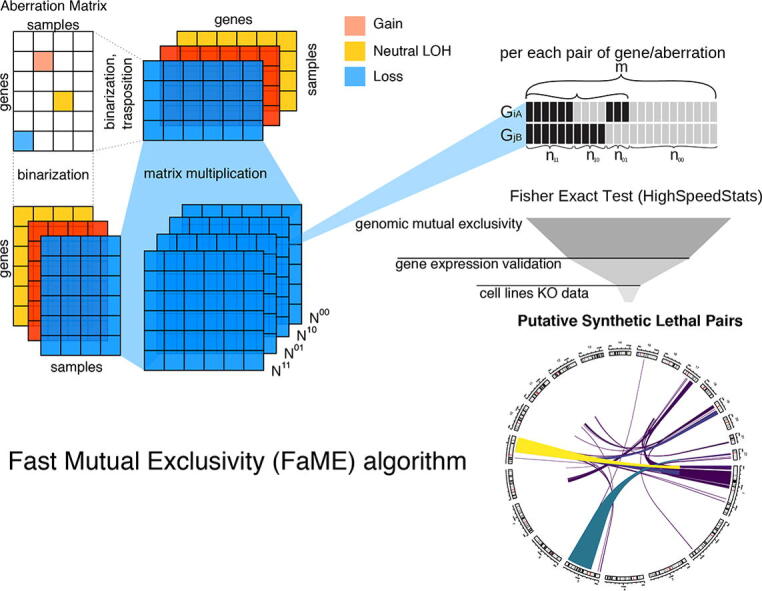
The ability of FaME to run mutual exclusivity tests for genomic aberrations on billions of gene pairs builds on few key elements. As testing for independence one gene pair at a time through the plain computation of a contingency table would require a prohibitive amount of time, we shifted the problem to a working framework that exploits matrix product computations (Fig. 1A) and leverages highly optimized libraries to considerably speed up such computation.
Fig. 1.

FaME schematics and performance (A) Example of computation according to the FaME method to compute independence tests between all possible gene pairs given a gene-based aberration matrix. Matrix multiplication is leveraged to compute the contingency tables upon binary transformations based on aberration type(s) of interest. A fast implementation of the Fisher’s exact test to compute the p-values is then applied. Direction indicates if the test returns a putative mutually exclusive pair (ME, OR < 1) or a co-occurrent pair (CO, OR > 1). (B) Visualization of the runtime of a naive implementation of the method core via sequential Fisher’s exact tests versus the FaME implementation (based on the number of analyzed genes). Each test has been run 3 times for the naive Fisher’s exact test and 10 for FaME. The points show the mean values. The curves behind the points are a loess regression fitted through the points. (C) Computation time per pair as a function of the number of tested genes and samples. By increasing the number of genes to test, the overhead of the computation is distributed among more pairs. (D) Visualization of the average and standard deviations of the execution time of 10 FaME runs (not considering I/O time) when either the number of tested samples or genes is changed.
Specifically, we first express the aberration status for a set of samples as a binary vector, where 1 indicates aberrant and 0 non-aberrant status. By computing the dot product of two such vectors, the result will correspond to the number of co-occurring elements (i.e. the number of samples in which both genes are aberrant). Formally, let be a binary variable that is equal to 1 when the k-th sample, with , harbors an aberration on the m-th gene and 0 otherwise and let be the vector of aberration statuses of all samples for the m-th gene. To count how many aberrations are co-occurring between genes and where , one can simply compute the dot product of the gene vectors, as , with product is equal to 1 only when both and are equal to 1. In order to compute the other components of a contingency table, we applied proper inversion of the aberration vectors. For instance, to compute the number of samples that are non-aberrant in either gene, we compute , whereby only samples where both and are equal to 0 will contribute to the result. Similarly, by computing and , we can obtain the number of samples where only either the first or the second gene is aberrant. In summary, we can compute all the elements of a contingency table by performing the following four dot-products of the gene aberration vectors:
where is a vector with the same length as the where all the elements are set to 1.
To further improve the computation speed (performance) of the algorithm, we used binary aberration matrices and their products instead of vectors; indeed, each element of a matrix product results from the dot product of the corresponding row and columns of two input matrices.
