

SUMMARY
The ubiquitous ribosome-associated complex (RAC) is a chaperone that spans ribosomes, making contacts near both the polypeptide exit tunnel and the decoding center, a position prime for sensing and coordinating translation and folding. Loss of RAC is known to result in growth defects and sensitization to translational and osmotic stresses. However, the physiological substrates of RAC and the mechanism(s) by which RAC is involved in responding to specific stresses in higher eukaryotes remain obscure. The data presented here uncover an essential function of mammalian RAC in the unfolded protein response (UPR). Knockdown of RAC sensitizes mammalian cells to endoplasmic reticulum (ER) stress and selectively interferes with IRE1 branch activation. Higher-order oligomerization of the inositol-requiring enzyme 1a (IRE1α) kinase/endoribonuclease depends upon RAC. These results reveal a surveillance function for RAC in the UPR, as follows: modulating IRE1α clustering as required for endonuclease activation and splicing of the substrate Xbp1 mRNA.
In brief
Wu et al. show that RAC modulates one of three branches of the unfolded protein response by specific effects on Xbp1 mRNA splicing. In spite of a ribosomal association similar to SRP, RAC does not influence substrate localization or pausing but rather formation of IRE1α foci required for enzyme activation.
Graphical Abstract
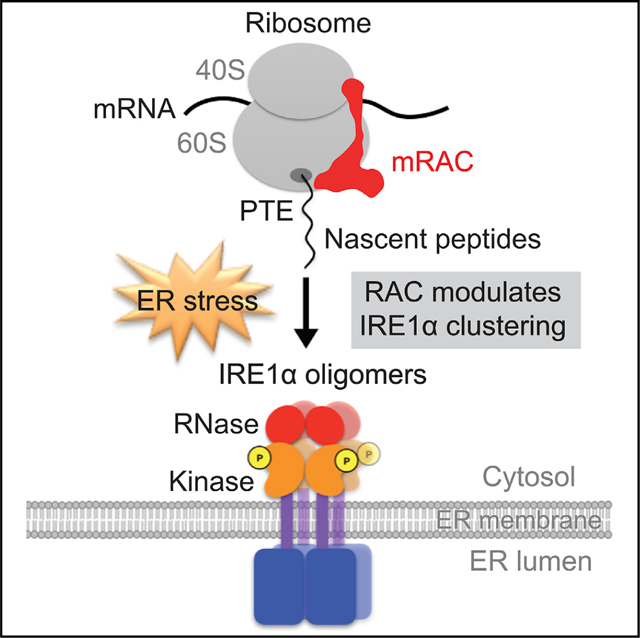
INTRODUCTION
Within the protein-dense interior of the cell, free molecular chaperones maintain protein homeostasis by facilitating post-translational folding and degradation of misfolded proteins under a wide range of environmental stresses (Balchin et al., 2016; Bukau et al., 2006; Hartl and Hayer-Hartl, 2009; Rosenzweig et al., 2019). Ribosome-associated chaperones, in addition to their probable roles in co-translational folding and degradation of defective nascent chains, occupy a position that enables them to preemptively influence the production of the protein (Deuerling et al., 2019; Kramer et al., 2009; Pechmann et al., 2013; Preissler and Deuerling, 2012; Zhang et al., 2017). As such, these complexes are at the forefront of quality control and homeostatic mechanisms. In contrast to post-translational chaperone mechanisms, chaperone actions on the ribosome remain less well understood. Elucidation of the details of ribosome-associated chaperone activity should offer insight into the known involvement of these systems in cancer, neurodegenerative disorders, and other human diseases (Hipp et al., 2014; Pechmann et al., 2013; Valastyan and Lindquist, 2014).
The ribosome-associated complex (RAC) is one of the two main ribosome-associated chaperones in eukaryotes. RAC is ubiquitous, is highly conserved, and directly binds to ribosomes near the polypeptide exit tunnel (PTE) where nascent chains are first exposed and near the decoding center where peptide bonds are formed with a 1:1 stoichiometry (Peisker et al., 2008; Yan et al., 1998). Mammalian RAC is a stable heterodimer composed of the non-canonical heat-shock protein 70 (Hsp70) homolog Hsp70L1 and its Hsp40 partner Mpp11 (Hundley et al., 2005; Jaiswal et al., 2011; Otto et al., 2005) (Ssz1 and Zuo1 in yeast) (Gautschi et al., 2001, 2002). Although Ssz1/Hsp70L1 lacks the C terminus domain of a canonical Hsp70, it retains the nucleotide-binding domain (NBD) that binds and hydrolyzes ATP as well as the substrate-binding domain (SBD) that occludes stretches of neutral and hydrophobic amino acids (Kampinga and Craig, 2010). Recent cross-linking data indicate that Ssz1 can directly interact with nascent polypeptides prior to Hsp70 chaperone Ssb1 contacts, indicating that Ssz1 is in position to be an active chaperone for co-translational folding (Zhang et al., 2020).
The core components of RACs are highly conserved and yet RAC has evolved functional diversity from yeast to higher eukaryotes. In yeast, growth defects in RAC depletion cells were only partially complemented by mammalian RAC under aminoglycoside stress (Jaiswal et al., 2011). Additionally, yeast RAC (Ssz1 and Zuo1) and its supporting Hsp70 chaperone Ssb1 are tethered to the ribosomes by forming a functional chaperone triad (Gautschi et al., 2001, 2002). Mammalian RAC also directly anchored to the ribosomes, but mammalian RAC recruits the cytosolic Hsp70 near the exit tunnel in a non-ribosome-associated manner. These differences between yeast and mammals imply some distinct regulatory mechanisms for mammalian RAC. In yeast, RAC stimulates the ATP hydrolysis of Ssb (Huang et al., 2005); in human, Mpp11 alone is efficient to stimulate modest levels of ATP hydrolysis by the cytosolic Hsp70 but requires the ATP binding of Hsp70L1 to fully activate the ATPase activity of RAC (Jaiswal et al., 2011). These data indicated that RAC has evolved distinct mechanisms to assist protein folding mediated by cytosolic Hsp70 chaperones in higher eukaryotes.
RAC, which associates with the ribosome near the PTE by its J domain partner Zuo1/Mpp11, spans the 60S and 40S ribosomal subunits (Peisker et al., 2008; Yan et al., 1998). Cross-linking and cryoelectron microscopy (cryo-EM) data indicate that Zuo1 interacts with eL31, close to the PTE, as well as eL22 and H24/H59 of 28S rRNA on the 60S subunit. On the 40S subunit, Zuo1 interacts with ES12 of H44 of 18S rRNA, which originates from the ribosome A-site base of the decoding center (Lee et al., 2016; Leidig et al., 2013; Zhang et al., 2014). Interestingly, Ssb1-bound substrates have an average lower translation rate than non-Ssb1 substrates (Willmund et al., 2013). Thus, the structural details of RAC’s position on the ribosome—near the nascent chain where it influences folding and near the decoding center where it influences translational rates—suggest it could coordinate de novo protein folding (Gautschi et al., 2001; Hundley et al., 2002; Pfund et al., 2001) and translation rates (Nelson et al., 1992). RAC’s ability to affect translation also modulates stop codon read through (Lee et al., 2016; Rakwalska and Rospert, 2004), −1 programmed ribosomal frameshifting (Muldoon-Jacobs and Dinman, 2006), and ribosome stalling and premature translation termination on luciferase reporter bearing C terminus poly-AAG/A sequences in yeast (Gribling-Burrer et al., 2019)—all by currently unknown mechanisms.
Consistent with these observations, the reduction of RAC expression is known to cause growth defects and sensitivity to aminoglycoside stress in both yeast and mammalian cells as well as to cold and osmotic stress in yeast (Gautschi et al., 2002; Hundley et al., 2005; Nelson et al., 1992; Yan et al., 1998). Yet, the mechanistic role of RAC in these stress responses remains obscure. Furthermore, there is a dearth of information regarding the physiological co-translational substrates of mammalian RAC. The homology of mammalian RAC to canonical Hsp70 chaperones and its presumed localization on cytosolic ribosomes led to the reasonable hypothesis that RAC supports co-translational folding of nascent cytosolic polypeptides (Otto et al., 2005). Here, we find that RAC is also associated with ER-bound ribosomes. Moreover, reduction in its expression does not activate the cytosolic heat shock response (HSR) as predicted by the hypothesis but rather indicates a central and unexpected role in modulating activation of the inositol-requiring enzyme 1α (IRE1α) branch of the unfolded protein response (UPR) at the ER.
RESULTS
Acute reduction of RAC selectively sensitizes cells to ER stress and attenuates the IRE1α arm of UPR
The hypothesis that RAC supports co-translational folding of nascent cytosolic polypeptides predicts that a reduction in RAC should lead to formation of incompletely folded cytosolic proteins and, thus, activation of the cytosolic HSR. To test this prediction, we reduced levels of RAC by transient transfection of small interfering RNA (siRNA) pools against Hsp70L1, a RAC component, for 48 h in HeLa cells and monitored the cytosolic HSR. As previously observed in yeast (Gautschi et al., 2001), reduction in Hsp70L1 led to a loss of its partner Mpp11 in HeLa cells (Figure 1A), suggesting it must be in complex with Hsp70L1 for stability. Unexpectedly, reduction of RAC by Hsp70L1 knockdown neither led to global protein aggregation (data not shown) nor sensitization to heat-shock-induced loss of viability after celastrol, which induces HSR by activating heat shock factor 1 (HSF1) (Westerheide et al., 2004; Figure 1B). Moreover, no measurable activation of the cytosolic HSR was observed after reduction of RAC by monitoring either phosphorylation of HSF1 (Figure 1C, top) or Hsp70 mRNA levels (Figure 1C, bottom), as would be expected if cytosolic proteins were misfolding.
Figure 1. RAC is required for activation of the IRE1α branch of the UPR by effects on Xbp1 splicing efficiency.

Effects of Hsp70L1 knockdown on the cellular manifestations and molecular correlates of the cytosolic heat shock response (HSR) and IRE1α branch of the UPR in the ER. Cytosolic stress was induced in HeLa cells by treatment with celastrol, which induces a HSR by activating heat shock transcription factor 1 (HSF1). ER stress was induced with thapsigargin, which inhibits the sarco-ER Ca2+ ATPase (SERCA) or DTT, which alters ER lumen redox and protein folding.
(A) Representative western blot (left) and quantification of RAC components in HeLa whole-cell lysates after treatment with siRNA against either control (C), Mpp11, or Hsp70L1 for 48 h. (right) Protein levels were normalized to GAPDH, and the si-control was set as 100%. White bar, Mpp11 protein level. Gray bar, Hsp70L1 protein level. ★, non-specific band.
(B and D) Cell viability was monitored using the MTS assay in HeLa cells pretreated with siRNA against either control (black diamond) or Hsp70L1 (red circle) for 48 h, followed by 24-h treatment with a DMSO negative control, celastrol (0.1, 1, 5, 10, 25 μM) (B), or thapsigargin (0.05, 0.1, 1, 5, and 50 μM) (D). (top) Cell viability (%) was normalized to DMSO-treated si-control cells. (bottom) Representative microscopic images of cell viability.
