

Abstract
A series of combretastatin A-4 (CA-4) sulfamate derivatives were synthesized and their structure–activity relationship on tubulin, arylsulfatase and tumor cell antiproliferation inhibition was studied. Among them, compound 16a showed excellent potency as well as CA-4 under the same conditions against six tumor cells including HTC-116, HeLa, HepG2, MGC803, MKN45 and MCF-7 cells, respectively. Molecular docking revealed that several important hydrogen bond interactions were formed between the sulfamate group of 16a and the colchicine binding site of tubulin and steroid sulfatase respectively. Although compound 16a was less active than CA-4 in regard to its in vitro activity as an inhibitor of tubulin polymerization, it was effective as an inhibitor of arylsulfatase. This novel combretastatin A-4 sulfamate derivative has the potential to be developed as a dual inhibitor of tubulin polymerization and arylsulfatase for cancer therapy.
A series of combretastatin A-4 (CA-4) sulfamate derivatives were synthesized and their structure–activity relationship on tubulin, arylsulfatase and tumor cell antiproliferation inhibition was studied.
The occurrence and development of cancer is a complex and long process, and cancer is one of the major causes for the increase in world mortality.1–3 In particular, breast cancer is the most common cancer with high incidence rates in women, making up about one third of global cancer diagnoses for women, and it is one of the major health problems in the world.4–7 In the past few decades, great progress was made in molecular-targeted anti-tumor drug research.8 Tubulin and arylsulfatase are promising targets in cancer therapy. Tubulin is the fundamental unit of microtubules in the cell, and it plays a critical role in maintaining the shape, movement and intracellular material transportation in the cell. Tubulin targeted inhibitors are effective in the treatment of cancer.9 There are three major binding pockets in tubulin, which were named after three different drugs, paclitaxel, vincristine and colchicine, respectively. Tubulin inhibitors binding to these pockets can arrest tumor cell mitosis and induce apoptosis.9,10 Paclitaxel and vincristine are effective chemotherapy drugs widely used in the clinic, and some marketed drugs were developed targeting the paclitaxel and vincristine binding sites.10 However, colchicine was not used in treating cancer due to its high toxicity, and studies on inhibitors targeting colchicine binding sites are one of the hotspots in antineoplastic drug research.11
Arylsulfatases are one kind of a protein family in charge of the hydrolysis of sulfonates, and they play a significant role in the hormonal regulation in cells, the degradation of cell components and signaling pathway regulation.12 The abnormal activity of arylsulfatases in the body is closely related to the growth of tumors. Currently, there are four extensively studied arylsulfatases, namely lysosomal arylsulfatases (ARS-A and ARS-B), endoplasmic reticulum arylsulfatase (ARS-C) and extracellular arylsulfatase (Hsulf-1). In the clinic, the activity of ARS-A and ARS-B has become an important indicator in tumor diagnosis as the ARS activity in urine would significantly increase in all types of tumor patients.13 ARS-C, also known as steroid sulfatase (STS), is majorly responsible for adjusting the balance of hormone levels (Scheme 1) by hydrolysis of estrone sulfate (E1S) and dehydroepiandrosterone sulfate (DHE-AS) in order to release estrone and dehydroepiandrosterone (DHEA). The abnormal state of STS will imbalance the hormone levels in the body as well as promote the growth and proliferation of cancer cells. Steroid sulfatase (STS) inhibitors are an effective therapy for the treatment of hormone dependent cancer such as breast and cervical cancer by inhibiting the hydrolysis of E1S.14 In the past few years, a lot of steroid sulfatase inhibitors (1–5, Fig. 1), including stilbene compounds, have been developed and they exhibit good inhibitory activity against breast cancer cells via inhibiting steroid sulfatase.15 The sulfamate group is an essential pharmacophore for potency, and estrone-3-O-sulfamate (EMATE, 1), the sulfamate derivative of estrone, is a typical representative of this kind of inhibitor.16
Scheme 1. The role of steroid sulfatase in hormone regulation.

Fig. 1. Chemical structures of 1–12.

