

To the Editor:
Large granular lymphocytic leukemia (LGL) is a lympho-proliferation of cytotoxic T cells (CTL) typically occurring in the elderly. LGL encompasses a spectrum of manifestations from semireactive oligoclonal to clonal CTL expansions, the latter frequently associated with the somatic STAT3 mutations. Clinical symptomatology ranges from silent to an indolent to chronic leukemia with cytopenias as a paraneoplastic manifestation. Asymptomatic LGL may represent an equivalent of T cell clonopathy of unclear significance (TCUS). Indeed, the demographics of LGL resembles that of TCUS, B cell clonopathy of unclear significance, clonal hematopoiesis of indeterminate potential (CHIP) and even monoclonal gammopathy of unclear significance (MGUS). As previously described, LGL is associated with B cell dyscrasias including MGUS [1], myelodysplastic syndrome (MDS; Table S1) [2–4], and various other neoplastic conditions [1, 5]. However, historical reports lack uniform diagnostic criteria, and most of them precede the advent of next generation sequencing to detect pathogenic somatic mutations. Nevertheless, a peculiar relationship between MDS and LGL does exist and may correspond to a common pathogenic role of aging in these disorders. Alternatively, the presence of LGL in MDS may indicate an overshooting cellular tumor surveillance response rather than a coincidental demographic relationship [2].
For diagnosis of LGL, our strict diagnostic criteria (>3/5) included: (i) presence of large granular lymphocytes (>500/ μL, by differential counts from complete blood count) for >6 months; (ii) abnormal cytotoxic T lymphocytes expressing CD2, CD56, and CD57 and lacking CD28; (iii) preferential usage of a TCR Vb family by flow cytometry; (iv) TCR gene rearrangement by PCR; and (v) Bone marrow infiltration with LGL is another diagnostic criterion but a designated marrow biopsy is often not performed in clear LGL which are otherwise asymptomatic. These criteria have been applied over the last 10 years and as a result we have compiled a large cohort of uniformly diagnosed and analyzed patients, n = 240, Tables S2–S4). In an earlier analysis of 204 patients (included in the current 240 patients), therapeutic options and outcomes were discussed in detail; 90% of the cases were successfully treated with at least >1 modalities, ~10% of the patients required salvage therapies (Table S3) [6].
Study of our LGL cohort yielded a series of 13 (13/240; 5.4%) cases with obvious coexistent MDS (Table 1 and S5). As a standard practice we thoroughly screened all our LGL patients for coincidental MDS. However, our earlier investigations along with recent reports by others have indicated that LGL equivalents (in form of excessive oligo/ clonal expansions) may be present in a proportion of patients with typical MDS [2]. In our MDS series, because of the thorough and systematic screen (morphologic and flow cytometric evaluation of the marrow and reflex specialized testing of TCR rearrangement, Vb flow cytometric clonotyping, and STAT3 mutational screening) it is unlikely that overt LGL may have been missed. Nevertheless, subclinical CTL lymphoproliferations are frequently encountered in MDS as previously demonstrated by intricate flow cytometric and molecular analyses of Vb expansions in MDS [7]. In these instances they may represent tumor immune surveillance and mediate some of cytopenias, particularly in less advanced MDS.
Table 1.