Formally let
be the binary matrix that describes the aberration status for all the genes to test, when considering aberration type A. Each row contains the aberration status for all the samples of a specific gene and each column contains the aberration status for all the genes in a specific sample. To compute the number of co-occurring samples when we compare aberration type to aberration type for all the combination of genes, we can compute the matrix multiplication .
The result will be the matrix:
Similarly, we can compute the contingency tables for all the pairs of genes using the following four matrix multiplications:
where denotes the matrix where each element has value equal to .
will correspond to the number of samples where both genes are aberrant, only the first gene is aberrant, only the second gene is aberrant, and both the genes are non-aberrant, respectively, for all the possible pairings of genes.
To account for occurrences where a specific gene aberration status is not possible to determine (i.e. NA), we redefine the so that when an aberration is undefined the binary variable is always equal to 0.
Formally
and, in order to properly exclude that instance from the contingency table calculation, similarly we set
These modified binary variables ensure that all the products of NA variables will be 0 and will never be counted.
To boost the performance of FaME in computing the matrix products, we use OpenBLAS (https://www.openblas.net/) instead of the default R BLAS library as it is highly optimized for the use of available hardware accelerations and parallelism to quickly compute the matrix multiplications. Further, the values (corresponding to a contingency table) are then used to compute the Fisher’s exact test p-values using the HighSpeedStats library, an R library that implements a very fast version of the Fisher’s exact test based on logarithm. Fisher's exact test is a statistical exact test used in the analysis of contingency tables: the test returns the sum of hypergeometric probabilities of all arrangements of the data that are equal or more extreme than the input contingency table, assuming the given marginal totals, on the null hypothesis that the two variables are independent. The test also returns the OR for each pair (denoted as “direction” in Fig. 1A), which can be used to discriminate ME pairs (OR < 1) from co-occurrent (CO) pairs (OR > 1).
Parts of the code of FaME are computed in parallel to accelerate the computation, although most of the computation time is due to memory allocation (large memory is required) that doesn’t benefit from multiple cores usage. The FaME’s method can be applied to any type of binary matrix, therefore testing for ME starting from any binary information such as gene SNVs, LOH state, epigenomic states. FaME speed will enable the exhaustive exploration of the entire genome and open the possibility to test different aberrations or combinations of aberrations. The current implementation of FaME does not have any command line parameter or different modes of execution and the algorithm is deterministic. The code of FaME is available at https://github.com/demichelislab/FaME.
4. Results
4.1. FaME computation performance
FaME provides a simple and fast solution to perform Mutual Exclusivity analysis of genomic aberrations based on the complete set of Fisher’s exact tests for all the possible combinations of input elements. The novelty of FaME is in the use of matrix multiplication to speed up the computation of the contingency tables thus providing powerful boost (about 100 fold) in terms of computational time with respect to the naive computational strategy of sequentially performing single Fisher’s exact tests (Fig. 1B). Moreover, exploiting matrix multiplication and vectorized operations, we are able to decrease the computation time for each pair by drastically reducing calculation overhead. For instance, upon increase of the number of tested genes from 10 to 10,000, the computing time goes from 139.6 to 0.0007 ms (on a set of 2,500 samples) (Fig. 1C). Further, the computation on 40 cores for 900 million gene pairs is comparable between 10 and 1,000 samples (612.9 s and 635.5 s) and increases less than double for 10,000 samples (1,032.4 s) (Fig. 1D). When increasing the number of samples, both matrix multiplication and Fisher’s exact tests become more computationally intensive (Fig. S2D), since the calculation of the Fisher’s exact test is based on a number of iterations that depends on the values in the contingency table. Compared with other tools for mutual exclusivity detection using synthetic data, as shown in Fig. S3, FaME produces comparable results when identifying ME events, at the same time outperforming the other tools in terms of calculation time. To our knowledge, FaME is the first tool based on matrix multiplication allowing for fast detection of mutually exclusive events.