Combretastatin A-4 (CA-4, 6a) and combretastatin A-1 (CA-1, 7) are cis-stilbene type natural products originally isolated from the South African bushwillow tree Combretum caffrum. They inhibit tubulin polymerization by interacting with the colchicine-binding sites of tubulin, thus arresting the mitosis of tumor cells and inducing their apoptosis. In addition, CA-4 disrupts tumor vasculaures at a nontoxic dose resulting in tumor cells starving to death without affecting the healthy cell's blood supply.17 Efforts directed toward the discovery of more potent and selective vascular disrupting agents continue on a global basis.18 In recent years, a large number of structure modifications were made to find more potent CA-4 analogues,19,20 and among them, AVE-8062A (10) and CA-4P (11) have entered phase II/III clinical trials (Fig. 1).20 Structure–activity relationship studies indicate that the cis-conformation is crucial for potency. Our groups also have made a lot of progress in finding more potent CA-4 analogues with improved properties by using the twin drug, prodrug and fluorine modification strategies.21
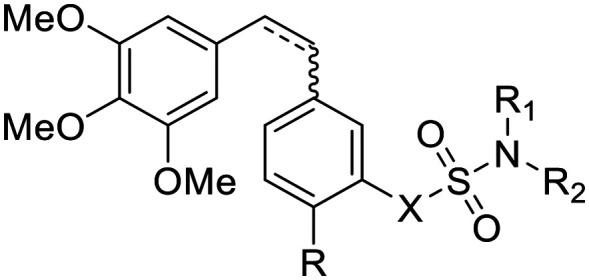
In addition to the strong arylsulfatase inhibition, the anti-tumor mechanism of EMATE is also related to its weak tubulin inhibition activity (IC50 = 25.90 μM, Table 2). 2-Methoxyestradiol (2-ME), an endogenous metabolite of 17-estradiol, is also a weak tubulin inhibitor (IC50 ≈ 40 μM) and it can inhibit microtubule assembly and induce G2/M arrest and apoptosis in many actively dividing cell types while sparing quiescent cells.22 2-ME can also bind to the colchicine site of tubulin. Therefore, it has potent antineoplastic activity as an apoptosis inducer and an angiogenesis inhibitor. However, 2-ME is ineffective for arylsulfatase inhibition as compared to EMATE.22 The sulfamide group is a widely used pharmacophore in medicinal chemistry as it possess both hydrogen bond donors and acceptors. There are many reports about sulfamate modification of other molecular structures as a potent dual inhibitor of carbonic anhydrase and steroid sulfatase.23 Inspired by the structure and the difference of activity in tubulin and sulfatase inhibition between EMATE and 2-ME, we speculated that sulfamate modification of CA-4 would lead to the foundation of effective dual inhibitors of both tubulin and steroid sulfatase. We hope to obtain a drug candidate with better potency, stability and pharmacokinetic profiles than CA-4 through the sulfamate derivatives of CA-4 and further structure–activity relationship (SAR) studies (Fig. 2). Herein, we reported the synthesis and biological evaluation of novel combretastatin analogues, while also discussing the molecular docking study and the SAR analysis.
The in vitro inhibitory activities of combretastatin A-4 sulfamate derivatives against arylsulfatase and tubulin.
| Entry | R1 | R2(R3)N | X | IC50 ± SD (μM) | |
|---|---|---|---|---|---|
| Arylsulfataseb | Tubulinc | ||||
| 16a (Z) | OMe | NH2 | O | 6.16 ± 0.55 | 6.60 ± 0.80 |
| 16b (E) | OMe | NH2 | O | 12.86 ± 0.25 | 55.70 ± 6.00 |
| 16c (Z) | OCF2H | NH2 | O | 4.64 ± 1.42 | 72.60 ± 10.7 |
| 16e (Z) | OEt | NH2 | O | 90.19 ± 7.83 | 3.10 ± 1.10 |
| 16g (Z) | H | NH2 | O | 0.47 ± 0.01 | >100 |
| 16i (Z) | OMe | NHMe | O | >100 | 1.80 ± 0.00 |
| 16j (Z) | OMe | N(Me)2 | O | >100 | 12.50 ± 1.50 |
| 17a a | OMe | NH2 | O | >100 | >100 |
| 19a (Z) | OMe | NH2 | NH | >100 | 86.20 ± 6.40 |
| EMATE | — | — | — | 5.01 ± 0.01 | 25.90 ± 7.10 |
| CA-4 | — | — | — | >100 | 1.00 ± 0.20 |
Saturated bond linked compound.
Steroid sulfatase.
Tubulin polymerization.
Fig. 2. The design of sulfamate derivatives of CA-4.

Firstly, compound 16a, the sulfamate modified CA-4 (6a), was synthesized and its antiproliferation activity was compared with that of CA-4 as the positive control. To our delight, compound 16a showed excellent anti-tumor activity with respect to CA-4 under the same conditions. Next, we designed a number of combretastatin A-4 sulfamate and sulfamide derivatives to explore their structure–activity relationship. The synthetic procedures are outlined in Schemes 2 and 3. The aldehyde intermediate (13) was purchased or prepared following a reported protocol.21b The hydroxyl group of 13 was protected with trityl to give 14. A (Z/E)-stilbene mixture was obtained from the aldehyde intermediates (14 or 13e, in Schemes 2 and 3) and triphenyl(3,4,5-trimethoxybenzyl)phosphonium bromide (15) via the Wittig reaction, followed by deprotection of the trityl group or reduction of the nitro group to give 3-hydroxyl or 3-amino substituted (on the B ring) stilbenes (6 and 9, Schemes 2 and 3). Their Z and E isomers were separated by flash column chromatography. Compounds 6 and 9 were further reacted with sulfamoyl chloride respectively to achieve the corresponding sulfamate (16) and sulfamide (19) derivatives of stilbene. The diphenylethane sulfamate and sulfamide derivatives (17 and 20) were obtained from the Pd/C catalyzed hydrogenation of 16 and 19.
Scheme 2. Synthetic route of combretastatin A-4 sulfamate derivatives. Reagents and conditions: a) Ph3CCl(TrCl), THF, Et3N, rt, 2 h; b) n-BuLi, THF, −78 °C, 30 min, then adding 14/THF solution, −78 °C–RT, overnight; c) 37% HCl (aq.), toluene, rt, 2 h; d) NaH, DMF, RSO2Cl (R = NH2, NHMe, N(Me)2), 0 °C to RT, overnight; e) H2 (1 atm), 10% Pd/C, EtOH, rt, 3 h.

