

Abstract
Developmental bioelectricity is the study of the endogenous role of bioelectrical signaling in all cell types. Resting potentials and other aspects of ionic cell physiology are known to be important regulatory parameters in embryogenesis, regeneration, and cancer. However, relevant quantitative measurement and genetic phenotyping data are distributed throughout wide-ranging literature, hampering experimental design and hypothesis generation. Here, we analyze published studies on bioelectrics and transcriptomic and genomic/phenotypic databases to provide a novel synthesis of what is known in three important aspects of bioelectrics research. First, we provide a comprehensive list of channelopathies—ion channel and pump gene mutations—in a range of important model systems with developmental patterning phenotypes, illustrating the breadth of channel types, tissues, and phyla (including man) in which bioelectric signaling is a critical endogenous aspect of embryogenesis. Second, we perform a novel bioinformatic analysis of transcriptomic data during regeneration in diverse taxa that reveals an electrogenic protein to be the one common factor specifically expressed in regeneration blastemas across Kingdoms. Finally, we analyze data on distinct Vmem signatures in normal and cancer cells, revealing a specific bioelectrical signature corresponding to some types of malignancies. These analyses shed light on fundamental questions in developmental bioelectricity and suggest new avenues for research in this exciting field.
Keywords: cancer, development, channelopathy, Vmem, resting potential, regeneration, ion channel
Introduction
Bioelectrical signaling is an ancient modality by which cells and tissues exchange information necessary for coordinating development, regeneration, and cancer suppression.1–4 Many details have now been uncovered about the ion channels and pumps that determine cell membrane resting potential (Vmem) and about the electrical synapses known as gap junctions that propagate those states across distances in vivo.5 Using a range of model species, modern developmental biology and genetics efforts have identified roles of bioelectrical signaling in cell migration,6,7 organ morphogenesis,8–10 regenerative axial polarity,11 size control,12 and many other important metazoan phenomena. The implications of these results stretch from basic evolutionary developmental biology13,14 to many aspects of biomedicine.15–19
However, this exciting emerging field has been hampered by the fact that, due to its highly interdisciplinary nature, important data are spread across publications in numerous subfields and have not been analyzed en masse. Thus, we undertook a synthesis and meta-analysis of data from published studies to ask several fundamental questions in three deeply related subfields: development, regeneration, and cancer.
How important is bioelectricity in embryogenesis? It is often thought that the roles of ion channels are only apparent from a handful of focused studies,10,20–22 and that genetics does not offer a convenient entry point to discover novel bioelectric controls. Thus, we analyzed databases of developmental phenotypes across a number of popular model systems to identify a comprehensive list of electrogenic genes that have been implicated in embryonic patterning.
Cancer can be thought of as a breakdown of the normal processes that orchestrate cells into cooperating toward production and maintenance of large-scale anatomical structures during development and adulthood.23–26 Interestingly, classical27,28 and recent29–32 functional data suggest that control of bioelectric signaling can not only induce cancer phenotypes but can also be used to suppress/normalize tumors. Can bioelectric parameters be used to distinguish between normal somatic cells and those that have defected back into a unicellular-like state (transformed cancer cells)? We thus attempted to identify every published study of resting potential in cells and analyzed them to derive a physiological signature of the transformed state across different tissue types.
Regeneration is a process by which complex organs and appendages are rebuilt to a specific target morphology from various starting states (injuries).33 The evolutionary origins of this capability across taxa are poorly understood,34 and it is unclear what molecular-genetic and biophysical components underlie regeneration of different types of body architectures. Thus, we analyzed transcriptomic datasets comparing regenerating versus intact adult tissues across Kingdoms, asking what types of regeneration-induced gene expression changes they might share. A small number of genes turned out to be a common feature of regeneration in all animals, from flatworms to mammals. Remarkably, analysis taking into account plants revealed only one component that all regenerating blastemas have in common: an ancient electrogenic protein.
Development, regeneration, and cancer have a profound connection. The first two concern the ability of cells to work together to, respectively, build and rebuild complex anatomical structures. The third, cancer, illustrates the effects of individual somatic cells abandoning multicellularity and reverting to a unicellular mode of existence that treats the rest of the body as its environment. In all of these cases, communication and interaction among cell groups are paramount. Evolution discovered very early how to exploit biophysical forces to coordinate information across space and time in living cells and tissues.35–37 It is now essential, for biomedicine and for evolutionary developmental biology, to understand how the molecular mechanisms of bioelectricity enable cooperative cell function in normal morphogenesis and its disruption during disease.38,39 Together, the following meta-analyses establish a rigorous starting point for novel work in this field, revealing a range of physiological and genetic targets for future investigation and shedding light on foundational concepts in developmental bioelectricity.
Materials and Methods
Literature search for Vmem values in somatic and cancerous cells
Literature search was completed in two distinct phases: first, the keywords “Vmem,” “Electrical Potential,” “Electrophysiological properties,” and “Membrane Potential” were used singly and in combination in PubMed searches. Published Vmem values and their sources were documented in a database to avoid duplication. After all potential papers that could be found using this method were exhausted, existing meta-analyses such as those found in previous studies40,41 were mined for any additional papers that contained Vmem values. These published studies were then themselves included in our analysis. We decided to only include data from rodent and human cells in our analysis because the vast majority of the papers focused on these models and data from other systems did not provide sufficient power for analysis.
Statistical analysis
Meta-analyses were performed to assess relationships between reported Vmem in cancerous and noncancerous tissues. A random-effects meta-analysis was performed to assess the relationship between reported mean Vmem in cancerous tissues and their corresponding somatic tissue type. Statistics were only performed on tissues for which there was both somatic and cancerous measurements taken in the same report. Analysis was performed with the rma function from the Metafor (v2.0-0) R package. p-value was considered significant at p < 0.05, and tests for heterogeneity were performed in each case.
To assess the relationship between the Vmem of a somatic tissue and the difference in Vmem between it and the Vmem of a corresponding cancerous tissue, a mixed-effect regression model was used. We modeled the change in voltage from somatic tissue to cancerous tissue as a linear function of the mean somatic voltage, and included all studies that provided estimates of both means and variability for both cell types. The rma function from the Metafor (v2.0-0) R package was used for this analysis. A least-squares regression was done as sensitivity analysis that included additional studies where estimates of within-study variation were not provided. All studies were equally weighted in this analysis. The lm function in the stats (v3.5.3) R package was used to estimate the linear regression line. p-value was considered significant at p < 0.05, and tests for heterogeneity were performed in each case.
Statistics were performed by the Tufts Biostatistics, Epidemiology, and Research Design Center. R version 3.5.3 (2019-03-1) run through R Studio v1.1.463 (c) 2009–2018, R Studio, Inc. was used to perform the meta-analyses. Raw p-values are reported without adjustment for multiple testing.
Channelopathy identification database and literature search
To identify as many ion channel-associated developmental and morphological phenotypes as possible, we searched species-specific databases that report phenotypes associated with genetic mutations. We primarily focused on phenotype databases to ensure that we had the broadest phenotype report possible, since these databases do not require that the phenotypes induced by genetic mutation be interesting enough to rise to the level of publication. In addition to ion pumps and channels, both of which establish bioelectrical potentials, gap junctions are important because they enable cells to communicate that bioelectric state to each other,5,42 and because their voltage-based and other gating modalities enable cell collectives to form networks, which process bioelectrical information in complex ways, such as forming feedback loops and even computational circuits.43,44 Thus, here we broaden the term “channelopathy” to include those induced by mutations in ion pumps and gap junctions, not only strictly ion channel proteins, because of the similar, crucial roles of these other two types of proteins in regulating the bioelectric state of cells.
Human channelopathies were identified from long-term, manual curation of publications that assess developmental phenotypes linked to genetic screens or clinically identified syndromes. Mouse channelopathies were identified through keyword searches in the MGI database45 for terms, including “channel,” “pump,” “transporter,” “antiporter,” “gap junction,” and “connexin” (and manually filtered to include only electrically relevant targets). Each entry was searched for morphogenesis and developmental phenotypes in the “Mutations, Alleles and Phenotypes” section, and the phenotype was verified in the reported primary literature. To identify zebrafish channelopathies, the database Zfin46 was used. Keyword searches for “Pannexin,” “Innexin,” “Ion Channel,” and “Ion Junction” were carried out to identify channelopathies, and morphogenesis phenotypes were identified in the “Disease Ontology” and “Gene Ontology” sections of each entry. Each phenotype identified was verified in the reported primary literature. Flybase47 was used to identify channelopathies in Drosophila. Keyword searches for “Ion Channel,” “Pannexin,” “Ion Pump,” and “Ion Transporter” were performed, and each entry was searched for developmental and morphogenesis phenotypes in the “Phenotype” section of the entry.
Additional genes mentioned in known papers (previously identified through PubMed searches) on ion channels in Drosophila were also included (and referenced accordingly), particularly, Smith et al.8 which contained a long list of wing-related Drosophila channelopathies. Each phenotype was verified in the reported primary literature. Genes identified in the Flybase search were also assessed using the Bristle Screen Online Database,48 which provided more detailed analyses of morphogenesis defects than Flybase alone. For the Caenorhabditis elegans, we created an XML code that searched Wormbase (release WS271) for patterning defects. The code was provided by one of the curators of the database, Christian A. Grove.
The results that gave multiple patterning defects for one gene were condensed into one entry. Identified entries were then individually assessed for ion channel activity and morphogenesis or developmental defects and confirmed in the reported primary literature.
Bioinformatics
Datasets containing lists of up- and downregulated transcripts in regeneration blastemas of axolotl (Ambystoma mexicanum, NCBI SRA064951),49 planarian head and tail fragments (Schmidtea mediterranea, NCBI SRP002478),50 deer antler (Cervus nippon, BioProject IDs: PRJNA397466 and PRJNA404007), and plant (Arabidopsis thaliana, NCBI GSE19863) were obtained from published supplementary data,49,50 NCBI SRA,51 or NCBI GEO.52 Annotation for the planaria dataset was obtained from eggNOG-mapper.53,54
Some of the datasets only provided F-tests for significant results but did not provide information about direction of fold change. Thus, all significantly altered transcripts, both up- and downregulated, were combined and considered as one pool. Genes with false discovery rate (FDR) <0.25 were considered significantly altered. All significantly altered genes in each dataset were then crossreferenced to identify any transcripts that were commonly differentially expressed across model systems.
For gene set enrichment analysis, “background” genes were assigned in each model organism based on protein homology to human protein-coding gene sequences obtained from BioMart. Gene Ontology sets were acquired from the MSigDB Collections.55 Gene sets related to ion channels and cell junctions or learning, memory, and cognition were selected, and Fisher's exact tests were performed to determine enrichment. FDR <0.25 was considered significant enrichment.
Results and Discussion
Numerous channelopathies reveal roles for bioelectrics in model systems
A number of studies have used gain-of-function approaches (misexpression of specific channel or pump proteins) to evaluate a role of bioelectric states in patterning processes such as development,22,56,57 regeneration,58,59 and tumor suppression/normalization.31,60–68 Some studies also identified endogenous roles for bioelectrical gradients.9,10,12,69,70 It is critical to identify the native conductances responsible for the salient bioelectrical prepatterns in vivo to place Vmem into pathways, understand the evolution of bioelectric control mechanisms, and provide candidate targets for biomedical intervention.
We searched databases of several popular model systems (mouse, zebrafish, Drosophila, and C. elegans) as well as literature, including human channelopathies, to identify known electrogenic genes with developmental phenotypes that include structural malformations. Tables 1–5 show channels, pumps, ion transporters, and gap junctions (and a few direct regulators of these) that are known to cause not just physiological disease conditions but also anatomical malformations (birth defects).
Table 1.
Human Channelopathies
| Protein (gene) | UniProt number | Channel type | Mutant phenotype | Reference |
|---|---|---|---|---|
| V-ATPase, (TCIRG1/VATB1) | Q13488/P15313 | Vacuolar proton pump | Facial dysmorphism, dense bones | 89,90 |
| Kir3.2 (kcnj6) | P48542 | Voltage-gated K+ channel | Keppen-Lubinsky syndrome—craniofacial defects and microcephaly | 91 |
| Kv10.1 (kcnh1) and VATB2 | O95259 and P21281 | K+ channel and vacuolar proton pump | Zimmermann-Laband and Temple-Baraitser syndrome—craniofacial and brain defects, dysplasia/aplasia of nails of thumb and great toe | 92,93 |
| GLRa4 | Q5JXX5 | Ligand-gated Cl− channel | Craniofacial defects | 94 |
| Kir6.1 (kcnj8) | Q15842 | K+ inwardly rectifying channel | Cantu syndrome—face, heart, skeleton, brain defects | 95–97 |
| NALCN | Q8IZF0 | Na+ leak channel | Freeman-Sheldon syndrome—congenital contractures of face and limbs, cerebral and cerebellar atrophy, small pituitary | 98 |
| CFTR | P13569 | Cl− channel transmembrane conductance regulator | Bilateral absence of vas deferens | 99,100 |
| SCN3A | Q9NY46 | Voltage-gated Na+ channel | Polymicrogyria | 101 |
| TRPV1, TRPV4 | Q8NER1, Q9HBA0 | Transient receptor potential cation channels | Temperature-induced face, heart defects | 102 |
| Kv3.1 (kcnc1) | P48547 | Voltage-gated K+ channel | Head/face dysmorphias | 103 |
| KT3.2 (kcnk9) | Q9NPC2 | K+ two-pore domain channel | Birk-Barel dysmorphism syndrome—craniofacial defects, cortical patterning defects | 104–106 |
| Kir6.2 (kcnj 11) | Q14654 | Inwardly rectifying K+ channel | Craniofacial defects, neural differentiation | 107 |
| Kv1.9 (kcnq1) (via epigenetic regulation) | P51787 | Voltage-gated K+ channel | Hypertrophy of tongue, liver, spleen, pancreas, kidneys, adrenals, genitalia | 108–111 |
| Kv1.9 (kcnq1) | P51787 | Voltage-gated K+ channel | Jervell and Lange-Nielsen syndrome—dysmorphism of inner ear | 112–114 |
| Kv3.3 (kcnc3) | O43525 | Voltage-gated K+ channel | Cerebellar dysplasia | 56 |
| Kir2.1 (kcnj2) | P63252 | Inwardly rectifying K+ channel | Andersen-Tawil syndrome—craniofacial defects, abnormal limb patterning, gracile ribs, and long bones | 115–117 |
| Nav1.2 (scn2a) | Q99250 | Voltage-gated Na+ channel | Laterality defects | 118 |
| Cav1.2 (cac1c) | Q13936 | Voltage-gated Ca2+ channel | Timothy syndrome-webbed fingers, dysmorphic facial features, heart development defects, small teeth | 119 |
| Cx43 (gja1) | P17302 | Gap junction | Heart defects (outflow tract and conotruncal), ODDD, visceroatrial heterotaxia | 120–122 |
ODDD, oculodentodigital dysplasia.
