

Abstract
Objectives
Developing a vaccine formula that alters the tumor‐infiltrating lymphocytes to be more immune active against a tumor is key to the improvement of clinical responses to immunotherapy. Here, we demonstrate that, in conjunction with E7 antigen‐specific immunotherapy, and IL‐10 and PD‐1 blockade, intratumoral administration of caerin 1.1/1.9 peptides improves TC‐1 tumor microenvironment (TME) to be more immune active than injection of a control peptide.
Methods
We compared the survival time of vaccinated TC‐1 tumor‐bearing mice with PD‐1 and IL‐10 blockade, in combination with a further injection of caerin 1.1/1.9 or control peptides. The tumor‐infiltrating haematopoietic cells were examined by flow cytometry. Single‐cell transcriptomics and proteomics were used to quantify changes in cellular activity across different cell types within the TME.
Results
The injection of caerin 1.1/1.9 increased the efficacy of vaccinated TC‐1 tumor‐bearing mice with anti‐PD‐1 treatment and largely expanded the populations of macrophages and NK cells with higher immune activation level, while reducing immunosuppressive macrophages. More activated CD8+ T cells were induced with higher populations of memory and effector‐memory CD8+ T subsets. Computational integration of the proteome with the single‐cell transcriptome supported activation of Stat1‐modulated apoptosis and significant reduction in immune‐suppressive B‐cell function following caerin 1.1 and 1.9 treatment.
Conclusions
Caerin 1.1/1.9‐containing treatment results in improved antitumor responses. Harnessing the novel candidate genes preferentially enriched in the immune active cell populations may allow further exploration of distinct macrophages, T cells and their functions in TC‐1 tumors.
Keywords: caerin peptide, CD8+ T cell, macrophage, quantitative proteomics, single‐cell RNA sequencing, TC‐1 tumor, tumor microenvironment
In this study, we demonstrate that the intratumoral injection of caerin peptides significantly increases the survival time of vaccinated and anti‐PD‐1‐treated TC‐1 tumor‐bearing mice, through modulating the heterogenicity of tumor‐infiltrating macrophages. The combined treatments result in more activated tumor‐infiltrating T cells.

Introduction
Human papillomavirus (HPV) infection accounted for 4.6% of 14 million new cancer cases reported worldwide in 2012, and HPV‐associated cancers comprise 29.1% of all 2.2 million infection‐related cancers, including nearly 100% of cervical cancers.1 Cervical cancers are the third‐most common cancer in women worldwide.2 Moreover, high‐risk HPV infection, especially HPV16 infection, is related to a fraction of head and neck epithelial carcinoma in both developed and undeveloped countries.3 HPV‐related cancers are most severe in developing countries where HPV prophylactic vaccination rates are low.
The use of immune checkpoint inhibitors has resulted in unprecedented rates of long‐lasting tumor responses in patients with a variety of solid tumors.4 However, immune checkpoint inhibitor monotherapy, such as PD‐12 and CTLA‐4 blockade,5 is less effective in the management of advanced cervical cancers.6 PD‐1 blockade combined with therapeutic vaccines synergises with each other to induce T‐cell‐mediated tumor control, as animal models have demonstrated,7 including HPV16 E6/7‐transformed TC‐1 tumor models.8 Clinical trials also showed synergetic results of immune checkpoint blockade and therapeutic vaccination.9
Cancer therapeutic vaccines aim at eliciting effector T cells, especially tumor antigen‐specific CD8+ T cells that target tumor cells, without affecting the normal cells or tissues as demonstrated in clinical trials, such as those against melanoma.10 However, vaccine‐induced regression of high‐risk HPV infection is related to high‐grade CIN lesions,11 but not established cervical cancers.12 Immunisation and simultaneous blocking of the cytokine interleukin‐10 (IL‐10) drastically increased vaccine‐induced antigen‐specific CD8+ T‐cell responses and improved tumor growth inhibition in a prophylactic setting compared with the same vaccination without IL‐10 signalling blockade.13 Tumor inhibition was also enhanced in a therapeutic setting by intraperitoneal administration of anti‐IL‐10 receptor antibodies.14
The immunosuppressive tumor microenvironment (TME) can dampen the function of tumor‐infiltrating effector T cells.15 The TME promotes the development of tumor‐associated macrophages, myeloid‐derived suppressive cells, B cells, and Th2 type T and regulatory T cells. TAMs are attracting increasing attention as they play key roles in tumor spread and in response to different therapies.16 TAMs can substantially accelerate the progression of untreated tumors and influence the efficacy of anticancer drugs, including checkpoint blockade immunotherapies.17, 18 Specifically, TAMs can assume opposing phenotypes and functions that are either tumoricidal (e.g. M1‐like MΦ) or tumor‐supportive (e.g. M2‐like MΦ).
Isolated from Australian amphibians, genus Litoria, host defence caerin peptides inhibited the proliferation of several different cancer cells, such as TC‐119 and HeLa,20, 21 and an additive effect was observed when they were used together. At concentrations non‐toxic to T cells, caerin 1.1 and 1.9 inhibited HIV‐infecting T cells within minutes post‐exposure, as well as the transfer of HIV from dendritic cells to T cells.22 Moreover, caerin 1.1 and 1.9 inhibited TC‐1 tumor growth in vivo when injected intratumorally, and the inhibition required an intact adaptive immune system.13, 20 The signalling of TNF‐α‐mediated apoptosis and T‐cell receptor was stimulated after HeLa cells were treated with the mixture of caerin 1.1 and 1.9.21 The activation of TCR pathway observed using proteomic analysis suggested that HeLa cells became more sensitive to T‐cell‐mediated killing.21
In this study, TC‐1 tumor‐bearing mice immunised with an HPV16 E7 peptide‐based vaccine containing anti‐IL‐10 receptor antibody and PD‐1 blockade were locally injected with a mixture of caerin 1.1 and 1.9 peptides. Tumor‐infiltrating lymphocytes (TILs) were isolated for scRNA‐seq analysis to reveal the cell types in TC‐1 tumor and the modulation of the TIL landscape by the immunotherapy containing caerin 1.1/1.9. Mass spectrometry‐driven quantitative proteomic analysis was performed to investigate the overall effect of the changes in TILs. Our study provides new insights into the heterogeneity of TILs and their functions in TC‐1 tumor, including novel markers to define immune‐activating macrophages and CD8+ T‐cell subpopulations, as well as the molecular mechanisms underlying TME modulation by caerin 1.1/1.9.
Results
Intratumoral injection of caerin 1.1/1.9 significantly increased the efficacy of a therapeutic vaccine combined with immune checkpoint inhibition therapy
In this study, we first found that the treatment with intratumoral injection of caerin 1.1/1.9 (molar ratio 1:1) in conjunction with PD‐1 blockade significantly increased the survival time of TC‐1 tumor‐bearing mice, compared with untreated mice or mice only treated with anti‐PD‐1 (Figure 1a, left). The weight of tumor was significantly reduced in mice receiving caerin 1.1/1.9 only or in combination with anti‐PD‐1, when compared to untreated mice or mice receiving anti‐PD‐1‐only treatment (Figure 1a, right).
Figure 1.
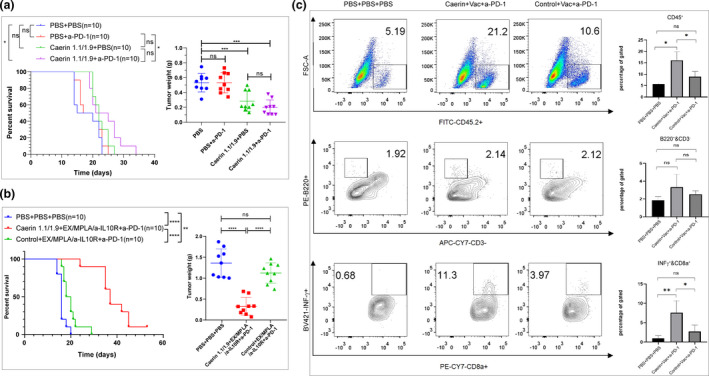
Groups of 5–10 C57BL/6 mice were subcutaneously transplanted with 5 × 105 TC‐1 tumor cells and subjected to different treatments. (a) On day 3, tumor‐bearing mice were treated with 300 µg of anti‐PD‐1, with 30 µg of caerin 1.1/1.9 (molar ratio 1:1) or with 300 µg of anti‐PD‐1 and 30 µg of caerin 1.1/1.9; a control group was administered with PBS only. Survival was monitored as described in the Methods. The experiment was performed twice, and the results shown are pooled from two independent experiments. Left: the survival rate of mice (no. of mice = 10). Right: on day 15, mice were sacrificed, and tumors were isolated and weighed (no. of mice = 9). (b) On day 3 after tumor challenge, mice were subcutaneously (s.c.) immunised with 40 µg of four overlapping HPV16 E7 peptides (EX) (10 µg per peptide), with 10 µg of monophosphoryl lipid A (MPLA), and with 300 µg of anti‐IL‐10R antibodies at days 3, 9 and 18, and with or without 300 µg of anti‐PD‐1 intraperitoneally (i.p.) on days 9 and 21 after tumor challenge. Additionally, tumor‐bearing mice were subjected to intratumoral injection of caerin 1.1/1.9 or a control peptide P3 from days 3 to 9. The experiment was performed twice, and the results shown are pooled from two independent experiments. Left: the survival curve of tumor‐bearing mice (no. of mice = 10). Right: On day 24, tumors were isolated and weighted (no. of mice = 9). (c) The flow cytometric results of tumor‐infiltrating CD45+ cells (upper panel), B cells (middle panel) and INF‐γ–secreting CD8+ T cells (lower panel). The results shown are from one of two independent experiments in (b).
Previously, we demonstrated in mice bearing an HPV16 E6/E7‐transformed TC‐1 tumor that blocking IL‐10 signalling at the time of immunisation, despite increasing antigen‐specific T‐cell numbers,23 did not affect the survival time of the mice.24 Here, 3 days post the tumor challenge with higher number (5 × 105) of TC‐1 cells, TC‐1 tumor‐bearing mice immunised with an HPV16 E7 peptide‐based vaccine containing anti‐IL‐10 receptor antibody and PD‐1 blockade were locally injected with a mixture of caerin 1.1 and 1.9 peptides (‘caerin’) or a control P3 peptide (‘control’). We found that the survival time of TC‐1 tumor‐bearing mice in the caerin group was significantly prolonged (Figure 1b, left) and the tumor weight was remarkably reduced by more than 50% (Figure 1b, right), compared with the untreated or the control group, respectively. Furthermore, the comparison of the three groups showed that CD45+ cells and interferon‐γ‐secreting CD8+ T cells were most abundant in the caerin group, while B cells were non‐significantly elevated (Figure 1c).
Single‐cell RNA‐seq identified six macrophage populations in TC‐1 tumor
Three groups of mice were subjected to tumor challenge and then treated for 30 days (Figure 2a, top). The two treatment groups showed significantly reduced tumor mass when compared to the untreated group, with an average reduction of 69% (caerin, P = 0.0011) and 42% (control, P = 0.0352). ELISpot results derived from the spleen and draining lymph nodes of individual mice (n = 3) in two groups demonstrated similar E7‐specific CD8+ T cells in the spleen and draining lymph nodes (Figure 2a, bottom).
Figure 2.