LGL/MDS patient series
| UPN number | Diagnosis | Age years | Cytogenetics | LGL Count K/µL | STAT3 mutation | TCR VB | Clone size % | Flow cytometry | TCR rearrangement | Treatment (Response) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | AML | 72 | 44,XY,del(7)(q22),−16, −17,−18,−20,+2mar[2]/45,idem,+8[10]/46,XY[8] | 634 | – | Vβ5.3 | 27 | + | + | CsA |
| 2. | RCMD | 82 | 45,XY,−7[10]/46,XY,del(5)(q13q33)[1]/46,XY[9] | 1776 | – | Vβ22 Vβ11 | 129 | + | − | Observation for LGL, darbepoetin, azacitidine |
| 3. | RA | 66 | 45,X,−X[20] | 129a | – | Vβ1 | 50 | + | + | CsA |
| 4. | RAEB-1 | 63 | 46, XY[20] | 990 | – | Vβ3 Vβ16 |
15 15 |
− | + | Observation for LGL, decitabine for MDS |
| 5. | RCMD | 71 | 46,XX[20] | 537 | – | Vβ2 | 51 | + | − | CsA+prednisone, darbepoetin, azacitidine |
| 6 | RCMD | 76 | 46,XY,del(20)(q11.2q13.1)[8]/46,XY[12] | 1120 | – | Vβ14 Vβ2 |
61 15 |
+ | + | CTx |
| 7. | CMML | 78 | 46, XX[20] | 680 | – | Vβ14 | 25 | + | + | CsA, tacrolimus, alemtuzumab |
| 8. | sAML | 62 | 46, XY[20] | 350a | – | Vβ2 | 18.1 | − | + | MTx |
| 9. | sAML | 69 | 46,XY,−7,+mar[2]/46,XY[19] | 2735 | p.D661V | N/A | N/A | + | + | CsA, Etanercept, Lenalidomide |
| 10. | RAEB-1 | 84 | 46,XX[20] | 6188 | p.D661V | Vβ21.3 | 88 | + | + | CsA, Tacrolimus, Tofacitinib, MTx +prednisone(NO) |
| 11. | ICUS | 72 | 47,XY,+mar?c[13]/46,XY[7] | 1600 | – | Vβ3 | 65 | + | + | CsA, MTx, Danazol, darbepoetin, low dose decitabine, lenalidomide(NO) |
| 12. | RA | 79 | 47,XY,+8[16]/46,XY[4] | 2100 | – | N/A | 87 | + | + | Epopoietin alfa, |
| 13. | t-AML | 36 | 48,XX,+1,der(1;7)(q10; p10),+8,+21[14]/46,XX[6] | 5780 | – | Vβ13.2 | 17 | + | + | CsA, MTx, tofacitinib, eltrombopag, pegfilgrastim, IVIG |
Detailed description of patients with MDS/LGL including cytogenetics, WHO staging and diagnostic criteria
Patient with LGL counts available from blood samples obtained post initiation of treatment and therefore explaining the low LGL counts
With the availability of mutational sequencing for MDS (Table S6), our LGL/MDS series inspired us to systematically investigate the coexistence of mutant myeloid clones in LGL. If detected, they could be consistent with the presence of subclinical MDS or coexistent CHIP in patients with LGL. In line with this theory our manifest MDS/LGL cases may potentially reflect the proverbial “tip of the ice-berg” pointing towards common pathogenic factors or the role of LGL in controlling clonal myeloid outgrowth.
Patients with coexistent MDS/LGL (13/240) displayed typical features of MDS including abnormal cytogenetics (8/13), dysmorphia of myeloid cells in the bone marrow (13/13), ringed sideroblasts (1/13) and the presence of highly clonal somatic myeloid mutations (8/13) with an average VAF of 28 ± 3% (Fig. 1a, b) [8]. When we compared LGL/MDS patients to those with LGL only (n = 227), somatic STAT3/STAT5 mutations were found in only 15% (2/13) versus 39% (87/221) patients (p = 0.14), respectively (Fig. 1c). However, the LGL counts, TCR rearrangement rate or mean age did not vary between LGL and LGL/MDS cases (Table S2). Patients with LGL/MDS had more thrombocytopenia than those with LGL (p = 0.014) in whom single-lineage cytopenias were more common, with neutropenia and anemia present in 43% and 53% of LGL. Similarly, pancytopenia or bi-lineage cytopenias were also less common in LGL versus LGL/MDS and MDS (not shown). When we systematically applied deep NGS for 36 most common mutant genes in MDS (Fig. 1d; Table S6), we did indeed detect the presence of mutant myeloid clones in 41/161 patients with pure LGL with an average VAF of 35 ± 1.6% and average 1.7 mutations per patient. As these mutations may present early in MDS or CHIP, we compared the mutational spectrum of LGL patients with those of typical MDS (as disease control; Fig. 1a) and CHIP (through meta-analysis; Table S7) [9]. Indeed, myeloid mutations were found in 26% of patients with otherwise typical LGL (Fig. 1e). Most of the carriers had a singular mutation 69% versus 29% in MDS, (p < 0.0001; Fig. 1a) but the allelic burden did not differ (Fig. 1b). The general distribution of the individual mutations was similar in patients with MDS/LGL, MDS and LGL. Notable differences include the overrepresentation of spliceosomal mutations, cohesin complex, PRC2 and RAS mutations in MDS and CHIP [8]. The typical CHIP mutations (e.g., TET2, ASXL1, and DNMT3A) were present in LGL suggesting that some of the cases may represent a coincidence between CHIP and LGL due to overlapping demographics or through a pathophysiologic link as stipulated above. Clonal burden in CHIP appears to be lower than that observed in LGL indicating a more advanced myeloid disease in LGL versus CHIP. It is worth mentioning that BCOR/BCORL1 hits were more common in LGL, a finding consistent with that made for aplastic anemia (AA) and possibly indicating an immune stress exerted on the hematopoietic stem cell compartment [10]. We stipulate that BCOR mutations in LGL are present in the myeloid fraction as previously described for AA and are, without specific molecular context, passenger lesions rather than drivers [10]. Alternatively, the presence of BCOR/BCORL1 hits are also reported in B cell lymphomas, and are thought to play a role in modulation of the immune system and interactions between germinal center (GC) B and GC B helper T cells [11], raising an interesting possibility that the BCOR/BCORL1 could be a lymphoid hit.