(C) (top) Representative phos-tag gel analysis of HSF1 phosphorylation, and (bottom) qRT-PCR of Hsp70 mRNA fold change in HeLa cells pretreated with either control (white bar) or Hsp70L1 (red bar) siRNA for 48 h, followed with or without a 3-h challenge with celastrol (2.5 μM). mRNA expression levels were normalized to the internal control HPRT and are shown as fold change relative to si-control (2−ΔΔCT).
(E) Effect of RAC on UPR activation was determined by monitoring the IRE1α-branch-mediated Xbp1 mRNA splicing using (top) RT-PCR and (bottom) qRT-PCR. Relative Xbp1 splicing efficacy (%) was calculated as [(spliced Xbp1/unspliced Xbp1) normalized to thapsigargin 4 h of vehicle si-control] × 100%. n = 3; ***p < 0.001, ****p < 0.0001.
(F) Dose-response curve of Xbp1 mRNA splicing efficiency (%) after treatment with individual siRNAs and the pool of all four against different targeting sequences of Hsp70L1. Hsp70L1 protein levels were determined by western blotting. Black circle is si-c; purple, navy, green, orange, and red are siHsp70L1–1, −2, −3, −4, and the pool, respectively. n = 5. Error bars, mean ± SD. (G) RAC knockdown on IRE1 activity as monitored by Xbp1 splicing efficacy (%) after DTT 1-h challenge.
We also monitored the UPR in the endoplasmic reticulum (ER), another stress pathway known to be activated by the accumulation of misfolded proteins in the ER. In contrast to the cytosolic HSR (Figure 1B), reduction in Hsp70L1 expression sensitized cells to treatment with an ER stress induced by thapsigargin (Figure 1D) that induces ER stress by inhibiting the sarco/ER Ca2+ ATPase (SERCA). Like the cytosolic HSR, the basal UPR pathway, as monitored by Xbp1 mRNA splicing, was not influenced by reduction in RAC in the absence of a UPR stimulus (Figure 1E). However, contrary to the prediction of a model in which RAC promotes folding of nascent polypeptides, reduction in RAC instead inhibited activation of UPR after a stimulus (Figure 1E).
To better understand the generality of the sensitization of cells to thapsigargin (0.5 μM) and reduction of the UPR response after Hsp70L1 and Mpp11 reduction, we monitored the activation of each of the three known arms of the UPR, namely, the IRE1α branch, the PRKR-like ER kinase (PERK) branch, and the activating transcription factor 6α (ATF6) branch. Rather than influencing all three arms as would be predicted for a generic role in ER co-translational folding, Hsp70L1 reduction selectively attenuated the activation of the IRE1α branch, as reflected in a reduced ability to splice Xbp1 mRNA as required for activation of transcription of many UPR responsive genes (Figure 1E) upon ER stress. Inhibition of Xbp1 mRNA splicing was also observed in RAC knockdown cells challenged with DTT (2 mM) for 1 h (Figure 1G), which induces ER stress by changing the luminal redox potential. This attenuation of Xbp1 splicing exhibited a clear dose dependence with the reduction in the amount of Hsp70L1 protein after treatment with siRNAs against different Hsp70L1 target sequences (Figure 1F). Altered RAC levels had no measurable effect on the PERK (Figure S1A) or the ATF6 (Figure S1B) branches of the UPR, either before or after activation of an ER stress response by thapsigargin for 4 h.
Xbp1 mRNA splicing is a non-canonical spliceosome-independent event (Cox and Walter, 1996; Mori et al., 1993), which takes place on the ER membrane (Yanagitani et al., 2009). There are three steps required for Xbp1 mRNA splicing. First, translation of uXbp1 mRNA is initiated and paused at a specific sequence (Yanagitani et al., 2009, 2011). Next, the stalled uXbp1 mRNA-ribosome-nascent chain (R-RNC) complex is targeted to the ER membrane by its hydrophobic region 2 (HR2) (Yanagitani et al., 2009). Then, upon ER stress, the IRE1α enzyme is activated by sequential steps of dimerization (Liu et al., 2000; Tirasophon et al., 1998), trans-autophosphorylation (Mori et al., 1993; Shamu and Walter, 1996; Zhou et al., 2006), and formation of high-order oligomers (Korennykh et al., 2009; Lee et al., 2008). These phosphorylated oligomers mediate the IRE1α endoribonuclease activity that excises the cryptic Xbp1 intron (Calfon et al., 2002; Yoshida et al., 2001). Once the 26-nucleotide (nt) intron is removed, the 5’ and 3’ fragments are re-joined by RtcB tRNA ligase (Jurkin et al., 2014; Kosmaczewski et al., 2014; Lu et al., 2014). Translation of the properly spliced Xbp1 mRNA is then terminated and the spliced mRNA released to the cytosol for reinitiation of translation and production of the stress responsive transcriptional factor sXbp1 (Cox and Walter, 1996). Understanding of the details of uXbp1 mRNA ribosome stalling and mechanisms by which oligomerization occurs and activates the nuclease remains incomplete. Because Xbp1 lacks a canonical signal sequence, novel mechanisms of targeting the stalled uXbp1 mRNA-ribosome-nascent chain complex (R-RNC) to the ER membrane are used (Yanagitani et al., 2011). The signal recognition particle (SRP) that recognizes canonical nascent chain signal sequences, pauses their translation, and targets them to the SRP receptor also plays a role in targeting of the stalled uXbp1 R-RNC to the ER membrane (Kanda et al., 2016; Plumb et al., 2015).
The position of RAC on the ribosome (Figure 2A), contacting both the 60S ribosomal subunit near the PTE and the 40S at the decoding center and thus overlapping with the SRP interaction surface, puts it in a position to potentially coordinate the co-translational regulation of Xbp1 mRNA splicing. This arrangement provided a structural basis for formulating hypotheses to explain the unexpected selective reduction of Xbp1 mRNA splicing in RAC knockdown cells. Three hypothetical mechanisms for RAC’s regulation of Xbp1 mRNA splicing were posed, as follows: (1) through modulation of substrate (uXbp1 mRNA) localization to the nuclease (IRE1α) on the ER prior to stress, (2) through direct activation of the nuclease activity (IRE1α phosphorylation, endoribonuclease activity, and high-order oligomerization) upon ER stress, and/or (3) through effects on substrate presentation or fitness (correct stalling) of the ribosome-associated mRNA substrate (Figure 2B).
Figure 2. RAC is not required for membrane targeting of the uXbp1 mRNA-ribosome substrate of the IRE1α-catalyzed splicing reaction.

Probable mechanisms of RAC regulation on Xbp1 mRNA splicing.
(A) (left) Cryo-EM density map of RAC with the 80S ribosome in S. cerevisiae by Chimera (EMD: 6103). RAC contacts both the 40S and 60S ribosomal subunits. RAC, red; 40S, gray; 60S, lavender; *, PTE, polypeptide tunnel exit. (right) Interaction sites of RAC with the 80S ribosome by PyMOL (PDB: 3J78). On the 60S subunits, RAC binds near the PTE by ribosomal protein eL22, uL22, and H24; H59 of 28S rRNA; as well as eL31 and H101. On the 40S subunits, RAC contacts the ES12 of H44 of 18S rRNA, which stems from the decoding center (DC) (Leidig et al., 2013; Yan et al., 1998; Zhang et al., 2014). RAC, black outline; *, PTE; eL31, green; H101, hot pink; H24, purple; H59, orange; eL22, cyan; uL22, yellow; H44, blue; PTC, the peptidyl transferase center.
(B) The Xbp1 splicing reaction. Under basal conditions, the IRE1α kinase-endonuclease E is in an inactive monomeric state on the ER. The substrate uXbp1 mRNA S requires several steps to be fit for the IRE1α enzyme and formation of the ES complex. First, translation must be initiated and then stalled at the proper location and the ribosome-uXbp1 is transported by SRP to the translocon on the ER membrane near where the IRE1α enzyme is located. The enzyme must also be activated. Upon ER stress (*), IRE1α is phosphorylated and then oligomerizes in foci. The active enzyme acts on the fit substrate to splice out the intron of uXbp1, releasing the product sXbp1 P. RAC could modulate Xbp1 splicing by affecting any of these steps.
(C) Subcellular fractionation of HeLa cells harvested after 4 h of DMSO control (blue bar) or thapsigargin (0.5 μM) (green bar) treatment was carried out using the sequential detergent extraction method. A total of 20% of total lysate was loaded; cytosol (Cyto)- and ER-membrane-bound fractions were collected and loaded in equal amounts. Quantification of western blots (left) for RAC and other components of the system used to determine a membrane localization value (ER/Cyto). Representative western blot images of subcellular fractionation (right). Rpl10 was used as a marker for ribosomes, BiP as a marker for ER lumen, Calnexin as marker for ER membrane, and GAPDH as a marker for cytosol. n = 3; *p < 0.05, **p < 0.01. Error bars, mean ± SD.
(D) RNA immunoprecipitation was performed in HeLa cells. Lysates were immunoprecipitated with antibody against IgG control or Mpp11, and the co-precipitated RNA was determined by qRT-PCR. (left) Representative western blot of bound and unbound proteins. A total of 1% total lysate was used as input. (right) uXbp1 mRNA is shown as Ct value. n = 3; **p < 0.01. Error bars, mean ± SD.
(E) Subcellular fractionation of RNA by the differential detergent method in HeLa cells pretreated with either vehicle control (white) or Mpp11 (blue) siRNA for 48 h.RNA was analyzed by qRT-PCR; samples were compared to vehicle si-control and calculated as 2−ΔCT. uXbp1 mRNA localization is shown as ER/cytosol. n = 2. Error bars, mean ± SEM.
RAC is not required for uXbp1 mRNA targeting to the ER membrane
Because Xbp1 mRNA splicing occurs on the ER membrane (Yanagitani et al., 2009), we predicted that RAC localizes to ER ribosomes in addition to its known presence in the cytosol (Otto et al., 2005). To further analyze the detailed subcellular localization of RAC in response to ER stress, we performed cell fractionation experiments by using differential detergent methodology (Jagannathan et al., 2011) after a challenge with 4 h of thapsigargin (0.5 mM) or DMSO negative control (Figure 2C) in HeLa cells. Mpp11 and Hsp70L1 are both enriched in the ER fraction under basal conditions in agreement with immunostaining of Mpp11 (data not shown). Concurrent ribosome distribution of 60% in the ER fractionation and 40% in the cytosol gave a membrane localization ratio (ER to cytosol) of nearly 1.6, suggesting no bias for cytosolic or ER ribosomes. These results suggest that RAC generally associates with ribosomes in both compartments but not all ribosomes, as it is substoichiometric to ribosomes (Kulak et al., 2014). After ER stress, both Mpp11 and ribosomes themselves are released from the ER to the cytosol with an attendant lowering of the membrane localization ratio (ER to cytosol ratio) to less than 1. The altered cellular localization of Mpp11 and ribosomes in response to ER stress may be due to several factors, including cells reducing global translation by regulated IRE1-dependent decay of mRNA (RIDD) as well as release of the small fraction of ribosomes on previously unspliced Xbp1 R-RNC. Interestingly, in contrast to the relocalization of Mpp11 with ribosome to the cytosol, Hsp70L1 remains on the ER after ER stress (Figure 2C).