Scheme 3. Synthetic route of combretastatin sulfamide derivatives. Reagents and conditions: a) n-BuLi, THF, −78 °C, 30 min, then adding 13e/THF solution, −78 °C–rt, overnight; b) Zn, CH3COOH, 2 h, rt; c) NaH, DMF, NH2SO2Cl, 0 °C–rt, overnight; d) H2 (1 atm), 10% Pd/C, EtOH, rt, 3 h.

The in vitro antiproliferative activity of these compounds were evaluated via the CCK-8 assay by using CA-4 and EMATE as the positive control. Six human tumor cell lines, including the HTC-116, HeLa, HepG2, MGC803, MKN45 and MCF-7, were tested. The results are summarized in Table 1. Among all the analogues, only compound 16i, the monomethylated 16a, showed comparable potency (IC50 = 3.4–11.3 nM) to 16a (IC50 = 3.6–9.5 nM) and CA-4 (IC50 = 2.9–8.1 nM) on the six tumor cell lines, except that its inhibitory activity (IC50 = 0.53 μM) on MCF-7 cells is slightly less potent than that of 16a (IC50 = 0.11 μM) and CA-4 (IC50 = 0.14 μM). As for compound 16j, the dimethyl substituted 16a, its potency was reduced although it showed stronger potency on HTC-116 cells (IC50 = 0.37 μM) than on the other five tumor cell lines (IC50 = 7.14–14.26 μM). The replacement of sulfamate with sulfamide (19a) also led to a potency decrease compared with 16a. 19a showed better potency (IC50 = 0.27, 0.75, 0.50 μM) on HeLa, HepG2, and MGC803 than on the other three cell lines (HCT-116, MKN45 and MCF-7, IC50 = 3.29, 9.56, 11.19 μM). The electronic and steric effects of R-substitution also has a great influence on potency. When the methoxyl group of 16a was replaced with a hydrogen (16g, R = H), difluoromethoxy (16c, R = OCF2H) or ethoxy (16e, R = OEt) group, their cellular potency decreased compared with that of 16a and CA-4. In addition, it was observed that the cis-double bond linked compounds (16a, 16c, 16e, 16g and 19a) showed better cellular potency than the trans-double bond (16b, 16d, 16f, 16h and 19e) and saturated bond linked compounds (17a–17d, 20). A CA-4 sulfate derivative (CA4S, Fig. 2) was identified as a phase II metabolite of CA4.24 We have also synthesized it as a control; however, it is ineffective in tumor cell cytotoxicity assays (IC50 > 100 μM).
The in vitro antiproliferation activities of combretastatin A-4 sulfamate and sulfamide derivatives against on six human tumor cellsa.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compounds | IC50 (mean ± SD, μM)a | ||||||||
| Entry | R | R1(R2)N | X | HTC-116 | HeLa | HepG2 | MGC803 | MKN45 | MCF-7 |
| CA-4 | — | — | — | 0.0047 ± 0.0008 | 0.0029 ± 0.0001 | 0.0029 ± 0.0007 | 0.0060 ± 0.0006 | 0.0081 ± 0.0004 | 0.14 ± 0.02 |
| EMATE | — | — | — | 0.90 ± 0.04 | 0.63 ± 0.06 | 0.85 ± 0.05 | 0.48 ± 0.09 | 0.54 ± 0.12 | 0.07 ± 0.10 |
| 16a (Z) | OMe | NH2 | O | 0.0050 ± 0.0005 | 0.0061 ± 0.0003 | 0.0036 ± 0.0002 | 0.0047 ± 0.0006 | 0.0095 ± 0.0004 | 0.11 ± 0.04 |
| 16b (E) | OMe | NH2 | O | 1.57 ± 0.07 | 4.50 ± 0.07 | 0.87 ± 0.09 | 2.98 ± 0.07 | 10.52 ± 0.04 | 5.44 ± 0.05 |
| 16c (Z) | OCF2H | NH2 | O | 9.11 ± 0.04 | 7.82 ± 0.05 | 4.81 ± 0.06 | 9.00 ± 0.05 | 12.52 ± 0.04 | 0.29 ± 0.05 |
| 16d (E) | OCF2H | NH2 | O | 12.84 ± 0.03 | 8.42 ± 0.04 | 5.54 ± 0.05 | 9.24 ± 0.04 | 13.84 ± 0.04 | 13.79 ± 0.03 |
| 16e (Z) | OEt | NH2 | O | 0.55 ± 0.07 | 0.48 ± 0.08 | 0.41 ± 0.05 | 0.27 ± 0.07 | 0.32 ± 0.07 | 0.30 ± 0.06 |
| 16f (E) | OEt | NH2 | O | 9.12 ± 0.04 | 5.64 ± 0.05 | 2.63 ± 0.07 | 7.63 ± 0.05 | 8.05 ± 0.05 | 13.71 ± 0.03 |
| 16g (Z) | H | NH2 | O | 9.50 ± 0.03 | 16.30 ± 0.03 | 9.36 ± 0.05 | 8.53 ± 0.04 | 15.32 ± 0.02 | 11.10 ± 0.04 |
| 16h (E) | H | NH2 | O | 40.30 ± 0.04 | 19.00 ± 0.03 | 11.85 ± 0.03 | 27.03 ± 0.02 | 25.04 ± 0.02 | 27.44 ± 0.02 |
| 16i (Z) | OMe | NHMe | O | 0.0113 ± 0.0003 | 0.0034 ± 0.0005 | 0.0039 ± 0.0010 | 0.0037 ± 0.0006 | 0.0073 ± 0.0003 | 0.53 ± 0.03 |
| 16j (Z) | OMe | N(Me)2 | O | 0.37 ± 0.07 | 14.26 ± 0.04 | 9.96 ± 0.03 | 7.14 ± 0.03 | 8.95 ± 0.37 | 11.06 ± 0.04 |
| 19a (Z) | OMe | NH2 | NH | 3.29 ± 0.77 | 0.27 ± 0.06 | 0.75 ± 0.06 | 0.50 ± 0.07 | 9.56 ± 0.09 | 11.19 ± 0.06 |
| 19b (E) | OMe | NH2 | NH | 8.65 ± 0.04 | 0.38 ± 0.05 | 7.53 ± 0.06 | 6.19 ± 0.06 | 18.87 ± 0.03 | 17.09 ± 0.04 |
| 17a b | OMe | NH2 | O | 2.63 ± 0.08 | 9.04 ± 0.04 | 1.06 ± 0.05 | 3.61 ± 0.08 | 11.87 ± 0.04 | 5.91 ± 0.06 |
| 17a b | OCF2H | NH2 | O | 26.48 ± 0.02 | 35.65 ± 0.01 | 15.88 ± 0.02 | 23.26 ± 0.02 | 71.38 ± 0.02 | 52.74 ± 0.03 |
| 17c b | OEt | NH2 | O | 10.16 ± 0.03 | 2.930 ± 0.07 | 3.65 ± 0.07 | 3.59 ± 0.06 | 7.67 ± 0.06 | 14.75 ± 0.02 |
| 17d b | H | NH2 | O | 52.49 ± 0.02 | 27.06 ± 0.03 | 12.03 ± 0.03 | 17.64 ± 0.04 | 25.70 ± 0.03 | >100 |
| 20 b | OMe | NH2 | NH | 30.63 ± 0.04 | 2.926 ± 0.07 | 8.10 ± 0.05 | 8.21 ± 0.04 | 73.88 ± 0.05 | 20.13 ± 0.03 |
The experiment was carried out using an EnoGeneCellTM counting Kit-8 (CCK-8) assay.
Saturated bond linked compound.
In order to verify the effectivity of these novel sulfamate derivatives of CA-4 on both arylsulfatase and tubulin, we tested their inhibitory activity on steroid sulfatase and tubulin polymerization with EMATE and CA-4 as the control, and the results are summarized in Table 2. The half inhibition rate (IC50) of EMATE on steroid sulfatase and tubulin polymerization is 5.01 μM and 25.90 μM, respectively. The test inhibitory potency of combretastatin A-4 on tubulin (IC50 = 1.0 μM) is consistent with literature reports; however, it is ineffective on steroid sulfatase. When the hydroxyl of CA-4 was modified by sulfamide (16a), it showed a slightly lower potency (IC50 = 6.60 μM) on tubulin polymerization inhibition than CA-4 together with a comparable potency (IC50 = 6.16 uM) on steroid sulfatase to that of EMATE. By comparing the anti-proliferation activities and the relationship of the steroid sulfatase and tubulin inhibition potencies of CA-4 and 16a, we can conclude that 16a is a dual inhibitor, with the additional steroid sulfatase inhibition compensating for the lost contribution of tubulin polymerization inhibition to its anti-tumor activity. From the SAR analysis (described in the ESI†) based on arylsulfatase and tubulin polymerization inhibition (Table 2), we can conclude the following: 1) the cis-isomers show better activity for arylsulfatase and tubulin polymerization inhibition than the trans-isomers and saturated bond linked analogues. 2) The electronic and steric effects of R-groups are different on the two targets. Electron withdrawing groups are favorable for arylsulfatase inhibition but are unfavorable for tubulin inhibition at the same time. Meanwhile, bulky groups are favorable for tubulin inhibition but unfavorable for arylsulfatase inhibition. 3) The amino group of 16a probably has a critical hydrogen bond interaction with the binding site of both targets. By comparing with 16a, monomethyl substitution increased the potency while dimethyl substitution decreased the potency for tubulin polymerization inhibition. However, the substitution of the amino group of 16a will lead to the loss of potency on arylsulfatase. 4) Replacing the sulfamate group with sulfamide decreases the potency on both targets. 5) Tubulin inhibition plays a dominant role over arylsulfatase inhibition in the anti-tumor activity in this kind of dual inhibition.
Compound (16a) was further selected for molecular docking studies in order to explore how the CA-4 sulfamate derivatives interact with tubulin and steroid sulfatase. The protein complexes tubulin (PDB: 5LYJ)24 and human placental estrone/DHEA sulfatase (PDB: 1P49)25 were used for simulation in Sybyl-X 2.0. The force field was Tripos with an 8 Å cutoff for non-bonded interactions, and the atomic point charges were also calculated with the Gasteiger–Huckel method. Minimization was achieved using the steepest descent method for the first 100 steps, followed by the Broyden–Fletcher–Goldfarb–Shanno (BFGS) method until the root-mean-square (RMS) of the gradient became <0.005 kcal/(mol Å). The Surflex-Dock module implemented in the Sybyl program was used for the docking study. The colchicine-binding site of tubulin was used and verified from the original ligand (CA-4) for docking (total score = 8.34, similarity = 0.73). The docking score and similarity of 16a is 6.27 and 0.45, respectively. As shown in Fig. 3, the A/B ring binding mode of 16a is almost identical to that of CA-4, both having van der Waals interaction with the same hydrophobic pocket formed by the amino acid residues VAL238, CYS241, LEU248, ALA250, LEU255, ALA316 and LYS352. The hydroxyl group of CA-4 formed a hydrogen bond with THR179, while the sulfamide group of 16a formed two hydrogen bonds with ALA180 and ASN349.
Fig. 3. The binding information of CA-4 in the co-crystal structure (A, PDB code: 5LYJ) and the docking result of 16a (B).

Both EMATE and 16a were docked into the same binding pocket of steroid sulfatase, and their docking scores were 6.16 and 5.34, respectively. As shown in Fig. 4, the hydrophobic part of EMATE and 16a were superimposable, having van der Waals interaction with the hydrophobic pocket formed by the residues VAL101, LEU103, VAL486, HIS485, PHE488, etc. The sulfamate group of 16a formed four important hydrogen bonds with the residues THR165, LYS134, ASP36 and LYS368 in the binding pocket. As for EMATE, there are six hydrogen bonds between the sulfamate group and the residues THR76, THR165, LYS134, ASP342 and ASP36. These important hydrogen bonding interactions explain why 16a is less potent than EMATE, and the methylation of the sulfamate group (16i and 16j) leads to the loss of potency for steroid sulfatase inhibition.
Fig. 4. The docking result of EMATE (A) and 16a (B) with steroid sulfatase (PDB code: 1P49).

In summary, we have synthesized a few novel combretastatin sulfamate derivatives, and evaluated their activities on steroid sulfatase, tubulin and tumor cell proliferation inhibition. Among them, compound 16a has a well-balanced sulfatase, tubulin and cellular inhibition potency. The SAR analysis and molecular docking study indicate that the sulfamate group is crucial for sulfatase inhibition and helpful to form additional hydrogen bonds with the colchicine-binding site of tubulin. These results highlighted that compound 16a is a promising anticancer agent, and may be valuable in finding more effective treatments for cancer. To the best of our knowledge, this is the first sulfamate analogue of CA-4. Further biological investigation is being conducted and the progress will be reported in the future.
Funding sources
National Natural Science Foundation of China; Innovation Program of Shanghai Municipal Education Commission; Shanghai Institute of Technology.
Abbreviations
- STS
Steroid sulfatase
- E1S
Estrone sulfate
- DHE-AS
Dehydroepiandrosterone sulfate
- DHEA
Dehydroepiandro-sterone
- CA-4
Combretastatin A-4
- EMATE
Estrone-3-O-sulfamate
- SAR
Structure–activity relationship
Author contributions
All the authors have given approval to the final version of this manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
Financial support from the National Natural Science Foundation of China (No. 21472126, 21302128, 21672151), the Innovation Program of Shanghai Municipal Education Commission (No. ZZZZyyx16006) and Shanghai Institute of Technology (No. YJ2016-41) is gratefully acknowledged. We are also thankful to Nanjing OGPharmaceutical Co. Ltd., Shanghai Research Institute of Chemical Industry Co. Ltd. and Shandong Huawei Pharmaceutical Co. Ltd. for their help in bioassays.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00372g
References
- Ahmedin J. Freddie B. Melissa M. C. Jacques F. Elizabeth W. David F. Global cancer statistics. Ca-Cancer J. Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Lindsey A. T. Freddie B. Rebecca L. S. Jacques F. Joannie L. T. Ahmedin J. Global cancer statistics, 2012. Ca-Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Glenn C. B. Lee M. E. Drug development: raise standards for preclinical cancer research. Nature. 2012;483:531–533. doi: 10.1038/483531a. [DOI] [PubMed] [Google Scholar]
- Jacques F. Isabelle S. Rajesh D. Sultan E. Colin M. Marise R. Donald M. P. David F. Freddie B. Cancer incidence and mortality worldwide: sources, methods and major patterns in Globocan 2012. Int. J. Cancer. 2015;136:e359–e386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- Nagi S. E. S. Firas Y. K. Sarsh E. B. Benjamin O. A. Management of locally advanced and metastatic breast cancer: guidelines, infrastructures and low resource settings. Breast Cancer Manage. 2016;5:69–77. doi: 10.2217/bmt-2016-0012. [DOI] [Google Scholar]
- Richard R. L. Adjuvant surgical oophorectomy plus tamoxifen in premenopausal women with operable hormone receptor-positive breast cancer: A global treatment option. Clin. Breast Cancer. 2016;16:233–237. doi: 10.1016/j.clbc.2016.03.003. [DOI] [PubMed] [Google Scholar]
- Lei F. Jun J. L. Kathrin S. W. Jessica S. L. Dianne F. M. Ke D. Y. Zhi M. S. Wan Q. C. Paul E. G. Breast cancer in China. Lancet Oncol. 2014;15:e279–e289. doi: 10.1016/S1470-2045(13)70567-9. [DOI] [PubMed] [Google Scholar]
- (a) Lu H. W. Jing Y. Z. Yao Y. Chang Y. W. Jian B. Z. Xiao H. S. Xiu L. S. Yan X. L. Ke X. L. Hong Y. Xiao D. M. Covalent binding design strategy: A prospective method for discovery of potent targeted anticancer reagents. Eur. J. Med. Chem. 2017;142:493–505. doi: 10.1016/j.ejmech.2017.09.024. [DOI] [PubMed] [Google Scholar]; (b) Lazo J. S. McQueeney Kelley E. Burnett James C. Wipf Peter. Sharlow E. R. Small molecule targeting of PTPs in cancer. Int. J. Biochem. Cell Biol. 2018;96:171–181. doi: 10.1016/j.biocel.2017.09.011. [DOI] [PubMed] [Google Scholar]; (c) Shah A. S. A. Joshi S. S. Ambhore A. G. Khandelwal S. S. Babhulkar R. P. Short review on recent approaches of cancer therapy. World J. Pharm. Pharm. Sci. 2020;9(8):1431–1444. [Google Scholar]
- (a) Allan J. John A. H. Nicholas J. L. Alan T. M. Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle. Med. Res. Rev. 1998;18:259–296. doi: 10.1002/(SICI)1098-1128(199807)18:4<259::AID-MED3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; (b) Eric K. R. Emiliano C. Novel agents that target tublin and related elements. Semin. Oncol. 2006;33:421–435. doi: 10.1053/j.seminoncol.2006.04.006. [DOI] [PubMed] [Google Scholar]; (c) Cheng Z. Lu X. Feng B. A review of research progress of antitumor drugs based on tubulin targets. Transl. Cancer Res. 2020;9(6):4020–4027. doi: 10.21037/tcr-20-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Haque A. Rahman M. D. A. Haque F. Serajul M. D. Khan M. S. Next Generation Antineoplastic Agents: A Review on Structurally Modified Vinblastine (VBL) Analogues. Curr. Med. Chem. 2018;25(14):1650–1662. doi: 10.2174/0929867324666170502123639. [DOI] [PubMed] [Google Scholar]; (b) Sinha D. A review on taxanes: an important group of anticancer compound obtained from Taxus sp. Int. J. Pharma Sci. Res. 2020;11(5):1969–1985. [Google Scholar]; (c) Imran M. Saleem S. Chaudhuri A. Ali J. Baboota S. Docetaxel: An update on its molecular mechanisms, therapeutic trajectory and nanotechnology in the treatment of breast, lung and prostate cancer. J. Drug Delivery Sci. Technol. 2020;60:101959. doi: 10.1016/j.jddst.2020.101959. [DOI] [Google Scholar]
- (a) Lu Y. Chen J. Xiao M. Li W. Miller D. D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Technol. 2012;29:2943–2971. doi: 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xia L. Y. Zhang Y. L. Yang R. Wang Z. C. Lu Y. D. Wang B. Z. Zhu H. L. Tubulin Inhibitors Binding to Colchicine-Site: A Review from 2015 to 2019. Curr. Med. Chem. 2020;27(40):6787–6814. doi: 10.2174/0929867326666191003154051. [DOI] [PubMed] [Google Scholar]; (c) McLoughlin E. C. O'Boyle N. M. Colchicine-binding site inhibitors from chemistry to clinic: a review. Pharmaceuticals. 2020;13(1):8. doi: 10.3390/ph13010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timo S. Ines S. Andreas K. Lutz F. Detection, production, and application of microbial arylsulfatases. Appl. Microbiol. Biotechnol. 2016;100:9053–9067. doi: 10.1007/s00253-016-7838-4. [DOI] [PubMed] [Google Scholar]
- Meng R. L. Rong W. Qing S. Advances in research of arysulfatase in tumor diagnosis and oncotherapy. Pharm. Biotechnol. 2014;21:81–85. [Google Scholar]
- (a) Stanway S. J. Delavault P. Purohit A. Woo L. W. L. Thurieau C. Potter B. V. L. Reed M. J. Steroid sulfatase: a new target for the endocrine therapy of breast cancer. Oncologist. 2007;12(4):370–374. doi: 10.1634/theoncologist.12-4-370. [DOI] [PubMed] [Google Scholar]; (b) Potter B. V. L. Steroid Sulfatase Inhibition by Aryl Sulfamates: Clinical Progress, Mechanism and Future Prospects. J. Mol. Endocrinol. 2018;61(2):T233–T252. doi: 10.1530/JME-18-0045. [DOI] [PubMed] [Google Scholar]
- (a) Woo L. W. L. Leblond B. Purohit A. Potter B. V. L. Synthesis and evaluation of analogues of estrone-3-O-sulfamate as potent steroid sulfatase inhibitors. Bioorg. Med. Chem. 2012;20(8):2506–2519. doi: 10.1016/j.bmc.2012.03.007. [DOI] [PubMed] [Google Scholar]; (b) Walter G. Liebl R. Angerer E. V. Stilbene-based inhibitors of estrone sulfatase with a dual mode of action in human breast cancer cells. Arch. Pharm. 2004;337(12):634–644. doi: 10.1002/ardp.200400904. [DOI] [PubMed] [Google Scholar]; (c) Woo L. W. L. Bubert C. Sutcliffe O. B. Smith A. Chander S. K. Mahon M. F. Purohit A. Reed M. J. Potter B. V. L. Dual Aromatase-Steroid Sulfatase Inhibitors. J. Med. Chem. 2007;50(15):3540–3560. doi: 10.1021/jm061462b. [DOI] [PubMed] [Google Scholar]