4.2. Application of FaME to literature-based SL gene pairs
We first sought to test the mutual exclusivity of genomic events corresponding to the set of 30 gene pairs previously reported in in vitro and/or in vivo studies as implicated in SL interactions (gene pairs and references in Table S6). Briefly, FaME was applied to the TCGA whole exome sequencing data upon in house processing; matched tumor/normal pairs of data were processed and adjusted for tumor ploidy and tumor purity to retain high quality data for downstream computations, while also including allele-specific copy number states such as CN-LOH. The complete cohort of tumor samples is depicted in Fig. 2A, which shows annotations for clinical and genomic variables, including the allele specific ploidy (asP) metric, which is used to stratify low ploidy (low asP), diploid and polyploid samples (high asP). The literature-based analysis resulted in the detection of mutual exclusivity evidence for 12 out of 30 pairs (40%), 7 of which at pan-diploid level (23.3% of tested pairs) (Fig. 2B, Fig. S4A). To verify the significance of this result, we computed one thousand iterations using random pairs of genes from pan-diploid samples while keeping alteration frequencies comparable to the set of 30 literature pairs and built the distribution of percentages of mutual exclusivity evidence. This showed that the results obtained by FaME on the 30 gene pairs result corresponded to the 99.99 percentile of the random distribution (Fig. S4B), largely supporting its significance beyond chance. Considering as input mutation matrices obtained from the combination of several mutational states, we compared the obtained results with SynLethDB [40], a database including evidences for synthetic lethal pairs, and observed that the introduction of LOH events improved the sensitivity of the tool ranging from 7% to 13%, while maintaining a good specificity (84 – 86% range) (Fig. S5).
Fig. 2.

Literature-based SL pairs analysis in the study cohort (A) Representation of main variables of 4,950 tumor samples across 27 tumor types from the study cohort. Processed data and variables as in Ciani et al. [25]. (B) Circos plot of literature-based synthetic lethal pairs; black lines highlight those scored as mutually exclusive by FaME in the study cohort (left). For each of the latter, a pan-cancer oncoprint color-coded based on tumor allele specific genomic status and significant tumor type (center) is shown. Boxplot report the statistics for mutual exclusivity of all genomic combinations in the specific tumor type or at the pan-cancer level (Sign. dataset column). Gain, hemi deletions, CN-LOH, SNVs were considered.
4.3. Genomic based detection of mutually exclusive gene pairs
We next leveraged the FaME algorithm to test for mutual exclusivity of gene pairs in different allele-specific aberrations combinations: a total of 4,836 runs were computed including all tumor types and all meaningful aberration combinations (Table S7). For each run all the combinations of genes were tested. Overall, 275 runs returned at least one significant ME genes pair at 10% FDR, 222 of which from diploid based analyses (Table S8, Table S9).
Only four significant results were detected that exclusively involved SNVs, in line with the modest frequency of non-synonymous point mutations within the overall landscape of genomic disruption of human tumors. Three and one ME SNV-based pairs were detected in Low Grade Glioma (LGG) and in Uveal Melanoma (UVM), respectively (Fig. S6); results included mutations of TP53 and CIC that have been recently observed in different subclones and involved in parallel evolution of LGG through multi-sampling analysis [41]. On the contrary, when including all types of genomic aberrations tested on a gene base, FaME returned more extensive significant results. As expected, based on the non-focal nature of the vast majority of somatic genomic lesions [42], we overall observed the involvement of genes that are adjacent to each other, therefore often contributing to signal that spans entire or sets of contiguous cytobands (Table S10). For instance, the mutual exclusivity search involving point mutations and/or partial or complete loss of DNA (i.e. combination Homo-del – Hemi-del - CN-LOH - SNV for loss-of-function events) returned significant (FDR < 10%) results for four tumor types, namely breast adenocarcinoma (BRCA), bladder adenocarcinoma (BLCA), glioblastoma (GBM), and low-grade glioma (LGG) (Fig. 3). While exploring the mutual exclusivity obtained for the loss of DNA/function aberrations, we observed the inclusion of certain gene pairs with published indications of SL. In the BLCA results, we observed loss-of-function mutations of RB1 and CDKN2A as mutually exclusive. These two genes are among the most frequently mutated in bladder cancer [43] and their SL relationship has been suggested in other tumor types, such as in lung cancer through knockdown of the RB1 gene in CKDN2A-mutant cell lines [44]. The BRCA significant pairs included mutual exclusivity between CDH1 and ROS1,CDH1 deficient tumors were suggested to carry SL relationships with inhibition of the tyrosine kinase ROS1 leading to new possible therapeutic strategies [45]. The GBM data analysis revealed mutual exclusivity between TP53 and CDKN2A,RB1 and CDKN2A and IDH1 and PTEN, as already observed using an information theoretic method to identify combinations of mutations that promote glioblastoma [46] (https://doi.org/10.1093/jmcb/mjv026). Notably, IDH1 mutations are mutually exclusive with loss of heterozygosity of chromosome 10q; the former associated with good prognosis and the latter to bad prognosis in glioblastoma and gliomas [47], [48].