Table 2.
Mouse Channelopathies
| Protein (Gene) | UniProt number | Channel type | Mutant phenotype | Reference |
|---|---|---|---|---|
| Kir7.1 (kcnj13) | P86046 | Inwardly rectifying K+ channel | Cleft palate, delayed lung development | 123 |
| HCN1 | O88704 | Hyperpolarization-activated cyclic nucleotide-gated K+ channel | Reduced brain size, loss of interneuron populations | 124,125 |
| CIC-3 (clcn3) | P51791 | H+/Cl− exchange transporter | Brain patterning defects | 126 |
| Kv3.1 (kcnc1) | P15388 | Voltage-gated K+ channel | Growth deficits | 124 |
| TWIK-1 (kcnk1) | O08581 | K+ two-pore domain channel | atrial dilation | 127 |
| Kv1.1 (kcna1) | P16388 | Voltage-gated K+ channel | Megencephaly | 128 |
| Kir6.2 (kcnj11) | Q61743 | Inwardly rectifying K+ channel | Craniofacial defects, negatively regulates neural differentiation | 129 |
| Kv1.9 (kcnq1) (via epigenetic regulation) | P97414 | Voltage-gated K+ channel | Hypertrophy of tongue, liver, spleen, pancreas, kidneys, adrenals, genitalia—Beckwith-Wiedemann syndrome; craniofacial and limb defects | 108–110 |
| Kv1.9 (kcnq1) | P97414 | Voltage-gated K+ channel | Jervell and Lange-Nielsen syndrome—dysmorphism of inner ear and limb, abnormalities in rectum, pancreas, and stomach | 108,112–114,130,131 |
| Kv3.3 (kcnc3) | Q63959 | Voltage-gated K+ channel | Cerebellar dysplasia (human), eye and wing defects (Drosophila) | 56 |
| Kir2.1 (kcnj2) | P35561 | Inwardly rectifying K+ channel | Andersen-Tawil syndrome—craniofacial defects (narrow maxilla, cleft secondary palate), abnormal limb patterning, gracile ribs, and long bones | 115–117,132 |
| GABA-A receptor (gabrb3) | P63080 | Ligand-gated Cl− channel | Angelman syndrome—cleft palate, cerebellar vermis hypoplasia, abnormal cochlear development and morphology | 133–137 |
| ANO1 | Q8BHY3 | Ca2+-activated Cl− channel | Tracheomalacia with cartilage ring defects, abnormal trachealis muscle development | 138 |
| Kir3.2 (kcnj6) | P48542 | G-protein coupled inwardly rectifying K+ channel | Cerebellar development defects | 139–142 |
| Kv11.1 (kcnh2) | O35219 | Voltage-gated inwardly rectifying K+ channel | Cardiac, craniofacial patterning defects | 143 |
| Kvb1.3 (kcnab1) | P63143 | Voltage-gated K+ channel subunit | Cardiac hypertrophy | 144 |
| 5-HT3B (htr3b) | Q9JHJ5 | Ligand-gated cation channel | Increased bone density and body length in females, decreased bone density and body length in males | 145 |
| CIC-2 (clcn2) | Q9R0A1 | Voltage-gated Cl− channel | Abnormal brain and eye morphology | 146 |
| CIC-5 (clcn5) | Q9WVD4 | H+/Cl− exchange transporter | Renal tubular defects, kyphosis, abnormal tooth development | 147 |
| nAChRα7 (chrna7) | P49582 | Ligand-gated cation channel | Abnormal bone structure, abnormal cerebral cortex morphology, abnormal myeloblast development | 148–150 |
| AchR (chrng) | P04760 | Ligand-gated cation channel | Abnormal skeletal muscle morphology, abnormal neuromuscular junction morphology | 151 |
| Cx50 (gja8) | P28236 | Gap junction | Abnormal lens development, micropthalmia | 152,153 |
| Cx45 (gjc1) | P28229 | Gap junction | Cardiac defects (cushion patterning) and impaired hematopoiesis | 154–157 |
| Cx43 (gja1) | P23242 | Gap junction | Deficits in myogenesis, abnormal osteoblast differentiation, left/right asymmetry randomization, heart defects (outflow tract and conotruncal), neural tube defects, delayed ossification of clavicles, ribs, vertebrae, and limbs, syndactyly and limb defects, craniofrontonasal syndrome | 158–164 |
| Cx37 (gja4) | P28235 | Gap junction | Lymphatic system patterning | 165,166 |
| Cx26 (gjb2) | Q00977 | Gap junction | Cochlear development defects | 167 |
| Cx40 (gja5) | Q01231 | Gap junction | Malformed bone in wrists, digits, and sternum joints | 168 |
| PANX3 | Q8CEG0 | Gap junction | Delayed hypertrophic chondrocyte and osteoblast differentiation and delayed initiation of bone mineralization | 169 |
| NMDAR2D (grin2d) | O15399 | Ligand-gated cation channel | Abnormal bone mineralization and structure, reduced heart size | IMPCa |
| GLRB | P48168 | Ligand-gated Cl− channel | Abnormal vertebral column morphology and abnormal intervertebral disk morphology | 170 |
| Kir7.1 (kcnj13) | P86046 | Inwardly rectifying K+ channel | Cleft palate, abnormal lung development, abnormal tracheal development | 123,171 |
| SCNN1B | Q9WU38 | Sodium-permeable, nonvoltage-sensitive Na+ channel | Abnormal pelvic girdle bone morphology, decreased rib number, abnormal kidney morphology | IMPCb 172 |
| Cav1.2 (cacna1c) | Q01815 | Voltage-gated Ca2+ channel | Abnormal brain morphology, enlarged lateral ventricles, cardiac hypertrophy | 173,174 |
https://www.mousephenotype.org/data/genes/MGI:95823
https://www.mousephenotype.org/data/genes/MGI:104696
Table 3.
Zebrafish Channelopathies
| Protein (gene) | UniProt number | Channel type | Mutant phenotype | Reference |
|---|---|---|---|---|
| PANX3 | E7F7V4 | Gap junction | Delayed hypertrophic chondrocyte and osteoblast differentiation and delayed initiation of bone mineralization | 169 |
| Nav1.4a (scn4aa) | A0A0R4IJX4 | Voltage-gated Na+ channel | Abnormal caudal fin structure, small head and trunk, disorganized skeletal muscle | 175 |
| Nav1.5 (scn12aa) | F1R3Q5 | Voltage-gated Na+ channel | Abnormal cardiac ventricle and atrium morphology | 176,177 |
| GluR2A (gria2a) | A0A0R4ING5 | Ligand-activated cation channel | Abnormal cranial cartilage, achondrogenesis, absent ethmoid cartilage, enlarged fourth ventricle | 178 |
| SLC8A4A | A4UQV6 | Ca2+/Na+ antiporter | Decreased length, abnormal digestive tract development, heart mislocalized abnormal liver development | 179,180 |
| SLC24A5 (nckx5) | B0V3S7 | Na+/K+/Ca2+ exchanger | Delayed development of melanin pigmentation (“golden” phenotype) | 181 |
| SLC26A2 | E7F9I7 | So2− transporter | Defective otolith patterning, abnormal semicircular canal morphology | 182 |
| MCU | F1QT29 | Ca2+ uniporter | Abnormal anterior/posterior axis specification; abnormal cell migration during gastrulation, abnormal notochord | 183 |
| CACNB2 | B2XY76 | Voltage-gated Ca2+ channel | Small cardiac ventricle, heart is malformed and edematous, fragile heart tube | 184 |
| P2RX3A | B3DG55 | ATP-gated cation channel, purinoceptor | Hypotrophic ceratobranchial cartilage, abnormal ceratohyal cartilage, pharyngeal and ventral mandibular arches malformed | 185 |
| PIEZO1 | E7FD74 | Mechanosensitive cation channel | Small head, caudal fin curled, erythrocyte deformities | 186,187 |
| SLC8A1 | Q32SG8 | Ca2+/Na+ antiporter | Small head, heart abnormalities | 188,189 |
| CLIC6 | A0A286YBT4 | Cl− intracellular channel | Abnormal otolith morphology, hydrocephalus, left/right defects, and curved body axis | 190 |
| TASK1 (kcnk3) | Q5TZ59 | K+ leak channel | Abnormal heart morphology | 191 |
| TASK2 (kcnk5) | A0A2R8PYU3 | K+ two-pore domain channel | Long anal fin; abnormal caudal fin | 12 |
| CNGA2A (cnga5) | Q0GFG2 | Cyclic nucleotide-gated cation channel | Malformed otolith, right/left symmetry defects | 192 |
| Kv2.1 (kcnb1) | A0A2R8Q685 | Voltage-gated K+ channel | Small fourth ventricle; disrupted gastrulation | 193 |
| Kir4.1 (kcjn10) | E7FD27 | Inwardly rectifying K+ channel | Swim bladder absent, pronephric duct dilated | 194 |
| Cav1.2 (cacna1c) | Q5TZF1 | Voltage-gated Ca2+ channel | Timothy syndrome, hydrocephalus; abnormal heart morphology, small heart, dilated pronephridic duct, cystic kidney, abnormal mandibular arch | 195–197 |
| TRPC1 | E1U7G1 | Transient receptor potential cation channel | Microphthalmia; disrupted angiogenesis, increased curvature in postvent region | 198 |
| Kir7.1 (kcnj13) | Q0KIZ7 | Inwardly rectifying K+ channel | Pigmentation issues on anal and caudal fins | 199,200 |
| MagT1 | Q7ZV50 | Mg2+ transporter | Small, malformed head | 201 |
| H+ V-ATPase | Multiple | Proton pump | Left–right asymmetry defects, muscle and nerve repair | 58,76 |
| CFTR | A0A0R4ID63 | cAMP-dependent Cl− channel | Primordial germ cell development | 202 |
| Cx41.8 (gja5) | F1QL21 | Gap junction | “leo” phenotype, pigmentation pattern defects | 203,204 |
| Cx39.4 (luchs) | Q1LWG0 | Gap junction | Loss of trunk striping, pigmentation pattern defects | 203 |
| Cx43 (gja1) | O57474 | Gap junction | Small fin size, failure of joint morphogenesis, and abnormal pattern regulation | 205–207 |
| FXYD6 | F1QK04 | Na+/K+ ATPase channel modulator | Abnormal Meckel's and ceratohyal cartilage, small head, abnormal skeletal muscle | 208 |
Table 4.