- (a) Mark P. T. Barry V. L. P. Discovery and development of the aryl o-sulfamate pharmacophore for oncology and women's health. J. Med. Chem. 2015;58:7634–7658. doi: 10.1021/acs.jmedchem.5b00386. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reed M. J. Purohit A. Woo L. W. L. Potter B. V. L. The development of steroid sulfatase inhibitors. Endocr.-Relat. Cancer. 1996;3:9–13. doi: 10.1677/erc.0.0030009. [DOI] [Google Scholar]; (c) Ramanpreet S. Jatinder S. Dhandeep S. Amteshwar S. J. Nirmal S. Sulfatase inhibitors for recidivist breast cancer treatment: A chemical review. Eur. J. Med. Chem. 2016;114:170–190. doi: 10.1016/j.ejmech.2016.02.054. [DOI] [PubMed] [Google Scholar]
- (a) Holmes T. Brown A. W. Suggitt M. Shaw L. A. Simpson L. Harrity J. P. A. Tozer G. M. Kanthou C. The influence of hypoxia and energy depletion on the response of endothelial cells to the vascular disrupting agent combretastatin A-4-phosphate. Sci. Rep. 2020;10(1):9926. doi: 10.1038/s41598-020-66568-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deng C. Zhao J. Zhou S. Dong J. Cao J. Gao J. Bai Y. Deng H. The Vascular Disrupting Agent CA4P Improves the Antitumor Efficacy of CAR-T Cells in Preclinical Models of Solid Human Tumors. Mol. Ther. 2020;28(1):75–88. doi: 10.1016/j.ymthe.2019.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Alghzzawy Z. M. Elmaghraby T. K. Hagag S. A. Awwad M. H. Effect of vascular disrupting agent (CA-4P) and ionizing radiation on tumor growth in rats. J. Basic Environ. Sci. 2019;6(2):97–103. [Google Scholar]
- (a) Lin C. M. Singh S. B. Chu P. S. Dempcy R. O. Schmidt J. M. Pettit G. R. Hamel E. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin: a structure-activity study. Mol. Pharmacol. 1988;34:200–208. [PubMed] [Google Scholar]; (b) Lippert J. W. Vascular disrupting agents. Bioorg. Med. Chem. 2007;15:605–615. doi: 10.1016/j.bmc.2006.10.020. [DOI] [PubMed] [Google Scholar]; (c) Katsetos C. D. Draber P. Tubulins as therapeutic targets in cancer: from bench to bedside. Curr. Pharm. Des. 2012;18:2778–2792. doi: 10.2174/138161212800626193. [DOI] [PubMed] [Google Scholar]; (d) Horsman M. R. Wittenborn T. R. Nielsen P. S. Elming P. B. Tumors resistant to checkpoint inhibitors can become sensitive after treatment with vascular disrupting agents. Int. J. Mol. Sci. 2020;21(13):4778. doi: 10.3390/ijms21134778. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Smolarczyk R. Czapla J. Jarosz-Biej M. Czerwinski K. Cichon T. Vascular disrupting agents in cancer therapy. Eur. J. Pharmacol. 2021;891:173692. doi: 10.1016/j.ejphar.2020.173692. [DOI] [PubMed] [Google Scholar]
- (a) Jing P. L. Yi L. C. Ming F. K. Chun W. C. Tseng H. Y. Chiung C. W. Yung N. Y. Jang Y. C. Shiow J. L. Hsing P. H. Concise synthesis and structure-activity relationships of combretastatin A-4 analogues, 1-aroylindoles and 3-aroylindoles, as novel classes of potent antitubulin agents. J. Med. Chem. 2004;47:4247–4257. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]; (b) Lin C. M. Ho H. H. Pettit G. R. Hamel E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry. 1989;28:6984–6991. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]; (c) Siebert A. Gensicka M. Cholewinski G. Dzierzbicka K. Synthesis of combretastatin A-4 analogs and their biological activities. Anti-Cancer Agents Med. Chem. 2016;16:942–960. doi: 10.2174/1871520616666160204111832. [DOI] [PubMed] [Google Scholar]; (d) Nikolay A. Z. Zefirova O. N. Heterocycles as classical and nonclassical ring B isosters in combretastatin A-4. Chem. Heterocycl. Compd. 2017;53:273–280. doi: 10.1007/s10593-017-2049-1. [DOI] [Google Scholar]; (e) Arora S. Gonzalez A. F. Solanki K. Combretastatin A-4 and its analogs in cancer therapy. Int. J. Pharm. Sci. Rev. Res. 2013;22:168–174. [Google Scholar]; (f) Tarade D. Ma D. Pignanelli C. Mansour F. Simard D. Berg S. V. D. Gauld J. McNulty J. Pandey S. Structurally simplified biphenyl combretastatin A4 derivatives retain in vitro anti-cancer activity dependent on mitotic arrest. PLoS One. 2017;12(3):e0171806. doi: 10.1371/journal.pone.0171806. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Jie C. Jun Y. Jin H. H. Yan Q. P. Lin H. Xing S. L. Synthesis, biological evaluation and mechanism study of chalcone analogues as novel anti-cancer agents. RSC Adv. 2015;5:68128–68135. doi: 10.1039/C5RA14888J. [DOI] [Google Scholar]
- (a) Sessa C. Lorusso P. Tolcher A. Farace F. Lassau N. Delmonte A. Braghetti A. Bahleda R. Cohen P. Hospitel M. et al. Phase I Safety, Pharmacokinetic and Pharmacodynamic Evaluation of the Vascular Disrupting Agent Ombrabulin (AVE8062) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013;19(17):4832–4842. doi: 10.1158/1078-0432.CCR-13-0427. [DOI] [PubMed] [Google Scholar]; (b) Eskens F. A. L. M. Tresca P. Tosi D. Van Doorn L. Fontaine H. Van der Gaast A. Veyrat-Follet C. Oprea C. Hospitel M. Dieras V. A phase I pharmacokinetic study of the vascular disrupting agent ombrabulin (AVE8062) and docetaxel in advanced solid tumours. Br. J. Cancer. 2014;110(9):2170–2177. doi: 10.1038/bjc.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bahleda R. Sessa C. Conte G. D. Gianni L. Capri G. Varga A. Oprea C. Daglish B. Hospitel M. Soria J. C. Phase I clinical and pharmacokinetic study of ombrabulin (AVE8062) combined with cisplatin/docetaxel or carboplatin/paclitaxel in patients with advanced solid tumors. Invest. New Drugs. 2014;32(6):1188–1196. doi: 10.1007/s10637-014-0119-0. [DOI] [PubMed] [Google Scholar]; (d) Garon E. B. Neidhart J. D. Gabrail N. Y. Oliveira M. R. D. Balkissoon J. Kabbinavar F. A randomized Phase II trial of the tumor vascular disrupting agent CA 4P (fosbretabulin tromethamine) with carboplatin, paclitaxel, and bevacizumab in advanced nonsquamous non-small-cell lung cancer. OncoTargets Ther. 2016;9:7275–7283. doi: 10.2147/OTT.S109186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Monk B. J. Sill M. W. Walker J. L. Darus C. J. Sutton G. Tewari K. S. Martin L. P. Schilder J. M. Coleman R. L. Balkissoon J. et al. Randomized phase II evaluation of bevacizumab versus bevacizumab plus fosbretabulin in recurrent ovarian, tubal, or peritoneal carcinoma: an NRG oncology/gynecologic oncology group study. J. Clin. Oncol. 2016;34(19):2279–2286. doi: 10.1200/JCO.2015.65.8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ma Z. L. Yan X. J. Zhao L. Zhou J. J. Pang W. Kai Z. P. Wu F. H. Combretastatin A-4 and their derivatives: potential fungicides targeting fungal tubulin. J. Agric. Food Chem. 2016;64:746–751. doi: 10.1021/acs.jafc.5b05119. [DOI] [PubMed] [Google Scholar]; (b) Zhao L. Zhou J. J. Huang X. Y. Cheng L. P. Pang W. Kai Z. P. Wu F. H. Design, synthesis and antiproliferative effects in tumor cells of new new combretastatin A-4 analogs. Chin. Chem. Lett. 2015;26:993–999. doi: 10.1016/j.cclet.2015.05.003. [DOI] [Google Scholar]; (c) Zhang S. W. Li T. Pang W. Wu J. J. Wu F. L. Liu Y. Y. Wu F. H. Synthesis, biological evaluation and molecular docking studies of Combretastatin A-4 phosphoramidates as novel anticancer prodrugs. Med. Chem. Res. 2020;29(12):2192–2202. doi: 10.1007/s00044-020-02632-2. [DOI] [Google Scholar]
- Kamath K. Tatiana O. Gary L. Dulal P. Leslie W. Mary A. J. 2-Methoxyestradiol suppresses microtubule dynamics and arrests mitosis without depolymerizing microtubules. Mol. Cancer Ther. 2006;5(9):2225–2233. doi: 10.1158/1535-7163.MCT-06-0113. [DOI] [PubMed] [Google Scholar]
- (a) Lloyd M. D. Thiyagarajan N. Ho Y. T. Woo L. W. L. Sutcliffe O. B. Purohit A. Reed M. J. Acharya K. R. Potter B. V. L. First Crystal Structures of Human Carbonic Anhydrase II in Complex with Dual Aromatase-Steroid Sulfatase Inhibitors. Biochemistry. 2005;44:6858–6866. doi: 10.1021/bi047692e. [DOI] [PubMed] [Google Scholar]; (b) Woo L. W. L. Jackson T. Putey A. Cozier G. Leonard P. Acharya K. R. Chander S. K. Purohit A. Reed M. J. Potter B. V. L. Highly Potent First Examples of Dual Aromatase-Steroid Sulfatase Inhibitors based on a Biphenyl Template. J. Med. Chem. 2010;53:2155. doi: 10.1021/jm901705h. [DOI] [PubMed] [Google Scholar]
- Roberto G. Andrea E. P. Katja B. Andrea C. Michel O. S. Structural basis of cis- and trans-combretastatin binding to tubulin. Chem. 2017;2:102. [Google Scholar]
- Debashis G. Three-dimensional structures of sulfatases. Methods Enzymol. 2005;400:273–293. doi: 10.1016/S0076-6879(05)00016-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.