To further understand the mechanism by which RAC regulates Xbp1 mRNA splicing, we monitored whether RAC and uXbp1 mRNA are in fact on the same ribosome-nascent chain complexes by RNA co-immunoprecipitation in HeLa cells. Cells were immunoprecipitated with antibody against either Mpp11 or IgG, and the co-precipitated RNA was analyzed by qRTPCR. Both Hsp70L1 and the ribosome were pulled down by Mpp11 (Figure 2D, left), consistent with an intact RAC-uXbp1 ribosome complex. Additionally, SRP54, a component of the SRP targeting secretory proteins to the ER, was not detected in the Mpp11 pulldown. This finding is to be expected considering that structural analysis indicates that SRP54 and RAC sterically clash when concomitantly modeled onto the ribosome, suggesting SRP54 and RAC may not bind to the same ribosome by known binding modes (Zhang et al., 2014). In summary, the results indicate that RAC, uXbp1 mRNA, and ribosomes are in the same macromolecular complex, as reflected in uXbp1 mRNA enrichment in the Mpp11 pulldown fraction (Figure 2D, right) as compared to the immunoglobulin G (IgG) control.
To directly test whether RAC regulates membrane targeting of uXbp1 mRNA as required for splicing, subcellular fractionation of uXbp1 mRNA (Figure 2E) from HeLa cells pretreated with either vehicle control (white) or Mpp11 (blue) siRNA for 48 h was performed. The localization of uXbp1 did not change in Mpp11 knockdown HeLa cells, indicating that RAC is not required for ER membrane targeting of uXbp1 mRNA. Consistent with this finding, recent studies demonstrate that although Xbp1 does not contain a canonical signal sequence, SRP54 targets uXbp1 mRNA to the SRP receptor on the ER membrane by binding to HR2 (Kanda et al., 2016; Plumb et al., 2015).
Reduction of RAC inhibits IRE1α oligomerization required for endoribonuclease activity
To determine whether RAC regulates Xbp1 mRNA splicing by modulating activation of the IRE1α endonuclease, we first monitored IRE1α clustering (Figures 3A–3E) by using an IRE1-GFP cassette whose expression is driven by a tetracycline-inducible CMV promoter in a stable T-REx293 cell line (Li et al., 2010). Consistent with prior reports (Tam et al., 2014), in HEK293 cells challenged with thapsigargin (0.5 μM), IRE1α formed multiple visible foci within 1 h, which over time condensed into larger foci at 4 h (Figures 3B and 3D) and then resolved by 8 h after the challenge. Reduction of RAC inhibited IRE1α foci formation as early as 1 h under thapsigargin (Figures 3B and 3D) and DTT (Figures 3C and 3E) treatment, in correlation with the observed reduction in Xbp1 splicing activity (Figures 3F and S2A). Previous biochemical data indicated that IRE1 oligomerization is required for Xbp1 mRNA splicing, whereas IRE1 dimer formation is adequate for RIDD activity due to the distinct substrate-binding interfaces in each multimer (Tam et al., 2014). Interestingly, reduction of RAC was without a measurable effect on RIDD activity, as shown by relative mRNA levels of Scavenger receptor class A member 3 (Scara3) (Figures 3G and S2B), and with only a very modest effect at time points longer than 4 h when monitoring Biogenesis of lysosome-related organelles complex-1 subunit 1 (Bloc1) (Figures S2C and S2D). These results demonstrate that reduction of RAC selectively inhibits IRE1α high-order oligomerization and activation of its endonuclease activity required for Xbp1 mRNA splicing.
Figure 3. RAC influences the formation of IRE1α foci involved in splicing.

(A–E) Representative fluorescent images of IRE1α clustering in the T-REx293 IRE1α-GFP cell line pretreated with either vehicle control (black diamond) or Hsp70L1 (red circle) siRNA for 48 h, followed by doxycycline (50 ng/μl) to induce tagged IRE1α expression for 24 h and DMSO control (A), thapsigargin for 1 and 4 h (B), or (C) DTT for 1 h. IRE1α, green; nuclei, Hoechst, blue. Scale bar, 10 μm. Quantification of IRE1α foci numbers and foci area size after challenge with thapsigargin (D) and DTT (E) by ImageJ. Results shown are compilation of two independent replicates. Each dot represents 1 field, 20 fields were analyzed in each repeat, and an average of 60 cells was analyzed per condition in each repeat. The mean is indicated by the line.
(F and G) Xbp1 mRNA splicing efficiency (%) was monitored using qRT-PCR (F), and RIDD activity was monitored (G) by mRNA fold change of Scavenger receptor class A member 3 (Scara3) over a 16-h thapsigargin time course in RAC knockdown HeLa cells under the same conditions used for generating the fluorescent images.
(H and I) (top) Representative western blot images of IRE1α kinase activity measured by phos-tag gel (H). HeLa cells were pretreated with either vehicle control (black diamond) or Hsp70L1 (red circle) siRNA for 48 h and challenged with thapsigargin (0.5 μM) for 0, 1, or 4 h (H) or with DTT (2 mM) for 0, 0.5, or 1 h (I). Hsp70 or Actin were used as loading controls. (bottom) p-IRE1 (%) was calculated as phosphorylated IRE1/total IRE1. n = 3; **p < 0.01. n = 3; *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars, mean ± SD.
IRE1α kinase activity was monitored by detecting IRE1α phosphorylation by using a phos-tag gel (Figure 3H). Acute reduction of RAC inhibited nearly 50% of IRE1α hyperphosphorylation activity compared to vehicle control after challenge with thapsigargin (0.5 μM) at 4 h but had no measurable change in phosphorylation of IRE1 after DTT treatment for up to 1 h.
Reduction of RAC increased translation and had only secondary effects on uXbp1 ribosome stalling under basal conditions
Careful studies by others have demonstrated that specific ribosome pausing of the Xbp1 mRNA is required for efficient Xbp1 mRNA splicing (Yanagitani et al., 2011). Although it is known that the C-terminal 26-residue region of uXbp1 is essential for translation pausing, the details of the stalling mechanism remain incompletely understood (Yanagitani et al., 2011). Yeast RAC has known effects on translation, and experiments to test whether mammalian RAC affects splicing efficiency by contributing to substrate “fitness,” as determined by translational stalling of Xbp1, were carried out using an engineered N-terminal FLAG tagged uXbp1 and sXbp1 mRNA (Figure 4A) similar to those reagents previously used (Yanagitani et al., 2011). These constructs, detected by an α-FLAG antibody, enabled determination of stalled translational intermediates and full-length product using NuPAGE Bis-Tris gels, under conditions that either preserve or remove the peptidyl-tRNA ester bond Xbp1. In vitro translation was performed in a mammalian cell-free lysate prepared from HEK293 cells pretreated with either vehicle control or Hsp70L1 siRNA for 48 h. Stalled intermediates of uXbp1 migrate at 47 kDa, confirmed by RNase A digestion to release the tRNA, whereas the full-length product migrates at 27 kDa (Figure 4B), as previously observed in the rabbit reticulocyte lysate cell-free system (Yanagitani et al., 2011). The hypothesis that RAC influences stalling by effects on the stability of the stalled complex predicts that more 47-kDa product will be observed upon loss of RAC. Interestingly, reduction of RAC increased both stalled and full-length products of uXbp1 and sXbp1 mRNA (Figures 4B, 4C, 4D, S3A, and S3B) due to its effects on translation but was without effects on the translation of exogenous, non-mammalian proteins GFP (Figure S3C) and Luciferase (Figure S3D) in the in vitro cell-free system. Contrary to the prediction, the reduction of RAC has a modest effect of a 30% increase on the uXbp1 stalled to full-length ratio (Figure 4E) and had no statistically significant effect on the stability of the uXbp1 stalled substrate measured as a ratio to the total Xbp1 translated (Figure 4F). Thus, the changes in the amount of the various products observed can largely be accounted for by increased translation rather than altered stalling.
Figure 4. RAC knockdown enhances translation.

(A) Workflow of cell-free protein synthesis. The in vitro transcription plasmids were designed containing the uXbp1 or sXbp1 human open reading frame, a N-terminal Flag epitope, and 30-nt poly-A sequence after the Xbp1 stop codon. mRNA templates were synthesized from linearized DNA templates by T7 RNA polymerase and 5′ capped. In vitro translation extracts were harvested after either vehicle control (black) or Hsp70L1 (red) siRNA treatment for 48 h. The reduction of Hsp70L1 to 36% and Mpp11 to 51% of control was confirmed by western blotting (bottom). Each translation reaction contained cell-free lysate, RNA templates, amino acids mixtures, energy generation system, and cofactors.
(B) A representative western blot of in vitro translation protein products of uXbp1 mRNA is shown. The reactions were incubated at 30°C for indicated incubation time. The samples were separated on NuPAGE Bis-Tris gels, which preserve the peptidyl-tRNA ester bond, and were detected by anti-FLAG Ab. White box, the shared domain of both uXbp1 and sXbp1. Blue box, the region of the spliced intron. Green and magenta box, the distinct segment of uXbp1 and sXbp1, respectively. In each experiment, RNase A was supplemented to confirm the identity of the stalled intermediate by cleaving the peptidyl-tRNA ester bond. ◆, uXbp1 stalling intermediates; ★, non-specific band; •, uXbp1 full-length product. n = 2.
(C and D) Gels were quantified for uXbp1 stalled (C) and full-length product (D). Symbols as above. (see Figure S3A for additional biological replicates and Figure S3B for sXbp1 in vitro translation protein products.
(E) The uXbp1 stalled to full-length ratio was normalized to the value at 10 min for the vehicle si-control.
(F) The uXbp1 stalled to total product ratio as a measure of stability was normalized to the 10-min reaction of vehicle si-control or si-Hsp70L1 in the same group. Protein half-life was determined using the initial rate as reflected in the 10- and 30-min time points. Si-c t 1/2 = 42 ± 8 (mins), si-Hsp70L1 t 1/2 = 59 ± 20 (mins). (G) Pulse labeling of nascent proteins with Click-IT AHA (L-azidohomoalanine), a methionine analog, for 0, 2, and 4 h in knockdown Mpp11 HeLa cells; pretreated with or without MG132 (5 μM) for 30 min; and separated by SDS-PAGE.
(H) Quantitation of the lane intensity of the gels. n = 3.
In this regard, significantly more de novo translated proteins were observed in the RAC knockdown (Mpp11) cells as monitored by pulse labeling using Click-it AHA (L-azidohomoalanine) (Figures 4G and 4H), a non-radioactive methionine analog. This effect was consistently observed with or without MG132, which blocks proteolytic activity of the 26S proteasome complex. Complicating interpretation is the fact that MG132 also transiently reduces translation at early time points (Cowan and Morley, 2004; Mazroui et al., 2007). Thus, enhancement of the general translation rate, rather than Xbp1-specific effects, in the knockdown appears to be responsible for the changes in the amounts of stalled and total Xbp1 protein and cannot readily explain the reduction in Xbp1 mRNA splicing.
Reduction of RAC increases uXbp1 ribosome stalling upon thapsigargin challenge
RAC’s position on the ribosome makes the substrate fitness hypothesis attractive, and it was also independently investigated using ribosome profiling (GEO: GSE173852). Under basal conditions of siRNA against vehicle control in HEK293 cells (Figure 5A, top), Xbp1 translating ribosomes stalled at codon Asn261, as expected (Shanmuganathan et al., 2019). Upon 4 h of thapsigargin treatment, the predominate ribosome density stalling at position Asn261 was diminished in the vehicle control and the overall increase in ribosome density occurred across uXbp1- and sXbp1overlapped 5’-end and M regions and the 3’-end region that encodes a distinct C terminus of the sXbp1 protein (Figure 5A, bottom), indicating the release of ribosome stalling subsequent to splicing and mRNA expression augmentation in both uXbp1 and sXbp1 upon the ER stress. Because IRE1α splices a 26-nt intron from uXbp1 mRNA, this results in an (+2)-nt frameshift in footprints, as shown in ribosome 3-nt periodicity analysis (Figure 5B), that encode the sXbp1 protein and with a C terminus distinct from uXbp1. To distinguish the ribosome footprint occupancy of the uXbp1 transcripts from the sXbp1 transcripts in the M region, we used the different 3-nt periodicity between the dominant form of uXbp1 (frame 0) under basal conditions and sXbp1 (frame 2) upon thapsigargin treatment (Figure 5B). The ribosome occupancy at Asn261 of uXbp1 reduced 24% upon thapsigargin treatment in siRNA vehicle control cells (Figure 5E), as a result of Xbp1 splicing.
Figure 5. RAC knockdown and ribosome occupancy on Xbp1 mRNA.

(A) Relative ribosome occupancy (reads per million[RPM]) within the Xbp1 transcript from siRNA against vehicle control in HEK293 cells with footprint displayed the position of ribosome A-site. The 5’ region, from the start codon to the splicing region; the middle region M is from the downstream splicing region to the stop codon of uXbp1; and the 3’ region, from the stop codon of uXbp1 to sXbp1, are indicated above the histogram. Under basal conditions (top histogram), ribosomes predominately occupied the Asn261 site (▾) of uXbp1. After 4 h of thapsigargin (bottom), the 26-nt intron is spliced out (gray box between 5’ and M regions), causing a (+2)-nt frameshift in footprints due to reinitiation and translation of the sXbp1 mRNA and an attendant substantial increase in occupancy in 3’-end region. The left-hand dotted line indicates the start site of Xbp1. The second dotted line indicates the stop codon of uXbp1, which is just 3’ to the Asn261 site. The right-hand dotted line indicates the stop codon of sXbp1. Gray box in the histogram indicates the Xbp1 splicing region. Scale bar, 200 nt.
(B) The 3-nt periodicity of the relative footprint (%) of the Xbp1 transcript from the 5’ region, the M region, and the 3’ region from siRNA against vehicle control under basal conditions (top) or 4 h of thapsigargin treatment (bottom). In the M region, the frame 0 footprints are predicted to be from uXbp1 transcripts and frame 2 footprints from sXbp1 transcripts. A clear shift in the periodicity in the M region is observed after thapsigargin treatment, as expected.
(C) Representative western blots of knockdown efficiency. Cells were harvested after pretreatment with the designated siRNA (vehicle control, white; Hsp70L1, red; Pelo, yellow; both Hsp70L1 and Pelo, orange) for 48 h and challenged with thapsigargin (0.5 μM) for 4 h.
(D) Effects of RAC on relative occupancy on uXbp1 and sXbp1 are reflected in the Log2 fold change of frame 2 (sXbp1)/frame 0(uXbp1) in the M region after thapsigargin treatment. These changes could be due to decreased IRE1 activity or in increased stalling. siRNA against vehicle control was set as 0. The line indicates the mean from two biological replicates. Δ and ⚬ represent the individual repeats.
(E) The relative ribosome occupancy at Asn261 (%) from frame 0 footprints in the M region of the uXbp1 transcript. Bars, mean from two biological replicates. Solid bars, DMSO. Hatched bars, after thapsigargin treatment. Δ and ⚬ indicate individual repeats.
As predicted, upon ER stress, reduction of RAC inhibited the frame 2 (sXbp1) to frame 0 (uXbp1) ratio in the M region under thapsigargin treatment, consistent with the Xbp1 splicing assay (Figure 1E). Contrary to the expectation that altered substrate stalling/fitness was contributing, no significant increase in ribosome stalling at the uXbp1 pausing site Asn261 was observed in knockdown RAC cells as compared to vehicle control under basal conditions (Figures 5E, S4A, S4B, and S4C). Notably, ribosomes accumulated at the uXbp1 stalling site in knockdown of RAC cells upon thapsigargin treatment with a 20% increase compared to DMSO. These results indicate that the inhibition of Xbp1 splicing is unlikely to be due to an abnormality of Xbp1 ribosome stalling but are consistent with a reduction of IRE1α endonuclease activity, as reflected in reduction of foci.
Because the Xbp1 stalled complex is not only subject to Xbp1 splicing by IRE1α upon ER stress but also subject to no-go decay (NGD) (Arribere and Fire, 2018; Guydosh and Green, 2014), disturbance of the mRNA surveillance pathway by manipulating Pelo expression was assessed. Knockdown of Pelo was expected to have an additive effect on ribosome stalling in knockdown RAC cells. Interestingly, the reduction in the frame 2 (sXbp1) to frame 0 (uXbp1) ratio in RAC knockdown was counteracted by Pelo knockdown (Figures 5C and 5D) and reflected in the Xbp1 splicing assay (Figure 6A).
Figure 6. RAC and translation dependence and Xbp1 mRNA independence of IRE1α clustering.

Cells were transiently transfected with siRNA against vehicle control, Hsp70L1, Pelo, or both Hsp70L1 and Pelo for 48 h.
(A) Quantification of Xbp1 mRNA splicing efficiency of designated siRNA knockdown by qRTPCR after 4 h of thapsigargin (0.5 μM) treatment in HEK293 cells. n = 3. Error bars, mean ± SD.
(B) Representative fluorescent image of IRE1α foci. IRE1α, green; nuclei, Hoechst blue.
(C) IRE1α foci were monitored after challenge with thapsigargin for 4 h in the designated knockdown cells. n = 2. Foci number (top) and foci area size (bottom) by ImageJ in T-REx293 IRE1-GFP cell line after treatments.
(D) Time course of IRE1α foci formation after thapsigargin treatment compared with vehicle control (left) or Xbp1 siRNA (right) for 48 h. n = 3. (E) The translation dependence of IRE1α foci formation was examined in cells pretreated with either control (harringtonine, 2 μg/ml) or cycloheximide (100 μg/ml) for 20 min, followed by 1 or 4 h of thapsigargin (0.5 μM) treatment. Each group was compared to untreated negative control. n = 2. Each dot represents 1 field, 20 fields were analyzed for each condition, and an average of 60 cells were analyzed per condition in each experiment. The mean was presented. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Scale bar, 10 μm.
Although the reduction of Pelo was predicted to trigger an increase in the ribosome occupancy at Asn261 of uXbp1, no significant change was observed with the messages used in this study. This result is consistent with the finding in Caenorhabditis elegans that depletion of Pelo alone has only mild effects on Xbp1 ribosome stalling on truncated mRNA (Arribere and Fire, 2018). A much more profound effect required a double mutant of Pelo and Ski2, a component of the ski exosome complex for RNA 3’ to 5’ degradation. Interestingly, double knockdown of RAC and Pelo increased ribosome occupancy at Asn261 of uXbp1 approximately 22% under basal conditions compared to siRNA vehicle control, and the increase of uXbp1 ribosome stalling in the reduction of RAC upon thapsigargin challenge was reverted in Pelo knockdown.
Reduction of Pelo counters the inhibition of Xbp1 mRNA splicing and IRE1α clustering in RAC knockdown during ER stress
Because both the in vitro translation assay and ribosome profiling suggest that the inhibition of Xbp1 splicing in knockdown RAC cells is unlikely due to aberrant Xbp1 ribosome stalling but may explained by a reduction of IRE1 endonuclease activity, Xbp1 mRNA splicing and IRE1α high-order oligomerization were monitored in Pelo and RAC double knockdown cells. In HEK293 cells, knockdown of Pelo increased Xbp1 mRNA splicing (Figure 6A), consistent with the concept that Dom34/Pelo evolved in the mRNA surveillance pathway of Hac1/Xbp1 in Saccharomyces cerevisiae (Guydosh and Green, 2014). Knockdown of Pelo countered the inhibition of Xbp1 mRNA splicing in RAC knockdown HEK293 cells (Figure 6A). Finally, knockdown of Pelo partly rescued the inhibition of RAC knockdown on IRE1α clustering in HEK293 cells (Figures 6B and 6C).
To examine whether the inhibition of IRE1α clustering in RAC knockdown is directly due to lack of the fitness of its Xbp1 substrate, we knocked down Xbp1 mRNA (Figure S5A) and monitored IRE1α clustering. After 1 h of thapsigargin treatment, there were no significant differences in IRE1α clustering when comparing vehicle control and knockdown Xbp1 cells and only modest differences after 4 h of thapsigargin treatment (Figures 6D and S5C), consistent with previous findings in yeast (Aragón et al., 2009), indicating that the presence of this substrate is not required for IRE1α phosphorylation (Figure S5B) or formation of the active high-order oligomeric complexes. Together, these data indicate that activation of IRE1α clustering does not require Xbp1 mRNA, suggesting that inhibition of IRE1α foci formation in RAC knockdown is not due to a lack of fitness of the Xbp1 substrate.
Because we also observed that RAC reduces translation rate, we hypothesized that the inhibition of IRE1α clustering could result from the slowdown of translation rate. To test this hypothesis, we pretreated cells with the translation inhibitors harringtonine and cycloheximide, which inhibit translation initiation and elongation, respectively, and then challenged with thapsigargin (0.5 mM). Strikingly, IRE1α clustering was dramatically inhibited as early as 1 h after ER stress in translation-inhibited cells (Figure 6E), whereas IRE1α phosphorylation was unperturbed in IRE1α-GFP cells (Figure S5F) and wild-type (WT) HEK293 cells (Figure S5G). This result suggests that translation of substrates other than Xbp1 are required for IRE1α clustering. The partly rescued inhibition effect on IRE1α clustering on double knockdown of Pelo and RAC also suggests that mRNA quality control may play a role in modulating IRE1α clustering.
Taken together, these results present a model in which RAC plays an essential role as a stress-responsive regulator (Figure 7). RAC modulates IRE1α activation, including IRE1α higher-order oligomerization, which is an Xbp1-independent and translation-dependent process required for activation of the attendant endonuclease activity.
Figure 7. A model for RAC’s role in IRE1α oligomerization and activation.

Schematic of the steps at which RAC influences the IRE1α branch of UPR. Under basal conditions, the monomeric IRE1α kinase-endonuclease is stabilized in an inactive form by binding to BiP. Translation of the IRE1α’s substrate uXbp1 mRNA is initiated and stalled on cytosolic ribosomes, presumably by SRP (magenta), which targets the substrate complex from the cytosol to the ER membrane. Upon ER stress, unfolded proteins compete with IRE1α for BiP and release of BiP allows IRE1α to dimerize, activating its kinase activity and mediating production of the autophosphorylation, which can catalyze RIDD. The dimerized IRE1α can further oligomerize, forming foci that correlate with the activation of the splicing endonuclease activity catalyzed by IRE1α. The cryptic exon is spliced from uXbp1, stalling relieved, translation terminated, and the sXbp1 released from the ribosome. The translation of sXbp1 mRNA is reinitiated, producing the Xbp1 transcription factor, which then activates expression of UPR genes. RAC (red) plays an unexpected role in activation of UPR by modulating the IRE1α high-order oligomerization required for activity. Activation depends upon translation but independently of Xbp1. The similarities in the RAC ribosome and SRP ribosome complex and their structural relationship to the nascent chain exit tunnel and the decoding center are shown in cryo-EM density maps of SRP (EMD: 1063) or RAC (EMD: 6103) with the 80S ribosome produced with Chimera. The IRE1α enzyme activation and the substrate preparation processes are indicated below the structure by the cartoons. IRE1α domains: red, RNase; orange, kinase; purple, transmembrane; blue, luminal domain.
DISCUSSION
RAC is a ubiquitous, highly conserved complex directly anchored to cytosolic and ER-bound ribosomes near both the PTE and the decoding center. Initially, homology to canonical cytosolic Hsp70 chaperones led to the suggestion that RAC was involved in cytosolic co-translational folding and response to cytosolic HSR. More recently, RAC’s involvement in translational activity has been reported in yeast (Gribling-Burrer et al., 2019; Lee et al., 2016; Muldoon-Jacobs and Dinman, 2006; Nelson et al., 1992; Rakwalska and Rospert, 2004). In spite of the structures revealing that RAC is uniquely positioned to govern protein homeostasis by coordinating co-translational protein folding and translational activity, to our knowledge, such coordination has not been demonstrated. The results presented here demonstrate RAC’s role in mediating communication between ER stress and translation activity. Moreover, the data reveal an unexpected and critical role of mammalian RAC in the UPR related to these activities. RAC acts as a stress-responsive element by modulating IRE1α clustering as required for splicing and translation of IRE1α’s substrate, namely, Xbp1, on the ribosome.
RAC is composed of the two subunits Hsp70L1 and Mpp11, which is a DNAJ homolog. Here, both subunits were acutely reduced by transient knockdown of Hsp70L1 with siRNA pools in HeLa and HEK293 cells, as Mpp11 stability depends on the presence of Hsp70L1. The use of this transient model reduced the influence of adaptive responses observed when RAC was reduced over a longer term. Knockout lines produced using CRISPRCas9 against Hsp70L1 by using two independent guide RNAs (gRNAs) in 293T cells (data not shown) dramatically impaired cell growth. However, the slow growth rate of the knockout RAC cells began to recover after continuous culture, suggesting that adaptations are occurring over extended culture times. Together, these results suggest that mammalian RAC is an essential gene product and that long-term knockout models may not be suitable for mechanistic studies.
Consistent with their homology, mammalian and yeast RAC share some common structural and functional features. Like the yeast RAC, human Hsp70L1 is associated with the ribosome by Mpp11 (data not shown). RAC-deficient human cells exhibited profound growth defects, consistent with the yeast system (Gautschi et al., 2001; Hundley et al., 2005; Jaiswal et al., 2011; Otto et al., 2005; Yan et al., 1998). Here, human RAC-deficient cells were sensitized to ER stress. Reduction of RAC sensitized both yeast and human cells to translation stress (Jaiswal et al., 2011; Otto et al., 2005). Unexpectedly, knockdown of RAC did not activate the cytosolic HSR as predicted by the hypothesis that RAC is required for cytosolic co-translational folding in human cells (Otto et al., 2005). Rather, the results indicate that one role RAC plays is a central part of the ER stress response—knockdown blunting activation of this pathway.
Rather than a general effect on the UPR, transient reduction of RAC specifically inhibited the activation of the IRE1 arm of the UPR in response to two ER-stress-promoting agents. The data indicate that RAC is required for IRE1α oligomerization but does not have significant effects on dimerization of the kinase. Although dimerization is required for the early autophosphorylation event, Xbp1 splicing is carried out by IRE1α oligomers (Aragón et al., 2009; Kimata et al., 2007; Li et al., 2010), which form subsequent to hyperphosphorylation. In contrast, RIDD substrates are spliced by IRE1α dimers (Tam et al., 2014), an activity unaffected by RAC knockdown. The unexpected role of RAC in modulating IRE1α oligomerization raises the question of how a ribosome-associated chaperone is capable of modulating association of IRE1α, an ER integral membrane protein whose dimerization is controlled by the luminal chaperone BiP, from the cytosol. The data also demonstrate that IRE1 clustering is a translation-dependent but Xbp1-independent process, suggesting that translation of substrates other than Xbp1 may be required for IRE1α clustering.
Notably, the effects of RAC on translation were restricted to endogenous mRNAs, as we did not detect any differential changes of GFP and Luc activities between WT and knockdown RAC in cell-free systems. One possibility is that cells have mechanisms for complex and precise regulation of natural cellular genes but not exogenous genes. Additionally, Xbp1 splicing data indicated reduced production of sXbp1 in RAC knockdown cells upon ER stress. Unexpectedly, this reduction was counteracted by knockdown of Pelo, suggesting that RAC and Pelo may act on related pathways.
In conclusion, RAC has an unexpected and central role in ER stress through its control of IRE1α clustering. The role of RAC in modulating the translation rate of natural substrate mRNAs and its presence on both cytosolic and ER ribosomes suggest that this activity is not restricted to Xbp1. The reciprocal effects of RAC and Pelo knockdown raise the possibility that RAC may also play a role in NGD. Thus, the ubiquitously expressed RAC, occupying an evocative position on ribosomes and required for optimal growth, may play a part in governing protein homeostasis by coordinating both protein and mRNA quality-control pathways.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Philip Thomas (philip.thomas@utsouthwestern.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The ribosome profiling datasets generated during this study are available at the Gene Expression Omnibus (GEO) database [GSE173852].
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HeLa Tet-on (Clontech), HEK293 (American Type Culture Collection, ATCC), HEK293T (ATCC), and T-REx293 IRE1-GFP cells (Li et al., 2010) were maintained at 37C, 5% CO2 in Dulbecco’s Modified Eagle’s Medium (Sigma) supplemented with 4500 mg/L glucose, L-glutamine, sodium pyruvate, sodium bicarbonate, 10% fetal bovine serum (Sigma), 100 U/ml penicillin, and 100 mg/ml streptomycin (Sigma). T-REx293 IRE1-GFP cells were kindly provided by Dr. Peter Walter (UCSF/HHMI). Transient transfection of plasmids were performed using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Transient gene knockdown was accomplished by transfection of small double-stranded interfering RNAs (siRNA) into cells using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. The siRNAs were synthesized by Dharmacon using the sequence information in the Key resources table. Silencer Negative Control No. 1 siRNA (Ambion) or ON-TARGET plus Non-targeting Control siRNA #1 (Dharmacon) were used as vehicle negative controls.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |||
|---|---|---|---|---|---|
|
| |||||
|
Antibodies
| |||||
| Mpp11 | Cell Signaling | Cat#12844, RRID: AB_2798042 | |||
| Hsp70L1 | Abcam | Cat#ab108612, RRID: AB_10866191 | |||
| GAPDH | Cell Signaling | Cat#2118S, RRID: AB_561053 | |||
| PERK | Cell Signaling | Cat#3192, RRID: AB_2095847 | |||
| Calnexin | Cell Signaling | Cat#2679; RRID: AB_2228381 | |||
| BiP | Cell Signaling | Cat#3177, RRID: AB_2119845 | |||
| Rpl10 | Abnova | Cat#PAB17331, RRID: AB_10643619 | |||
| SRP54 | BD | Cat#610940, RRID:AB_398253 | |||
| IRE1α | Cell Signaling | Cat#3294, RRID: AB_823545 | |||
| Hsp70 | Cell Signaling | Cat#4872, RRID: AB_2279841 | |||
| Pelo | Proteintech | Cat#10582-1-AP, RRID: AB_2236833 | |||
| Flag | Sigma | Cat#F1804, RRID: AB_262044 | |||
| IRDye 680RD secondary antibodies goat anti-mouse IgG |
Li-Cor | Cat#926-68070, RRID: AB_10956588 | |||
| IRDye 680RD secondary antibodies goat anti-rabbit IgG |
Li-Cor | Cat#926-68071, RRID: AB_10956166 | |||
|
| |||||
|
Chemicals, peptides, and recombinant proteins
| |||||
| Thapsigargin | Sigma | Cat#T9033 | |||
| Celastrol | Sigma | Cat#C0869 | |||
| Cycloheximide | Sigma | Cat#C7698 | |||
| Homoharringtonine | Sigma | Cat#SML1091-10MG | |||
| Doxycycline | Sigma | Cat#D9891 | |||
| Digitonin | Millipore | Cat#300410 | |||
|
| |||||
|
Critical commercial assays
| |||||
| CellTiter 96 AQueous one solution cell proliferation assay (MTS) | Promega | Cat#G3582 | |||
| High-capacity cDNA reverse transcription kit | ThermoFisher Scientific | Cat#4368814 | |||
| Click-iT protein reaction buffer kit | ThermoFisher Scientific | Cat#C10276 | |||
|
| |||||
|
Deposited data
| |||||
| Ribosome profiling | This paper | GEO: GSE173852 | |||
|
| |||||
|
Experimental models: Cell lines
| |||||
| HeLa Tet-on | Clontech | N/A | |||
| HEK293 | ATCC | CRL-1573 | |||
| HEK293T | ATCC | CRL-11268 | |||
| T-REx293 IRE1-GFP | Li et al., 2010 | N/A | |||
|
| |||||
|
Oligonucleotides
| |||||
| siRNA Hsp70L1 target sequence-1: CAGAAAUACAUCGCGGAAA |
Dharmacon | M-021084-01-0005 | |||
| siRNA Hsp70L1 target sequence-2: UAACAUCGGUGGUGCACAU |
Dharmacon | M-021084-01-0005 | |||
| siRNA Hsp70L1 target sequence-3: GGAAAUGCGCGAGCCAUGA |
Dharmacon | M-021084-01-0005 | |||
| siRNA Hsp70L1 target sequence-4: GUAUUGGGCUCAGAUGCAA |
Dharmacon | M-021084-01-0005 | |||
| siRNA Mpp11 target sequence-1: GAACCAAGAUCAUUAUGCA |
Dharmacon | M-025435-02-0005 | |||
| siRNA Mpp11 target sequence-2: GAAAUCAACUGGUGGAGGU |
Dharmacon | M-025435-02-0005 | |||
| siRNA Mpp11 target sequence-3: GAACUUGUCGAGAUGGUAA |
Dharmacon | M-025435-02-0005 | |||
| siRNA Mpp11 target sequence-4: AGGACUGCAUGAAACGAUA |
Dharmacon | M-025435-02-0005 | |||
| ON-TARGETplus Non-targeting Control siRNAs #1: UGGUUUACAUGUCGACUAA |
Horizon | D-001810-01-20 | |||
| Additional primer sequences | Tables S1 and S2 | ||||
|
| |||||
|
Recombinant DNA
| |||||
| Plasmid: pcDNA3.1(+)-Flag-uXbp1-A | GenScript | N/A | |||
| Plasmid: pcDNA3.1(+)-Flag-sXbp1-A | GenScript | N/A | |||
| Plasmid: pJI204-T7-GFP-A | Yang et al., 2019 | N/A | |||
| Plasmid: pJI204-T7-Luc-A | Yang et al., 2019 | N/A | |||
|
| |||||
|
Software and algorithms
| |||||
| Fiji/ImageJ | NIH image | https://imagej.net/Fiji | |||
| Prism 8 | GraphPad | https://www.graphpad.com/scientificsoftware/prism/ | |||
METHOD DETAILS
Cell viability
HeLa cells were transiently transfected with siRNA against either control or Hsp70L1 for 48 hr. Cells were plated at a density of 5,000 cells per well in triplicate in a 96-well plate. Cells were treated with either DMSO control, Celastrol (0.1, 1, 5, 10, 25 μM) or Thapsigargin (0.05, 0.1, 1, 5, 50 μM) for 24 hr. Cell viability was measured using CellTiter 96 (Promega), which is an MTS-based assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium), according to the manufacturer’s instructions.
