

2.1. Introduction: Importance of the Tumor Microenvironment
The premise of this book is the importance of the tumor microenvironment (TME). Until recently, most research on and clinical attention to cancer biology, diagnosis, and prognosis were focused on the malignant (or premalignant) cellular compartment that could be readily appreciated using standard morphology-based imaging. More current approaches using fine-needle aspirates, tumor disaggregation, or examination of circulating tumor cells have also typically interrogated just the obviously malignant cell population, not only disregarding other potential cellular constituents, but also the spatial and structural context. Now, thanks to long-standing but only recently fully appreciated work by researchers focusing on the host-malignant cell interface, attention has finally shifted to the study and clinical application of these complex interactions. Evaluation of the TME and tumor immune microenvironment (TIME) in and around cancers has been repeatedly shown to be important in stratification and classification as well as for determining prognosis and predictive response to therapy. To date, specific analysis targets that have been shown, for example, to predict a response to therapy have followed using limited-scope assays adapted for standard methodologies such as traditional single-marker 3,3′-diaminobenzidine (DAB) chromogenic immunohistochemistry (IHC). Several emerging, clinically relevant TME/TIME biologies cannot be effectively analyzed with these one- or two-at-a-time methods, however, which has driven adoption of new technologies in clinical diagnostics, the topic of this chapter.
In addition to specific, molecularly defined biomarkers, attention has also turned to the recognition that changes in the stroma around tumors appear to have real prognostic significance. The term, “desmoplasia”, that describes a common feature seen adjacent to most carcinomas (characterized by a decreased eosinophilia and texture difference as compared to normal or benign reactive stroma) has until recently been noted through exclusively a subjective recognition step performed at the microscope by experienced pathologists. This too is being studied with structural and molecular precision using optics and artificial intelligence (AI) rather than molecular probes, as will be described.
The immune microenvironment was shown to be the most highly predictive feature derived from a total tumor (including tumor cells and TME/TIME) gene expression analysis in breast cancer [1]. In the case of colon cancer, the location (intratumoral versus peripheral) of specific immune cells, chiefly CD8 T cells and macrophages, proved to stratify tumors into high and low risk for mortality [2]. Using unbiased AI-assisted image analysis, cellular presence and spatial relationships in the extracellular matrix (ECM) in and around tumors was shown to have independent prognostic value [3]. Detection of specific patterns of inflammation, along with evaluation of immune checkpoint activation, can predict responses to new classes of immunomodulatory therapies for cancer [4–7].
Such phenomena can be studied or detected in the intact patient (or preclinical animal model) with imaging methods that can provide some insight into host-tumor interactions in situ and (semi-) non-invasively. These techniques include positron-emission tomography (PET) and magnetic resonance (MR) imaging, coupled with reagents that, for example, can highlight immune cell populations in and around a tumor site [8–12]. Optical coherence tomography (OCT) and acousto-optics-based in-vivo imaging can characterize tumor vasculature, another revealing host-tumor interface [13, 14], while mammography [15] and MR imaging [16] can reveal certain structural clues to stromal properties. That said, this chapter will focus on techniques applied to tissue specimens removed and kept more or less structurally intact. In other words, we will examine almost exclusively microscopy-based techniques, as these currently combine high-resolution, even “super-resolution”, morphology, along with new capabilities for multiplexed molecular specificity to help dissect immune cell repertoire as well as the presence and activity state of tumor-cell molecules of interest. In combination, these then allow for exquisite understanding of how and where cell-cell interactions may be playing a role, along with a refined notion of how structural molecules such as collagens or elements such as adjacent blood vessels and nerves may be involved in either determining or revealing tumor properties and outcomes.
IHC has long been the gold standard for detecting expression patterns of therapeutically relevant proteins to identify prognostic and predictive biomarkers as well to enable patient selection for targeted therapy in oncology. This has historically consisted of specific primary antibodies being detected by secondary antibodies, raised against the IgG domain of the host species the primary antibody was generated in, and developed by the reaction of horseradish peroxidase with DAB to demark where the protein is expressed on the section of tissue via precisely located deposition of the resulting brown precipitate. This platform has been ubiquitously used and has shown great utility and reproducibility in the detection of a single protein target on a section of tissue. In the new age of immuno-oncology (IO) there has been a revolution in the fundamental application of IHC; one protein on each section no longer provides enough information to draw effective conclusions about the cell types or biomarkers present in the TME. Accurate characterization of the various immune cell populations can require a large number of cell surface markers, resident cytoplasmic proteins and transcription factors, and has been historically been reserved for flow cytometry analysis. However, recent discoveries have highlighted the importance of demonstrating spatial relationship between certain biomarkers, such as PD-1 and PD-L1, in understanding a patient’s likelihood to respond to checkpoint inhibitor therapy [17–19]. These spatially informed analyses cannot be conducted using cells dissociated from tumor specimens and require examination of intact tissue sections. The new demands being placed on such tissue specimens, with an emphasis on getting more information from increasingly small biopsy material, have spurred the development of advanced multiplex IHC strategies for target discovery and the development of novel, commercially available kits to run on clinical samples for patient stratification and therapy guidance (recently also reviewed by [20, 21]).
We will discuss not only the underlying basis of each highlighted methodology, but also suggest whether it is challenging or straightforward to implement, sufficiently sensitive, practical, affordable, and potentially suitable for clinical use. Even if technically compelling, it is likely that some, possibly most, of the methods discussed here will, when the dust settles, remain research-use-only (RUO) tools rather than being implemented in clinical trial settings or ultimately becoming adopted as a companion/complementary diagnostic.
2.2. Imaging Methods for Evaluating the Extracellular Matrix (ECM)
2.2.1. Characterization of the Structural Components of the TME
The stroma surrounding and investing the malignant epithelial cells play a central and increasingly appreciated role in cancer biology. In addition to the immune cells under great scrutiny, the other structural materials, including loose and dense collagen and other extracellular matrix components, as well as more well-defined anatomic components such as blood vessels, nerves and fat cells, merit increased attention [22, 23]. While the multiplexing techniques touched upon previously rely on labeling of tissue samples with primary antibodies and the detection of these antibodies via fluorescent or enzymatic tags coupled with some form of signal amplification, there are also platforms that can perform molecular imaging by directly sensing the concentration of inherent chemical or molecular constituents using spectroscopy-based tools independent of any exogenous stains, molecular probes, or amplification systems. One of the most significant advantages of these tools is that they are, or can be, stain-free, allowing the subsequent use of traditional histochemical techniques, if desired.
The most prominent detection techniques include vibrational spectroscopic imaging, mass spectroscopic imaging, and magnetic resonance spectroscopic imaging. Of these, vibrational spectroscopic imaging appears particularly promising, and consists of two types of vibrational spectroscopic imaging: mid-infrared imaging and Raman. The former was initially implemented via Fourier-transform infrared (FTIR) spectroscopy, which offered a combination of spatial, spectral, and chemical detail that could be advantageous in the characterization of the TME [24–27]. FTIR is used to obtain an infrared spectrum of absorption and emission across a wide spectral range. Certain molecular species can be recognized—and spatially resolved—based on absorption properties arising from quantized vibrations of chemical bonds.
Originally, the resolution of FTIR was too coarse to provide details at the cellular or subcellular level, but recent advances in the imaging platforms as well as computational tools allow for this platform to be used to understand expression of biomarkers (using this term in a broad sense) in the TME. Subsequent work, still based on FTIR, led to the development of benchtop instrumentation capable of resolving single lymphocytes and subclassifying them, via random-forest tools machine-learning tools, into B-cell and T-cell subtypes, a task not possible with previous instrumentation [28]. More recently, FTIR techniques were replaced by tunable laser methodology with improved speed, spatial resolution, signal-to-noise, and speed [29]. However, scanning time per biopsy-sized specimen was still approximately 1 h, so this work remains best suited to the clinical research space. It thus appears that mid-IR imaging has the potential to reveal increasingly more complex information about cellular details and tumor and immune cells in response to therapy or defining inflammatory status. IHC-based approaches use predetermined markers to understand the dynamic state of the immune cells; IR interrogations are (relatively) unbiased techniques that may be able to classify cell types using criteria other than surface-marker expression. Remaining challenges to broader adoption include slow run and acquisition times, high sensitivity of results to pre-analytic variables, and inconsistency of data processing and analysis [30]. These limitations, coupled with the wide variety of how data is analyzed, result in a paucity of comparison studies across institutions.
Raman microscopy is another very promising but technically challenging approach to obtaining highly informative, subcellularly resolved detail (more detail than IR-spectroscopy), with molecularly specific contrast. There are a variety of technologies that can be deployed, including CARS (coherent anti-Stokes Raman scattering, [31]) and SRS (stimulated Raman spectroscopy, [32]), discussion of whose relative merits is beyond the scope of this chapter. A recent review [33] described a number of results and potential applications, noting that a wide range of relevant tissue phenotypes can be resolved, including identification of cell type and distribution, and organelle content. Functional information is also available, such as nucleic-acid-to-protein ratios in nuclei, cell cycle, and activation status of immune cells. Clearly, such information, especially if combined with more conventional immunostain-based readouts, could be of great value. Again, however, instrumentation cost and complexity, along with relatively slow imaging speeds, remain obstacles to wider deployment.
Other approaches to obtaining additional information on components of the TME include traditional special stains for collagen and for elastin, which is being recognized as an important tool for detection of occult vascular invasion [34]. These require extra microscope sections to be prepared and stained. There are also non-destructive methods for collagen detection that can be performed on unstained or H&E-stained slides; these include second-harmonic generation (SHG) microscopy [35, 36] as well as birefringence-based approaches [37]. The interaction between structural proteins in the TME and events involved in mobilizing or excluding inflammatory cells from the tumoral region is proving to be a rewarding area of investigation (reviewed in [38]). Overall, non-antibody-based methods for characterizing the tumor and in particular the physical microenvironment should become increasingly recognized as being sources of non-duplicative information that can be used on its own, or combined with probe-based multiplex studies, to improve cancer characterization, and maybe particularly valuable when machine-learning-based tools are deployed for detection, characterization, and prognosis.
2.3. Labeling Approaches
2.3.1. Chromogens
Bright-field microscopy-based chromogenic detection of proteins has been the most widely accepted and utilized immunohistochemical assay. This platform utilizes various enzymes to convert soluble substrates to insoluble chromogenic products that can be readily viewed by eye or captured with an imager and whose presence signals the localization of the target of interest on the tissue section [39]. The two enzymes most commonly used are horseradish peroxidase (HRP) and alkaline phosphatase (AP); however, novel enzymatic-based detection systems are currently being developed for advanced applications [40, 41]. HRP is more commonly used than AP for IHC applications as a result of it being a smaller and more stable enzyme, as well as being less expensive [42]. The most commonly used HRP-substrate chromogen in IHC is 3,3′-diaminobenzidine (DAB), a derivative of benzidine, which can be oxidized by hydrogen peroxide to produce a dark brown color. This is the most heavily utilized chromogen due to the reliable chemistry involved, the distinctness of its color from the commonly deployed blue counterstain of hematoxylin as well as the impressive stability of the product [43]. However, there are other chromogens that can be used in this type of assay that, when activated by the appropriate enzyme, generate insoluble chromogens in a variety of colors, including red, green, yellow, blue, orange, purple, and teal. Variations on these chromogens are commercially available from different vendors including Biocare Medical, Enzo Life Sciences, Ventana Medical Systems, and Leica Biosystems; most of these colors can be formed by both HRP- and AP-based reactions. These alternative color options provide the ability to develop multiplex approaches in bright-field-based IHC. Bright-field multiplex assays provide a few critical advantages over their immunofluorescence counterparts, but also suffer from significant limitations. They are typically completed sequentially with a stripping of the primary/secondary complex following each chromogen development, so as to remove the remaining HRP-labeled secondary antibodies and reduce the likelihood of contamination and false positives in the assay. However, when an assay has two primary antibodies raised in different host species, such as mouse and rabbit, and the chromogens used to visualize each marker require different enzymes, such as HRP and AP, the primary antibodies can be simultaneously incubated on the tissue section to save time and reduce stress to the tissue section [44, 45].
Downstream analysis of most multiplex IHC-stained slides is now frequently realized using digital imaging and image-analysis-based quantitative tools that depend on the development of reliable and easy-to-use software-based algorithms to count, stratify, and capture the spatial localization of the cells or cellular compartments labeled by the chromogens. This presents one of the largest challenges to chromogenic multiplexing: if more than two colors are being deployed and run the risk of colocalization, the spectral absorption features of the labels may overlap and cause difficulties in detection and unmixing at the pixel level [46]. A chromogenic duplex, utilizing DAB and a red chromogen with a hematoxylin counterstain, while sometimes challenging to resolve visually, can be separated spectrally—with the caveat that (light-absorbing) chromogens, if co-localized and amplified to high density, can essentially result in such dark signals that reliable unmixing or quantitation is impossible. (This represents an advantage of fluorescence-based methods, in which overlapping signals get brighter, not darker.) Another triple-color chromogen combination that has been identified as spectrally distinguishable is a blue and yellow combination, with a nuclear fast red counterstain. However, once additional chromogens are added, difficulties mount [46]. Evaluating an assay that has three or more markers can become very challenging, with or without the assistance of image analysis. Each marker needs to be spectrally distinct from one another so that the imaging system and coupled computer algorithms can be deployed to identify it uniquely from the other chromogens in one assay. When selecting the chromogens to be used in each assay, one must consider the durability of each reagent, specifically in regard to heat and pH sensitivity. Typically, yellow, blue, and green chromogens are heat- or pH-sensitive while red and DAB are more stable. This results in the placement of these sensitive chromogens in the last position in the processing sequence, thereby restricting each assay to only using one of these more sensitive chromogens if a heat- or pH-based stripping step is involved. The most effective utilization of chromogenic multiplexing is when the proteins of interest are expressed on exclusive cell types or unique subcellular localizations, thereby systematically avoiding pixel-level multiple stains. This, of course, assumes excellent staining methodology with little or no non-specific staining, as well as cooperative biology: not every specimen follows the textbook.
Although some of these chromogens will merge and result in unique colors to identify cells that express both proteins of interest, such as red and green (=purple) or blue and yellow (=green or black, depending on which chromogen is applied first), this can often complicate the analysis of these assays. The characterization of the TME in the context of immuno-oncology (IO) often requires identification of multiple markers expressed on the same cell population. The need to identify co-localized proteins in bright-field multiplex IHC assays has led to the development of a novel light-mergeable chromogen platform, as developed by Ventana Medical Systems (a member of the Roche group). This platform utilizes peroxidase-catalyzed oxidation, similar to the previously mentioned HRP and DAB reaction, to covalently bind dyes in the visible spectrum to tissue sections localized at the site of primary antibody binding [47]. This is accomplished through the use of tyramide signal amplification (TSA)-modified dyes and oxidation of tyramide moieties by HRP (free radical tyramide species), which will covalently bind to tyrosines in proteins within the fixed tissue specimen; therefore, anything that is conjugated to these tyramide moieties will also be covalently bound to the tissue sections.
Tyramide conjugation is used by multiple platforms, and the general technology is referred to as tyramide signal amplification. This approach overcomes the challenges faced by more traditional chromogens when antibodies are co-localized, namely, lack of differentiation between the color of a double-positive cell and a single-positive cell (for example, moderately dense brown plus red can resemble a single dark brown signal). The platform utilizes dyes that have narrow spectral signatures, unlike the more traditional IHC chromogens, as to take up less of the visible spectra with each stain; this allows for the formation of visually distinct colors when two markers are expressed on the same cell. Currently, Ventana offers three reagents that comprise the platform, DISCOVERY Yellow, DISCOVERY Purple, and DISCOVERY Teal (there are other DISCOVERY chromogens, but these are the only tyramide-based reagents). When DISCOVERY Purple is co-localized with DISCOVERY Yellow, an orange color is created. Similarly, when DISCOVERY Purple is co-localized with DISCOVERY Teal, and when DISCOVERY Yellow is co-localized with DISCOVERY Teal, dark blue and green colors result, respectively.
An outline of a workflow for TSA assays (which are applied in both chromogenic as well as fluorescence-based techniques) is as follows: Primary antibodies are incubated and detected with species-specific secondary antibodies, labeled with HRP. Oxidation of fluorescently labeled tyramide molecules results in covalent conjugation to the tissue section directly around the primary/secondary antibody complex. The primary/secondary complex is stripped off the tissue section, using heat or pH, leaving the covalently bound chromogenic-deposition enzymes or fluorescently labeled tyramide molecules where the antibodies bound. This type of staining approach requires sequential application of the primary antibodies, with a heat- or pH-strip of the primary and secondary complexes and quenching of the remaining peroxidase enzymes between each detection. This results in lengthy protocol times, as well as repeated exposure of the tissue to high temperature or pH conditions, steps that can impact the tissue quality or the chromogen performance. The covalent labeling of tissue sections through tyramide reactions is not unique to Ventana; other platforms that have the same working principle will be discussed below. The Ventana DISCOVERY Chromogens are currently the only system that utilizes tyramide technology to improve bright-field multiplex capabilities.