Fig. 3.

Mutual exclusivity search results for loss of function related aberration types in diploid tumors. The combination Homo-del, Hemi-del, CNLOH, SNV, returned significant results (FDR < 10%) for four tumor types, namely breast adenocarcinoma (BRCA), bladder adenocarcinoma (BLCA), glioblastoma (GBM), and low grade glioma (LGG, not shown). Red highlighted lines correspond to the gene pairs reported for each tumor type. Full genomic pairs data is available in Table S8. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Although, this analysis certainly includes a wide set of signals related to the presence of tumor subtypes and therefore not related to potential SL interactions, we reasoned that mutual exclusivity analysis of genomic aberrations can serve as backbone for the nomination of yet unreported potential SL events and that data integration through multiple omics layers can help shortlist and prioritize the best candidates.
4.4. Integrated characterization of nominated mutually exclusive gene pairs
We built FaME to perform exhaustive fast ME analyses of genomic data. As combination with other sources of biological information could guide the filtering and prioritization of the results, we here opted to pursue a case study focused on the integrated analysis on TS and druggable gene (Table S3 and S4) pairs. Such events could in principle be exploited in therapeutic frameworks and, given the relatively high frequency of TS loss, become relevant for a significant fraction of patients. In particular DNA repair genes, a specific class of TS genes, have gained attention for their therapeutic potential after the identification of the synthetic lethality between BRCA1/2 loss and PARP inhibition [11], [49].
First, through a diploid samples based analysis of LOH events (CN-LOH and Hemi-del) of TS and of alterations in druggable genes, FaME identified 23,080 (Fisher’s exact test, FDR < 0.05; 0.2% of total analyzed combinations) study-specific mutually exclusive relationships. Of note, 1,714 emerged only by leveraging CN-LOH status data (Table S11). As ME gene pairs related to SL interactions rarely show concomitant impairment at the transcript levels [21], we queried the corresponding transcript levels to ultimately shortlist gene pairs where concomitant expression impairment is rarely/never observed (Table S12, S13 and S14) and found evidence for mutual exclusivity at both genomic and the transcriptional levels for 1,027 gene pairs (4.6% of the genomic level combinations) (Fig. 4A).
Fig. 4.

Allele-specific status elicits the identification of mutually exclusive alterations (A) FaME workflow of mutually exclusive pairs of tumor suppressors and druggable genes at the genomic and transcriptomic level in the study cohort. Circos plot of significant mutually exclusive pairs at the genomic level; colors indicate whether a pair is detected using either Hemi-del or CN-LOH lesions or both. (B) Validation of FaME output in independent cell line database using Project Achilles data. (C) A representative pair is shown based on OR and significance. The rainfall plot of the dependency scores (CERES scores) upon KO of the druggable gene in diploid cells color coded by the tumor suppressor genomic copy number status (top) and gene expression level density plot across samples (log scaled RPKM values) (bottom) is shown; dotted lines delineate the quadrant of concomitant low expression (lower left quadrant) based on FaME mutually exclusive genomic events.
To seek orthogonal support towards SL involvement for the nominated pairs, we turned to a totally independent source of experimental cell line-based knockouts data from Project Achilles [50]. Specifically, for each pair we focused on the dependency results for the druggable gene, while stratifying for the genomic status of the corresponding TS (Table S15). We detected independent significant support (Chi-squared, FDR < 0.1, OR > 1) for 20 of the pairs initially nominated by FaME at both genomic and transcriptomic level (Fig. 4B, Fig. S7, Table S16). The shortlist includes the TP53 and CAMK2G pair with both genes previously associated with survival and time to progression in glioblastoma patients in multiple studies [51], [52], [53] and identified as key genes characterizing colon and rectal cancer [54].