Briefly, 20 μl of CellTiter reagent was added into 100 μl of cell medium at the indicated time point, incubated at 37°C for 1 hr, and absorbance at 490 nm was monitored using a SpectraMax plus 384 microplate reader (Molecular Devices). Cell viability (%) was normalized to DMSO treated vehicle si-control cells.
RNA isolation, cDNA synthesis and quantitative real time-PCR
Total RNA was isolated from cells using the NucleoSpin RNA kit (Machery-Nagel) according to the manufacturer’s instructions. RNA was quantified using a NanoDrop 2000c Spectrophotometer. cDNAs were synthesized from 2 μg of total RNA template using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific). cDNA synthesis conditions were as follows: 25°C for 10 min, 37°C for 120 min, 85°C for 5 min. qPCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) on a 7900HT Fast Real-Time PCR System with a 384 well block module (Applied Biosystems). qPCR conditions were as follows: 50°C for 2 min, 95°C for 10 min, 95°C 15 s, 60°C for 1 min, with 40 cycles of amplification. The primers used for qPCR amplifications are shown in the Key resources table. Fold change mRNA expression levels were determined by the 2−ΔΔCT method (Livak and Schmittgen, 2001). The Ct value in each condition was normalized to an HPRT internal control and then normalized to siRNA vehicle control sample. Each cDNA was measured in triplicate per sample for each primer pair.
Xbp1 mRNA splicing assay
Xbp1 mRNA splicing was measured either by PCR amplification flanking the intron splicing site or by qPCR using spliced and unspliced specific primers. cDNA were produced and used as template to amplify the Xbp1 fragments. For PCR amplification, flanking primers (in the Key resources table) were used to generate a 474 bp amplicon from unspliced Xbp1 or a 448bp amplicon from spliced Xbp1. PCR conditions were as follows: 98°C for 30 s, 98°C for 10 s, 60°C for 30 s, 72°C for 30 s, with 30 cycles of amplification followed by 72°C 10 min. The fragments were separated on a 2% Agarose/1X TBE gel (Lonza, MetaPhor Agarose), visualized by ethidium bromide staining, and detected using a GelDoc-It2 imager (UVP). For qPCR, specific primers (in the Key resources table) were used. Xbp1 splicing efficacy (%) was calculated as [(spliced Xbp1/unspliced Xbp1) normalized to thapsigargin 4h si-control] X 100%.
Protein analysis by immunoblotting
Cells were lysed in radio-immunoprecipitation assay buffer (RIPA) (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% (v/v) NP-40, 0.5% (w/v) Sodium deoxycholate, 0.1% SDS) supplemented with 1X cOmplete EDTA-free protease and phosphatase inhibitor cocktail (Roche). Total cell lysates were cleared by centrifugation at 16,000 g for 10 min at 4°C. Total supernatant protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific). 18 μg of cleared total cell lysates were supplement with 5X Laemmli sample buffer (10% SDS, 250 mM Tris-HCl pH 6.8, 0.1% (w/v) Bromophenol blue, 50% Glycerol, 500 mM DTT), heated at 95°C for 10 min, and separated on a 10% Tris-glycine gel prior to being transferred onto 0.45 μm PVDF membranes (Millipore Immobilon-FL). Membranes were blocked in 5% (w/v) nonfat dry milk in 1X TBS with 0.1% Tween-20 for 1 hr in room temperature. Blots were incubated with the indicated primary antibodies at 4°C overnight in 1X TBS with 0.1% Tween-20 with gentle rocking, followed by indicated secondary antibodies at room temperature for 1 h in 1X TBS with 0.1% Tween-20 and visualized using a LI-COR Odyssey CLx.
Phos-tag mobility shift assay
For the phos-tag assay, samples were collected in RIPA buffer as described for total cell lysates above and heated at 95°C for 5 min. 25 μg of total cell lysates were analyzed by 6% Tris-glycine gel supplemented with 25 μM Phos-tag acrylamide (Wako) and 50 μM MnCl2. SDS-PAGE was run at a constant 100 V for 3 hr prior to transfer onto 0.45 μm PVDF membranes (Millipore Immobilon-FL) at 400 mA for 2 hr and visualized using a LI-COR Odyssey CLx.
Subcellular fractionation
Cell fractionation was performed by sequential detergent extraction method (Jagannathan et al., 2011) with critical technical advice from Dr. Christopher Nicchitta at Duke University. Briefly, HeLa cells were plated onto 6-well plates one day prior to cell harvest and were grown to 70%–80% confluency on the harvest day. Cells were washed with 2 mL of cold PBS and treated with 100 μg/ml cycloheximide in PBS for 10 min on ice. Cells were then permeabilized using 200 μl Permeabilization buffer (110 mM KOAc, 25 mM K-HEPES, pH 7.2, 2.5 mM Mg(OAc)2, 1 mM EGTA, 0.03% Digitonin, 1 mM DTT, 50 μg/ml cycloheximide, 1x cOmplete EDTA-free protease inhibitor cocktail (Roche), 100 U/ml RNasin ribonuclease inhibitors (Promega) and incubated on ice for 5 min. Plates were tilted to drain the soluble material, which was collected as the cytosolic fraction. Cells sans cytosol were then washed with 200 μl Wash buffer (110 mM KOAc, 25 mM K-HEPES, pH 7.2, 2.5 mM Mg(OAc)2, 1 mM EGTA, 0.004% Digitonin, 1 mM DTT, 50 μg/ml cycloheximide, 1x cOmplete EDTA-free protease inhibitor cocktail, 100 U/ml RNasin ribonuclease inhibitors). Next, cells were treated with 200 μl Lysis buffer (400 mM KOAc, 25 mM K-HEPES, pH 7.2, 15 mM Mg(OAc)2, 1% NP-40, 1 mM DTT, 50 μg/ml cycloheximide, 1x cOmplete EDTA-free protease inhibitor cocktail, 100 U/ml RNasin ribonuclease inhibitors) and incubated on ice for 5 min. Plates were tilted to drain the soluble material which was collected as membrane fraction. Both cytosolic and membrane fractions were subjected to centrifugation at 7,500 g for 10 min to remove insoluble debris. Samples were dissolved in 5x Laemmli sample buffer and DTT, heated at 95°C for 10 min, and separated by SDS-PAGE. For analysis of uXbp1 RNA localization, each fraction was subjected to RNA purification by NucleoSpin RNA (Machery-Nagel) followed by cDNA synthesis and analysis by qRT-PCR.
RNA immunopreciptation
HeLa cells were treated with 100 μg/ml cycloheximide in growth medium at 37°C for 10 min to inhibit translation. RNA immunopreciptation was performed based on a previously described method (Sanz et al., 2009) with slight modifications. Cells were washed with cold PBS, solubilized in polysome buffer (50 mM Tris-HCl, pH 7.5, 12 mM MgCl2, 100 mM KCl, 1% NP-40, 1 mM DTT, 100 μg/ml cycloheximide, 1X EDTA-free protease inhibitor cocktail and 200 U/ml ribonuclease inhibitor and incubated on ice for 20 min. Samples were subjected to a 10,000 g centrifugation step for 10 min at 4°C and the post-mitochondrial supernatant collected. The resulting supernatants were quantified by BCA assay. 2 mg of supernatants were incubated with either rabbit monoclonal anti-Mpp11 antibody or a normal rabbit IgG as negative control rotating at 4°C. After 3 hr, 40 μl of Protein G Dynabead (Thermo Fisher Scientific) were washed once with polysome buffer and supplement to the supernatants and incubation with rotation at 4°C for 16 hr. The beads were placed in a magnetic stand on ice to separate the bound and unbound fractions. The beads were washed in high salt buffer (50 mM Tris-HCl, pH 7.5, 12 mM MgCl2, 300 mM KCl, 1% NP-40, 1 mM DTT, 100 μg/ml cycloheximide, 1X EDTA-free protease inhibitor cocktail and 200 U/ml ribonuclease inhibitor) four times for 5 min each. The bound and unbound materials were divided into two equal portions for either protein or RNA analysis. For protein analysis, the beads were directly released from the beads with 5X Laemmli sample buffer and heated at 99°C for 10 min prior to SDS-PAGE. For RNA analysis, RNA was harvested from the beads by NucleoSpin RNA kit (Machery-Nagel), subjected to cDNA synthesis, and analyzed by qPCR.