Fig. 1.
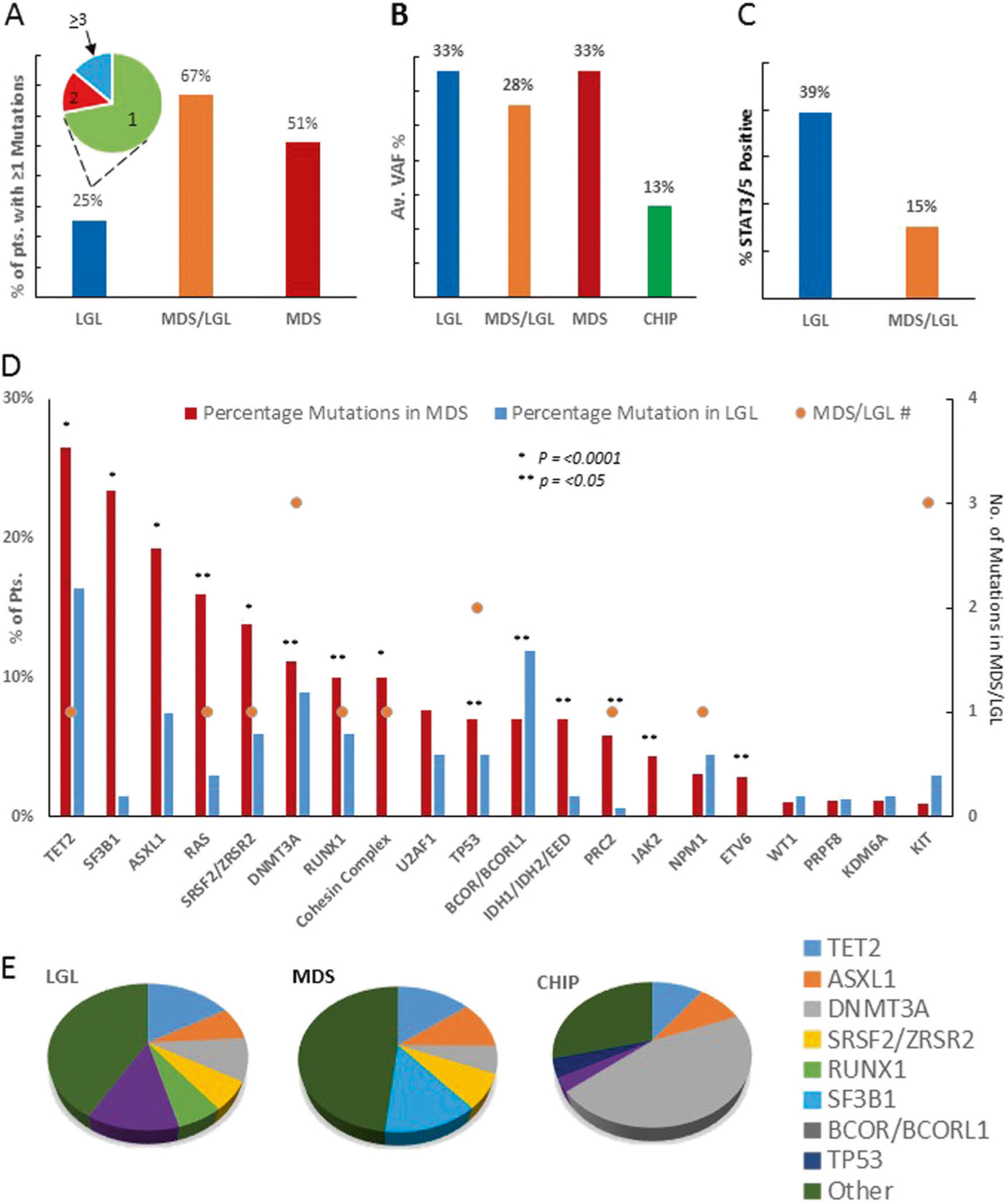
Comparative data analysis of LGL, LGL/MDS, MDS, and CHIP. a Bar chart representation of percentage of patients observed to have >1 target somatic mutation in LGL, MDS/LGL, and MDS patients. The Pie chart in a depicts the distribution of LGL patients, annotated numbers reflect the number of observed mutations (1, 2, and >3); b bar chart data describes the average variant allelic frequency for LGL, MDS/LGL, MDS, and CHIP (CHIP average VAF% was calculated by averaging the reported median VAF from meta-analysis in ASH [8], c bar chart data for STAT3/5b positive patients in LGL and MDS/LGL as a percentage of patient cohort; d bar chart data describes the percentage of target somatic mutation seen in LGL and MDS. Colored circles represent the number of mutations plotted against the target gene (some genes with very low frequencies have been omitted, genes part of; e most common mutations observed as a color coded pie chart
Theoretically, in paroxysmal nocturnal hematuria (PNH) PIGA defects acquired by hematopoietic stem cells (HSCs) lead to clonal escape in lieu of immune attack. However, in contrast to AA, target cells in LGL likely include committed progenitor (resulting in single-lineage cytopenias) and thus the selection process would not be operative due to a limited life span of progenitor versus HSC. In contrast to these assumptions, there is a case report of a PNH patient within LGL in the literature [12], and we have also described LGL like clonal expansions in PNH [13]. In our series of LGL we detected 3 AA/PNH patients, but because of the lack of complete records we have not included one of these cases in our LGL cohort.