Furthermore, it includes the ATM and PPIH druggable gene pair (Fig. 4C), a potential novel vulnerability for cancer patients with ATM deficiency (mutated in 5–20% of human tumors based on TCGA data). ATM is a central regulator of the response to DNA damage, that demonstrated synthetically lethal interaction with PARP [55], although not all cancer patients with ATM loss respond to PARP inhibition [56]. It has been suggested that ATM mutated gene copies proportion, protein expression levels, and epigenetic regulation should also be considered when defining the selection criteria for PARP inhibitor-based therapy [57]. PPIH, a member of the protein family of cyclophilins with prolyl isomerase activity, interacts with spliceosomal proteins and is necessary for proper spliceosome assembly and catalytic function in vitro [58], [59]. Interestingly, ATM-dependent DNA repair pathway is also activated by transcription-blocking DNA lesions [60]. Given their function in transcription-related processes, we might speculate that impairment (either by mutation or pharmacological inhibition) of PPIH might reduce transcription processivity and induce DNA lesions, which would not be timely and correctly repaired by the cell in absence of ATM, thereby generating genotoxic stress and potentially inducing cell death.
These results highlight the potential of our approach that combines high-quality allele-specific data and correspondent transcript levels with fast and comprehensive genome-wide mutual exclusivity calculation.
5. Discussion
We propose a framework for the fast computation of mutual exclusivity that allows to efficiently test billions of genomic aberration pairs. We here demonstrate that through the application of FaME on allele-specific genomic data we recapitulate previously reported SL gene pairs and provide proof of concept that an exhaustive mutual exclusivity genomic analysis coupled with matched transcriptomics can potentially shortlist novel SL candidates, although requiring ad hoc experimental validation. Multiple biological scenarios not related to SL could also lead to the detection of mutual exclusive genomic aberrations. These include instances of distinct cancer subtypes, each characterized by specific genomic features resulting from different cells of origin or initiating mechanisms that drove tumorigenesis. Furthermore, in the presence of a true positive SL pair, a brute force gene-based analysis might also nominate a set of false positives due to genomic proximity and aberrations that span multiple genes. Further, any data driven search for mutual exclusivity is limited by the size of the cohort and by the frequencies of the inspected aberrations. When a large set of tests is performed, the multiple hypothesis correction can penalize the nomination of SL related pairs. One such example of pairs that did not emerge through the integrated characterization analysis due to thresholds on FDR is the case of SPOP mutation and ERG translocation (proxied by interstitial deletions; (p-value 0.00236, FDR at 13%)) that was recently reported in prostate cancer [13]. Although a brute force genomic based approach likely returns multiple hits that do not relate to SL events per se, we expect that the ability to exhaustively inquire large well characterized genomic datasets provides the backbone for the identification of therapeutic targets for anticancer therapy [5], [11], [49], [61]. Interestingly, the application of FaME can be potentially extended to any binary data representing gene states thus allowing for fast and efficient integration of multi-omics information.
6. Conclusions
We introduce FaME, a computational framework that allows for brute force exploration of mutual exclusivity across multiple fine-grained genomic alterations. Importantly, this strategy opens up to the interrogation of all gene pairs without requiring a priori selection regarding, for instance, the type of alterations considered, in order to relieve the computational burden. Thus, FaME empowers the nomination of mutually exclusive events in a previously uncharted territory and represents a valuable tool towards discovery of synthetic lethality interactions.