Drosophila Channelopathies
| Protein (gene) | UniProt number | Channel type | Misexpression phenotype | Reference |
|---|---|---|---|---|
| LD30634p (ATP6AP2) | Q9VHG4 | V-ATPase proton pump | Abnormal wing hair and bristle patterning, pigmentation and brain patterning, small size, small head size, blistered wings | 90,209–211 |
| BEST1 | Q9V3J6 | Ca2+-activated Cl− channel | Bifurcation of posterior crossvein | 8 |
| BEST2 | Q9VRW4 | Ca2+-activated Cl− channel | Wings small and severely malformed | 8 |
| BEST3 | Q9VUM7 | Ca2+-activated Cl− channel | Small narrow wings (male), missing anterior and incomplete posterior crossover vein, L2 vein bifurcated | 8 |
| BIB | P23645 | Nonselective cation channel, aquaporin | Defects in neural development, hyperplasia of neuroblasts, sensillum precursors and peripheral glia, small wing size, failure of heart differentiation, increased macrochaetae and supernumary bristles, notum malformation and color defects | 48,212–215 |
| BRV2 | M9PFT4 | Transient receptor potential channel | Bifurcation of posterior crossvein | 8 |
| GluClα | Q94900 | Glutamate-gated Cl− channel | Bristle defects | 8 |
| GluRIIB | Q9VMP3 | Ionotropic glutamate receptor | Bifurcation of posterior crossvein | 8 |
| GPHR | Q3ZAN1 | Voltage-gated Cl− channel | Abnormally small body and wing blades | 216 |
| iINAF-A; B; C | A8JUT0, A8WH7, Q6IIF2 | Transient receptor potential channel modulator | Bifurcation of posterior crossvein | 8 |
| INX2 | Q9V427 | Gap junction | Small eye size, failure of epithelial patterning, loss of bristles, bristle morphology defects, abnormal foregut development, failed germline differentiation, and spermatogenesis | 48,217–221 |
| INX3 | Q9VAS7 | Gap junction | Cuticle defects, and irregular denticle belts, loss of epithelial organization, polarity defects in epidermis, dorsal closure defects, incomplete formation of posterior crossvein and L5 vein, bifurcation of L4 and L5 veins | 219,222 |
| INX4 | Q9VRX6 | Gap junction | Failure of germline differentiation and spermatogenesis | 221 |
| IR67A | Q9VT09 | Ionotropic glutamate receptor | Thick veins, bifurcation of posterior crossvein, and L3 vein | 8 |
| IR76A | Q9VW39 | Ionotropic receptor | Incomplete posterior crossvein, bifurcated L5 vein | 8 |
| IR7B | Q9W3P4 | Ionotropic receptor | Incomplete or bifurcated posterior crossvein, bifurcated L5 vein | 8 |
| Ir84a | Q9VIA5 | Ionotropic glutamate receptor | Thick veins, bifurcated posterior crossvein | 8 |
| Ir92a | Q9VDN3 | Ionotropic glutamate receptor | Bristle defects, abnormal vein pigment | 8 |
| Ir94g | Q9VCM1 | Ionotropic receptor | Bristle defects, abnormal vein pigment | 8 |
| Ir94h | Q9VCM0 | Ionotropic receptor | Bristle defects | 8 |
| Kir2.1 (Irk1) | Q95UP7 | Inwardly rectifying K+ channel | Thick veins, loss of anterior crossvein, L5 and L4 bifurcation, notum malformation, bristle morphology defects | 8,48 |
| Kir2.2 (Irk2) | Q8WQ82 | Inwardly rectifying K+ channel | Wing patterning defects, bristle defects, L5 and L4 bifurcations, loss of anterior crossvein, thick veins | 116 |
| Kir2.3 (Irk3) | X2JAW9 | Inwardly rectifying K+ channel | Small wings, severe wing vein defects L5 and L4 bifurcations, loss of anterior crossvein, thick veins | 116 |
| JYα | A8QI34 | Cation-transporting P-type ATPase | Defects in spermatogenesis and sperm motility | 223 |
| KCNQ | Q8IT87 | Voltage-gated K+ channel | Slow rate of development, enclosure failure, abnormal heart function, posterior crossvein bifurcation | 111,224 |
| α1U (na) | Q8I877 | Na+ leak channel | Small size, long cylindrical abdomen, bristle defects, ectopic vein, notum malformation | 8,48,225 |
| nAChRα5 (CHRNA5) | Q8T5F5 | Ligand-gated cation channel | Bifurcated posterior crossvein, notum malformation | 8,48 |
| nAChRα6 (CHRNA6) | M9PFD8 | Ligand-gated cation channel | Bifurcated posterior crossvein | 8 |
| nAChRα7 (CHRNA7) | Q86MN7 | Ligand-gated cation channel | Bifurcated posterior crossvein | 8 |
| NaCP60E | Q9W0Y8 | Voltage-gated Na+ channel | Bifurcated posterior crossvein, pigment abnormality | 8 |
| Nan | Q9VUD5 | Transient receptor potential channel | Incomplete or bifurcated L5 wing vein | 8 |
| Nhe2 | Q9NGZ4 | Na+/H+ exchanger | Epithelial patterning, abnormal gut morphology, defects in fat body development, defects in eye morphology | 93,226 |
| nrv2 | Q24048 | Na+/K+-transporting P-type ATPase | Cardiac lumen collapse, defective blood–brain barrier formation, tracheal tube size defects, bristle loss | 48,227,228 |
| Ogre | P27716 | Gap junction | Small, disorganized optic lobes | 229 |
| olf186-F | Q9U6B8 | Ca2+ release-activated Ca2+ channel | Small body size, wing defects, abnormal abdominal dorsal multidendritic neurons | 230–232 |
| Or47a | P81921 | Ligand-gated cation channel | Posterior crossvein bifurcation | 8 |
| ppk | O44940 | Degenerin/epithelial Na+ channel | Large body size, bifurcated posterior crossvein | 8,233 |
| ppk17 | Q9VJI4 | Degenerin/epithelial Na+ channel | Bifurcated posterior crossvein, L2 vein, and L3 vein | 8 |
| ppk25 | A1Z6S4 | Degenerin/epithelial Na+ channel | Bifurcated posterior crossvein and L5 vein | 8 |
| ppk30 | Q9VAJ5 | Degenerin/epithelial Na+ channel | Bifurcated L4 and L5 veins, incomplete posterior crossvein and L5 vein | 8 |
| Rdl | P25123 | GABA-gated Cl− channel | L2 incomplete, ectopic bristles, pigment defect | 8 |
| rpk | O46342 | Degenerin/epithelial Na+ channel | Small wings, incomplete L5 formation, bifurcation of L4 and L5 veins | 8,234 |
| SERCA | Q8STG9 | Ca2+-transporting P-Type ATPase | Ectopic veins, ectopic bristles, pigment defect, cardiac dilation, wing notches, malformed legs, reduced eye number, abnormal eyes | 8,235,236 |
| sh | P08510 | Voltage-gated K+ channel | Abnormal pigmentation, abnormal wing expansion, abnormal abdominal muscles, head eversion defects, foreshortened wings and legs, bifurcated posterior crossvein | 8,237,238 |
| SLO2 | A8DY93 | Ca2+-activated K+ channel | Incomplete posterior crossvein | 8 |
| Stim | P83094 | CRAC channel regulator | Loss of bristles, hair cell duplication, small larval size, small abnormal eyes, notched wings, small wings, blistered wings, ectopic wing veins | 8,48,232,239 |
| Task6 | Q9VFS9 | Two-pore domain K+ channel | Bifurcated posterior crossvein, notum malformation | 8,48 |
| Teh1 | Q9VH54 | Voltage-gated Na+ channel | Black spots on wing below L5 vein | 8 |
| THADA | Q9VWB9 | sarco(endo)plasmic reticulum Ca2+-ATPase regulator | Gain/loss of bristles, malformed notum | 48 |
| TRPM | A8DYE2 | Transient receptor potential channel | Decreased cell size, small salivary glands, shortened malpighian tubules | 240,241 |
| Trpml | Q9VW35 | Transient receptor potential channel | Reduced neuromuscular junction boutons, crumpled wings, gain of bristles, notum malformation | 48,242,243 |
| unc79 | Q06AJ1 | Na+ leak channel complex regulator | Bifurcated posterior crossvein, cylindrical abdomen | 8,225 |
| unc80 | Q9VB11 | Na+ leak channel complex regulator | Bifurcated posterior crossvein, ectopic veins, pigment defects | 8 |
| wtrw | Q9VHY7 | Transient receptor potential channel | Incomplete posterior crossvein | 8 |
| CG18549 | Q9VG64 | Ion channel regulatory protein | Posterior crossover vein bifurcation, bristle morphology defects, notum malformation | 8,48 |
CRAC, calcium release-activated channel; TRP, transient receptor potential channel.
Table 5.
Caenorhabditis elegans Channelopathies
| Protein (gene) | UniProt number | Channel type | Mutant phenotype | Reference |
|---|---|---|---|---|
| acr-2 | P48182 | Ligand-gated cation channel | Increased length and girth, abnormal gonad morphology | 71 |
| acr-6 | Q9N4M3 | Ligand-gated cation channel | Increased girth | 71 |
| acr-7 | P45963 | Ligand-gated cation channel | Increased length | 71 |
| acr-9 | Q18556 | Ligand-gated cation channel | Reduced girth | 71 |
| acr-10 | Q21645 | Ligand-gated cation channel | Increased girth | 71 |
| acr-14 | Q22224 | Ligand-gated cation channel | Increased girth | 71 |
| acr-15 | O16926 | Ligand-gated cation channel | Reduced girth and length | 71 |
| acr-18 | G5EG72 | Ligand-gated cation channel | Reduced girth and length | 71 |
| acr-19 | B3WFZ2 | Ligand-gated cation channel | Decreased body length and increased girth | 71 |
| acr-21 | A0A3P6MY71 | Ligand-gated cation channel | Increased girth | 71 |
| acr-23 | G5EG88 | Ligand-gated cation channel | Increased girth | 71 |
| che-6 | O61827 | Cyclic nucleotide-gated channel | Abnormal sensillum morphology | 244 |
| eat-4 | P34644 | Glu/Na+ symporter | Abnormal pharynx morphology | 245 |
| eat-6 | P90735 | Na+/K+ ATPase | Abnormal sarcomere morphology, decreased girth and body length, abnormal pharynx | 245–247 |
| egl-2 | A0A0K3AVF4 | Voltage-gated K+ channel | Increased length and girth, abnormal chemosensory neurons | 71,248 |
| egl-19 | Q8MQA1 | Voltage-gated Ca2+ channel | Variable body length, decreased girth, blocked anus, tail morphology defects, pigmentation defects | 71,245,246,249–251 |
| egl-23 | C0Z3L1 | K+ two-pore domain channel | Decreased body length and increased girth | 71,246 |
| egl-36 | G5EFC3 | Voltage-gated K+ channel | Increased body length and girth | 71 |
| exc-4 | Q8WQA4 | Voltage-gated Cl− channel | Defects in excretory canal, pigmentation defects | 252 |
| exp-2 | H2KZQ6 | Voltage-gated K+ channel | Abnormal pharyngeal muscles, pharyngeal disorganization | 246,253, |
| flr-1 | G5EGI5 | Degenerin/epithelial Na+ channel | Pigmentation defects, reduced body length, reduced girth | 71,249 |
| lev-1 | Q27218 | Ligand-gated cation channel | Reduced girth and body length | 71 |
| lev-8 | Q93329 | Ligand-gated cation channel | Increased body length and girth | 71 |
| lov-1 | Q09624 | Transient receptor potential channel | Increased girth | 71,254 |
| mec-10 | P34886 | Epithelial Na+ channel | Variable body length and increased girth | 71 |
| mod-1 | Q9GQ00 | Ligand-gated Cl− channel | Increased body length and girth | 71 |
| unc-77 | V6CKM5 | Na+ leak channel | Decreased body length and girth | 71 |
| ocr-3 | Q22374 | Transient receptor potential channel | Increased girth | 71 |
| ocr-4 | Q9N3Y9 | Transient receptor potential channel | Decreased body length and increased girth | 71 |
| osm-9 | G5EBV8 | Transient receptor potential channel | Reduced girth and decreased body length, abnormal uterine seam cells | 71,255 |
| sup-9 | O17185 | K+ two-pore domain channel | Increased girth and length | 71 |
| tax-4 | Q03611 | Cyclic nucleotide-gated channel | Failure of left/right asymmetric neuronal fate specification, abnormal sensory neurons | 256,257 |
| trp-1 | P34586 | Transient receptor potential channel | Increased girth | 71 |
| trp-2 | Q8I6Y9 | Transient receptor potential channel | Decreased body length and increased girth | 71 |
| trp-4 | Q9GRV5 | Transient receptor potential channel | Increased girth | 71 |
| unc-110 | Q18120 | Outwardly rectifying K+ channel | Reduced girth | 71 |
| unc-2 | A0A3B1E663 | Voltage-gated Ca2+ channel | Reduced girth and length, abnormal axon branching, failure of left/right asymmetric neuronal fate specification | 71,246,248,258 |
| unc-7 | Q03412 | Gap junction | Variable body width, short body length | 71 |
| unc-8 | Q21974 | Epithelial stretch-gated Na+ channel | Reduced girth and body length, swollen ventral nerve cord | 71,259 |
| unc-9 | O01393 | Gap junction | Small male fan, short body length, reduced girth | 71 |
| unc-17 | P34711 | VAChT | Reduced body size and girth | 246,260 |
| unc-32 | P30628 | V-Type ATPase | Abnormal body wall muscle morphology, pigmentation defects, small size and increased girth, abnormal pharynx, narrow rachis | 71,245,246,261 |
| unc-36 | P34374 | Voltage-gated Ca2+ channel | Increased length, reduced girth, loss of left/right asymmetry | 245,246,262 |
| unc-38 | Q23022 | Ligand-gated cation channel | Reduced girth and decreased body length | 71 |
| unc-49 | G5ECD3 | Ligand-gated Cl− channel | Reduced body size | 263 |
| unc-58 | Q22271 | K+ two-pore domain channel | Reduced body length and girth | 264,265 |
| unc-63 | Q9N587 | Ligand-gated cation channel | Decreased body length and increased girth | 71 |
| unc-68 | A0A2C9C3E8 | Ca2+ release channel | Reduced girth and body length, abnormal pharyngeal axons | 71,266 |
| unc-77 | V6CKM5 | Na+ leak channel | Reduced girth, decreased body length. Abnormal vulva morphogenesis | 71,267 |
| unc-80 | Q9XV66 | Na+ leak channel | Small eggs | 268 |
| unc-103 | G5EFJ9 | Voltage-gated K+ channel | Reduced girth, variable body length, abnormal sarcomere morphology, abnormal body wall musculature | 71,265,268 |
| unc-105 | Q09274 | Epithelial Na+ channel | Animals are shorter and thinner, abnormal body wall musculature, abnormal body morphology, protruding vulva | 71,246,265 |
| trpa-1 | Q18297 | Transient receptor potential channel | Variable body length and increased girth | 71 |
| delm-1 | O45402 | Degenerin/epithelial Na+ channel | Reduced girth and decreased body length | 71 |
| trpa-2 | Q21517 | Transient receptor potential channel | Increased girth and decreased body length | 71 |
| acc-4 | Q9U358 | Ligand-gated Cl− channel | Reduced girth and body length | 71 |
| asic-2 | Q22851 | Epithelial Na+ channel | Decreased body length and increased girth | 71 |
| egas-3 | Q9XTS9 | Epithelial Na+ channel | Abnormal body proportions | 71 |
| egas-2 | Q9U1T8 | Epithelial Na+ channel | Reduced girth and body length | 71 |
| del-9 | Q18077 | Acid-sensing ion channel | Abnormal body proportions | 71 |
| delm-2 | P91103 | Degenerin/epithelial Na+ channel | Reduced girth and body length, abnormal ventral nerve cord patterning | 71,269 |
| acd-2 | P91100 | Degenerin/epithelial Na+ channel | Reduced girth | 71 |
| del-7 | Q18651 | Epithelial Na+ channel | Decreased body length | 71 |
| acd-5 | O01664 | Degenerin/epithelial Na+ channel | Reduced girth and body length compared | 71 |
| asic-1 | O01635 | Epithelial Na+ channel | Reduced girth and body length | 71 |
A large number of channelopathies were identified across taxa (Fig. 1A), with an outsize representation of channelopathies in the less complex organisms, particularly Drosophila melanogaster and C. elegans (Fig. 1B). While the large number of channelopathies identified may reflect the use of these model organisms in broad mutation screens (e.g., wing and bristle formation in Drosophila,8 or length and girth in C. elegans71), it may also be the case that the large number of identified mutations in these species is due to smaller individual-to-individual variation than is seen in mammals, allowing for more narrowly defined “normal” phenotypes.