IRE1α foci imaging
T-REx293 IRE1-GFP cells were seeded at 3×105/6 well and reverse transfected with siRNA against control, Hsp70L1, Pelo, or both Hsp70L1 and Pelo. Transfections were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. For imagining, cells were re-plated onto Poly-L-Lysine coated 12 mm coverslips (Corning) at 6×104/24 well. Cells were treated with doxycycline (Sigma) to induce IRE1-GFP expression for 24h, and then treated with thapsigargin (0.5 mM) (Sigma) for 0, 1 or 4 hr or DTT (2 mM) for 1 hr to induce a UPR response. Treated cells were fixed with 4% paraformaldehyde for 20 min at room temperature at indicated time points. Nuclei were stained with Hoechst (Thermo Fisher Scientific) for 10 min at room temperature. Coverslips were mounted with ProLong diamond antifade mountant (Thermo Fisher Scientific). Images were captured with LSM 880 laser scanning confocal microscope (Zeiss). A Plan Apochromat 100x/1.4 numerical aperture (NA) oil objective was used. Images were analyzed for foci per cell count and size using ImageJ (Fiji).
Plasmids
To generate the uXbp1 and sXbp1 in vitro transcription plasmids, the uXbp1 and sXbp1 human open reading frame (ORF) were designed containing a N-terminal Flag epitope and 30 nt poly-A sequence after the Xbp1 stop codon, which is synthesized by GenScript, and inserted into pcDNA3.1 (+) downstream of a T7 promoter at HindIII and KpnI sites. The GFP and luciferase plasmids, which both containing a T7 promoter and 30 nt poly-A sequence after stop codon, recapitulated those previously described (Yang et al., 2019). The GFP plasmid was designed containing three tandem Flag-tag at the N terminus and the luciferase gene was codon optimized in the luciferase plasmid.
In vitro transcription
uXbp1 and sXbp1 templates were generated from plasmids linearized with BamHI and eGFP. Luc templates were produced from plasmids linearized with EcoRI. Templates were purified by phenol/chloroform extraction and ethanol precipitation. RNA was synthesized by T7 RNA polymerase (NEB), capped by 3’-O-Me-m7G(5’)ppp(5’)G RNA Cap Structure Analog (NEB) and purified using RNA Cleanup Kit (NEB). The quality of RNA transcripts was monitored by denaturing RNA electrophoresis in 1X TAE agarose gels and quantified with a NanoDrop 2000c Spectrophotometer.
In vitro translation by mammalian cell-free lysate
In vitro translation extracts were harvested at 48 hr post-transfection of siRNA against either control or Hsp70L1 in HEK293T cells. Mammalian cell-free lysate was harvested as previously described (Rakotondrafara and Hentze, 2011) with modifications. Cells were released from the plates with trypsin, then washed with cold DPBS, harvested by centrifugation at 1000 g for 5 min at 4°C. The cell pellet was resusspended in fresh ice-cold hypotonic buffer (10 mM HEPES-KOH, pH 7.6, 10 mM potassium acetate, 0.5 mM magnesium acetate, 5 mM DTT, proteasome inhibitor cocktail) in a 1:1 volume ratio and incubated on ice for 30 min. Cells were then homogenized by passing through a 1 mL syringes with a 27 G 3/4 needle for 10–20 times until > 95% cells had ruptured as monitored by trypan blue staining. Potassium acetate was adjusted to a final centration of 50 mM. The lysates were clarified by centrifugation at 16,000 g for 10 min at 4°C, and the supernatant was snap frozen in liquid nitrogen and stored at −80°C before use. Each translation reaction contained 66% of the in vitro translation lysate, 180 ng of RNA templates and buffer supplemented with 1 mM ATP (NEB), 0.2 mM GFP (Sigma), 8 mM Creatine phosphate (Sigma), 0.13 units/μl Creatine phosphokinase (Sigma), 20 mM HEPES-KOH pH 7.6, 2 mM DTT, 0.83 mM Mg (OAc)2, 0.1 M KOAc, 20 μM amino acid mixtures (Promega), 500 μM Spermidine (Sigma), 0.4 units/ul RNase inhibitor (Thermo Fisher Scientific). The reactions were incubated in a 30°C water bath for indicated time. To preserve peptidyl-tRNA ester bonds, in vitro translation products were denatured with 4X Native PAGE sample Buffer (Thermo Fisher Scientific), run on NuPAGE Bis-Tris gels (Invitrogen) with MES-SDS running buffer at 170 V for 2 hr and transferred onto 0.45 μm PVDF membranes (Millipore Immobilon-FL #IPFL00010) at 35 V for 1 h. For the RNase A treatment to release tRNA, 1 μl of 10 mg/ml RNase A (Thermo Fisher Scientific) was added to each reaction after indicated translation time and incubated for 15 min at 37°C. For luciferase assay, each reaction contained Luc RNA template, 0.12 μl of Steady-Glo luciferase assay substrate (Promega) in the mammalian cell free translation buffer system. Real-time monitoring of luciferase activity was carried out on a FLUOstar OPTIMA microplate reader (BMG LABTECH) at 30°C.
Pulse labeling global nascent proteins with bioorthogonal non-canonical amino-acid tagging
Cell lysates were harvested at 48 hr post-transfection of siRNA against either control or Mpp11 in HeLa cells. Cells were washed twice with PBS, incubated in DMEM high glucose without methionine (GIBCO) with 10% dialyzed FBS, pretreated with or without MG132 (5 μM) at 37°C for 30 min, and replaced with medium containing a final concentration of 50 μM Click-IT L-Azidohomoalanine (AHA) (Thermo Fisher Scientific). After 0, 2, or 4 hr, cells were washed twice with PBS, lysed in lysis buffer (1% SDS in 50 mM Tris-HCl, pH 8.0, 1X protease inhibitor cocktail, 1X phosphatase inhibitor cocktail), incubated on ice for 15 min, vortex for 5 min followed by a 16,000 g centrifugation step for 5 min at 4°C. The supernatants were collected and subjected to azide-alkyne ligation (click chemistry) using Biotin-alkyne (Thermo Fisher Scientific) and the Click-iT Protein Reaction Buffer Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Proteins were collected following methanol/chloroform precipitation. Samples were separated on 10% SDS-PAGE, transferred to 0.45 μm PVDF membranes (Millipore Immobilon-FL), which were then blocked with 5% (w/v) bovine serum albumin in 1X TBS with 0.1% Tween-20 for 1 hr at room temperature. Blots were incubated with IRDye 800CW Streptavidin at 4°C overnight in 1X TBS with 0.1% Tween-20, and visualized using a LI-COR Odyssey CLx.
Ribosome footprint profiling - library preparation
HEK293 cells transiently transfected with siRNA against vehicle control, Hsp70L1, Pelo, or both Pelo and Hsp70L1 were grown in standard grown media in the absence of Pen-Strep for 48 hr, and followed by 4 hr thapsigargin (0.5 μM) treatment. Cells were rapidly cooled and washed in ice-cold PBS, lysed in lysis buffer (20 mM Tris-HCl/pH7.4, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 1% (v/v) Triton X-100, and 25 U/ml Turbo DNase I), and incubated on ice for 10 min. Cells were then triturated through a 26-G needle ten times and subjected to centrifugation at 16,000 g for 10 min at 4°C. Cell lysates were digested with 100 U RNase I (Ambion) per A260 lysate at room temperature for 45 min with gentle agitation and followed by the addition of 200 U RiboLock RNase Inhibitor (Thermo Scientific). Ribosome pellets containing mRNA fragments were isolated by 1 M sucrose cushion in polysome buffer (20 mM Tris-HCl/ pH7.4, 150 mM NaCl, 7.5 mM MgCl2, 0.5 mM DTT, 20 U/ml RiboLock RNase Inhibitor) and centrifuged at 70,000 rpm for 2 hr at 4°C by Beckmen TLA-110 rotor. Ribosome protected mRNA fragments were isolated using TRIzol and separated by denaturing 12% polyacrylamide gel containing 8M urea. RNA was visualized by SYBR Gold (Invitrogen), and the size of the fragments ranging from 18 to 34 nt were isolated to generate the ribosome-protected mRNA fragment library. 3’ oligonucleotide adaptor ligation, reverse transcription, circularization, and rRNA depletion using biotinylated rRNA depletion oligos were performed as previously described (Ingolia et al., 2012; Lueck et al., 2019). Libraries were barcoded using indexing primers for each sample during PCR amplification. Barcoded libraries were then pooled and sequenced in an Illumina NextSeq 500 as per manufacturer protocol to typically generate 40–70 million reads per sample.
Ribosome footprint profiling – data analysis
Data files for each barcoded sample (minus adaptor sequence at 3’ end) were first mapped to four rRNA sequences (RNA5S1, RNA58SN5, RNA18SN5, and RNA28SN5) using HISAT (Kim et al., 2015) to eliminate rRNA contaminant reads. The remaining reads were aligned to the transcript variant with the longest coding sequence (CDS) of each human gene (GRCh38) using HISAT. Multi-mapped reads were discarded, and only reads with the size ranging from 18 to 34 nt were utilized for further analysis. The 5’ end position from each read was annotated on each transcript, and the 16th position from the 5’ end of each read was used to infer the position of the ribosome on transcripts (Guydosh and Green, 2014; Ingolia et al., 2009). Footprint occupancy at each nucleotide position of Xbp1 was calculated by RPM (footprint Reads Per total Million-mapped reads). Triplet periodicity of ribosome footprints was calculated using reads mapped in each of three subregions of uXbp1 transcript: 5’ end region (upstream CDS region of the 26-mer splicing region of uXbp1; nucleotides 80–565), M region (downstream CDS region of the 26-mer splicing region of uXbp1; nucleotides 596–859), and 3’end region (early region of 3’UTR of uXbp1 that overlaps CDS of sXbp1; nucleotides 860–1230). Among reads aligned at the first base of a codon (footprints at Frame 0) in the M subregion, the proportion of the reads at Asp261 was calculated to show the relative ribosome occupancy at the stall site in uXbp1. The sequencing data was analyzed using Galaxy platform (Afgan et al., 2016). Graphs were generated using Prism 9 (GraphPad Software).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using the unpaired two-tailed Student’s t test as implemented in the GraphPad Prism 8 software. Statistically significant was considered differences with P values less than 0.05. Data points are the means and error bars the standard deviation (SD). The number of independent experiments is designated as n in each figure legend.
Supplementary Material
Highlights.
RAC knockdown sensitizes cells to ER stress by reducing IRE1α splicing of Xbp1
RAC reduction does not influence substrate Xbp1 mRNA localization or pausing
RAC affects clustering of IRE1α required for activation of its splicing activity
ACKNOWLEDGMENTS
Authors thank the following collaborators for kindly providing reagents and technical support: Dr. Elizabeth Craig’s lab at the University of Wisconsin-Madison for the knockout RAC yeast strains; Dr. Peter Walter’s lab at the UCSF for the T-REx293 IRE1-GFP cell line; Dr. Sabine Rospert’s lab at the University of Freiburg, Germany, for anti-Mpp11 antibody; Dr. Ineke Braakman’s Lab at the University Utrecht, the Netherlands, for one of the knockout Hsp70L1 cell lines (data not shown); Dr. Christopher Nicchitta at Duke University for critical technical advice regarding the differential detergent cell fractionation experiments; Dr. Benjamin Tu and his lab at UTSW for yeast technical support; Dr. Jen Liou’s lab at UTSW for microscope technical support; and the microscope imaging core at the physiology department at UTSW. This research was supported by the cancer biology program and the mechanisms of disease and translational science (MoDTS) graduate track at UTSW and HHMI Med into grad initiative to I.-H.W., National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health (grant R37DK49835) and Ruth S. Harrell Professorship to P.T., and National Institutes of Health (R35GM118118) and the Welch Foundation (I-1560) to Y.L.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109217.
INCLUSION AND DIVERSITY
While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
REFERENCES
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, et al. (2016). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44, W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragón T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, and Walter P. (2009). Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 457, 736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, and Fire AZ (2018). Nonsense mRNA suppression via nonstop decay. eLife 7, e33292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Bukau B, Weissman J, and Horwich A. (2006). Molecular chaperones and protein quality control. Cell 125, 443–451. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, and Ron D. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. [DOI] [PubMed] [Google Scholar]
- Cowan JL, and Morley SJ (2004). The proteasome inhibitor, MG132, promotes the reprogramming of translation in C2C12 myoblasts and facilitates the association of hsp25 with the eIF4F complex. Eur. J. Biochem 271, 3596–3611. [DOI] [PubMed] [Google Scholar]
- Cox JS, and Walter P. (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404. [DOI] [PubMed] [Google Scholar]
- Deuerling E, Gamerdinger M, and Kreft SG (2019). Chaperone Interactions at the Ribosome. Cold Spring Harb. Perspect. Biol 11, a033977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi M, Lilie H, Fünfschilling U, Mun A, Ross S, Lithgow T, Rücknagel P, and Rospert S. (2001). RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc. Natl. Acad. Sci. USA 98, 3762–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautschi M, Mun A, Ross S, and Rospert S. (2002). A functional chaperone triad on the yeast ribosome. Proc. Natl. Acad. Sci. USA 99, 4209–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribling-Burrer AS, Chiabudini M, Zhang Y, Qiu Z, Scazzari M, Wölfle T, Wohlwend D, and Rospert S. (2019). A dual role of the ribosome-bound chaperones RAC/Ssb in maintaining the fidelity of translation termination. Nucleic Acids Res. 47, 7018–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh NR, and Green R. (2014). Dom34 rescues ribosomes in 3’ untranslated regions. Cell 156, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, and Hayer-Hartl M. (2009). Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol 16, 574–581. [DOI] [PubMed] [Google Scholar]
- Hipp MS, Park SH, and Hartl FU (2014). Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514. [DOI] [PubMed] [Google Scholar]
- Huang P, Gautschi M, Walter W, Rospert S, and Craig EA (2005). The Hsp70 Ssz1 modulates the function of the ribosome-associated J-protein Zuo1. Nat. Struct. Mol. Biol 12, 497–504. [DOI] [PubMed] [Google Scholar]
- Hundley H, Eisenman H, Walter W, Evans T, Hotokezaka Y, Wiedmann M, and Craig E. (2002). The in vivo function of the ribosome-associated Hsp70, Ssz1, does not require its putative peptide-binding domain. Proc. Natl. Acad. Sci. USA 99, 4203–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundley HA, Walter W, Bairstow S, and Craig EA (2005). Human Mpp11 J protein: ribosome-tethered molecular chaperones are ubiquitous. Science 308, 1032–1034. [DOI] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan S, Nwosu C, and Nicchitta CV (2011). Analyzing mRNA localization to the endoplasmic reticulum via cell fractionation. Methods Mol. Biol 714, 301–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal H, Conz C, Otto H, Wölfle T, Fitzke E, Mayer MP, and Rospert S. (2011). The chaperone network connected to human ribosome-associated complex. Mol. Cell. Biol 31, 1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, Hoffmann T, Busslinger M, and Martinez J. (2014). The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J. 33, 2922–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampinga HH, and Craig EA (2010). The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol 11, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda S, Yanagitani K, Yokota Y, Esaki Y, and Kohno K. (2016). Autonomous translational pausing is required for XBP1u mRNA recruitment to the ER via the SRP pathway. Proc. Natl. Acad. Sci. USA 113, E5886–E5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, and Kohno K. (2007). Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol 179, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, and Walter P. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, Hesselberth JR, Wolin SL, and Hammarlund M. (2014). The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep. 15, 1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer G, Boehringer D, Ban N, and Bukau B. (2009). The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol 16, 589–597. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, and Mann M. (2014). Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324. [DOI] [PubMed] [Google Scholar]
- Lee KP, Dey M, Neculai D, Cao C, Dever TE, and Sicheri F. (2008). Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Sharma R, Shrestha OK, Bingman CA, and Craig EA (2016). Dual interaction of the Hsp70 J-protein cochaperone Zuotin with the 40S and 60S ribosomal subunits. Nat. Struct. Mol. Biol 23, 1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidig C, Bange G, Kopp J, Amlacher S, Aravind A, Wickles S, Witte G, Hurt E, Beckmann R, and Sinning I. (2013). Structural characterization of a eukaryotic chaperone–the ribosome-associated complex. Nat. Struct. Mol. Biol 20, 23–28. [DOI] [PubMed] [Google Scholar]
- Li H, Korennykh AV, Behrman SL, and Walter P. (2010). Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 107, 16113–16118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Schröder M, and Kaufman RJ (2000). Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem 275, 24881–24885. [DOI] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lu Y, Liang FX, and Wang X. (2014). A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 55, 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueck JD, Yoon JS, Perales-Puchalt A, Mackey AL, Infield DT, Behlke MA, Pope MR, Weiner DB, Skach WR, McCray PB Jr., and Ahern CA (2019). Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun 10, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazroui R, Di Marco S, Kaufman RJ, and Gallouzi IE (2007). Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 18, 2603–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Ma W, Gething MJ, and Sambrook J. (1993). A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756. [DOI] [PubMed] [Google Scholar]
- Muldoon-Jacobs KL, and Dinman JD (2006). Specific effects of ribosome-tethered molecular chaperones on programmed −1 ribosomal frameshifting. Eukaryot. Cell 5, 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson RJ, Ziegelhoffer T, Nicolet C, Werner-Washburne M, and Craig EA (1992). The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell 71, 97–105. [DOI] [PubMed] [Google Scholar]
- Otto H, Conz C, Maier P, Wölfle T, Suzuki CK, Jenö P, Rücknagel P, Stahl J, and Rospert S. (2005). The chaperones MPP11 and Hsp70L1 form the mammalian ribosome-associated complex. Proc. Natl. Acad. Sci. USA 102, 10064–10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Willmund F, and Frydman J. (2013). The ribosome as a hub for protein quality control. Mol. Cell 49, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisker K, Braun D, Wölfle T, Hentschel J, Fünfschilling U, Fischer G, Sickmann A, and Rospert S. (2008). Ribosome-associated complex binds to ribosomes in close proximity of Rpl31 at the exit of the polypeptide tunnel in yeast. Mol. Biol. Cell 19, 5279–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfund C, Huang P, Lopez-Hoyo N, and Craig EA (2001). Divergent functional properties of the ribosome-associated molecular chaperone Ssb compared with other Hsp70s. Mol. Biol. Cell 12, 3773–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumb R, Zhang ZR, Appathurai S, and Mariappan M. (2015). A functional link between the co-translational protein translocation pathway and the UPR. eLife 4, e07426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, and Deuerling E. (2012). Ribosome-associated chaperones as key players in proteostasis. Trends Biochem. Sci 37, 274–283. [DOI] [PubMed] [Google Scholar]
- Rakotondrafara AM, and Hentze MW (2011). An efficient factor-depleted mammalian in vitro translation system. Nat. Protoc 6, 563–571. [DOI] [PubMed] [Google Scholar]
- Rakwalska M, and Rospert S. (2004). The ribosome-bound chaperones RAC and Ssb1/2p are required for accurate translation in Saccharomyces cerevisiae. Mol. Cell. Biol 24, 9186–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Nillegoda NB, Mayer MP, and Bukau B. (2019). The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol 20, 665–680. [DOI] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamu CE, and Walter P. (1996). Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15, 3028–3039. [PMC free article] [PubMed] [Google Scholar]
- Shanmuganathan V, Schiller N, Magoulopoulou A, Cheng J, Braunger K, Cymer F, Berninghausen O, Beatrix B, Kohno K, von Heijne G, and Beckmann R. (2019). Structural and mutational analysis of the ribosome-arresting human XBP1u. eLife 8, e46267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam AB, Koong AC, and Niwa M. (2014). Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 9, 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, and Kaufman RJ (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan JS, and Lindquist S. (2014). Mechanisms of protein-folding diseases at a glance. Dis. Model. Mech 7, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, and Morimoto RI (2004). Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem 279, 56053–56060. [DOI] [PubMed] [Google Scholar]
- Willmund F, del Alamo M, Pechmann S, Chen T, Albanése V, Dammer EB, Peng J, and Frydman J. (2013). The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 152, 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Schilke B, Pfund C, Walter W, Kim S, and Craig EA (1998). Zuotin, a ribosome-associated DnaJ molecular chaperone. EMBO J. 17, 4809– 4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagitani K, Imagawa Y, Iwawaki T, Hosoda A, Saito M, Kimata Y, and Kohno K. (2009). Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell 34, 191–200. [DOI] [PubMed] [Google Scholar]
- Yanagitani K, Kimata Y, Kadokura H, and Kohno K. (2011). Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1u mRNA. Science 331, 586–589. [DOI] [PubMed] [Google Scholar]
- Yang Q, Yu CH, Zhao F, Dang Y, Wu C, Xie P, Sachs MS, and Liu Y. (2019). eRF1 mediates codon usage effects on mRNA translation efficiency through premature termination at rare codons. Nucleic Acids Res. 47, 9243– 9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, and Mori K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ma C, Yuan Y, Zhu J, Li N, Chen C, Wu S, Yu L, Lei J, and Gao N. (2014). Structural basis for interaction of a cotranslational chaperone with the eukaryotic ribosome. Nat. Struct. Mol. Biol 21, 1042–1046. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sinning I, and Rospert S. (2017). Two chaperones locked in an embrace: structure and function of the ribosome-associated complex RAC. Nat. Struct. Mol. Biol 24, 611–619. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Valentín Gesé G, Conz C, Lapouge K, Kopp J, Wölfle T, Rospert S, and Sinning I. (2020). The ribosome-associated complex RAC serves in a relay that directs nascent chains to Ssb. Nat. Commun 11, 1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, and Kaufman RJ (2006). The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 103, 14343–14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ribosome profiling datasets generated during this study are available at the Gene Expression Omnibus (GEO) database [GSE173852].