Our results may indicate LGL is often associated with the presence of myeloid mutations, generally consistent with CHIP. Some of the notable differences such as lower frequency of DNMT3A mutations (Fig. 1), higher allelic burden and the occasional presence of more penetrant hits may indicate that LGL identifies with a more advanced disease. It is possible that common age-associated pathogenic mechanisms, including mutagenic stress, inflammation, metabolic changes could also be contributing to both LGL and CHIP outgrowth [4]. Conversely, LGL may, in fact, evolve as a consequence of immune surveillance reaction to CHIP or myeloid neoplasms with one of three possible outcomes; (1) the aberrant clone is completely eradicated; (2) a partial eradication, e.g., in form of persistent CHIP; (3) further myeloid clonal expansion, mutational diversification with disease evolution to overt LGL/MDS cases to be viewed as a failure of immune surveillance. Driver antigens have been suspected in the past, but it has not been established whether these are shared antigens between patient groups or are patient specific and correspond to auto- or neo-antigens induced by a breach of tolerance or molecular mimicry e.g., a viral antigen. Though WT-1 and cyclin D have been proposed previously, neither has been successfully identified as auto-antigens, unless the neo-antigens are derived from mutant proteins. However, given the prevalence of myeloid clones in LGL, one would stipulate that the expected rate of MDS evolution should be higher (54/174 myeloid clones versus 13 MDS cases in LGL), if LGL-mediated tumor surveillance was absent (Fig. 2). Alternatively, it can also be hypothesized that the MDS evolution may indeed be the result of recurrent DNA damages provoked by the excessive LGL proliferation, immune inhibition of myelopoietic evolution and persistence of a myeloid escape clone (Fig. 2) [14]. It is unclear why the LGL proliferation persists in cases where the obvious insult has been eradicated (no aberrant myeloid clone).
Fig. 2.