FIG. 1.

Channelopathies across model systems. (A) The number of known morphogenetic phenotypes arising from ion channel mutations (as obtained from their respective databases) is shown for human, mouse, zebrafish, fruit fly, and nematode models. These data reflect possible differences in the density with which each model system's genome has been examined for anatomical channelopathies. (B) The ratio of channelopathies per total number of genes in each organism is shown; plotting the number of morphogenetic channelopathies relative to the genome size enables estimates of the size-corrected prevalence of ion channel genes that impact anatomy. The disproportionate number of anatomical channelopathies found in Drosophila may suggest that its genome is especially reliant on ion channels for patterning, or could be due to higher density of analysis having been done in the fruit fly model system. (C) Channelopathy phenotypes were broadly categorized, and relative proportions are compared in a pie chart. The majority of developmental channelopathies affect size or patterning, although changes to pigmentation, germline mutations, and missing structures were also identified. (D) Tissue-specific channelopathy phenotypes were considered in combined rodent and human models, and the relative proportion of tissue-specific effects were compared. Brain, heart, and craniofacial defects were most commonly affected by ion channel mutations, with limb and skeletal defects also prevalent. The disproportionate representation of brain, heart, face, and limb defects in channelopathies suggests that ion channels play an important role in patterning of these organs.
Channelopathies appear to primarily affect size and organization of tissues. The phenotypes created by channelopathies that we identified were mostly limited to alterations in size (31.6% of identified phenotypes), body patterning (i.e., left/right asymmetry, craniofacial patterning, limb patterning, etc., 31.9% of identified phenotypes), or tissue patterning (i.e., organ structure, 26.1% of identified phenotypes) (Fig. 1C). Due to variation in organ structure and function between species, we could not directly compare organs between all organisms, but we did detect a wide variety of organs affected in mammals (Fig. 1D). Craniofacial defects (14.7%), neural defects (16%), skeletal defects (12%), limb defects (10%), and heart defects (10%) were particularly common (Fig. 1D). These data reveal that numerous aspects of pattern formation, especially including the skeletal, neural, craniofacial, and cardiac systems, depend on the function of gene products that regulate bioelectric state.
Thus, the unexpected identification of ion channels in unbiased screens is a means by which many laboratories are brought into the field of bioelectricity. It should be noted that the lists in these Tables are a significant underestimate of the true prevalence of bioelectrical controls in development because single-gene knockouts/mutants are often masked by compensation and redundancy of functionality among channel family members or even completely different types of conductances.72,73 Future work will examine the phenotypes of combinatorial knockouts and knock-ins of dominant negative proteins targeting entire subclasses of electrogenic proteins. Despite the difficulty of probing physiological mechanisms with gene targeting approaches, it is seen that classical genetic strategies have already revealed a striking number of bioelectric components in development of model systems with widely diverging developmental architectures, including man.
Electrogenic proteins are an ancient common factor in regeneration across Kingdoms
Regeneration is a process that implements morphogenesis in complex body organs or appendages. It is similar to development, except that unlike embryogenesis, which always begins with the same reliable state (fertilized egg), regeneration builds species-specific target morphologies from diverse starting states (injury conditions with variable amounts and locations of missing tissue). A critical tissue in all instances of regeneration is the blastema—early cells at the site of injury, which must make decisions about differentiation, proliferation, and spatial patterning that determine the fate of the wound (scarring or functional regeneration). We thus used bioinformatics to compare published transcriptomic analyses of transcriptional changes occurring in regeneration blastemas (Fig. 2A). We specifically sought to compare extremely distant taxa (crossing Kingdoms of life), to identify components that were associated with regeneration per se, and not the specifics of any one body-plan architecture or physiological/ecological lifestyle.
FIG. 2.

Bioelectric signature of transcriptional profile in regeneration. (A) A schematic representation of the analysis performed: published transcriptomic profiles of regeneration blastemas from planaria, axolotl, deer antlers, and plants (Arabidopsis) were compared to determine common DEGs across these highly diverse samples. (B) Twenty-three differentially expressed genes are common to regeneration among taxa as diverse as mammals (deer antlers), amphibians (axolotl), and invertebrate flatworms (planaria). These are listed in (B′), and include two subunits of the proton pump complex and a voltage-gated chloride channel. (C) Remarkably, when a plant (Arabidopsis) dataset is included, only one DEG is common in regeneration across Kingdoms, and thus across independent origins of multicellularity: an evolutionarily ancient and highly conserved protein that is a key subunit of both, the V-ATPase proton pump and gap junctions (C′), which have both been implicated in regeneration, wound healing, and developmental patterning.58,74,77,78,205,270–277 DEGs, differentially expressed genes.
Common to animal regeneration (planarian body fragments, frog limb, axolotl limb, and deer antler) were 10 transcripts listed in Table 6 and Figure 2B. These 10 genes define a regenerative signature across regeneration modes, including those based on adult stem cells, dedifferentiation, and tissue renewal.
Table 6.
Transcripts Common to Blastemas Across Kingdoms
| Gene name | Type of protein |
|---|---|
| POLA1 | DNA polymerase alpha 1, catalytic subunit |
| DPP10 | Dipeptidyl peptidase like 10 |
| GRN | Granulin precursor |
| TPM1 | Tropomyosin 1 |
| PSAP | Prosaposin |
| CTSB | Cathepsin B |
| ITGB2 | Integrin subunit beta 2 |
| CD151 | CD151 molecule (Raph blood group) |
| ATP6V0C | ATPase H+-transporting V0 subunit c |
| PLD3 | Phospholipase D family member 3 |
Remarkably, when crossreferenced with transcriptomes from plant (Arabidopsis) regeneration, one common gene remains: the transmembrane ring protein component of the proton-transporting V-ATPase (Fig. 2C). This fascinating structure is related to both gap junctions (a.k.a., electrical synapses)74 and ion pumps that polarize cells, and has already been functionally implicated in embryonic left–right patterning in chick, zebrafish, and frog,75,76 zebrafish eye morphogenesis,77 frog tail regeneration,58 wound healing in Drosophila,78 and stem cell regulation in the mouse brain.79 In some of these cases (e.g., chick and frog), the V-ATPase pump is known to function on the cell surface, generating a significant hyperpolarization of cells by the efflux of positive charges.58,76,80 It is also a regulator of pH, at both the cellular and organelle level,81 and the conserved c subunit has been proposed to function as a gap junction.74,82–86
It is a remarkable fact that an ancient precursor to widely conserved proteins that set resting potential (ion pumps) and distribute it across tissue networks (gap junctions) is the one common factor in regeneration across Kingdoms. Moreover, because of the distinct origins of multicellularity in plants and animals, it is interesting to note that evolution pressed this highly versatile protein complex into service for tissue regeneration at least twice independently. This finding is consistent with the central importance of bioelectrical cellular states and communication in regenerative patterning processes.
Vmem differences among normal and transformed cell types in human and rodents
Seminal work by Binggeli and Weinstein41 building on the work of Cone27,28,87,88 hypothesized that resting membrane potential, Vmem, predictably varied across cell types according to cell type and cell cycle stage. They performed a meta-analysis of literature that reported Vmem in a variety of cell types and stages, showing that proliferative cells and cancer cells both were more depolarized than differentiated cells, and that a boundary existed at around −36 mV that differentiated proliferating and nonproliferating cells.41
Binggeli and Weinstein were only able to find Vmem values for a few examples in each cell type, however, in the ensuing 34 years electrophysiological determination of Vmem has become a standard laboratory practice, providing ample examples for further assessment of Vmem as a function of cell cycle state. Thus, we revisited the Binggeli and Weinstein study,41 and analyzed all of the Vmem measurements we could find in a thorough literature search. We found Vmem values for 41 cancer cell types across 3 species from 18 publications and Vmem values for 70 noncancer cell types across 9 species from 24 publications (Supplementary Table S1). Our list did also include the publications described in Binggeli and Weinstein.41
We found that Vmem was generally more hyperpolarized in normal differentiated tissues than in their cancerous counterparts for both rodent and human tissues (Fig. 3A), a finding consistent with the conclusions in Binggeli and Weinstein. A meta-analysis using a random-effects model comparing data which had both cancer and somatic tissue Vmem values in the same study showed a significant decrease in Vmem in somatic tissue versus cancer (human cancer-somatic delta = 16.0, 95% confidence interval [CI]: 1.1–30.8, p = 0.0343, rodent cancer-somatic delta = 32.3, 95% CI: 8.8–55.7, p = 0.0068), although significant heterogeneity between studies was observed in both human and rodent studies (Tables 7 and 8).
FIG. 3.

Meta-analysis of bioelectric data in cancer. (A) Violin plots show membrane potential (Vmem) values for somatic and cancerous tissues in human and rodent cell types. Rodent cancerous cells were significantly more depolarized than somatic cells (95% CI for change in voltage: −30.3 to −11.2, p < 0.0001), but Vmem in human cells overall was not significantly different in somatic and cancerous cells. Data were collected from reports in the literature (Supplementary Table S1) and analyzed by meta-regression. Papers that contained values for both somatic and cancerous tissues are shown as paired, with a line connecting the values. When only considering paired samples, a random-effects model found that somatic cells were significantly more hyperpolarized than cancerous cells derived from the same cell type. For human somatic cells, the average was 16 mV more hyperpolarized than comparable cancer cells (95% CI: 1.1–30.8, p = 0.0343). Rodent somatic cells were on average 32.3 mV more hyperpolarized than the corresponding cancer cells (95% CI: 8.8–55.7, p = 0.0068). Heterogeneity between the studies was significant (p < 0.05) for both human and rodent studies. *p < 0.05, **p < 0.01. (B) There is an inverse relationship between the Vmem of somatic cells and the amount of change between the somatic and cancerous cells of the same type, suggesting that cells that start more hyperpolarized have larger increases in depolarization than those that start with Vmem closer to 0. A mixed-effects model identified this relationship as significant, with a 0.85-fold reduction in Vmem change for every 1 mV change in the somatic Vmem. p < 0.0001. (C, D) When considered in broad tissue-specific categories, the majority of cancerous cells appear to be more depolarized than the somatic tissues from which they are derived in rodent model systems (C) and human model systems (D). In this analysis, only data for which both somatic and cancerous cell types were represented in our dataset could be used, (grey shaded data in Table 7 for human cells and 8 for rodents) resulting in an insufficient number of studies that could be included in a meta-analysis. Thus, we were unable to determine the significance of this relationship. (E–H) Data were grouped into broad categories to show the types of somatic (E, G) and cancer (F, H) tissues from which we were able to collect Vmem values in both human and rodent model systems. CI, confidence interval.
Table 7.
Human Cell Vmem Measurements
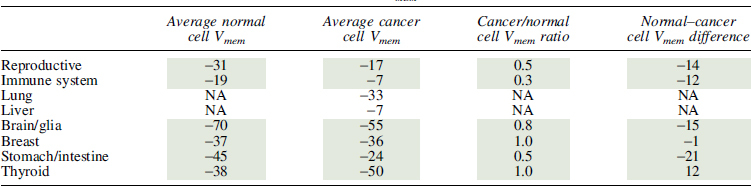 |
NA, not available.
Table 8.
Rodent Cell Vmem Measurements
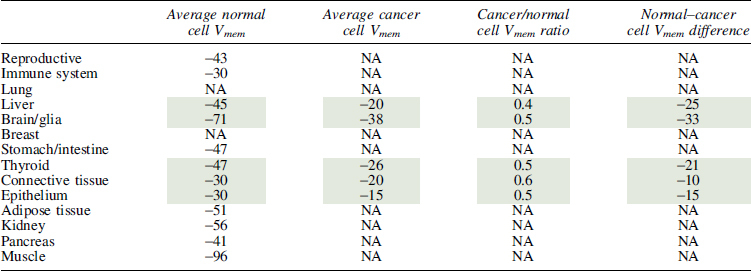 |
Furthermore, cancer cells seemed to fall within a narrower Vmem range than somatic cells such that the cancer cells derived from more hyperpolarized somatic cells showed a greater change in Vmem than those that were derived from less hyperpolarized cells (Fig. 3B, mixed-effects model y = −16.9 − 0.85x, where y is the difference between cancer and somatic Vmem and x is the somatic Vmem, p < 0.0001). While this relationship appears to be maintained when the cells are grouped by tissue type, we did not have a sufficient number of studies to determine whether this is a statistically significant relationship or not. The studies report that Vmem values in rodent cancer lines between −38 and −15 mV (a 23 mV difference) compared with a −96 to −30 mV variance in somatic tissues (a 66 mV difference) (Fig. 3C).
Interestingly, while the variance was broad, all rodent cancer cells had about half the Vmem of their corresponding somatic cells (Table 8), suggesting a linear relationship between somatic and cancerous cell Vmem. In human tissues, cancer cells may have a more depolarized Vmem (Fig. 3D), but insufficient published data are available to give significance. Regardless, the relationship between cancer and somatic cells was complicated, with some tissue types showing hyperpolarization in cancer (thyroid) (Fig. 3D) and more variation in the fold change over somatic Vmem (Table 7). While it is as yet unknown why thyroid cancer contradicts the general trend, such exceptions could be informative; for example, it may be due to the fact that most thyroid cancers are differentiated cancers, which may be expected to exhibit the more hyperpolarized values of differentiated cells. Overall, we find that a comprehensive analysis of Vmem measurements reveal a consistent hyperpolarized relationship between somatic tissue and cancer, with variation dependent on tissue type and model system.