CRediT authorship contribution statement
Tarcisio Fedrizzi: Conceptualization, Methodology, Software, Data curation, Visualization, Writing – original draft. Yari Ciani: Conceptualization, Methodology, Data curation, Writing – original draft, Visualization, Writing – review & editing. Francesca Lorenzin: Conceptualization, Data curation, Writing – original draft. Thomas Cantore: Testing and Benchmarking, Writing – original draft, Writing – review & editing. Paola Gasperini: Conceptualization, Data curation, Writing – review & editing. Francesca Demichelis: Conceptualization, Methodology, Writing – original draft, Visualization, Writing – review & editing, Supervision, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank Francesco Orlando and all members of the Demichelis laboratory for fruitful input, Davide Prandi for technical input during the design phase of the study, and Alberto Inga for discussions on the study results. This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 648670; SPICE) to F.D.; from the Italian Ministry of University and Research (FARE Programme) to F.D.; from the National Cancer Institute (SPORE P50-CA211024) to F.D.; and from the Prostate Cancer Foundation Young Investigator Award (19YOUN16) to F.L..
Footnotes
Supplemental Information includes 8 figures and 17 tables. The FaME code is available at https://github.com/demichelislab/FaME. Table S9 contains the links to complete FaME results from the anayzed 27 TCGA datasets.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.08.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.O'Neil N.J., Bailey M.L., Hieter P. Synthetic lethality and cancer. Nat Rev Genet. 2017;18(10):613–623. doi: 10.1038/nrg.2017.47. [DOI] [PubMed] [Google Scholar]
- 2.Dobzhansky T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics. 1946;31:269–290. doi: 10.1093/genetics/31.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartwell L.H., Szankasi P., Roberts C.J., Murray A.W., Friend S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 4.Kaelin W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 5.Huang A., Garraway L.A., Ashworth A., Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov. 2020;19(1):23–38. doi: 10.1038/s41573-019-0046-z. [DOI] [PubMed] [Google Scholar]
- 6.McCann K.E., Hurvitz S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context. 2018;7:1–30. doi: 10.7573/1740439810.7573/dic.212540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirza M.R. The forefront of ovarian cancer therapy: update on PARP inhibitors. Ann Oncol: Official Journal of the European Society for Medical Oncology / ESMO. 2020;31:1148–1159. doi: 10.1016/j.annonc.2020.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Fedoriw A., Rajapurkar S.R., O'Brien S., Gerhart S.V., Mitchell L.H., Adams N.D. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell. 2019;36(1):100–114.e25. doi: 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 9.Kryukov G.V., Wilson F.H., Ruth J.R., Paulk J., Tsherniak A., Marlow S.E. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351(6278):1214–1218. doi: 10.1126/science:aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etemadmoghadam D., Weir B.A., Au-Yeung G., Alsop K., Mitchell G., George J. Synthetic lethality between CCNE1 amplification and loss of BRCA1. PNAS. 2013;110(48):19489–19494. doi: 10.1073/pnas.1314302110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farmer H., McCabe N., Lord C.J., Tutt A.N.J., Johnson D.A., Richardson T.B. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 12.Helming K.C., Wang X., Wilson B.G., Vazquez F., Haswell J.R., Manchester H.E. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. 2014;20(3):251–254. doi: 10.1038/nm.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernasocchi T., El Tekle G., Bolis M., Mutti A., Vallerga A., Brandt L.P. Dual functions of SPOP and ERG dictate androgen therapy responses in prostate cancer. Nat Commun. 2021;12(1) doi: 10.1038/s41467-020-20820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan E.M. WRN helicase is a synthetic lethal target in microsatellite unstable cancers. Nature. 2019;568:551–556. doi: 10.1038/s41586-019-1102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ciriello G., Cerami E., Sander C., Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22(2):398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R. Link synthetic lethality to drug sensitivity of cancer cells. Briefings in Bioinformatics. 2019;20:1295–1307. doi: 10.1093/bib/bbx172. [DOI] [PubMed] [Google Scholar]
- 17.Sinha S., Thomas D., Chan S., Gao Y., Brunen D., Torabi D. Systematic discovery of mutation-specific synthetic lethals by mining pan-cancer human primary tumor data. Nat Commun. 2017;8(1) doi: 10.1038/ncomms15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dao P. BeWith: A Between-Within method to discover relationships between cancer modules via integrated analysis of mutual exclusivity, co-occurrence and functional interactions. PLoS Comput Biol. 2017;13 doi: 10.1371/journal.pcbi.1005695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gatto F., Miess H., Schulze A., Nielsen J. Flux balance analysis predicts essential genes in clear cell renal cell carcinoma metabolism. Sci Rep. 2015;5:10738. doi: 10.1038/srep10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilsson A., Nielsen J. Genome scale metabolic modeling of cancer. Metab Eng. 2017;43:103–112. doi: 10.1016/j.ymben.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 21.Jerby-Arnon L., Pfetzer N., Waldman Y., McGarry L., James D., Shanks E. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell. 2014;158(5):1199–1209. doi: 10.1016/j.cell.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 22.Mina M., Raynaud F., Tavernari D., Battistello E., Sungalee S., Saghafinia S. Conditional selection of genomic alterations dictates cancer evolution and oncogenic dependencies. Cancer Cell. 2017;32(2):155–168.e6. doi: 10.1016/j.ccell.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Gao B., Zhao Y., Li Y., Liu J., Wang L., Li G. Prediction of driver modules via balancing exclusive coverages of mutations in cancer samples. Adv Sci (Weinh) 2019;6(4):1801384. doi: 10.1002/advs.v6.410.1002/advs.201801384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S. MEScan: a powerful statistical framework for genome-scale mutual exclusivity analysis of cancer mutations. Bioinformatics. 2021;37:1189–1197. doi: 10.1093/bioinformatics/btaa957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciani, Y. et al. Allele-specific genomics is an orthogonal feature in the landscape of primary tumors phenotypes., 10.2139/ssrn.3779554 (2021).
- 26.Nichols C.A., Gibson W.J., Brown M.S., Kosmicki J.A., Busanovich J.P., Wei H. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat Commun. 2020;11(1) doi: 10.1038/s41467-020-16399-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A. Toward a shared vision for cancer genomic data. The New England J Med. 2016;375(12):1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demichelis F. SNP panel identification assay (SPIA): a genetic-based assay for the identification of cell lines. Nucleic Acids Res. 2008;36:2446–2456. doi: 10.1093/nar/gkn089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prandi D., Demichelis F. Ploidy- and purity-adjusted allele-specific DNA analysis using CLONETv2. Curr Protoc Bioinformatics. 2019;67(1) doi: 10.1002/cpbi.v67.110.1002/cpbi.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collado-Torres L., Nellore A., Kammers K., Ellis S.E., Taub M.A., Hansen K.D. Reproducible RNA-seq analysis using recount2. Nat Biotechnol. 2017;35(4):319–321. doi: 10.1038/nbt.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao M, Kim P, Mitra R, Zhao J, Zhao Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Research 44, D1023-1031, 10.1093/nar/gkv1268 (2016). [DOI] [PMC free article] [PubMed]
- 33.Whirl-Carrillo M., McDonagh E.M., Hebert J.M., Gong L., Sangkuhl K., Thorn C.F. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cotto K.C. DGIdb 3.0: a redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018;46:D1068–D1073. doi: 10.1093/nar/gkx1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenman C.D. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics. 2010;11:164–175. doi: 10.1093/biostatistics/kxp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyers R.M. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng Y. Identifying mutual exclusivity across cancer genomes: computational approaches to discover genetic interaction and reveal tumor vulnerability. Briefings Bioinf. 2019;20:254–266. doi: 10.1093/bib/bbx109. [DOI] [PubMed] [Google Scholar]
- 38.Kim Y.A., Madan S., Przytycka T.M. WeSME: uncovering mutual exclusivity of cancer drivers and beyond. Bioinformatics. 2017;33:814–821. doi: 10.1093/bioinformatics/btw242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leiserson M.D.M., Reyna M.A., Raphael B.J. A weighted exact test for mutually exclusive mutations in cancer. Bioinformatics. 2016;32(17):i736–i745. doi: 10.1093/bioinformatics/btw462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo J., Liu H., Zheng J. SynLethDB: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acids Res. 2016;44:D1011–D1017. doi: 10.1093/nar/gkv1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abstracts from the 20th International Conference on Brain Tumor Research and Therapy, July 20-22, 2014, Lake Tahoe, California. Neuro Oncol 16 Suppl 3, iii1-52, 10.1093/neuonc/nou206.1 (2014). [DOI] [PMC free article] [PubMed]
- 42.Consortium, I. T. P.-C. A. o. W. G. Pan-cancer analysis of whole genomes. Nature 578, 82-93, 10.1038/s41586-020-1969-6 (2020). [DOI] [PMC free article] [PubMed]
- 43.Cancer Genome Atlas Research, N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315-322, 10.1038/nature12965 (2014). [DOI] [PMC free article] [PubMed]
- 44.Kim N. Differential regulation and synthetic lethality of exclusive RB1 and CDKN2A mutations in lung cancer. Int J Oncol. 2016;48:367–375. doi: 10.3892/ijo.2015.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bajrami I., Marlow R., van de Ven M., Brough R., Pemberton H.N., Frankum J. E-Cadherin/ROS1 Inhibitor Synthetic Lethality in Breast Cancer. Cancer Discov. 2018;8(4):498–515. doi: 10.1158/2159-8290.CD-17-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Melamed R.D., Wang J., Iavarone A., Rabadan R. An information theoretic method to identify combinations of genomic alterations that promote glioblastoma. J Mol Cell Biol. 2015;7(3):203–213. doi: 10.1093/jmcb/mjv026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daido S. Loss of heterozygosity on chromosome 10q associated with malignancy and prognosis in astrocytic tumors, and discovery of novel loss regions. Oncol Rep. 2004;12:789–795. [PubMed] [Google Scholar]
- 48.Deng L., Xiong P., Luo Y., Bu X., Qian S., Zhong W. Association between IDH1/2 mutations and brain glioma grade. Oncol Lett. 2018 doi: 10.3892/ol10.3892/ol.2018.9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 50.Tsherniak A. et al. Defining a Cancer Dependency Map. Cell 170, 564-576 e516, 10.1016/j.cell.2017.06.010 (2017). [DOI] [PMC free article] [PubMed]
- 51.Reddy S.P., Britto R., Vinnakota K., Aparna H., Sreepathi H.K., Thota B. Novel glioblastoma markers with diagnostic and prognostic value identified through transcriptome analysis. Clin Cancer Res. 2008;14(10):2978–2987. doi: 10.1158/1078-0432.CCR-07-4821. [DOI] [PubMed] [Google Scholar]
- 52.Serao N.V., Delfino K.R., Southey B.R., Beever J.E., Rodriguez-Zas S.L. Cell cycle and aging, morphogenesis, and response to stimuli genes are individualized biomarkers of glioblastoma progression and survival. BMC Med Genomics. 2011;4:49. doi: 10.1186/1755-8794-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van den Boom J., Wolter M., Kuick R., Misek D.E., Youkilis A.S., Wechsler D.S. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163(3):1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li J.N. Differences in gene expression profiles and carcinogenesis pathways between colon and rectal cancer. J Dig Dis. 2012;13:24–32. doi: 10.1111/j.1751-2980.2011.00551.x. [DOI] [PubMed] [Google Scholar]
- 55.Mateo J. DNA-repair defects and olaparib in metastatic prostate cancer. The New England J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marshall C.H., Sokolova A.O., McNatty A.L., Cheng H.H., Eisenberger M.A., Bryce A.H. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur Urol. 2019;76(4):452–458. doi: 10.1016/j.eururo.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang J, Beckta JM, Bindra RS. Re: Catherine H. Marshall, Alexandra O. Sokolova, Andrea L. McNatty, et al. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur Urol 2019;76:452-8. Eur Urol 76, e109-e110, 10.1016/j.eururo.2019.04.041 (2019). [DOI] [PMC free article] [PubMed]
- 58.Rajiv C., Davis T.L. Structural and functional insights into human nuclear cyclophilins. Biomolecules. 2018;8 doi: 10.3390/biom8040161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajiv C., Jackson S.R., Cocklin S., Eisenmesser E.Z., Davis T.L. The spliceosomal proteins PPIH and PRPF4 exhibit bi-partite binding. Biochem J. 2017;474:3689–3704. doi: 10.1042/BCJ20170366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tresini M. The core spliceosome as target and effector of non-canonical ATM signalling. Nature. 2015;523:53–58. doi: 10.1038/nature14512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chan D.A., Giaccia A.J. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov. 2011;10(5):351–364. doi: 10.1038/nrd3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.