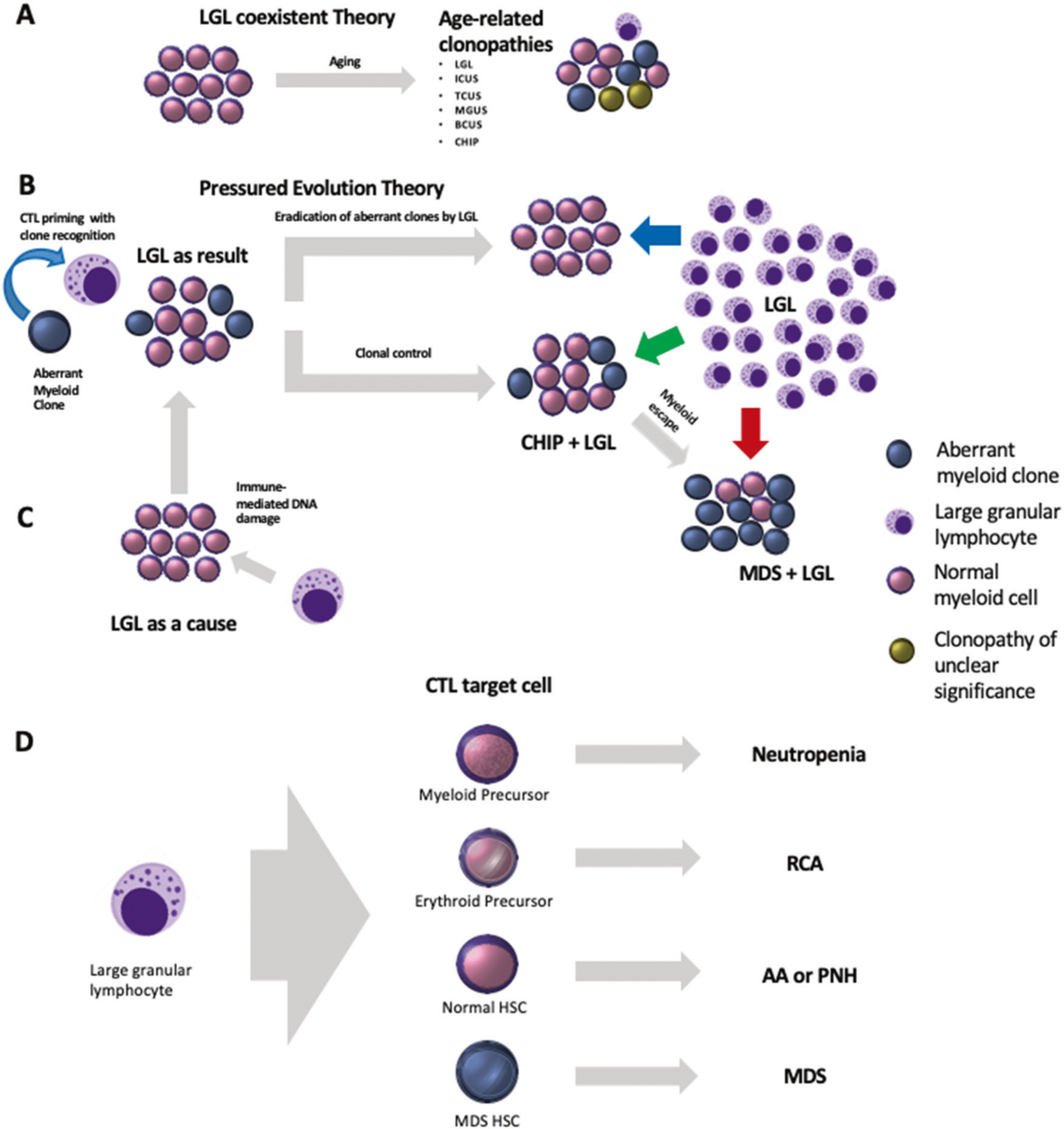
Pathogenesis model for LGL leukemia. Activation resulting in the oligoclonal/purely clonal cytotoxic T cell expansion. a LGL coexistent theory confers to the development of clonopathies and persistence as the result of age-related changes. LGL may coexist with ICUS, BCUS, MGUS, CHIP, and TCUS. b, c Pressured evolution theory, however, suggests that LGL evolution is either the result of myeloid aberrancies or may indeed be the cause of myeloid aberrancy occurrence. Further, LGL may play an integral role in either (1) Eradicating or (2) Controlling the aberrancy or completely failing to harness the escape clone leading to disease advancement to MDS. d Fate of precursor (myeloid and erythroid) and hematopoietic stem cells (Normal and MDS) when targeted by LGL and the associated clinic-pathological conditions
In sum, LGL may represent an extreme spectrum of CTL responses involved in the tumor surveillance of malignant hematopoiesis. To that end, interestingly, LGL may be very analogous to genetically engineered CAR-T cells shown to display predominance of a single or few most effective clone [15].