Because this is a meta-analysis, we are limited by the types of Vmem characterization performed in the literature. Moreover, these values were determined by workers using diverse electrophysiological techniques, which may not be uniform across the dataset. Figure 2E–H shows the distribution of cell subtypes from which we were able to collect Vmem information. The rodent dataset lacks Vmem values for many cancer subtypes that have corresponding somatic tissue values and thyroid is over-represented, but otherwise the cell types cover a broad range of cancerous and somatic tissues. The values found for human somatic and cancerous tissues were distributed across many tissue types, and we found similar representation between somatic and cancerous tissue types, indicating that the data in human tissues might be the most reflective of general cancer versus somatic tissue phenotypes. It is clear however that comprehensive profiling of diverse cancer types, from multiple patients/cell lines in each category, using identical electrode types and extracellular media, will be an important component of future work.
Conclusion
Developmental bioelectricity is currently at a very exciting point, because physiology data are beginning to be integrated with molecular genetics. This is enabling a better understanding of the controls of growth and form at many scales, from single-cell behavior to multicell cooperation in organ morphogenesis.
The meta-analysis presented here uncovered some important patterns. First, a large number of channelopathies reveal the genetic basis for some bioelectric disorders. This list will certainly continue to grow as more model systems' genomes receive better coverage, but this will always be an underestimate because of the rich ability of physiological networks to drive complex dynamics even when the protein profile of cells is not altered. A comparison of phenotypes arising from genetic versus physiological stress will shed welcome light on the relationship between genome and anatomy. Many other genomic and profiling large datasets should be mined for interesting patterns of ion channel gene involvement.
Second, we reveal the remarkable fact that a small core of genes is strongly conserved with respect to expression in the regeneration blastema across Phyla. One target, the V-ATPase, which is conserved even across Kingdoms, is likely to be a critical part of repair. It was either harnessed at least twice by independent origins of multicellularity, or was already utilized by the unicellular ancestor for similar functions. This aspect, and the V-ATPase function in unicellular and multicellular morphogenesis, represents an important area for future research.
Finally, we report a higher resolution study of the bioelectric signature of cancer. The patterns we identified point to the urgent need for more data: many more normal and transformed cell types need to be profiled for their bioelectric state, to better understand the physiological controls of cellular cooperation in morphogenesis and defection by cancer cells. It is clear that physiomic profiling is a gap and opportunity for inclusion in genomic, transcriptomic, proteomic, and epigenetic global profiling efforts.
Because of its central role in neural function and embryogenesis, progress in bioelectricity has huge implications not only for basic understanding of evolution and developmental biology but also for regenerative medicine and synthetic bioengineering that impact numerous areas of the biosciences.
Supplementary Material
Author Contributions
P.S., A.K., C.H., and M.L. analyzed data, wrote text, and made figures. All coauthors have reviewed and approved the article before submission.
Author Disclosure Statement
The authors confirm there is no conflict of interest, actual or potential, for each listed author. No competing financial interests exist.
Funding Information
The authors gratefully acknowledge support of the Allen Discovery Center program through The Paul G. Allen Frontiers Group (12171) and the Barton Family Foundation.
Supplementary Material
References
- 1.Funk R.Ion gradients in tissue and organ biology. Biol Syst 2013;2:105 [Google Scholar]
- 2.Bates E.Ion channels in development and cancer. Annu Rev Cell Dev Biol 2015;31:231–247 [DOI] [PubMed] [Google Scholar]
- 3.McLaughlin KA, Levin M. Bioelectric signaling in regeneration: Mechanisms of ionic controls of growth and form. Dev Biol 2018;433:177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin M, Pezzulo G, Finkelstein JM. Endogenous bioelectric signaling networks: exploiting voltage gradients for control of growth and form. Annu Rev Biomed Eng 2017;19:353–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathews J, Levin M. Gap junctional signaling in pattern regulation: Physiological network connectivity instructs growth and form. Dev Neurobiol 2017;77:643–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funk RH.Endogenous electric fields as guiding cue for cell migration. Front Physiol 2015;6:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao M.Electrical fields in wound healing—An overriding signal that directs cell migration. Semin Cell Dev Biol 2009;20:674–682 [DOI] [PubMed] [Google Scholar]
- 8.George LF, Pradhan SJ, Mitchell D, et al. Ion channel contributions to wing development in Drosophila melanogaster. G3 (Bethesda) 2019;9:999–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pai VP, Lemire JM, Pare JF, et al. Endogenous gradients of resting potential instructively pattern embryonic neural tissue via notch signaling and regulation of proliferation. J Neurosci 2015;35:4366–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams DS, Uzel SG, Akagi J, et al. Bioelectric signalling via potassium channels: A mechanism for craniofacial dysmorphogenesis in KCNJ2-associated Andersen-Tawil Syndrome. J Physiol 2016;594:3245–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durant F, Morokuma J, Fields C, et al. Long-term, stochastic editing of regenerative anatomy via targeting endogenous bioelectric gradients. Biophys J 2017;112:2231–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perathoner S, Daane JM, Henrion U, et al. Bioelectric signaling regulates size in zebrafish fins. PLoS Genet 2014;10:e1004080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levin M, Martyniuk CJ. The bioelectric code: An ancient computational medium for dynamic control of growth and form. Biosystems 2018;164:76–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin M.Morphogenetic fields in embryogenesis, regeneration, and cancer: Non-local control of complex patterning. Biosystems 2012;109:243–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mathews J, Levin M. The body electric 2.0: Recent advances in developmental bioelectricity for regenerative and synthetic bioengineering. Curr Opin Biotechnol 2018;52:134–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez-Diaz S, Levin M. Alteration of bioelectrically-controlled processes in the embryo: A teratogenic mechanism for anticonvulsants. Reprod Toxicol 2014;47:111–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin M.The wisdom of the body: Future techniques and approaches to morphogenetic fields in regenerative medicine, developmental biology and cancer. Regen Med 2011;6:667–673 [DOI] [PubMed] [Google Scholar]
- 18.Reid B, Zhao M. The electrical response to injury: Molecular mechanisms and wound healing. Adv Wound Care (New Rochelle) 2014;3:184–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bates EA.A potential molecular target for morphological defects of fetal alcohol syndrome: Kir2.1. Curr Opin Genet Dev 2013;23:324–329 [DOI] [PubMed] [Google Scholar]
- 20.Dahal GR, Pradhan SJ, Bates EA. Inwardly rectifying potassium channels influence Drosophila wing morphogenesis by regulating Dpp release. Development 2017;144:2771–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitcairn E, Harris H, Epiney J, et al. Coordinating heart morphogenesis: A novel role for Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels during cardiogenesis in Xenopus laevis. Commun Integr Biol 2017;10:e1309488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin M, Thorlin T, Robinson KR, et al. Asymmetries in H+/K+-ATPase and cell membrane potentials comprise a very early step in left-right patterning. Cell 2002;111:77–89 [DOI] [PubMed] [Google Scholar]
- 23.Moore D, Walker SI, Levin M. Cancer as a disorder of patterning information: Computational and biophysical perspectives on the cancer problem. Converg Sci Phys Oncol 2017;3:043001 [Google Scholar]
- 24.Rubin H.Cancer as a dynamic developmental disorder. Cancer Res 1985;45:2935–2942 [PubMed] [Google Scholar]
- 25.Sonnenschein C, Soto AM. The Society of Cells: Cancer Control of Cell Proliferation. Oxford, New York: Springer, 1999: xiv, 154 [Google Scholar]
- 26.Burr HS.Biologic organization and the cancer problem. Yale J Biol Med 1940;12:277–282 [PMC free article] [PubMed] [Google Scholar]
- 27.Cone CD, Tongier M. Control of somatic cell mitosis by simulated changes in the transmembrane potential level. Oncology 1971;25:168–182 [DOI] [PubMed] [Google Scholar]
- 28.Cone CD.Unified theory on the basic mechanism of normal mitotic control and oncogenesis. J Theor Biol 1971;30:151–181 [DOI] [PubMed] [Google Scholar]
- 29.Lobikin M, Chernet B, Lobo D, et al. Resting potential, oncogene-induced tumorigenesis, and metastasis: The bioelectric basis of cancer in vivo. Phys Biol 2012;9:065002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Djamgoz MB, Coombes RC, Schwab A. Ion transport and cancer: From initiation to metastasis. Philos Trans R Soc Lond B Biol Sci 2014;369:20130092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chernet BT, Adams DS, Lobikin M, et al. Use of genetically encoded, light-gated ion translocators to control tumorigenesis. Oncotarget 2016;7:19575–19588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentile S.hERG1 potassium channel in cancer cells: A tool to reprogram immortality. Eur Biophys J 2016;45:649–655 [DOI] [PubMed] [Google Scholar]
- 33.Birnbaum KD, Alvarado AS. Slicing across kingdoms: Regeneration in plants and animals. Cell 2008;132:697–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maden M.The evolution of regeneration—Where does that leave mammals? Int J Dev Biol 2018;62:369–372 [DOI] [PubMed] [Google Scholar]
- 35.Larkin JW, Zhai X, Kikuchi K, et al. Signal percolation within a bacterial community. Cell Syst 2018;7:137–145.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Martinez-Corral R, Prindle A, et al. Coupling between distant biofilms and emergence of nutrient time-sharing. Science 2017;356:638–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prindle A, Liu J, Asally M, et al. Ion channels enable electrical communication in bacterial communities. Nature 2015;527:59–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levin M, Selberg J, Rolandi M. Endogenous bioelectrics in development, cancer, and regeneration: Drugs and bioelectronic devices as electroceuticals for regenerative medicine. iScience 2019;22:519–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whited JL, Levin M. Bioelectrical controls of morphogenesis: From ancient mechanisms of cell coordination to biomedical opportunities. Curr Opin Genet Dev 2019;57:61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marino AA, Iliev IG, Schwalke MA, et al. Association between cell membrane potential and breast cancer. Tumour Biol 1994;15:82–89 [DOI] [PubMed] [Google Scholar]
- 41.Binggeli R, Weinstein RC. Deficits in elevating membrane potential of rat fibrosarcoma cells after cell contact. Cancer Res 1985;45:235–241 [PubMed] [Google Scholar]
- 42.Pereda AE, Curti S, Hoge G, et al. Gap junction-mediated electrical transmission: Regulatory mechanisms and plasticity. Biochim Biophys Acta 2013;1828:134–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palacios-Prado N, Bukauskas FF. Heterotypic gap junction channels as voltage-sensitive valves for intercellular signaling. Proc Natl Acad Sci U S A 2009;106:14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manicka S, Levin M. Modeling somatic computation with non-neural bioelectric networks. Sci Rep 2019;9:18612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bult CJ, Blake JA, Smith CL, et al. Mouse Genome Database (MGD) 2019. Nucleic Acids Res 2019;47:D801–D806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howe DG, Bradford YM, Conlin T, et al. ZFIN, the Zebrafish Model Organism Database: Increased support for mutants and transgenics. Nucleic Acids Res 2013;41:D854–D860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thurmond J, Goodman JL, Strelets VB, et al. FlyBase 2.0: The next generation. Nucleic Acids Res 2019;47:D759–D765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mummery-Widmer JL, Yamazaki M, Stoeger T, et al. Genome-wide analysis of Notch signalling in Drosophila by transgenic RNAi. Nature 2009;458:987–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu CH, Tsai MH, Ho CC, et al. De novo transcriptome sequencing of axolotl blastema for identification of differentially expressed genes during limb regeneration. BMC Genomics 2013;14:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kao D, Felix D, Aboobaker A. The planarian regeneration transcriptome reveals a shared but temporally shifted regulatory program between opposing head and tail scenarios. BMC Genomics 2013;14:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leinonen R, Sugawara H, Shumway M, et al. The sequence read archive. Nucleic Acids Res 2011;39:D19–D21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res 2013;41:D991–D995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huerta-Cepas J, Forslund K, Coelho LP, et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol Biol Evol 2017;34:2115–2122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huerta-Cepas J, Szklarczyk D, Forslund K, et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 2016;44:D286–D293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberzon A, Birger C, Thorvaldsdottir H, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khare S, Nick JA, Zhang Y, et al. A KCNC3 mutation causes a neurodevelopmental, non-progressive SCA13 subtype associated with dominant negative effects and aberrant EGFR trafficking. PLoS One 2017;12:e0173565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pai VP, Aw S, Shomrat T, et al. Transmembrane voltage potential controls embryonic eye patterning in Xenopus laevis. Development 2012;139:313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adams DS, Masi A, Levin M. H+ pump-dependent changes in membrane voltage are an early mechanism necessary and sufficient to induce Xenopus tail regeneration. Development 2007;134:1323–1335 [DOI] [PubMed] [Google Scholar]