2.3.2. Fluorescent Dyes
Standard Multiplexing
Standard immunofluorescence (IF) multiplexing is similar in many ways to chromogenic multiplexing but provides critical improvements that allow for much more complex analysis (see Table 2.1 for a summary of current chromogenic and IF multiplexing methods). Standard IF relies on the same principles of target-specific primary antibodies, and species-specific secondary antibodies as previously described. However, in this platform, the secondary antibodies are directly conjugated with a fluorescent dye instead of a reactive enzyme (in some instances, the primary antibodies themselves are conjugated to dyes, so removing a requirement for species-specific secondary antibodies). These fluorescent tags do not catalyze a subsequent reaction as is done with HRP, but they themselves are the detection platform. Each fluorescent tag has an absorption spectrum, and when struck by excitation light in the appropriate range will emit a spectrally distinct signature. Most standard IF is captured using conventional wide-field, or alternatively, confocal, microscopes; we will discuss the mechanisms for acquiring these images later in this section. The utilization of such fluorescent dyes allows for better efficacy in detection of co-localized signals compared to chromogenic labeling techniques. Each dye after it is excited will emit energy that can be captured from the specific localization of the complex of the primary and secondary antibodies.
Table 2.1.
Summary and review of the multiplex staining methods
| Multiplex staining method | Company | Platform | Advantages | Disadvantages |
|---|---|---|---|---|
| Chromogenic IHC | Leica Biocare Ventana Enzo Dako |
Bright-field chromogens | Standardized platform Protocols are easily validated and reproducibly Established workflow for image acquisition and analysis |
Sections are exposed to heat or pH stripping Chromogens have broad spectral signatures; limited to 2–3 targets Challenging to (sub)phenotype one cell type |
| Standard IF | Thermo Fisher BioLegend eBioscience |
Fluorophore-tagged primary and secondary antibodies | All markers can be detected simultaneously; short assay time No repeated heat or pH stripping steps |
Primary antibodies need to be raised in unique host species to allow for accurate detection Choice of commercial fluorophores limits assays to 4–5 markers |
| Tyramide signal amplification | Akoya Ventana |
OPAL DISCOVERY™ chromogens | Antibodies can be selected independent of host species IgG | Sections are exposed to heat or pH stripping Long protocols as a result of subsequent antibody incubation and detection Challenging to phenotype one cell type |
| Hapten-tagged primary antibodies | Cell IDx | UltraPlex | All markers can be labeled and detected simultaneously; short assay time Antibodies can be selected independent of host species IgG No repeated heat or pH stripping steps |
No amplification of primary signal Primary and secondary antibodies need to be specifically conjugated as a pair Some potential for steric hindrance when phenotyping one cell type |
| DNA-barcoded secondary antibodies | Ultivue | UltiMapper | All markers can be detected simultaneously; short assay time Antibodies can be selected independent of host species IgG No repeated heat or pH stripping steps |
Primary and secondary antibodies need to be specifically conjugated as a pair Amplification and hybridization reactions must perform identically to ensure staining consistency |
| Cycling IHC platforms | NeoGenomics Akoya t-CyCIF |
MultiOmyx CODEX |
All markers can be detected simultaneously Antibodies can be selected independent of host species IgG No repeated heat or pH stripping steps |
No amplification of primary signal Long sample acquisition times; stains and images only 2–4 markers per cycle |
| Mass-tagged primary antibodies | Fluidigm IONpath |
CyTOF Hyperion MIBI |
All markers can be detected simultaneously Antibodies can be selected independent of host species IgG No repeated heat or pH stripping steps Significantly decreases the crosstalk between antibody labels; removes challenges around auto-fluorescence |
Long sample acquisition times Images produced are representations of the actual subcellular localization of primary antibody binding Challenging to obtain the material required to label the antibodies |
This standard multiplexing approach does present an initial challenge of requiring primary antibodies to be raised in distinct host species, so that the secondary reagents would not bind to primary antibodies against the other targets, resulting in a false-positive signal. There are protein engineering techniques that do allow for the antibody-binding domain of reactive and specific antibodies to be attached to the Fc region of unique species. This is to say, if a primary antibody was generated in a mouse, the antibody-binding domain could be isolated and attached to the Fc region of goat IgG. There are reagents that target the IgG domain of a wide variety of species such as mouse, rabbit, rat, goat, chicken, and even two distinct types of hamster. However, these engineering efforts can be technically challenging and do not ensure that the detection would be identical to the original antibody. Another possibility is to generate secondary reagents specific to unique isoforms of antibodies raised in the same species, such as mouse antibodies can be formed with IgG1, IgG2a, IgG2b, and IgG3. If the primary reagents had distinct Fc regions, even if they were raised in the same species, they could still be discriminated using the appropriate secondary. If such engineering efforts were completed and secondary reagents were identified, a high-level multiplex assay could be attained with a low level of false positives. This type of assay is not an open platform, as all reagents would need to be engineered and paired to the appropriate secondary before the assay could be optimized for staining conditions.
One example of a demonstration of high-level multiplexing was accomplished by Dr. Dragan Maric and colleagues [48] (and several posters at scientific conferences). In a tour-de-force, these investigators performed simultaneous 10-plex imaging with various species/isotypes primary antibodies and lectins, followed by specific secondary antibodies (e.g., goat anti-rabbit, goat anti-mouse IgG1, goat anti-mouse IgG2a), accompanied by 10 different fluorophores (DyLight 405, Alexa Fluor 350, Alexa Fluor 430, Alexa Fluor 546, Alexa Fluor 594, Alexa Fluor 647, Alexa Fluor 700, IRDye 800CW, PerCP), optimized single-filter cubes, sCMOS camera, and XY stage tiling and stitching (with Z-series acquisitions if needed). Imaging and signal disambiguation could thus be performed using a standard fluorescence microscope, equipped with multiple filter cubes. This heroic method could then be iterated through further rounds of 10-plex per round (20, 30, etc.), and made more efficient by employing either: (i) both short- and long-Stokes-shift fluorophores (e.g., Pacific Blue and Pacific Orange) with multiple cameras; (ii) adopting new-generation “flow cytometry” fluorophores, such as the BD Biosciences Brilliant Violets, Brilliant Ultraviolets, and Brilliant Blues, and/or ThermoFisher SuperBrights [49]; see also [50–52]; or quantum dots (discussed below).
In summary, the approaches described above can—in principle—allow all primary reagents to be simultaneously incubated followed by a second, single incubation with the corresponding secondaries. The difficulties encountered with these standard strategies have led to the development of other multiplexing techniques, described below.
Sequential Fluorescence Staining
Sequential staining involves incubation with a probe, deposition of an insoluble label via enzymatic polymerization, followed by a heat- or pH-stripping step to remove the catalyzing enzyme, and then repeating the labeling process to deposit a spectrally distinct second layer, and so on, for as many labels that can be distinguished via fluorescence detection. While long and laborious, this approach has compensating benefits that include more accessible labeling strategies and simplified imaging demands. One example is the approach commercially developed as OPAL, offered by Akoya Bioscience, formerly the quantitative pathology branch of Perkin Elmer. However, this is not a novel approach, having been described previously by various labs [53–55]. This platform utilizes the conjugation of fluorescent dyes to tyramide moieties, as described previously with the DISCOVERY chromogens, and the covalent binding of these fluorescent tyramide moieties to the tissue section; in fact, Ventana has described a TSA-based approach similar to OPAL that runs on their DISCOVERY platform. This approach allows the user to select any primary antibody and pair it with any fluorescent dye, interchanging the pairing of primary and secondary dye flexibly. The most significant advantage of this approach is the ability to use primary antibodies from the same species. Challenges include those previously described with the DISCOVERY chromogens: lengthy protocol times and the requirement of sequential antibody incubation with stripping between each detection. An additional limitation of this platform is the loss of sensitivity as the number of markers deployed increases in the same cell or subcellular compartment. As described previously, this approach results in covalent attachment of tyramide moieties to the tissue surrounding the primary and secondary antibody complex. This deposition of tyramide can potentially prevent the ability of sequential antibodies to bind in close proximity to targets previously detected (steric hindrance), and so the order in which targets are detected becomes important, as will be described below. Collectively the field has recognized many of the challenges associated with both the chromogenic and standard IF approaches for accurate and easy multiplex detection of factors in the TME. There have been many recent approaches to resolve some of these outstanding challenges that have resulted in the next generation of multiplex IHC.
2.3.3. Hapten-Based Secondaries (Cell IDx)
A new commercial platform that attempts to address the problems facing chromogenic and standard IF multiplexing for IHC involves the UltraPlex technology developed by Cell IDx. The UltraPlex still uses primary and secondary antibody complexes to detect the antigens of interest as in the standard IF assay but relies on a unique modification of the primary antibodies to overcome the usual requirement for antibody type diversity (Fig. 2.1). This platform uses primary antibodies labeled with peptide-based hapten tags recognized by high-affinity rabbit monoclonal anti-hapten secondary antibodies—rather than being based on antibody Fc-region targeting used in the traditional approach, allowing the use of a panel of primary antibodies with any combination of species of origin. This then permits an optimal choice of antibodies without having to consider primary and secondary compatibility. Haptens are the smallest chemical moieties of an epitope that can bind effectively to an antigen-binding site of an antibody [56]; these molecules can elicit an immune response when attached to a “carrier” protein. Haptens have been used in a wide variety of biological assay such as protein purification, biomarker detection, and the production of monoclonal antibodies [57]. However, for multiplex biomarker detection, their use until recently had been limited by the paucity of available hapten and anti-hapten antibody pairs, mostly featuring just fluorescein, biotin, and digoxigenin. Cell IDx offers four-plex panels for researchers with standard fluorescence microscopes or scanners, since the typical fluorescent tags attached to the secondary antibody panel were selected to display low-to no spectral overlap in typical band-pass spectral windows; they thus avoid the need for complicated and pricey multispectral imaging systems (covered later in this section). Cell IDx offers custom six-plex panels to researchers with imagers that include spectral unmixing capabilities. As a result of the modularity of hapten design it will be possible going forward to incorporate more markers per imaging step, and then, if necessary, employ multispectral imaging or other detection technology to resolve the resulting signals. With this approach, only two labeling steps are required: incubation with the primary antibody cocktail followed (after a wash) with incubation with the secondary cocktail. This technique thus significantly reduces the overall time of the assay and eliminates the need for the repeated heat- or pH-based stripping steps previously described.
Fig. 2.1.
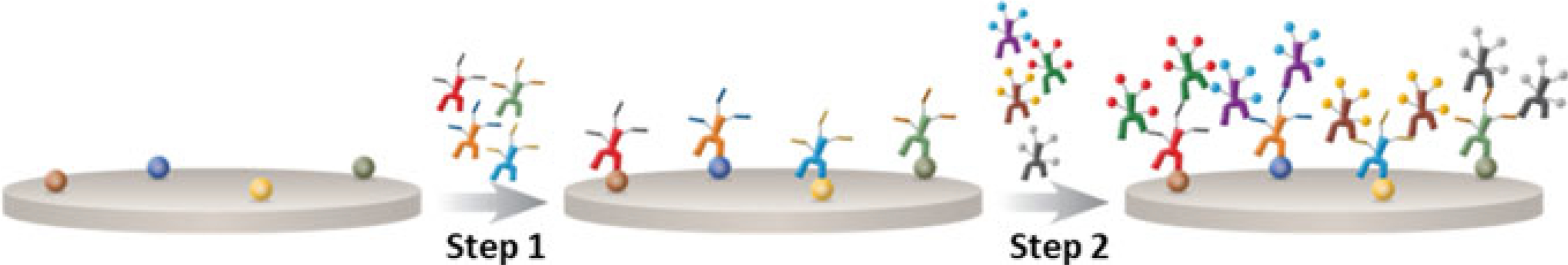
Systematic workflow for Cell IDx UltraPlex. Step 1: A cocktail of primary antibodies, previously conjugated to unique hapten tags, is incubated with the tissue sections. Step 2: A cocktail of fluorescently labeled secondary antibodies, specific to the intended primary antibody’s hapten tag, is used to detect the localization of the primary antibody (Modified from “UltraPlex Technology”, by Cell IDx, Inc., [https://cellidx.com/technology/technology]. Copyright 2016–2019 by Cell IDx. Reprinted with permission)
It can thus take advantage of a serial detection strategy in which an initial multiplexed combination of labels would be applied, imaged, and then primary and secondary antibody complex stripped off and the process repeated—depending on the number of discriminable labels, this could result in, say, multiple targets visualized in a 2-stage process, and the process could then be repeated with another set of markers to increase further the multiplexed signals that could be resolved on a single tissue section. In common with all of the techniques described in this chapter, serial histological sections (slides) can be stained, imaged, and overlaid with different multiplexed panels; an example demonstrating the quantification of 11 markers on 3 serial tissue sections was recently published [58]. The image registration approach would be more beneficial in the preclinical or exploratory space; however, in clinical application, this would be less attractive as the main benefit of multiplexed IHC is to reduce the overall number of slides required to obtain large amounts of histological data. This is especially important when tissue samples are limited, as in the case of core needle biopsies.
Although this platform has made significant progress to address the challenges of multiplex IHC, it still faces a few potential challenges of its own. The first and potentially most significant is the simultaneous primary and secondary antibody complex formation. In the previously described platforms, there were also primary and secondary antibody complex formations; however, they were stripped between each labeling cycle, only leaving the covalently bound tyramide. The UltraPlex platform does not remove the primary and secondary antibody complex when detecting the antigens of interest, with the potential for steric hindrance to occur if there are multiple co-localized proteins expressed at high levels. However, in practice, the steric issue seems manageable, as the non-amplified indirect labeling system does not involve deposits of polymers, and triple-co-expressors are readily visualized (David Schwartz, Cell IDx, personal communication). Another significant challenge that faces the UltraPlex as well as oligonucleotide-based platforms described below is their primary antibody conjugation. The reproducibility of these platforms depends on where the hapten or oligonucleotide tags are conjugated with the primary antibody of choice, where they bind and how consistently they bind to the primary antibody. If there are lot-to-lot variations of the number of hapten or oligonucleotide tags conjugated to the primary antibody, this could result in varied quantification of expression levels of proteins, even if applied to the same tissue at the same concentration. As part of the QC process, each lot could be titrated on a control tissue to ensure consistency, but this again would not be ideal for clinical application. As this platform does not amplify the fluorescent signal produced, as other enzyme-based multiplex IHC approaches do, difficulties may be encountered when attempting to detect proteins expressed at a low level, or proteins like PD-1, whose expression may involve a large dynamic range.
2.3.4. DNA-Tagged Primaries
Another commercial platform attempting to resolve the challenges presented by chromogenic and standard multiplex IHC is based on the UltiMapper™ detection kits and InSituPlex® technology from Ultivue (Fig. 2.2). This platform utilizes primary antibodies directly labeled with specific oligonucleotide sequences, or DNA barcodes, that are then amplified and hybridized to complementary strands of oligonucleotides labeled with fluorescent tags (Walter, Manesse, Mohammed, and Natan, US Patent US20180164308A1). This platform is very similar in concept to the use of DNA-labeled primary antibodies for increasing the sensitivity of immuno-PCR assays [59], except now applied to multiplex IHC.
Fig. 2.2.
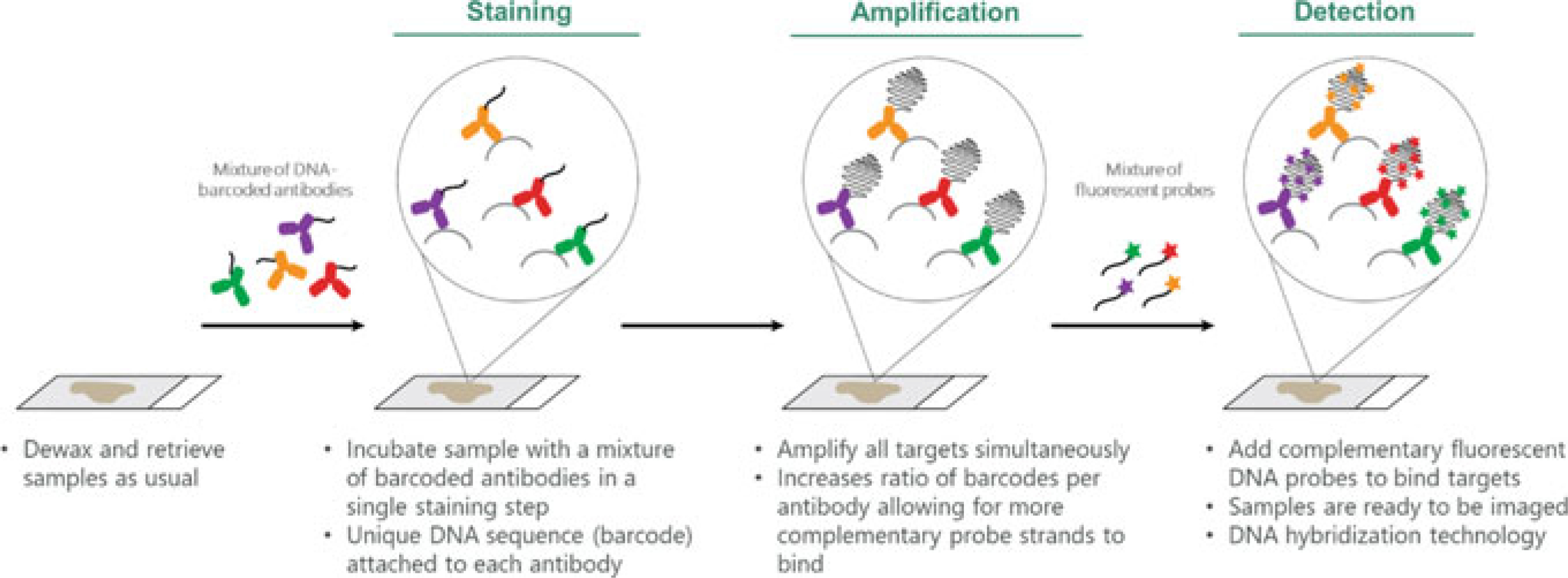
Systematic workflow for Ultivue UltiMapper™. Primary antibodies, labeled with unique DNA barcodes, are incubated on tissue sections. The strands of DNA conjugated to the primary antibodies are amplified, and then hybridized to complementary strands of DNA. The complementary strands of DNA are labeled with a fluorescent tag. (Modified from “InSituPlex® Technology,” by Ultivue, Inc., 2017 [https://www.ultivue.com/technology/]. Copyright 2019 by Ultivue, Inc. Reprinted with permission)
Currently, the InSituPlex platform offers four commercially available kits, each designed to detect up to four markers, looking at mechanisms such as T-cell activation, antigen cross-presentation, T-cell exhaustion, and immunogenicity in the TME. Since the DNA-labeled primary antibody will only be specifically detected by the fluorescent tag conjugated to the complementary oligonucleotide sequence, as in the case with the hapten-based approach, all the primary antibodies can be applied to the specimen simultaneously, even if they are derived from the same species. An advantage of this approach is that the use of oligonucleotide components allows for an amplification step that increases signal intensity. Currently, each individual UltiMapper kit is limited to detection of four markers because the fluorescent tags associated with the complementary oligonucleotides have no spectral overlap, and do not require complicated multispectral unmixing. However, this does not mean that the Ultivue platform is limited to detecting 4 markers; InSituPlex technology can enable higher levels of multiplexing on the same tissue slide through the use of repeat cycles of oligo-exchanging using a mild probe removal step. For example, the workflow for an 8-plex assay would include staining all 8 antibodies, amplifying the barcodes on all 8 antibodies, hybridizing labeled-oligonucleotides to barcodes corresponding to targets 1–4, and imaging. A second cycle would include dehybridizing the complimentary labeled oligonucleotides, hybridizing new labeled oligos to barcodes corresponding to targets 5–8, and imaging. The images from this platform can be captured in the same way that standard IF multiplex images are captured with filters designed for the specific fluorescent tags used in each assay. The UltiMapper platform offers the potential to generate high-level multiplex IHC assays for studying the TME in the context of IO.
2.3.5. Cyclic Labeling and Imaging
There is a class of multiplexed IHC platforms that involves the use of a limited number of markers per imaging session, followed by a label quench and repeated incubations with new probes; these are referred to as cycling platforms. Multi-epitope-ligand-cartography (MELC), now known as Toponome imaging system (TIS), incubates with two antibodies at a time, labels them with phycoerythrin and fluorescein, captures the images, photo-bleaches the section, and repeats for up to 100 markers [60–63]. This platform was one of the first cycling IHC methodology for multiplexing but comes with a number of limitations. Due to the photobleaching-based method of quenching, it can realistically only capture one field of view at a time.
Sequential immunoperoxidase labeling and erasing (SIMPLE) is another cycling-based multiplex platform. This approach utilizes the alcohol-soluble peroxidase substrate, 3-amino-9-ethylcarbazole (a red chromogen). With this platform each cycle consists of detection of a single antigen, imaging, and removal of 3-amino-9-ethylcarbazole by graded alcohol washes, an antibody wash; the cycles are repeated for as many markers as desired [64]. These two platforms have since been largely replaced by more effective cycling multiplex platforms, which operate on the same fundamental principle but utilize more agile technologies.
MultiOmyx is another cycling approach to multiplex IHC, developed by GE Healthcare and currently marketed by NeoGenomics (Fig. 2.3). This platform uses fluorescently labeled primary antibodies, up to 4 per round; after imaging, the signals are quenched, and the cycle can be repeated to capture staining behavior for over 60 antigens per slide [65, 66]. This platform uses alkaline oxidation reactions to quench the fluorescent signal and can be applied to the whole section simultaneously. Directly labeled primary antibodies are used to detect the antigens of interest, an approach that typically results in poor sensitivity for proteins expressed at a low level, or that can encounter difficulties with targets expressed over a large dynamic range.
Fig. 2.3.
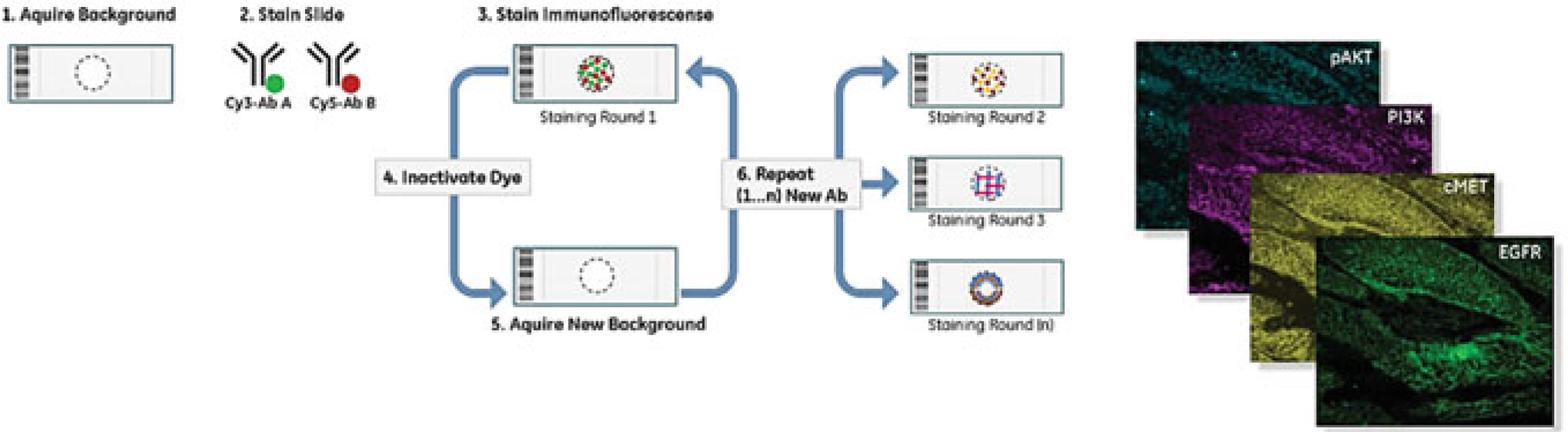
Systematic workflow for MultiOmyx (also known as MxIF). Primary antibodies, labeled with either FITC or PE, are incubated on the tissue sections. The slides are imaged, the dyes are inactivated via alkaline oxidation, and the cycle is repeated. Images are registered after they are scanned and a composite image, as well as the individual images, can be used to study marker expression. Not shown here, the platform is commonly used with Cy3 and Cy5 tyramide signal amplification (TSA) HRP-antibodies (2 cycles), and the “CyDyes” inactivated by a GE patented method (licensed to NeoGenomics) (Modified from “MultiOmyx™ Hyperplexed Immunofluorescence Assay” by NeoGenomics Laboratories, Inc. [https://neogenomics.com/pharma-services/lab-services/multiomyx/technology/hyperplexed-immunofluorescence-assay]. Copyright 2019. Reprinted with permission)
Akoya Biosciences has developed another platform for the generation of highly multiplexed IHC panels, CODEX2 (Co-Detection by Indexing, version 2, see Fig. 2.4 for workflow). Their CODEX2 platform is a major reengineering of the academic CODEX1 [67]. CODEX2 also uses unique DNA barcodes to label their primary antibodies, differing from Ultivue in that they do not amplify this sequence; instead, each primary antibody is designed to have a unique DNA oligonucleotide duplex with designated 5’ overhangs. The CODEX2 platform simultaneously incubates all primary antibodies in one initial staining step, then the slide is transferred to the CODEX system where the slides are subject to secondary reagents, namely, fluorescently labeled complementary oligonucleotide sequences to specific primary antibody 5′ overhangs. Once on the CODEX instrument, the secondary reagents are applied, the slide is imaged, and then stripped with alcohol washes; the cycle repeats again with a new round of secondary reagents labeled with complementary oligonucleotides to a different set of primary antibodies. The antibodies to be revealed first typically have the shortest DNA overhangs and these overhang regions get longer as the detection of that primary antibody moves back in the cycling protocol. This platform only stains three primary antibodies at one time, using spectrally distinct fluorescent markers, and currently can stain, image, quench, and repeat for, typically, ~30 markers. The CODEX platform can be integrated with any three-color fluorescence microscope.
Fig. 2.4.
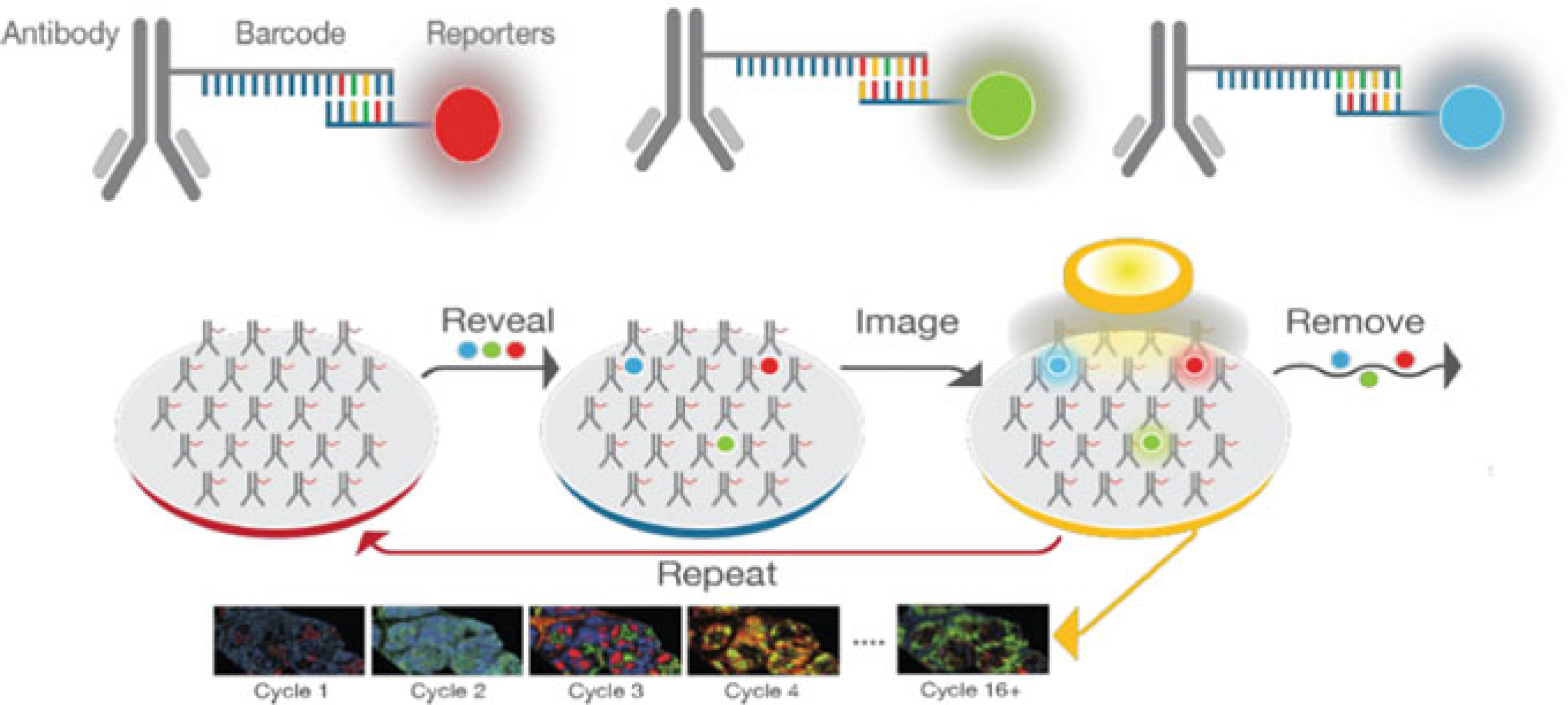
Cyclic workflow for CODEX2 high multiplexing. Primary antibodies, labeled with unique DNA barcode tags are incubated on tissue samples. Primary antibodies are then detected by CODEX reporters (fluorescently labeled DNA strands complementary to the DNA barcode tags). Three CODEX reporters are assayed during each imaging cycle. First, tissues are stained with the full panel of CODEX antibodies. Iterative cycles of labeling, imaging, and removing reporters are performed via a fully automated fluidics system and images are compiled across cycles to produce data with single-cell resolution. Unlike the “academic” approach of CODEX1, the commercialization of CODEX2 uses proprietary method(s). (Modified from Dakshinamoorthy et al., “Highly multiplexed single-cell spatial analysis of tissue specimens using CODEX®”, [https://www.akoyabio.com/application/files/9315/5553/3117/Poster-Akoya_CODEX_AACR-Mar_2019.pdf] Copyright 2019, Akoya Biosciences. Reprinted with permission)
This platform requires that the primary and secondary reagent pairs be predetermined and conjugated specifically for each panel. As with the Cell IDx UltraPlex and NeoGenomics MultiOmyx platforms, there is no amplification of the secondary signal as there is with the UltiMapper and TSA platforms, and thus it could run into the same risks with targets that are expressed at low levels or with a large dynamic range. The CODEX platform, like the UltiMapper, does not encounter the challenges of insoluble tyramide deposits that could potentially limit the sensitivity of looking at multiple targets on the same subcellular localization of the same cell. This platform does not employ rigorous stripping steps, as each fluorescent staining reaction is quenched with washes in graded alcohol. The speed of this platform depends largely on the size of the sample, as after each round of staining the whole section needs to be imaged, and depending on the number of markers being evaluated; smaller tissue sections with smaller panels would provide more reasonable assay run times than if large areas or high multiplicity were required. If core needle biopsy samples were to be evaluated for a triplex using CODEX, there might be clinical utility. However, there is also utility of this approach for more discovery-level questions, in which time is less of a critical factor, and the potential to look a large number of targets on a single tissue specimen is a priority.
Akoya Biosciences is positioning CODEX2 to accomplish around a 50-plex performance, useful for discovery; this offering is complemented by their recent acquisition of technologies combining tyramide signal amplification reagents and the Vectra Polaris multispectral slide scanner that together could be deployed for preclinical studies or even clinical applications.
Finally, t-CyCIF (tissue-based cyclic immunofluorescence), another cyclic immunolabeling and detection system was recently described by Lin et al. [68] (see Fig. 2.5). The method takes advantage of an effective and low-impact strategy for elimination of fluorescent labels after each 4-color imaging round. The technique is based on the use of a high-pH hydrogen peroxide solution (4.5% H2O2 and 24 mM NaOH in PBS for 1 h at RT) combined with white-light illumination to induce fluorophore bleaching. The authors point out that t-CyCIF uses widely available reagents, conventional slide scanners and microscopes, manual or automated slide processing and simple protocols, and could be readily deployed in most clinical or research laboratories using standard equipment and processes.
Fig. 2.5.
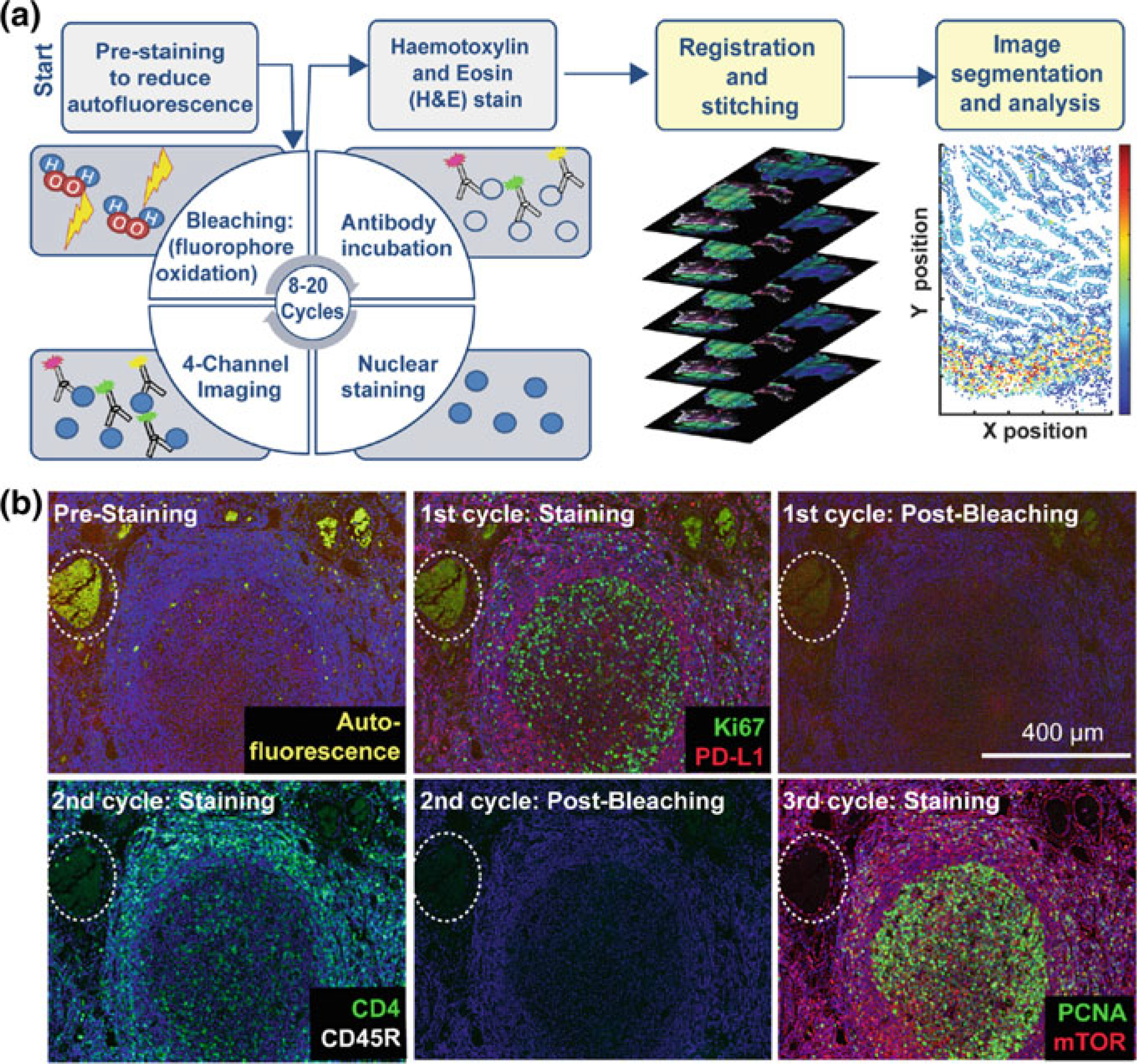
Steps in the t-CyCIF process. a Schematic of the cyclic process whereby t-CyCIF images are assembled via multiple rounds of four-color imaging. b Image of human tonsil prior to pre-staining and then over the course of three rounds of t-CyCIF. The dashed circle highlights a region with auto-fluorescence in both green and red channels (used for Alexa-488 and Alexa-647, respectively) and corresponds to a strong background signal. With subsequent inactivation and staining cycles (three cycles are shown here), this background signal becomes progressively less intense; the phenomenon of decreasing background signal and increasing signal-to-noise ratio as cycle number increases was observed in several staining settings (Modified from [68] DOI:https://doi.org/10.7554/eLife.31657. © 2018, Lin et al. Reprinted under the terms of the Creative Commons Attribution License)
2.4. Lanthanide-Based Mass Spectrometry Imaging
Mass spectrometry imaging (MSI) is a technique used to visualize the spatial distribution of chemical compositions by their molecular masses. MSI techniques can vaporize molecules from within specific regions of tissues into gas-phase ions and then measure their mass. In similar fashion to laser-scanning confocal microscopy, an ionization beam is scanned across the tissue section to build up an image of the molecules present [69].
Such methods directly detect molecules of interest, but the use of specific labeled probes can greatly augment sensitivity and specificity. For example, antibodies used to detect proteins of interest can be labeled with metal ion tags of specific mass, and the tag identity is resolved using coupled mass spectrometry instrumentation, typically based on time of flight. This approach allows for significantly more antibodies to be used simultaneously than with the usual IF methods, with limited crosstalk between species, a major problem with standard fluorescence detection.
CyTOF, as developed by Fluidigm, is one of the platforms that utilizes MSI technology to conduct highly multiplexed flow-based analysis of single cells. The workflow to conduct CyTOF experiments are similar to traditional flow cytometry but has some critical differences that allow for significantly higher levels of multiplexing. Antibodies, labeled with distinct transition element isotopes and added cell suspensions are vaporized as single-cell droplets introduced into an argon plasma; the resulting elemental ions are sampled by a time of flight (TOF) mass spectrometer. This type of approach eliminates many of the challenges faced by the multiplexing platforms previously described, while presenting some new challenges. The main advantage of this approach is the use of elemental isotopes to label antibodies, resulting in the simultaneous detection of over 30 markers on each cell [70]. Because cells (and tissues) typically contain undetectable background levels of the lanthanides used for labeling, this approach is not affected by background signals analogous to the autofluorescence that can interfere in IF analyses, particularly of FFPE tissues [71]. In this approach, there is no amplification, but this is offset by the high level of sensitivity and linear dynamic range afforded by the MS technique. Flow-based CyTOF methods do not provide information on antigen spatial distributions. However, the approach was extended with the addition of a scanning ablation system that allowed the advantages of CyTOF to translate into tissue-level imaging. This type of platform is known as imaging mass cytometry (IMC), of which there are currently two commercially available variants. The Hyperion Imaging System, as developed by Fluidigm, and the Multiplex Ion Beam Imaging System (MIBI), as developed by IONpath both offer the same fundamental technology, differing in choice of scanning beam. The Hyperion system builds upon the foundation of the CyTOF platform: the labels and antibody pairings are the same, while the scanning laser vaporization beam records the coordinates of each laser pulse to capture the spatial localization of the signals being collected on the TOF spectrometer. The data is integrated into a reconstructed approximation of the tissue architecture and cellular localization. It has been reported that this technology can accurately replicate the tissue architecture, and the spatial interaction on the cellular level all the way down to antigen-presenting cells interacting with CD8 T cells while capturing over 40 markers [72]. Image reconstruction and cell segmentation are significantly improved in this platform with the inclusion of nuclear markers to demark each distinct cell. This approach completely obliterates the tissue specimen during the scan, which may be viewed as a disadvantage in clinical setting, and involves long capture times; it may, however, prove an effective discovery platform. Recently, the Hyperion system has been extended to allow the simultaneous detection of transcriptomic (RNA) as well as protein analytes, a major procedural advance that can provide information on otherwise difficult-to-detect soluble antigens [73].
The MIBI approach differs from the Hyperion platform primarily in that it uses a primary ion beam rather than a laser. As a consequence, the tissue section is not completely obliterated in the course of capturing the images and achievable x–y resolution provided by the ion beam can considerably exceed that of the laser. Moreover, repeated scans can provide axial distribution information for high-resolution 3D imaging (see Fig. 2.6 for general workflow). As with the Hyperion system, antibodies deployed in the MIBI platform are labeled with stable lanthanides, each highly enriched for a single isotope. Primary antibodies are combined in solution and simultaneously incubated on the tissue section which is then subjected to a rasterized ion beam. When the primary antibodies are exposed to the ion beam, the lanthanide adducts bound to the antibodies are liberated and detected via mass cytometry. When the MIBI platform was directly compared to conventional mass cytometry, as well as with an FDA-approved QIA IHC assay, the results were highly concordant [74]. A later report indicated the value of multiplexed imaging in the detailed characterization of tumor-immune cell interactions in TNBC with remarkable depth and precision [6]. Both the Hyperion and MIBI system offer similar advantages over traditional multiplexing IHC approaches: reduced background, increased sensitivity, higher level multiplexing, increased linearity of detection to expression, and increase tag stability. However, the sample acquisition time for each of these platforms likely restricts them to discovery use.
Fig. 2.6.
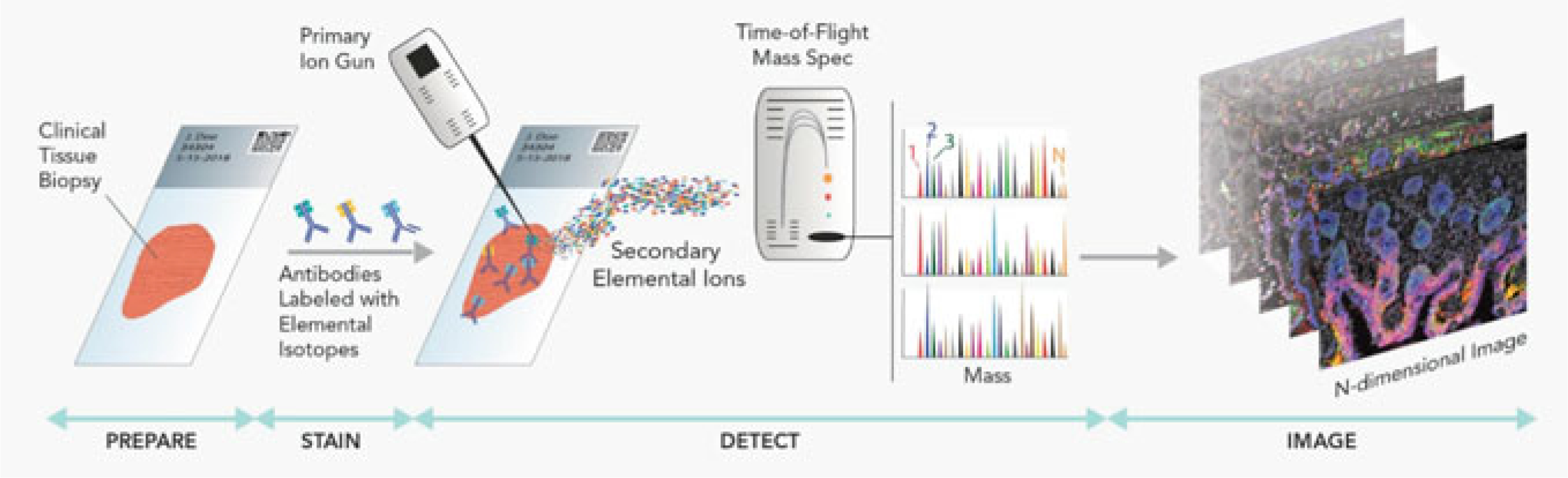
Systematic workflow for MIBI. Primary antibodies, labeled with stable lanthanides enriched for specific metal isotopes, are incubated on the tissue section. The lanthanide molecules are liberated when exposed to a rasterized primary ion beam. The labels are then acquired on a TOF spectrometer and the exact coordinates of the signal are recorded. These data are used to reconstruct the cellular localization on the tissue section as well as the subcellular expression patterns. (Modified from “MIBI™ Technology,” [https://www.ionpath.com/mibi-technology/]. Copyright 2019 by IONpath, Inc. Reprinted with permission)
2.5. Amplification Strategies
Many of the multiplex IHC approaches described previously rely on the amplification of the fluorescent signal to increase sensitivity. There are a few amplification strategies that are utilized by more than one platform. The first type of amplification strategy is, in fact, no amplification. This approach utilizes directly labeled primary antibodies that are detected without any type of amplification. One of the platforms we described previously that took advantage of this strategy was mass spectrometry imaging, both Hyperion and MIBI. The lack of amplification serves to improve the linear relationship between detection and expression levels; since the platform captures specific characteristics of the isotopes, the intensity of the signal comes from the number of particles associated with a cell, not the intensity of the signal detected on the cell. Another approach that uses no amplification is the MultiOmyx platform that employs primary antibodies directly labeled with fluorescent tags. Historically, these reagents have not been sensitive enough to provide a sufficient signal for detection, but with novel conjugation techniques [75] and commercially available reagents, this platform can achieve performance equivalent to that seen with DAB staining.
2.5.1. Tyramide Signal Amplification (TSA)
TSA is one of the more commonly used approaches to increase the signal of the IHC staining pattern. The TSA approach is utilized by both OPAL/Ventana for IF and Ventana DISCOVERY chromogens for bright field. TSA utilizes enzymatic oxidation, through HRP, of labeled tyramide molecules. This enzymatic reaction results in a covalent binding of the tyramide to the tissue in the FFPE section directly around the site of antibody binding. Platforms that utilize this type of amplification will not have predictable linear relationships between detection and expression of antibodies; one primary antibody detected by one HRP-labeled secondary will result in the covalent binding of many more tyramide molecules. Another limitation of this amplification strategy is encountered when trying to fine-grain phenotype a particular cell type. Attempting to detect four or more markers co-localized in one subcellular localization on one cell type (as seen, for example, in activation of CD8 T cells) the repeated deposition of tyramide will result in decreased sensitivity during sequential staining steps.
HRP immunoperoxidase tyramide labeling details, and innovative assays, have been published by several authors [76–78]. Alternatives to HRP exist [79], and in particular, soybean peroxidase has been engineered for live cell labeling with biotin-tyramine by Ting and colleagues, for use in microscopy and mass spectrometry proteomics [80–86].
Krieg and Halbhuber [87] published “self-anchoring” peroxidase substrates; these never really took off. More recently, Sato, Nakamura and colleagues [40, 41] have reported that N-methylated luminol derivatives enhanced with tyrosine-specific chemical modifications, when reacted with HRP and H2O2, result in 10-fold better labeling efficiency than achievable with the equivalent tyramide derivative.
The initial tyramide patents have expired [88, 89], for early peer-reviewed publication; patents are available at Google Patents and FreePatentsOnline. This is relevant for basic research labs (clinical labs are unlikely to take the risk) to synthesize tyramide-fluorophores “in house”. Buchwalow and Bocker [90] cite a published academic synthesis and include their protocol and indicate that with modest investment (less than €50.00 in 1998, excluding antibodies), sufficient reagents can be prepared to support staining of 100,000 slides [91].
Another cost saving may be achieved by recognizing that tyramide signal amplification works best when the primary antibody is diluted 10× to 100× [92–94]. That is, if the primary antibody is $200 for 500 uL, and usually 10 uL ($4) is used, a 10× dilution results in use of $0.40 in reagents (autostainers often use large volumes of diluted antibody and were designed for hematoxylin, eosin, and DAB, not for commercial fluorescent tyramides). Labs that can be frugal on precious primary antibody need to also optimize the storage and use of antibodies by aliquoting and (probably) storing frozen (−80 C is better than −20 C, also more expensive; any freezer is at risk of failing and should have battery backup, wireless/cloud-based alert systems, since building electrical power is also a failure point).
Many of the immunoperoxidase tyramide protocols include heating and/or stripping steps, and in some cases, pre-packaged reagents that are tied to specific commercial instruments for autostaining and imaging. Endogenous peroxidases can be inactivated by several methods. Ichii et al. [95] obtained excellent results using PeroxAbolish reagent (Biocare Medical) to inactive peroxidases for 2-plex fluorescent tyramide signal amplification.
2.5.2. DNA-Enabled Amplification
This strategy involves the use of DNA-barcode amplification as exemplified with the Ultivue UltiMapper platform. This platform has unique DNA barcodes conjugated to each primary antibody, and secondary reagents labeled with fluorescent tags that are complementary sequences to the primary barcode. To increase the number of fluorescent tags that would be able to bind to the primary antibody barcode, the UltiMapper platform utilizes an enzymatic reaction to amplify the length of the DNA barcode in repetitive units. This method ensures that the secondary reagents will be able to bind more frequently and increase the overall signal of each primary antibody binding. As with TSA, this approach increases the overall signal strength but does not preserve a strong linear relationship between detection and expression. The DNA amplification does present a potential challenge for the platform. While providing increased signal strength, it may result in inconsistent results even within the same tissue section. The signal intensity is completely dependent upon the amplification and hybridization of the DNA barcode; if these reactions are not totally reproducible, either inter- or intra-run, variability in detected signal strengths will be encountered. On balance, there appear to be many advantages of amplifying the signal of an IHC-based multiplex platform, but as noted, caution (i.e., stringent QC) is indicated.
2.6. More on Labels
Each of the platforms described up to this point has been dependent on some sort of label to facilitate multiplexed detection of biologically significant features in the TME. Most of the assays described employ fluorescent tags and there is a large and growing selection to choose from. Three of the commercial platforms discussed here use fluorescent tags to label their primary or secondary antibodies, but the actual fluorophore, and the characteristics associated with them are kept proprietary: OPAL, UltiMapper, and UltraPlex dyes are not publicly disclosed. Commonly, when secondary antibodies are used for standard IF staining, Alexa Fluor (AF) reagents are used. These reagents are highly stable and robust in FFPE tissue staining. AF dyes are available across a wide spectral range with the excitation peak indicated by the numeric component of the dye name, as denoted here: AF350, AF405, AF430, AF488, AF532, AF546, AF555, AF568, AF594, AF610, AF633, AF635, AF647, AF660, AF680, AF700, AF750, and AF790. This breadth of choice provides flexibility for investigators in choosing their laser and filter cube settings using conventional fluorescence microscopy, which can be used with up to 6 simultaneous fluors (or as described above, even up to 10). Using multispectral instrumentation allows for semi-routine unmixing of up to about 9 simultaneous labels, although spectral tools and software are not magic bullets. Typically, excitation and emission spectra for similar spectral region dyes provided by different suppliers are comparable—choices may come down to performance based on brightness and stability (resistance to photobleaching). Each variation on these dyes can generate different intensities of emitted light, at the same expression levels. The AF488 reagent has similar excitation and emission properties to fluorescein as does the Brilliant Violet (BV) 510 dye from BioLegend. Fluorescein has the lowest intensity of these three dyes in this spectral range, while BV510 is the brightest. However, the performance of BV510 and other Brilliant dyes on fluorescence tissue sections may benefit from optimization of mounting media, objective lens, illumination intensity, exposure time, detector performance, and user. We suggest that Brilliant dyes (and the limited 5-plex Super Bright dyes from ThermoFisher Inc.) will be provide a solid reagent choice for extensive multiplexing.
Another species of fluorescent tags that behaves very different from the AF reagents is quantum dots (QD), a class of very small semiconductor particles. QDs will emit light of specific frequencies with appropriate excitation—and differently colored QDs can be excited with a common excitation source if necessary. Emission bands are available across a wide spectral range and have sharper and narrower peaks than typical organic dyes, a property favorable to multiplexed detection. There are a number of methods for conjugating QD to antibodies [96] and each has been applied to the platform of multiplex IHC. They can also be used in repeated stain, image, and release cycles leading to high levels of multiplexing [97]. However, quantum dots have to date failed to make much of an impact on tissue-based fluorescence imaging methods, although some labs continue to develop the methods with promising results [98]. On the whole, these reagents have been more widely implemented in in-vivo imaging studies than in the labeling of antibodies for multiplex IHC [99, 100]. Other nanoparticle-based probes have also been developed, some extremely bright. For example, phosphor-integrated dots [101] are reported to be as much as 100-fold greater in brightness than quantum dots. However, robust multicolored nanoparticle competition for quantum dots has not yet emerged.
The last type of labels that were described in multiplex IHC platforms were the lanthanide-based distinct transition element isotopes used in the ICM platforms. These labels are distinct from the other tags described here. The distinct transition element isotopes tags allow for a much higher number of channels to be used, and therefore targets to be detected; the mass peaks can be detected with tremendous precision—the challenge being not the separation of different isotope species, but the preparation of labels with sufficient purity that the contaminants don’t contribute to erroneous signal attribution. These tags also significantly reduce the challenges presented by autofluorescence (always present but enhanced by formalin fixation of tissue samples).
2.7. Instrumentation for Fluorescence-Based Multiplexing
The foundation of fluorescence microscopy is based on the concept of absorption and subsequent reradiation of light by organic and inorganic constituents and labels. Each fluorescent dye has specific excitation and emission values that allow scientists to evaluate the specific emission from one fluorophore as a readout of expression. Fluorophores are excited by light of various wavelengths and emit light of a different wavelength. When a microscope is properly configured, only the emission light will reach the photodetector (which can be the operator’s eyes, or an electronic sensor). In a typical epifluorescence configuration, light in one or more spectral bands passes through a wavelength-selective excitation filter, and are then reflected off by a chromatic mirror to pass through the microscope objective and illuminate the specimen. Emitted light is then collected by the objective, and passing through the chromatic mirror and an emission barrier filter, is transmitted to the observer and/or photosensor. In most microscopes, the excitation filter, chromatic mirror, and barrier filter are incorporated into one optical block, referred to as a cube (Fig. 2.7).
Fig. 2.7.
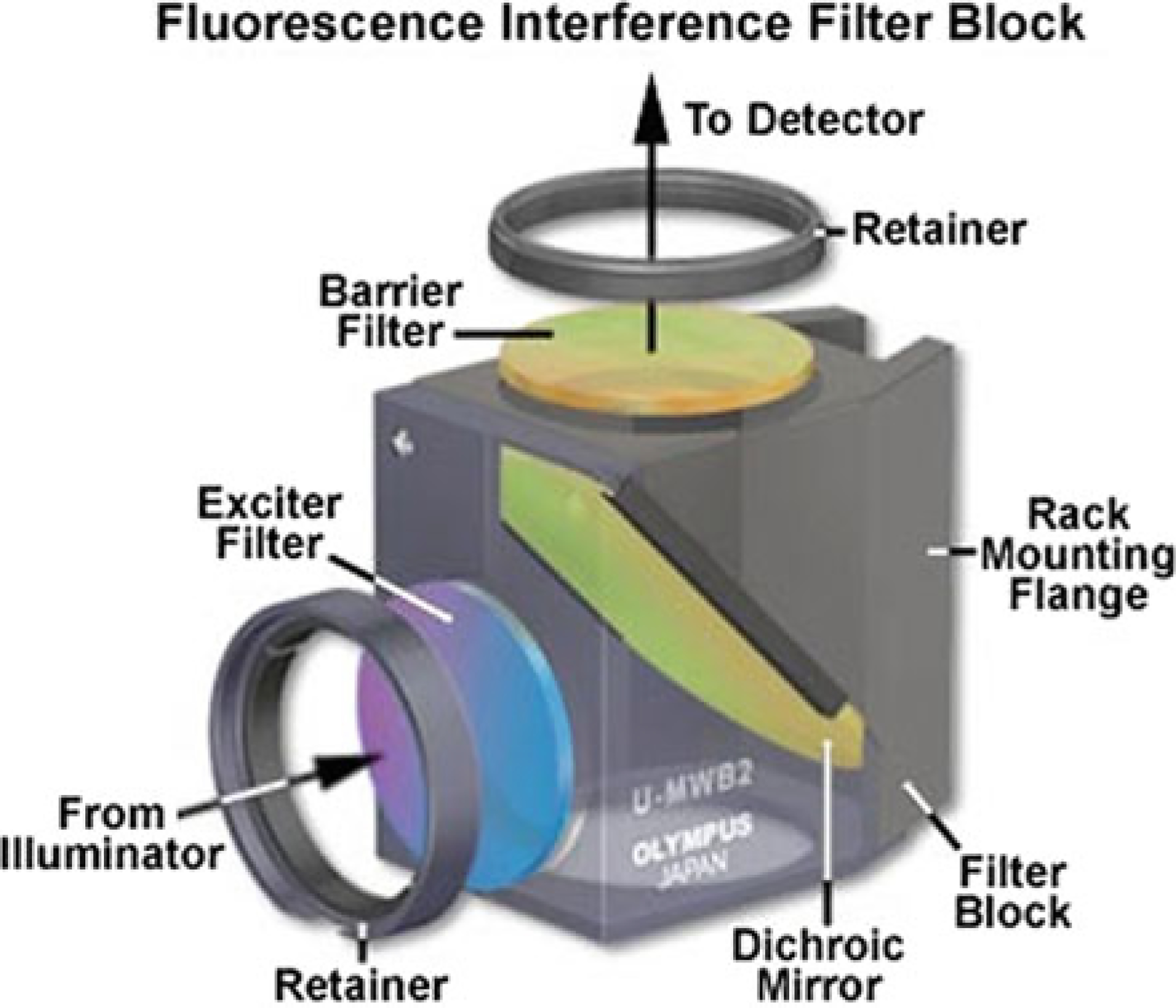
Fluorescent Filter Cube. Components typically contained in a fluorescent cube; Exciter filter, Dichroic Mirror, and Barrier Filter. These work together to ensure that the only frequency that makes it to the photo recorder is that of the fluorescent tag being excited, plus whatever autofluorescence is present in that channel (Modified from ‘Fluorescence Filters’, [https://www.olympus-lifescience.com/en/microscope-resource/primer/techniques/fluorescence/filters/]. Image courtesy of Olympus Corporation. Reprinted with permission)
There are two classes of fluorescent filters that can be used in this context: single-band and multiband. The latter provides the ability to image multiple different fluorescent signals simultaneously, rather than requiring cycling through a number of single-band configurations; this is accomplished at the cost of potentially increased noise, crosstalk, and decreased signal. Multiplexing is usually accomplished by employing multiple filter wheels housed in a turret, or the use of filter wheels that can permit more flexibility and excitation-emission pairings but add to cost and complexity.
There are many options for fluorescent tags that can be added to either the primary or secondary antibody, as previously described. Each of these different fluorophores has optimal fluorescent cubes and light settings. Companies such as Akoya have built custom fluorescent cubes to work optimally with their proprietary OPAL dyes. Other labeling platforms, because they don’t have optimized filter pairings, may be limited in the number of markers that can be imaged simultaneously, unless spectral unmixing is employed (see below). In order for fluorescent signals to be detected simultaneously without unmixing, the spectral emissions from each channel should overlap minimally so that the signal obtained from each label is distinct. With commercially available fluorophores and judicious selection of fluorescent filter pairs, it would be realistic to achieve 4–5-color multiplexing with relatively simple capture and quantitation tools.
2.7.1. Multispectral Imaging (MSI)
Multispectral imaging (MSI) refers to the acquisition of spectrally resolved information at each pixel of an image. There are a variety of instrumental approaches available, including multispectral confocal microscopes from a number of suppliers. One of the more widely used is the Vectra and related products based on liquid-crystal tunable filters. These acquire spectral datasets by capturing a number of wide-field images at different (tuned) wavelength bands with single or multiple excitation sources (leading to the acquisition of excitation-emission matrices). MSI can be applied to either bright field or IF stains to help resolve multiplexed staining into accurate single-component images [102]. In bright-field (chromogenic) staining, multiplexing has more limitations than is the case with IF. For one thing, chromogenic stains are subtractive—that is, the more stains are co-localized, the fewer photons are transmitted, with the limit going to zero (a.k.a., black) as individual or combined stain intensities increase. Furthermore, some chromogens have broad and variable absorption spectra—in particular, DAB behaves not as an ideal absorber but also as a scatterer, and thus does not display Beer–Lambert law behavior—though optical density can be computed and used adequately (in practice, the pathologist’s eye does not estimate optical density, and many pathology image acquisition and analysis software do not bother computing optical density.) Depending on how variable the staining intensity is, even in a single specimen more than one unmixing spectral “endmember” for DAB may have to be deployed for proper unmixing (Levenson, personal communication). Consequently, while unmixing in bright field is doable, it is most successful when stains are deployed to have limited colocalization [103, 104]. DAB/HRP and other chromogens also have issues with respect to dynamic range, compared to fluorescence [105].
More commonly, MSI approaches are applied to IF. MSI alleviates two major challenges associated with IF assay development: reduction of the impact of autofluorescence and proper separation of fluorophores that have overlapping spectral signatures. Autofluorescence is endogenously found in most tissues and is enhanced by formalin fixation; it is elicited by all excitation wavelengths but is stronger with UV or blue light excitation. Sample preparation techniques intended to reduce autofluorescence prove only partially successful, only resulting in about 50% reduction in background signal [106], while MSI has been shown to achieve over 95% reduction in favorable circumstances [102, 107]. Of course, autofluorescence is not physically eliminated, and still contributes deleterious shot-noise that can compromise results, especially for dim signals.
The most common form of spectral unmixing used by software programs is referred to as linear unmixing; this is a linear regression of a number of given spectral shapes into a spectrum of a sample. Central to the accurate completion of this approach is the generation of spectral libraries, whose members are used to fit the measured, mixed spectra. Spectral libraries can be created by staining a control tissue with a marker that has a strong signal, for example, CD3 or CD20 on human tonsil, repeatedly, using each of the fluorescent labels that will be employed. An unstained slide (that has, however, been treated exactly as if it were being stained with a fluorescent reagent—only minus the reagent) can be used to generate an accurate autofluorescence signal. This can then be used to calculate a “pure” spectrum for the fluorescent dyes by subtracting the AF spectrum from the measured AF plus dye spectrum. While simple in theory, there are a number of potential pitfalls; the subtraction process is affected by the presence of noise, and parameters have to be adjusted depending on whether the desired fluorescent signal is brighter or dimmer than the autofluorescence channel(s). Once the computed “pure” spectra are accurately determined, linear unmixing with them and one (or more) authentic AF spectra will generally provide good results [100, 108]. There are other more complicated methods of defining spectra and unmixing that address issues such as whether non-negativity and sum-to-100% constraints are invoked [109], but these will not be discussed in detail here.
MSI is a powerful technique that has significantly improved the ability of IF stains to be effectively quantified by image-analysis platforms. This approach could be applied to many of the multiplex IHC platforms discussed previously whose multiplexing capabilities are currently limited by the use of fluorophores that are spectrally distinct (as opposed to being significantly overlapping); examples of systems that could be assisted by MSI include, for example, Ultivue, Cell IDx, CODEX, and MultiOmyx.
One platform that has capitalized on incorporating MSA is OPAL; achieving up to 9-color multiplex IHC through the use of spectrally overlapping fluorophores that are unmixed post-staining (see www.akoyabio.com for examples). Importantly, the complex and laborious labeling process has been adapted for use on the Leica BOND RX automated stainer platform. The developers of the OPAL platform have also produced a user-friendly fluorescence and chromogenic multispectral scanner that is integrated with MSI software to facilitate this process. The most recent iteration of these scanners is the Vectra Polaris, paired with their own inForm software. Once a composite image is generated by InForm, consisting of a multilayer file with individual planes reflecting the contribution of each label, resulting data can be imported into a variety of image-analysis platforms for quantitative evaluation; these include, but are not limited to, inForm, HALO, Definiens, Visiopharm, and QuPath (open source).
2.8. Assay and Antibody Validation
The accuracy and reproducibility of each IHC assay are fundamental to the value of the platform as a whole. Without a properly validated assay, whether single plex or high-level multiplex, the resulting data are at best suggestive, and at worst, simply wrong. Moreover, as the complexity of the system increases, opportunities to engage in wishful thinking go up proportionately. Rigor is essential—not just for scientific discovery, but especially for pharma-related work, as the results may be used to undergird drug discovery and testing and may eventually form the basis for an FDA-regulated companion/complementary diagnostic.
One example of recommended steps is given here. Antibody validation can begin with a DAB-based detection system—even for assays destined for fluorescence-based implementation. Known positive and negative control cell types in normal tissues, as well as positive and negative tumor cell lines as determined by mRNA expression, can be employed as part of the antibody screening process. Once one or more monoclonal or polyclonal reagents are identified, further specificity testing should be completed, including protein competition assays, sh/siRNA or CRISPR knockdown, and serial-section comparison to ISH staining patterns. After a primary antibody is validated and optimized using DAB, the resulting staining pattern should be considered the gold standard against which subsequent multiplex assays are compared. If an assay is going to be converted from DAB to IF multiplex, first the monoplex IF stain needs to be performed and compared directly against serial sections prepared using the DAB method. This evaluation can be at either the qualitative or quantitative level, though the latter is preferable; higher level of confidence in the assay can be obtained if quantitative analyses are completed at this point. Once the DAB protocol is satisfactorily converted to IF, and signal intensity and staining patterns appear to be consistent, it is possible to build up the multiplex assay.
Each of the platforms described previously would entail different requirements for multiplex assay development; the TSA-based approach used in OPAL and the Discovery chromogens would be the most time-intensive as the repeated stripping impacts antigen presentation or preservation as well as the resulting intensity of the chromogens or fluorescent tags. Most of the multiplex assay platforms that simultaneously detect all the primary antibodies are less affected by multiplex optimization per se; the biggest challenge for these platforms is staining intensity. The TSA-based platforms require each antigen-detecting antibody (and the integrity of the target antigen) to be confirmed at each stage of the multiplex assay to ensure that the signal is preserved—or at least is reproducibly affected as a function of assay stage. Particular attention needs to be paid to determining the effect of reagent sequence-order on the resulting multiplexed image data. Consideration also has to be paid to selecting the appropriate label-target pairs (low-abundance target, high-brightness label and vice versa), as well as trying to ensure that dyes that might be hard to distinguish spectrally are directed as much as possible to targets that are spatially distinct.
When the multiplex assay is developed and the staining pattern and intensity reflect that of the monoplex IF, quantitative analysis should be performed for accuracy verification. Serial sections of the monoplex IF and multiplex IHC assay need to be stained, acquired, and analyzed. The type of analysis could vary, as different labs will used different approaches; metrics to be monitored may include total number of positive cells, total stain intensity across the slide, or simply the brightest or dimmest signal from known positive regions. Ideally, once the analysis methodology is selected, the monoplex IF and multiplex IHC should be concordant to above 80% of one another, however, measured. A rule of thumb: if more than 20% of the signal is lost when converting the monoplex IF to multiplex IHC, further method development is indicated. If the assay passes the accuracy test, and concordance of each marker is satisfactory, the reproducibility of the assay then needs to be evaluated. A precision test accounts for inter- and intra-run variability; for example, three sections of one tissue are run each day for three days. All nine sections are then scanned and evaluated for consistency of staining intensity and frequency of the populations, again looking for at least 80% concordance between slides run in the same run and across multiple days. Once an assay has passed accuracy and precision testing it can be considered validated, and there will be a high level of confidence that the result generated will be are accurate and reproducible—assuming nothing changes [110]. As can be imagined, assay optimization can be a long and tricky process.
2.9. RNA
Francis Crick’s molecular biology central dogma, as incorrectly restated by James Watson as, “DNA makes RNA and RNA makes protein” [111], is massively simplistic and glosses over a lot of details: time, space, fate, mRNA splicing, alleles, closed, poised, and/or open chromatin, microRNAs, RNAi, and more [112–116]. Messages and proteins may be produced by transcriptional and/or translational bursting, respectively, where transcription and/or translation alternate between an active and inactive state [117, 118]. Long noncoding RNAs (lncRNAs) can be sponges for microRNAs or have open reading frames coding for proteins. Some human mRNAs are polycistronic with secondary structures and RNA binding proteins dictating which open reading frame is translated into proteins (see also microproteins). RNA splicing is usually performed in the nucleus, very close to the transcription site, but some introns have zip code(s) to traffic to distant sites, such as neuronal synapses, before being spliced to mature mRNA (and splicing machinery is on site too). The number of protein molecules translated from each mRNA (or lncRNA) varies considerably (see [119, 120])—and may not account for transcripts that are never translated.
Detection of any specific RNA is typically directed at single-stranded sequences, though denaturation of double-stranded RNA can be performed. Usually, the temperature and formamide and other buffers are controlled so that ~20 base oligonucleotides (optionally with extended handles) bind to their target sequence with (near) perfect complementarity. This is usually best done by selecting binder and target sequences with 45–55% GC ratio and optimizing conditions for this range. Specialty reagents, such as locked nucleic acids (LNAs) or peptide nucleic acids (PNAs), can be used with shorter sequences. Such probes have non-standard components (standard are A, T, C, G, sometimes U, ribose or deoxyribose, phosphate) making them more expensive—and risk binding to non-unique targets. Some researchers may assume that all messenger RNA is identical to their favorite canonical “full length” cDNA, and their favorite protein is the simple translation of said message, posted from NCBI GenBank (to say nothing about post-translational modifications, PTMs). This is wishful thinking: biology is rarely as elementary as Watson’s (mis)quote above. A nice test case is FoxP3, the canonical Treg transcription factor, which the PubMed search: foxp3 alternative splicing, suggests is not so simple (it may regulate some alternative splicing, in addition to its own alternative splicing and PTMs).
Some companies sell probes with proprietary sequences (see [121] for ACDbio’s statement). While literal replication of experiments would be possible by purchasing identical reagents (assuming the vendor does not change any components), independent studies directed at the same target may be impossible without knowledge of the specific sequences employed. It is advisable, then, that all users of RNA-targeting products: (i) only purchase products that have known sequences and copyright pre-approval to include in publications; and (ii) include the sequences, products, lot numbers, and specific hybridization conditions (including buffer composition), that applies to each publication. The analogous situation for antibodies should be to only purchase and use recombinant antibodies and include the sequences in each publication.
Pardue and Gall were the first to demonstrate histologic detection of mRNA using in-situ hybridization with radioactive DNA hybridization probes [122–124]. Femino et al. [125] achieved single RNA molecule fluorescence in-situ hybridization (smFISH). The Femino et al. approach was not commercialized (to our knowledge). Raj, Tyagi, and colleagues used a “more is better” approach to tile, typically, 48 oligonucleotides (20 bases each, usually unique sequence, one fluorophore on one end), on an mRNA (smFISH also works on long noncoding RNA), and after denaturation, formerly double-stranded DNA or RNA) [126–130] which was commercialized by Biosearch Technologies. Examples shown in Figs. 2.8 (both raw and GPU deconvolution) and 2.9 (DAPI counterstain).
Fig. 2.8.
Widefield image of RNA FISH (fluorescence in situ hybridization), labeling TOP1 (DNA topoisomerase). Single slice of a 1024 × 1024 × 39 image, a before deconvolution and, b after deconvolution, 6 s (Image courtesy of Dane Maxfield and Microvolution, [https://www.microvolution.com/gallery]. Reprinted with permission)
Fig. 2.9.
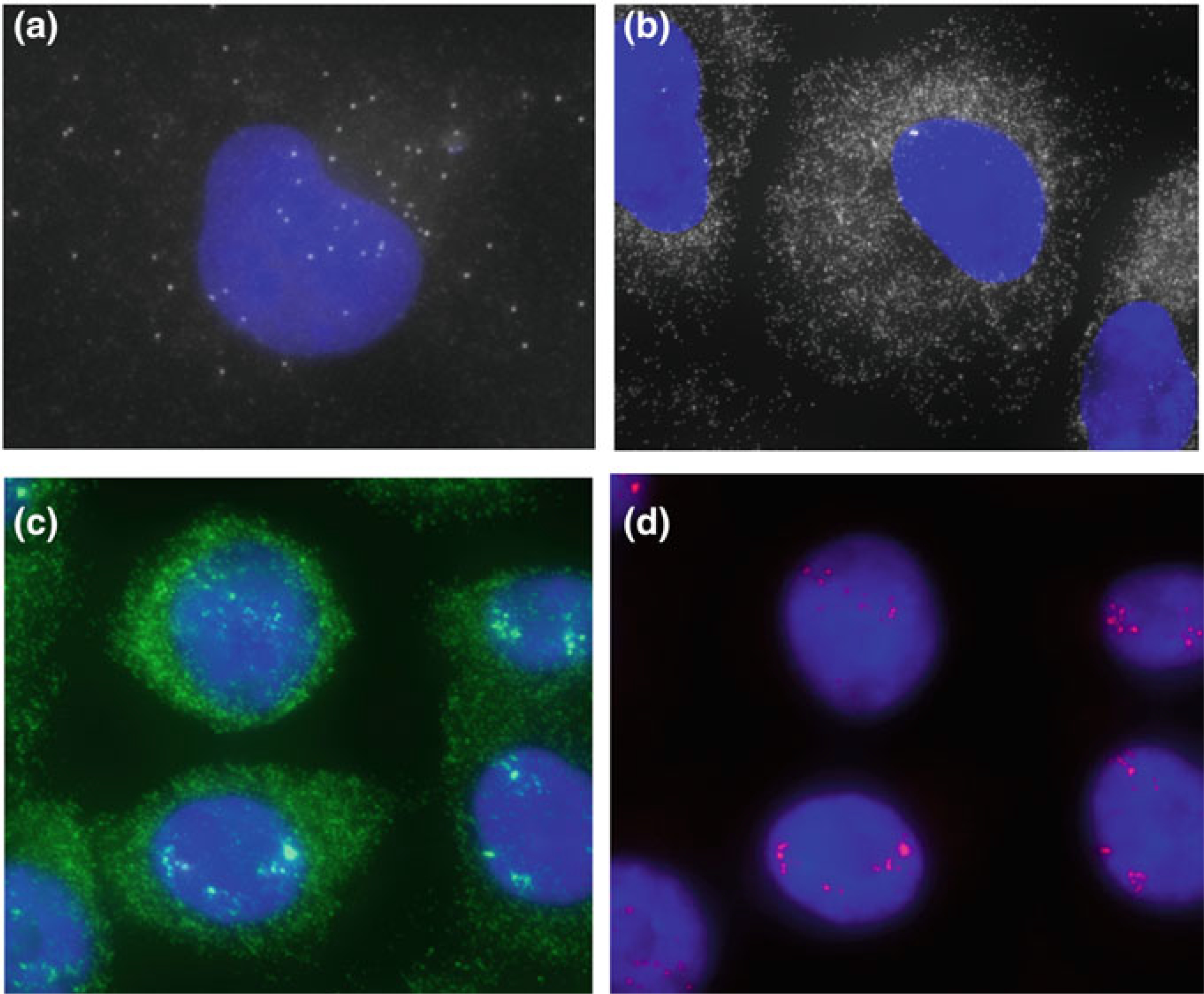
Single-molecule RNA FISH data showing transcriptional burst sites. Stellaris® FISH Probes recognizing human a POLR2A (labeled with Quasar®570, # VSMF-2294-5), b GAPDH (labeled with Quasar®570, # VSMF-2026-1), c ERBB2 (Her-2/neu) exons (mRNA, green, labeled with Quasar®570, # VSMF-2102-5), and d intron specific (red, Quasar®670, # ISMF-2103-5) in human A549 cells (Images courtesy of LGC Biosearch Technologies)
DNA topoisomerase 1 (TOP1, see Fig. 2.8) is a sparse mRNA (and pre-mRNA). POLR2A RNA appears to be a positive control, both for its sparseness, and its place in the Central Dogma of molecular biology. The widely used GAPDH may be less useful, due to its high abundance per cell.
Raj and colleagues have contributed additional refinements to smFISH, including Turbo FISH, where 10× higher probe concentrations enables 30 s hybridization and 5 min total processing time [131], intron chromosomal expression FISH (iceFISH), to measure simultaneously multiple genes expression along chromosomes (transcriptional bursts painted by combinatorial colors) [132], and single nucleotide variants (polymorphisms) (SNP FISH), that is, maternal and paternal alleles, as well as mutant alleles, by painting the constant “backbone” of the RNA with a probe set of one fluorophore, and each SNV in a different color(s) [133].
2.10. RNAscope® v2
RNAscope is licensed to two companies, ThermoFisher (by acquisition of Affymetrix/eBioscience, which had acquired Panomics) and Biotechne/ACDbio. The original format was published by Wang et al. [121], ACDbio, who generously estimate 8000 fluorophores per typical 20 ZZ’s trees when fully decorated), is still available in 2019, and less expensive than “v2” (and less auto-plexable). ACDbio’s new (mid-2019) RNAscope® Multiplex Fluorescent v2, which combines RNAscope branched-DNA (bDNA) is comparable to Akoya Biosciences OPAL TSA (early bDNA methods and publications reviewed by Player et al. [134]). The latter approach is compatible with automated immunostainers in an automated workflow that can be expected to be Clinical Laboratory Improvement Amendments (CLIA)-approvable.
The RNAscope Multiplex v2 (from Advanced Cell Diagnostics, Inc.) is schematized in Fig. 2.10. ZZ is the “double Z probe” pair for specific target hybridization (e.g., a pair of nearby 20-base sequences on the target RNA). These are followed by preamplifiers, amplifiers, and dyes. The dyes are illustrated as being coupled to the branch structures, but in fact the chemistry of activated tyramides is to diffuse a short distance and react covalently with tyrosine (i.e., exposed on nearby protein or substratum). Since OPAL reagents are fluorescent tyramides, amplification is by HRP/H2O2, and can be automated on an immunohistochemical stainer.
Fig. 2.10.
Schematic of RNAscope Multiplex v2. Up to four RNA targets can be detected simultaneously using four targets ZZ RNA-hybridizing probes. Each probe has a specific TSA-linked fluorophore channel for detection (Image courtesy of Bio-Techne Corporation. Reprinted with permission)
RNAscope v2, typically 4-plex with Opal 520, Opal 570, Opal 620, Opal 690 (typical filter sets for conventional fluorescence microscopes, namely, fluorescein/GFP, Cy3, Texas Red, Cy5/Alexa Fluor 647, respectively). ACDbio encourages the use of Vectra “multispectral” slide scanner product line form Akoya Biosciences. In Spring 2019 Akoya Biosciences introduced 8-plex OPAL kits (see Tyramide signal amplification section), so in principle this could go an additional 4-plex for RNA and/or immunofluorescence protein detections.
In theory, a single ZZ probe pair risks missing RNA molecules if these are occluded by RNA secondary structures and/or RNA binding proteins. This risk could be mitigated—at greater probe set price—by using multiple pairs.
2.10.1. Quantitative Hybridization Chain Reaction and Similar
Choi et al. [135] unveiled their third-generation hybridization chain reaction (HCRv3) with modes including “analog” relative RNA expression profiling, and “digital” single-molecule counting options. The methods were developed for whole mount optically cleared embryo developmental studies and should image equally well in thin tissue sections and thicker optically cleared cancer tissues. Diehl and colleagues [136, 137] developed a multiplex method similar to both DNA-PAINT and hybridization chain reaction.
2.10.2. Spatial Transcriptomics, MERFISH, SeqFISH+
RNA sequencing (RNAseq) and single-molecule RNA fluorescence in-situ hybridization (smFISH), can be used to inform (and sometimes correct) each other. This has been termed spatial transcriptomics [138–143]. Moor et al. [144, 145] used laser microdissection and smFISH to examine gut epithelial cells. This work in many ways is complemented by Lau, Coffey, and colleagues using single-cell RNA sequencing (scRNAseq) and multiplex immunofluorescence imaging (MxIF, aka MultiOmyx), and mass cytometry (CyTOF flow cytometry) [146, 147]. MxIF had previously been examined in the context of colon cancer (61-plex, 747-patient tissue microarrays) [65, 148].
The dueling acronyms, MERFISH [149] and seqFISH+ [150] each seek to detect and visualize hundreds to thousands of RNAs in large-scale tissue context. Slide-seq [151], and “spatial transcriptomics” use barcoded microbeads coated with capture reagents, applied to a tissue section, and then their barcodes are sequenced in situ to provide spatial encoding. Following this, the beads are released from the tissue and subjected to next-generation sequencing (NGS) [140, 141, 152], with the detected barcodes allowing the eluted RNA species to be correctly assigned to their originating location.
2.10.3. Digital Spatial Profiling (DSP) for Combined Protein and RNA Multiplexing (NanoString Technologies)
Decalf et al. [153] and the GeoMx™ DSP brochure (from NanoString Technologies, [154]) describe the NanoString digital spatial profiling (DSP) method for in-situ high multiplexing of both protein and RNA in either FFPE or fresh-frozen tissue sections. With this method, tissues are stained with “imaging” reagents (up to four fluorescently tagged markers for region of interest (ROI) delineation) and “profiling” reagents (antibodies or nucleic acid probes conjugated to oligonucleotide barcodes via a UV-cleavable linker). The approach has some similarities to laser capture microdissection, but the output is the applied UV laser shot in each ROI frees up the spatially defined barcoded targets and leaves the original sample intact. The NanoString instrument maps collected barcodes back to tissue location, thereby providing a spatial readout of target RNA/protein abundance, with resolution capabilities down to even the single-cell level.
2.11. Conclusions
The era of highly multiplexed, spatially resolved TME profiling is upon us and by the time you read this chapter, it will already be out of date. Nevertheless, understanding the principle methods described here should provide a basis for evaluating the emerging technologies. What should be evident, however, is that numerous challenges remain. In first place is the need to maintain and increase vigilance in terms of quality control. It is vital to be alert to the potential for pre-analytical variables, reagent misadventures including complex interactions between labels, steric interference, target lability in the face of prolonged staining and imaging protocols, photobleaching, usability issues posed by complex and potentially mysterious software tools, all to interfere with achieving reliable and reproducible results by investigators and eventually by clinical personnel. The techniques need to be performed properly across multiple institutions and national locations. Moreover, statisticians will have to be ready to make sense of the mass of spatial and quantitative information that will cascade out once multiplexed data acquisition is routinized. Beyond these issues, costs of instrumentation and reagents will have to be addressed; some of the more advanced optical and mass-spec-based tools will price out in the high 6-to-7-figure range. Fortunately, some of the tools take advantage of simple reagents and conventional fluorescence microscopes, which, for slide-scanning-enabled instruments, might put equipment cost (just for the imager, not any automated stainers required) at around $200,000, a more tractable number. In all, further development, as emphasized by Parra et al. will require teams consisting of pathologists, oncologists, immunologists, engineers, and skilled technicians. Reliable automation accompanied by a focus on quality will be key to success.
Beyond these more technical concerns, regulatory and practical clinical utility issues must also be navigated. In all cases, methods and assays must meet a much more stringent list of requirements for use in clinical diagnostics. This may require proceeding through the difficult and expensive FDA approval process. Even for (for now non-FDA-regulated) laboratory-developed tests (LDTs) there are important considerations. Tests must have known sensitivity and specificity as compared to a reference standard, must be reproducible, must use adequately sourced reagents, and undergo ongoing quality checks. The bar is high, in other words, for adoption in clinical diagnostics. As an aside, the fact that these considerations are not required for research grants and publications is clearly one of the sources of lack of reproducibility of a great deal of peer-reviewed work published in even the highest quality journals.
In order to warrant clinical application, there must be a clear need for the method. In the case of cancer prognostics, while this seems a worthy goal, there is really very little impact on clinical decision-making if a new test adds additional relative recurrence/mortality risk stratification because these are rarely actionable on an individual basis. It may help to know that one cancer is slightly better or worse than another, but in most cases, this does not change the treatment. There are a few examples of present utility in situations in which a clinical decision is already on a borderline. For example, it was known that early-stage ER+ breast cancer patients had an absolute survival benefit from adjuvant chemotherapy that was almost the same as the risk of lethality of the treatment. This was the opportunity that gave rise to the OncotypeDX test (an RT-PCR method for determining proliferative rate) that proved to stratify this subset into a lower risk group, for whom adjuvant chemotherapy was clearly worse than the benefit, and a higher risk group, for whom adjuvant chemotherapy had some demonstrable benefit. It has now been shown that any method for assessing proliferative rate can perform equally well in this stratification. Given the high bar for development of such a test, is there a future for multianalyte spatially resolved/microanatomically-defined testing? The emerging field of immunotherapy is a clear area where this might be immediately important. Here, the immune cells must be defined with more than one marker, in many cases, and their location and proximity to one another, and to areas of tumor cells have already proven to be revealing with respect to clinical choices about immunotherapy. Spatially resolved multiplexing technologies will contribute to the future of these exciting developments. Internet resources are available [155–167].
Contributor Information
George McNamara, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Justin Lucas, Bristol-Myers Squibb, Princeton, NJ, USA.
John F. Beeler, Bristol-Myers Squibb, Princeton, NJ, USA
Ajay Basavanhally, Bristol-Myers Squibb, Princeton, NJ, USA.
George Lee, Bristol-Myers Squibb, Princeton, NJ, USA.
Cyrus V. Hedvat, Bristol-Myers Squibb, Princeton, NJ, USA
Vipul A. Baxi, Bristol-Myers Squibb, Princeton, NJ, USA
Darren Locke, Bristol-Myers Squibb, Princeton, NJ, USA.
Alexander Borowsky, UC Davis Health, Sacramento, CA, USA.
Richard Levenson, UC Davis Health, Sacramento, CA, USA.
References
- 1.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H et al. (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14(5):518–527 Epub 2008/04/29 [DOI] [PubMed] [Google Scholar]
- 2.Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C et al. (2014) Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol 232 (2):199–209 Epub 2013/10/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck AH, Sangoi AR, Leung S, Marinelli RJ, Nielsen TO, van de Vijver MJ et al. (2011) Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci Transl Med 3(108):108ra13. Epub 2011/11/11 [DOI] [PubMed] [Google Scholar]
- 4.Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M et al. (2014) Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest 94(1):107–116 Epub 2013/11/13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rehman JA, Han G, Carvajal-Hausdorf DE, Wasserman BE, Pelekanou V, Mani NL et al. (2017) Quantitative and pathologist-read comparison of the heterogeneity of programmed death-ligand 1 (PD-L1) expression in non-small cell lung cancer. Mod Pathol 30(3):340–349 Epub 2016/11/12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S et al. (2018) A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell 174(6):1373–1387 e19. Epub 2018/09/08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, Kulangara K et al. (2017) PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 ihc assay comparison project. J Thorac Oncol. 12(2):208–222 Epub 2016/12/04 [DOI] [PubMed] [Google Scholar]
- 8.Perez-Medina C, Tang J, Abdel-Atti D, Hogstad B, Merad M, Fisher EA et al. (2015) PET imaging of tumor-associated macrophages with 89Zr-labeled high-density lipoprotein nanoparticles. J Nucl Med Off Publ, Soc Nucl Med 56(8):1272–1277 Epub 2015/06/27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tavare R, McCracken MN, Zettlitz KA, Knowles SM, Salazar FB, Olafsen T et al. (2014) Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc Natl Acad Sci U S A 111(3):1108–1113 Epub 2014/01/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seo JW, Tavare R, Mahakian LM, Silvestrini MT, Tam S, Ingham ES et al. (2018) CD8(+) T-cell density imaging with (64)Cu-labeled cys-diabody informs immunotherapy protocols. Clin Cancer Res 24(20):4976–4987 Epub 2018/07/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McConnell HL, Schwartz DL, Richardson BE, Woltjer RL, Muldoon LL, Neuwelt EA (2016) Ferumoxytol nanoparticle uptake in brain during acute neuroinflammation is cell-specific. Nanomed-Nanotechnol 12(6):1535–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shu CJ, Guo S, Kim YJ, Shelly SM, Nijagal A, Ray P et al. (2005) Visualization of a primary anti-tumor immune response by positron emission tomography. Proc Natl Acad Sci USA 102(48):17412–17417 Epub 2005/11/19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vakoc BJ, Fukumura D, Jain RK, Bouma BE (2012) Cancer imaging by optical coherence tomography: preclinical progress and clinical potential. Nat Rev Cancer 12(5):363–368 Epub 2012/04/06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin R, Chen J, Wang H, Yan M, Zheng W, Song L (2015) Longitudinal label-free optical-resolution photoacoustic microscopy of tumor angiogenesis in vivo. Quant Imaging Med Surg 5(1):23–29 Epub 2015/02/20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyd NF, Guo H, Martin LJ, Sun L, Stone J, Fishell E et al. (2007) Mammographic density and the risk and detection of breast cancer. N Engl J Med 356(3):227–236 Epub 2007/01/19 [DOI] [PubMed] [Google Scholar]
- 16.Jones EF, Sinha SP, Newitt DC, Klifa C, Kornak J, Park CC et al. (2013) MRI enhancement in stromal tissue surrounding breast tumors: association with recurrence free survival following neoadjuvant chemotherapy. PLoS One 8(5):e61969. Epub 2013/05/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A et al. (2017) Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 8:15095. Epub 2017/04/28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giraldo NA, Nguyen P, Engle EL, Kaunitz GJ, Cottrell TR, Berry S et al. (2018) Multidimensional, quantitative assessment of PD-1/PD-L1 expression in patients with Merkel cell carcinoma and association with response to pembrolizumab. J Immunother Cancer 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng Z (2014) Utilizing quantitative immunohistochemistry for relationship analysis of tumor microenvironment of head and neck cancer patients. J Immunother Cancer 2(3) [Google Scholar]
- 20.Parra ER, Francisco-Cruz A, Wistuba II (2019) State-of-the-art of profiling immune contexture in the era of multiplexed staining and digital analysis to study paraffin tumor tissues. Cancers (Basel). 11(2). Epub 2019/02/23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parra ER (2018) Novel platforms of multiplexed immunofluorescence for study of paraffin tumor tissues. J Cancer Treat Diagn 2(1):43–53 [Google Scholar]
- 22.Mao Y, Keller ET, Garfield DH, Shen K, Wang J (2013) Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev 32(1–2):303–315 Epub 2012/11/02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whiteside TL (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene 27(45):5904–5912 Epub 2008/10/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker MJ, Trevisan J, Bassan P, Bhargava R, Butler HJ, Dorling KM et al. (2014) Using Fourier transform IR spectroscopy to analyze biological materials. Nat Protoc 9(8):1771–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhargava R (2007) Towards a practical Fourier transform infrared chemical imaging protocol for cancer histopathology. Anal Bioanal Chem 389(4):1155–1169 [DOI] [PubMed] [Google Scholar]
- 26.Bhargava R, Levin IW (2001) Fourier transform infrared imaging: Theory and practice. Anal Chem 73(21):5157–5167 [DOI] [PubMed] [Google Scholar]
- 27.Bhargava R, Levin IW (2005) Spectrochemical analysis using infrared multichannel detectors preface. Sheff Analy Chem 2005:Xv–Xvi [Google Scholar]
- 28.Leslie LS, Wrobel TP, Mayerich D, Bindra S, Emmadi R, Bhargava R (2015) High definition infrared spectroscopic imaging for lymph node histopathology. PLoS One 10(6): e0127238. Epub 2015/06/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mittal S, Yeh K, Leslie LS, Kenkel S, Kajdacsy-Balla A, Bhargava R (2018) Simultaneous cancer and tumor microenvironment subtyping using confocal infrared microscopy for all-digital molecular histopathology. P Natl Acad Sci USA 115(25):E5651–E5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goormaghtigh E (2016) Infrared imaging in histopathology: is a unified approach possible? Biomed Spectrosc Imag 5(4):325–346 [Google Scholar]
- 31.Dravid UA, Mazumder N (2018) Types of advanced optical microscopy techniques for breast cancer research: a review. Lasers Med Sci 33(9):1849–1858 Epub 2018/10/13 [DOI] [PubMed] [Google Scholar]
- 32.Min W, Freudiger CW, Lu S, Xie XS (2011) Coherent nonlinear optical imaging: beyond fluorescence microscopy. Annu Rev Phys Chem 62:507–530 Epub 2011/04/02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cicerone MT, Camp CH (2017) Histological coherent Raman imaging: a prognostic review. The Analyst 143(1):33–59 Epub 2017/11/04 [DOI] [PubMed] [Google Scholar]
- 34.Kirsch R, Messenger DE, Riddell RH, Pollett A, Cook M, Al-Haddad S et al. (2013) Venous invasion in colorectal cancer: impact of an elastin stain on detection and interobserver agreement among gastrointestinal and nongastrointestinal pathologists. Am J Surg Pathol 37 (2):200–210 Epub 2012/10/31 [DOI] [PubMed] [Google Scholar]
- 35.Natal RA, Vassallo J, Paiva GR, Pelegati VB, Barbosa GO, Mendonca GR et al. (2018) Collagen analysis by second-harmonic generation microscopy predicts outcome of luminal breast cancer. Tumour Biol 40(4):1010428318770953. Epub 2018/04/18 [DOI] [PubMed] [Google Scholar]
- 36.Chen X, Nadiarynkh O, Plotnikov S, Campagnola PJ (2012) Second harmonic generation microscopy for quantitative analysis of collagen fibrillar structure. Nat Protoc 7(4):654–669 Epub 2012/03/10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shribak M (2015) Polychromatic polarization microscope: bringing colors to a colorless world. Sci Rep 5:17340 Epub 2015/11/28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen IJ, Blasberg R (2017) Impact of the tumor microenvironment on tumor-infiltrating lymphocytes: focus on breast cancer. Breast Cancer (Auckl) 11:1178223417731565 Epub 2017/10/06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veitch NC (2004) Horseradish peroxidase: a modern view of a classic enzyme. Phytochemistry 65(3):249–259 [DOI] [PubMed] [Google Scholar]
- 40.Sato S, Nakamura K, Nakamura H (2015) Tyrosine-specific chemical modification with in situ hemin-activated luminol derivatives. ACS Chem Biol 10(11):2633–2640 [DOI] [PubMed] [Google Scholar]
- 41.Sato S, Nakamura K, Nakamura H (2017) Horseradish-peroxidase-catalyzed tyrosine click reaction. Chembiochem: a Eur J Chem Biol 18(5):475–478 Epub 2016/12/23 [DOI] [PubMed] [Google Scholar]
- 42.Beyzavi K, Hampton S, Kwasowski P, Fickling S, Marks V, Clift R (1987) Comparison of horseradish peroxidase and alkaline phosphatase-labelled antibodies in enzyme immunoassays. Ann Clin Biochem 24(Pt 2):145–152 Epub 1987/03/01 [DOI] [PubMed] [Google Scholar]
- 43.Schwenecke H, Benzidine MD, Benzidine Derivatives (2005) Ullmann’s encyclopedia of industrial chemistry, 7th edn. Wiley, Inc., New York, p 18 [Google Scholar]
- 44.Nakane PK, Pierce GB Jr (1966) Enzyme-labeled antibodies: preparation and application for the localization of antigens. J Histochem Cytochem 14(12):929–931 Epub 1966/12/01 [DOI] [PubMed] [Google Scholar]
- 45.Mason DY, Sammons R (1978) Alkaline-phosphatase and peroxidase for double immunoenzymatic labeling of cellular constituents. J Clin Pathol 31(5):454–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Loos CM (2010) Chromogens in multiple immunohistochemical staining used for visual assessment and spectral imaging: the colorful future. J Histotechnol 33(1):31–40 [Google Scholar]
- 47.Day WA, Lefever MR, Ochs RL, Pedata A, Behman LJ, Ashworth-Sharpe J et al. (2017) Covalently deposited dyes: a new chromogen paradigm that facilitates analysis of multiple biomarkers in situ. Lab Invest 97(1):104–113 [DOI] [PubMed] [Google Scholar]
- 48.Bogoslovsky T, Bernstock JD, Bull G, Gouty S, Cox BM, Hallenbeck JM et al. (2018) Development of a systems-based in situ multiplex biomarker screening approach for the assessment of immunopathology and neural tissue plasticity in male rats after traumatic brain injury. J Neurosci Res 96(4):487–500 Epub 2017/05/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McNamara G, Difilippantonio M, Ried T, Bieber FR (2017) Microscopy and image analysis. Curr Protoc Hum Genet 94:4 1–4 89. Epub 2017/07/12 [DOI] [PubMed] [Google Scholar]
- 50.McNamara G (2019) Fluorescence spectra graphs https://www.linkedin.com/pulse/fluorescence-spectra-graphs-george-mcnamara
- 51.McNamara G (2019) 18plex flow cytometry from Brilliants—when will fluorescence microscopes catch up? 2018. https://www.linkedin.com/pulse/18plex-flow-cytometry-from-brilliants-when-catch-up-george-mcnamara/
- 52.McNamara G (2019) “Resolution Blues” meets 21plex salute fluorescence microscopy for immuno-oncology and basic biomedical research 2017. https://www.linkedin.com/pulse/resolution-blues-meets-21plex-salute-fluorescence-basic-mcnamara
- 53.Kerstens HM, Poddighe PJ, Hanselaar AG (1995) A novel in situ hybridization signal amplification method based on the deposition of biotinylated tyramine. J Histochem Cytochem 43(4):347–352 Epub 1995/04/01 [DOI] [PubMed] [Google Scholar]
- 54.vanGijlswijk RPM, Zijlmans HJMAA, Wiegant J, Bobrow MN, Erickson TJ, Adler KE et al. (1997) Fluorochrome-labeled tyramides: use in immunocytochemistry and fluorescence in situ hybridization. J Histochem Cytochem 45(3):375–82 [DOI] [PubMed] [Google Scholar]
- 55.Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC et al. (2017) Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 35(10):936–939 Epub 2017/08/31 [DOI] [PubMed] [Google Scholar]
- 56.Rodgers JR, Rich RR (2013) Antigens and antigen presentation. In: Clinical immunology, 4th edn, pp 77–89 [Google Scholar]
- 57.Lemus R, Karol MH (2008) Conjugation of haptens. Methods Mol Med 138:167–182 Epub 2008/07/10 [DOI] [PubMed] [Google Scholar]
- 58.Levin M, Lingen M, Schwartz D, Snyder H (2017) Multiplex immunofluorescence profiling of tumor infiltrating immune subsets in HNSCC biopsies provides a powerful tool when combined with patient outcome data. Cancer Res 77 [Google Scholar]
- 59.Joerger RD, Truby TM, Hendrickson ER, Young RM, Ebersole RC (1995) Analyte detection with DNA-labeled antibodies and polymerase chain-reaction. Clin Chem 41 (9):1371–1377 [PubMed] [Google Scholar]
- 60.Schubert W, Bonnekoh B, Pommer AJ, Philipsen L, Bockelmann R, Malykh Y et al. (2006) Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol 24(10):1270–1278 Epub 2006/10/03 [DOI] [PubMed] [Google Scholar]
- 61.Friedenberger M, Bode M, Krusche A, Schubert W (2007) Fluorescence detection of protein clusters in individual cells and tissue sections by using toponome imaging system: sample preparation and measuring procedures. Nat Protoc 2(9):2285–2294 [DOI] [PubMed] [Google Scholar]
- 62.Schubert W, Gieseler A, Krusche A, Serocka P, Hillert R (2012) Next-generation biomarkers based on 100-parameter functional super-resolution microscopy TIS. New Biotechnol 29 (5):599–610 Epub 2012/01/03 [DOI] [PubMed] [Google Scholar]
- 63.Schubert W (2015) Advances in toponomics drug discovery: Imaging cycler microscopy correctly predicts a therapy method of amyotrophic lateral sclerosis. Cytom Part A J Int Soc Anal Cytol 87(8):696–703 Epub 2015/04/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Glass G, Papin JA, Mandell JW (2009) SIMPLE: a sequential immunoperoxidase labelling and erasing method. J Histochem Cytochem 58(10):899–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A et al. (2013) Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. P Natl Acad Sci USA 110(29):11982–11987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hollman-Hewgley D, Lazare M, Bordwell A, Zebadua E, Tripathi P, Ross AS et al. (2014) A single slide multiplex assay for the evaluation of classical hodgkin lymphoma. Am J Surg Pathol 38(9):1193–1202 [DOI] [PubMed] [Google Scholar]
- 67.Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G et al. (2018) Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174(4):968–981 e15. Epub 2018/08/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin JR, Izar B, Wang S, Yapp C, Mei SL, Shah PM et al. (2018) Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matros A, Mock HP (2013) Mass spectrometry based imaging techniques for spatially resolved analysis of molecules. Front Plant Sci 4:89 Epub 2013/04/30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bendall SC, Simonds EF, Qiu P, el Amir AD, Krutzik PO, Finck R et al. (2011) Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332(6030):687–696 Epub 2011/05/10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dempsey LA (2017) CyTOF analysis of anti-tumor responses. Nat Immunol 18(3):254. Epub 2017/02/16 [DOI] [PubMed] [Google Scholar]
- 72.Singh M, Chaudhry P, Gerdtsson E, Maoz A, Cozen W, Hicks J et al. (2017) Highly multiplexed imaging mass cytometry allows visualization of tumor and immune cell interactions of the tumor microenvironment in FFPE tissue sections. Blood 130 [Google Scholar]
- 73.Schulz D, Zanotelli VRT, Fischer JR, Schapiro D, Engler S, Lun XK et al. (2018) Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst 6(4):531. Epub 2018/04/27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD et al. (2014) Multiplexed ion beam imaging of human breast tumors. Nat Med 20(4):436–442 Epub 2014/03/04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li ZY, Theile CS, Chen GY, Bilate AM, Duarte JN, Avalos AM et al. (2015) Fluorophore-conjugated holliday junctions for generating super-bright antibodies and antibody fragments. Angew Chem Int Ed 54(40):11706–11710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Krieg R, Halbhuber KJ (2010) Detection of endogenous and immuno-bound peroxidase—the status Quo in histochemistry. Prog Histochem Cytochem 45(2):81–139 [DOI] [PubMed] [Google Scholar]
- 77.Kotani N, Gu J, Isaji T, Udaka K, Taniguchi N, Honke K (2008) Biochemical visualization of cell surface molecular clustering in living cells. Proc Natl Acad Sci USA 105(21):7405–7409 Epub 2008/05/23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, O’Shea CC (2017) ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Sci. 357 (6349). Epub 2017/07/29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ryan BJ, Carolan N, O’Fagain C (2006) Horseradish and soybean peroxidases: comparable tools for alternative niches? Trends Biotechnol 24(8):355–363 [DOI] [PubMed] [Google Scholar]
- 80.Han Y, Branon TC, Martell JD, Boassa D, Shechner D, Ellisman MH et al. (2019) Directed evolution of split APEX2 peroxidase. ACS Chem Biol 14(4):619–635 Epub 2019/03/09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaewsapsak P, Shechner DM, Mallard W, Rinn JL, Ting AY (2017) Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 6. Epub 2017/12/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martell JD, Deerinck TJ, Lam SS, Ellisman MH, Ting AY (2017) Electron microscopy using the genetically encoded APEX2 tag in cultured mammalian cells. Nat Protoc 12(9):1792–1816 Epub 2017/08/11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK et al. (2017) Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife 6. Epub 2017/04/26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K et al. (2016) Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11 (3):456–475 Epub 2016/02/13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Han S, Udeshi ND, Deerinck TJ, Svinkina T, Ellisman MH, Carr SA et al. (2017) Proximity biotinylation as a method for mapping proteins associated with mtDNA in living cells. Cell Chem Biol 24(3):404–414 Epub 2017/02/28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK et al. (2015) Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12(1):51–54 Epub 2014/11/25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krieg R, Halbhuber KJ (2004) Novel oxidative self-anchoring fluorescent substrates for the histochemical localization of endogenous and immunobound peroxidase activity. J Mol Histol 35(5):471–487 [DOI] [PubMed] [Google Scholar]
- 88.Bobrow MN, Harris TD, Shaughnessy KJ, Litt GJ (1989) Catalyzed reporter deposition, a novel method of signal amplification application to immunoassays. J Immunol Methods 125 (1–2):279–285 Epub 1989/12/20 [DOI] [PubMed] [Google Scholar]
- 89.Bobrow MN, Litt GJ, Shaughnessy KJ, Mayer PC, Conlon J (1992) The use of catalysed reporter deposition as a means of signal amplification in a variety of formats. J Immunol Methods 150(1–2):145–149 [DOI] [PubMed] [Google Scholar]
- 90.Buchwalow IB, Böcker W (2010) Immunostaining enhancement in Immunohistochemistry. In: Basica and methods. Springer, New York, pp 47–59 [Google Scholar]
- 91.Hopman AHN, Ramaekers FCS, Speel EJM (1998) Rapid synthesis of biotin-, digoxigenin-, trinitrophenyl-, and fluorochrome-labeled tyramides and their application for in situ hybridization using CARD amplification. J Histochem Cytochem 46(6):771–777 [DOI] [PubMed] [Google Scholar]
- 92.Kaplan D (2003) Enzymatic amplification staining for single cell analysis: applied to in situ hybridization. J Immunol Methods 283(1–2):1–7 Epub 2003/12/09 [DOI] [PubMed] [Google Scholar]
- 93.Kaplan D, Meyerson H, Lewandowska K (2001) High resolution immunophenotypic analysis of chronic lymphocytic leukemic cells by enzymatic amplification staining. Am J Clin Pathol 116(3):429–436 Epub 2001/09/14 [DOI] [PubMed] [Google Scholar]
- 94.Kaplan D, Smith D (2000) Enzymatic amplification staining for flow cytometric analysis of cell surface molecules. Cytometry 40(1):81–85 Epub 2000/04/08 [DOI] [PubMed] [Google Scholar]
- 95.Takahashi H, Ruiz P, Ricordi C, Delacruz V, Miki A, Mita A et al. (2012) Quantitative in situ analysis of FoxP3+ T regulatory cells on transplant tissue using laser scanning cytometry. Cell Transplant 21(1):113–125 Epub 2011/09/21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xing Y, Chaudry Q, Shen C, Kong KY, Zhau HE, WChung L et al. (2007) Bioconjugated quantum dots for multiplexed and quantitative immunohistochemistry. Nat Protoc 2 (5):1152–1165 [DOI] [PubMed] [Google Scholar]
- 97.Zrazhevskiy P, True LD, Gao X (2013) Multicolor multicycle molecular profiling with quantum dots for single-cell analysis. Nat Protoc 8(10):1852–1869 Epub 2013/09/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu B, Jiang B, Zheng ZP, Liu TC (2019) Semiconductor quantum dots in tumor research. J Lumin 209:61–68 [Google Scholar]
- 99.Gao X, Cui Y, Levenson RM, Chung LW, Nie S (2004) In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol 22(8):969–976 Epub 2004/07/20 [DOI] [PubMed] [Google Scholar]
- 100.Levenson RM, Mansfield JR (2006) Multispectral imaging in biology and medicine: slices of life. Cytometry Part A J Int Soc Anal Cytol 69(8):748–758 Epub 2006/09/14 [DOI] [PubMed] [Google Scholar]
- 101.Gonda K, Watanabe M, Tada H, Miyashita M, Takahashi-Aoyama Y, Kamei T et al. (2017) Quantitative diagnostic imaging of cancer tissues by using phosphor-integrated dots with ultra-high brightness. Sci Rep 7(1):7509. Epub 2017/08/10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mansfield JR, Hoyt C, Levenson RM (2008) Visualization of microscopy-based spectral imaging data from multi-label tissue sections. Curr Protoc Mol Biol Chapter 14:Unit 14 9. Epub 2008/10/31 [DOI] [PubMed] [Google Scholar]
- 103.Walker RA (2006) Quantification of immunohistochemistry–issues concerning methods, utility and semiquantitative assessment I. Histopathology 49(4):406–410 Epub 2006/09/19 [DOI] [PubMed] [Google Scholar]
- 104.Taylor CR, Levenson RM (2006) Quantification of immunohistochemistry–issues concerning methods, utility and semiquantitative assessment II. Histopathology 49(4):411–424 Epub 2006/09/19 [DOI] [PubMed] [Google Scholar]
- 105.Rimm DL (2006) What brown cannot do for you. Nat Biotechnol 24(8):914–916 [DOI] [PubMed] [Google Scholar]
- 106.Oliveira VC, Carrara RC, Simoes DL, Saggioro FP, Carlotti CG Jr, Covas DT et al. (2010) Sudan Black B treatment reduces autofluorescence and improves resolution of in situ hybridization specific fluorescent signals of brain sections. Histol Histopathol 25(8):1017–1024 Epub 2010/06/17 [DOI] [PubMed] [Google Scholar]
- 107.Mansfield JR (2010) Distinguished photons: a review of in vivo spectral fluorescence imaging in small animals. Curr Pharm Biotechnol 11(6):628–638 Epub 2010/05/26 [DOI] [PubMed] [Google Scholar]
- 108.Dickinson ME, Bearman G, Tille S, Lansford R, Fraser SE (2001) Multi-spectral imaging and linear unmixing add a whole new dimension to laser scanning fluorescence microscopy. Biotechniques 31(6):1272, 4–6, 8. Epub 2002/01/05 [DOI] [PubMed] [Google Scholar]
- 109.Megjhani M, Correa de Sampaio P, Leigh Carstens J, Kalluri R, Roysam B (2017) Morphologically constrained spectral unmixing by dictionary learning for multiplex fluorescence microscopy. Bioinformatics 33(14):2182–2190. Epub 2017/03/24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bordeaux J, Welsh A, Agarwal S, Killiam E, Baquero M, Hanna J et al. (2010) Antibody validation. Biotechniques 48(3):197–209 Epub 2010/04/03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moran L (2007) Basic concepts: the central dogma of molecular biology. https://sandwalk.blogspot.com/2007/01/central-dogma-of-molecular-biology.html
- 112.Li JJ, Bickel PJ, Biggin MD (2014) System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li JJ, Biggin MD (2015) Statistics requantitates the central dogma. Science 347 (6226):1066–1067 [DOI] [PubMed] [Google Scholar]
- 114.Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK et al. (2015) Genomic variation. Impact of regulatory variation from RNA to protein. Science 347(6222):664–667. Epub 2015/02/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R et al. (2015) Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Sci 347(6226):1259038. Epub 2015/03/07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hausser J, Mayo A, Keren L, Alon U (2019) Central dogma rates and the trade-off between precision and economy in gene expression. Nat Commun 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Symmons O, Chang M, Mellis IA, Kalish JM, Park J, Susztak K et al. (2019) Allele-specific RNA imaging shows that allelic imbalances can arise in tissues through transcriptional bursting. Plos Genet 15(1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Caveney PM, Norred SE, Chin CW, Boreyko JB, Razooky BS, Retterer ST et al. (2017) Resource sharing controls gene expression bursting. ACS Synth Biol 6(2):334–343 Epub 2016/10/04 [DOI] [PubMed] [Google Scholar]
- 119.Li JJ, Bickel PJ, Biggin MD (2014) System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2:e270 Epub 2014/04/02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Eraslan B, Wang D, Gusic M, Prokisch H, Hallstrom BM, Uhlen M et al. (2019) Quantification and discovery of sequence determinants of protein-per-mRNA amount in 29 human tissues. Molecular systems biology. 15(2):e8513. Epub 2019/02/20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A et al. (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn: JMD 14(1):22–29 Epub 2011/12/15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gall JG, Pardue ML (1969) Formation and detection of Rna-DNA hybrid molecules in cytological preparations. P Natl Acad Sci USA 63(2):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pardue ML, Gall JG (1969) Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proc Natl Acad Sci USA 64(2):600–604 Epub 1969/10/01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pardue ML, Gall JG (1970) Chromosomal localization of mouse satellite DNA. Science 168 (3937):1356–1358 Epub 1970/06/12 [DOI] [PubMed] [Google Scholar]
- 125.Femino AM, Fay FS, Fogarty K, Singer RH (1998) Visualization of single RNA transcripts in situ. Science 280(5363):585–590 Epub 1998/05/09 [DOI] [PubMed] [Google Scholar]
- 126.Vargas DY, Raj A, Marras SAE, Kramer FR, Tyagi S (2005) Mechanism of mRNA transport in the nucleus. P Natl Acad Sci USA 102(47):17008–17013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S (2006) Stochastic mRNA synthesis in mammalian cells. PLoS Biol 4(10):1707–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual RNA molecules using multiple singly labeled probes. Nat Methods 5(10):877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Raj A, Tyagi S (2010) Detection of individual endogenous Rna transcripts in situ using multiple singly labeled probes. In: Methods in enzymology, vol 472: Single Molecule Tools, Pt A: Fluorescence Based Approaches 472:365–386 [DOI] [PubMed] [Google Scholar]
- 130.Batish M, Raj A, Tyagi S (2011) Single molecule imaging of RNA in situ. Methods Mol Biol 714:3–13 Epub 2011/03/25 [DOI] [PubMed] [Google Scholar]
- 131.Shaffer SM, Wu MT, Levesque MJ, Raj A (2013) Turbo FISH: a method for rapid single molecule RNA FISH. PLoS One. 8(9):e75120. Epub 2013/09/26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Levesque MJ, Raj A (2013) Single-chromosome transcriptional profiling reveals chromosomal gene expression regulation. Nat Methods 10(3):246–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Levesque MJ, Ginart P, Wei YC, Raj A (2013) Visualizing SNVs to quantify allele-specific expression in single cells. Nat Methods 10(9):865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Player AN, Shen LP, Kenny D, Antao VP, Kolberg JA (2001) Single-copy gene detection using branched DNA (bDNA) in situ hybridization. J Histochem Cytochem 49(5):603–611 [DOI] [PubMed] [Google Scholar]
- 135.Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J et al. (2018) Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145(12). Epub 2018/06/28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schweller RM, Zimak J, Duose DY, Qutub AA, Hittelman WN, Diehl MR (2012) Multiplexed in situ immunofluorescence using dynamic DNA complexes. Angew Chem Int Ed Engl 51(37):9292–9296 Epub 2012/08/16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zimak J, Schweller RM, Duose DY, Hittelman WN, Diehl MR (2012) Programming in situ immunofluorescence intensities through interchangeable reactions of dynamic DNA complexes. Chembiochem: A Eur J Chem Biol 13(18):2722–2728 Epub 2012/11/21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X (2015) RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348(6233):aaa6090. Epub 2015/04/11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Moor AE, Itzkovitz S (2017) Spatial transcriptomics: paving the way for tissue-level systems biology. Curr Opin Biotechnol 46:126–133 Epub 2017/03/28 [DOI] [PubMed] [Google Scholar]
- 140.Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J et al. (2016) Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353(6294):78–82 Epub 2016/07/02 [DOI] [PubMed] [Google Scholar]
- 141.Strell C, Hilscher MM, Laxman N, Svedlund J, Wu C, Yokota C et al. (2019) Placing RNA in context and space—methods for spatially resolved transcriptomics. FEBS J 286(8):1468–1481 Epub 2018/03/16 [DOI] [PubMed] [Google Scholar]
- 142.Burgess DJ (2019) Spatial transcriptomics coming of age. Nat Rev Genet 20(6):317. Epub 2019/04/14 [DOI] [PubMed] [Google Scholar]
- 143.Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu DN et al. (2014) Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510(7505):363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Moor AE, Golan M, Massasa EE, Lemze D, Weizman T, Shenhav R et al. (2017) Global mRNA polarization regulates translation efficiency in the intestinal epithelium. Science 357 (6357):1299–1303 Epub 2017/08/12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Moor AE, Harnik Y, Ben-Moshe S, Massasa EE, Rozenberg M, Eilam R et al. (2018) Spatial Reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 175(4):1156–1167 e15. Epub 2018/10/03 [DOI] [PubMed] [Google Scholar]
- 146.McKinley ET, Sui Y, Al-Kofahi Y, Millis BA, Tyska MJ, Roland JT et al. (2017) Optimized multiplex immunofluorescence single-cell analysis reveals tuft cell heterogeneity. JCI Insight. 2(11). Epub 2017/06/02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Herring CA, Banerjee A, McKinley ET, Simmons AJ, Ping J, Roland JT et al. (2018) Unsupervised trajectory analysis of single-cell RNA-Seq and imaging data reveals alternative tuft cell origins in the gut. Cell Syst 6(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li CX, Ma HT, Wang Y, Cao Z, Graves-Deal R, Powel AE et al. (2014) Excess PLAC8 promotes an unconventional ERK2-dependent EMT in colon cancer. J Clin Inv 124 (5):2172–2187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD et al. (2018) Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science. 362(6416). Epub 2018/11/06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y et al. (2019) Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568(7751):235–239 Epub 2019/03/27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR et al. (2019) Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363(6434):1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lein E, Borm LE, Linnarsson S (2017) The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358(6359):64–69 Epub 2017/10/07 [DOI] [PubMed] [Google Scholar]
- 153.Decalf J, Albert ML, Ziai J (2019) New tools for pathology: a user’s review of a highly multiplexed method for in situ analysis of protein and RNA expression in tissue. J Pathol 247(5):650–661 Epub 2018/12/21 [DOI] [PubMed] [Google Scholar]
- 154.NanoString Technologies I. GeoMxTM digital spatial profiler
Internet Resources
- 155. https://www.cellsignal.com/contents/resources-applications/fluorescent-multiplex-immunohistochemistry/fluoresence-mihc.
- 156. https://www.ultivue.com/technology/
- 157. https://cellidx.com/technology/technology.
- 158. https://neogenomics.com/pharma-services/lab-services/multiomyx/technology/hyperplexed-immunofluorescence-assay.
- 159. https://www.olink.com/data-you-can-trust/technology/
- 160. https://www.ionpath.com/mibi-technology/
- 161. https://www.olympus-lifescience.com/en/microscope-resource/primer/techniques/fluorescence/filters/
- 162. https://www.akoyabio.com/
- 163.McNamara (2019). https://www.linkedin.com/pulse/fluorescence-spectra-graphs-george-mcnamara
- 164.McNamara (2018). https://www.linkedin.com/pulse/18plex-flow-cytometry-from-brilliants-when-catch-up-george-mcnamara/
- 165.McNamara Fluorescence Data Tables. http://www.geomcnamara.com/data
- 166.McNamara (2017). https://www.linkedin.com/pulse/resolution-blues-meets-21plex-salute-fluorescence-basic-mcnamara/
- 167.McNamara (2012) PubSpectra (data download site). https://works.bepress.com/gmcnamara/9