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute grants R01HL118281, R01HL123904, R01HL132071, and R35HL135795.
Footnotes
Supplementary information The online version of this article (https://doi.org/10.1038/s41375–019-0601-y) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Viny AD, Lichtin A, Pohlman B, Loughran T, Maciejewski J. Chronic B-cell dyscrasias are an important clinical feature of T-LGL leukemia. Leuk Lymphoma 2008;49:932–8. [DOI] [PubMed] [Google Scholar]
- 2.Roe C, Alali N, Padron E, Burnette PK, Sweet KL, Sallman D, et al. Characterization of Myelodysplastic Syndromes (MDS) with T-Cell Large Granular Lymphocyte Proliferations (LGL). Blood 2016;128:113. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Sokol L, Bennett JM, Moscinski LC, List A, Zhang L. T-cell large granular lymphocyte proliferation in myelodysplastic syndromes: Clinicopathological features and prognostic significance. Leuk Res 2016;43:18–23. [DOI] [PubMed] [Google Scholar]
- 4.Saunthararajah Y, Molldrem JL, Rivera M, Williams A, Stetler-Stevenson M, Sorbara L, et al. Coincident myelodysplastic syndrome and T-cell large granular lymphocytic disease: clinical and pathophysiological features. Br J Haematol 2001;112:195–200. [DOI] [PubMed] [Google Scholar]
- 5.Pawarode A, Baer M. T/B and not T/B: high frequency of B-cell dyscrasias in T-LGL leukemia. Leuk lymphoma 2008;49:845–6. [DOI] [PubMed] [Google Scholar]
- 6.Sanikommu SR, Clemente MJ, Chomczynski P, Afable MG 2nd, Jerez A, Thota S, et al. Clinical features and treatment outcomes in large granular lymphocytic leukemia (LGLL). Leuk Lymphoma 2018;59:416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geng S, Weng J, Du X, Lai P, Huang X, Chen S, et al. Comparison of the distribution and clonal expansion features of the T-cell gammadelta repertoire in myelodysplastic syndrome-RAEB and RAEB with progression to AML. DNA cell Biol 2012; 31:1563–70. [DOI] [PubMed] [Google Scholar]
- 8.Malcovati L, Cazzola M. The shadowlands of MDS: idiopathic cytopenias of undetermined significance (ICUS) and clonal hematopoiesis of indeterminate potential (CHIP). Hematol Am Soc Hematol Educ Program 2015;2015:299–307. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014;371:2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med 2015;373:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hatzi K, Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med 2014;20:343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyer T, Grardel N, Copin MC, Roumier C, Touzart A, Buchdahl AL, et al. Paroxysmal nocturnal hemoglobinuria (PNH) and T cell large granular lymphocyte (LGL) leukemia-an unusual association: another cause of cytopenia in PNH. Ann Hematol 2015; 94:1759–60. [DOI] [PubMed] [Google Scholar]
- 13.Risitano AM, Maciejewski JP, Muranski P, Wlodarski M, O’Keefe C, Sloand EM, et al. Large granular lymphocyte (LGL)-like clonal expansions in paroxysmal nocturnal hemoglobinuria (PNH) patients. Leukemia 2005;19:217–22. [DOI] [PubMed] [Google Scholar]
- 14.Sloand EM, Melenhorst JJ, Tucker ZC, Pfannes L, Brenchley JM, Yong A, et al. T-cell immune responses to Wilms tumor 1 protein in myelodysplasia responsive to immunosuppressive therapy. Blood 2011;117:2691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scherer LD, Brenner MK, Mamonkin M. Chimeric antigen receptors for T-cell malignancies. Front Oncol 2019;9:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.