- 59.Tseng AS, Beane WS, Lemire JM, et al. Induction of vertebrate regeneration by a transient sodium current. J Neurosci 2010;30:13192–13200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chernet BT, Fields C, Levin M. Long-range gap junctional signaling controls oncogene-mediated tumorigenesis in Xenopus laevis embryos. Front Physiol 2015;5:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chernet BT, Levin M. Transmembrane voltage potential of somatic cells controls oncogene-mediated tumorigenesis at long-range. Oncotarget 2014;5:3287–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chernet BT, Levin M. Transmembrane voltage potential is an essential cellular parameter for the detection and control of tumor development in a Xenopus model. Dis Model Mech 2013;6:595–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37–51 [DOI] [PubMed] [Google Scholar]
- 64.Gomez-Varela D, Zwick-Wallasch E, Knotgen H, et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res 2007;67:7343–7349 [DOI] [PubMed] [Google Scholar]
- 65.Isbilen B, Fraser SP, Djamgoz MB. Docosahexaenoic acid (omega-3) blocks voltage-gated sodium channel activity and migration of MDA-MB-231 human breast cancer cells. Int J Biochem Cell Biol 2006;38:2173–2182 [DOI] [PubMed] [Google Scholar]
- 66.Ohkubo T, Yamazaki J. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int J Oncol 2012;41:267–275 [DOI] [PubMed] [Google Scholar]
- 67.Wang H, Zhang Y, Cao L, et al. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res 2002;62:4843–4848 [PubMed] [Google Scholar]
- 68.Warnier M, Roudbaraki M, Derouiche S, et al. CACNA2D2 promotes tumorigenesis by stimulating cell proliferation and angiogenesis. Oncogene 2015;34:5383–5394 [DOI] [PubMed] [Google Scholar]
- 69.Beane WS, Morokuma J, Adams DS, et al. A Chemical genetics approach reveals H,K-ATPase-mediated membrane voltage is required for planarian head regeneration. Chem Biol 2011;18:77–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vandenberg LN, Morrie RD, Adams DS. V-ATPase-dependent ectodermal voltage and pH regionalization are required for craniofacial morphogenesis. Dev Dyn 2011;240:1889–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yemini E, Jucikas T, Grundy LJ, et al. A database of Caenorhabditis elegans behavioral phenotypes. Nat Methods 2013;10:877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koni PA, Khanna R, Chang MC, et al. Compensatory anion currents in Kv1.3 channel-deficient thymocytes. J Biol Chem 2003;278:39443–39451 [DOI] [PubMed] [Google Scholar]
- 73.Kim EZ, Vienne J, Rosbash M, et al. Non-reciprocal homeostatic compensation in Drosophila potassium channel mutants. J Neurophysiol 2017;117:2125–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levin M.Isolation and community: A review of the role of gap-junctional communication in embryonic patterning. J Membr Biol 2002;185:177–192 [DOI] [PubMed] [Google Scholar]
- 75.Gokey JJ, Dasgupta A, Amack JD. The V-ATPase accessory protein Atp6ap1b mediates dorsal forerunner cell proliferation and left-right asymmetry in zebrafish. Dev Biol 2015;407:115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adams DS, Robinson KR, Fukumoto T, et al. Early, H+-V-ATPase-dependent proton flux is necessary for consistent left-right patterning of non-mammalian vertebrates. Development 2006;133:1657–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nuckels RJ, Ng A, Darland T, et al. The vacuolar-ATPase complex regulates retinoblast proliferation and survival, photoreceptor morphogenesis, and pigmentation in the zebrafish eye. Invest Ophthalmol Vis Sci 2009;50:893–905 [DOI] [PubMed] [Google Scholar]
- 78.Fraire-Zamora JJ, Simons M. The vacuolar ATPase is required for ERK-dependent wound healing in the Drosophila embryo. Wound Repair Regen 2018;26:102–107 [DOI] [PubMed] [Google Scholar]
- 79.Lange C, Prenninger S, Knuckles P, et al. The H(+) vacuolar ATPase maintains neural stem cells in the developing mouse cortex. Stem Cells Dev 2011;20:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harvey WR, Wieczorek H. Animal plasma membrane energization by chemiosmotic H+ V-ATPases. J Exp Biol 1997;200 (Pt 2):203–216 [DOI] [PubMed] [Google Scholar]
- 81.Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflugers Arch 2009;457:589–598 [DOI] [PubMed] [Google Scholar]
- 82.Finbow ME, Harrison M, Jones P. Ductin—A proton pump component, a gap junction channel and a neurotransmitter release channel. Bioessays 1995;17:247–255 [DOI] [PubMed] [Google Scholar]
- 83.Dunlop J, Jones PC, Finbow ME. Membrane insertion and assembly of ductin: A polytopic channel with dual orientations. EMBO J 1995;14:3609–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bruzzone R, Goodenough DA. Gap junctions: Ductin or connexins—Which component is the critical one? Bioessays 1995;17:744–745 [DOI] [PubMed] [Google Scholar]
- 85.Finbow ME, Goodwin SF, Meagher L, et al. Evidence that the 16 kDa proteolipid (subunit c) of the vacuolar H(+)-ATPase and ductin from gap junctions are the same polypeptide in Drosophila and Manduca: Molecular cloning of the Vha16k gene from Drosophila. J Cell Sci 1994;107 (Pt 7):1817–1824 [DOI] [PubMed] [Google Scholar]
- 86.Finbow ME, Pitts JD. Is the gap junction channel—The connexon—Made of connexin or ductin? J Cell Sci 1993;106:463–471 [DOI] [PubMed] [Google Scholar]
- 87.Cone CD, Cone CM. Induction of mitosis in mature neurons in central nervous system by sustained depolarization. Science 1976;192:155–158 [DOI] [PubMed] [Google Scholar]
- 88.Cone CD.The role of the surface electrical transmembrane potential in normal and malignant mitogenesis. Ann NY Acad Sci 1974;238:420–435 [DOI] [PubMed] [Google Scholar]
- 89.Borthwick KJ, Kandemir N, Topaloglu R, et al. A phenocopy of CAII deficiency: A novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J Med Genet 2003;40:115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tamirisa S, Papagiannouli F, Rempel E, et al. Decoding the regulatory logic of the Drosophila male stem cell system. Cell Rep 2018;24:3072–3086 [DOI] [PubMed] [Google Scholar]
- 91.Masotti A, Uva P, Davis-Keppen L, et al. Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K(+) channel encoded by KCNJ6. Am J Hum Genet 2015;96:295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kortum F, Caputo V, Bauer CK, et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet 2015;47:661–667 [DOI] [PubMed] [Google Scholar]
- 93.Simons C, Rash LD, Crawford J, et al. Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet 2015;47:73–77 [DOI] [PubMed] [Google Scholar]
- 94.Labonne JD, Graves TD, Shen Y, et al. A microdeletion at Xq22.2 implicates a glycine receptor GLRA4 involved in intellectual disability, behavioral problems and craniofacial anomalies. BMC Neurol 2016;16:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hiraki Y, Miyatake S, Hayashidani M, et al. Aortic aneurysm and craniosynostosis in a family with Cantu syndrome. Am J Med Genet A 2014;164A:231–236 [DOI] [PubMed] [Google Scholar]
- 96.Cooper PE, Reutter H, Woelfle J, et al. Cantu syndrome resulting from activating mutation in the KCNJ8 gene. Hum Mutat 2014;35:809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brownstein CA, Towne MC, Luquette LJ, et al. Mutation of KCNJ8 in a patient with Cantu syndrome with unique vascular abnormalities—Support for the role of K(ATP) channels in this condition. Eur J Med Genet 2013;56:678–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chong JX, McMillin MJ, Shively KM, et al. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet 2015;96:462–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uzun S, Gokce S, Wagner K. Cystic fibrosis transmembrane conductance regulator gene mutations in infertile males with congenital bilateral absence of the vas deferens. Tohoku J Exp Med 2005;207:279–285 [DOI] [PubMed] [Google Scholar]
- 100.Wilschanski M, Dupuis A, Ellis L, et al. Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med 2006;174:787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smith RS, Kenny CJ, Ganesh V, et al. Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral motor development. Neuron 2018;99:905–913.e907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hutson MR, Keyte AL, Hernandez-Morales M, et al. Temperature-activated ion channels in neural crest cells confer maternal fever-associated birth defects. Sci Signal 2017;10:pii: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Poirier K, Viot G, Lombardi L, et al. Loss of Function of KCNC1 is associated with intellectual disability without seizures. Eur J Hum Genet 2017;25:560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Veale EL, Hassan M, Walsh Y, et al. Recovery of current through mutated TASK3 potassium channels underlying Birk Barel syndrome. Mol Pharmacol 2014;85:397–407 [DOI] [PubMed] [Google Scholar]
- 105.Barel O, Shalev SA, Ofir R, et al. Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am J Hum Genet 2008;83:193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bando Y, Hirano T, Tagawa Y. Dysfunction of KCNK potassium channels impairs neuronal migration in the developing mouse cerebral cortex. Cereb Cortex 2014;24:1017–1029 [DOI] [PubMed] [Google Scholar]
- 107.Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Eng J Med 2004;350:1838–1849 [DOI] [PubMed] [Google Scholar]
- 108.Lee MP, Ravenel JD, Hu RJ, et al. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J Clin Invest 2000;106:1447–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weksberg R, Nishikawa J, Caluseriu O, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet 2001;10:2989–3000 [DOI] [PubMed] [Google Scholar]
- 110.Moore ES, Ward RE, Escobar LF, et al. Heterogeneity in Wiedemann-Beckwith syndrome: Anthropometric evidence. Am J Med Genet 2000;90:283–290 [PubMed] [Google Scholar]
- 111.Wen H, Weiger TM, Ferguson TS, et al. A Drosophila KCNQ channel essential for early embryonic development. J Neurosci 2005;25:10147–10156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rivas A, Francis HW. Inner ear abnormalities in a Kcnq1 (Kvlqt1) knockout mouse: A model of Jervell and Lange-Nielsen syndrome. Otol Neurotol 2005;26:415–424 [DOI] [PubMed] [Google Scholar]
- 113.Casimiro MC, Knollmann BC, Yamoah EN, et al. Targeted point mutagenesis of mouse Kcnq1: Phenotypic analysis of mice with point mutations that cause Romano-Ward syndrome in humans. Genomics 2004;84:555–564 [DOI] [PubMed] [Google Scholar]
- 114.Chouabe C, Neyroud N, Guicheney P, et al. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J 1997;16:5472–5479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bendahhou S, Donaldson MR, Plaster NM, et al. Defective potassium channel Kir2.1 trafficking underlies Andersen-Tawil syndrome. J Biol Chem 2003;278:51779–51785 [DOI] [PubMed] [Google Scholar]
- 116.Dahal GR, Rawson J, Gassaway B, et al. An inwardly rectifying K+ channel is required for patterning. Development 2012;139:3653–3664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yoon G, Oberoi S, Tristani-Firouzi M, et al. Andersen-Tawil syndrome: Prospective cohort analysis and expansion of the phenotype. Am J Med Genet A 2006;140:312–321 [DOI] [PubMed] [Google Scholar]
- 118.Fakhro KA, Choi M, Ware SM, et al. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning. Proc Natl Acad Sci U S A 2011;108:2915–2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004;119:19–31 [DOI] [PubMed] [Google Scholar]
- 120.Britz-Cunningham SH, Shah MM, Zuppan CW, et al. Mutations of the Connexin43 gap-junction gene in patients with heart malformations and defects of laterality. N Engl J Med 1995;332:1323–1329 [DOI] [PubMed] [Google Scholar]
- 121.Debeer P, Van Esch H, Huysmans C, et al. Novel GJA1 mutations in patients with oculo-dento-digital dysplasia (ODDD). Eur J Med Genet 2005;48:377–387 [DOI] [PubMed] [Google Scholar]
- 122.Pizzuti A, Flex E, Mingarelli R, et al. A homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phenotype. Hum Mutat 2004;23:286. [DOI] [PubMed] [Google Scholar]
- 123.Villanueva S, Burgos J, Lopez-Cayuqueo KI, et al. Cleft palate, moderate lung developmental retardation and early postnatal lethality in mice deficient in the Kir7.1 inwardly rectifying K+ channel. PLoS One 2015;10:e0139284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zheng J, Trudeau MC. Handbook of Ion Channels. Boca Raton, FL: CRC Press, 2015; xx, 671 [Google Scholar]
- 125.Duque A, Gazula VR, Kaczmarek LK. Expression of Kv1.3 potassium channels regulates density of cortical interneurons. Dev Neurobiol 2013;73:841–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stobrawa SM, Breiderhoff T, Takamori S, et al. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 2001;29:185–196 [DOI] [PubMed] [Google Scholar]
- 127.Christensen AH, Chatelain FC, Huttner IG, et al. The two-pore domain potassium channel, TWIK-1, has a role in the regulation of heart rate and atrial size. J Mol Cell Cardiol 2016;97:24–35 [DOI] [PubMed] [Google Scholar]
- 128.Petersson S, Persson AS, Johansen JE, et al. Truncation of the Shaker-like voltage-gated potassium channel, Kv1.1, causes megencephaly. Eur J Neurosci 2003;18:3231–3240 [DOI] [PubMed] [Google Scholar]
- 129.Zhou Y, Zhu J, Lv Y, et al. Kir6.2 deficiency promotes mesencephalic neural precursor cell differentiation via regulating miR-133b/GDNF in a Parkinson's disease mouse model. Mol Neurobiol 2018;55:8550–8562 [DOI] [PubMed] [Google Scholar]
- 130.Takagi T, Nishio H, Yagi T, et al. Phenotypic analysis of vertigo 2 Jackson mice with a Kcnq1 potassium channel mutation. Exp Anim 2007;56:295–300 [DOI] [PubMed] [Google Scholar]
- 131.Than BL, Goos JA, Sarver AL, et al. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene 2014;33:3861–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Belus MT, Rogers MA, Elzubeir A, et al. Kir2.1 is important for efficient BMP signaling in mammalian face development. Dev Biol 2018;444Suppl 1:S297–S307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Culiat CT, Stubbs LJ, Woychik RP, et al. Deficiency of the beta 3 subunit of the type A gamma-aminobutyric acid receptor causes cleft palate in mice. Nat Genet 1995;11:344–346 [DOI] [PubMed] [Google Scholar]
- 134.Wee EL, Zimmerman EF. GABA uptake in embryonic palate mesenchymal cells of two mouse strains. Neurochem Res 1985;10:1673–1688 [DOI] [PubMed] [Google Scholar]
