

Abstract
Go-Ichi-Ni-San 2 (GINS2), as a newly discovered oncogene, is overexpressed in several cancers. However, the specific role of GINS2 in the development of pancreatic cancer (PC), to our knowledge, is poorly understood. We systematically explored the potential role of GINS2 in epithelial–mesenchymal-transition (EMT)-stimulated PC in vitro and vivo. GINS2 was overexpressed in human PC specimens, which was positively associated with tumor size (P = 0.010), T stage (P = 0.006), vascular invasion (P = 0.037), and the poor prognosis (P = 0.004). Interestingly, a close correlation between GINS2, E-cadherin, and Vimentin (P = 0.014) was found in human PC specimens and cell lines that coordinately promoted the worse survival of PC patients (P = 0.009). GINS2 overexpression stimulated EMT in vitro, including promoting EMT-like cellular morphology, enhancing cell motility, and activating EMT and ERK/MAPK signal pathways. However, PD98059, a specific MEK1 inhibitor, reversed GINS2 overexpression-stimulated EMT in vitro. Conversely, GINS2 silencing inhibited EMT in PANC-1 cells, which was also rescued by GINS2-GFP. Moreover, GINS2 was colocalized and co-immunoprecipitated with ERK in GINS2 high-expression Miapaca-2 and PANC-1 cells, implying a tight interaction of GINS2 with ERK/MAPK signaling. Meanwhile, GINS2 overexpression inhibited distant liver metastases in vivo, following a tight association with EMT and ERK/MAPK signaling, which was reversed by MEK inhibitor. Overexpression of GINS2 contributes to advanced clinical stage of PC patient and promotes EMT in vitro and vivo via specifically activating ERK/MAPK signal pathway.
Subject terms: Cell biology, Paediatric cancer
Introduction
Pancreatic cancer (PC) is predicted to be the second most frequent cause of death within the next 10 years in North America and Europe [1]. It ranks as the first morbidity and the second age-standard fatality in Chinese male population [2]. Most (85%) patients are diagnosed with advanced local stage or metastatic progression because of its aggressive biology [3]. Epithelial-to-mesenchymal transition (EMT) is regarded as a key stimulator in accelerating the above progression. The key epithelial markers, E-cadherin (E-cad), is lost during the EMT process, leading to the damage of cell–cell adhesion and the gain of mesenchymal properties, which finally contributes to the strong cell invasiveness and metastasis of PC [4].
The Go-Ichi-Ni-San (GINS) family, including four subunits (GINS1–4), has a specific function in DNA replication and cell cycle [5], and plays a vital role in the development of chromosome and DNA replication forks [6]. GINS2 situates in human chromosome 16q24 with 1196 bp mRNA and ~21 kDa of molecular weight. It is identified as a critical regulator in DNA replication and cell cycle process [7], and an oncogene in various cancers, such as non-small-cell lung [8], breast [9], cervical [10], and thyroid cancers [11]. However, its definite role in PC, to our knowledge, has not been reported.
ERK/mitogen-activated protein kinase (MAPK) signaling is a key modulator in cell biology (proliferation, differentiation, motility, apoptosis, and survival) via cell membrane receptor signals transmission [12]. It promotes a malignant phenotype in cancers when facing multiple intrinsic and extrinsic stimuli [12, 13]. The activation of ERK/MAPK signaling promotes EMT progression in several cancers. For example, Calreticulin promoted epidermal growth factor (EGF)-induced EMT in PC through activating EGFR-ERK/MAPK signal [14]; Musashi2 promoted EGF-stimulated EMT in PC via activating ZEB1-ERK/MAPK signal pathway [15]. Up to now, only one study show a close association of GINS2 with EMT in non-small-cell lung cancer (NSCLC) [8]. Based on the above studies, we intend to investigate the potential role and molecular mechanism of GINS2 in EMT, which offers a novel and promising gene target for PC intervention.
Materials and methods
Clinical specimens and PC cells
The present research was authorized by the academic committee from the Fourth People’s Hospital in Shenyang city and the first hospital of China Medical University. All patients accepted the specimen consent and the study methodology has been admitted by the China Medical University. Seventy-four paraffin-embedded PC and paired pancreas were picked up from surgical treatment patients from 2007 to 2017, which were definitely diagnosed as pancreatic ductal adenocarcinoma. Patients with endocrine carcinoma, acinar cell carcinoma, and invasive intraductal papillary mucinous carcinoma were excluded from this study. Twenty and 18 PC specimens were additionally collected for western blotting (WB) and PCR, respectively. Aspc-1, Bxpc-3, PANC-1, and Miapaca-2 cells were purchased from the cell culture collection in Chinese Academy of Sciences in Shanghai.
Immunohistochemistry
Our study was not a prospective research and 74 samples were enough for immunohistochemical (IHC) assays. IHC was conducted according to our previous study [10, 11]. Our study was not a prospective research. IHC was conducted according to our previous study [14, 15]. In detail, PC sections were deparaffinized and dehydrated first. They were next incubated with H2O2, subjected to high-pressure repair, and blocked with bovine serum albumin (BSA). Anti-GINS2 (Abcam, Cambridge, UK, dilution, 1 : 200), E-cad (Abcam, dilution, 1 : 500), and Vimentin (Proteintech, Chicago, IL, dilution, 1 : 1000) sections were incubated overnight. After washing with phosphate-buffered saline (PBS), slices were covered with the secondary antibody, detected with DAB, co-stained with haematoxylin, and evaluated by two pathologists.