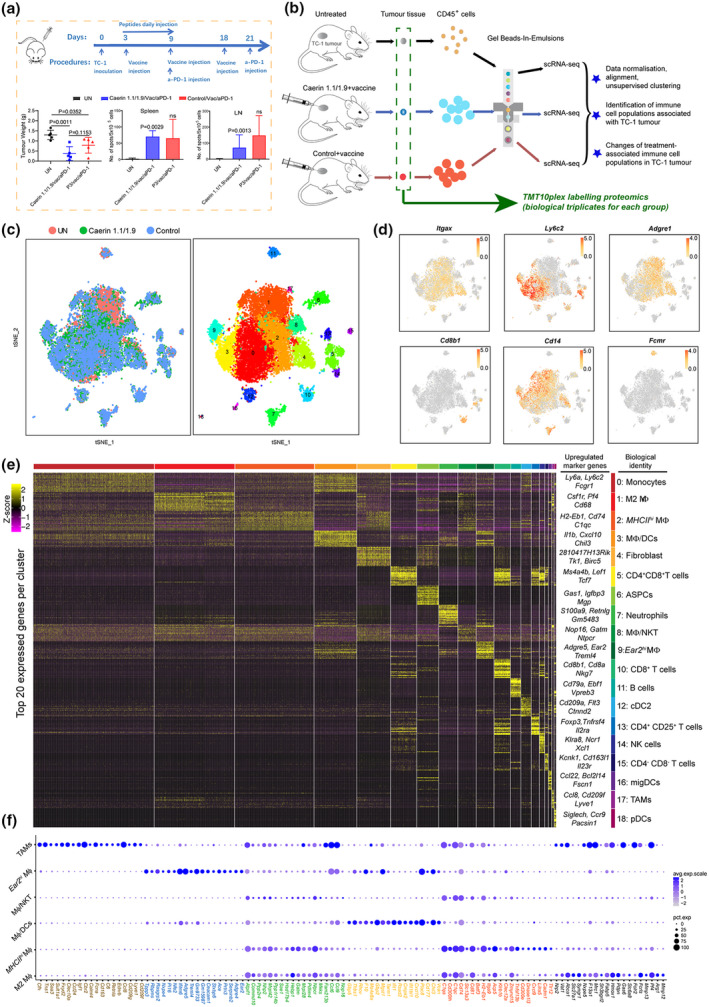
Identification of immune cell populations of TC‐1 tumor by single‐cell RNA sequencing. (a) Timeline of immunisation of TC‐1‐bearing mice. 30 days post‐tumor challenge, spleens and draining lymph nodes from control and immunised, and PD‐1‐blocked TC‐1 tumor‐bearing mice, treated with caerin 1.1/1.9 or a control peptide (P3), were isolated, and single cells were prepared and subsequently cultured in the presence of HPV16 E7 CTL epitope RAHYNIVTF overnight. ELISPOT assay was performed as described in the Methods. Left: Tumor weight; middle: splenic HPV16 E7‐specific CD8+ T cells; and right: draining lymph node HPV16 E7‐specific CD8+ T cells. The results shown are from one of two independent experiments (no. of mice = 5 in each group). (b) Schematic diagram of the experimental design (including injection procedure) and data processing. scRNA‐seq analyses were performed using pooled samples (n = 5 in each group). (c) t‐Stochastic neighbour embedding (t‐SNE) representation of aligned gene expression data in single cells extracted from untreated (n = 4011), vaccinated plus caerin 1.1/1.9 (n = 5685)‐treated or control (n = 5415) TC‐1‐bearing mice showing cellular origin (top) and partition into 19 distinct clusters (bottom). (d) Gene expression patterns projected onto t‐SNE plots of Itgax, Ly6C2, Adgre1, Cd8b1, Cd14 and Fcmr (scale: log‐transformed gene expression). (e) Heatmap showing the 20 most upregulated genes (ordered by decreasing P‐value) in each cluster defined in (c) and selected enriched genes used for biological identification of each cluster (scale: log2 fold change). MΦ represents macrophage; ASPC, adipogenic stem and precursor cell; NKT, natural killer T cell; cDC, conventional dendritic cell; NK, natural killer cell; migDC, migratory dendritic cell; TAM, tumor‐associated macrophage; and pDC, plasmacytoid dendritic cell. (f) Bubble map of marker gene expression distribution in different macrophage populations, including M2 MΦ, MHCIIhi MΦ, MΦ/DCs, MΦ/NKT, Ear2hi MΦ and TAM. The bubble size represents the ratio of the sum of the expression levels of the marker genes in a certain subpopulation to the sum of their expression levels in all cells, while the bubble colour represents the average expression of the marker genes in the cell population.
Total viable CD45+ leucocytes were isolated from tumors (Figure 2b and Supplementary table 1). Gene expression data from cells extracted from three groups were aligned and projected in a 2‐dimensional space through t‐stochastic neighbour embedding (t‐SNE) to identify tumor‐associated immune cell populations and the overlapping patterns (Figure 2c and Supplementary figure 1). There were 19 distinct cell clusters (clusters 0–18) detected by using the unsupervised graph‐based clustering method (see Methods) (Figure 2c through 2e; Supplementary figure 2). The presence of lymphocyte lineages was supported by the established canonical markers, such as Nkg7, Cd19, Fcmr, Cd8b1 and Cd79a, as well as myeloid cells that were determined by the identifications of Cd11c, Cd14, Cd68, Cd209a, Adgre1, Itgax, Csf1r, Lgals3, Ccr2 and Ly6c2.25, 26 The expression patterns and distribution of a few marker genes are shown in Figure 2d and e (see Supplementary table 2 for the full list of all marker genes detected).
Non‐macrophage cell populations included monocytes (cluster 0; marker genes: Ly6a, Ly6c2, Fcgr1 and Dpep2), neutrophils (cluster 7; Retnlg, S100a8, Cxcl2 and Hdc), B cells (cluster 11; Cd79a, Fcmr, Ly6d and Mzb1), conventional DC type 2 (cDC2) (cluster 12; Cd209a, Flt3, Ctnnd2 and Epcam),27 natural killer (NK) cells (cluster 14; Gzma, Xcl1, Ncr1 and Klrb1c), migratory DCs (migDC) (cluster 16; Ccl22, Bcl2l14, Fscn1 and Cacnb3),28 plasmacytoid dendritic cells (pDCs) (cluster 18; D13Ertd608e, Siglech, Ccr9 and Pacsin1)27 and four clusters with gene signatures suggesting various phenotypes of T cells (clusters 5, 10, 13 and 15). Fibroblasts (cluster 4; 2810417H13Rik, Tk1, Birc5 and Cdca3), and adipogenic stem and precursor cells (ASPCs) (cluster 6; Gas1, Igfbp3, Col3a1, Mgp and Cyr61)29 were identified as contaminants (Supplementary table 2).
The macrophage populations were examined in more detail, and the expression of the top 20 marker genes of each macrophage population was compared with their expression across all macrophage cell populations (Figure 2f). Specific gene expression patterns differentiating these MΦ populations and certain overlaps could be determined. Many of the top 20 marker genes of cluster 1, including Pf4, Arg1, Fabp5 and Mmp12, were associated with M2 MΦ, and similar numbers of these cells were detected in tumors treated with caerin peptides or control peptide (Supplementary figure 2c and d). The top three highly expressed genes with significance in cluster 2 were H2‐Eb1, Cd74 and C1qc (Supplementary table 2), confirming the MΦ signature. These genes marked MHCIIhi border‐associated MΦ in mouse brain.28 The high expressions of several H2 (MHCII) members, including H2‐Eb1, H2‐Ab1, H2‐Aa, H2‐DMb1 and H2‐DMa, were also confirmed in cluster 2; thus, we considered these as MHCIIhi MΦ hereafter. Cluster 3 had a mixed cell phenotype, including proinflammatory MΦ (Cxcl10, Gbp2 and Thbs1), Ly6chi ‐infiltrating MΦ (Chil3 and Plac8) and dendritic cells (Rsad2, Ifit1, Ifit2 and Ifi205), which was was labelled as MΦ/DCs. Cluster 8 was mainly composed of macrophages (Nop16, Gatm and Pf4) and NKT cells (Ntpcr, Mrpl28 and Commd10), and was identified as MΦ/NKT. Because of the exclusive high expression of Ear2, cluster 9 was assigned as Ear2hi MΦ (Chil3, Adgre5, Ace and Ifitm6).30, 31 Besides the signatures of MΦ (Adgre1, Csf1r, Fcgr1 and Cd68), cluster 17 also exhibited upregulated expression of chemokines (Ccl8, Ccl24, Ccl6 and Ccl9), suggesting the identity of TAMs, as Ccl8 and Siglec1 were detected as markers for TAMs in breast cancer,32 and both were highly expressed with the former being exclusive. The presence of other significantly upregulated genes, for example Cd209f, Clec10a and Cd163, indicated phenotype of activated M2 MΦ in TAMs.
We found that, in comparison with the untreated group, the two treatment groups had significantly increased populations of monocytes, MHCIIhi MΦ, MΦ/DCs, Ear2hi MΦ, CD8+ T cells and pDCs and substantially reduced M2 MΦ and B‐cell populations (Supplementary table 1; Supplementary figure 2c and d). CD8+ T cells appeared nearly exclusive to tumors from immunised animals, and the M2 MΦ population was reduced in these tumors, possibly because of the use of anti‐IL‐10 antibody in the immunisation, since it directly associates with the secretion of IL‐10.33 The numbers of MHCIIhi MΦ and NK cells were greatest in the caerin group. Notably, B cells were almost depleted in the caerin group (15/5,685 cells), when compared to the untreated group (194/4,011 cells) and the control group (87/5,415 cells) (Supplementary table 1).
Intratumor injection of caerin peptides significantly increased Arg1− tumor‐infiltrating macrophages
The six macrophages represented the largest cell populations (Supplementary figure 3), constituting 49.58%, 47.47% and 42.00% of total cells in the untreated, the caerin and the groups, respectively (Supplementary table 2). The correlation between each group of MΦs was derived based on the expression of significantly upregulated genes (Figure 3a). Notably, M2 MΦ and MHCIIhi MΦ were highly correlated with MΦ/NKT, suggesting close development of these MΦ lineages. The normalised cell numbers of different MΦs in the tumors were compared, and the M2 MΦ populations were reduced in the treatment groups by nearly 80% (Figure 3b). Injection of caerin largely induced MHCIIhi and Ear2hi MΦs in tumors, while reducing TAMs, when compared to the other two groups. M2 MΦ was reduced from 73.2% of macrophages in untreated tumor to 16.2% and 18.9% in the caerin and control groups, respectively (Figure 3c).
Figure 3.
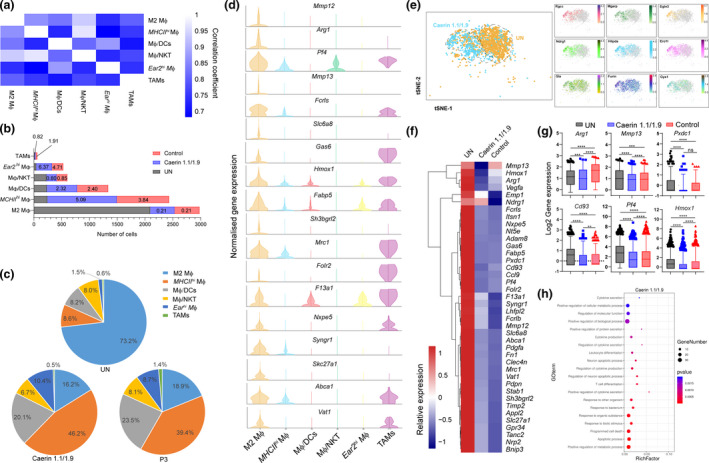
Immunisation increased immuno‐active macrophage populations. (a) Correlation analysis amongst six macrophage populations. (b) The comparison of the number of cells assigned to each macrophage in different groups (untreated, caerin 1.1/1.9 and control), and the fold change in cell numbers of two treatments relative to the untreated group was displayed on the bars. (c) The proportions of different macrophages in each group. (d) The comparison of the normalised expression of selected M2 MΦ marker genes and other macrophages. (e) t‐SNE representation of aligned gene expression data of M2 MΦ and selected genes (including Rgcc, Mgarp, Egln3, Ndrg1, Hilpda, Ero1l, Gla, Furin and Gys1) from untreated and caerin 1.1/1.9 groups. (f) Relative expression comparison of the top 40 marker genes of M2 MΦ amongst three groups. (g) Statistical analysis of the expression (log2 value) of Arg1, Mmp13, Pxdc1, Cd93, Pf4 and Hmox1. (h) Biological processes enriched in M2 MΦ population post the treatment containing caerin 1.1/1.9.
We next sought to unravel the phenotype and functions associated with the specific gene expression patterns of each macrophage. The expressions of key lineage‐associated genes of M2 MΦ were compared in parallel with their expressions in other macrophages (Figure 3d). Several marker genes appeared exclusive to M2 MΦ, such as Mmp12, Arg1, Mmp13 and Slc6a8, which were found to correlate with tumor angiogenesis and invasiveness.34, 35, 36 The role of Arg1 in immunosuppression has been described.37 Some marker genes of M2 MΦ were also highly expressed in TAMs, suggesting a similarity between these two MΦs in terms of cellular function.
The distribution of M2 MΦ in the untreated and the caerin groups was compared in Figure 3e. The expression of some marker genes, such as Rcgg, Ndrg1 and Egln3, aligned in a much greater extent with M2 MΦ in the caerin group. The relative expression of the top 40 marker genes of M2 MΦ was hierarchically clustered and compared amongst groups (Figure 3f). All genes were significantly downregulated in the caerin and control groups, except Mmp13 and Ndrg1. To evaluate the significance of this observation, the expression values of selected genes were further compared, where significant downregulation of Arg1, Mmp13, Pf4 and Hmox1 was present in the caerin group when compared to the control (Figure 3g). Most of the biological processes enriched in M2 MΦ represented by the marker genes unique to the caerin group were related to immune responses including apoptosis, responses to stimulus with organic substance, cytokine production and secretion, and T‐cell differentiation (Figure 3h). Different gene expression patterns relating to metabolism and transport of macromolecules (Supplementary figure 4b), and responses to heat and wounding (Supplementary figure 4c) were enriched in the untreated and the control groups.
MHCIIhi MΦ was significantly increased in the control and caerin groups, more significantly with the latter. The expression of selected marker genes of MHCIIhi MΦ was compared across six MΦs (Figure 4a), confirming Lira5, Cxcl9, Dnase1l3 and Cd300e as the signatures exclusive to MHCIIhi MΦ. The expression of Cadm1, Cxcl9 and Cd300e was increased in macrophages, and Dnase1l3 has been reported as a signature of CD141+CLEC9A+ DCs.38 In addition, Clec12a was recently found to be highly expressed in myeloid cells including macrophages and DC subsets.39 Thus, the MHCIIhi MΦ cluster displayed a characteristic of mixing phenotypes and its subpopulations were further investigated.
Figure 4.