- 135.Homanics GE, DeLorey TM, Firestone LL, et al. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci U S A 1997;94:4143–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maison SF, Rosahl TW, Homanics GE, et al. Functional role of GABAergic innervation of the cochlea: Phenotypic analysis of mice lacking GABA(A) receptor subunits alpha 1, alpha 2, alpha 5, alpha 6, beta 2, beta 3, or delta. J Neurosci 2006;26:10315–10326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.DeLorey TM, Sahbaie P, Hashemi E, et al. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: A potential model of autism spectrum disorder. Behav Brain Res 2008;187:207–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rock JR, Futtner CR, Harfe BD. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev Biol 2008;321:141–149 [DOI] [PubMed] [Google Scholar]
- 139.Rakic P, Sidman RL. Sequence of developmental abnormalities leading to granule cell deficit in cerebellar cortex of weaver mutant mice. J Comp Neurol 1973;152:103–132 [DOI] [PubMed] [Google Scholar]
- 140.Rakic P, Sidman RL. Weaver mutant mouse cerebellum: Defective neuronal migration secondary to abnormality of Bergmann glia. Proc Natl Acad Sci U S A 1973;70:240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hatten ME, Liem RK, Mason CA. Weaver mouse cerebellar granule neurons fail to migrate on wild-type astroglial processes in vitro. J Neurosci Nurs 1986;6:2676–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Patil N, Cox DR, Bhat D, et al. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet 1995;11:126–129 [DOI] [PubMed] [Google Scholar]
- 143.Teng GQ, Zhao X, Lees-Miller JP, et al. Homozygous missense N629D hERG (KCNH2) potassium channel mutation causes developmental defects in the right ventricle and its outflow tract and embryonic lethality. Circ Res 2008;103:1483–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tur J, Chapalamadugu KC, Padawer T, et al. Deletion of Kvbeta1.1 subunit leads to electrical and haemodynamic changes causing cardiac hypertrophy in female murine hearts. Exp Physiol 2016;101:494–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dickinson ME, Flenniken AM, Ji X, et al. High-throughput discovery of novel developmental phenotypes. Nature 2016;537:508–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Blanz J, Schweizer M, Auberson M, et al. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci 2007;27:6581–6589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wang SS, Devuyst O, Courtoy PJ, et al. Mice lacking renal chloride channel, CLC-5, are a model for Dent's disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum Mol Genet 2000;9:2937–2945 [DOI] [PubMed] [Google Scholar]
- 148.Koval LM, Zverkova AS, Grailhe R, et al. Nicotinic acetylcholine receptors alpha4beta2 and alpha7 regulate myelo- and erythropoiesis within the bone marrow. Int J Biochem Cell Biol 2008;40:980–990 [DOI] [PubMed] [Google Scholar]
- 149.Lin H, Hsu FC, Baumann BH, et al. Cortical parvalbumin GABAergic deficits with alpha7 nicotinic acetylcholine receptor deletion: Implications for schizophrenia. Mol Cell Neurosci 2014;61:163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mallon AM, Iyer V, Melvin D, et al. Accessing data from the International Mouse Phenotyping Consortium: State of the art and future plans. Mamm Genome 2012;23:641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Takahashi M, Kubo T, Mizoguchi A, et al. Spontaneous muscle action potentials fail to develop without fetal-type acetylcholine receptors. EMBO Rep 2002;3:674–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chang B, Wang X, Hawes NL, et al. A Gja8 (Cx50) point mutation causes an alteration of alpha 3 connexin (Cx46) in semi-dominant cataracts of Lop10 mice. Hum Mol Genet 2002;11:507–513 [DOI] [PubMed] [Google Scholar]
- 153.White TW.Unique and redundant connexin contributions to lens development. Science 2002;295:319–320 [DOI] [PubMed] [Google Scholar]
- 154.Frank M, Eiberger B, Janssen-Bienhold U, et al. Neuronal connexin-36 can functionally replace connexin-45 in mouse retina but not in the developing heart. J Cell Sci 2010;123:3605–3615 [DOI] [PubMed] [Google Scholar]
- 155.Kumai M, Nishii K, Nakamura K, et al. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 2000;127:3501–3512 [DOI] [PubMed] [Google Scholar]
- 156.Nishii K, Kumai M, Egashira K, et al. Mice lacking connexin45 conditionally in cardiac myocytes display embryonic lethality similar to that of germline knockout mice without endocardial cushion defect. Cell Commun Adhes 2003;10:365–369 [DOI] [PubMed] [Google Scholar]
- 157.Nishii K, Kumai M, Shibata Y. Regulation of the epithelial-mesenchymal transformation through gap junction channels in heart development. Trends Cardiovasc Med 2001;11:213–218 [DOI] [PubMed] [Google Scholar]
- 158.Araya R, Eckardt D, Riquelme MA, et al. Presence and importance of connexin43 during myogenesis. Cell Commun Adhes 2003;10:451–456 [DOI] [PubMed] [Google Scholar]
- 159.Civitelli R.Cell-cell communication in the osteoblast/osteocyte lineage. Arch Biochem Biophys 2008;473:188–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dobrowolski R, Hertig G, Lechner H, et al. Loss of connexin43-mediated gap junctional coupling in the mesenchyme of limb buds leads to altered expression of morphogens in mice. Hum Mol Genet 2009;18:2899–2911 [DOI] [PubMed] [Google Scholar]
- 161.Ewart JL, Cohen MF, Meyer RA, et al. Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development 1997;124:1281–1292 [DOI] [PubMed] [Google Scholar]
- 162.Lecanda F, Warlow PM, Sheikh S, et al. Connexin43 deficiency causes delayed ossification, craniofacial abnormalities, and osteoblast dysfunction. J Cell Biol 2000;151:931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Reaume AG, de Sousa PA, Kulkarni S, et al. Cardiac malformation in neonatal mice lacking connexin43. Science 1995;267:1831–1834 [DOI] [PubMed] [Google Scholar]
- 164.Davy A, Bush JO, Soriano P. Inhibition of gap junction communication at ectopic Eph/ephrin boundaries underlies craniofrontonasal syndrome. PLoS Biol 2006;4:e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kanady JD, Dellinger MT, Munger SJ, et al. Connexin37 and Connexin43 deficiencies in mice disrupt lymphatic valve development and result in lymphatic disorders including lymphedema and chylothorax. Dev Biol 2011;354:253–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kanady JD, Munger SJ, Witte MH, et al. Combining Foxc2 and Connexin37 deletions in mice leads to severe defects in lymphatic vascular growth and remodeling. Dev Biol 2015;405:33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chang Q, Tang W, Kim Y, et al. Timed conditional null of connexin26 in mice reveals temporary requirements of connexin26 in key cochlear developmental events before the onset of hearing. Neurobiol Dis 2015;73:418–427 [DOI] [PubMed] [Google Scholar]
- 168.Pizard A, Burgon PG, Paul DL, et al. Connexin 40, a target of transcription factor tbx5, patterns wrist, digits, and sternum. Mol Cell Biol 2005;25:5073–5083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Oh SK, Shin JO, Baek JI, et al. Pannexin 3 is required for normal progression of skeletal development in vertebrates. FASEB J 2015;29:4473–4484 [DOI] [PubMed] [Google Scholar]
- 170.Meier H, Chai CK. Spastic, an hereditary neurological mutation in the mouse characterized by vertebral arthropathy and leptomeningeal cyst formation. Exp Med Surg 1970;28:24–38 [PubMed] [Google Scholar]
- 171.Yin W, Kim HT, Wang S, et al. The potassium channel KCNJ13 is essential for smooth muscle cytoskeletal organization during mouse tracheal tubulogenesis. Nat Commun 2018;9:2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pradervand S, Wang Q, Burnier M, et al. A mouse model for Liddle's syndrome. J Am Soc Nephrol 1999;10:2527–2533 [DOI] [PubMed] [Google Scholar]
- 173.Bett GC, Lis A, Wersinger SR, et al. A mouse model of Timothy syndrome: A complex autistic disorder resulting from a point mutation in Cav1.2. N Am J Med Sci (Boston) 2012;5:135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Fu Y, Westenbroek RE, Yu FH, et al. Deletion of the distal C terminus of CaV1.2 channels leads to loss of beta-adrenergic regulation and heart failure in vivo. J Biol Chem 2011;286:12617–12626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Patel N, Smith LL, Faqeih E, et al. ZBTB42 mutation defines a novel lethal congenital contracture syndrome (LCCS6). Hum Mol Genet 2014;23:6584–6593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chopra SS, Stroud DM, Watanabe H, et al. Voltage-gated sodium channels are required for heart development in zebrafish. Circ Res 2010;106:1342–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shu X, Cheng K, Patel N, et al. Na,K-ATPase is essential for embryonic heart development in the zebrafish. Development 2003;130:6165–6173 [DOI] [PubMed] [Google Scholar]
- 178.Li IC, Chen YC, Wang YY, et al. Zebrafish Adar2 Edits the Q/R site of AMPA receptor Subunit gria2alpha transcript to ensure normal development of nervous system and cranial neural crest cells. PLoS One 2014;9:e97133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Doganli C, Kjaer-Sorensen K, Knoeckel C, et al. The alpha2Na+/K+-ATPase is critical for skeletal and heart muscle function in zebrafish. J Cell Sci 2012;125:6166–6175 [DOI] [PubMed] [Google Scholar]
- 180.Shu X, Huang J, Dong Y, et al. Na,K-ATPase alpha2 and Ncx4a regulate zebrafish left-right patterning. Development 2007;134:1921–1930 [DOI] [PubMed] [Google Scholar]
- 181.Lamason RL, Mohideen MA, Mest JR, et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 2005;310:1782–1786 [DOI] [PubMed] [Google Scholar]
- 182.Liu F, Xia W, Hu J, et al. Solute carrier family 26 member a2 (slc26a2) regulates otic development and hair cell survival in zebrafish. PLoS One 2015;10:e0136832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Prudent J, Popgeorgiev N, Bonneau B, et al. Bcl-wav and the mitochondrial calcium uniporter drive gastrula morphogenesis in zebrafish. Nat Commun 2013;4:2330. [DOI] [PubMed] [Google Scholar]
- 184.Chernyavskaya Y, Ebert AM, Milligan E, et al. Voltage-gated calcium channel CACNB2 (beta2.1) protein is required in the heart for control of cell proliferation and heart tube integrity. Dev Dyn 2012;241:648–662 [DOI] [PubMed] [Google Scholar]
- 185.Kucenas S, Cox JA, Soto F, et al. Ectodermal P2X receptor function plays a pivotal role in craniofacial development of the zebrafish. Purinergic Signal 2009;5:395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Delcourt N, Quevedo C, Nonne C, et al. Targeted identification of sialoglycoproteins in hypoxic endothelial cells and validation in zebrafish reveal roles for proteins in angiogenesis. J Biol Chem 2015;290:3405–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Faucherre A, Kissa K, Nargeot J, et al. Piezo1 plays a role in erythrocyte volume homeostasis. Haematologica 2014;99:70–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ebert AM, Hume GL, Warren KS, et al. Calcium extrusion is critical for cardiac morphogenesis and rhythm in embryonic zebrafish hearts. Proc Natl Acad Sci U S A 2005;102:17705–17710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shimizu H, Langenbacher AD, Huang J, et al. The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. Elife 2017;6:pii: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Choksi SP, Babu D, Lau D, et al. Systematic discovery of novel ciliary genes through functional genomics in the zebrafish. Development 2014;141:3410–3419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liang B, Soka M, Christensen AH, et al. Genetic variation in the two-pore domain potassium channel, TASK-1, may contribute to an atrial substrate for arrhythmogenesis. J Mol Cell Cardiol 2014;67:69–76 [DOI] [PubMed] [Google Scholar]
- 192.Shim H, Kim JH, Kim CY, et al. Function-driven discovery of disease genes in zebrafish using an integrated genomics big data resource. Nucleic Acids Res 2016;44:9611–9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shen H, Bocksteins E, Kondrychyn I, et al. Functional antagonism of voltage-gated K+ channel alpha-subunits in the developing brain ventricular system. Development 2016;143:4249–4260 [DOI] [PubMed] [Google Scholar]
- 194.Mahmood F, Mozere M, Zdebik AA, et al. Generation and validation of a zebrafish model of EAST (epilepsy, ataxia, sensorineural deafness and tubulopathy) syndrome. Dis Model Mech 2013;6:652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Andersen ND, Ramachandran KV, Bao MM, et al. Calcium signaling regulates ventricular hypertrophy during development independent of contraction or blood flow. J Mol Cell Cardiol 2015;80:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jin X, Muntean BS, Aal-Aaboda MS, et al. L-type calcium channel modulates cystic kidney phenotype. Biochim Biophys Acta 2014;1842:1518–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ramachandran KV, Hennessey JA, Barnett AS, et al. Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J Clin Invest 2013;123:1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yu PC, Gu SY, Bu JW, et al. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ Res 2010;106:1221–1232 [DOI] [PubMed] [Google Scholar]
- 199.Inaba M, Yamanaka H, Kondo S. Pigment pattern formation by contact-dependent depolarization. Science 2012;335:677. [DOI] [PubMed] [Google Scholar]
- 200.Iwashita M, Watanabe M, Ishii M, et al. Pigment pattern in jaguar/obelix zebrafish is caused by a Kir7.1 Mutation: Implications for the regulation of melanosome movement. PLoS Genet 2006;2:e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Zhou H, Clapham DE. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci U S A 2009;106:15750–15755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Liao H, Chen Y, Li Y, et al. CFTR is required for the migration of primordial germ cells during zebrafish early embryogenesis. Reproduction 2018;156:261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Irion U, Frohnhofer HG, Krauss J, et al. Gap junctions composed of connexins 41.8 and 39.4 are essential for colour pattern formation in zebrafish. Elife 2014;3:e05125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Watanabe M, Iwashita M, Ishii M, et al. Spot pattern of leopard Danio is caused by mutation in the zebrafish connexin41.8 gene. EMBO Rep 2006;7:893–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hoptak-Solga AD, Nielsen S, Jain I, et al. Connexin43 (GJA1) is required in the population of dividing cells during fin regeneration. Dev Biol 2008;317:541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Iovine MK, Higgins EP, Hindes A, et al. Mutations in connexin43 (GJA1) perturb bone growth in zebrafish fins. Dev Biol 2005;278:208–219 [DOI] [PubMed] [Google Scholar]
- 207.Sims K Jr., Eble DM, Iovine MK.. Connexin43 regulates joint location in zebrafish fins. Dev Biol 2009;327:410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Robu ME, Larson JD, Nasevicius A, et al. p53 activation by knockdown technologies. PLoS Genet 2007;3:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hermle T, Guida MC, Beck S, et al. Drosophila ATP6AP2/VhaPRR functions both as a novel planar cell polarity core protein and a regulator of endosomal trafficking. EMBO J 2013;32:245–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hermle T, Saltukoglu D, Grunewald J, et al. Regulation of Frizzled-dependent planar polarity signaling by a V-ATPase subunit. Curr Biol 2010;20:1269–1276 [DOI] [PubMed] [Google Scholar]
- 211.Rujano MA, Cannata Serio M, Panasyuk G, et al. Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J Exp Med 2017;214:3707–3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Djiane A, Krejci A, Bernard F, et al. Dissecting the mechanisms of Notch induced hyperplasia. EMBO J 2013;32:60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Doherty D, Jan LY, Jan YN. The Drosophila neurogenic gene big brain, which encodes a membrane-associated protein, acts cell autonomously and can act synergistically with Notch and Delta. Development 1997;124:3881–3893 [DOI] [PubMed] [Google Scholar]
- 214.Hartenstein AY, Rugendorff A, Tepass U, et al. The function of the neurogenic genes during epithelial development in the Drosophila embryo. Development 1992;116:1203–1220 [DOI] [PubMed] [Google Scholar]
- 215.Rao Y, Bodmer R, Jan LY, et al. The big brain gene of Drosophila functions to control the number of neuronal precursors in the peripheral nervous system. Development 1992;116:31–40 [DOI] [PubMed] [Google Scholar]
- 216.Charroux B, Royet J. Mutations in the Drosophila ortholog of the vertebrate Golgi pH regulator (GPHR) protein disturb endoplasmic reticulum and Golgi organization and affect systemic growth. Biol Open 2014;3:72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Bauer R, Lehmann C, Fuss B, et al. The Drosophila gap junction channel gene innexin 2 controls foregut development in response to Wingless signalling. J Cell Sci 2002;115:1859–1867 [DOI] [PubMed] [Google Scholar]
- 218.Bauer R, Lehmann C, Martini J, et al. Gap junction channel protein innexin 2 is essential for epithelial morphogenesis in the Drosophila embryo. Mol Biol Cell 2004;15:2992–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lehmann C, Lechner H, Loer B, et al. Heteromerization of innexin gap junction proteins regulates epithelial tissue organization in Drosophila. Mol Biol Cell 2006;17:1676–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Richard M, Hoch M. Drosophila eye size is determined by Innexin 2-dependent Decapentaplegic signalling. Dev Biol 2015;408:26–40 [DOI] [PubMed] [Google Scholar]
- 221.Smendziuk CM, Messenberg A, Vogl AW, et al. Bi-directional gap junction-mediated soma-germline communication is essential for spermatogenesis. Development 2015;142:2598–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Giuliani F, Giuliani G, Bauer R, et al. Innexin 3, a new gene required for dorsal closure in Drosophila embryo. PLoS One 2013;8:e69212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Masly JP, Jones CD, Noor MA, et al. Gene transposition as a cause of hybrid sterility in Drosophila. Science 2006;313:1448–1450 [DOI] [PubMed] [Google Scholar]
- 224.Ocorr K, Reeves NL, Wessells RJ, et al. KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci U S A 2007;104:3943–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Humphrey JA, Hamming KS, Thacker CM, et al. A putative cation channel and its novel regulator: Cross-species conservation of effects on general anesthesia. Curr Biol 2007;17:624–629 [DOI] [PubMed] [Google Scholar]
- 226.Simons M, Gault WJ, Gotthardt D, et al. Electrochemical cues regulate assembly of the Frizzled/Dishevelled complex at the plasma membrane during planar epithelial polarization. Nat Cell Biol 2009;11:286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Paul SM, Ternet M, Salvaterra PM, et al. The Na+/K+ ATPase is required for septate junction function and epithelial tube-size control in the Drosophila tracheal system. Development 2003;130:4963–4974 [DOI] [PubMed] [Google Scholar]
- 228.Schwabe T, Bainton RJ, Fetter RD, et al. GPCR signaling is required for blood-brain barrier formation in drosophila. Cell 2005;123:133–144 [DOI] [PubMed] [Google Scholar]
- 229.Lipshitz HD, Kankel DR. Specificity of gene action during central nervous system development in Drosophila melanogaster: Analysis of the lethal (1) optic ganglion reduced locus. Dev Biol 1985;108:56–77 [DOI] [PubMed] [Google Scholar]
- 230.Hattori Y, Usui T, Satoh D, et al. Sensory-neuron subtype-specific transcriptional programs controlling dendrite morphogenesis: Genome-wide analysis of Abrupt and Knot/Collier. Dev Cell 2013;27:530–544 [DOI] [PubMed] [Google Scholar]
- 231.Pathak T, Agrawal T, Richhariya S, et al. Store-operated calcium entry through Orai is required for transcriptional maturation of the flight circuit in Drosophila. J Neurosci 2015;35:13784–13799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pathak T, Trivedi D, Hasan G. CRISPR-Cas-induced mutants identify a requirement for dSTIM in larval dopaminergic cells of Drosophila melanogaster. G3 (Bethesda) 2017;7:923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wegman LJ, Ainsley JA, Johnson WA. Developmental timing of a sensory-mediated larval surfacing behavior correlates with cessation of feeding and determination of final adult size. Dev Biol 2010;345:170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hevia CF, Lopez-Varea A, Esteban N, et al. A search for genes mediating the growth-promoting function of TGFbeta in the Drosophila melanogaster Wing Disc. Genetics 2017;206:231–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Abraham DM, Wolf MJ. Disruption of sarcoendoplasmic reticulum calcium ATPase function in Drosophila leads to cardiac dysfunction. PLoS One 2013;8:e77785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Periz G, Fortini ME. Ca(2+)-ATPase function is required for intracellular trafficking of the Notch receptor in Drosophila. EMBO J 1999;18:5983–5993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Peabody NC, Diao F, Luan H, et al. Bursicon functions within the Drosophila CNS to modulate wing expansion behavior, hormone secretion, and cell death. J Neurosci 2008;28:14379–14391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Luan H, Lemon WC, Peabody NC, et al. Functional dissection of a neuronal network required for cuticle tanning and wing expansion in Drosophila. J Neurosci 2006;26:573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Eid JP, Arias AM, Robertson H, et al. The Drosophila STIM1 orthologue, dSTIM, has roles in cell fate specification and tissue patterning. BMC Dev Biol 2008;8:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Georgiev P, Okkenhaug H, Drews A, et al. TRPM channels mediate zinc homeostasis and cellular growth during Drosophila larval development. Cell Metab 2010;12:386–397 [DOI] [PubMed] [Google Scholar]
- 241.Hofmann T, Chubanov V, Chen X, et al. Drosophila TRPM channel is essential for the control of extracellular magnesium levels. PLoS One 2010;5:e10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Sandstrom DJ.Extracellular protons reduce quantal content and prolong synaptic currents at the Drosophila larval neuromuscular junction. J Neurogenet 2011;25:104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wong CO, Palmieri M, Li J, et al. Diminished MTORC1-dependent JNK activation underlies the neurodevelopmental defects associated with lysosomal dysfunction. Cell Rep 2015;12:2009–2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Smith HK, Luo L, O'Halloran D, et al. Defining specificity determinants of cGMP mediated gustatory sensory transduction in Caenorhabditis elegans. Genetics 2013;194:885–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Avery L.The genetics of feeding in Caenorhabditis elegans. Genetics 1993;133:897–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Shephard F, Adenle AA, Jacobson LA, et al. Identification and functional clustering of genes regulating muscle protein degradation from amongst the known C. elegans muscle mutants. PLoS One 2011;6:e24686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Watson E, MacNeil LT, Arda HE, et al. Integration of metabolic and gene regulatory networks modulates the C. elegans dietary response. Cell 2013;153:253–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Troemel ER, Sagasti A, Bargmann CI. Lateral signaling mediated by axon contact and calcium entry regulates asymmetric odorant receptor expression in C. elegans. Cell 1999;99:387–398 [DOI] [PubMed] [Google Scholar]
- 249.Kamath RS, Fraser AG, Dong Y, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003;421:231–237 [DOI] [PubMed] [Google Scholar]
- 250.Trent C, Tsuing N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics 1983;104:619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kwok TC, Hui K, Kostelecki W, et al. A genetic screen for dihydropyridine (DHP)-resistant worms reveals new residues required for DHP-blockage of mammalian calcium channels. PLoS Genet 2008;4:e1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Berry KL, Bulow HE, Hall DH, et al. A C. elegans CLIC-like protein required for intracellular tube formation and maintenance. Science 2003;302:2134–2137 [DOI] [PubMed] [Google Scholar]
- 253.Davis MW, Fleischhauer R, Dent JA, et al. A mutation in the C. elegans EXP-2 potassium channel that alters feeding behavior. Science 1999;286:2501–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hu J, Barr MM. ATP-2 interacts with the PLAT domain of LOV-1 and is involved in Caenorhabditis elegans polycystin signaling. Mol Biol Cell 2005;16:458–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ghosh S, Sternberg PW. Spatial and molecular cues for cell outgrowth during C. elegans uterine development. Dev Biol 2014;396:121–135 [DOI] [PubMed] [Google Scholar]
- 256.Sarin S, O'Meara MM, Flowers EB, et al. Genetic screens for Caenorhabditis elegans mutants defective in left/right asymmetric neuronal fate specification. Genetics 2007;176:2109–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Komatsu H, Mori I, Rhee JS, et al. Mutations in a cyclic nucleotide-gated channel lead to abnormal thermosensation and chemosensation in C. elegans. Neuron 1996;17:707–718 [DOI] [PubMed] [Google Scholar]
- 258.Chen CH, Lee A, Liao CP, et al. RHGF-1/PDZ-RhoGEF and retrograde DLK-1 signaling drive neuronal remodeling on microtubule disassembly. Proc Natl Acad Sci U S A 2014;111:16568–16573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Tavernarakis N, Shreffler W, Wang S, et al. unc-8, a DEG/ENaC family member, encodes a subunit of a candidate mechanically gated channel that modulates C. elegans locomotion. Neuron 1997;18:107–119 [DOI] [PubMed] [Google Scholar]
- 260.Zhu H, Duerr JS, Varoqui H, et al. Analysis of point mutants in the Caenorhabditis elegans vesicular acetylcholine transporter reveals domains involved in substrate translocation. J Biol Chem 2001;276:41580–41587 [DOI] [PubMed] [Google Scholar]
- 261.Green RA, Kao HL, Audhya A, et al. A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell 2011;145:470–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Chuang CF, Vanhoven MK, Fetter RD, et al. An innexin-dependent cell network establishes left-right neuronal asymmetry in C. elegans. Cell 2007;129:787–799 [DOI] [PubMed] [Google Scholar]
- 263.Bellemer A, Hirata T, Romero MF, et al. Two types of chloride transporters are required for GABA(A) receptor-mediated inhibition in C. elegans. EMBO J 2011;30:1852–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Chen S, Spence AM, Schachter H. Isolation of null alleles of the Caenorhabditis elegans gly-12, gly-13 and gly-14 genes, all of which encode UDP-GlcNAc: Alpha-3-D-mannoside beta1,2-N-acetylglucosaminyltransferase I activity. Biochimie 2003;85:391–401 [DOI] [PubMed] [Google Scholar]
- 265.Park EC, Horvitz HR. Mutations with dominant effects on the behavior and morphology of the nematode Caenorhabditis elegans. Genetics 1986;113:821–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Axang C, Rauthan M, Hall DH, et al. Developmental genetics of the C. elegans pharyngeal neurons NSML and NSMR. BMC Dev Biol 2008;8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Yeh E, Ng S, Zhang M, et al. A putative cation channel, NCA-1, and a novel protein, UNC-80, transmit neuronal activity in C. elegans. PLoS Biol 2008;6:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sedensky MM, Meneely PM. Genetic analysis of halothane sensitivity in Caenorhabditis elegans. Science 1987;236:952–954 [DOI] [PubMed] [Google Scholar]
- 269.Schmitz C, Kinge P, Hutter H. Axon guidance genes identified in a large-scale RNAi screen using the RNAi-hypersensitive Caenorhabditis elegans strain nre-1(hd20) lin-15b(hd126). Proc Natl Acad Sci U S A 2007;104:834–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Takayama K, Muto A, Kikuchi Y. Leucine/glutamine and v-ATPase/lysosomal acidification via mTORC1 activation are required for position-dependent regeneration. Sci Rep 2018;8:8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Monteiro J, Aires R, Becker JD, et al. V-ATPase proton pumping activity is required for adult zebrafish appendage regeneration. PLoS One 2014;9:e92594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Finbow ME, Pitts JD. Structure of the ductin channel. Biosci Rep 1998;18:287–297 [DOI] [PubMed] [Google Scholar]
- 273.Pietrantonio PV, Gill SS. Ductin, a component of the V-ATPase, is developmentally regulated in Heliothis virescens midgut, and anti-ductin antibodies label lateral membranes. Cell Tissue Res 1997;289:97–108 [DOI] [PubMed] [Google Scholar]
- 274.Banerji R, Eble DM, Iovine MK, et al. Esco2 regulates cx43 expression during skeletal regeneration in the zebrafish fin. Dev Dyn 2016;245:7–21 [DOI] [PubMed] [Google Scholar]
- 275.Meier C, Rosenkranz K. Cx43 expression and function in the nervous system-implications for stem cell mediated regeneration. Front Physiol 2014;5:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Oviedo NJ, Morokuma J, Walentek P, et al. Long-range neural and gap junction protein-mediated cues control polarity during planarian regeneration. Dev Biol 2010;339:188–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Oviedo NJ, Levin M. smedinx-11 is a planarian stem cell gap junction gene required for regeneration and homeostasis. Development 2007;134:3121–3131 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.