Western blotting
The total proteins extracted from whole-cell lysates and PC specimens were inserted into 10% SDS-polyacrylamide gels, transmitted to wet transfer, blocked with 5% skimmed-milk (diluted with TBST) and incubated with GINS2 (Abcam, dilution: 1 : 1000; ab197123), E-cad (Abcam, dilution: 1 : 1000; ab40772), ZO-1 (Proteintech, Chicago, IL, dilution: 1 : 1000; 21773-1-AP), N-cadherin (Proteintech, dilution: 1 : 500; 13769-1-AP), Vimentin (Proteintech, dilution: 1 : 2000; 10366-1-AP), MMP9 (Proteintech, dilution: 1 : 500; 10375-2-AP), Fibronectin (Proteintech, dilution: 1 : 1000; 66042-1-Ig), pERK/ERK (Cell Signaling Technology, Beverly, USA, dilution: 1 : 500/1 : 1000, respectively; #9101/#4695), p-p38/p38 (Cell Signaling Technology, 1 : 1000 and 1 : 1000, respectively; #4511/#8690), p-JNK/JNK (Cell Signaling Technology, 1 : 1000 and 1 : 1000, respectively; #4668/#9252), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Proteintech, dilution: 1 : 3000; 60004-1-Ig) antibodies. The secondary antibodies (Proteintech) were incubated and all the bands were detected with the ECL instrument (Bio-Rad, California, USA). Cells were pretreated MEK1 inhibitor PD98059 (20 μM, Cell Signaling Technology, #9903) for 2 h prior to WB according to our previous study [16]. WB was repeated three times.
Immunofluorescence staining
PC cell lines were implanted into 24-well culture plates covered with slices, fixed in 4% paraformaldehyde, permeabilized with Triton X-100 (0.25%), and incubated with 5% BSA. Then, plates were stained with the following primary antibodies: GINS2 (Abcam, 1 : 50), E-cad (Abcam, 1 : 100), Vimentin (Proteintech, 1 : 100), and ERK (Cell Signaling Technology, 1 : 50). The secondary antibodies (Vector Laboratories, California, USA) were conjugated with fluorescein isothiocyanate for GINS2 and tetramethylrhodamine for E-cad, Vimentin, and ERK. Hoechst 33258 (Vector Laboratories) were used for nuclear visualizing. Aspc-1 and Bxpc-3 cells were used to stain with E-cad and Vimentin in green fluorescent protein (GFP), GINS2-GFP, and GINS2-GFP plus PD98059 groups, whereas Miapaca-2 and PANC-1 cells were used to stain with GINS2 and ERK, respectively.
Immunoprecipitation
According to our previous study [10, 11, 16], lysates from normal cells were extracted in the immunoprecipitation (IP) lysis buffer. In detail, the antibodies of GINS2, ERK, and GAPDH were incubated with magnetic-beads (Thermo Scientific, Rockford, IL, USA) for at least 4 h. Antibody–beads complex was treated with the supernatants of RIPA-treated lysates overnight after washing with IP lysis buffer. The final immunocomplex was boiling with 5× loading buffer for WB analysis with GINS2 and ERK antibodies, respectively. Input and IgG groups were used as a positive and negative control, respectively.
Real-time quantitative PC
According to our previous study [16], the mRNA of target genes was detected in SYBR Premix Ex TaqTMII regent (Takara Biology, Dalian, China) as below: 95 °C for 40 s and 45 cycles of 95 °C for 10 s and 58 °C for 40 s. The primers were summarized in Supplementary Table 1. Quantification of amplification products was calculated with post-PCR melt dissociation following the ΔΔCt method.
siRNA and lentivirus vector-mediated GINS2 overexpression
Two effective sequence (3′-untranslated region) of GINS2siRNA were followed:
1. 5′-CATGCAATGGATAGAAGGTTCTGTA-3′;
2. 5′-CATCATGCAATGGATAGAAGGTTCT-3′.
siRNA transfections and Oligofectamine-3000 (Invitrogen, USA) were mixed together for transfection according to the protocol. One of above siRNA sequences was used for the late experiment in vitro. Lentivirus vector-mediated GINS2 overexpression (GINS2-GFP) and empty vector (GFP) were purchased from Genechem (Shanghai, China). Aspc-1 and Bxpc-3 cells with low GINS2 expression were available for constructing GINS-overexpressing stable PC cell lines after puromycin treatment. GINS2 silencing and overexpressing effect was detected by WB shown in the “Results” section.
EMT construction
PC cells with GINS2-GFP and GFP were pre-cultured with medium containing 1% fetal bovine serum (FBS) twice within 4 days, to enhance EMT induction. The transformation involving EMT-like cellular morphology, EMT epithelial and mesenchymal markers, and cell motility were evaluated to reflect the EMT development model. Prior to morphology assays, PC cells were pretreated with PD98059 (20 μM) twice, combining with 1% FBS medium within 4 days. DMSO was used for diluting PD98059 and was added as a control.
Transwell assays
Based on our previous study [16], GINS2siRNA- and siRNAcontrol (siRNActrl)-transfected PANC-1 cells, and GINS2-GFP- and GFP-transfected AsPC-1 cells pretreated with PD98059 (20 μM for 2 h twice) were implanted into membrane inserts (BD Biosciences, USA) covered with Matrigel (Corning, Bedford, MA, USA) in 24-well plates with free serum medium. Medium combining 10% FBS was put into the bottom as the stimulus. The migrated cells at the underside of the inserts were fixed and co-stained with crystal violet (Sigma). The final migrated cell number were calculated in at least five random fields/each well. The invasion assay was conducted in the similar way without Matrigel. We repeated three times for this experiment.
Liver metastatic model
Animals were kept under the Institutional Animal Care of China Medical University according to the national guidelines for animal studies. As PD98059 was not prevalently used in vivo previously, we used another MEK1 inhibitor U0126 (Cell Signaling Technology) instead of PD98059 in vivo according to previous studies [17–20].
We estimated five mice in each group is enough in vivo for the final statistics analysis. Total 15 nude mice (BALB/c Nude, female, 6–8 weeks old) were acclimatized for a week and were randomly divided into GFP, GINS2-GFP, and GINS2-GFP plus U0126 groups (n = 5 in each group). The investigator was blinded to the groups during the experiment when assessing the outcome. According to the protocol of our previous studies [15], GINS2-GFP- and control (GFP)-transfected Aspc-1 cells (1 × 107 ml−1) mixed with pre-cold PBS (150 μl) were slowly injected into the lower-middle part of spleen. A cotton swab was pressed toward the injection site to avoid bleeding and leakage. Three days after injection, U0126 was injected intraperitoneally (100 μl at the dose of 50 μmol/kg) every 3 days in GINS2-GFP plus U0126 group according to protocol of previous studies [17–20]. All mice were killed after 3 weeks. We calculated liver metastases number and took the samples for the late hematoxylin and eosin (HE), and IHC assays.
Statistical analysis
Nonparametric paired, χ2-, and Spearman’s relation tests were used for the statistical analyses in IHC assays. The Kaplan–Meier curve, log-rank, and Cox regression tests were used to calculate the prognosis data. Other data including WB, quantitative reverse-transcriptase PCR, transwell, and the comparing of liver metastatic number were identified as means ± SD and were compared via paired or independent t-test. P-value is regarded statistically significant when *P < 0.05 and **P < 0.01.
Results
Overexpression of GINS2 was tightly associated with the clinicopathological significance of PC patients
GINS2 showed cytoplasmic and nuclear expression in adjacent pancreas (Fig. 1a, d) and PC (high expression in Fig. 1b and low expression in Fig. 1c). It is well known that E-cad and Vimentin are the critical epithelial and mesenchymal hallmarks in EMT, respectively [20]. E-cad membrane expression in the pancreas (Fig. 1e) and PC (Fig. 1k) was regarded as normal expression, whereas E-cad cytoplasmic or negative expression in PC samples (Fig. 1h) were identified as abnormal expression. Vimentin showed membrane and cytoplasmic staining in the pancreas (Fig. 1f) and PC (Fig. 1i, l). IHC showed GINS2 was overexpressed in human PC specimens in contrast with the paired pancreas (40/74, 54% vs. 19/74, 25.6%, P < 0.01) (Fig. 1a, b). PC specimens with GINS2 overexpression (Fig. 1g) was associated with E-cad abnormal (Fig. 1h) and Vimentin-positive expression (Fig. 1i) in most serial sample slices and vice versa (Fig. 1j–l). Spearman’s correlation test further confirmed that GINS2 showed a negative association with E-cad and a positive relationship with Vimentin in 74 PC specimens (r = −0.287, P = 0.014; r = 0.270, P = 0.020) (Table 1).
Fig. 1. The expression of GINS2, E-cad, and Vimentin in human PC and adjacent pancreas by IHC.

a–c GINS2 expression in paired pancreas (a) and PC specimens (b, c). d–f GINS2 (d), E-cad (e), and Vimentin (f) expression in adjacent pancreas as a control. g–i GINS2 (g), E-cad (h), and Vimentin expression (i) in one PC tissue. j–l GINS2 (j), E-cad (k), and Vimentin expression (l) in another sample (×200 magnification).
Table 1.
GINS2 was negatively associated with E-cad in clinical samples.
| Parameters | GINS2 | r | P | ||
|---|---|---|---|---|---|
| Negative | Positive | Total | |||
| E-cad | |||||
| Abnormal | 17 | 31 | 48 | −0.287 | 0.014 |
| Normal | 17 | 9 | 26 | ||
| Total | 34 | 40 | 74 | ||
| Vimentin | |||||
| Positive | 4 | 14 | 18 | 0.270 | 0.020 |
| Negative | 30 | 26 | 56 | ||
| Total | 34 | 40 | 74 | ||
GINS2 overexpression was positively related with tumor size (P = 0.010), T stage (P = 0.006), and vascular invasion (P = 0.037), but had no relationship with other characters (Table 2). Patients with GINS2 overexpression had a poor survival (P = 0.004) (Fig. 2a). Both E-cad and Vimentin were not associated with the prognosis (P = 0.162; P = 0.180) (Fig. 2b, c), but patients with high GINS2 combining E-cad abnormal expression or Vimentin-positive expression coordinately contributed to the worse survival of PC patients (P = 0.004; P = 0.016) (Fig. 2d, e) even though we compared the total survival data based on the four different combinations together into the survival plots (Fig. 2d, e). In multivariate analysis, GINS2 was an independent unfavorable prognostic index (P = 0.045) (Table 3).
Table 2.
The clinical significance of GINS2 expression in human PC samples.
| Parameters | No. of patients | GINS2 | P | |
|---|---|---|---|---|
| Low | High | |||
| Cases | 74 | 34 | 40 | |
| Age(years) | ||||
| ≤65 | 53 | 22 | 31 | 0.224 |
| >65 | 21 | 12 | 9 | |
| Gender | ||||
| Male | 48 | 22 | 26 | 0.979 |
| Female | 26 | 12 | 14 | |
| Tumor location | ||||
| Head | 51 | 25 | 26 | 0.429 |
| Body tail | 23 | 9 | 14 | |
| Tumor size(cm) | ||||
| <3 | 38 | 23 | 15 | 0.010 |
| ≥3 | 36 | 11 | 25 | |
| Differentiation | ||||
| Well | 22 | 9 | 13 | 0.132 |
| Moderate | 28 | 10 | 18 | |
| Poor | 24 | 15 | 9 | |
| T stage | ||||
| T1 + T2 | 33 | 21 | 12 | 0.006 |
| T3 + T4 | 41 | 13 | 28 | |
| Lymph nodes metastasis | ||||
| N0 (negative) | 51 | 25 | 26 | 0.429 |
| N1 (positive) | 23 | 9 | 14 | |
| Vascular permeation | ||||
| Absent | 36 | 21 | 15 | 0.037 |
| Present | 38 | 13 | 25 | |
| Pre-therapeutic CA19-9 level | ||||
| <37 U/ml | 21 | 10 | 11 | 0.856 |
| ≥37 U/ml | 53 | 24 | 29 | |
Fig. 2. The association of GINS2, E-cad, and Vimentin with the survival of PC patients.

a High (+) and low (−) expression of GINS2 against prognosis. b Normal (+) and abnormal (−) expression of E-cad against prognosis. c Positive (+) and negative (−) expression of Vimentin against prognosis. d Combination of GINS2 and E-cad against prognosis. e Combination of GINS2 and Vimentin against prognosis.
Table 3.
Survival data in univariate and multivariate analysis.
| Parameters | Median survival (months) | Univariate analysis P (log-rank) | Multivariate analysis hazard ratio (95% CI) | P |
|---|---|---|---|---|
| Age (<65/≥65 years) | 29/21 | 0.204 | ─ | |
| Gender (male/female) | 21/28 | 0.186 | ─ | |
| Tumor location (head/body tail) | 27/18 | 0.079 | ─ | |
| Tumor size (<3/≥3 cm) | 28/19 | 0.078 | ─ | |
| Well /poor/moderate differentiation | 28/25/19 | 0.086 | ─ | |
| T stage (T1 + T2/T3 + T4) | 27/20 | 0.123 | ─ | |
| Lymph node metastasis (N0/N1) | 28/16 | 0.001 | 2.138 (1.124–4.067) | 0.021 |
| Vascular permeation (absent/present) | 28/19 | 0.003 | 1.960 (0.977–3.935) | 0.058 |
| CA19-9 level (<37 U/ml/≥37 U/ml) | 26/24 | 0.290 | ─ | |
| GNIS2 (high/low) | 17/30 | 0.004 | 1.976(1.017–3.843) | 0.045 |
| E-cad (normal/abnormal) | 26/22 | 0.162 | ─ | |
| Vimentin (positive/negative) | 17/26 | 0.180 | ─ |
Similarly, GINS2 protein and mRNA expression was much higher in PC specimens in contrast with paired pancreas (P < 0.01 and P < 0.01, respectively) by WB and PCR (Fig. 3a, b). In vitro, the protein and mRNA expression of GINS2 and Vimentin was increased, but E-cad was decreased in PANC-1 and Miapaca-2 cells compared with that in Aspc-1 and Bxpc-3 cells (Fig. 3c, d). The tight association of GINS2 with E-cad and Vimentin in human PC specimens and cell lines has driven us to focus on its specific function in EMT in vitro and in vivo.
Fig. 3. The expression of GINS2, E-cad, and Vimentin in PC specimens and cell lines by WB and qRT-PCR.

a GINS2 protein level in 18 PC and paired pancreas (9 cases were shown here; N: paired pancreas; C: PC). b GINS2 mRNA level in 20 PC and paired pancreas (N: paired pancreas; C: PC). Paired-sample t-test was used for the data of a and b. c, d. GINS2, E-cad, and Vimentin protein (c) and mRNA (d) levels in PC cells. Bars indicate ± SE. *P < 0.05; **P < 0.01 compared with the control.
GINS2-mediated EMT and ERK/MAPK signaling in vitro
Aspc-1 cells with low GINS2 were available to construct GINS2-overexpressing stable cell lines, whereas PANC-1 cells with high GINS2 was used for GINS2 silencing. GINS2 protein expression in GINS2siRNA group was significantly downregulated in contrast with siRNActrl group (Fig. 4a, b), whereas it was overexpressed in GINS2-GFP group (Fig. 4c, d). GINS2 silencing upregulated EMT epithelial markers E-cad and ZO-1, and downregulated mesenchymal markers Vimentin and MMP9 (Fig. 4a, b). Meanwhile, pERK expression was also inhibited by GINS2 silencing (Fig. 4a, b). Conversely, GINS2 overexpression upregulated pERK, Vimentin, and MMP9 but downregulated E-cad and ZO-1. However, p-p38 and p-JNK, as the critical and parallel targets in MAPK pathway, were not affected by GINS2 silencing and overexpression (Fig. 4), implying a specific interaction of GINS2 with ERK/MAPK signaling. In addition, GINS2 overexpression-reversed GINS2 silencing induced the change of EMT and ERK/MAPK signaling (Supplementary Fig. 1). In detail, GINS2 overexpression-inhibited GINS2 silencing induced the decrease of Vimentin, MMP9, and pERK, and the increase of E-cad and ZO-1. Based on above results, GINS2 mediated EMT and ERK/MAPK signal pathway in vitro.
Fig. 4. GINS2 specifically regulated EMT and ERK/MAPK signaling in vitro.
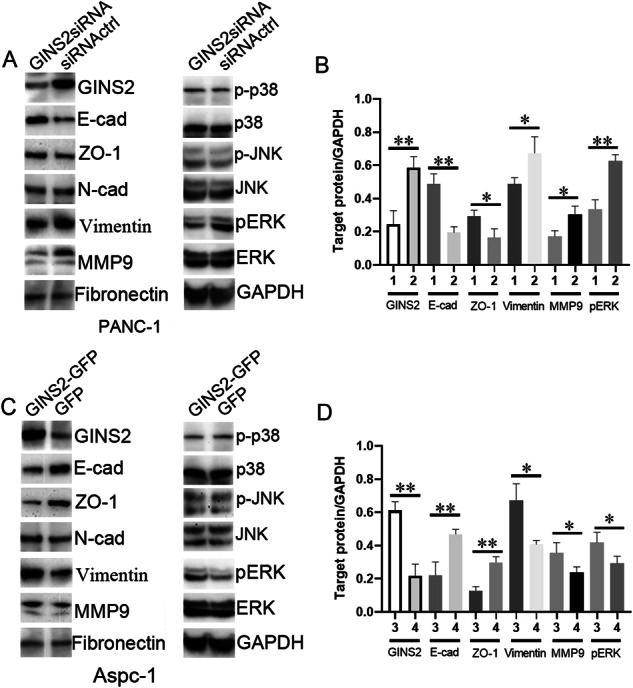
a The protein level of GINS2, E-cad, ZO-1, N-cad, vimentin, MMP9, Fibronectin, p-p38, p-JNK, and pERK in GINS2siRNA- and siRNA-transfected PANC-1 cells. b The quantification of WB data (a). c The protein level of the above proteins in GINS2-GFP- and GFP-transfected Aspc-1 cells. d The quantification of WB data (c). E-cad: E- cadherin; N-cad: N-cadherin. 1: GINS2siRNA; 2: siRNActrl; 3. GINS2-GFP; 4: GFP. Bars indicate ± SE. *P < 0.05; **P < 0.01 in contrast with the control.
GINS2 promoted EMT in vitro via specifically stimulating ERK/MAPK signaling
PD98059 is a highly selective inhibitor of MEK1 (also named MAPK or ERK kinases) [21]. GINS2 overexpression induced EMT-like cellular morphology in Aspc-1 cells: the majority of cells (70–80%) lacked of their epithelial characters (tight and adhesive junction) and exhibited a spindle-shaped/fibroblast-like status (Fig. 5a). However, PD98059-reversed GINS2 overexpression stimulated an EMT-like cellular shape. Little spindle-shaped/fibroblast-like cellular morphology (30–45%) was found in GINS2-GFP plus PD98059 group in contrast with GINS2-GFP group (70–80%) (Fig. 5a). A similar experiment was also repeated in GINS2-overexpressing Bxpc-3 cells (Fig. 5b). Taken together, GINS2 overexpression induced EMT-like cellular morphology in vitro via activating ERK/MAPK signaling.
Fig. 5. Cellular morphology (×100 magnification) and IF assays (×200 magnification) in vitro.
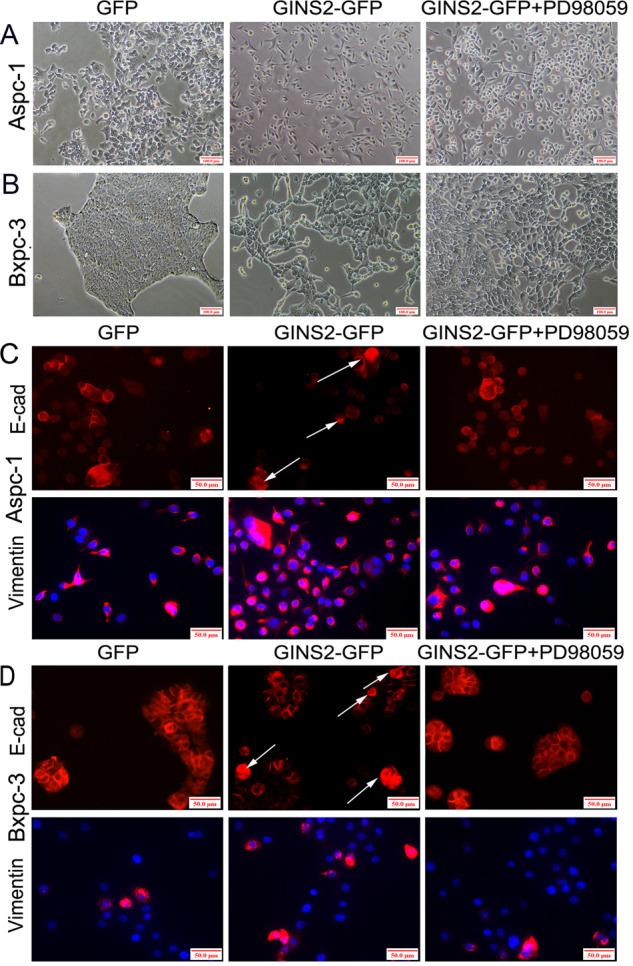
a, b Cellular morphology in GFP, GINS2-GFP, and GINS2-GFP combining PD98059 groups in Aspc-1 (a) and Bxpc-3 cells (b). c, d E-cad and Vimentin staining in GFP, GINS2-GFP, and GINS2-GFP combining PD98059 groups in Aspc-1 (c) and Bxpc-3 cells (d).
We next investigated whether PD98059 inhibited GINS2 overexpression-induced EMT signaling. Immunofluorescence (IF) showed that GINS2 overexpression downregulated E-cad and upregulated Vimentin in Aspc-1 and Bxpc-3 cells, which was reversed by PD98059 (Fig. 5c, d). Meanwhile, certain E-cad cytoplasmic expression was observed in GINS2-GFP in vitro (as arrows shown in Fig. 5c, d) compared with GFP and GINS2-GFP plus PD98059 groups, which was consistent with the results in IHC assays (Fig. 1). WB confirmed that GINS2 overexpression-activated EMT and ERK/MAPK signaling in Aspc-1 cells was significantly inhibited by PD98059 (Fig. 6a, b). Moreover, GINS2 was co-immunoprecipitated and colocalized with ERK in GINS2 high-expression Miapaca-2 and PANC-1 cells by IP (Fig. 6c) and IF (Fig. 6d), respectively. Therefore, a tight interaction of GINS2 with ERK/MAPK pathway coordinately mediated EMT signaling in vitro.
Fig. 6. PD98059 inhibited GINS2 overexpression-induced EMT signal pathway.
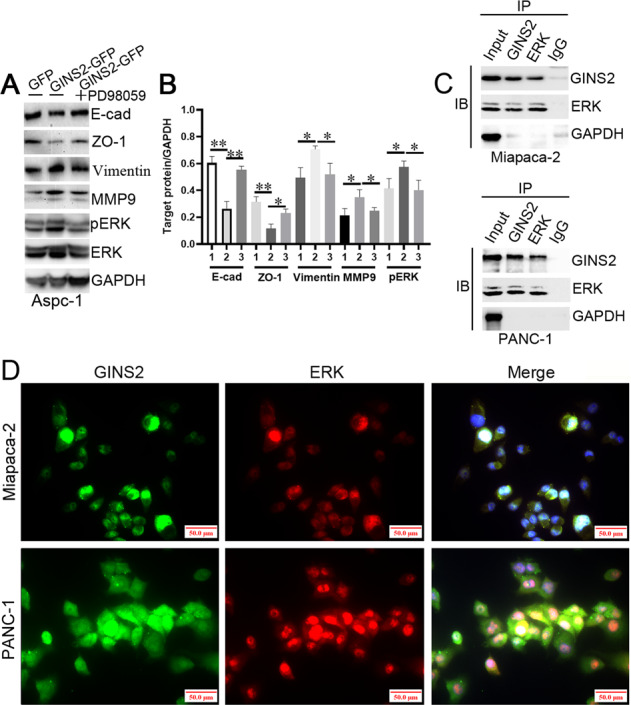
a The protein level of E-cad, ZO-1, vimentin, MMP9, and pERK in GFP, GINS2-GFP, and GINS2-GFP combining PD98059 groups. b The quantification of WB data (a). c GINS2 was co-immunoprecipitated with ERK in Miapaca-2 and PANC-1 cells. d GINS2 was colocalized with ERK in Miapaca-2 and PANC-1 cells by IF. 1: GFP, 2: GINS2-GFP, 3: GINS2-GFP plus PD98059. Bars indicate ± SE. *P < 0.05; **P < 0.01 in contrast with the control.
EMT is a critical stimulator in cell motility and metastasis, and finally aggregates the advanced stages of cancer [22, 23]. In the present study, cell invasion and migration were obviously decreased in GINS2siRNA group in contrast with siRNActrl group in PANC-1 cells (Fig. 7a). Conversely, GINS2 overexpression promoted cell motility in Ascp-1 cells (Fig. 7b). However, PD98059 significantly inhibited GINS2 overexpression-enhanced cell motility in vitro (Fig. 7b). Therefore, GINS2 promoted cell motility via activating ERK/MAPK signal.
Fig. 7. PD98059 inhibited GINS2 overexpression-enhanced cell motility in vitro.
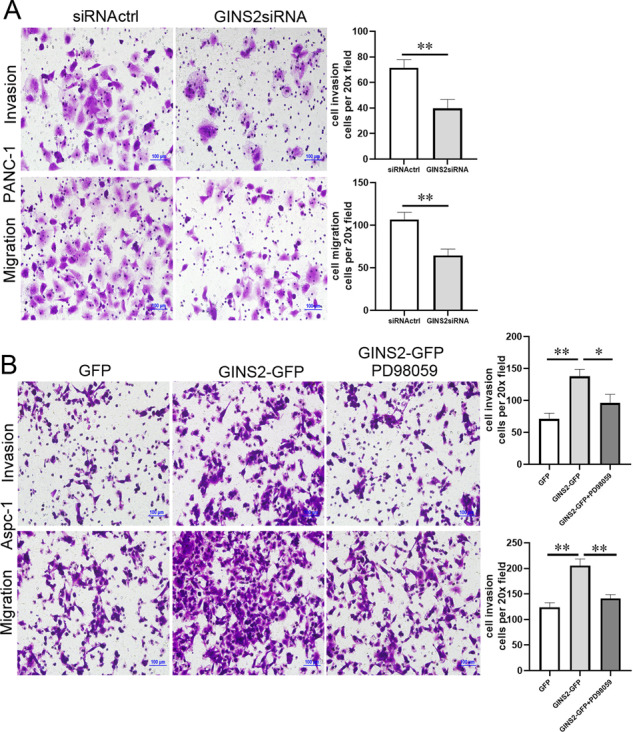
a Cell invasion and migration in siRNActrl and GINS2siRNA groups in PANC-1 cells. b Cell invasion and migration in GFP, GINS2-GFP, and GINS2-GFP combine PD98059 groups in Aspc-1 cells. Bars indicate ± SE. *P < 0.05; **P < 0.01 in contrast with the control.
MEK inhibitor-inhibited GINS2 overexpression promoted liver metastasis in vivo
The liver metastatic number in GINS2-GFP group was significantly increased in contrast with GFP groups (P = 0.001). However, U0126, another MEK inhibitor that was prevalently used in vivo compared with PD98059, significantly inhibited GINS2 overexpression-promoted liver metastasis (Fig. 8a, b). A large serial area of liver metastasis in GINS2-GFP group was also shown in HE staining in contrast with GFP and GINS2-GFP plus U0126 groups (Fig. 8a). IHC further showed that GINS2 overexpression promoted pERK and Vimentin, but inhibited E-cad expression in contrast with the scramble GFP group (Fig. 8c–e), which was reversed by U0126 (Fig. 8c, e, f). Consistent with the results in human PC tissues and cell lines, E-cad abnormal (cytoplasm) expression was prevalently observed in GINS2-GFP group, whereas it exhibited normal (membrane) expression in GFP and GINS2 + U0126 groups. Taken together, MEK inhibitor inhibited GINS2 overexpression-promoted liver metastasis in vivo.
Fig. 8. MEK inhibitor-inhibited GINS2 overexpression promoted liver metastasis in vivo.

a Liver metastases and HE staining (×100 magnification) in GFP, GINS2-GFP, and GINS2-GFP + U0126 groups. b The statistical analysis of liver metastatic number between GFP, GINS2-GFP, and GINS2-GFP + U0126 groups. c The statistic results in IHC assays. d–f The representative images of target proteins expression in GFP (d), GINS2-GFP (e), and GINS2-GFP + U0126 (f) groups in vivo. Bars indicate ± SE. *P < 0.05; **P < 0.01 in contrast with the control.
Discussion
GINS2, as a novel oncogene, is poorly understood in cancer, especially in PC. The current study offers convincing evidence that overexpression of GINS2 contributes to advanced clinical stage of PC patients in coordination with EMT signaling. Meanwhile, GINS2 promotes EMT in PC in vitro and vivo via specifically activating ERK/MAPK signal, which has not been studied yet, to our knowledge.
Overexpression of GINS2 in PC was positively associated with tumor size, T stage, vascular invasion, and bad survival of PC patients, which was consistent with the results of a previous study. For example, overexpression of GINS2 in NSCLC was tightly related with lymph node and distant metastasis, T and clinical stage, and poor prognosis of NSCLC patients [8]. GINS2 overexpression was also associated with multiple features in cervical and breast cancer [9, 10]. Interestingly, a close correlation of GINS2 with E-cad and Vimentin was shown in PC samples, which cooperatively promoted worse prognosis of PC patients. Meanwhile, GINS2 was closely associated with E-cad in four PC cell lines, which are the key biomarkers of EMT signaling [24]. For example, DNMT3A repressed E-cad via DNA methylation involved with EMT-stimulated metastasis in gastric cancer [25]. Gli1 promoted transforming growth factor (TGF)-β1/EGF-induced EMT in PC through inhibiting E-cad [26]. miR-151a induced EMT by targeting E-cad in NSCLC cells [27]. Based on the above study, we next investigated the potential role of GINS2 in EMT in vitro and in vivo.
