

SUMMARY
A general approach for heritably altering gene expression has the potential to enable many discovery and therapeutic efforts. Here, we present CRISPRoff—a programmable epigenetic memory writer consisting of a single dead Cas9 fusion protein that establishes DNA methylation and repressive histone modifications. Transient CRISPRoff expression initiates highly specific DNA methylation and gene repression that is maintained through cell division and differentiation of stem cells to neurons. Pairing CRISPRoff with genome-wide screens and analysis of chromatin marks establishes rules for heritable gene silencing. We identify sgRNAs capable of silencing the large majority of genes including those lacking canonical CpG islands (CGIs) and reveal a wide targeting window extending beyond annotated CGIs. The broad ability of CRISPRoff to initiate heritable gene silencing even outside of CGIs expands the canonical model of methylation-based silencing and enables diverse applications including genome-wide screens, multiplexed cell engineering, enhancer silencing, and mechanistic exploration of epigenetic inheritance.
Keywords: CRISPR, epigenetics, DNA methylation, cell therapy
In Brief:
CRISPRoff programs heritable epigenetic memory and is able to heritably silence most genes, including genes without CpG islands. This heritable silencing is highly specific and persists through differentiation of iPSCs into neurons.
Graphical Abstract
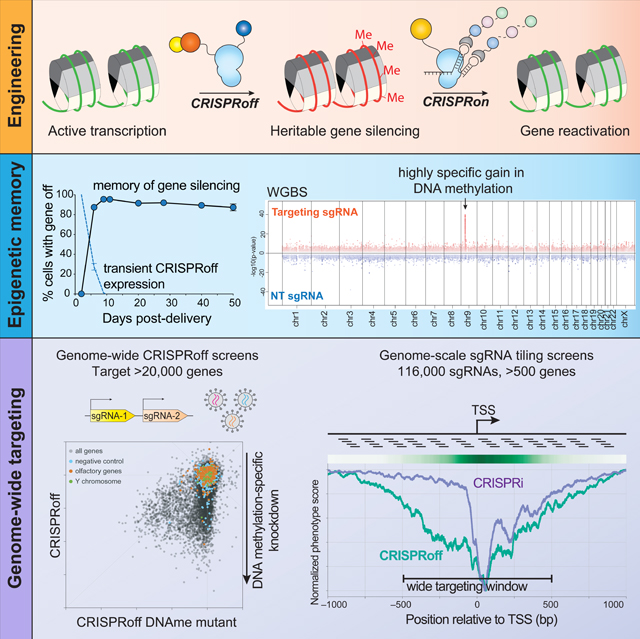
INTRODUCTION
Advances in gene editing have transformed our ability to modify the human genome. In particular, CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 (CRISPR-associated protein 9) and other CRISPR systems can be programmed with a single guide (sg)RNA to introduce DNA breaks at a specified site to inactivate gene function or to stimulate precise DNA editing by homology-directed repair (Knott and Doudna, 2018). Additionally, base and prime editing strategies allow for precise DNA sequence modifications but generally rely on one or more DNA single strand nicks (Anzalone et al., 2020). These technologies have been optimized for targeted changes in the underlying DNA sequence and are therefore ideally suited for repairing or introducing pathogenic mutations. However, the reliance on endogenous DNA repair machinery presents challenges as the complexity of these pathways can make it difficult to limit the outcome to a single desired change (Yeh et al., 2019).
An alternative modality for modulating gene function is to rewrite the epigenetic landscape to control gene expression without changing the underlying DNA sequence. We and others have shown that fusing protein scaffold or enzyme domains to catalytically inactive dCas9 can enhance (CRISPRa) or repress (CRISPRi) transcription in mammalian cells (Holtzman and Gersbach, 2018; Xu and Qi, 2019). Programmable epigenome editing is tunable, reversible, and does not require DNA breaks, effectively bypassing the cellular toxicity associated with gene editing (Jost et al., 2020). However, current programmable epigenome editing technologies typically rely on constitutive expression of dCas9-fusion proteins to maintain transcriptional control. As such, these modalities remain less suitable for therapeutic cell and organismal engineering.
Recent work has demonstrated that it is possible for epigenome editing to write a stable transcriptional program that is remembered and propagated by human cells without constitutive expression of the programmable epigenetic modulators (Amabile et al., 2016; Bintu et al., 2016; Park et al., 2019; Van et al., 2021). In particular, Amabile et al. showed that it was possible to heritably silence human genes by recruitment of a cocktail of DNA methyltransferase and KRAB domains. However, to date only a small number of endogenous human loci have been tested for silencing by epigenetic memory writers (Amabile et al., 2016; O’Geen et al., 2019; Tarjan et al., 2019). Moreover, previous designs of programmable epigenetic silencers utilize either two or three fusion proteins for each target gene, which is experimentally cumbersome—especially for multiplexed gene targeting—and complicates an optimal gene targeting strategy. Furthermore, a TALE-based fusion of KRAB and the DNMT3A and DNMT3L domains resulted in low efficacy long-term gene silencing (Mlambo et al., 2018). Thus, it is unclear how generalizable these approaches are for establishing heritable gene silencing and whether there are genomic features that are required for writing and maintaining heritable epigenetic silencing. We hypothesized that an epigenetic editor composed of a single dead Cas9 fusion would enable us to broadly explore the biology and utility of heritable epigenetic gene silencing.
Here, we present the design, development, and characterization of CRISPRoff, a programmable epigenetic memory writer protein that can durably silence gene expression. Transient expression of CRISPRoff writes an epigenetic program that human cells maintain for more than 450 cell divisions, highlighting that this form of gene silencing is stable and heritable. CRISPRoff epigenetic memories can be reversed robustly using a multi-partite epigenetic editor we call CRISPRon, which removes DNA methylation and recruits transcriptional machinery. Using genome-wide CRISPRoff screens, we show that this approach can durably and specifically silence the large majority of protein-coding genes and has a wide targeting window across gene promoters. Surprisingly, canonical CpG island annotations are not necessary for stable gene silencing by CRISPRoff. Lastly, we demonstrate that CRISPRoff can be used for silencing enhancers and engineering gene silencing programs in human stem cells that persist through differentiation to neurons. More generally, this system allows us to broadly explore the biological rules underlying epigenetic silencing and provides a robust tool for controlling gene expression, targeting enhancers, and exploring the principles of epigenetic inheritance.
RESULTS
Rational design of a single fusion epigenome memory editor
We designed a CRISPR-based programmable epigenome editor protein, termed CRISPRoff-V1, composed of ZNF10 KRAB, Dnmt3A (D3A), and Dnmt3L (D3L) protein domains fused to catalytically inactive S. pyogenes dCas9 (Figure 1A). To test whether a transient pulse of CRISPRoff epigenetic editing could silence gene expression durably, we transiently co-transfected HEK293T cells stably expressing a DNA methylation-sensitive GAPDH-Snrpn GFP reporter with either CRISPRoff-V1, dCas9-KRAB (CRISPRi), or dCas9-D3A-3L, along with sgRNAs targeting the GAPDH-Snrpn synthetic promoter (Liu et al., 2016; Stelzer et al., 2015) (Figure 1B). All three epigenetic editor proteins transiently silenced the GFP reporter (Figure 1C). As expected for a transient transfection, expression of each epigenetic editor protein was lost over time, which for dCas9-KRAB and dCas9-D3A-3L resulted in restored expression of GFP. By contrast, for CRISPRoff-V1, gene silencing memory and CpG island (CGI) methylation was maintained long after CRISPRoff expression was lost (Figure 1C).
Figure 1. Durable and multiplexed gene silencing by CRISPRoff.

(A) A schematic of dCas9 epigenetic editor fusion proteins that were tested for gene silencing activity. 3L denotes Dnmt3L.
(B) Plasmids encoding dCas9 fusions and sgRNAs were co-transfected into HEK293T cells stably expressing a DNA methylation-sensitive Snrpn-GFP reporter. Transfected cells were sorted 2 days after transfection and GFP silencing was monitored over time.
(C) A time course comparing GFP silencing activities of CRISPRoff-V1, dCas9–3A-3L, and dCas9-KRAB.
(D) Bisulfite PCR analysis of the Snrpn locus before or after CRISPRoff targeting. The white circles indicate unmethylated CpG dinucleotides and black circles represent methylated CpG dinucleotides. Each row represents one sequencing read. The red square denotes the sgRNA binding site.
(E) A comparison of CRISPRoff-V1 (black) and CRISPRoff-V2 (blue) editors in silencing the endogenously GFP-tagged H2B gene. The dotted lines represent protein expression of CRISPRoff-V1 and -V2.
(F) A representative flow cytometry plot of H2B-GFP expression of cells at 50 days post-transfection of CRISPRoff V2.
(G) Bisulfite sequencing analysis of a 126 bp region of the H2B CpG island. The red square denotes the sgRNA binding site.
(H) Quantification of cells with ITGB1, CD81, or CD151 silenced 3 weeks post-transfection (p.t.) of CRISPRoff-V1 or -V2 with individual sgRNAs (a-c) or a pool of three sgRNAs (a, b, c).
(I) Quantification of cells with ITGB1, CD81, CD151 silenced 30 days p.t. from single or double gene targeting experiments.
(J) Quantification of multiplexed triple gene silencing by either gating on ITGB1-off cells then gating for CD81- and CD151-off cells (left bar) or by first gating on ITGB1-off cells, then CD151-off cells, and finally CD81-off cells (right bar). The asterisks denote the population of cells with the marked gene turned off.
(K) A representative flow cytometry plot of cells targeted for ITGB1, CD81, and CD151 silencing. Cells were first gated on ITGB1 silencing and the represented population displays CD81 and CD151 silencing.
(L) A histogram plot of CLTA expression at 15 months p.t. showing 38 clones that retained CLTA repression and one clone that reactivated CLTA expression.
The mean values in C, E, H-J were measured from three independent experiments. Error bars represent SD of the mean.
Silencing by CRISPRoff-V1 appeared to be meta-stable as gene expression of the reporter gradually increased with time (Figure 1C). To stabilize gene silencing memory, we encoded CRISPRoff with proteolysis-resistant linkers to minimize proteolysis that could result in untethered D3A-D3L and off-target DNA methylation as previously reported (Galonska et al., 2018; Hofacker et al., 2020). CRISPRoff variants programmed variably durable gene silencing (Figures S1A and S1B). Second, we hypothesized that positioning D3A-3L at the N-terminus of dCas9 would allow Dnmt3A optimal access to CpG sites for DNA methylation (Zhang et al., 2018) (Figure S1C). The CRISPRoff-V2 epigenetic editors we engineered each had similar gene silencing stability so we used CRISPRoff-V2.1 in all subsequent experiments (Figures S1D–S1F).
Transient expression of CRISPRoff-V2 programmed a durable memory of gene silencing for at least 50 days post-transfection, with over 80% of transfected cells silencing the Snrpn-GFP reporter and over 90% silencing the endogenously GFP-tagged gene HIST2H2BE (H2B) (Figures 1E, 1F, and S1G). H2B silencing was accompanied by CGI methylation (Figure 1G). Notably, starting at 10 days post-transfection, no CRISPR-off protein was detected (Figure 1E).
Transfection of CRISPRoff-V2 mRNA also silenced expression of the endogenously GFP-tagged gene CLTA, supporting that transient expression of CRISPRoff leads to effective gene silencing (Figure S1H). These results demonstrate that CRISPRoff epigenetic memory does not depend on sustained transgene expression.
To further compare CRISPRoff-V1 and V2, we silenced three cell surface-localized proteins (ITGB1, CD81, CD151) that are not required for cell proliferation or survival. Transfection of CRISPRoff-V1 with one sgRNA silenced each target gene in a fraction of cells and a pool of three sgRNAs improved silencing of ITGB1 and CD81 (Figure 1H). CRISPRoff-V2 improves silencing of each gene, with at least 80% silencing at 3 weeks post-transfection (Figure 1H).
Durable and multiplexed silencing of endogenous genes
We demonstrated the efficacy of CRISPRoff-V2 in a variety of cell types, namely induced pluripotent stem cells (iPSCs), HeLa, U2OS, and K562 (as a doxycycline-inducible system) (Figures S2A–S2D). We further show that CRISPRoff can be programmed by orthogonal DNA binding proteins: dCas9 from S. aureus (dSauCas9) and dCas12a from Lachnospiracea (dLbCas12a) (Figures S2E and S2F). Silencing with dLbCas12a was improved when three crRNAs (CRISPR RNAs) were encoded as a single transcript that can be processed by dLbCas12a into individual crRNAs, suggesting a route to multiplexed gene silencing.
To explore multiplexed silencing of endogenous human genes with S. pyogenes-based CRISPRoff, we targeted ITGB1, CD81, and CD151 in two, three, and four gene combinations (Figures 1I–1K and S1I–S1K). At 30 days post-transfection, we observed robust multiplexed gene silencing of each gene combination (Figure 1I). We observed that cells that silenced one gene have a higher likelihood of silencing the other targeted genes (Figures 1I–1K, S1I, and S1K). For example, when co-targeting ITGB1 and CD81, cells that successfully silenced ITGB1 had a 25-fold higher percentage of cells that also silenced CD81 compared to cells that failed to silence ITGB1 (Figure 1I).
To measure long term maintenance of epigenetic memory, we targeted the endogenous CLTA gene and followed CLTA expression in single cell clones (Leonetti et al., 2016a). Remarkably, at 15 months post-transfection or after ~450 cell divisions, 38 out of 39 clones maintained silencing of CLTA (Figure 1L).
Epigenome editing is highly specific
To assess the specificity of CRISPRoff, we performed RNA-seq of cells 33 days post-transfection of CRISPRoff and sgRNAs targeting ITGB1, CD81, and CD151 or a negative control sgRNA. Comparison of untransfected cells with cells transfected with CRISPRoff show minimal off-target gene knockdown (Figure 2A). CRISPRoff targeting of ITGB1, CD81, and CD151 were highly specific and showed near complete repression of the targeted gene (Figures 2B–2D and S3A). RNA-seq analyses of three other cell lines with an endogenously GFP-tagged gene repressed by CRISPRoff (RAB11A, CLTA, and H2B) also showed robust and highly specific transcript knockdown (Figures S3B–S3D). Analysis of neighboring genes within a 1 megabase window from the target gene showed no significant changes in gene expression (Figures S3E and S3F). When analyzing the datasets from CRISPRoff targeting of ITGB1, CD81, and CD151, we observed 1–3 non-target transcripts with a log2 fold-change > 2 and adjusted p-value < 0.5 in each gene knockdown experiment, albeit at much lower magnitude compared to the targeted gene (Table S1). Differential expression of non-targeted transcripts may be due to indirect effects associated with target gene knockdown or off-target CRISPRoff activity.
Figure 2. Highly specific and robust transcriptional silencing by CRISPRoff.

(A–D) RNA-seq plots of HEK293T cells transfected with CRISPRoff and non-targeting (NT) sgRNAs compared to sgRNAs targeting (B) ITGB1, (C) CD81, or (D) CD151. A comparison of untransfected cells and CRISPRoff with NT sgRNA is shown in (A. The volcano plots (bottom) display the targeted genes as the most significantly repressed transcripts globally. The data are representative of the average of two independent replicates.
(E) A Manhattan plot displaying differentially methylated CpGs between cells treated with CRISPRoff and CLTA-targeting or NT sgRNAs (30 days post-transfection) analyzed by WGBS. Red dots represent CpGs that gained DNA methylation in targeting sgRNA cells and blue dots represent CpGs that gained DNA methylation in NT sgRNA cells. The arrow denotes the genomic position of CLTA.
(F) A comparison of CpG methylation along a 55 kb window that includes the CLTA locus. Tracks labelled ‘Untr.’ represent untransfected cells; the ‘NT’ tracks represent cells transfected with CRISPRoff and non-targeting sgRNA; the ‘T’ tracks represent cells transfected with CRISPRoff and targeting sgRNA. R1 and R2 represent two technical replicates. Red marks represent methylated (beta-value >0.5) and the blue marks represent unmethylated (<0.5) CpG dinucleotides. CpG islands are shown in green.
(G) A comparison of H3K9me3 ChIP-seq signal across the H2B gene in cells transfected with CRISPRoff and H2B-targeting (purple) or NT sgRNAs (blue) taken at 5 days and 30 days p.t. The sgRNA binding site is denoted along with the CpG islands and neighboring genes. The BOLA1 gene contains two annotated transcriptional start sites, labeled TSS1 and TSS2.
(H) Volcano plot comparing H3K9me3 ChIP-seq data between CRISPRoff transfected with either H2B-targeting or NT sgRNAs. Red dots highlight the genes proximal to the H2B target.
We assessed CRISPRoff DNA methylation specificity by whole genome bisulfite sequencing (WGBS) 30 days post-silencing of CLTA (Figure S3G). We detected a single dominant gain in DNA methylation at the CLTA promoter, in a 1.5 kb window across the CLTA promoter, highlighting the high specificity of CRISPRoff (Figure 2E). We did not detect spreading of DNA methylation into the closest neighboring genes (Figure 2F).
Consistent with previous analyses of DNA methylation in cells treated with DNMT-based epigenetic editors, we observed modestly higher global DNA methylation in CRISPRoff-transfected cells (<2%; Figures S3H and S3I) (Galonska et al., 2018; O’Geen et al., 2019). However, global DNA methylation also varied between cell clones not exposed to CRISPRoff to a similar degree and the differences in local DNA methylation patterns at non-target DNA sites between cell clones was greater than any of the modest, non-specific changes in methylation seen following expression of CRISPRoff (Figure S3I and S3J). To examine whether possible CRISPRoff sgRNA dependent or independent off-target DNA methylation could alter gene expression, we inspected the top 10 most differentially methylated DNA regions (Table S2). We did not detect any transcriptional differences for genes at or near the top 10 non-target differentially methylated regions (Figures S3K and S3L and Table S2). Thus, our WGBS data suggest that any off-target methylation differences are infrequent and unlikely to modulate gene expression or cellular phenotypes.
Lastly, we used ChIP-seq to profile changes in repressive H3K9me3 modifications after CRISPRoff targeting of the HIST2H2BE (H2B) gene. We detected a strong increase in H3K9me3 within a ~5 kb region across the H2B promoter at 5 days post CRISPRoff transfection that was maintained at 30 days, demonstrating the stable propagation of H3K9me3 and DNA methylation as discussed further below (Figure 2G). Comparing H2B-targeting and non-targeting sgRNA conditions showed that the most significant gain of H3K9me3 occurred at the H2B locus and three neighboring genes: HIST2H2AC, HIST2H2AB, and BOLA1 (Figure 2H). We detected knockdown of HIST2H2AC expression whereas sequencing reads mapping to HIST2H2AB were not detected in our RNA-seq data (Table S1). We did not detect transcriptional knockdown of BOLA1 (Table S1). These data, coupled with whole genome bisulfite sequencing data that showed confinement of CpG methylation, highlight the transcriptional and epigenomic specificity of CRISPRoff, while also documenting local epigenetic spreading and maintenance from the target site of establishment.
Gene silencing is reversible by targeted DNA demethylation
An attractive property of epigenome editing is the potential for reversibility (Amabile et al., 2016; Holtzman and Gersbach, 2018; Liu et al., 2016; Xu and Qi, 2019). To test the reversibility of CRISPRoff-mediated gene silencing, we used Cas9-mediated gene editing to inactivate DNMT1—the main DNA methylation maintenance enzyme in mammalian cells—in cells where we had previously silenced H2B, CLTA, or the GAPDH-Snrpn GFP reporter. At 9 days post DNMT1 knockout, 60–80% of cells reactivated gene expression (Figure S4A). Similarly, treatment of cells with a small molecule inhibitor of DNMT1, 5-aza-2′-deoxycytidine (5-aza-dC), reactivated CLTA gene expression (Figure S4B). These results demonstrate that depletion of DNA methylation is sufficient to reverse CRISPRoff-mediated gene silencing and motivated us to optimize programmable tools for reactivation of CRISPRoff-silenced genes.
DNA methylation of a cytosine within a cytosine-guanine dyad can be actively removed by the TET (ten-eleven translocation) family enzymes, which have been repurposed for programmable demethylation of human gene promoters for gene activation (Holtzman and Gersbach, 2018; Maeder et al., 2013; Xu et al., 2016). We tested whether we could reactivate CRISPRoff-silenced genes by targeted DNA demethylation of CLTA. Initially, we used a previously reported dCas9 fusion to the TET1 DNA demethylase catalytic domain (TETv1) (Liu et al., 2016). We co-transfected plasmids expressing TETv1 and sgRNAs targeting the CLTA promoter, measured GFP over time, and observed reactivation of gene expression in 20% of cells (Figures 3A–C and S4C). To improve reactivation, we optimized the fusion protein by repositioning TET1 at the N-terminus of dCas9 and encoding XTEN linkers between TET1 and dCas9. Separating TET1 and dCas9 with an 80 amino acid XTEN80 linker (TETv4) resulted in stable CLTA reactivation in more than 70% of cells (Figures 3C and 3D). Gene reactivation was achieved in up to 60% of TETv4-transfected cells with one sgRNA sequence and was improved by pooling three sgRNAs across the promoter (Figure S4C). Bisulfite sequencing of the CLTA locus before and after dCas9-TET-mediated reactivation showed that the CLTA CGI was nearly completely demethylated after CLTA reactivation (Figures 3E and S4D).
Figure 3. CRISPRon reverses silenced genes by combining DNA demethylation and transcriptional activators.

(A) A schematic of the four TET1 catalytic domain fusions to dCas9 (TETv1-v4) that were tested for reactivation of CRISPRoff-silenced genes.
(B) CRISPRoff-silenced CLTA-GFP cells were transfected with plasmids encoding TETv1–4 and targeting or NT sgRNAs to assess reactivation.
(C) A time course of CLTA reactivation after transfection of each of the four TET fusions in (A). The mean values were measured from three independent experiments. Error bars represent SD.
(D) A representative flow cytometry plot of CLTA reactivation measured at 28 days p.t. of TETv4 and targeting sgRNAs.
(E) Bisulfite-PCR analysis of the CLTA CGI after reactivation by TET1v1 shows high levels of demethylated CpG cytosines (white circles) compared to CRISPRoff-silenced cells.
(F) A schematic of the TETv4 and transactivator ribonucleoprotein complex mediated by a sgRNA encoding two MS2 RNA aptamers. Transactivator domains include monopartite, bipartite, and tripartite architectures of VP64, p65, and Rta.
(G) Fold change in the fraction of CLTA-GFP reactivated cells compared to TETv4 alone, measured two days p.t. The data are calculated from the mean of eight technical replicates from three independent experiments.
(H) Comparison of the expression of CLTA-GFP in single cells, measured by cytometry 28 days p.t. with CRISPRon. The data are aggregated from three technical replicates. * p value < 1e-4, ** p value < 1e-20, *** p value < 1e-100, **** p value = 0, relative to the GFP positive population in the TETv4 condition by the Wilcoxon rank-sum test. Unsilenced CLTA-GFP cells are provided as a benchmark for wild type expression levels.
We observed that CLTA reactivation consistently peaks then stabilizes starting at 9 days post-TET1 transfection (Figure 3C). In an effort to modulate the kinetics of reactivation, we designed a system called CRISPRon, composed of TETv4, a previously reported modified sgRNA that encodes two MS2 stem loop sequences, and the MS2 coat protein (MCP) fused to various combinations of the transactivator domains VP64, p65-AD, and Rta (Chavez et al., 2015; Konermann et al., 2015) (Figures 3F and S4E). We first confirmed that co-expression of dCas9 and MCP-transactivator fusion proteins could increase transcription of the endogenously expressed CLTA gene, indicating that these fusion proteins are functional for recruiting the transcriptional machinery (Figure S4F).
We then expressed negative control (NT) or CLTA-targeting sgRNAs with various CRISPRon combinations or TETv4 only in CLTA silenced cells and monitored CLTA expression over time. We observed that select CRISPRon combinations, such as TETv4 with p65-Rta and TETv4 with VPR, robustly reactivated CLTA expression within 2 days (Figure 3G). At later time points, a CRISPRon combination of TETv4 and Rta reactivated CLTA expression in a larger fraction of cells relative to TETv4 (Figure S4G). By 28 days post-transfection, the median fluorescence of reactivated CLTA-GFP was significantly higher with CRISPRon combinations of TETv4 with Rta and TETv4 with p65-Rta compared to TETv4 only (Figure 3H). We did not detect TETv4 or MCP fusion protein expression at this time point. As a further control, co-expressing the MCP transactivator fusions with dCas9 (no TET), or a single fusion dCas9-VPR, showed only transient activation of CLTA and by 10 days post-transfection, CLTA levels reverted to the silenced state (Figure S4H). Together, these results show that our optimized TET1-dCas9 fusion proteins can robustly reactivate CRISPRoff-silenced genes and that co-recruitment of various transactivator domains modulates the kinetics of reactivation.
Genome-wide targeting of CRISPRoff
The simple design of CRISPRoff motivated us to perform pooled, genome-wide screens to determine its generalizability for silencing genes in the human genome. We designed a sgRNA library that targets over 20,000 protein-coding genes and includes ~1,000 non-targeting sgRNAs (Horlbeck et al., 2016a; Replogle et al., 2020). We constructed the sgRNA library to encode two unique protospacers targeting the same gene per lentiviral vector, as our experiments show improvement in CRISPRoff activity when using multiple sgRNAs targeting the same gene (Replogle et al., 2020) (Figure 4A and Table S3).
Figure 4. Genome-wide gene silencing by CRISPRoff.

(A) A schematic of the dual sgRNA lentiviral vector used in the CRISPRoff genome-wide screens that contains two unique sgRNAs targeting the same gene.
(B) A schematic of a pooled genome-wide screen to determine the targeting landscape of CRISPRoff.
(C) A time course of CLTA expression in HEK293T after transfection of dCas9-KRAB (gray), CRISPRoff-V2 (black), or mutant CRISPRoff-D3AE765A (orange).
(D) A comparison of phenotype scores (γ) between CRISPRoff (y-axis) and CRISPRoff mutant (x-axis) screens. Three types of expected negative controls are highlighted as negative control pseudo-genes (blue), olfactory genes (orange), and Y chromosome genes (green).
(E) A violin plot of the phenotype scores (γ) for genes defined as essential or nonessential from DepMap. Each replicate screen is plotted for CRISPRoff (green) and CRISPRoff mutant (orange).
(F) A plot of true and false positive rates of genes defined as essential by DepMap.
(G) A plot illustrating the distance of an essential gene hit, defined as having a γ ≤ −0.2, from the nearest essential gene hit. Each dot corresponds to a gene hit’s nearest neighboring essential gene, with the x-axis showing the distance between the two genes and the y-axis as the neighboring gene’s phenotype score.
We performed growth-based pooled screens since gene essentiality datasets are available from previous functional genomics efforts, allowing us to compare the performance of CRISPRoff to other genome-wide dropout screens. To perform a CRISPRoff pooled screen, we packaged the sgRNA library into lentiviral particles then transduced and selected HEK293T cells such that on average each cell expresses one sgRNA vector. We then transiently transfected this pool of cells with plasmids encoding CRISPRoff and sorted cells that expressed the CRISPRoff protein. We harvested a population of CRISPRoff-transfected cells as a time zero (T0) sample and continued to passage a population of CRISPRoff-transfected cells for at least 10 cell doublings (T10), followed by deep sequencing of genomic DNA at both time points to read out and quantify the sgRNA sequences as a proxy for cell count. We inferred that sgRNAs that were depleted in the T10 population relative to T0 are active, as these sgRNAs effectively silenced the expression of essential genes and drop out of the population (Figure 4B). As a control, we performed in parallel an identical screen with a CRISPRoff variant encoding the Dnmt3AE765A mutation, which is catalytically inactive and thus unable to maintain durable gene silencing and instead mirrors the transient silencing effect of CRISPRi (Figure 4C). By comparing these two screens, we identified sgRNAs in the CRISPRoff screen that silenced gene expression in a manner that is DNA methylation dependent.
Analysis of the phenotype score (γ, with a more negative score indicating a stronger growth defect) for each gene showed that CRISPRoff expression reproducibly led to a more pronounced growth defect phenotypes compared to the CRISPRoff mutant (Figures 4D, 4E, S5A–S5C, and Table S3). A large set of genes showed drastic growth defects that were specific to CRISPRoff-mediated knockdown, highlighting the durable gene silencing effect of CRISPRoff. We also detected a subset of genes with comparable phenotypes between the two screens, likely due to their essentiality upon transient knockdown by the CRISPRoff mutant (Figure 4D). We evaluated the specificity of silencing across the screens by analyzing the phenotype scores of control sgRNAs. Almost all negative control sgRNAs or sgRNAs targeting unexpressed genes (olfactory or Y chromosome genes) had little to no measured phenotype (1% with γ<−0.2) (Figure 4D).
To evaluate the generality of CRISPRoff for gene silencing, we assessed the phenotype scores of genes that we expect to have growth phenotypes upon knockdown. Analysis of genes associated with DNA replication and the ribosome, which are predicted to be highly essential for cell proliferation, were among the most severe growth phenotypes (Figures S5D and S5E). We then analyzed the phenotypes for common essential genes that are required for cell proliferation or survival in most cancer cell lines (Meyers et al., 2017). The growth defects of these genes were far more pronounced in CRISPRoff (median γ=−0.2) compared to the CRISPRoff mutant (median γ=−0.05), whereas the majority of nonessential genes did not produce growth phenotypes (Figure 4E). The CRISPRoff mutant resulted in weak phenotype scores due to the lack of DNA methylation-dependent durable gene silencing. Collectively, the CRISPRoff screen resulted in high positive rate of calling essential genes with low false positive gene hits, suggesting that CRISPRoff has the ability to silence the majority of genes in the human genome (Figure 4F).
Programmable epigenome editors can initiate epigenetic marks that spread from the site of establishment (Hathaway et al., 2012; Stepper et al., 2017). We wondered whether some CRISPRoff gene hits were due to a “neighboring gene effect” caused by DNA methylation spreading from a nearby essential gene. We catalogued gene “hits” (defined by genes with phenotype scores of −0.2 or lower) and determined their linear distance on the genome from the nearest gene hit. Although a subset of gene hits were within a 1 kb window distance, the majority of CRISPRoff hits were over 10 kb from the nearest gene hit (Figure 4G). Since CpG islands are largely restricted within a 1–2 kb window (Deaton and Bird, 2011), we postulate that the majority of our observed gene hits are specific to the targeted gene promoter, consistent with the specificity demonstrated in our CRISPRoff RNA-seq, WGBS, and ChIP-seq experiments.
CRISPRoff silencing of genes that lack CGI annotations
It is estimated that ~30% of human genes are not associated with a promoter CpG island (CGI) (Deaton and Bird, 2011). Given the observed generality of CRISPRoff for gene silencing, we wondered whether genes that lack CGI annotations can be silenced durably by CRISPRoff. Surprisingly, we found over 300 genes without CGI annotations with growth defects upon knockdown (γ≤−0.1) by CRISPRoff, with 160 producing growth phenotypes γ≤−0.2 (Figure 5A). The majority of these genes had weak to no phenotype in the CRISPRoff mutant screen, indicating that their knockdown is DNA methylation dependent despite the absence of an annotated CGI.
Figure 5. CRISPRoff-mediated silencing of genes without promoter CpG island annotations.

(A) A plot comparing the phenotype score of genes between the CRISPRoff and CRISPRoff mutant screens with genes that lack a CGI annotation highlighted in red.
(B) Histograms of mNeonGreen fluorescence of five HEK293T cell lines, each with the indicated gene endogenously tagged with split mNeonGreen.
(C) Quantification of cells with CALD1, DYNC2LI1, LAMP2, MYL6, or VPS25 silenced after CRISPRoff or CRISPRoff mutant treatment. The data were measured at 14 days p.t., except for VPS25 which was collected at 11 days p.t. due to a growth defect upon gene knockdown.
(D) Quantification of percent of cells with DYNC2LI1 or LAMP2 reactivated after TETv4 treatment with targeting or non-targeting sgRNAs, obtained at 14 days p.t.
(E) CpG methylation profiling within the LAMP2, DYNC2LI1, and MYL6 promoters after CRISPRoff treatments. White circles represent the CpG methylation status of untransfected HEK293T cells. Each dot is an average of eight independent clones.
(F, G, H) Time course plots of DYNC2LI1 (F), LAMP2 (G), and MYL6 (H) expression after transfection of either CRISPRoff or CRISPRoff mutant. Error bars represent the SD of three independent replicates.
(I) A histogram of DYNC2LI1 expression in 33 clonal lines, measured at 50 days p.t. A positive control of untransfected cells is labeled.
(J) A Manhattan plot displaying differentially methylated CpGs between cells treated with CRISPRoff and either DYNC2LI1-targeting or NT sgRNA, as analyzed by WGBS. The arrow points to the genomic location of DYNC2LI1.
(K) A view of a 10 kb genomic window containing the DYNC2LI1 locus, highlighting gain of CpG methylation (red) at the promoter in cells transfected with CRISPRoff and DYNC2LI1-targeting sgRNAs.
To validate our observation that CRISPRoff can silence genes without annotated CGIs, we endogenously tagged five genes with no annotated CGI – CALD1, DYNC2LI1, LAMP2, MYL6, and VPS25 – in HEK293T with mNeonGreen (mNG) and assessed durable silencing by CRISPRoff (Figure 5B). At 14 days post-transfection of CRISPRoff, we detected a large percentage of cells that turned off DYNC2LI1, LAMP2, MYL6, and VPS25 (Figure 5C). We did not detect stable silencing of CALD1 at 14 days, potentially due to its promoter being almost completely devoid of CpG dinucleotides or non-optimal sgRNAs used in the experiment (Figure 5C). Transfection of the CRISPRoff mutant did not silence gene expression durably. Treatment of DYNC2LI1 and LAMP2-off cells with TETv4 led to reactivation of gene expression in ~70% of cells (Figure 5D).
We isolated LAMP2, DYNC2LI1, and MYL6 silenced cells and profiled the DNA methylation status of the gene promoter by bisulfite sequencing. Cytosines within a CG context were highly methylated in silenced cells (Figure 5E). We passaged DYNC2LI1, LAMP2, and MYL6-silenced cells for 30 days and observed stable silencing of DYNC2LI1 and LAMP2 (Figures 5F–5I). We also followed 33 single cell clones with DYNC2LI1 silenced and all clones repressed the gene by 50 days post-transfection with CRISPRoff (Figure 5I). Although MYL6 underwent silencing associated with DNA methylation at an early time point, gene expression reactivated to near pre-CRISPRoff level by day 30. Future studies are needed to understand biological features associated with stable versus metastable epigenetic memory for genes without annotated CGIs.
Lastly, to probe the extent of DNA methylation across a non-CGI annotated gene, we performed WGBS of cells with DYNC2LI1 silenced after 30 days. We detected a single dominant gain in DNA methylation at the DYNC2LI1 promoter (Figure 5J) consisting of a ~1.2 kb region of the promoter (Figure 5K). Together, these data establish that epigenetic editing using CRISPRoff is not limited to genes with canonical CGI annotations and can be targeted to most genes encoded in the human genome. Moreover, based on these findings, we propose that the theoretical framework of CGI gene annotation does not always predict the presence of functional CpG sites, bolstering the power of CRISPRoff and CRISPRon for functional testing of CpG methylation in modulating gene expression.
Targeting rules for CRISPRoff platform
We next explored the targeting landscape of CRISPRoff within gene promoters. Previously, we and others used sgRNA tiling screens and machine learning approaches to show that active sgRNAs for CRISPRi are localized in a narrow window at gene promoters, particularly at a nucleosome-depleted region immediately downstream of the transcription start site (TSS) (Gilbert et al., 2014). Despite successfully using these CRISPRi rules to design the genome-wide CRISPRoff essentiality screens, it remained untested whether the location of effective guides for CRISPRoff was similarly limited to this narrow window.
To empirically determine the targeting window of CRISPRoff, we designed a pooled sgRNA promoter tiling library against a subset of genes that are essential for cell growth based on previous CRISPRi screens in K562 cells and our genome-scale CRISPRoff screen (Figure S6A). The library tiles PAM-containing sequences +/− 1 kb from the TSS of 520 genes (425 with one annotated CGI, 56 with multiple CGIs, and 39 with no annotated CGI; defined by the presence of a CGI within 2.5 kb of the TSS), totaling ~116,000 unique sgRNAs (Figures 6A, 6B, and Table S4). We performed the CRISPRoff screens in HEK293T cells using the same experimental workflow as the genome-wide screens, in parallel with the CRISPRoff D3AE765A methyltransferase mutant. We also transduced the sgRNA tiling library into K562 cells stably expressing dCas9-KRAB and performed a CRISPRi screen to compare gene silencing mediated by dCas9-KRAB alone.
Figure 6. Pooled sgRNA tiling screens reveal a wide targetable window of CRISPRoff-mediated gene repression.

(A) A schematic of the sgRNA library that tiles PAM-containing sgRNAs within a +/− 1 kb window from annotated transcription start sites (TSS).
(B) A summary of the number of genes per indicated category that comprise the tiling sgRNA library.
(C) A comparison of the phenotype score (γ) for genes with annotated CGI between CRISPRoff (y-axis) and CRISPRoff mutant (x-axis). Each dot is the average of the three most active sgRNAs for each gene. The red dots highlight genes that lack a promoter CGI annotation.
(D) An aggregate plot comparing the normalized phenotype score for each sgRNA targeting genes with one annotated CGI. The green line represents screen data from CRISPRoff in HEK293Ts, orange from CRISPRoff mutant in HEK293Ts, and purple from CRISPRi in K562s.
(E, F, G) Representative sgRNA activity score profiles for DKC1, GPN2, and ZCCHC9 from the indicated screen (y-axis). The green bar depicts the annotated CGI obtained from UCSC Genome Browser.
(H) Representative sgRNA activity score profile for ORC5 from the indicated screen (y-axis).
(I) An aggregate plot comparing the normalized phenotype score for each sgRNA for genes without annotated CGIs.
(J) An overlay of normalized sgRNA phenotype score from the CRISPRoff screen (green) with MNase signal that represents nucleosome occupancy (gray). The plot is an aggregate of genes with one annotated CGI.
(K) An overlay of normalized sgRNA phenotype score from the CRISPRoff screen (green) with MNase signal that represents nucleosome occupancy (gray). The plot is an aggregate of the 39 genes with no annotated CGI.
(L) A plot of sgRNA activity along with MNase signal for H2B, derived from the sgRNA tiling screen outlined in Figure S6F.
To evaluate the screens, we calculated the phenotype score (γ) for the three most active sgRNAs per gene and compared phenotypes across the three screens. We first focused on the 425 genes with one annotated CGI, as these were predicted to be canonical targets for CRISPRoff-mediated silencing. The phenotypes for the CRISPRoff screen (median γ = −0.33) were more pronounced compared to the CRISPRoff mutant screen phenotypes (median γ = −0.15), establishing that strong phenotypes observed in the CRISPRoff screen are DNA methylation-dependent (Figure 6C).
To compare the optimal sgRNAs between CRISPRoff and CRISPRi, we normalized the phenotypes across the screens and generated an aggregate plot of sgRNA activities relative to the TSS (Figure 6D). Consistent with our previous work, highly active sgRNAs for CRISPRi were centered on a narrow window (~75 bp) directly downstream of the TSS. Similarly, active sgRNAs for the CRISPRoff mutant mirrored CRISPRi, which we expected because the KRAB domain remains functional in this fusion protein despite the lack of DNA methylation activity. In contrast, the active CRISPRoff sgRNAs were broadly distributed across the TSS, notably within a 1 kb window centered on the TSS. Representative gene analysis of DKC1, GPN2, and ZCCHC9 shows that even within a single promoter, active sgRNAs for CRISPRoff are distributed across −500 to +500 bp from the TSS—a greatly widened targeting window for silencing compared to CRISPRi (Figures 6E–6G). We also observed effective sgRNAs outside the CGI that were DNA methylation-dependent (Figure 6G), suggesting that functional CpGs are not necessarily confined to canonical CGIs as observed in our WGBS data. Our aggregate plot analysis of active sgRNAs targeting the 56 genes with multiple annotated CGIs also shows a broad targeting window, similar to genes with one annotated CGI, and centered at the TSS (Figures S6B and S6C).
Analysis of the 39 genes without promoter CGIs showed many highly active sgRNAs of comparable phenotype strength to genes with annotated CGIs (Figure 6C, colored red) and the phenotypes were strongly diminished in the CRISPRoff mutant screen. A representative gene plot of ORC5 shows that similarly to genes with annotated CGIs, active CRISPRoff sgRNAs are spread across −500 to +500 bp from the TSS (Figure 6H). Moreover, we observed that CRISPRoff has a broadened targeting window despite the lack of an annotated CGI for these 39 genes (Figure 6I). Our experiments demonstrate that the optimal window for CRISPRoff gene silencing is similarly broad for genes with and without annotated CGIs, likely due to low density CpG sites that are functional for methylation-dependent gene silencing as we demonstrated for DYNC2LI1, LAMP2, MYL6, and VPS25 (Figure 5C).
We observed that active sgRNAs are not evenly distributed but instead appear in a periodic pattern within the –500 to +500 bp window, as shown for DKC1, GPN2, and ZCCHC9 (Figures 6E–6G). Overlaying nucleosome occupancy with CRISPRoff sgRNA activity scores for all genes showed that the most active sgRNAs are located in nucleosome-depleted regions of gene promoters, as we and others have shown previously for Cas9 and dCas9-based tools (Figures 6J, 6K, S6D and S6E) (Horlbeck et al., 2016b; Isaac et al., 2016).
To validate that the CRISPRoff targeting window is similar for genes that do not have a growth phenotype upon knockdown, we designed tiling sgRNA libraries spanning +/− 2.5 kb from the TSS to target four endogenous genes: CLTA, H2B, RAB11A, and VIM. For each custom sgRNA library screen, we utilized the corresponding HEK293T cell line that expresses the endogenously GFP-tagged gene (Leonetti et al., 2016b). Each cell line was transduced with the respective sgRNA library, transfected with CRISPRoff, and the cells were passaged for 4 weeks to ensure that gene silencing was durable (Figure S6F). We then sorted GFP positive and GFP negative cell populations for each screen and processed the samples as described above. We calculated sgRNA efficacy by identifying sgRNAs in the gene-off (GFP-) population compared to the gene-on (GFP+) population (Table S5).
Similar to the growth screens, active sgRNAs for CLTA, H2B, and VIM spanned a large window across the TSS (Figures 6L and S6G–S6I). Active CRISPRoff sgRNAs for CLTA were within two distinct regions, with one region upstream of the TSS outside of the annotated CGI (Figure S6G). Unexpectedly, sgRNAs targeting ~2 kb upstream of the H2B TSS were highly active (Figure 6L). Similarly, for VIM, active sgRNAs spanned a 2 kb window +/− 1 kb from the TSS. By contrast, active sgRNAs for RAB11A were constricted to a narrow window at the TSS. Overlaying nucleosome occupancy with sgRNA activity showed that the RAB11A promoter is nucleosome-dense (Figure S6H). From these data, we interpret that CRISPRoff accessibility is restricted by nucleosomes; however, once bound, CRISPRoff can silence gene expression even when distal to the TSS.
Durable gene silencing is dependent on H3K9me3 and DNA methylation maintenance
To explore the mechanism underlying CRISPRoff-mediated heritable memory, we made use of the wide targeting window across the H2B promoter to investigate the establishment, spreading, and maintenance of H3K9me3 histone modifications and CpG methylation marks. We targeted CRISPRoff to the TSS (sgRNA-A) and to a distal site ~2 kb upstream of the TSS (sgRNA-B) (Figure S7A). At 30 days post CRISPRoff transfection, 89% of cells maintained H2B silencing when sgRNA-A was delivered compared to 76% with sgRNA-B (Figure S7B). Using ChIP-seq, we showed that both sgRNAs induced deposition at day 5 and maintenance at day 30 of H3K9me3 across the locus despite the ~2 kb distance between the sgRNA binding sites (Figure S7C). The acquired H3K9me3 modifications in CRISPRoff-treated cells overlapped with the unmethylated CpG region in untreated parental cells (bottom track in Figure S7C). In contrast, deposition and maintenance of H3K9me3 was far weaker with CRISPRoff bearing the D3A mutation, consistent with the failure to sustain gene repression with the mutant (Figure S7D).
We next profiled CRISPRoff-mediated CpG methylation at the targeted distal and TSS regions. At day 5, we detected establishment and spreading of CpG methylation from the site of initiation to a ~2 kb site from the sgRNA binding site (labeled site 1 and site 3, Figures S7E and S7F). By day 30, we detected stable maintenance of DNA methylation at both sgRNA binding sites (Figures S7G and S7H). We also detected a high degree of CpG methylation between the sgRNA binding sites, suggesting a linear movement of spreading from the site of initiation (site 2, Figure S7I). These data, together with our WGBS data, highlight the orchestration of histone and DNA methylation deposition, spreading, and maintenance and suggest that there are underlying regulatory principles that likely depend on the genomic context.
CRISPRoff gene silencing in iPSCs and iPSC-derived neurons
Due to the utility of stem cells for studying the development and function of specific cell types, we employed CRISPRoff in induced pluripotent stem cells. We transfected iPSCs with CRISPRoff and sgRNAs targeting CD81 or a non-targeting control and found that at 30 days post-transfection, many iPSCs had stably silenced CD81 (Figures 7A and 7B). Thus, CRISPRoff-encoded memory of silencing is stably maintained in stem cells.
Figure 7. CRISPRoff gene silencing in iPSCs, iPSC-derived neurons, and enhancers.

(A) An experimental workflow of CD81 knockdown by CRISPRoff in iPSCs, followed by NGN2-mediated differentiation of edited cells into neurons.
(B) Quantification of cells with CD81 silenced by CRISPRi or CRISPRoff with CD81-targeting or NT sgRNAs, measured at 30 days p.t. The error bars represent SD from three independent experiments.
(C) Quantification of cells with CD81 silenced at the indicated time points from (A). The gray bars indicate the percent of iPSC-edited cells with CD81 silenced that were not differentiated during the experiment. The red bars represent cells that were carried through the neuronal differentiation protocol. The error bars represent SD from three independent experiments.
(D) A representative histogram of CD81 expression at days 8 of neuronal differentiation of parental-unedited (gray) or CD81-edited iPSCs (red).
(E) Bisulfite PCR of a 140 bp region of the CD81 promoter in cells transfected with CRISPRoff and NT or CD81-targeting sgRNA.
(F) Representative bright field microscopy images of differentiated neurons derived from iPSCs transfected with CRISPRoff and MAPT-targeting or NT sgRNA.
(G) Quantification of cells with Tau-off in cells transfected with CRISPRoff and NT or MAPT-targeting sgRNA, measured at 10 days post-differentiation.
(H) Representative flow cytometry plots of Tau protein staining in iPSC-derived neurons after CRISPRoff transfection with NT or MAPT-targeting sgRNA. The gates are based on unperturbed iPSC-derived neurons.
(I) A schematic of the PVT1 locus with the promoter and four enhancer elements (E1-E4) labeled with the distance from the TSS.
(J) Plots of normalized PVT1 transcript levels from quantitative RT-qPCR of cells treated with CRISPRoff (left) or CRISPRoff D3A mutant (right) and sgRNAs targeting either the promoter (Pr.) or the four enhancer elements (E1-E4), normalized to control sgRNAs. Asterisks denote statistical significance by t-test and each technical replicate is shown as red dots.
We isolated CD81-off iPSCs and differentiated the cells into neurons, as previously described (Tian et al., 2019) (Figures 7A). We then measured CD81 protein levels at the neural precursor cell stage (day 0 of differentiation) and after cells had differentiated into neurons (8 days post-differentiation). We observed that CD81 remained silenced during and after neuronal differentiation in 90% of cells (Figures 7C and 7D). A similar fraction of undifferentiated iPSCs maintained CD81 silencing during the same time course, suggesting that the reactivation of CD81 in ~10% of cells was not due to the differentiation process. We harvested genomic DNA from CD81-off neurons and detected heavily methylated promoter CpG dinucleotides compared to neurons treated with CRISPRoff and a non-targeting sgRNA (Figure 7E).
We next applied CRISPRoff-mediated editing of iPSC-derived neurons to silence MAPT, a gene that encodes the Tau protein and is implicated in various neurological diseases (Iqbal et al., 2016). MAPT is transcriptionally repressed in iPSCs by H3K27me3 rather than by DNA methylation and H3K9me3 and its expression increases substantially during neuronal differentiation (Guenther et al., 2010). We hypothesized that CRISPRoff could write an epigenetic memory of silencing at the MAPT locus that would persist through neuronal differentiation to silence MAPT in neurons. We transiently transfected CRISPRoff into iPSCs along with sgRNAs targeting MAPT or a non-targeting control (Figure 7F). At day 10 of the differentiation protocol, we measured Tau protein levels and found ~30% of cells with reduced Tau expression compared to a non-targeting control (Figures 7F-H). Together, these data support CRISPRoff-mediated epigenome editing as an applicable technology for rewriting gene expression programs in iPSC-derived cells, especially for modulating gene expression in cells where delivery of gene editing platforms remains a challenge.
CRISPRoff targeting of enhancer elements
Finally, we explored the potential utility of CRISPRoff for silencing promoter-distal elements by targeting enhancers that control the expression of the PVT1 long noncoding RNA (Cho et al., 2018; Fulco et al., 2016). We transiently expressed CRISPRoff in the MDA-MB-231 breast cancer cell line with sgRNAs targeting either the PVT1 promoter or at four previously identified enhancer elements downstream of the PVT1 promoter: E1 (+15.5 kb), E2 (+60 kb), E3 (+105 kb), and E4 (+113 kb) (Figure 7I). We detected a significant reduction of PVT1 transcript levels with sgRNAs targeting E1, E3, and E4 (~40–60%) compared to 80% knockdown with promoter-targeting sgRNAs (Figure 7J, left). In contrast, parallel experiments using the CRISPRoff-Dnmt3AE765A mutant resulted in less robust knockdown (Figure 7J, right). These results highlight the potential use of CRISPRoff for mapping and dissecting the functions of enhancer elements and noncoding regulatory elements in the human genome.
DISCUSSION
Here, we present CRISPRoff and CRISPRon, two technologies for programmably writing and erasing epigenetic memories to control gene expression programs. Transient expression of CRISPRoff writes a robust, specific, and multiplexable gene silencing program that is memorized by cells through cell division and differentiation, which can be rapidly reversed by CRISPRon. We show that CRISPRoff can specifically and robustly silence the large majority of human genes. Our initial experiments demonstrate CRISPRoff can perturb enhancers, opening the potential to target genome elements that control tissue-specific gene expression (Fulco et al., 2016; Tarjan et al., 2019).
Our finding that targeted DNA methylation outside of annotated CGIs can lead to robust memorized gene silencing extends the canonical model of methylation-based gene silencing, which posits that a high density of CpG methylation is a requirement for stable propagation of silencing (Boyes and Bird, 1992). Although CRISPRoff-mediated writing of DNA methylation and histone modification are artificially programmed, our results motivate the need to define functional DNA methylation and dissect regulatory DNA elements and other host factors that mediate initiation, spreading, and maintenance of histone and DNA methylation marks. For example, targeting CRISPRoff 2 kb upstream of the H2B TSS leads to acquisition and maintenance of H3K9me3 and DNA methylation marks at the same genomic positions as targeting CRISPRoff directly proximal to the TSS, pointing to the existence of preexisting boundaries that restrict epigenetic spreading. By allowing the initiation of silencing at a defined time and genomic location, CRISPRoff provides a unique tool for addressing these and other fundamental questions regarding the mechanism and biological role of heritable gene silencing in mammalian cells (Audergon et al., 2015; Iglesias et al., 2018; Ragunathan et al., 2015; Yu et al., 2018).
CRISPRoff provides a valuable complement to existing CRISPRi and CRISPR nuclease approaches (Doench, 2018; Hanna and Doench, 2020; Shalem et al., 2015). CRISPRoff gene silencing can lead to effectively complete nulls without inducing a DNA damage response facilitating multigene targeting screens or therapeutic cell engineering (Ihry et al., 2018). The ability to target CRISPRoff to a large window upstream of the TSS allows access to promoter SNPs that could be utilized for allele-specific targeting of disease-associated mutations. This could broadly enable approaches to silence dominant negative alleles. Similarly, silencing of long noncoding RNAs and regulatory RNAs provides a new avenue for stable reprograming of gene expression. Silencing of inhibitory elements such as antisense transcripts, could result in a heritable increase in expression of some genes, potentially enabling therapeutic efforts to mitigate haploinsufficiency or imprinting disorders (Buiting et al., 2016). More broadly, heritable epigenetic silencing provides a general tool for rewiring human gene expression programs.
Limitations of Study
Most of our CRISPRoff experiments, including the genome-wide screens, were performed with sgRNAs previously predicted to be optimal for CRISPRi (Horlbeck et al., 2016a; Replogle et al., 2020). We envision that future efforts to design optimal sgRNAs for CRISPRoff will further improve gene silencing activity. Moreover, we performed our experiments in polyclonal cell lines, which inherently have clonal variations in DNA methylation. It will be valuable to explore potential clonal differences in methylation and the effects on CRISPRoff activity and potential off-target sites. Lastly, we highlight MYL6 as a non-CGI gene that has metastable silencing over time. It remains unknown exactly how many genes are amenable to stable versus metastable silencing and future efforts are needed to identify the regulatory features that dictate the stability of programmed epigenetic memory.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contacts, Luke Gilbert (luke.gilbert@ucsf.edu) and Jonathan Weissman (weissman@wi.mit.edu).
Materials Availability
The CRISPRoff and CRISPRon plasmids are available from Addgene: #167981 for CRISPRoff-V2.1 and #167983 for TETv4.
Data and Code Availability
The RNA-seq, ChIP-seq, and whole genome bisulfite sequencing data are available on GEO (GSE168012).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture, DNA transfections, and flow cytometry
All cell lines were cultured at 37°C with 5% CO2 in tissue culture incubators. HEK293T (female), HeLa (female), and U2OS (female) cells were cultured in Dulbecco’s modified eagle medium (DMEM) in 10% FBS (HyClone or VWR), 100 units/mL streptomycin, 100 μg/ml penicillin, and 2 mM glutamine. K562 (female) cells were maintained in RPMI-1640 with 25 mM HEPES and 2.0 g/L NaHCo3 in 10% FBS, 2 mM glutamine, 100 units/mL streptomycin, and 100 mg/mL penicillin. WTC Gen1c iPSCs (male) were cultured in mTESR media (STEMCELL Technologies) under feeder-free conditions on growth factor-reduced Matrigel (BD Biosciences). Cells were passaged using Accutase (STEMCELL Technologies) and seeded on Matrigel coated plates with mTESR media supplemented with p16-Rho-associated coiled-coil kinase (ROCK) inhibitor Y-27632 (10 μM; Selleckchem).
Lentiviral particles were produced by transfecting standard packaging vectors into HEK293T using TransIT-LT1 Transfection Reagent (Mirus, MIR2306). Media was changed 24 hours post-transfection with complete DMEM supplemented with 15 mM HEPES. Viral supernatants were harvested 48–60 hours after transfection and filtered through a 0.45 μm PVDF syringe filter. Lentiviral infections included polybrene (8 μg/ml).
METHOD DETAILS
Plasmid Design and Construction
The dCas9 and KRAB sequences were obtained from a previous CRISPRi construct (Gilbert et al., 2013). The D3A and D3L sequences, including the D3A-D3L fusion, originated from (Stepper et al., 2017) and were assembled with dCas9 and KRAB DNA sequences into a CAG-expression plasmid using NEBuilder® HiFi DNA Assembly (NEB). The D3A domain consists of the N612-V912 of the Homo sapiens DNMT3A protein (N608-V908 of the Mus musculus Dnmt3a protein) and the D3L domain consists of G208-L421 of the Mus musculus Dnmt3l protein. The TETv1 design was constructed by PCR amplification of the dCas9-TET1CD sequence from Fuw-dCas9-Tet1CD (Addgene #84475) and assembled into a CAG-expression plasmid. XTEN linker sequences were previously published (Schellenberger et al., 2009). All CRISPRoff and TET1 fusion proteins include BFP as either a direct fusion or with a P2A-cleavage sequence to measure transfection efficiency by flow cytometry. The dSaCas9 (D10A, N508A) sequence was PCR amplified from pX603 (Addgene #61594) and the dLbCas12a sequence was PCR amplified from (Tak et al., 2017). VP64, p65, and Rta were PCR amplified from SP-dCas9-VPR (Addgene #63798). The GAPDH-Snrpn-GFP lentiviral reporter originated from Addgene #70148 (Liu et al., 2016; Stelzer et al., 2015).
The sgRNA plasmids were constructed by restriction cloning of protospacers downstream of a U6 promoter using BstXI and BlpI cut sites, as previously described. The sgRNA expression plasmids also express a T2A-mCherry marker to measure transfection efficiency. The sgRNA sequences used for CRISPRoff and CRISPRon experiments are listed in Table S6. The sgRNA sequences were chosen based on our previous algorithm to predict active CRISPRi sgRNAs (Horlbeck et al., 2016a).
The MS2 plasmids were constructed by first transferring the mU6 promoter-sgRNA-EF1a-puromycin-T2A-mCherry cassette into a non-lentiviral vector by restriction cloning. The MCP-XTEN80-NLS-VPR-2xP2A cassette was ordered as four gBlocks (IDT) and cloned into the aforementioned non-lentiviral plasmid by Gibson assembly. The sgRNA-MS2 loop sequence was designed based on the SAM system (Konermann et al., 2015) with the BstXI and BlpI restriction sites incorporated from our previous mU6 sgRNA expression design (Addgene #84832). The DNA sequence encoding the MS2-sgRNA scaffold is 5’-GTTTAAGAGCTAaGCCAACATGAGGATCACCCATGTCTGCAGGGCaTAGCAAGTTTA AATAAGGCTAGTCCGTTATCAACTTGGCCAACATGAGGATCACCCATGTCTGCAGGG CCAAGTGGCACCGAGTCGGTGCTTTTTTT-3’. For construction of the transactivator plasmids, each domain or combination of domains was PCR amplified and cloned by Gibson assembly into a plasmid that encodes the sgRNA and MS2 coat protein (MCP). Guide sequences were cloned by double digest of the vector and ligation of annealed oligos, as previously described.
All mRNA constructs were synthesized using the mMESSAGE mMachine™ T7 Ultra Transcription Kit (Thermo Fisher Scientific). The T7 promoter sequence (5’-TAATACGACTCACTATAGG-3’) was first cloned upstream of the CRISPRoff sequence. The T7-CRISPRoff sequence was PCR amplified and used as template for in vitro synthesis reactions. Following the manufacturer protocol for synthesis, the reactions were cleaned by chloroform extraction and isopropanol precipitation.
CRISPRon and CRISPRoff transfections and analysis
Transient transfection experiments in HEK293T were performed in 24-well plates using TransIT-LT1 Transfection Reagent (Mirus) and Opti-MEM™ I Reduced Serum Medium (Thermo Fisher Scientific). Cells at 70–80% confluency were transfected with 300 ng of plasmid encoding CRISPRoff, dCas9-KRAB, or dCas9-D3A-3L and 150 ng of plasmids encoding sgRNAs. CRISPRoff experiments in HeLa and U2OS cells were performed by nucleofection of plasmids using the SE Cell Line 96-well Nucleofector Kit (Lonza) and a 96-well Shuttle™ Device (Lonza), per manufacturer protocol. Transfected cells were sorted 2 days after transfection using a BD FACSAria II or FACSAria Fusion and sorted cells were passaged every 2–3 days to measure durability of gene silencing. Experiments that compare the silencing activity of different CRISPRoff fusions (Figures 1E, 4C, S1B, and S1F) were performed in cells that stably express the targeting sgRNA to normalize sgRNA expression. To generate cell lines stably expressing sgRNAs, cells were transduced with lentiviral particles that express the sgRNAs and sorted for sgRNA-positive cells 2–3 days after transduction.
Quantification of ITGB1, CD81, and CD151 protein levels were measured by cell surface antibody staining of live cells. Cells were incubated with APC- or PE-labeled antibody (BioLegend) for ~30 min in the dark at RT, washed twice with PBS containing 10% FBS, and protein expression was measured on a BD LSR II flow cytometer.
For CRISPRon experiments, 1×105 CLTA-GFP-silenced HEK293T cells were seeded in each well of a 24-well plate. When cells reached 60–80% confluency the next day, cells were transfected with 500 ng of dCas9 plasmid (dCas9 or TETv1–4) and 300 ng of sgRNA-transactivator plasmid (sgRNA only, VP64, p65, Rta, VP64-p65, p65-Rta, or VPR) using TransIT-LT1 Transfection Reagent (Mirus) and Opti-MEM™ I Reduced Serum Medium (Thermo Fisher Scientific). Cells were monitored for BFP (dCas9 or TETv1–4) and mCherry (guide-transactivator) expression 24 hours after transfection. Two days post-transfection, 7.5×104 BFP and mCherry double positive cells were sorted using a BD FACSAria Fusion. Cells were allowed to recover for 4 days after the sort and were subsequently analyzed by flow cytometry every 2–3 days on an Attune NxT cytometer (Thermo Fisher Scientific).
All flow cytometry data were analyzed using FlowJo and the raw FACS plots presented in the figures are in log10 scale.
PVT1 enhancer targeting
Quantitative RT-PCR quantification of PVT1 expression was done as described in (Cho et al., 2018). Briefly, MB-MDA-231 cells were transfected with CRISPR DNA together with a sgRNA vector using Neon (1400 volt, 10 ms, 4 pulse). Double positive cells were sorted after 2 days and continued to culture for 3 days. RNA were extracted with Zymo spin column and gene expression was quantified with SYBR qPCR mix (LightCycler 480) using 45 ng of RNA. The expression of PVT1 were normalized to GAPDH gene in ddCt method. T-test was used to calculate the statistical significance based on 3–5 biological replicates per condition. The primer sequence used are: PVT1 forward (CGAGCTGCGAGCAAAGAT), PVT1 reverse (CGTGTCTCCACAGGTCACAG), GAPDH forward (GGGCTCTCCAGAACATCATC), GAPDH reverse (CCTGCTTCACCACCTTCTTG). The sgRNAs targeting PVT1 enhancers 1–4, promoter, and lambda (controls) were from (Cho et al., 2018) and listed on Table S6.
RNA sequencing
HEK293T cells that have maintained stable silencing of target genes were harvested 33 days (ITGB1, CD81, and CD151) or 28 days (CLTA, HIST2H2BE, RAB11A, and VIM) post CRISPRoff transfection. Cells were dislodged from plates with PBS, centrifuged at 500 × g for 5 min and washed again with PBS. Total RNA was extracted using Direct-zol RNA MiniPrep (Zymo R2051). Library preparations were carried out using TruSeq Stranded mRNA Library Preparation Kit (Illumina RS-111–2101), starting with 1000 ng total RNA. Final libraries were assessed using a 2100 Bioanalyzer (Agilent), quantified using Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific), and sequenced as single end 50 base pair reads on a HiSeq 4000 (Illumina). For processing the sequencing reads, linker sequences (AGATCGGAAGAGCACACGTCTGAACTC) were removed using FASTX-clipper (FASTX-Toolkit). The reads were then aligned to the human genome (GRCh37) using the STAR (Spliced Transcripts Alignment to a Reference, version 2.5) aligner against the Gencode Gene V24lift37 transcriptome annotation. Read quantification was carried out with featureCounts (Liao et al., 2014). All downstream analyses were performed with Python (version 2.7) using a combination of Numpy (v1.12.1), Pandas (v0.17.1), and Scipy (v0.17.0) libraries. Knockdown efficiency was calculated by normalizing gene Transcripts per Million (TPM) for the experimental samples with the mean TPM of the control (non-targeting) samples. Differential expression analysis was performed using DESeq2 (Love et al., 2014). We note that non-target differentially expressed transcripts are lowly expressed genes.
Chromatin immunoprecipitation and analysis
At 30 days post transfection, 10×106 cells were crosslinked with 1% formaldehyde for 10 min at room temperature and quenched with 1.25 M glycine. Crosslinked cells were washed twice with cold PBS containing 1% Halt™ protease inhibitors (Thermo Fisher Scientific) and the cell pellets were flash frozen at −80 °C until sample preparation. Cells were lysed in lysis buffer (5 mM PIPES pH 8, 85 mM KCl, 1% Igepal, 1% protease inhibitors) for 10 min on ice. Nuclei were isolated after spinning the suspension at 2000 rpm at 4 °C for 5 min. Nuclei were lysed at 4 °C for 10 min in nuclei lysis buffer (50 mM Tris pH 8, 10 mM EDTA, 1% SDS, 1% protease inhibitors). Chromatin shearing was performed at 4 °C using a Diagenode Bioruptor® Pico sonication device in 1.5 ml Bioruptor® Pico Microtubes. The shearing program was optimized to obtain 200–700 bp fragments (30 seconds on, 30 seconds off for 10 cycles). The sonicated samples were centrifuged at 13,000 rpm at 4 °C for 10 min and the supernatant was collected and diluted 5-fold in IP dilution buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Igepal, 0.25% deoxycholate, 1% protease inhibitors). A fraction of the input was saved and frozen (4% of total) prior to proceeding to immunoprecipitation. Immunoprecipitations were performed overnight at 4 °C using 5 μg of anti-H3K9me3 antibody (abcam ab8898). The washing steps were performed with Pierce™ Protein A/G magnetic beads (Thermo Fisher Scientific) using the following protocol: once with IP wash buffer 1 (20 mM Tris pH 8, 2 mM EDTA, 50 mM KCl, 1% Triton X-100, 0.1% SDS), twice with high salt buffer (20 mM Tris pH 8, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.01% SDS), once with IP wash buffer 2 (10 mM Tris pH 8, 1 mM EDTA, 0.25 lithium chloride, 1% Igepal, 1% deoxycholate), and twice with TE buffer (10 mM Tris pH 8, 1 mM EDTA). Samples were eluted in elution buffer (50 mM sodium bicarbonate, 1% SDS) at 65 °C for 1 hour and reversed crosslinked initially in 300 mM NaCl and RNase A for 1 hour at 37 °C, followed by 63 °C overnight with Proteinase K (Thermo Fisher). The DNA samples were purified using a Zymo Clean & Concentrator kit and libraries were prepared using the NEBNext® Ultra™ II DNA Library Prep kit (NEB).
Reads were aligned to the human genome (hg19) using bowtie v2.3.2 (Langmead and Salzberg, 2012). Alignments were processed using deepTools2 bamCoverage (Ramírez et al., 2016), normalizing reads to 1x average coverage. The resulting bigWig files were visualized on the Integrative Genomics Viewer (IGV). Peak analysis was performed using MACS (https://github.com/macs3-project/MACS). Downstream analysis and enrichment at promoter regions, which were defined as +/− 2 kb of each TSS (with the TSSs based on previously published annotations (Horlbeck et al., 2016) were performed using Python 3.6 with the deepTools2 package. Differential H3K9me3 enrichment was analyzed using DESeq2 (Love et al., 2014), treating distinct sgRNAs against the same TSS as replicate samples. We note that BOLA1 contains two TSS annotations and our ChIP-seq data show enrichment for H3K9me3 near TSS1, the closest to the H2B promoter, and no enrichment for TSS2 located 15 kb away (Figures 2G and 2H). We do not detect transcriptional knockdown of BOLA1 in our RNA-seq data (Table S1).
Western blotting
Western blots of CRISPRoff constructs were performed by harvesting HEK293T cells 2 days post-transfection of CRISPRoff constructs. Cells were washed with cold PBS and lysed with RIPA buffer (Thermo Fisher Scientific) supplemented with Halt™ Protease Inhibitor Cocktail (Thermo Fisher Scientific). After 30 min of lysis at 4 °C, the samples were centrifuged at 20,000 × g for 20 min at 4 °C. The soluble fractions were collected and protein concentrations were quantified by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). 40 μg of total protein was mixed and heated with SDS loading buffer, separated on Bolt™ 4–12% Bis-Tris Plus Gels (Thermo Fisher Scientific), and wet transferred into PVDF membrane in buffer containing 1× MOPS and 10% methanol. Membranes were blocked with Odyssey® Blocking Buffer (LI-COR), incubated with antibodies against S. pyogenes Cas9 (Active Motif 61577) and calnexin (Abcam ab22595) at 4 °C overnight. Membranes were washed at least 3 times with blocking buffer before incubation with IRDye® secondary antibodies against Cas9 and calnexin. Blots were imaged using Odyssey® CLx (LI-COR).
Bisulfite sequencing PCR
Genomic DNA was extracted from ~1–2×106 cells according to manufacturer’s instructions using the PureLink Genomic DNA Mini Kit (Invitrogen). For each condition, 1 μg genomic DNA underwent bisulfite conversion and cleanup according to manufacturer’s instructions using the EpiTect Bisulfite kit (Qiagen). Purified bisulfite-converted DNA was amplified using EpiMark Hot Start Taq (NEB). Amplicons were gel purified using a Gel DNA Recovery Kit (Zymo) and PCR amplified again using EpiMark Hot Start Taq. Amplicons were cloned into the pCR2.1 TOPO vector according to manufacturer’s instructions using the TOPO TA Cloning Kit (Invitrogen). Cloning products were transformed into Stellar E. coli cells (Takara) and plated on carbenicillin plates with X-gal for blue-white screening. Colonies were picked per condition and sequenced by Sanger sequencing. Primer sequences for bisulfite-PCR amplification are listed in Table S7. The primer sequences for amplifying the GAPDH-Snrpn fragment was obtained from (Liu et al., 2016).
Whole genome bisulfite sequencing and analysis
We generated whole genome bisulfite sequencing (WGBS) libraries for 12 samples, corresponding to WT (untreated), NT (non-targeting), and T (targeting) for CLTA and DYNC2LI1 experiments and profiled in two replicates. After DNA extraction and RNAse A treatment using the PureLink Genomic DNA Mini Kit (Invitrogen), 1 μg of DNA was diluted to 7.7 ng/μL in 130 uL and sheared to 450–550 bp length using a Covaris E220 evolution with intensifier for 50s at 140V, 7°C, 10% amplitude, 200 cycles. The sonicated DNA was recovered and concentrated using Ampure XP beads and sizes of the sheared DNA were checked on an Agilent TapeStation device with a D1000 HS DNA ScreenTape. Next, the sheared DNA was bisulfite converted using the EZ DNA Lightning kit (Zymo, Cat. No. D5030) according to the manufacturer’s instructions and a desulphonation step of 16 minutes. Then, 500 ng of sheared and converted DNA was subjected to library preparation using a Swift Accel®-NGS Methyl-Seq DNA Library Kit (Cat. No. 30024) and Methyl-Seq Unique Dual Indexing Primers (Cat. No. 39096). The prepared libraries were quantified using a KAPA Library Quantification kit (Roche, Cat. No. KK4873) and sequenced using paired-end 150bp reads (300 cycles) on an Illumina NovaSeq6000 instrument with an S4 flow cell and a 35% spike-in from another non-WGBS library to diversify the sample pools. We obtained on average 707M paired-end reads (range 629–835M) across all 12 libraries.
Prior to alignment, sequencing reads were trimmed using fastp (version 0.21.0, (Chen et al., 2018)) and the following parameters: --adapter_sequence=AGATCGGAAGAGCACACGTCTGAACTCCAGTCA -adapter_sequence_r2=AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT --trim_front1=0 --trim_tail1=20 --trim_front2=20 --trim_tail2=0. Trimming is required to remove bases that are added during the Adaptase reaction that could affect alignment and DNA methylation calling. Processed reads were aligned to the hg38 reference genome using methylCtools (version 1.0.0, https://github.com/hovestadt/methylCtools, (Hovestadt et al., 2014)) and bwa mem (version 0.7.17, arXiv: 1303.3997v1) using default parameters. Over 98% of reads were aligned as proper pairs across samples. After marking of PCR duplicates using sambamba (version 0.8.0, (Tarasov et al., 2015)), genome-wide CpG methylation values were called using methylCtools using the --trimPE parameter. Average CpG coverage was ~25-fold across samples. Bisulfite conversion efficiency was estimated to be greater than 99.5% based on non-CpG methylation.
Downstream analyses were performed in R (version 4.0.2, https://www.r-project.org/) using the bsseq (version 1.24.4, (Hansen et al., 2012)) and DSS (version 2.36.0, (Park and Wu, 2016)) packages. Specifically, we applied the DMLtest function to first call differentially methylated loci (using 500bp smoothing windows) between treatments, and then the callDMR function to define differentially methylated regions. Results were visualized by plotting log10-transformed p-values associated with individual loci (positive values for loci that gained methylation, negative values for loci that lost methylation). Loci were colored by their difference in beta-values (−1: blue, 0: white, +1: red). Close-ups of genomic regions were generated by visualizing beta-values of individual loci in IGV (Robinson et al., 2011). Data was displayed as bar charts (min/0: blue, mid/0.5, max/1: red).
Cas9 genome editing and 5-aza-dC treatments
Lentiviral particles expressing Cas9 from S. pyogenes were transduced into HEK293T cells that have CRISPRoff-silenced Snrpn-GFP or GFP-tagged CLTA and H2B. Cas9-expressing cells, marked by BFP fluorescence in the lentivirus vector, were FACS-sorted. To inactive DNMT1, lentiviral particles expressing a sgRNA that targets DNMT1 were infected into the cell lines. Reactivation of the silenced genes was assessed by GFP expression, measured by flow cytometry. The last time point was taken at 9 days post sgRNA infection, as cell viability was severely reduced past this time point.
For 5-aza-dC treatment, 1×105 CRISPRoff-silenced CLTA-GFP HEK293T cells were seeded in each well of a 24-well plate. 24 hours later, the media was replaced with media supplemented with aqueous 5-aza-2’-deoxycytidine (5-aza-dC) (MP Biomedicals). The following day, 5-aza-dC-containing media was aspirated, cells were detached and analyzed for viability and GFP expression on an Attune NxT flow cytometer (Thermo Fisher Scientific). Cells were subsequently passaged with fresh media every 2–3 days and analyzed by flow cytometry.
Genome-wide CRISPRoff screen and analysis
For genome-wide CRISPRoff screens, we constructed a compact library to maximize on-target knockdown while minimizing overall library size. We targeted each gene in the human genome with two unique sgRNAs expressed from tandem U6 expression cassettes in a single vector. To select the optimal sgRNAs targeting each gene, we relied on our previously published hCRISPRi v2.1 library (Horlbeck et al., 2016a). A three tiered approach was used to balance empirical data with computational predictions and select the most active sgRNA pair for each gene. First, for strong essential genes (p-value<0.001 and gamma<−0.2 in hCRISPRi v2 growth screen), sgRNAs were ranked by growth. Next, for genes that were identified as a significant hit in previous CRISPRi screens, sgRNAs were ranked by the sum of Z-scored phenotypes across screens. Finally, for all other genes, sgRNAs were ranked by the regression scores in hCRISPRi v2.1. Using this ranking scheme, we designed a genome-wide library consisting of only 21378 elements (20360 targeting elements plus 1018 non-targeting controls). A list of protospacer sequences in the genome-wide library is available in Table S3.
To clone our libraries, we began by generating a modified single sgRNA lentiviral expression vector, pJR104, from the parental pJR85 (Addgene 140095) by: (i) replacing the BFP fluorescent marker with a BsmBI-negative GFP, (ii) replacing the sgRNA constant region with an unmodified constant region (i.e. without a Perturb-seq capture sequence), and (iii) incorporating a UCOE element upstream of the EF1alpha promoter to prevent silencing. Dual-sgRNA oligos were synthesized as an oligonucleotide pool (Twist Biosciences) with the structure: 5’- PCR adapter - CCACCTTGTTG - protospacer A - gtttcagagcgagacgtgcctgcaggatacgtctcagaaacatg - protospacer B - GTTTAAGAGCTAAGCTG - PCR adapter-3’. Oligo pools were PCR-amplified, digested with BstXI/BlpI, gel extracted, ligated into the sgRNA lentiviral vector pJR104, and transformed to generate an intermediate library as previously described (Replogle et al., 2020). An insert, pJR98, consisting of a sgRNA constant region variant 3 (Adamson et al., 2016) and a hU6 promoter was synthesized (IDT), BsmBI-digested, and ligated into the BsmBI-digested intermediate library. The final dual-guide library was then transformed for amplification and sequenced by next-generation sequencing to ensure library representation and uniformity.
Pooled tiling sgRNA screens in HEK293T cells were performed by first transducing cells with lentiviral particles encoding the sgRNA library. The infection efficiency was measured 2 days post-infection by flow cytometry, aiming for 20–30% sgRNA-positive cells. The screens were performed with two technical replicates and each sgRNA was represented by at least 1000 cells throughout the duration of the screens. Two days post transduction, cells were treated with puromycin until the cell population was 90% sgRNA positive, as marked by mCherry encoded in the lentiviral vector. For transient transfection of CRISPRoff, ~8×106 cells were first seeded on 15 cm2 plates. About 20–24 hr later (70–80% confluency), each 15 cm2 plate of cells were transfected with 20 μg of plasmids encoding CRISPRoff or CRISPRoff-Dnmt3AE765A catalytic mutant. Two days post-transfection, cells were sorted for CRISPRoff expression (BFP) and plated on 15 cm2 plates. Four days post-sorting, cells were trypsinized and an aliquot of cells (~110×106) was harvested as an initial time point T(0) and the rest of the cell population was passaged for at least 10 more cell doublings. Cells were then collected as a final time point (T10).
DNA libraries of T(0) and T(10) were prepared for deep sequencing essentially as previously described (Jost et al., 2020). Briefly, genomic DNA was isolated using a NucleoSpin Blood XL kit (Macherey–Nagel). Then, isolated gDNA was directly amplified by 23 cycles of PCR using NEBNext Ultra II Q5 PCR MasterMix (NEB), appending Illumina adaptors and unique sample indices (oJR234 forward primer: 5’-AATGATACGGCGACCACCGAGATCTACACCGCGGTCTGTATCCCTTGGAGAACCAC CT-3’; index primers 5’-CAAGCAGAAGACGGCATACGAGATnnnnnGCGGCCGGCTGTTTCCA GCTTAGCTCTTAAA-3’). Sequencing was performed on a NovaSeq 6000 (Illumina) using a 19 bp read 1, 19 bp read 2, and 5 bp index read 1 with custom sequencing primers oJR326 (custom read 1, 5’- CGCGGTCTGTATCCCTTGGAGAACCACCTTGTTGG-3’), oJR328 (custom read 2, 5’- GCGGCCGGCTGTTTCCAGCTTAGCTCTTAAAC-3’), and oJR327 (custom index read 1, 5’- GTTTAAGAGCTAAGCTGGAAACAGCCGGCCGC-3’).
Sequencing counts from CRISPR screens were processed to calculate gene phenotypes using a custom Python script, similar to as previously described (Horlbeck et al., 2016a), except now two protospacer sequences are matched instead of just one. In this case, gene phenotype is the same as sgRNA phenotype, and is defined by log2 sgRNA enrichment / cell doublings. The calculated phenotype scores are reported in Table S3. All additional CRISPR screen data analyses and plotting were performed in Python 3.6 using a combination of Numpy (v1.16.2), Pandas (v0.23.4), Scipy (v1.4.1), and sklearn (v0.22.2). The DepMap essential and nonessential genes were downloaded from DepMap Public 20Q2 at https://depmap.org/portal/download/ (Blomen et al., 2015; Hart et al., 2014). Gene set enrichment analysis (GSEA) was performed using GSEAPY (v0.9.19) in Python using the 2019 Human KEGG Pathway database.
Tiling screen library design, experimental specifications, and analysis
For the growth-based screen, a tiling sgRNA library targeting essential genes was designed based on our previously published genome-wide CRISPRi screen in K562s. For genes with no canonical CGIs (as defined by no CGIs within 2.5 kb of TSS, with the TSSs based on previously published annotations (Horlbeck et al., 2016a) and CGI annotations from the UCSC Genome Browser), all genes with an average growth phenotype score less than −0.2 were picked. For genes with one or multiple CGIs (also defined as within 2.5 kb of TSS), genes with an average growth phenotype score between −0.2 and −0.4 were selected. In total, 39 genes with no canonical CGIs, 425 genes with one annotated CGI, and 56 genes with multiple CGIs were chosen. A list of protospacer sequences comprising the library is available in Table S4.
For each gene, all sequences +/−2.5 kb (or +/− 1 kb depending on the position and length of the CGI) of the TSS containing 19 bp followed by an NGG PAM were extracted as potential sgRNAs. All sequences were prepended with a 5’ G to enable robust transcription from the U6 promoter, whether or not this base was present in the genomic sequence. The sgRNAs were scored for off-target sites using weighted Bowtie, as previously described (Horlbeck et al., 2016a). Briefly, sgRNAs were scored by uniqueness in the genome, as determined by an empirically derived and experimentally verified scoring metric: PAM G1 = 40, PAM G2 = 19, PAM N = 0, the next 7 bases from the PAM = 28, the next 5 bases = 19, and the last 7 bases = 10. A mismatch score was then calculated by the sum of the mismatches with the scoring metric. This mismatch score was implemented using the Phred score threshold feature of Bowtie using the --nomaqround, -n 3, -l 15, -a, and --best flags. For the most stringent threshold, sgRNAs were required to have no more than 1 alignment (the sgRNA target site itself) in the genome with a mismatch score of 39. Control non-targeting sgRNAs were extracted from a previously tested list of control sgRNAs (Horlbeck et al., 2016a).
The tiling libraries for endogenously GFP-tagged CLTA, HIST2H2BE (H2B), RAB11A, and VIM were designed similarly, selecting for sgRNAs +/− 2.5 kb from the TSS and yielding ~600 sgRNAs per gene. The protospacer sequences for each GFP-tagged gene library are available in Table S5.
Oligonucleotide pools were designed with flanking PCR and restriction sites (BstXI and BlpI), synthesized by Agilent Technologies, and cloned into the sgRNA expression vector pCRISPRia-v2 (Addgene #84832), as described previously (Horlbeck et al., 2016a). The expression vector contains a U6 promoter driving the sgRNA expression, as well as an EF1α promoter driving puromycin T2A-mCherry.
The tiling screens in HEK293Ts were performed in a similar workflow as the genome-wide CRISPRoff screens. To perform the tiling screen in K562s, cells stably expressing dCas9-KRAB were first transduced with lentiviral particles of the tiling sgRNA library. Two days post transduction (20–30% infection), cells were treated with puromycin until the population consisted of 90% sgRNA-expressing cells. A T(0) time point was then collected and cells were continued to passage for 10 more cell doublings to obtain the T(10) time point.
Sequencing counts from CRISPR screens were processed using the Python-based ScreenProcessing pipeline (https://github.com/mhorlbeck/ScreenProcessing), as previously described (Horlbeck et al., 2016a) to calculate sgRNA phenotypes. sgRNA phenotype score is defined by log2 sgRNA enrichment / cell doublings. All additional CRISPR screen data analyses and plotting were performed in Python 2.7 using a combination of Numpy (v1.12.1), Pandas (v0.17.1), and Scipy (v0.17.0). K562 and GM12878 MNase-seq data was obtained from the ENCODE consortium as processed continuous signal data (BigWig file format; Michael Snyder lab, Stanford University). The average of the K562 and GM12878 MNase-seq data was used. The sequencing counts of each protospacer are available in Table S4 and the calculated phenotype scores are available in Table S4. We note that the phenotypes in K562 CRISPRi (median γ = −0.46) are more pronounced compared to the HEK293T CRISPRoff screen (median γ = −0.33). However, as we have previously demonstrated, because the genes were chosen based on essentiality in K562s, this difference likely can be attributed to cell type variability between K562 and HEK293T.
GFP-tagged sgRNA tiling screen
The tiling sgRNA screens in HEK293T GFP-tagged cell lines were performed in a similar workflow as the growth-based screens described above. The previously published endogenously GFP-tagged cell lines (CLTA, HIST2H2BE, RAB11A, VIM) were further FACS sorted to yield >99% GFP-positive cells to minimize background GFP-negative cells. After generating cell lines that stably express the respective sgRNA library, plasmids expressing CRISPRoff were transfected. Two days later, the transfected cells were sorted and subsequently passaged for 4 weeks by trypsinization every 2–3 days. At the 4 weeks time point, each cell line had the following detectable GFP-silenced population: 21.8% CLTA, 22.7% HIST2H2BE, 3.05% RAB11A, and 24.7% VIM. The GFP-on and GFP-off populations were FACS sorted into separate bins, collecting ~2×106 cells per population for each cell line. The log2 fold change in sgRNA abundance was quantified by the presence of each sgRNA in the GFP-on population compared to the total population. Analysis was performed using Python 2.7, similar to the other tiling screens described previously. The deep sequencing counts from each screen and the calculated phenotype scores are available in Table S5.
iPSC manipulation and neuronal differentiation
Transient transfections of iPSCs were performed in 6-well plates using TransIT-LT1 Transfection Reagent (Mirus). First, a mixture of 0.5 ml of mTeSR1 and 2 μl of 10 mM Y-27632 ROCK inhibitor were added to each well of a Matrigel coated 6-well plate. Then, a mixture of plasmids encoding dCas9-KRAB or CRISPRoff (1 μg), 1 μg of sgRNA plasmids, and 200 ng of plasmid encoding BCL-XL (Li et al., 2018) were added to 0.4 ml of Opti-MEM™. TransIT-LT1 (12 μl) was added to the DNA-Opti-MEM™ mixture and added to each well of a 6-well plate. Cells at 70–80% confluence were lifted with Accutase, washed with DPBS, and counted using a Countess (Thermo Fisher AMQAX1000). About 1.5×106 cells were resuspended in 1 ml of mTeSR1 and added to each well containing the transfection mixtures. Transfected cells were sorted 3 days post-transfection on a BD FACS Fusion and plated in mTeSR media supplemented with 10 μM Y-27632 ROCK inhibitor.
Neuronal differentiations were performed on passage number 46 iPSCs using doxycycline-inducible NGN2 (Tian et al., 2019). On day −3, cells at 70–80% confluency were lifted with Accutase and washed with DPBS. About 7.5×105 cells were resuspended in 2 ml of N2 Pre-differentiation Media each well of a Matrigel coated 6-well plate. On day 0, cells were lifted with Accutase and washed with DPBS. About 5×105 cells were resuspended in 2 ml classic N2/B27 Differentiation Media and plated onto Poly-D-Lysine coated plates (Corning 354413). On day 3, the media in each well were aspirated and replaced with 2 ml of fresh N2/B27 Differentiation Media. On day 7, 1 ml of media was removed from each well and replaced with 1 ml of fresh N2/B27 Differentiation Media. N2 Pre-differentiation Media was made with 1X Knockout DMEM/F12 (Thermo Fisher 11320–033), 1X NEAA (Thermo Fisher 11140–050), 1X N2 Supplement (Thermo Fisher 17502–048), 10 ng/ml NT-3 (PreproTech 450–03), 10 ng/ml BDNF (PreproTech 450–02), 1 μg/ml Mouse Laminin (Thermo Fisher 23017–015), 10 nM Y-27632 ROCK inhibitor, and 2 μg/ml doxycycline (Sigma-Aldrich D3447–500MG). Classic N2/B27 Differentiation Media was made with 0.5X DMEM/F12 (Thermo Fisher 10888–033), 0.5X Neurobasal-A (10888–022), 1X NEAA, 0.5X GlutaMAX (Thermo Fisher 35050–061), 0.5X N2 Supplement, 0.5X B27-VA Supplement (Thermo Fisher 12587010), 10 ng/ml NT-3, 10 ng/ml BDNF, 1 μg/ml Mouse Laminin, and 2 μg/ml doxycycline.
We used iNeuron RNA-Seq (https://kampmannlab.ucsf.edu/ineuron-rna-seq) to support the activation of MAPT gene expression through neuronal differentiation of iPS cells.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical tests and number of independent replicates per experiment are indicated in the figure legends. The statistical significance from RNA-seq, ChIP-seq, and whole genome bisulfite sequencing experiments are detailed in the Methods section.
ADDITIONAL RESOURCES
Not applicable
Supplementary Material
(A) A schematic of the CRISPRoff-V1 construct and various linker sequences used to generate protein variants.
(B) A time course of CLTA-GFP silencing after transfection of CRISPRoff-V1 variants or controls dCas9-KRAB (gray) and dCas9-D3A-D3L (orange).
(C) A crystal structure of DNMT3A (orange) and DNMT3L (yellow) in complex with CpG-containing DNA (PDB 5YX2). The arrows point to the dCas9 attachment positions for CRISPRoff-V1 and CRISPRoff-V2.
(D) A schematic of four CRISPRoff-V2 constructs that varies BFP as a linker between dCas9 and KRAB or separated from CRISPRoff by a P2A sequence. The V2.3 and V2.4 constructs encode NLS sequences at the amino and carboxyl termini of dCas9.
(E) A western blot of dCas9, dCas9-KRAB, CRISPRoff construct protein expression.
(F) A time course of CLTA-GFP silencing after transfection of the V1 and V2.1, V2.3, V2.4 constructs, along with dCas9-KRAB and dCas9 only controls.
(G) A time course of Snrpn-GFP silencing after transfection of dCas9-D3A-3L (orange), dCas-9KRAB (gray), or CRISPRoff-V1 (black) and V2 (blue).
(H) A representative flow cytometry plot of HEK293T CLTA-GFP cells 6 days after transfection of mRNA encoding CRISPRoff.
(I) Representative flow cytometry plots of multiplexing gene targeting of two genes simultaneously, measured at 30 days post-transfection.
(J) Quantification of gene silencing measured at 31 days post-transfection of CRISPRoff with four simultaneous sgRNAs targeting ITGB1, CLTA, CD81, and CD151.
(K) Quantification of cells with CLTA, CD81, and CD151 silenced in cells that have ITGB1 either silenced (blue) or unsilenced (gray) in the four gene knockdown experiment.
The error bars in B, F, G, J, and K represent SD from three independent experiments.
(A) Flow cytometry histograms of CRISPRoff expression (BFP) before and after doxycycline (dox) treatment. After 24 hours of dox administration, the media was replaced with media without doxycycline (1d and 2d post dox-wash) to turn off CRISPRoff expression.
(B) Quantification of K562 cells with CD81 silenced 10 days after initial dox-induction of CRISPRoff expression. Doxycycline was included in the media for either 3 days (middle) or 4 days (right) prior to washing cells to remove doxycycline.
(C) Quantification of cells with ITGB1, CD81, or CD151 silenced in HeLa and U2OS measured at 18 days post-CRISPRoff-V1 transfection with sgRNAs targeting the indicated genes.
(D) A representative flow cytometry plot of CD81 expression in iPS cells after transfection of CRISPRoff with either non-targeting sgRNAs or sgRNAs targeting CD81.
(E, F) A comparison of cells with CLTA (E) and H2B (F) silenced 10 days after transfection of CRISPRoff with dCas9 from S. pyogenes (dSpyCas9) or S. aureus (dSauCas9) or dCas12a from Lachnospiraceae bacterium (dLbCas12a).
The error bars in B, C, E, and F are SD from three independent experiments.
(A) An RNA-sequencing TPM (transcripts per kilobase million) plot for HEK293T cells transfected with CRISPRoff and NT (non-targeting) sgRNAs compared to untransfected HEK293T cells.
(B, C, D) RNA-sequencing TPM (transcripts per kilobase million) are plotted for HEK293T cells transfected with CRISPRoff and either NT sgRNAs compared to sgRNAs targeting CLTA (B), HIST2H2BE (C), or RAB11A (D). The data are representative of the average of two independent replicates.
(E, F) Representation of gene expression changes +/– 1000 kb from the annotated targeted gene for CD81, CD151, ITGB1 (E) and RAB11, HIST2H2BE, CLTA (F). Each box represents a gene.
(G) A barplot of genome-wide average CpG coverage obtained from whole genome bisulfite sequencing. WT indicates untransfected cells; NT indicates CRISPRoff delivered with non-targeting sgRNAs; T indicates CRISPRoff delivered with sgRNAs targeting CLTA or DYNC2LI1. The experiments were performed in two replicates. CLTA experiments are presented in Figure 2, DYNC2LI1 experiments are presented in Figure 5.
(H) A barplot of genome-wide average CpG methylation (beta-values) in the samples described in Figure S3G.
(I) Heatmap plots comparing average CpG methylation (beta-values) of 20-CpG sliding windows between untransfected (WT) replicates (left), WT and NT (middle), and WT and targeting sgRNAs (right) for CLTA (top row) and DYNC2LI1 (bottom row) experiments. Red color indicates highest density and blue color indicates lowest density. White areas indicate the absence of windows with the respective average methylation levels.
(J) A heat map showing pairwise correlations between genome-wide CpG methylation profiles of the samples described in Figure S3G, including replicates. Samples are sorted by unsupervised hierarchical clustering. Dark brown color indicates highest correlation, light yellow color indicates lowest correlation. This analysis highlights the variations in global CpG methylation between samples for CLTA and DYNC2LI1 experiments.
(K) A close up of a 17 kb genomic region containing the ZSCAN16 gene. DNA methylation profiles for untransfected, NT, and targeting sgRNA cells from the CLTA experiment are shown. The box highlights a differentially methylated region at the gene promoter, indicating a gain of CpG methylation in the targeting sgRNA cells compared to the control cells.
(L) A close up of a 30 kb genomic region containing the RPS6KA6 gene. The box highlights a gain of CpG methylation at the promoter in the targeting sgRNA cells. A CpG island located in the promoter of the gene is indicated in green.
(A) A bar plot of HEK293T cells reactivating CRISPRoff-silenced H2B (black), Snrpn-GFP (gray), or CLTA (blue) 9 days after Cas9-mediated knockout of DNMT1.
(B) Time course measurements of cells with reactivated CLTA expression after increasing doses of 5-aza-dC in HEK293T cells with CLTA silenced by CRISPRoff.
(C) A comparison of CLTA-GFP expression 28 days post transfection of the four TET fusions in Figure 3A co-transfected with one sgRNA sequence or a pool of three sgRNAs. Error bars represent the range of two technical replicates.
(D) A schematic of the CLTA CGI (green) with sgRNA binding sites annotated (a, b, c). The lollipop plot shading represents the percent of CpG methylation, as measured by bisulfite-PCR of CRISPRoff-silenced cells. Gene annotations were obtained from UCSC Genome Browser.
(E) A schematic of vectors that express a CLTA-targeting sgRNA and MS2 coat protein (MCP) fusion to various transcriptional activators.
(F) Violin plots that represent median CLTA-GFP fluorescence 2 days post-transfection of sgRNAs targeting CLTA and either dCas9 or dCas9 and MCP-fused transactivators into cells with endogenously expressed CLTA-GFP.
(G) A time course of HEK293T cells with CLTA-GFP reactivation after transfection of sgRNAs targeting CLTA and either TETv4 only, or TETv4 along with various MCP-fused transactivator domains into cells with CRISPRoff-silenced CLTA. Untreated cells are represented in white circles.
(H) A time course of HEK293T cells with CLTA-GFP reactivation after transfection of sgRNAs targeting CLTA and either dCas9-VPR or dCas9 along with various MCP-fused transactivator domains, or untransfected cells. The transfections were performed in the absence of TETv4 to measure persistent gene activation in the absence of DNA demethylation.
The error bars in G and H are SD from three independent experiments.
(A) A plot comparing the phenotype score (γ) of genes between technical replicates of the CRISPRoff (left) and CRISPRoff D3A methyltransferase mutant (right) genome-wide screens. The negative control sgRNAs are highlighted in blue.
(B) Violin plots of the phenotype score (γ) of all genes from each screen.
(C) A histogram of the number of genes with the indicated phenotype score (γ) from the CRISPRoff and CRISPRoff mutant screens. The light green and light orange lines correspond to the phenotype scores of negative control sgRNAs.
(D, E) Gene set enrichment analysis (GSEA) for genes associated with DNA replication and ribosome, confirming enrichment of expected essential genes. Genes are ranked from lowest (red) to highest (blue) phenotype scores.
(A) The genes chosen for the sgRNA tiling screens are highlighted on a volcano plot depicting gene phenotype scores from previous genome-wide CRISPRi screens in K562 cells (Horlbeck et al. 2016). The colors represent genes with one (orange), multiple (purple), or no annotated CGI (green).
(B) An aggregate plot comparing the normalized phenotype score for each sgRNA for genes with multiple CGIs. The green line represents screen data from CRISPRoff transfection into HEK293T cells, orange from CRISPRoff mutant into HEK293T, and purple from K562 CRISPRi.
(C) Representative sgRNA activity score profile for TBCD from the three screens. The green bar depicts the annotated CGIs obtained from UCSC Genome Browser.
(D, E) Plots overlaying the sgRNA phenotype scores and MNase signals for GFER and IMMT.
(F) An experimental workflow of tiling sgRNA screens to determine optimal sgRNAs for four endogenously GFP-tagged genes: CLTA, H2B, RAB11A, and VIM. The indicated population in the histograms represent the population of cells that have maintained gene silencing 4 weeks after CRISPRoff transfection.
(G, H, I) Overlay plots of sgRNA activity and MNase signal for CLTA, RAB11A, and VIM from the sgRNA tiling screen.
(A) A schematic of the H2B promoter with two sgRNA sites annotated: sg-A at the TSS and sg-B located ~2 kb upstream of the TSS. The CpG island spans 1.4 kb. Sites 1, 2, and 3 represent regions probed for CpG methylation by bisulfite PCR as described in Figures S7E–S7I.
(B) A time course of H2B silencing after transfection of CRISPRoff with sg-A or sg-B.
(C) A comparison of H3K9me3 profiles at the H2B (HIST2H2BE, colored red) promoter at day 5 (green tracks) and day 30 (purple tracks) post-transfection of CRISPRoff with sg-A, sg-B, or non-targeting (NT) sgRNA. The sgRNA binding sites are labeled, along with CpG island annotations (green), and the basal unmethylated CpG region of the H2B promoter prior to transfection obtained from WGBS of WT untransfected cells in Figure 2.
(D) A comparison of H3K9me3 profiles at the H2B promoter, as described in C, except in experiments using CRISPRoff with a D3A methyltransferase mutation.
(E, F) Quantification of CpG methylation at site 1 and site 3 of the H2B promoter (labeled in S7A) in cells transfected with CRISPRoff and either sg-A (blue), sg-B (orange), or non-targeting (gray) sgRNA. The cells were harvested for bisulfite PCR at day 5 post transfection.
(G, H, I) Quantification of CpG methylation at sites 1, 2, and 3 of the H2B promoter, obtained at 30 days post transfection.
Table S1. A list of statistics from RNA-seq experiments, related to Figure 2. The first tab contains differentially expressed genes in the ITGB1, CD81, and CD151 RNA-seq datasets. The second tab contains RNA-seq statistics for HIST2H2BE and the neighboring genes.
Table S2. A list of differentially methylated CpG regions from whole genome bisulfite sequencing, related to Figure 2. The list consists of data comparing CRISPRoff delivered with CLTA-targeting or non-targeting sgRNA. The table also contains statistics from the CLTA RNA-seq data that pertain to differentially methylated regions.
Table S3. A list of sgRNA protospacer sequences and calculated phenotype scores from the genome-wide CRISPRoff screens, related to Figure 4.
Table S4. A list of sgRNA protospacer sequences, deep sequencing counts, and calculated phenotype scores from the genome scale sgRNA tiling screens, related to Figure 5. Each tab contains the counts from the three genome scale sgRNA tiling screens: HEK293T CRISPRoff, HEK293T CRISPRoff mutant, and K562 CRISPRi.
Table S5. A list of sgRNA protospacer sequences, deep sequencing counts, and calculated phenotype scores from the tiling screens across the promoters of GFP-tagged CLTA, HIST2H2BE (H2B), RAB11A, and VIM, related to Figure 6.
Table S6. A list of sgRNA sequences used for CRISPRoff and CRISPRon experiments, related to Figures 1–7.
HIGHLIGHTS.
CRISPRoff is a single fusion protein that programs heritable epigenetic memory
CRISPRoff can heritably silence most genes, including genes without CpG islands
CRISPRoff is highly specific and has a broad targeting window across gene promoters
CRISPRoff epigenetic memory persists through differentiation of iPSCs into neurons
ACKNOWLEDGMENTS
We are grateful to Alex Ge, Matt Laurie, Christina Liem, Chris Hsiung, and Albert Xu for technical assistance, and Tessa Bertozzi, Bruce Conklin, Sandy Johnson, Barbara Panning, Fyodor Urnov, and members of the Weissman and Gilbert labs for helpful discussions. Funding: DARPA Safe Genes and PREPARE programs (LAG, JSW), the HHMI Hanna H. Gray Fellows Program (JKN), the Jane Coffin Childs Memorial Fund (JC), NIH K99/R00 GM134154 (JC), NIH F31 NS115380 (JMR), NSF Graduate Research Fellowship (GNR and AC), NIH F32 AG063487 (AJS), NIH RM1-HG007735 (HYC), NIH DP1CA216873 (BEB), NIH 1RM1HG009490 (JSW), NIH DP2 CA239597 (LAG), NIH DP2 GM119139 (MK). LAG is supported by a Goldberg-Benioff Endowed Professorship. JSW and HYC are HHMI Investigators. BEB is the Bernard and Mildred Kayden Endowed MGH Research Institute Chair, and an American Cancer Society Research Professor. This study was supported in part by UCSF HDFCCC Laboratory for Cell Analysis Shared Resource Facility (NIH P30CA082103) and by the Gene Regulation Observatory at the Broad Institute. The IGI supported oligonucleotide pools.
Footnotes
DECLARATION OF INTERESTS
JKN, JC, GCP, LA, and JSW have filed patent applications related to CRISPRoff, CRISPRon, and CRISPRi/a screening. JMR consults for Maze Therapeutics. LAG, JSW, HYC, and BEB consult for and hold equity in Chroma Medicine. JSW declares outside interest in KSQ Therapeutics, Maze Therapeutics, Amgen, and Tessera Therapeutics. MK serves on the Scientific Advisory Boards of Engine Biosciences, Casma Therapeutics, and Cajal Neuroscience. BEB declares outside interests in Fulcrum Therapeutics, Arsenal Biosciences, HiFiBio, and Cell Signaling Technologies. HYC is a co-founder of Accent Therapeutics, Boundless Bio, and is an advisor for 10x Genomics, Arsenal Biosciences, and Spring Discovery.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile A, Migliara A, Capasso P, Biffi M, Cittaro D, Naldini L, and Lombardo A. (2016). Inheritable Silencing of Endogenous Genes by Hit-and-Run Targeted Epigenetic Editing. Cell 167, 219–232.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Koblan LW, and Liu DR (2020). Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844. [DOI] [PubMed] [Google Scholar]
- Audergon PNCB, Catania S, Kagansky A, Tong P, Shukla M, Pidoux AL, and Allshire RC (2015). Restricted epigenetic inheritance of H3K9 methylation. Science 348, 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bintu L, Yong J, Antebi YE, McCue K, Kazuki Y, Uno N, Oshimura M, and Elowitz MB (2016). Dynamics of epigenetic regulation at the single-cell level. Science 351, 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Boyes J, and Bird A. (1992). Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl-CpG binding protein. EMBO J. 11, 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buiting K, Williams C, and Horsthemke B. (2016). Angelman syndrome — insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 12, 584–593. [DOI] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer E, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. (2015). Highly-efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinforma. Oxf. Engl. 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Xu J, Sun R, Mumbach MR, Carter AC, Chen YG, Yost KE, Kim J, He J, Nevins SA, et al. (2018). Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, and Bird A. (2011). CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG (2018). Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 19, 67–80. [DOI] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galonska C, Charlton J, Mattei AL, Donaghey J, Clement K, Gu H, Mohammad AW, Stamenova EK, Cacchiarelli D, Klages S, et al. (2018). Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Frampton GM, Soldner F, Hockemeyer D, Mitalipova M, Jaenisch R, and Young RA (2010). Chromatin Structure and Gene Expression Programs of Human Embryonic and Induced Pluripotent Stem Cells. Cell Stem Cell 7, 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RE, and Doench JG (2020). Design and analysis of CRISPR–Cas experiments. Nat. Biotechnol. 38, 813–823. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, and Irizarry RA (2012). BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Brown KR, Sircoulomb F, