

Abstract
Preclinical data suggest that medroxyprogesterone acetate (MPA) has both anti-metastatic and anti-angiogenic activity in the absence of hormone receptors (HR). This phase II trial assessed the activity of MPA alone or in combination with low-dose chemotherapy in patients with metastatic HR-negative breast cancer. Postmenopausal women with HR-negative disease were eligible if they had not received more than 3 chemotherapy regimens for metastatic disease. All patients were treated with MPA 1,000–1,500 mg/day orally; patients in cohort two also received low-dose oral cyclophosphamide and methotrexate (ldCM, 50 mg/day and 2.5 mg twice daily on Days 1 and 2 each week). Tissue and circulating biomarkers were assessed serially. The primary endpoint was clinical benefit response defined as objective response or stable disease ≥6 months. Thirty patients were enrolled (14 MPA monotherapy; 16 MPA + ldCM); median age was 55 (35–80); nearly all had visceral involvement. Despite dose escalation in 90 % of patients, only 17 (57 %) patients ever achieved MPA trough concentrations >50 ng/ml. One patient developed grade 4 renal failure in the setting of rapid disease progression and dehydration. There were no objective responses. One patient in each cohort (~7 %) had stable disease for > 6 months. Skin Nm23 expression increased after 4 weeks of MPA + ldCM, but there were no significant changes in TSP-1, PAI-1 antigen, or PAI-1 activity. MPA had limited activity and does not warrant further development in patients with HR-negative advanced breast cancer. Poor bioavailability limited exposure despite dose escalation.
Keywords: Glucocorticoid receptor, Metastasis suppressor gene, Angiogenesis, Thrombospondin-1, Nm023, Plasminogen activator inhibitor type I
Critical features of malignancy include uncontrolled proliferation, insensitivity to negative growth regulation, evasion of apoptosis, lack of senescence, invasion and metastasis, angiogenesis, and genomic elasticity [1, 2]. Our most successful therapies to date inhibit proliferation via the estrogen receptor (ER) and HER2 pathways or induce apoptosis via cytotoxic chemotherapy or ionizing radiation. Targeted therapeutic advances for patients with tumors that lack ER and HER2 will require identification of novel ‘driver’ mutations or new strategies that attack other hallmarks of malignancy.
As most patients succumb to metastatic disease, there is well-justified interest in targeting the metastatic cascade. Nm23, the prototype metastasis suppressor gene (MSG) first described in 1988 [3], was identified on the basis of its reduced expression in highly metastatic tumors or cell lines, as compared to tumorigenic but poorly metastatic specimens. Unlike classical tumor suppressor genes, MSGs suppress in vivo metastatic capacity without impacting the growth of the primary tumor [4, 5]. Rather than simply inhibiting invasion, which has often been completed by the time a patient walks into the clinic, MSGs inhibit colonization and outgrowth of tumors at distant metastatic sites [6, 7]. This later window of progression provides a substantial therapeutic opportunity.
But how does one restore the function (in this case the suppressive effect) of a gene or protein that has been lost? Reduced Nm23 protein expression, not allelic deletion or mutations, was correlated with poor patient survival [8], prompting a search for methods to restore expression. Molecular analyses of the Nm23 promoter identified transcription factor binding sites regulated by glucocorticoid response elements, suggesting that Nm23 expression might be partly regulated by the glucocorticoid receptor (GR) [4, 9]. Indeed in preclinical studies, the atypical glucocorticoid medroxyprogesterone acetate (MPA) increased Nm23 expression in vitro and inhibited metastasis in vivo in aggressive HR-negative breast cancer models in a GR-dependent manner [10].
MPA has a long clinical history but was largely abandoned in favor of tamoxifen over three decades ago [11]. Although initially developed as an alternative hormonal therapy, several studies documented responses in patients with ER-negative tumors [12-15]. No optimal dose or schedule was identified with several studies finding similar response rates with doses ranging from 400 to 1,400 mg per day [16-18]. Studies incorporating pharmacokinetic analysis found higher MPA trough concentrations in responding versus non-responding patients. The lack of clear dose–response relationship may be partially explained by the significant inter-patient variation in bioavailability and metabolism. A retrospective analysis of 380 patients treated with varying doses of MPA confirmed a relationship between systemic exposure and efficacy [19].
Additionally, MPA increased expression of anti-angiogenic peptides thrombospondin (TSP-1) and plasminogen activator inhibitor type I (PAI-1) (unpublished data); culture supernatant from MPA treated HR-negative breast cancer cells did not support angiogenesis in an ex vivo aortic ring assay [20, 21]. Interestingly, prolonged in vitro exposure of endothelial cells to low concentrations of several different chemotherapeutic agents also markedly induces gene and protein expression of TSP-1. Although the contribution of the antiangiogenic versus direct antitumor effect of low-dose chemotherapy has been debated, low-dose cyclophosphamide (but not high-dose intermittent cyclophosphamide) was ineffective in xenograft models implanted in TSP-1-null mice [22], suggesting that induction is TSP-1 is required for efficacy. As both MPA and low-dose cyclophosphamide induce TSP-1, this phase II study was designed to explore the clinical efficacy and biologic activity of MPA, alone and in combination with low-dose cyclophosphamide and methotrexate (ldCM), in patients with metastatic HR-negative breast cancer.
Patients and methods
Trial eligibility
Postmenopausal women with HR-negative (both estrogen and progesterone) metastatic breast cancer were eligible if they had received no more than three prior chemotherapy regimens for metastatic disease. In addition, patients were required to have a performance status ≤2 on the Eastern Cooperative Oncology Group (ECOG) scale with adequate renal (creatinine ≤2.5 mg/dL), hepatic (total bilirubin ≤2.0 mg/dL; AST and ALT <2.0 times upper limit of normal or ≤5 × normal in patients with liver involvement), and hematologic (ANC ≥1,000/mm3; platelets ≥75,000/mm3) function. Patients were excluded if they had experienced a deep venous thrombosis or pulmonary embolism within 12 months of study entry or if they had extensive pleural effusion or ascites. Patients with asymptomatic, treated central nervous system metastasis were eligible provided chronic steroid therapy was not required. Each participating Institutional Review Board approved the protocol, and all patients provided individual written informed consent prior to screening and study entry.
Treatment plan
MPA was obtained from Spectrum Chemicals (New Brunswick, NJ) and compounded into 250 mg capsules (Chao Center for Industrial Pharmacy and Contract Manufacturing, West Lafayette, Indiana). Patients in cohort one received MPA 1,000 mg continuously as a single daily oral dose; 28 days was considered one cycle. As previous studies had reported significant variability in bioavailability and metabolism, we assessed drug exposure after 10–14 days of therapy. If the trough MPA serum concentration obtained on day 10–14 was <50 ng/ml (the target concentration based on preclinical data and prior clinical trials), the MPA dose was increased to 1,500 mg/day. Additional trough PK samples were obtained to monitor drug exposure, but no further dose adjustments were made based on those subsequent MPA levels.
As both MPA and low-dose cyclophosphamide increase TSP-1 expression in vivo, we hypothesized synergistic activity with low-dose cyclophosphamide in combination with MPA. Therefore, patients in cohort two also received low-dose oral cyclophosphamide 50 mg/day continuously with methotrexate 2.5 mg twice daily on Days 1 and 2 (ldCM) of each week; 28 days was considered one cycle. MPA treatment and dose escalation were identical to cohort one.
Safety and efficacy assessments
Patients were evaluated clinically every 4 weeks. Laboratory assessments including complete blood count and serum chemistry were assessed at each evaluation. Toxicity was assessed based on the National Cancer Institute Common Toxicity Criteria (NCI-CTC v3.0). MPA could be held or reduced for Grade 3 or greater thromboembolism, weight gain, or peripheral edema. ldCM could be held or reduced for Grade 3 or greater neutropenia, mucositis, or hepatic dysfunction. Disease status was assessed according to the Response Evaluation Criteria for Solid Tumors (RECIST) [23] criteria every 8 weeks or more frequently when clinically indicated.
Drug exposure and pharmacodynamics
Serum for determination of MPA trough concentration was obtained prior to MPA administration once between Days 10–14 and after 4 and 8 weeks of therapy. Samples were obtained in a standard 10 ml red top tube and allowed to clot at room temperature for 30 min. Serum was separated by centrifugation at 3,000 × g for 30 min and stored at −20 °C in 1 ml aliquots for later analysis; triplicate aliquots were preserved for each patient at each time point. MPA was quantified using norgestimate (NGM) as the internal standard, liquid–liquid extraction, and HPLC–MS/MS (ABSciex4000, Applied Biosystems). MPA and NGM were separated by gradient mobile phase (acetonitrile:formic acid) and HPLC using a biphenyl column (Restek 5 μm 50 × 4.6 mm). The Q1/Q3s for MPA and NGM were 387/327 and 370/124, respectively. The lower limit of quantification (LOQ) is 1 ng/mL using 100 μL of serum.
Plasma samples to measure TSP-1, PAI-1 antigen, and PAI-1 activity were obtained at baseline, after 2, 4, and 8 weeks of therapy. Samples were obtained in a heparinized green top tube, separated by centrifugation at 3,000 g for 30 min within 2 hours, and stored at −20 °C in 1 ml aliquots for later analysis; triplicate aliquots were preserved for each patient at each time point. All samples were measured in duplicate using commercially available enzyme linked immunosorbant assays (ELISA, Chemicon, Temecula, CA and DiaPharma, West Chester, OH). All samples with a coefficient of variation >10 % were repeated. The assays have the following limits of detection: TSP-1 9.77 ng/ml, PAI-1 antigen 0.5 ng/ml, and PAI-1 activity 0.5 ng/ml.
Archived primary tumor and serial skin biopsy samples (baseline and C2D1) were stained for Nm23 using an affinity purified polyclonal rabbit anti-Nm23-H1/H2 antibody (Cymbus, Southampton, United Kingdom). An isotype-matched control antibody (goat anti-rabbit immunoglobulin G; Santa Cruz Biotechnology, Santa Cruz, CA) was used on a second independent section to control for nonspecific binding. In brief, sections are deparaffinized in xylene, dehydrated in 100 % ethanol, rehydrated in 95 % ethanol and phosphate-buffered saline (PBS), and blocked sequentially with hydrogen peroxide and goat serum. Antigen retrieval was accomplished by incubating the slides in 10 m M citrate buffer, pH 3.0, for 30 min at 37 °C. Slides were incubated with primary antibody (diluted 1:5 in PBS with 1 % goat serum) in a humidified chamber overnight at room temperature. Staining was visualized using the Vectastain ABC kit and the DAB Substrate kit (Vector Laboratories, Burlingame, CA). Staining intensities were graded (0–3+) by a single pathologist (MM) blinded to treatment group, response, and timing of skin biopsy.
Statistical analyses
Cohorts one and two were analyzed independently for toxicity and efficacy; there were no plans for formal comparison between the treatment groups. A regimen that produced at least a 20 % clinical benefit rate (CR + PR + SD ≥ 6 months) would be considered worthy of further study in this patient population. A serial optimal two-stage design for each regimen with 3 % type I error probability for H0:p = .1 and 80 % power for H1:p = .3 required between 15 and 25 patients per treatment cohort. 15 patients were treated in the first stage of each cohort. If fewer than 2 patients among the 15 derived clinical benefit, then regimen would be rejected. If 2 or more patients derived clinical benefit, then an additional 10 patients would be enrolled. If 5 or fewer patients among the total 25 derived clinical benefit, then regimen would be rejected. Patients were enrolled sequentially beginning with cohort one. Enrollment to cohort 2 began upon completion of cohort one.
Secondary endpoints included characterization of MPA exposure based on trough levels, impact of MPA on circulating angiogenic peptides and Nm23 expression in skin. The change in angiogenic peptides from baseline to C1D15 and C2D1 was assessed using the repeated measures ANOVA and Bonferroni’s multiple comparison test when appropriate; Nm23 skin expression between baseline and C2D1 was compared using the Wilcoxin signed rank test. Additionally, progression free survival (PFS) was analyzed using the Kaplan–Meier method.
Results
Patient population
Thirty patients were enrolled, 14 in cohort 1 and 16 in cohort 2. Patient characteristics are shown in Table 1. Patients were heavily pre-treated with the majority having visceral disease at study entry. All patients were evaluable for toxicity. Seven patients (2 in cohort 1 and 5 in cohort 2) had symptomatic decline and clinical evidence of rapid progression that was not confirmed radiographically.
Table 1.
Patient Characteristics
| Cohort 1 MPA monotherapy (n = 14) |
Cohort 2 MPA + ldCM (n = 16) |
Total (n = 30) |
|
|---|---|---|---|
| Median age | 59 (35–79) | 53 (39–80) | 55 (35–80) |
| Race | |||
| Caucasian | 9 (64 %) | 12 (75 %) | 21 (70 %) |
| African American | 5 (36 %) | 3 (19 %) | 8 (26 %) |
| Asian | 0 | 1 (6 %) | 1 (3 %) |
| ECOG PS* | |||
| 0 | 4 (29 %) | 8 (50 %) | 12 (40 %) |
| 1 | 8 (57 %) | 6 (38 %) | 14 (46 %) |
| 2 | 1 (7 %) | 1 (6 %) | 2 (7 %) |
| Visceral disease | 8 (57 %) | 15 (94 %) | 23 (77 %) |
| Prior therapy | |||
| Anthracycline | 14 (100 %) | 13 (81 %) | 27 (90 %) |
| Taxane | 13 (93 %) | 13 (81 %) | 26 (87 %) |
| Platinum | 7 (50 %) | 10 (62 %) | 17 (57 %) |
| Bevacizumab | 10 (71 %) | 7 (44 %) | 17 (57 %) |
| Capecitabine | 5 (36 %) | 10 (62 %) | 15 (50 %) |
| Gemcitabine | 4 (29 %) | 6 (37 %) | 10 (33 %) |
| Vinorelbine | 6 (43 %) | 3 (19 %) | 9 (30 %) |
Eastern Cooperative Oncology Group performance status missing in 2 patients
Toxicity and treatment delivered
MPA was generally well tolerated with few patients experiencing Grade 3 or 4 toxicities (Table 2). Two patients in cohort 1 required dose reductions due to grade 2 nausea and rash. ldCM did not induce significant myelosuppression even in this heavily pretreated population. Three patients in cohort 2 required dose reduction of ldCM and MPA due to toxicity. Treatment was held due to nausea, emesis, and dehydration in one patient who later developed grade 4 renal failure. Evaluation found rapidly progressive disease, but a possible relationship to study therapy could not be excluded. One patient had increased transaminases that improved with dose reduction. Doses were reduced due to grade 2 mucositis and fatigue in one patient. There were no thromboembolic events, fluid retention, pleural effusions, or weight gain to suggest significant estrogenic exposure.
Table 2.
Treatment related toxicity
| Cohort 1 MPA monotherapy |
Cohort 2 MPA + ldCM |
Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| Grade |
Grade |
Grade |
|||||||
| 2 | 3 | 4 | 2 | 3 | 4 | 2 | 3 | 4 | |
| Nausea | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Dysgeusia | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Mucositis | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| Dysphagia | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| Fatigue | 0 | 0 | 0 | 2 | 1 | 0 | 2 | 1 | 0 |
| Pain | 3 | 0 | 0 | 1 | 0 | 0 | 4 | 0 | 0 |
| Dyspnea | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 0 |
| Hot flashes | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Irregular menses | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Insomnia | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Diaphoresis | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Rash | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 0 |
| Hypokalemia | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Transaminase elevated | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Alkaline phosphatase elevated | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| Renal failure | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Dehydration | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
Worst toxicity reported per patient based on NCI-CTC v3.0
Efficacy
There were no objective responses. Five patients (3 in cohort 1 and 2 in cohort 2) had stable disease, but only 2 patients remained stable for ≥6 months. One patient with prior anthracycline and capecitabine therapy had a stable lung lesion for 7 months on MPA monotherapy. One patient with prior anthracycline, taxane, capecitabine, gemcitabine, and vinorelbine exposure had stable lymph node metastases with ldCM + MPA for 8 months. Overall median PFS was approximately 1.8 months in both cohorts.
Drug exposure and pharmacodynamics
Trough MPA concentrations are shown in Table 3. Although the sample size is too small for definitive conclusions, there was no obvious difference in MPA exposure with concomitant ldCM. Nearly all patients required dose escalation. Overall exposure was less than anticipated with just over half of patients ever achieving concentrations >50 ng/mL. Both patients with prolonged stable disease had MPA concentrations >50 ng/mL after initial dose escalation. Prior studies had suggested a decrease in MPA exposure at the time of disease progression [24], but MPA remained >50 ng/mL at progression in both patients who derived clinical benefit.
Table 3.
MPA trough concentrations
| Cohort 1 MPA monotherapy |
Cohort 2 MPA + ldCM |
Total | |
|---|---|---|---|
| Mean MPA (ng/ml) | 14.5 ± 8.9 | 42.1 ± 66.4 | 26.7 ± 45.6 |
| Cycle 1, Day 10–14 | |||
| Patients requiring dose escalation | 14/14 | 13/16 | 27/20 |
| Mean MPA (ng/ml) | 52.6 ± 65.3 | 66.4 ± 71.5 | 58.6 ± 66.8 |
| Cycle 2, Day 1 | |||
| Patients achieving MPA ≥ 50 ng/ml at any time | 10/14 | 7/16 | 17/30 |
Serial plasma samples for measurement of TSP-1, PAI-1 antigen, and PAI-1 activity were available for 12 patients in cohort 1 and 13 patients in cohort 2 (Fig. 1). PAI-1 antigen increased after MPA monotherapy (p < 0.001), but the difference was not significant when corrected for multiple testing. No other changes in plasma angiogenic peptides were identified. Archived primary tumor samples were obtained in 19 patients; Nm23 was absent or expressed at low levels (0 or 1 +) in 63 % of tumors. Serial skin biopsies were evaluable for 7 patients in cohort 1 and 6 patients in cohort 2 (Fig. 2). Nm23 expression in skin was unchanged during MPA monotherapy but increased significantly after 4 weeks of treatment with MPA + ldCM (p = 0.03).
Fig. 1.
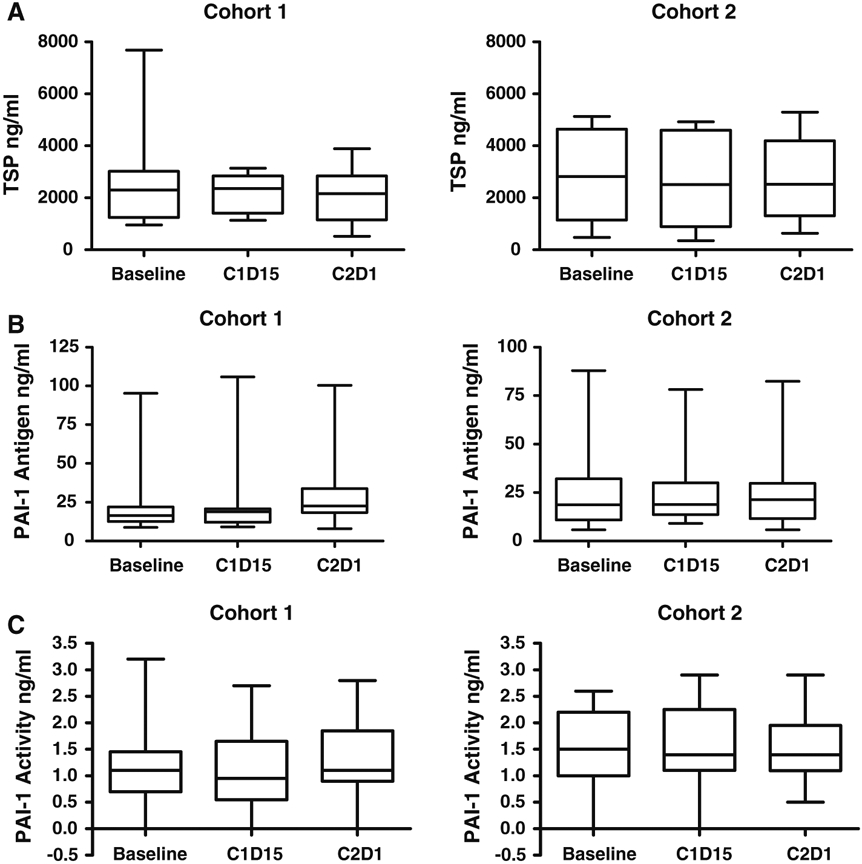
Plasma concentration of TSP-1 (a), PAI-1 antigen (b), and PAI-1 activity (c) was assessed in 12 patients in cohort 1 and 13 patients in cohort 2. PAI-1 antigen increased after MPA monotherapy (p < 0.001), but the difference was not significant when corrected for multiple testing. (C1D15 = Cycle 1, Day 15, C2D1 = Cycle 2, Day 1)
Fig. 2.
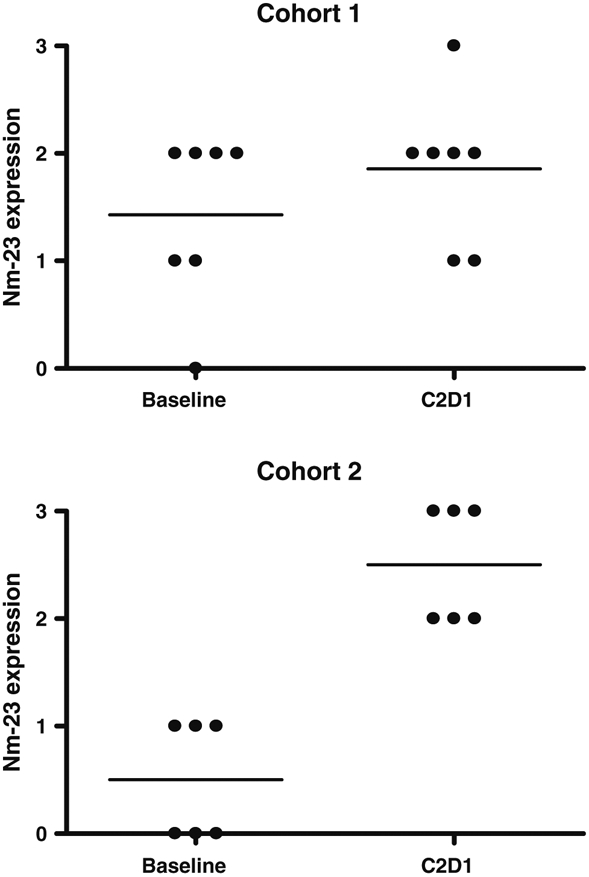
Punch skin biopsies were assessed for Nm-23 expression at baseline and after 4 weeks of therapy. Nm23 expression in skin was unchanged during MPA monotherapy (a) but increased significantly after 4 weeks of treatment with MPA + ldCM (B, p = 0.03). (C2D1 = Cycle 2, Day 1)
Discussion
Despite a sound preclinical foundation, MPA monotherapy had little clinical or biologic activity in patients with advanced HR-negative metastatic breast cancer in this prospective, phase II trial. The activity of ldCM in our study was also less than that previously reported. Colleoni had reported an objective response rate of 19 % with 31 % of patients achieving clinical benefit [25]. However, the patients in Colleoni’s trial were less heavily pretreated, and the majority of responding patients (4 of 12) had hormone sensitive disease. Colleoni had also reported a reduction in serum vascular endothelial growth factor (VEGF) after 2 months of ldCM therapy, though the change in serum VEGF did not correlate with response or clinical benefit. As release of VEGF stored in platelets complicates analysis of circulating VEGF levels [26-28] and changes in TSP-1 were more closely associated with activity of MPA and ldCM in preclinical models, we did not measure VEGF in our patients.
Several important limitations may have contributed to the limited efficacy we observed. First, the complexity of HR-negative breast cancer has increasingly been recognized [29-31]. Although most HR-negative breast cancer cell lines [32] and at least 25 % of HR-negative primary breast cancers express glucocorticoid receptor (GR) [33], levels vary significantly. We did not require GR expression for study entry, and the limited number of archived samples available precluded any assessment of benefit based on GR expression. Second, despite real-time assessment of MPA trough concentration and mid-cycle dose escalation, few patients reached concentrations in the potentially therapeutic range. Interestingly, both patients with clinical benefit had concentrations above the potential therapeutic threshold 50 ng/ml based on previous preclinical and clinical studies. Third, we did not assess changes in tumor Nm-23 levels. We were concerned that requiring serial tumor biopsy would be unacceptable to some patients and would place patients with disease limited to less accessible sites (i.e., lung) at greater risk. In preclinical studies, MPA altered Nm23 expression in skin biopsies, providing rationale for including skin biopsies as a correlate in this trial. We intended the skin biopsies as only a correlate of biologic activity and potentially effective drug exposure rather than as a direct correlate of clinical activity. Given the limited drug exposure, the lack of modulation of Nm23, TSP, and PAI-1 is not surprising. The apparent increase in Nm23 expression after combined MPA + ldCm must be interpreted with caution given the small sample size and multiple correlatives assessed. In addition, while changes in skin expression correlated with changes in tumor Nm23 expression in preclinical models (unpublished data), the correlation in patients remains unknown.
Finally, metastasis is a complicated multistep process, and reintroduction of one MSG may not be sufficient to alter tumor growth. Since the initial identification of Nm-23 as the prototype metastasis suppressor gene, it has become clear that Nm-23 (and other MSGs) has more functions than were originally anticipated [34]. Although reduced Nm23 expression has been generally correlated with increased metastatic potential, this correlation does not hold true for all cancer types. Neuroblastoma provides a notable exception, where increased Nm23 expression is correlated with more aggressive disease. In neuroblastoma, a mutant nm23 has been identified [35, 36], unlike in solid tumors such as breast cancer, where decreased Nm23 expression has not been linked to mutations in the Nm23-H1 gene [37].
Importantly, preliminary study was never intended to be definitive but rather to provide an initial exploration of the biological and clinical activity, deciding a priori that a clinical benefit rate of 20 % or greater with either regimen (MPA monotherapy or MPA + ldoCM) would be worthy of further study. In retrospect, the level of activity was an ambitious goal in this pretreated population. Although negative in the sense that the primary endpoint was not reached, this trial highlights the difficulty in studying anti-metastatic agents. The development of the anti-metastatic therapies, like that of other anti-proliferative rather than cytotoxic compounds, poses a challenge in clinical trial design. The reinduction of MSGs implies reduction or delay in the development of new metastatic lesions rather than time to progression or objective tumor response as the proper primary endpoint. Unfortunately, delay in the development of new lesions cannot be determined with certainty in uncontrolled trials, and the benefit of preventing outgrowth of new lesions while existing lesions continue to progress is uncertain. Intuitively, the impact of reinduction of metastases suppressor gene (MSG) expression is expected to be greatest in patients with micrometastatic disease—an intuition that will require commitment of substantial human and financial resources to a randomized trial, in the adjuvant or post-neoadjuvant therapy settings, to test [38]. Alternatively, trials in patients who have received limited treatment for metastatic disease could be conducted with the primary endpoint being development of a new, rather than further growth of a preexisting metastasis, as the primary endpoint [39]. The negative results we report here should not dampen enthusiasm for the study of other anti-metastatic agents in patients with less advanced disease.
Acknowledgments
Supported by NIH/NCI P30 CA082709-AV-133 (Avon Partners for Progress Supplement) and the Breast Cancer Research Foundation (KDM). In addition, we are grateful for the funding support to the Translational Breast Cancer Research Consortium (TBCRC) from The AVON Foundation, The Breast Cancer Research Foundation, and Susan G. Komen for the Cure.
Footnotes
Conflict of Interest None of the authors have any conflicts to disclose
Contributor Information
Kathy D. Miller, Indiana University Melvin and Bren Simon Cancer Center, 535 Barnhill Drive, RT 473, Indianapolis, IN 46202, USA
Sandra K. Althouse, Indiana University Melvin and Bren Simon Cancer Center, 535 Barnhill Drive, RT 473, Indianapolis, IN 46202, USA
Lisle Nabell, University of Alabama Birmingham, Birmingham, USA.
Hope Rugo, University of California San Francisco, San Francisco, USA.
Lisa Carey, University of North Carolina, Chapel Hill, USA.
Gretchen Kimmick, Duke University Medical Center, Durham, USA.
David R. Jones, Indiana University Melvin and Bren Simon Cancer Center, 535 Barnhill Drive, RT 473, Indianapolis, IN 46202, USA
Maria J. Merino, Laboratory of Pathology, National Cancer Institute, Bethesda, USA
Patricia S. Steeg, Women’s Malignancies Branch, National Cancer Institute, Bethesda, USA
References
- 1.Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70 [DOI] [PubMed] [Google Scholar]
- 2.Sledge GW Jr, Miller KD (2003) Exploiting the hallmarks of cancer: the future conquest of breast cancer. Eur J Cancer 39(12):1668–1675 [DOI] [PubMed] [Google Scholar]
- 3.Steeg PS, Bevilacqua G, Pozzatti R, Liotta LA, Sobel ME (1988) Altered expression of NM23, a gene associated with low tumor metastatic potential, during adenovirus 2 Ela inhibition of experimental metastasis. Cancer Res 48(22):6550–6554 [PubMed] [Google Scholar]
- 4.Ouatas T, Halverson D, Steeg P (2003) Dexamethasone and medroxyprogesterone acetate elevate Nm23-H1 metastasis suppressor expression in metastatic human breast carcinoma cells: New uses for old compounds. Clin Cancer Res 9:3763–3772 [PubMed] [Google Scholar]
- 5.Yoshida B, Sokoloff M, Welch D, Rinker-Schaeffer C (2000) Metastasis-suppressor genes: a review and perspective on an emerging field. J Natl Cancer Inst 92:1717–1730 [DOI] [PubMed] [Google Scholar]
- 6.Leone A, Flatow U, King C, Sandeen M, Margulies I, Liotta L, Steeg P (1991) Reduced tumor incidence, metastatic potential, and cytokine responsiveness of nm23-transfected melanoma cells. Cell 65:25–35 [DOI] [PubMed] [Google Scholar]
- 7.Leone A, Flatow U, VanHoutte K, Steeg P (1993) Transfection of human nm23-H1 into the human MDA-MB-435 breast carcinoma cell line: effects on tumor metastatic potential, colonization, and enzymatic activity. Oncogene 8:2325–2333 [PubMed] [Google Scholar]
- 8.Cropp C, Lidereau R, Leone A, Liscia D, Cappa A, Campbell G, Barker E, Doussal V, Steeg P, Callahan R et al. (1994) NME1 protein expression and loss of heterozygosity mutations in primary human breast tumors. J Natl Cancer Inst 86:1167–1169 [DOI] [PubMed] [Google Scholar]
- 9.Ouatas T, Clare S, Hartsough M, DeLaRosa A, Steeg P (2002) MMTV-associated transcription factor binding sites increase nm23-H1 metastasis suppressor gene expression in human breast carcinoma cell lines. Clin Exp Metast 19:35–42 [DOI] [PubMed] [Google Scholar]
- 10.Palmieri D, Halverson D, Ouatas T, Salerno M, Johnson J, Figg W, Hollingshead M, Hursting S, Berigan D, Steinberg S et al. (2005) Medroxyprogesterone acetate elevation of Nm23-H1 metastasis suppressor expression in hormone receptor—negative breast cancer. J Natl Cancer Inst 97:632–642 [DOI] [PubMed] [Google Scholar]
- 11.Van Veelen H, Willemse P, Tjabbes T et al. (1986) Oral high-dose medroxyprogesterone acetate versus tamoxifen. Cancer 58:7–13 [DOI] [PubMed] [Google Scholar]
- 12.Nishimura R, Nagao K, Matsuda M, Baba K, Matsuoka Y, Yamashita H, Fukuda M, Higuchi A, Ikeda K (1997) Predictive value of serum medroxyprogesterone acetate concentration for response or recurrent breast cancer. Eur J Cancer 33(9):1407–1412 [DOI] [PubMed] [Google Scholar]
- 13.Tashiro H, Nomura Y (1995) Mitomycin C, methotrexate, and vincristine with medroxyprogesterone acetate or prednisolone for doxorubicin resistant advanced breast cancer–a randomized control study. Anticancer Res 15(5B):2229–2237 [PubMed] [Google Scholar]
- 14.Koyama H, Tominaga T, Asaishi K, Abe R, Iino Y, Enomoto K, Miura S, Nomura Y, Nakazato H, Abe O (1999) A randomized controlled comparative study of oral medroxyprogesterone acetate 1,200 and 600 mg in patients with advanced or recurrent breast cancer. Oncology 56(4):283–290 [DOI] [PubMed] [Google Scholar]
- 15.Byrne MJ, Gebski V, Forbes J, Tattersall MH, Simes RJ, Coates AS, Dewar J, Lunn M, Flower C, Gill PG et al. (1997) Medroxyprogesterone acetate addition or substitution for tamoxifen in advanced tamoxifen-resistant breast cancer: a phase III randomized trial. Australian-New Zealand Breast Cancer Trials Group. J Clin Oncol 15(9):3141–3148 [DOI] [PubMed] [Google Scholar]
- 16.Hortobagyi GN, Buzdar AU, Frye D, Yap HY, Hug V, Pinnamaneni K, Fraschini G, Halvorson HC, Blumenschein GR (1985) Oral medroxyprogesterone acetate in the treatment of metastatic breast cancer. Breast Cancer Res Treat 5(3):321–326 [DOI] [PubMed] [Google Scholar]
- 17.Gallagher CJ, Cairnduff F, Smith IE (1987) High dose versus low dose medroxyprogesterone acetate: a randomized trial in advanced breast cancer. Eur J Cancer Clin Oncol 23(12):1895–1900 [DOI] [PubMed] [Google Scholar]
- 18.Davila E, Vogel CL, East D, Cairns V, Hilsenbeck S (1988) Clinical trial of high-dose oral medroxyprogesterone acetate in the treatment of metastatic breast cancer and review of the literature. Cancer 61(11):2161–2167 [DOI] [PubMed] [Google Scholar]
- 19.Pannuti F, Camaggi CM, Strocchi E, Martoni A (1986) MPA at high doses in advanced breast cancer: a statistical evaluation. Chemioterapia 5(3):159–163 [PubMed] [Google Scholar]
- 20.Zhou L, Isenberg J, Cao Z, Roberts D (2006) Type I collagen is a molecular target for inhibition of angiogenesis by endogenous thrombospondin-1. Oncogene 25:536–545 [DOI] [PubMed] [Google Scholar]
- 21.Isenberg J, Calzada M, Zhou L, Guo N, Lawler J, Wang XQ, Frazier W, Roberts D (2005) Endogenous thrombospondin-1 is not necessary for proliferation but is permissive for vascular smooth muscle cell responses to platelet-derived growth factor. Matrix Biol 24:110–123 [DOI] [PubMed] [Google Scholar]
- 22.Bocci G, Francia G, Man S, Lawler J, Kerbel RS (2003) Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci U S A 100(22):12917–12922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC et al. (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(3):205–216 [DOI] [PubMed] [Google Scholar]
- 24.Nishimura R, Nagao K, Matsuda M, Baba K, Matsuoka Y, Yamashita H, Fukuda M, Higuchi A, Ikeda K (1997) Predictive value of serum medroxyprogesterone acetate concentration for response in advanced or recurrent breast cancer. Eur J Cancer 33(9):1407–1412 [DOI] [PubMed] [Google Scholar]
- 25.Colleoni M, Rocca A, Sandri MT, Zorzino L, Masci G, Nole F, Peruzzotti G, Robertson C, Orlando L, Cinieri S et al. (2002) Low-dose oral methotrexate and cyclophosphamide in metastatic breast cancer: antitumor activity and correlation with vascular endothelial growth factor levels. Ann Oncol 13(1):73–80 [DOI] [PubMed] [Google Scholar]
- 26.Adams J, Carder PJ, Downey S, Forbes MA, MacLennan K, Allgar V, Kaufman S, Hallam S, Bicknell R, Walker JJ et al. (2000) Vascular endothelial growth factor (VEGF) in breast cancer: comparison of plasma, serum, and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res 60(11):2898–2905 [PubMed] [Google Scholar]
- 27.Verheul HM, Hoekman K, Luykx-de Bakker S, Eekman CA, Folman CC, Broxterman HJ, Pinedo HM (1997) Platelet: transporter of vascular endothelial growth factor. Clin Cancer Res 3(12 Pt 1):2187–2190 [PubMed] [Google Scholar]
- 28.Wynendaele W, Derua R, Hoylaerts MF, Pawinski A, Waelkens E, de Bruijn EA, Paridaens R, Merlevede W, van Oosterom AT (1999) Vascular endothelial growth factor measured in platelet poor plasma allows optimal separation between cancer patients and volunteers: a key to study an angiogenic marker in vivo? Ann Oncol 10(8):965–971 [DOI] [PubMed] [Google Scholar]
- 29.Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT, Pergamenschikov A, Williams CF, Zhu SX, Lee JC et al. (1999) Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A 96(16):9212–9217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121(7):2750–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perou C, Sorlie T, Eisen M et al. (2000) Molecular portraits of human breast tumours. Nature 406:747. [DOI] [PubMed] [Google Scholar]
- 32.Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD (2001) Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem 276(20):16649–16654 [DOI] [PubMed] [Google Scholar]
- 33.Pan D, Kocherginsky M, Conzen SD (2011) Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res 71(20):6360–6370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steeg PS, Zollo M, Wieland T (2011) A critical evaluation of biochemical activities reported for the nucleoside diphosphate kinase/Nm23/Awd family proteins: opportunities and missteps in understanding their biological functions. Naunyn-Schmiedeberg’s Arch Pharmacol 384(4–5):331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang CL, Zhu XX, Thoraval DH, Ungar D, Rawwas J, Hora N, Strahler JR, Hanash SM, Radany E (1994) Nm23-H1 mutation in neuroblastoma. Nature 370(6488):335–336 [DOI] [PubMed] [Google Scholar]
- 36.Leone A, Seeger RC, Hong CM, Hu YY, Arboleda MJ, Brodeur GM, Stram D, Slamon DJ, Steeg PS (1993) Evidence for nm23 RNA overexpression, DNA amplification and mutation in aggressive childhood neuroblastomas. Oncogene 8(4):855–865 [PubMed] [Google Scholar]
- 37.Leone A, Flatow U, VanHoutte K, Steeg PS (1993) Transfection of human nm23-H1 into the human MDA-MB-435 breast carcinoma cell line: effects on tumor metastatic potential, colonization and enzymatic activity. Oncogene 8(9):2325–2333 [PubMed] [Google Scholar]
- 38.Steeg PS (2012) Perspective: the right trials. Nature 485(7400):S58–59 [DOI] [PubMed] [Google Scholar]
- 39.Brabletz T, Lyden D, Steeg PS, Werb Z (2013) Roadblocks to translational advances on metastasis research. Nat Med 19(9):1104–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]