GINS2 overexpression stimulated EMT in vitro, including promoting EMT-like cellular morphology, enhancing cell motility, and inducing EMT signaling in vitro. Conversely, GINS2 silencing inhibited EMT in PANC-1 cells. The promotion of GINS2 in EMT was only observed in NSCLC previously [8]. It is well known that ERK activation plays a significant role in several critical events of EMT, including promoting the loss of adherent junctions and stimulating the gain of mesenchymal properties [28, 29]. For example, ERK signaling plays a key role in directing the mesenchymal character of NSCLC cells [30]. Meanwhile, ERK is required for the disassembly of cell adherent junctions and the induction of cell motility by TGF-β. Treatment of PANC-1 cells by TGF-β leads to the increased pERK, which is indispensable for the EMT induction by TGF-β [31]. In addition, ERK/MAPK is a central molecule stimulator driving EMT in PC via various signaling. HNRNPA2B1 regulated EMT in PC cells through the ERK-snail pathway [32] CD133 promoted EMT through interacting with the ERK signaling in PC metastasis [33]. Here we first found that GINS2 had a specific regulation targeting ERK/MAPK signaling. MEK1 inhibitor PD98059-reversed GINS2 overexpression-promoted EMT in vitro. Moreover, GINS2 was colocalized and co-immunoprecipitated with ERK in GINS2 high-expression PC cells. Finally, MEK inhibitor inhibited GINS2 overexpression-promoted distant liver metastases and GINS2 overexpression-activated ERK/MAPK and EMT signaling in vivo. Taken together, a tight interaction of GINS2 with ERK/MAPK signaling coordinately regulated EMT in PC. In previous studies, GINS2 acted as an important modulator in apoptosis via the p38/MAPK signal pathway in leukemia cells [34]. GINS2 silencing inhibited DNA replication and G2/M phase in Leukemic HL60 cells [35], and disrupted the transition of glioma cells at the G1/S stage [36]. GINS2 silencing inhibited cell proliferation, tumorigenic ability, and cell motility in early-stage cervical cancer [10], and promoted cell proliferation in thyroid cancer via regulating CITED2 and LOXL2 [11]. For the first time, we reported a novel oncogenic role of GINS2 in PC via mediating EMT and ERK/MAPK signaling, which was not shown previously.
In conclusion, overexpression of GINS2 contributes to advanced clinical stage of PC patients and promotes EMT in vitro and in vivo via specifically activating the ERK/MAPK signal. A tight interaction of GINS2 with ERK-MPAK signaling coordinately promotes EMT in PC, which supplies a novel direction in revealing the malignant biology of PC and offers a promising gene target treatment in PC intervention. EMT and ERK signaling also play a vital role in promoting drug resistance [37, 38]. GINS2 silencing is predicted to induce cell apoptosis and inhibit cell proliferation by decreasing CITED2 and subsequently regulating p53 and p21 axis in thyroid cancer [11]. GINS2 might promote chemotherapy resistance in PC via EMT, ERK, and p53/p21 signaling, which would be investigated in our future study. Meanwhile, EGF, as a stimulus in Kras/Raf/MEK/ERK signaling (a key pathway in PC) [39, 40], plays a significant role in EMT as shown in our previous study [14, 15]. GINS2 might promote EGF-induced EMT via regulating the phosphorylation of EGFR, which would supply a new sight in targeting Kras-driven PC.
Supplementary information
Supplemental Table 1. The target prime sequences
Acknowledgments
Funding
This work was supported by the Chinese National Science Foundation for youth scholar (number 81401941 to W.S.).
Author contributions
W.S. took charge of the study design and concept. Experiments were conducted by L.H. and S.C. H.F. and D.J. finished the statistical data. L.H., C.C., and W.S. was in responsible for the data analysis and explanation. All authors approved the final manuscript and contributed equally to this study.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41417-020-0206-7) contains supplementary material, which is available to authorized users.
References
- 1.Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol. 2020. 10.1038/s41575-019-0245-4. [DOI] [PubMed]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–32. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Rawla P, Sunkara T, Gaduputi V. Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J Oncol. 2019;10:10–27. doi: 10.14740/wjon1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaianigo N, Melisi D, Carbone C. EMT and treatment resistance in pancreatic cancer. Cancers (Basel) 2017;12:9.. doi: 10.3390/cancers9090122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroni M, De March M, Medagli B, Krastanova I, Taylor IA, Amenitsch H, et al. New insights into the GINS complex explain the controversy between existing structural models. Sci Rep. 2017;10:40188. doi: 10.1038/srep40188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubota Y, Takase Y, Komori Y, Hashimoto Y, Arata T, Kamimura Y, et al. A novel ring-like complex of Xenopus proteins essential for the initiation of DNA replication. Genes Dev. 2003;17:1141–52. doi: 10.1101/gad.1070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacNeill SA. Structure and function of the GINS complex, a key component of the eukaryotic replisome. Biochem J. 2010;425:489–500. doi: 10.1042/BJ20091531. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Sun L, Zhang S, Zhang S, Li W. GINS2 facilitates epithelial-to-mesenchymal transition in non-small-cell lung cancer through modulating PI3K/Akt and MEK/ERK signaling. J Cell Physiol. 2019. 10.1002/jcp.29381. [DOI] [PubMed]
- 9.Peng L, Song Z, Chen D, Linghu R, Wang Y, Zhang X, et al. GINS2 regulates matrix metallopeptidase 9 expression and cancer stem cell property in human triple negative Breast cancer. Biomed Pharmacother. 2016;84:1568–74. doi: 10.1016/j.biopha.2016.10.032. [DOI] [PubMed] [Google Scholar]
- 10.Ouyang F, Liu J, Xia M, Lin C, Wu X, Ye L, et al. GINS2 is a novel prognostic biomarker and promotes tumor progression in early-stage cervical cancer. Oncol Rep. 2017;37:2652–62. doi: 10.3892/or.2017.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye Y, Song YN, He SF, Zhuang JH, Wang GY, Xia W. GINS2 promotes cell proliferation and inhibits cell apoptosis in thyroid cancer by regulating CITED2 and LOXL2. Cancer Gene Ther. 2019;26:103–13. doi: 10.1038/s41417-018-0045-y. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35:600–4. doi: 10.3109/10799893.2015.1030412. [DOI] [PubMed] [Google Scholar]
- 13.Salaroglio IC, Mungo E, Gazzano E, Kopecka J, Riganti C. ERK is a pivotal player of chemo-immune-resistance in cancer. Int J Mol Sci. 2019;20:2505. [DOI] [PMC free article] [PubMed]
- 14.Sheng W, Chen C, Dong M, Wang G, Zhou J, Song H, et al. Calreticulin promotes EGF-induced EMT in pancreatic cancer cells via Integrin/EGFR-ERK/MAPK signaling pathway. Cell Death Dis. 2017;8:e3147. doi: 10.1038/cddis.2017.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheng W, Shi X, Lin Y, Tang J, Jia C, Cao R, et al. Musashi2 promotes EGF-induced EMT in pancreatic cancer via ZEB1-ERK/MAPK signaling. J Exp Clin Cancer Res. 2020;39:16. doi: 10.1186/s13046-020-1521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L, Chen S, Fan H, Ai F, Sheng W. BZW2 promotes the malignant progression of colorectal cancer via activating the ERK/MAPK pathway. J Cell Physiol. 2019. 10.1002/jcp.29361. [DOI] [PubMed]
- 17.Flores LG, Yeh HH, Soghomonyan S, Young D, Bankson J, Hu Q, et al. Monitoring therapy with MEK inhibitor U0126 in a novel Wilms tumor model in Wt1 knockout Igf2 transgenic mice using 18F-FDG PET with dual-contrast enhanced CT and MRI: early metabolic response without inhibition of tumor growth. Mol Imaging Biol. 2013;15:175–85. doi: 10.1007/s11307-012-0588-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marampon F, Bossi G, Ciccarelli C, Di Rocco A, Sacchi A, Pestell RG, et al. MEK/ERK inhibitor U0126 affects in vitro and in vivo growth of embryonal rhabdomyosarcoma. Mol Cancer Ther. 2009;8:543–51. doi: 10.1158/1535-7163.MCT-08-0570. [DOI] [PubMed] [Google Scholar]
- 19.Marampon F, Gravina GL, Di Rocco A, Bonfili P, Di Staso M, Fardella C, et al. MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by downregulating growth and DNA repair signals. Mol Cancer Ther. 2011;10:159–68. doi: 10.1158/1535-7163.MCT-10-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venhuizen JH, Jacobs FJC, Span PN, Zegers MM. P120 and E-cadherin: double-edged swords in tumor metastasis. Semin Cancer Biol. 2019;S1044-579X:30106–3. doi: 10.1016/j.semcancer.2019.07.020. [DOI] [PubMed] [Google Scholar]
- 21.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–32. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 22.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Song Y, Ye M, Zhou J, Wang Z, Zhu X. Targeting E-cadherin expression with small molecules for digestive cancer treatment. Am J Transl Res. 2019;11:3932–44. [PMC free article] [PubMed] [Google Scholar]
- 25.Cui H, Hu Y, Guo D, Zhang A, Gu Y, Zhang S, et al. DNA methyltransferase 3A isoform b contributes to repressing E-cadherin through cooperation of DNA methylation and H3K27/H3K9 methylation in EMT-related metastasis of gastric cancer. Oncogene. 2018;37:4358–71. doi: 10.1038/s41388-018-0285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Q, Sheng W, Dong M, Dong X, Dong Q, Li F. Gli1 promotes transforming growth factor-beta1- and epidermal growth factor-induced epithelial to mesenchymal transition in pancreatic cancer cells. Surgery. 2015;158:211–24. doi: 10.1016/j.surg.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Daugaard I, Sanders KJ, Idica A, Vittayarukskul K, Hamdorf M, Krog JD, et al. miR-151a induces partial EMT by regulating E-cadherin in NSCLC cells. Oncogenesis. 2017;6:e366. doi: 10.1038/oncsis.2017.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santamaria PG, Nebreda AR. Deconstructing ERK signaling in tumorigenesis. Mol Cell. 2010;38(no. 1):3–5. doi: 10.1016/j.molcel.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 29.Zuo JH, Zhu W, Li MY, et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem. 2011;112(no. 9):2508–17. doi: 10.1002/jcb.23175. [DOI] [PubMed] [Google Scholar]
- 30.Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74:309–19. doi: 10.1158/0008-5472.CAN-12-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zavadil J, Bitzer M, Liang D, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc Natl Acad Sci USA. 2011;98(no. 12):6686–91. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai S, Zhang J, Huang S, Lou B, Fang B, Ye T, et al. HNRNPA2B1 regulates the epithelial-mesenchymal transition in pancreatic cancer cells through the ERK/snail signalling pathway. Cancer Cell Int. 2017;17:12. doi: 10.1186/s12935-016-0368-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding Q, Miyazaki Y, Tsukasa K, Matsubara S, Yoshimitsu M, Takao S. CD133 facilitates epithelial-mesenchymal transition through interaction with the ERK pathway in pancreatic cancer metastasis. Mol Cancer. 2014;13:15. doi: 10.1186/1476-4598-13-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Y, Wang S, Liu B, Zhong L. Roles of GINS2 in K562 human chronic myelogenous leukemia and NB4 acute promyelocytic leukemia cells. Int J Mol Med. 2013;31:1402–10. doi: 10.3892/ijmm.2013.1339. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Zhong L, Liu BZ, Gao YJ, Gao YM, Hu XX. Effect of GINS2 on proliferation and apoptosis in leukemic cell line. Int J Med Sci. 2013;10:1795–804. doi: 10.7150/ijms.7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen YL, Li HZ, Hu YW, Zheng L, Wang Q. Loss of GINS2 inhibits cell proliferation and tumorigenesis in human gliomas. CNS Neurosci Ther. 2019;25:273–87. doi: 10.1111/cns.13064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du B, Shim JS. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21:965. doi: 10.3390/molecules21070965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abrams SL, Steelman LS, Shelton JG, Wong EW, Chappell WH, Bäsecke J, et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle. 2010;9:1781–91. doi: 10.4161/cc.9.9.11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014;13:928–42. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 40.Yang K, Li Y, Lian G, Lin H, Shang C, Zeng L, et al. KRAS promotes tumor metastasis and chemoresistance by repressing RKIP via the MAPK-ERK pathway in pancreatic cancer. Int J Cancer. 2018;142:2323–34. doi: 10.1002/ijc.31248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. The target prime sequences