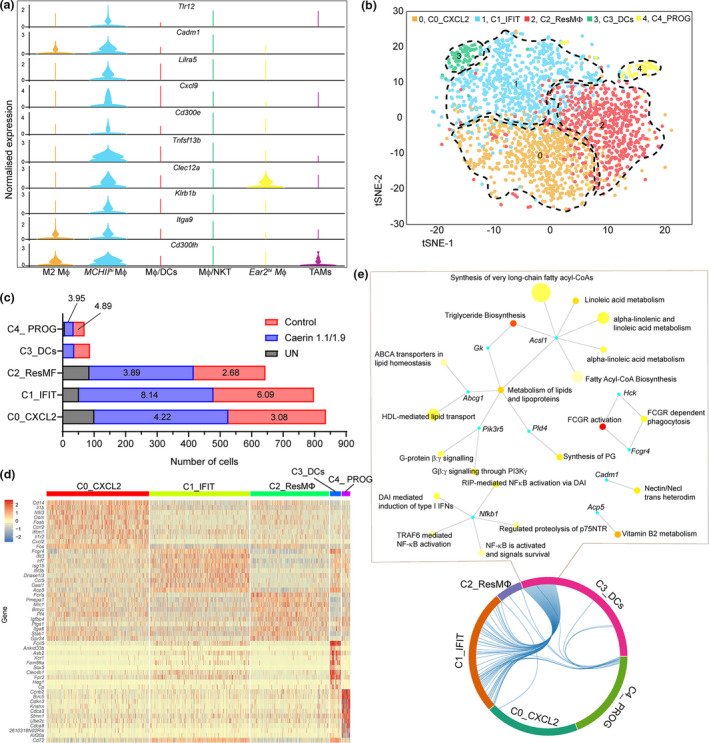
Immunisation in combination with PD‐1 and IL‐10 blockade, and the injection of caerin 1.1/1.9 or control changed the heterogeneity of MHCIIhi MΦ population. (a) Comparison of normalised expression of selected marker genes of MHCIIhi MΦ in all MΦ populations. (b) 2D t‐SNE distributions of five subpopulations of MHCIIhi MΦ. (c) Comparison of the cell numbers of subpopulations in the untreated, caerin or control groups. The fold changes of cell number in treatment groups with respect to the untreated group were displayed on the bars. (d) Heatmap showing the 10 most upregulated genes (ordered by decreasing P‐value) in each cluster defined in (b) and selected enriched genes used for biological identification of each cluster (scale: log2 fold change). (e) Biological process network based on genes that are commonly upregulated between subpopulations C1_MHCIIhi ‐IFIT and C3_MHCIIhi ‐DCs.
A total of five subpopulations (C0 to C4) of MHCIIhi MΦ were identified, with the C0, C1 and C2 possessing the highest cell numbers (Figure 4b). C3 was largely present in the control and caerin groups with similar cell numbers, while C4 was absent from the untreated group (Supplementary table 3). The normalised cell numbers of five subpopulations were compared (Figure 4c), and caerin peptide injection stimulated a much higher number of C0, C1 and C2 with respect to the other two groups. The expressions of the top 10 marker genes of each subpopulation were compared across all five subpopulations (Figure 4d). The first subpopulation, C0_MHCIIhi ‐CXCL2, characterised by Cxcl2, Nfil3 and Osm, had the phenotype of activated macrophages.40, 41, 42 The subpopulation C1 showed significant expressions of Ifit3b, Ifit3 and Ifit2, signatures of polarised M1 macrophages,43 suggesting proinflammatory function. In addition, C1 also showed significant upregulation of several other interferon‐induced protein‐relevant genes, which thus was labelled as C1_MHCIIhi ‐IFIT.
The third subpopulation had the lowest number of significantly upregulated genes compared with the other four subpopulations and was characterised by the expression of Fcrls, Ptgs1, Mrc1 and Igfbp4, the signature of resident‐like macrophages.28, 44, 45 Thus, this subpopulation is referred to as C2_MHCIIhi ‐ResMΦ. The fourth subpopulation, named C3_MHCIIhi ‐DCs, possessed the highest number of marker genes of DCs, such as Ankrd33b, Xrc1 and Asb2. Also, the signature of B cells, Fcrl5, was significantly expressed in C3_MHCIIhi ‐DCs, suggesting antigen‐presenting capacity. The fifth subpopulation was characterised by several marker genes of progenitor cells, such as Birc5,46 Cdkn3,47 Ccnb2 48 and Kif20a,49 which was referred to as C4_MHCIIhi ‐PROG. The H2 (MHCII) members were mainly elevated in C1_MHCIIhi ‐IFIT and C4_MHCIIhi ‐PROG.
The correlations amongst these subpopulations were evaluated based on the upregulated genes (Figure 4e). C1_MHCIIhi ‐IFIT and C3_MHCIIhi ‐DCs were correlated with a much higher degree. Genes that were shared between C1_MHCIIhi ‐IFIT and C3_MHCIIhi ‐DCs revealed biological processes mutually exerted by these two subpopulations, including the metabolism of lipids and lipoproteins, G protein signalling and FCGR activation. Reactome pathways based on unique marker genes were analysed (Supplementary figure 5). C0_MHCIIhi ‐CXCL2 showed enrichment in caspase‐mediated cleavage of cytoskeletal proteins, immune system, apoptotic cleavage of cellular proteins and apoptotic execution phase. The signalling of interferon, interferon gamma and cytokine in immune system were highly enriched in C1_MHCIIhi ‐IFIT. The pathways found in C2_MHCIIhi ‐ResMΦ were less significant (relatively high P‐values), such as GPCR ligand binding, chemokine receptors bind chemokines and collagen formation. C3_MHCIIhi ‐DCs showed enrichment in haemostasis, GPVI‐mediated activation cascade and adaptive immune system. Many cell cycle‐related pathways were found enriched in C4_MHCIIhi ‐PROG, which were less relevant to activating immune response (Supplementary table 3).
The populations of Ear2hi MΦ were remarkably elevated in the caerin group (Supplementary figure 6a). The distribution of cells expressing selected proinflammatory marker genes appeared to align well with Ear2hi MΦ in the caerin group, compared with untreated and the control groups. Significant upregulation of Ear2, Ace, Adgre4, Serpinb2 and Prtn3 in Ear2hi MΦ was identified in the caerin group (Supplementary figure 6b and c). A high degree of gene expression concordance was present amongst different groups, yet distinct biological processes were found (Supplementary figure 6d). Ear2hi MΦ showed the suppression of many biological processes, such as transferase activity, phosphorylation and cellular protein metabolism in untreated tumors, while the caerin group had activated cellular structure remodelling and immune response genes, including those suggesting myeloid cell differentiation.
Two treatments increased the population of MΦ/DCs
The MΦ/DCs population was significantly stimulated by the two treatments, which had 543 (9.55%) and 535 (9.88%) in the caerin and control groups, respectively, compared with only 165 (4.11%) in the untreated group (Supplementary table 1). We investigated the functions of MΦ/DCs by further dividing them into a total of four subpopulations (Supplementary table 4). The first subpopulation, characterised by Ccl5, Ccl12 and Slamf7, were strongly related to M1 polarisation macrophages50, 51, 52; thus, it was referred to as C0_MΦ/DC‐CCL5. The second subpopulation had the characteristic of stem/progenitor cells, supported by Cd24a,53 Htra3,29 Nr4a2 54 and Zfhx3 55; thus, we named it as C1_MΦ/DC‐CD24A. The third subpopulation had Prtn3,56 Il15 57 and Gstm1 58 as its marker genes, suggesting potential functions with similarity to neutrophils, which was referred to as C2_MΦ/DC‐PRTN3. The last subpopulation was named C3_MΦ/DC‐SLC7A11, since the marker genes, including Slc7a11,59 S100a8 60 and Wfdc21,61 were associated with DCs. C0_MΦ/DC‐CCL5 appeared highly enriched in immune response‐relevant biological processes, such as immune system process, innate immune response and defence response, many of which were also significant in C2_MΦ/DC‐PRTN3 (Supplementary table 4). The enrichment of response to wounding, inflammatory response and regulation of cytokine production was implied by the genes in C3_MΦ/DC‐SLC7A11. The stem/progenitor cell‐associated processes, such as ribonucleoprotein complex biogenesis, ribosome biogenesis and positive regulation of I‐κB kinase/NF‐κB cascade, were exclusively present in C1_MΦ/DC‐CD24A.
More CD8+ T cells infiltrated to TC‐1 tumor following vaccination and PD‐1 blockade, and CD8+ T cells were more activated in the caerin 1.1/1.9 treatment group
In the control and caerin groups, the population of CD8+ T cells infiltrating to TC‐1 tumor was 3.73% (caerin) and 4.58% (control) of the total CD45+ cells compared with only 0.27% in untreated tumors (Supplementary table 1). With peptide treatments, CD8+ T cells were more separated from the other three T‐cell populations in the 2D t‐SNE space (Figure 5a), indicating a possible variation in function. Out of all T‐cell types, the proportions of CD8+ T were 40% (caerin) and 37% (control) compared with 3.3% in the untreated group (Figure 5b). Most marker genes of CD8+ T cells showed much higher expression than other T‐cell populations (Figure 5c; see Supplementary table 5 for the expression of marker genes in other T cells), including genes that enhance the activation of CD8+ T cells, such as Ucp2, Fth1, Apoe, Fcer1g and Calm3.
Figure 5.
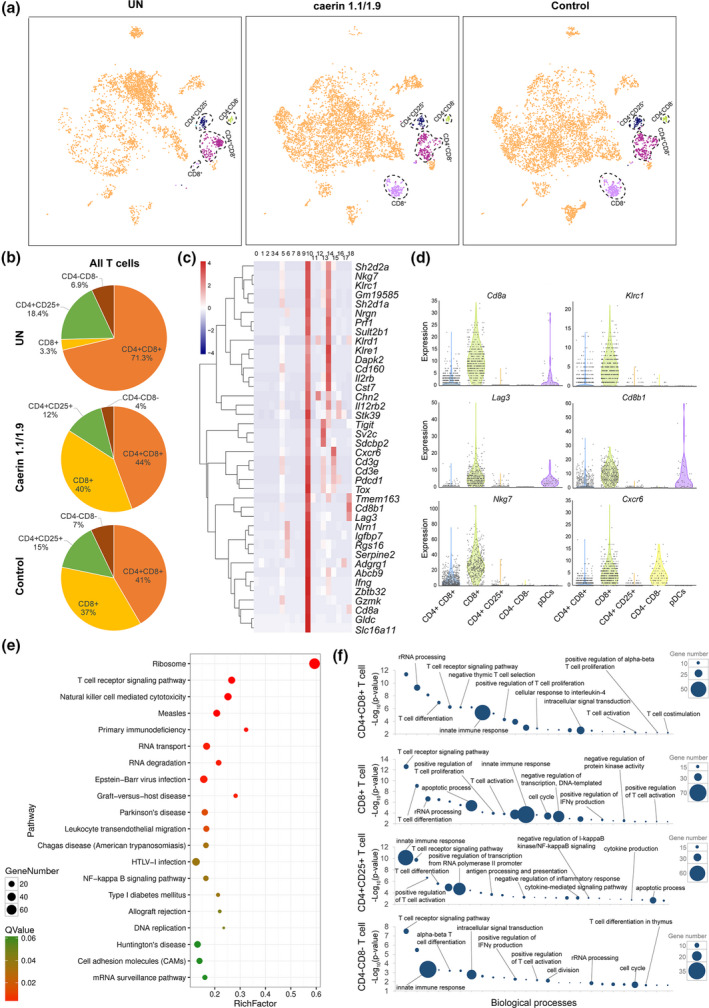
Comparison of TC‐1 tumor‐infiltrating T‐cell populations with the caerin 1.1/1.9 or control peptide treatments. (a) 2D visualisation of cell populations inferred from RNA‐seq data for all CD45+ cells in untreated and treated with caerin 1.1/1.9 or control; cell types with significant changes because of the treatments were featured. Colour coded as indicated. (b) Proportions of CD4+CD8+, CD8+, CD4+CD25+ and CD4‐CD8‐ cells within T cells extracted from the TC‐1 tumor. (c) Hierarchy clustering of the top 40 marker genes of CD8+ T cells in comparison with other types of T cells. (d) Violin plots compare the gene expression of selected genes showing statistically significant upregulation in CD8+ T cells to other T‐cell populations and pDCs, including Cd8a, Klrc1, Lag3, Cd8b1, Nkg7 and Cxcr6. (e) The top 20 KEGG pathways enriched in CD8+ T cells. (f) Gene ontology enrichment analysis of biological processes in T cells in the TC‐1 tumor. The top 25 enriched biological processes in four T‐cell subsets were compared in terms of P‐value and gene numbers, respectively.
Since pDCs present antigens (Ag) and induce immunogenic T‐cell responses through differentiation of cytotoxic CD8+ T cells and effector CD4+ T cells,62, 63 we compared the gene expression of signature genes of CD8+ T cells across four types of T cells and pDCs (Figure 5d). It shows that Cd8a, Klrc1 and Lag3 were almost exclusive to CD8+ T cells, while comparable expression of Cxcr6 was observed in CD4−CD8− T cells, and lower expression of Cd8a, Cd8b1 and Lag3 was observed in pDCs. Ribosome was determined to be the most enriched KEGG pathway, followed by T‐cell receptor signalling and natural killer cell‐mediated cytotoxicity (Figure 5e). Translation was found to be the most enriched process in all T‐cell populations except CD4+CD25+ and was thus excluded to highlight the difference amongst other enriched processes (Supplementary table 5). The top 25 enriched biological processes in CD4+CD8+, CD8+, CD4+CD25+ and CD4−CD8− T cells were compared (Figure 5f). Since these cells share similar T‐cell lineage development, overlaps of certain biological processes were observed, such as T‐cell differentiation and T‐cell receptor signalling, and innate immune response was observed as expected. However, T‐cell‐relevant processes were more enriched in CD8+ T cells suggested by lower P‐values than in the other three subtypes. Furthermore, there were a set of processes only enriched in CD8+ T cells, such as positive regulation of histone deacetylation, activation of cysteine‐type endopeptidase activity involved in apoptotic process and regulation of cytokine production (Figure 5f and Supplementary table 5).
The expression of marker genes, including Cd8a, Cd8b1, Tox, Lag3, Ifng, Nkg7, Nrgn, Gldc, Prf1, Abcb9, Nrn1 and Rgs16, was compared amongst three groups (Figure 6a), where significant upregulations of these genes induced by two peptide treatments were observed, except Nrn1 in the control group. In addition, Cd8a, Cd8b1, Tox, Ifng, Prf1 and Rgs16 were significantly elevated in the caerin compared with the control groups, suggesting that CD8+ T cells were more activated. The subpopulations of CD8+ T cells were further investigated to reveal the changes in heterogeneity because of peptide treatment, and five subpopulations (C0 to C4) were identified (Figure 6b and Supplementary figure 7). We found that the signatures representing naïve T cells, including Sell, Lef1 and Tcf7, had higher expression in untreated tumor (Figure 6c). Peptide treatment caused elevation of signatures for exhausted T cells, such as Tigit, Lag3, Tox and Pdcd1, while the effector T cells were stimulated suggested by the upregulation of Gzmb and Prf1. The average expression of the top 20 markers genes of five subpopulations was compared (Supplementary figure 7e), where the marker genes of subpopulations 2 and 3 were more exclusive.
Figure 6.
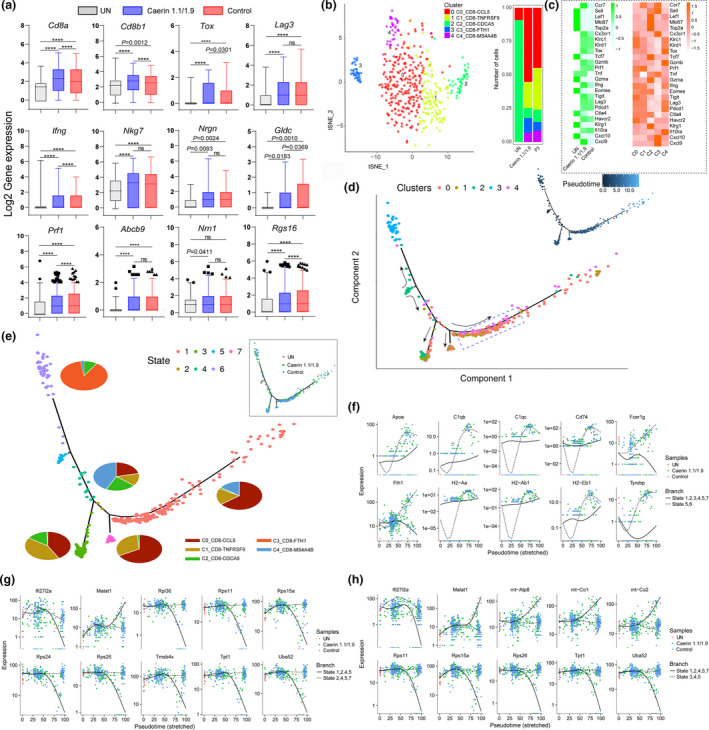
Comparison of CD8+ T‐cell marker genes with two treatments and state transition analysis based on integrated expression in subpopulations. (a) The expressions (log2 value) of Cd8a, Cd8b1, Tox, Lag3, Ifng, Nkg7, Nrgn, Gldc, Prf1, Abcb9, Nrn1 and Rgs16 in untreated tumors, treatments containing caerin 1.1/1.9 or control. (b) The analysis of subpopulations of CD8+ T cells and the number of cells under the aforementioned three conditions. (c) The heatmaps compared the average expressions of selected genes in the untreated and treated tumors, and in five subpopulations. (d) The ordering of CD8+ T‐cell subpopulations along pseudotime in a two‐dimensional state space is defined by Monocle3. Cell orders are inferred from the expression of most dispersed genes across CD8+ T‐cell subpopulations. Each point corresponds to a single cell, and each colour represents a T‐cell subpopulation. Cells on the same or neighbouring branches are expected to be more hierarchically related. (e) Developmental states of CD8+ T cells inferred by pseudotime. The space distribution of cells is defined in (b). Each colour represents a distinct state on the trajectory generated by Monocle. Pie charts show the proportion of cell clusters at the state when multiple clusters are involved. The top 10 genes differentially expressed in different states on branches 1 (f), 2 (g) and 3 (h) along with stretched pseudotime with respect to different treatments.
The first cluster C0_CD8‐CCL5 cells characterised by marker genes Ccl5,64 Cd3e,65 Cxcr6 66 and Gzmk 67 were considered as memory T cells. Most of the top 20 highly expressed genes in the second cluster were various ribosomal proteins, such as Rpl32, Rpl26, Rpl23 and Rpl28. It has been reported that translation is upregulated during effector CD8+ T‐cell expansion.68 In addition, Tnfrsf9 69 and Prf1 70 appeared to highly express in this subpopulation. Thus, these were likely effector CD8+ T cells and were named C1_CD8‐TNFRSF9. The third cluster, C2_CD8‐CDCA5, was characterised by significant upregulation of Cdca5, Cdc6 and Ccna2, commonly associated with dividing T cells.71 Additionally, several histones and regulators, including Tmsb10 and Ptma, were amongst those genes with the highest expression in C2_CD8‐CDCA5. The fourth cluster possessed more than 1,500 genes showing significant upregulation and was characterised by Fth1, Cd74 and Ifitm3. The relevance of Fth1 to CD8+ effector T‐cell response was reported, which revealed that it played an immunomodulatory role in cytokine signalling, adaptive immunity and cell death.72 The high expression of Cd74 and Ifitm3 was detected in memory T cell.67, 73 Thus, this cluster was composed of effector‐memory T cells, referred to as C3_CD8‐FTH1 hereafter. The remaining cell cluster, C4_CD8‐MS4A4B, was characterised by MS4a4B, Ly6a, Cd8b1 and Ly6e, and was naïve T cells. Notably, the CD8+ T cells of untreated tumors only contained two of five subpopulations, that is C0_CD8‐CCL5 and C2_CD8‐CDCA5, while the control and caerin groups induced all five subpopulations (Figure 6b; Supplementary figure 7a; Supplementary table 5). The caerin group possessed a higher number of C0_CD8‐CCL5 and C3_CD8‐FTH1 than the control.
We then projected CD8+ T cells onto the two‐dimensional state space defined by Monocle3 for pseudotime analysis, to obtain the information inferring lineage trajectories from expression data (Figure 6d). Most cells from each subpopulation aggregated based on expression similarities, and different clusters formed into a relative process in pseudotime that began with C2_CD8‐CDCA5 (dividing CD8+ T cells), then developed in separate directions, with one direction developing to C3_CD8‐FTH1 cells (effector‐memory CD8+ T cells). It appeared that C0_CD8‐CCL5 (memory CD8+ T cells), C1_CD8‐TNFRSF9 (effector CD8+ T cells) and C4_CD8‐MS4A4B (naïve CD8+ T cells) started to emerge at approximately similar pseudotime on the other direction, gradually overlapping on three branches along the pseudotime trajectory, two of which also included certain amount of C2_CD8‐CDCA5 cells, indicating functional divergence of this subpopulation. On these two branches, C2_CD8‐CDCA5 aggregated with C0_CD8‐CCL5 and C1_CD8‐TNFRSF9, which suggested a close correlation between regulatory, effector and memory CD8+ T cells, and different functions might be executed. C4_CD8‐ MS4A4B was diversely present together with C0_CD8‐CCL5 and C1_CD8‐TNFRSF9 along the pseudotime, especially at the middle area, implying their close association.
Seven states were thus identified based on pseudotime analysis (Figure 6e), where cells in transitional state 2 and state 5 exclusively corresponded to C0_CD8‐CCL5 and C2_CD8‐CDCA5, respectively. Most cells of states 1, 3 and 7 were C0_CD8‐CCL5, C1_CD8‐TNFRSF9 and C2_CD8‐CDCA5. A transitional state 4 was identified, which consisted of all clusters except C3_CD8‐FTH1. The predicted developmental trajectory was also confirmed by the marker genes with similar expression patterns, which hierarchically clustered these markers along the pseudotime in each state (Supplementary figure 8). The potential divergence of cell functions in different state cells was investigated (Supplementary table 5). Notably, state 1 showing a significantly elevation of genes enriched in the signalling pathways of IL‐2 and IL‐3, G protein and G13 at a later stage, and the caerin group had a higher population in this state than the other two groups (Figure 6d and Supplementary table 5). Apoptosis was the only pathway enriched in state 3, and signalling by EGFR1, chemokine and TGF‐β was present in state 7. The transition state 2 between states 3 and 7 had very different functions, such as the enrichments of macrophage markers, ApoE and miR‐146 in inflammation and atherosclerosis, and antigen processing and presentation. The presence of more state 6 cells at late pseudotime potentially in the caerin group correlated with the marker genes playing roles in TNF‐α, NF‐kB signalling and inflammatory response.
The genes significantly differentiating the branches were also analysed, with expression variation of top 10 genes along the pseudotime trajectory compared amongst different groups (Figure 6f–h). Most of these genes were expressed near pseudotime zero in the untreated tumors but had a significantly prolonged expression in the control or caerin group. During the first transition, the genes highly associated with the immune system, and their expression, declined at an early stage in states 5 and 6 possibly because of low cell numbers, then increased sharply onwards at pseudotime 25, where more cells expressing these genes were observed in the caerin group. There was a slow increase in expression of Apoe, C1qb, Cd74, H2‐Aa, H2‐Ab1 and H2‐Eb1 along the pseudotime on the branch involving states 1, 2, 3, 4, 5 and 7, which also correlated well with higher cell number stimulated in the caerin group (Figure 6f). The expression of Rps11, Rps15a, Rps36, Rps24 and Rps26 on the branch appeared downregulated along the pseudotime in states 1, 2, 4 and 5 (Figure 6g), indicating the deactivation of translation, which was also the case on the state 1, 2, 4, 5 and 7 branch (Figure 6h). In addition, the expression trend of these genes in states 3, 4 and 5 aligned well with the cell distribution in the caerin group.
Caerin 1.1/1.9 treatment remarkably reduced the numbers of tumor‐infiltrating B cells
We observed a cluster of B cells as a major tumor‐associated cell population, representing 4.84% (194 out of 4011 cells, the 5th highest population) of total CD45+ cells isolated from the untreated TC‐1 tumor (Supplementary figure 13a; Supplementary table 1). In contrast, the proportion of B cells decreased to 1.61% (87 cells) and 0.26% (15 cells) in P3 treatment and caerin 1.1/1.9‐treated groups, respectively (Supplementary table 1). The GO term analysis of B cells revealed enrichment of biological processes such as the activation (corrected P = 9.95E‐15), differentiation (corrected P = 8.27E‐07) and proliferation of B cells (corrected P = 3.49E‐10), regulation of B‐cell receptor signalling (corrected P = 3.53E‐04), regulation of antigen receptor‐mediated signalling (corrected P = 1.69E‐05) and regulation of immune system process (corrected P = 1.51E‐13) (Supplementary figure 13b). The KEGG enrichment analysis also revealed that the B‐cell receptor signalling pathway was the second most significant pathway (P = 5.76E‐06) following ribosome, while antigen processing and presentation was also enriched (P = 0.0282) (Supplementary figure 13c). The expressions of the marker genes in untreated and treated TC‐1 tumor were compared (Supplementary figure 13d), and indicated a significantly lower number of cells expressing these genes with two treatments, especially with the injection of caerin 1.1/1.9. The total expression of genes known to induce activation and/or proliferation of B cells, such as Blk, Cd19, Bcl11a and Cxcr5, was compared across three groups (Supplementary figure 13e); many of these genes seemed largely downregulated by the injection of caerin 1.1/1.9. The biological processes related to the upregulated genes uniquely present with caerin 1.1/1.9‐containing treatment mainly included a network‐centred endocytosis (Supplementary figure 13f), where the interactions with apoptotic process, immune system process and protein transport were identified.
Caerin 1.1/1.9 enhanced the infiltration of NK cells into TC‐1 tumor
The population of NK cells was elevated significantly by the treatment containing caerin 1.1/1.9, which had a cell number of 89, compared with that of 20 in untreated and 38 in P3 groups, respectively (Supplementary table 2). There was a certain extent of overlap in the distribution of marker genes between NK, CD4+CD8+ and CD8+ T cells (Supplementary figure 14a). The distribution of cells expressing selected marker genes with respect to different treatments was compared in Supplementary figure 14b, where significantly higher cell numbers were detected in caerin 1.1/1.9‐containing treatment than other two conditions. The pathways enriched in the marker genes of NK cells were highly relevant to immune response, such as signalling pathways of natural killer cell‐mediated cytotoxicity and the T‐cell receptor, and differentiation of Th1, Th2 and Th17 (Supplementary figure 14c). The expressions (log2 value) of selected marker genes in three samples were compared in Supplementary figure 14d, where significant upregulations of these genes were detected in NK cells of the caerin group. The identifications of Sell, Klrc1 and Gata3 amongst the upregulated genes with significance indicated CD56bright NK cells existing in this population74, 75; caerin further induced the expression of other marker genes of CD56bright NK, such as Impdh1, Inpp4b and Cd69. The ‘mature NK’ was present in this population, as supported by CTSW, Hpox and Manf. The presence of Pik3r1, Cd27 and Pdcd4 in this population also suggested a characteristic of ‘transitional NK’,76 potentially associated with the transition from CD56bright to CD56dim NK cells. Klrc2 has recently been discovered as the key marker for ‘adaptive NK’,76 and the treatment with caerin significantly upregulated the expression of Klrc2; other signatures of ‘adaptive NK’, including Id2, Cd7 and Klrd1, were also present in this population. Notably, the marker genes of ‘active’, ‘terminal’ and ‘inflamed’ NK cells reported previously were not confidently detected here. Thus, this population of NK cells represent a mixture of CD56bright, mature, transitional and adaptive NK cells, a sign of functional NK cells with minimal inflammation. The expression of Klrc2 in NK cells under different conditions (displayed in Supplementary figure 14d) clearly showed that it was expressed in much more cells induced by caerin 1.1/1.9. In addition, we found a higher percentage of Klrc2 in cells treated by caerin 1.1/1.9. The Reactome pathways enriched in NK cells of the caerin group included apoptosis, intrinsic pathway for apoptosis and a few CD28 stimulation‐relevant pathways (Supplementary figure 14f), implying active immune response. These pathways were either not enriched or with much lower P‐values in untreated or P3 groups.
TMT10plex labelling quantitative proteomics revealed a higher immune response induced by the injection of caerin 1.1/1.9
To validate our scRNA‐seq data and capture treatment‐dependent alterations in protein content, quantitative proteomic analysis of tumors was performed using the TMT labelling method (details of protein quantitation and annotations summarised in Supplementary table 6). The pairwise comparison showed that significantly more proteins were regulated in the caerin group than in the control (Supplementary figure 9a and b). The hierarchical clustering of quantified proteins implied consistency between biological triplicates of each group. A total of 238 proteins were uniquely upregulated in the caerin group (Supplementary figure 9c).
The gene ontology enrichment of upregulated proteins with each treatment was analysed (Supplementary table 6; Supplementary figures 10 and 11). In the caerin group, the enrichment of many biological processes was more significant than in the control, for instance immune system process (caerin, FDR = 8.10E‐28; control, FDR = 1.29E‐12) and innate immune response (caerin, FDR = 9.85E‐22; control, FDR = 3.60E‐05). The top 60 proteins significantly regulated in the caerin group with respect to the untreated group are shown in Figure 7a, the proteins involved in immune response and regulation processes, such as Gzma, Gzmc, Irf5, Tgtp1, Prg2 and Ighg1, were present. The quantities of selected proteins uniquely regulated in the caerin group are shown in Figure 7b.
Figure 7.
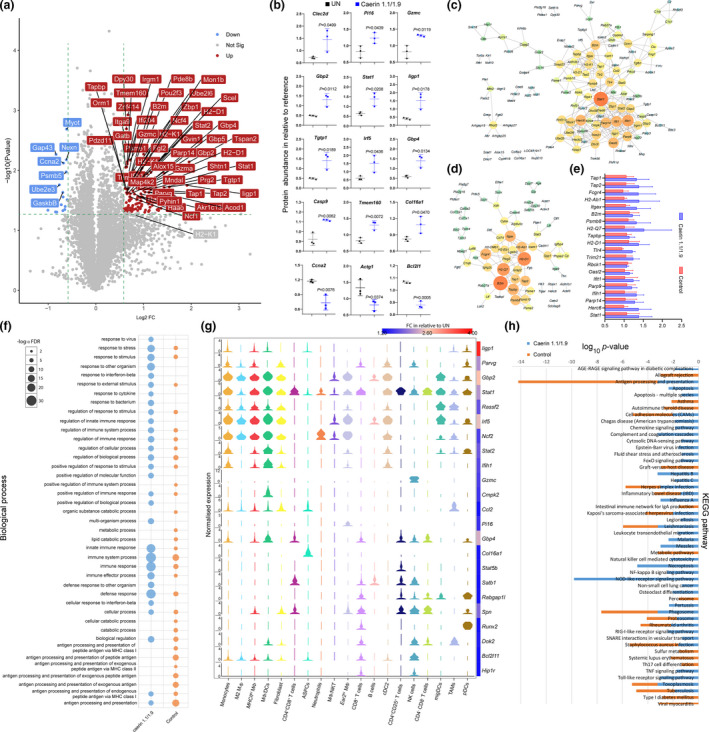
Quantitative proteomic analysis of TC‐1 tumor in different groups (untreated, caerin or control). The samples were isolated from the same tumor tissues used for scRNA‐seq analysis (no. of mice = 3 in each group), and the proteomic experiment was performed once. (a) The volcano graph shows proteins significantly regulated (FC > 1.5, P < 0.05) only in the caerin group with respect to the untreated group. (b) Comparison of the abundance of selected proteins in the untreated and caerin groups relative to the reference. A two‐tailed Student's t‐test was used to evaluate the significance. PPIs of significantly upregulated proteins in the caerin group (c) and control group (d); the size and the colour of the nodes represent the interaction degree of the protein. (e) Comparison of fold change (with respect to internal references) of the top 10 highest degree values on the PPIs in caerin and control groups. (f) Comparison of biological processes enriched in the significantly elevated proteins in the caerin and control groups, respectively, with respect to the untreated group. (g) The correlation between the gene expressions (in 19 cell populations) of the proteins showing significant upregulation only in the caerin group, and the fold change of these proteins in relation to the untreated group. (h) The KEGG pathways enriched in the caerin and control groups (P < 0.05).
The protein–protein interaction (PPI) analysis of upregulated proteins identified intensive interactions in both treatments (Figure 7c and d; the complete predicted PPIs are recorded in Supplementary table 6). There were 337 interactions identified amongst the 285 proteins, forming a highly connected network (P < 1.0E‐16). The distribution analysis of path length of the network indicated that most interactions could occur with fewer than 4 steps and the frequency peaked at 3 steps, which was also reflected by the high average neighbourhood connectivity associated with more neighbours (Supplementary figure 12). In addition, the relatively low betweenness (close to zero) and closeness (most between 0.20 and 0.35) centrality values further implied the rigorous interplay amongst the proteins. Similar nodes can be found on two networks with different interaction degrees, with Stat1 and B2m being the node with the highest degrees in the caerin and control groups, respectively. The fold changes of the top 10 most interacting protein nodes on two PPIs were compared with respect to internal references (Figure 7e). Most of these proteins showed higher abundance in the caerin group. Stat1 was upregulated by 2.2‐fold compared with the untreated tumor (Supplementary table 6) and detected as a marker gene in monocytes, MΦ/DCs and CD4+CD25+ T cells with caerin treatment (Supplementary table 6) with no significant change in protein or mRNA with control peptide treatment.
The top 40 enriched biological processes were compared in Figure 7f. Caerin treatment caused significant upregulation of proteins involved in immune response and regulation processes in the tumor, while many proteins upregulated appeared to play important roles in the processes related to antigen processing and presentation in the control group. This was in accordance with the finding that the injection of caerin largely reduced the population of B cells as suggested by the scRNA‐seq. A correlation was observed between proteins significantly upregulated only in the caerin group and cell populations identified in scRNA‐seq (Figure 7g). Of those proteins showing a fold change greater than 2, many were closely correlated with normalised expressions of genes in the populations of monocytes, MΦ and DCs, such as Iigp1, Gbp2, Irf5 and Parvg. There were a few proteins more closely associated with their gene expression in T‐cell and NK populations, including Satb1, Spn, Dok2 and Hip1r. Marker gene Gzmc appeared exclusive to NK cells, while the protein upregulation was only considered significant in the caerin group. Stat1 was more abundant with caerin treatment, which was detected as an upregulated gene in nearly all cell populations except B cells, suggesting that Stat1 was largely regulated by caerin injection.
The KEGG pathways enriched (P < 0.05) in upregulated proteins were compared (Figure 7h), and more pathways were significantly identified in the caerin group, including apoptosis, natural killer cell‐mediated cytotoxicity, necroptosis, the signalling of nod‐like receptor (NLR), TNF, chemokine, NF‐κB, RIG‐I‐like receptor and Toll‐like receptor and several disease‐related pathways. Amongst these KEGG pathways, NLR signalling was determined as the most enriched pathway (P = 1.55E‐10), supported by the significantly increased contents of Gbp2, Gbp5, Nlrc4, Ccl2, Tlr4 and so forth in the caerin group; many of their genes were detected as signatures for MΦ/DCs. Notably, the antigen processing and presentation pathway was less significantly changed in the caerin group (P = 6.0E‐4) than in the control (P = 1.0E‐11), in accordance with the observation that the population of B cells was remarkably reduced with the injection of caerin peptides.
Discussion
Intratumoral injection of caerin 1.1/1.9 inhibited TC‐1 growth but did not prolong the survival of TC‐1 tumor‐bearing mice (Figure 1b),13 whereas anti‐PD‐1 treatment did not inhibit TC‐1 tumor growth or increase the survival time of TC‐1 tumor‐bearing mice.8 Intratumoral injection of caerin 1.1/1.9 mixture significantly prolonged survival time after immunisation with an HPV16 E7 peptide‐based vaccine along with IL‐10 and PD‐1 blockade, via the modulation of the TME, when compared to intratumoral injection of a control peptide. The scRNA‐seq of CD45+ tumor‐infiltrating cells and proteomic analysis of tumor uncovered that caerin 1.1/1.9 changed heterogeneity and function of multiple tumor‐infiltrating leucocytes, especially macrophage populations (largely elevated the populations of M1, MHCIIhi and Ear2hi MΦs), reducing protumorigenic B cells and inducing more active CD8+ T cells, which thus modulated the TME to a proinflammatory environment that may favor tumor rejection.
The numbers of tumor‐infiltrating CD8+ T cells are similar between the treatment groups, but significantly more than those of the untreated group. The antigen‐specific CD8+ T cells in the spleen and draining lymph nodes were similar in the two treatments, suggesting caerin peptides did not influence the generation of vaccine‐induced CD8+ T cells; however, the CD8+ T cells in the caerin group were significantly more activated than those in the control group, which seemed translated to increased survival time in this group (Figure 1c).
Most of our knowledge of TAMs comes from histological examinations and in vitro profiling using flow cytometry.77 There remains a significant knowledge gap on how TAMs function in vivo and how these cellular activities can be harnessed to improve anticancer therapy. Recently, it has been shown that macrophages, as well as other tumor‐infiltrating cells, have heterogeneous phenotypes in the TME by single‐cell RNA‐sequencing technology.78 Similar results were observed here that six populations with distinct gene expression patterns and opposing functions were identified (Figures 3 and 4). The immunosuppressive TAM phenotype (‘M2’) is generally believed to be positive for Arg1,79 because of the production of interleukin‐4 and lactic acid by tumor cells. The M2 MΦ was abundant in the untreated group, while the treatment groups displayed a substantial reduction in this type of MΦ. Furthermore, Arg expression in macrophage was significantly reduced in the caerin group, so were Pf4, Mmp13 and Hmox1, known to promote tumorigenesis and metastasis.80, 81, 82 On the contrary, MΦs with immune‐activating functions, including MHCIIhi MΦ, MΦ/DCs and Ear2hi MΦ, were amplified by the two treatments, especially in the caerin group. Therefore, caerin 1.1/1.9 injection further expanded macrophages with immune‐activating function while reducing the immune‐suppressive macrophage of vaccinated and PD‐1‐blocked TC‐1 tumor‐bearing mice, through mechanisms yet to be identified. Currently, we are attempting to confirm the findings of the scRNA‐seq and proteomic results by flow cytometry and real‐time PCR, and to investigate whether depleting the MΦ changes the outcome of the triple therapy.
It has been found that B cells significantly promoted tumor growth in E6/E7‐expressing TC‐1 tumor‐bearing mice, while the reduction in B cells was found to stimulate a more efficient antitumor T‐cell response, suggesting immunosuppressive functions of B cells in this model.83 B cells suppress the antitumor immunity through complicated interplay with tumor tissues and other lymphocytes, such as T cells, antigen‐presenting cells (APCs), Tregs and myeloid‐derived suppressor cells (MDSCs).84, 85, 86 The scRNA‐seq showed that the treatment containing caerin 1.1/1.9 nearly depleted B‐cell population, with the expression of marker genes largely downregulated in general, including many genes regulating B‐cell differentiation and activation, such as Ms4a1 (Cd20), Fcer2a (Cd23), Cd79a and Ebf1.87 This was also reflected in proteomic analysis that antigen processing and presentation pathway was remarkably less enriched than the control group, and the biological processes associated with MHCI and MHCII were insignificantly modulated in the caerin groups with respect to untreated or control groups. Meanwhile, the proteasome pathway was largely inhibited, because of the downregulations of Psmb5, Psmb6, Psmb7, Psmd9 and H2afz, in the caerin group as suggested by the proteomic analysis (Supplementary table 6), which might suppress the depletion of actin at the centrosome, and thus impair cell polarity and organisation of B cells because of being incapable of separating the centrosome from the nucleus.88 In addition, the proteomic analysis also found that proteoglycans (PGs) in cancer were inhibited by caerin 1.1/1.9‐containing treatment (Supplementary table 6), which might also negatively affect B‐cell development.89 However, the flow cytometric estimation of B‐cell numbers was similar amongst the three groups, which may need further experimentation to confirm the scRNA‐sequencing results.
Previously, we found that caerin 1.1/1.9 attracted NK cells to the TC‐1 tumor.20 Even though the cell numbers were relatively low in the current study, NK cells were elevated significantly in the caerin group. The overlapping distribution of its marker genes with CD4+CD8+ and CD8+ T cells suggested certain similarities in their cellular function. We found that Cd56bright NK cells were largely activated only in the caerin group. Cd56bright NK cells were reported to have a regulatory role in early immune response because of the capability of producing different cytokines and shaping adaptive response.90 Notably, scRNA‐seq determined Gzmc as the marker gene with the second‐highest upregulation and the largest mean expression, and there were more NK cells showing higher expression of Gzmc in the caerin group. This was consistent with the proteomic analysis that the content of Gzmc was increased and natural killer cell‐mediated cytotoxicity pathway activated only in the caerin group, suggesting higher cytotoxicity might be induced to cause more efficient cell death, which was reflected as the enrichment of apoptosis and intrinsic pathway for apoptosis only in this group. It has been found that Gzmc can support CTL‐mediated killing via the granule exocytosis pathway during late primary alloimmune responses,91 potentially related to the mutual function shared between NK and two T‐cell populations. The upregulation of Gzmc activated caspase‐independent cell death with a mitochondrial phenotype,92 which was in accordance with our observation that activation and myristylation of BID and translocation to mitochondria was the second most enriched process in the caerin group.
Nod‐like receptor signalling activation was indicated by both scRNA‐seq and proteomic analyses. Nlrc4 acts as a downstream transcriptional target of p53 and shows potential antitumorigenic functions.93 The upregulation of Nlrp3 was shown to induce Stat1 phosphorylation through IFN‐γ and thus promote an antitumorigenic environment.94 The proteomic analysis identified Stat1 only upregulated in the caerin group with significance, and it appeared as the node with the highest degree of interactions with other upregulated proteins. Stat1 was found to act as an essential mediator of the antitumor response by inhibiting MDSC accumulation and promoting T‐cell‐mediated immune responses in murine head and neck squamous cell carcinoma.95 The NF‐κB pathway was significantly activated with the injection of caerin 1.1/1.9 (Figure 7g), which might function synergistically with Stat1 to induce more iNOS and Il12,96 thus triggering recruitment of NK cells and CTLs. This was consistent with the observation that the NK population and the activation of CD8+ T cells were largely promoted, and much higher upregulation of Il12b was detected in migDCs by scRNA‐seq in the caerin 1.1/1.9 group. These NK cells and CD8+ T cells joined the C0_MΦ/DC‐CCL5 to elicit apoptosis in the TC‐1 tumor, which was highly activated exclusively in the caerin group suggested by proteomic analysis. In addition, the co‐activation of Stat1 and Rela (significantly upregulated in neutrophils and migDCs) potentially triggered the expression of iNOS, consequently producing nitric oxide that could contribute to tumor elimination.97 Here, insignificant upregulation (about a 1.3‐fold change relative to the untreated or control group) of Nos3 in the caerin group was identified by proteomic analysis.
Conclusions
Therapeutic vaccination with an HPV16 E7 peptide‐based vaccine incorporating IL‐10 receptor antibody plus PD‐1 blockade induces antitumor responses, caerin 1.1/1.9 injection further increased the efficacy of the treatment and expanded immune‐activating macrophages (MHCIIhi and Ear2hi MΦs) and NK cells, reduced immune‐suppressive B cells and resulted in more activated CD8+ T cells, which might directly mediate proinflammatory apoptosis of tumor cells with Stat1 playing a key role with the regulators on multiple pathways. We demonstrated that caerin 1.1/1.9‐containing treatment results in improved antitumor responses. Harnessing the novel candidate genes preferentially enriched in the immune active cell populations may allow further exploration of distinct macrophages, T cells and their functions in TC‐1 tumors, providing a valuable resource for researchers in the field.
Methods
Mice
Six‐ to eight‐week‐old, specific pathogen‐free (SPF) adult female C57BL/6 (H2b) mice were ordered from the Animal Resource Centre of Guangdong Province and kept at the Animal Facility, the first affiliated hospital of Guangdong Pharmaceutical University, Guangdong, China. Experiments were approved by and then performed in compliance with the guidelines of Animal Experimentation Ethics Committee (Ethics Approval Number: FAHGPU20160316). All mice were kept at SPF condition on a 12‐h light/12‐h dark cycle at 22°C, and the humidity was 75%. Five mice were kept in each cage, and were provided with sterilised standard mouse food and water.
Cell line, peptide synthesis and antibodies
A murine TC‐1 cell line transformed with HPV16 E6/E7 was obtained from Shanghai Institute for Cell Resources Centre, Chinese Academy of Sciences, and cultured following the procedure described previously.20
Caerin 1.1 (GLLSVLGSVAKHVLPHVLPHVVPVIAEHL‐NH2), caerin 1.9 (GLFGVLGSIAKHVLPHVVPVIAEKL‐NH2), control peptide P3 (GTELPSPPSVWFEAEFK‐OH), HPV16 E7 CTL epitope RAHYNIVTF, and four overlapping peptides representing the entire HPV 16 E7 protein, EX (MHGDTPTLHEYMLDLQPETTDLYCYEQLNDSSEEE, LNDSSEEEDEIDGPAGQAEPDRAHYNIVTFCCKC, DRAHYNIVTFCCKCDSTLRLCVQSTHVDIR and CVQSTHVDIRTLEDLLMGTLGIVCPICSQKP), were synthesised (purity > 99%) by Mimotopes Proprietary Limited (Wuxi, China). The concentration of lipopolysaccharide in caerin 1.1, caerin 1.9 and P3 was 0.03, 0.03 and 0.44 EU mL−1, respectively, as measured by Kinetic Turbidimetric Assay (Xiamen Bioendo Technology Co., Ltd).
Rat anti‐mouse anti‐IL‐10 receptor (1B1.3), anti‐PD‐1 (J43) hamster anti‐mouse monoclonal antibody (MAb) and IgG isotype control antibody (LTF‐2) were purchased from BioXcell, USA, and stored at −80°C till further use.
Tumor challenge
TC‐1 cells, at approximately 70% confluency, were harvested with 0.25% trypsin and washed repeatedly with PBS. 5 × 105 cells/mouse in 0.2 mL of PBS were injected subcutaneously into the left flank. Tumor sizes were assessed every 3 days using callipers to determine the average diameter of each tumor, calculated as width × width × length. Mice were given 1% sodium pentobarbital by i.p. injection when treatment was performed. Mice were sacrificed when the tumor diameter reached 15 mm (for survival curve) or 20 mm (for scRNA‐seq), by CO2 inhalation at the end of each experiment.
Immunisation of mice
Three days post‐TC‐1 challenge, when tumors were palpable, seven mice were immunised intramuscularly (i.m.) with the vaccine containing 40 µg of four overlapping HPV16 E7 peptides (EX) (10 µg per peptide), 10 µg of monophosphoryl lipid A (MPLA; Sigma‐Aldrich), and 300 µg of anti‐IL‐10R antibodies, dissolved in 100 µL of PBS, at days 3, 9 and 18, and with or without vaccine containing 300 µg of anti‐PD‐1 intraperitoneally (i.p.) on days 9 and 21.
Tumor local administration of caerin peptide
Three days post‐TC‐1 challenge, when the tumors’ diameters reached 3–5 mm, the mice either immunised or unimmunised were intratumorally injected with 30 µg of caerin peptides (caerin 1.1 and 1.9) or PBS for six consecutive days.
Flow cytometry
TC‐1 tumors were dissected and homogenised using a tumor dissociation kit (Miltenyi Biotec GmbH, Germany) to obtain single cells. Single cells were stained with FITC‐CD45.2 (clone: 104), APC‐Cy7‐CD3 (clone: 145‐2C11), PE‐Cy7‐CD8a (clone: 53‐6.7), BV421‐INF‐γ (clone: XMG1.2), PE‐B220 (clone: RA3‐6B2) and FVS510 to separate dead cells from viable cells before analysing on a flow cytometer (FACSAria II; BD Biosciences, San Jose, CA, USA). Results were analysed with FlowJo v10.0 software (Tree Star Inc., Ashland, OR, USA).
ELISPOT
Single spleen cell suspensions were added to membrane base 96‐well ELISPOT plate (Millipore, Bedford, MA) coated with anti‐IFN‐γ (BD Harlingen, San Diego, CA). HPV16 E7 CTL epitope RAHYNIVTF was added at various concentrations, and cells were incubated with the peptide overnight at 37°C with 5% CO2. The plate was incubated with detection antibody (a biotinylated anti‐mouse IFN‐γ antibody; BD Harlingen, San Diego, CA) after extensive washing in wash buffer for 2 h at room temperature. Antigen‐specific IFN‐γ‐secreting cells were detected by sequential exposure of the plate to avidin–horseradish peroxidase (Sigma‐Aldrich) and DAB (Sigma‐Aldrich). The plate was washed and allowed to air‐dry overnight, and foci of staining were counted by the ELISPOT reader system (CTL, Germany).
Isolation of tumor‐infiltrating CD45+ cells
TC‐1 tumor from untreated, vaccinated and PD‐1 blockade mice in conjunction with the injection of caerin 1.1/1.9 (molar ratio 1:1, at 39 µg mL−1) or control peptide P3 was pooled (3 tumors/group), cut into 2 × 2 mm pieces and digested by adding 2.35 mL of RPMI 1640, 100 µL of enzyme D, 50 µL of enzyme R and 12.5 µL of enzyme A into a gentleMACS C Tube, followed by disassociation using gentleMACS Dissociator from Miltenyi (Gladbach, Germany). After removal of dead cell and cell debris, the remaining cells were labelled with CD45 microbeads (130‑110‑618) and passed through the LS column. The purity and viability of the CD45+ cells were more than 80% confirmed by flow cytometry and trypan blue staining.
GEM generation, barcoding and cDNA amplification
The 10×™ GemCode™ Technology was used to generate a pool of ˜750 000 barcodes to separately index each cell’s transcriptome. Libraries were generated and sequenced from the cDNA, and the 10× barcodes were used to associate individual reads back to the individual partitions. Upon dissolution of the Single Cell 3′ Gel Bead in a GEM, primers containing (i) an Illumina® R1 sequence (read 1 sequencing primer), (ii) a 16 nt 10× barcode, (iii) a 10 nt Unique Molecular Identifier (UMI) and (iv) a poly‐dT primer sequence were released and mixed with cell lysate and Master Mix. Silane magnetic beads were used to remove leftover biochemical reagents and primers from the post‐GEM reaction mixture. Full‐length, barcoded cDNA was then amplified by PCR to generate sufficient mass for library construction.
Sequencing and genome alignment
The Single Cell 3′ Protocol produced Illumina‐ready sequencing libraries. The Single Cell 3′ 16 bp 10× barcode and 10 bp UMI were encoded in Read 1, while Read 2 was used to sequence the cDNA fragment. Sample index sequences were incorporated as the i7 index read. Cell Ranger (http://support.10xgenomics.com/single‐cell/software/overview/welcome) was used to do the genome alignment. A read was exonic if at least 50% of it intersected an exon, intronic if it was non‐exonic and intersected an intron, and intergenic otherwise. Cell Ranger further aligned exonic reads to annotated transcripts. A read that was compatible with the exons of an annotated transcript, and aligned to the same strand, was considered mapped to the transcriptome. Only reads that were confidently mapped to the transcriptome were used for UMI counting.
Depth normalisation, expression QC and data normalisation
The read depth between libraries was equalised before merging, to reduce the batch effect introduced by sequencing. Seurat was used to explore QC metrics and filter cells with the following filtration: (i) 500 < gene counts < 4000 per cell; (ii) UMI counts < 8000 per cell; and (iii) the percentage of mitochondrial genes < 10%. We employed a global‐scaling normalisation method ‘LogNormalise’ to normalise the gene expression measurements, where a gene expression level = log10(1 + (UMI A ÷ UMI Total) × 10 000).
Clustering cells and differentially expressed gene analysis
Distances between the cells were calculated based on previously identified PCs using Seurat. To cluster the cells, we applied modularity optimisation techniques SLM,98 to iteratively group cells together. Then, we used likelihood‐ratio test to find differential expression for a single cluster, compared with all other cells, with following criteria: (i) P‐value ≤ 0.01; (ii) log FC ≥ 0.360674. log FC means log fold change of the average expression between the two groups; and (iii) the percentage of cells where the gene is detected in specific cluster > 25%.
Marker gene analysis
We further selected the top 20 genes as the marker genes according to the result of differentially expression genes. Then, the expression distribution of each marker gene was demonstrated by using heat map and bubble diagram. The putative biological identity of each cluster was assigned by using a murine gene expression atlas25 and the database of cell markers.26
Constructing single‐cell trajectories and differential expression analysis
Single‐cell trajectory was analysed using the matrix of cells and gene expressions by Monocle (version 2.6.4). Once the cells were ordered, we visualised the trajectory in the reduced dimensional space. The key genes related to the development and differentiation process with FDR < 1E‐5 and grouped genes with similar trends in expression were analysed. The BEAM module of Monocle was used to test for branch‐dependent gene expression by formulating the problem as a contrast between two negative binomial GLMs.
Protein extraction, TMT10plex labelling and high pH reversed‐phase fractionation
Tumor samples were collected and directly frozen in liquid nitrogen prior to protein extraction and sample preparation. Biological triplicates were collected for each treatment. These tissues were the same biological triplicates from which the CD45+ cells were extracted for scRNA‐seq. The tissue samples were homogenised thoroughly in SDT buffer (4%(w/v) SDS, 100 mM Tris‐HCl, pH 7.6, and 0.1 M DTT) at 4°C, with the total protein contents quantified using the Pierce BCA protein assay on a NanoDrop 2000 (Thermo Fisher Scientific, Bremen, Germany). Certain amounts of samples containing 500 µg of protein were subjected to trypsin digestion by the filter‐aided proteome preparation (FASP) described elsewhere.99 Tryptic peptides were desalted on Sep‐Pak C18 columns (Waters, Milford, MA), lyophilised and quantified on the NanoDrop 2000 using OD280 nm.
The samples containing 100 μg peptides were transferred to Eppendorf tubes and labelled by TMT10plex following the manufacturer’s instruction (Thermo Scientific, Waltham, MA, USA). In this study, 126, 127N and 127C were used to label untreated tumor samples; 128N, 128C and 129N were used to label the treatments with caerin peptides; the treatments with the addition of P3 were labelled using 129C, 130N and 130C, respectively. The labelled samples were mixed and fractionated using a Pierce™ High pH Reversed‐Phase Peptide Fractionation Kit (Thermo Fisher Scientific, IL, USA) according to the manufacturer’s instruction. All fractions were lyophilised on a SpeedVac and resuspended in 12 μL 0.1%FA for LC‐MS/MS analysis.
Easy nLC tandem Q Exactive MS/MS analyses
A 10 μL of sample solution was loaded onto a two‐dimensional EASY nLC1000 system coupled to a Q Exactive Hybrid Quadrupole‐Orbitrap Mass Spectrometer (Thermo Scientific). In the nanoLC separation system, mobile phase A solution contains 0.1% formic acid in water, and mobile phase B solution contains 84% acetonitrile and 0.1% formic acid. Before peptide loading, the columns were pre‐equilibrated with 95% mobile phase A for 30 min. The samples were first injected into the sample loading column (Thermo Scientific Acclaim PepMap100, 100 μm × 2 cm, nanoViper C18), then fractionated by the analytic column (Thermo Scientific EASY column, 10 cm, ID75 μm, 3 μm, C18‐A2) and Thermo EASY SC200 trap column (RP‐C18, 3 μm, 100 mm × 75 μm). The phosphopeptides were first transferred to the Thermo Scientific EASY column (2 cm × 100 μm 5 μm‐C18) and then separated via the trap column using a gradient of 0–55% mobile phase B for 220 min. Then, the columns were rinsed with 100% mobile phase B for 8 min and re‐equilibrated to the initial conditions for 12 min. The flow rate of the above procedures was 300 nL/min. The samples were then analysed on a Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The ion spray voltage was set to 5500 V, the declustering potential was set to 100 V, the curtain gas flow was set at 30, ion source gas 1 was set at 40, the ion source gas 2 was set at 50, and spray temperature was set at 450°C. The mass spectrometer acquired the mass spectral data in an Information Dependent Acquisition (IDA) mode. Full‐scan MS data were acquired over the mass range 300–1800 with a resolution of 70 000 at 200 m/z. AGC (automatic gain control) target was set at 106, the maximum IT was set at 50 ms, and the dynamic exclusion was set at 60.0 s. In every full scan, twenty MS/MS spectra were obtained. The MS/MS activation type was HCD, and the isolation window was 2 m/z. The resolution of MS/MS was 35 000 at 200 m/z, the normalised collision energy was set at 30, and the underfill was set at 0.1%.
Protein identification and quantification
The MS/MS data were searched against SwissProt Mouse (76 413 sequences, downloaded on 12 December 2014) database for protein identification using Mascot2.2 (Matrix Science, London, UK) and Proteome Discoverer1.4 software (Thermo Fisher Scientific, Waltham, MA, USA) with the following search settings: enzyme trypsin; two missed cleavage sites; precursor mass tolerance 20 ppm; fragment mass tolerance 0.1 Da; fixed modifications – carbamidomethyl (C), TMT10plex (N‐term) and TMT10plex (K); and variable modifications – oxidation (M) and TMT10plex (Y). The results of the search were further submitted to generate the final report using a cut‐off of 1% FDR on peptide levels, and only unique peptides were used for protein quantitation. All peptide ratios were normalised by the median protein ratio, and the median protein ratio was 1 after the normalisation. The protein showing a fold change ≥ 1.2 compared with the untreated group and the P < 0.05 were considered significantly regulated by the treatment and included in further analysis.
Protein–protein interaction (PPI) analysis
Interactions amongst significantly regulated proteins were predicted using STRING.100 STRING provides a critical assessment and integration of protein–protein interactions from multiple resources, including direct (physical) and indirect (functional) associations. A spring model was used to generate the network images. All resources were selected to generate the network, and ‘confidence’ was used as the meaning of network edges, and the required interaction score of 0.700 was selected for all PPI, to highlight the most confident interactions. Neither the 1st nor 2nd shell of the PPI was included in this study. Protein without any interaction with other proteins was excluded from displaying in the network.
Biological process and pathway analysis
The enrichment of biological processes, WikiPathways,101 KEGG pathways102 and Reactome pathways103 in the significantly upregulated genes (P < 0.05) of different cell populations/subpopulations determined by scRNA‐seq, or proteins significantly upregulated (fold change > 1.2 and P < 0.05) by the treatments with respective to untreated group derived from quantitative proteomics, was analysed by Enrichr (http://amp.pharm.mssm.edu/Enrichr/). In addition, the proteins determined to be differentially expressed were analysed by the online tool Innate DB (https://www.innatedb.ca/index.jsp), to further investigate the modulation of immune response‐relevant pathways. In the analysis, the up/downregulation of key regulators identified in different treatments, with a fold change ≥ 2 and P < 0.05, was used to predict the activation/inhibition of signalling pathways.
Conflict of interest
The authors declare no competing interests.
Author contributions
Guoying Ni: Data curation; Investigation; Methodology; Writing‐original draft. Xiaodan Yang: Data curation; Investigation; Visualization. Junjie Li: Data curation; Investigation. Xiaolian Wu: Investigation; Project administration; Resources; Supervision. Ying Liu: Investigation; Methodology. Hejie Li: Investigation; Writing‐review & editing. Shu Chen: Investigation; Project administration; Resources; Supervision. Conor E Fogarty: Investigation; Writing‐review & editing. Ian Frazer: Investigation; Writing‐review & editing. Guoqiang Chen: Funding acquisition; Project administration; Resources; Supervision. Xiaosong Liu: Conceptualization; Formal analysis; Funding acquisition; Methodology; Project administration; Resources; Supervision; Validation; Writing‐original draft; Writing‐review & editing. Tianfang Wang: Conceptualization; Data curation; Formal analysis; Funding acquisition; Resources; Software; Supervision; Visualization; Writing‐original draft; Writing‐review & editing.
Supporting information
Supplementary figures 1–14
Supplementary table 1
Supplementary table 2
Supplementary table 3
Supplementary table 4
Supplementary table 5
Supplementary table 6
Supplementary table 7
Supplementary table captions
Acknowledgments
We thank Professor Abigail Elizur for her valuable advice and support. We are grateful for the sequencing platform and/or bioinformation analysis and proteomic experiment of Gene Denovo Biotechnology Co., Ltd (Guangzhou, China) and Applied Protein Technology, Co. Ltd (Shanghai, China).
Data availability statement
The scRNA‐seq data are available on the Institute Single Cell Portal (https://singlecell.broadinstitute.org/single_cell) under accession number SCP1093. The mass spectrometry proteomic data have been deposited to the ProteomeXchange Consortium via the PRIDE104 partner repository with the data set identifier PXD021264.
Contributor Information
Xiaosong Liu, Email: xiaosongl@yahoo.com.
Tianfang Wang, Email: twang@usc.edu.au.
References
- 1.Zhou C, Tuong ZK, Frazer IH. Papillomavirus immune evasion strategies target the infected cell and the local immune system. Front Oncol 2019; 9: 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tuyaerts S, Van Nuffel AMT, Naert Eet al. PRIMMO study protocol: a phase II study combining PD‐1 blockade, radiation and immunomodulation to tackle cervical and uterine cancer. BMC Cancer 2019; 19: 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ni G, Huang K, Luan Yet al. Human papillomavirus infection among head and neck squamous cell carcinomas in southern China. PLoS One 2019; 14: e0221045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018; 359: 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saglam O, Conejo‐Garcia J. PD‐1/PD‐L1 immune checkpoint inhibitors in advanced cervical cancer. Integr Cancer Sci Ther 2018; 5: 10.15761/ICST.1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borcoman E, Le Tourneau C. Pembrolizumab in cervical cancer: latest evidence and clinical usefulness. Ther Adv Med Oncol 2017; 9: 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tondini E, Arakelian T, Oosterhuis Ket al. A poly‐neoantigen DNA vaccine synergizes with PD‐1 blockade to induce T cell‐mediated tumor control. Oncoimmunology 2019; 8: 1652539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandra J, Dutton JL, Li Bet al. DNA vaccine encoding HPV16 oncogenes E6 and E7 induces potent cell‐mediated and humoral immunity which protects in tumor challenge and drives E7‐expressing Skin Graft rejection. J Immunother 2017; 40: 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Curran MA, Glisson BS. New hope for therapeutic cancer vaccines in the era of immune checkpoint modulation. Annu Rev Med 2019; 70: 409–424. [DOI] [PubMed] [Google Scholar]
- 10.Ott PA, Hu Z, Keskin DBet al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017; 547: 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kenter GG, Welters MJ, Valentijn ARet al. Vaccination against HPV‐16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 2009; 361: 1838–1847. [DOI] [PubMed] [Google Scholar]
- 12.van Poelgeest MI, Welters MJ, van Esch EMet al. HPV16 synthetic long peptide (HPV16‐SLP) vaccination therapy of patients with advanced or recurrent HPV16‐induced gynecological carcinoma, a phase II trial. J Transl Med 2013; 11: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ni G, Chen S, Yuan Jet al. Comparative proteomic study reveals the enhanced immune response with the blockade of interleukin 10 with anti‐IL‐10 and anti‐IL‐10 receptor antibodies in human U937 cells. PLoS One 2019; 14: e0213813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llopiz D, Aranda F, Diaz‐Valdes Net al. Vaccine‐induced but not tumor‐derived Interleukin‐10 dictates the efficacy of Interleukin‐10 blockade in therapeutic vaccination. Oncoimmunology 2016; 5: e1075113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gasser S, Lim LHK, Cheung FSG. The role of the tumour microenvironment in immunotherapy. Endocr Relat Cancer 2017; 24: T283–T295. [DOI] [PubMed] [Google Scholar]
- 16.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer 2016; 16: 447–462. [DOI] [PubMed] [Google Scholar]
- 17.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017; 14: 399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon SR, Maute RL, Dulken BWet al. PD‐1 expression by tumour‐associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017; 545: 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni G, Liang D, Cummins SFet al. Comparative proteomic study of the antiproliferative activity of frog host‐defence peptide caerin 1.9 and its additive effect with caerin 1.1 on TC‐1 cells transformed with HPV16 E6 and E7. Biomed Res Int 2018; 2018: 7382351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma B, Yuan J, Chen Set al. Topical application of temperature‐sensitive caerin 1.1 and 1.9 gel inhibits TC‐1 tumor growth in mice. Am J Transl Res 2020; 12: 191–202. [PMC free article] [PubMed] [Google Scholar]
- 21.Ni G, Chen S, Chen Met al. Host‐defense peptides caerin 1.1 and 1.9 stimulate TNF‐alpha‐dependent apoptotic signals in human cervical cancer HeLa cells. Front Cell Dev Biol 2020; 8: 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.VanCompernolle SE, Taylor RJ, Oswald‐Richter Ket al. Antimicrobial peptides from amphibian skin potently inhibit human immunodeficiency virus infection and transfer of virus from dendritic cells to T cells. J Virol 2005; 79: 11598–11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Ni G, Wu Xet al. Blocking IL‐10 signalling at the time of immunization renders the tumour more accessible to T cell infiltration in mice. Cell Immunol 2016; 300: 9–17. [DOI] [PubMed] [Google Scholar]
- 24.Pan X, Ma B, You Xet al. Synthesized natural peptides from amphibian skin secretions increase the efficacy of a therapeutic vaccine by recruiting more T cells to the tumour site. BMC Complement Altern Med 2019; 19: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hume DA, Summers KM, Raza S, Baillie JK, Freeman TC. Functional clustering and lineage markers: insights into cellular differentiation and gene function from large‐scale microarray studies of purified primary cell populations. Genomics 2010; 95: 328–338. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Lan Y, Xu Jet al. Cell Marker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 2019; 47: D721–D728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guilliams M, Dutertre CA, Scott CLet al. Unsupervised high‐dimensional analysis aligns dendritic cells across tissues and species. Immunity 2016; 45: 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Hove H, Martens L, Scheyltjens Iet al. A single‐cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 2019; 22: 1021–1035. [DOI] [PubMed] [Google Scholar]
- 29.Schwalie PC, Dong H, Zachara Met al. A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 2018; 559: 103–108. [DOI] [PubMed] [Google Scholar]
- 30.Han JH, Lee S, Park YSet al. IFITM6 expression is increased in macrophages of tumor‐bearing mice. Oncol Rep 2011; 25: 531–536. [DOI] [PubMed] [Google Scholar]
- 31.Lin JD, Nishi H, Poles Jet al. Single‐cell analysis of fate‐mapped macrophages reveals heterogeneity, including stem‐like properties, during atherosclerosis progression and regression. JCI Insight 2019; 4: e124574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassetta L, Fragkogianni S, Sims AHet al. Human tumor‐associated macrophage and monocyte transcriptional landscapes reveal cancer‐specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 2019; 35: 588–602.e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qi L, Yu H, Zhang Yet al. IL‐10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget 2016; 7: 71673–71685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kudo Y, Iizuka S, Yoshida Met al. Matrix metalloproteinase‐13 (MMP‐13) directly and indirectly promotes tumor angiogenesis. J Biol Chem 2012; 287: 38716–38728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He MK, Le Y, Zhang YFet al. Matrix metalloproteinase 12 expression is associated with tumor FOXP3(+) regulatory T cell infiltration and poor prognosis in hepatocellular carcinoma. Oncol Lett 2018; 16: 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Boniface J, Mao Y, Schmidt‐Mende J, Kiessling R, Poschke I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early‐stage breast cancer patients. Oncoimmunology 2012; 1: 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grzywa TM, Sosnowska A, Matryba Pet al. Myeloid cell‐derived arginase in cancer immune response. Front Immunol 2020; 11: 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villani AC, Satija R, Reynolds Get al. Single‐cell RNA‐seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017; 356: eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Álvarez B, Nieto‐Pelegrín E, Martínez de la Riva Pet al. Characterization of the porcine CLEC12A and analysis of its expression on blood dendritic cell subsets. Front Immunol 2020; 11: 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torossian F, Guerton B, Anginot Aet al. Macrophage‐derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI Insight 2017; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Plaen IG, Han XB, Liu X, Hsueh W, Ghosh S, May MJ. Lipopolysaccharide induces CXCL2/macrophage inflammatory protein‐2 gene expression in enterocytes via NF‐kappaB activation: independence from endogenous TNF‐alpha and platelet‐activating factor. Immunology 2006; 118: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roy S, Schmeier S, Arner Eet al. Redefining the transcriptional regulatory dynamics of classically and alternatively activated macrophages by deepCAGE transcriptomics. Nucleic Acids Res 2015; 43: 6969–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang C, Lewis C, Borg NAet al. Proteomic identification of interferon‐induced proteins with tetratricopeptide repeats as markers of M1 macrophage polarization. J Proteome Res 2018; 17: 1485–1499. [DOI] [PubMed] [Google Scholar]
- 44.Poczobutt JM, De S, Yadav VKet al. Expression profiling of macrophages reveals multiple populations with distinct biological roles in an immunocompetent orthotopic model of lung cancer. J Immunol 2016; 196: 2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cochain C, Vafadarnejad E, Arampatzi Pet al. Single‐cell RNA‐Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res 2018; 122: 1661–1674. [DOI] [PubMed] [Google Scholar]
- 46.Hrdinka M, Yabal M. Inhibitor of apoptosis proteins in human health and disease. Genes Immun 2019; 20: 641–650. [DOI] [PubMed] [Google Scholar]
- 47.Nalepa G, Barnholtz‐Sloan J, Enzor Ret al. The tumor suppressor CDKN3 controls mitosis. J Cell Biol 2013; 201: 997–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grinenko T, Eugster A, Thielecke Let al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat Commun 2018; 9: 1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geng A, Qiu R, Murai Ket al. KIF20A/MKLP2 regulates the division modes of neural progenitor cells during cortical development. Nat Commun 2018; 9: 2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keophiphath M, Rouault C, Divoux A, Clément K, Lacasa D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler Thromb Vasc Biol 2010; 30: 39–45. [DOI] [PubMed] [Google Scholar]
- 51.Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol 2006; 35: 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulz D, Severin Y, Zanotelli VRT, Bodenmiller B. In‐depth characterization of monocyte‐derived macrophages using a mass cytometry‐based phagocytosis assay. Sci Rep 2019; 9: 1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Challen GA, Bertoncello I, Deane JA, Ricardo SD, Little MH. Kidney side population reveals multilineage potential and renal functional capacity but also cellular heterogeneity. J Am Soc Nephrol 2006; 17: 1896–1912. [DOI] [PubMed] [Google Scholar]
- 54.Chen S, Luo GR, Li T, Liu TX, Le W. Correlation of Nr4a2 expression with the neuron progenitors in adult zebrafish brain. J Mol Neurosci 2013; 51: 719–723. [DOI] [PubMed] [Google Scholar]
- 55.Ma G, Gao A, Yang Yet al. Zfhx3 is essential for progesterone/progesterone receptor signaling to drive ductal side‐branching and alveologenesis in mouse mammary glands. J Genet Genomics 2019; 46: 119–131. [DOI] [PubMed] [Google Scholar]
- 56.Tusi BK, Wolock SL, Weinreb Cet al. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 2018; 555: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verri WA Jr, Cunha TM, Ferreira SHet al. IL‐15 mediates antigen‐induced neutrophil migration by triggering IL‐18 production. Eur J Immunol 2007; 37: 3373–3380. [DOI] [PubMed] [Google Scholar]
- 58.Giladi A, Paul F, Herzog Yet al. Single‐cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat Cell Biol 2018; 20: 836–846. [DOI] [PubMed] [Google Scholar]
- 59.Merckx E, Albertini G, Paterka Met al. Absence of system xc ‐ on immune cells invading the central nervous system alleviates experimental autoimmune encephalitis. J Neuroinflammation 2017; 14: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia C, Braunstein Z, Toomey AC, Zhong J, Rao X. S100 proteins as an important regulator of macrophage inflammation. Front Immunol 2017; 8: 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xie Z, Guo Z, Liu J. Whey acidic protein/four‐disulfide core domain 21 regulate sepsis pathogenesis in a mouse model and a macrophage cell line via the Stat3/toll‐like receptor 4 (TLR4) signaling pathway. Med Sci Monit 2018; 24: 4054–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kool M, Geurtsvankessel C, Muskens Fet al. Facilitated antigen uptake and timed exposure to TLR ligands dictate the antigen‐presenting potential of plasmacytoid DCs. J Leukoc Biol 2011; 90: 1177–1190. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Raper A, Sugita Net al. Characterization of Siglec‐H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood 2006; 107: 3600–3608. [DOI] [PubMed] [Google Scholar]
- 64.Marçais A, Coupet CA, Walzer T, Tomkowiak M, Ghittoni R, Marvel J. Cell‐autonomous CCL5 transcription by memory CD8 T cells is regulated by IL‐4. J Immunol 2006; 177: 4451–4457. [DOI] [PubMed] [Google Scholar]
- 65.Pizzolato G, Kaminski H, Tosolini Met al. Single‐cell RNA sequencing unveils the shared and the distinct cytotoxic hallmarks of human TCRVδ1 and TCRVδ2 γδ T lymphocytes. Proc Natl Acad Sci USA 2019; 116: 11906–11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wein AN, McMaster SR, Takamura Set al. CXCR6 regulates localization of tissue‐resident memory CD8 T cells to the airways. J Exp Med 2019; 216: 2748–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weng NP, Araki Y, Subedi K. The molecular basis of the memory T cell response: differential gene expression and its epigenetic regulation. Nat Rev Immunol 2012; 12: 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Araki K, Morita M, Bederman AGet al. Translation is actively regulated during the differentiation of CD8+ effector T cells. Nat Immunol 2017; 18: 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cannons JL, Lau P, Ghumman Bet al. 4–1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol 2001; 167: 1313–1324. [DOI] [PubMed] [Google Scholar]
- 70.Verdeil G, Puthier D, Nguyen C, Schmitt‐Verhulst AM, Auphan‐Anezin N. STAT5‐mediated signals sustain a TCR‐initiated gene expression program toward differentiation of CD8 T cell effectors. J Immunol 2006; 176: 4834–4842. [DOI] [PubMed] [Google Scholar]
- 71.Cao X, Leonard K, Collins LIet al. Interleukin 12 stimulates IFN‐gamma‐mediated inhibition of tumor‐induced regulatory T‐cell proliferation and enhances tumor clearance. Cancer Res 2009; 69: 8700–8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu NQ, De Marchi T, Timmermans AMet al. Ferritin heavy chain in triple negative breast cancer: a favorable prognostic marker that relates to a cluster of differentiation 8 positive (CD8+) effector T‐cell response. Mol Cell Proteomics 2014; 13: 1814–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wakim LM, Gupta N, Mintern JD, Villadangos JA. Enhanced survival of lung tissue‐resident memory CD8⁺ T cells during infection with influenza virus due to selective expression of IFITM3. Nat Immunol 2013; 14: 238–245. [DOI] [PubMed] [Google Scholar]
- 74.Luetke‐Eversloh M, Killig M, Romagnani C. Signatures of human NK cell development and terminal differentiation. Front Immunol 2013; 4: 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vosshenrich CA, García‐Ojeda ME, Samson‐Villéger SIet al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA‐3 and CD127. Nat Immunol 2006; 7: 1217–1224. [DOI] [PubMed] [Google Scholar]
- 76.Yang C, Siebert JR, Burns Ret al. Heterogeneity of human bone marrow and blood natural killer cells defined by single‐cell transcriptome. Nat Commun 2019; 10: 3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gentles AJ, Newman AM, Liu CLet al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015; 21: 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arlauckas SP, Garren SB, Garris CSet al. Arg1 expression defines immunosuppressive subsets of tumor‐associated macrophages. Theranostics 2018; 8: 5842–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mantovani A, Allavena P. The interaction of anticancer therapies with tumor‐associated macrophages. J Exp Med 2015; 212: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010; 141: 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pucci F, Rickelt S, Newton APet al. PF4 promotes platelet production and lung cancer growth. Cell Rep 2016; 17: 1764–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen LH, Liao CY, Lai LC, Tsai MH, Chuang EY. Semaphorin 6A attenuates the migration capability of lung cancer cells via the NRF2/HMOX1 axis. Sci Rep 2019; 9: 13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang A, Dadaglio G, Oberkampf Met al. B cells promote tumor progression in a mouse model of HPV‐mediated cervical cancer. Int J Cancer 2016; 139: 1358–1371. [DOI] [PubMed] [Google Scholar]
- 84.Tedder TF. B10 cells: a functionally defined regulatory B cell subset. J Immunol 2015; 194: 1395–1401. [DOI] [PubMed] [Google Scholar]
- 85.Bodogai M, Moritoh K, Lee‐Chang Cet al. Immunosuppressive and prometastatic functions of myeloid‐derived suppressive cells rely upon education from tumor‐associated B cells. Cancer Res 2015; 75: 3456–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity 2015; 42: 607–612. [DOI] [PubMed] [Google Scholar]
- 87.Park J, Shrestha R, Qiu Cet al. Single‐cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 2018; 360: 758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ibañez‐Vega J, Del Valle Batalla F, Saez JJ, Soza A, Yuseff MI. Proteasome dependent actin remodeling facilitates antigen extraction at the immune synapse of B cells. Front Immunol 2019; 10: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reijmers RM, Spaargaren M, Pals ST. Heparan sulfate proteoglycans in the control of B cell development and the pathogenesis of multiple myeloma. FEBS J 2013; 280: 2180–2193. [DOI] [PubMed] [Google Scholar]
- 90.Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology 2009; 126: 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Getachew Y, Stout‐Delgado H, Miller BC, Thiele DL. Granzyme C supports efficient CTL‐mediated killing late in primary alloimmune responses. J Immunol 2008; 181: 7810–7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chowdhury D, Lieberman J. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu Rev Immunol 2008; 26: 389–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sadasivam S, Gupta S, Radha V, Batta K, Kundu TK, Swarup G. Caspase‐1 activator Ipaf is a p53‐inducible gene involved in apoptosis. Oncogene 2005; 24: 627–636. [DOI] [PubMed] [Google Scholar]
- 94.Zaki MH, Vogel P, Body‐Malapel M, Lamkanfi M, Kanneganti TD. IL‐18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol 2010; 185: 4912–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ryan N, Anderson K, Volpedo Get al. STAT1 inhibits T‐cell exhaustion and myeloid derived suppressor cell accumulation to promote antitumor immune responses in head and neck squamous cell carcinoma. Int J Cancer 2020; 146: 1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dai R, Phillips RA, Karpuzoglu E, Khan D, Ahmed SA. Estrogen regulates transcription factors STAT‐1 and NF‐kappaB to promote inducible nitric oxide synthase and inflammatory responses. J Immunol 2009; 183: 6998–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Farlik M, Reutterer B, Schindler Cet al. Nonconventional initiation complex assembly by STAT and NF‐kappaB transcription factors regulates nitric oxide synthase expression. Immunity 2010; 33: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Blondel VD, Guillaume J‐L, Lambiotte R, Lefebvre E. Fast unfolding of communities in large networks. J Stat Mech 2008; 2008: P10008. [Google Scholar]
- 99.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6: 359–362. [DOI] [PubMed] [Google Scholar]
- 100.Szklarczyk D, Franceschini A, Wyder Set al. STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43: D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kutmon M, Riutta A, Nunes Net al. WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res 2016; 44: D488–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Croft D, Mundo AF, Haw Ret al. The Reactome pathway knowledgebase. Nucleic Acids Res 2014; 42: D472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Perez‐Riverol Y, Csordas A, Bai Jet al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019; 47: D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures 1–14
Supplementary table 1
Supplementary table 2
Supplementary table 3
Supplementary table 4
Supplementary table 5
Supplementary table 6
Supplementary table 7
Supplementary table captions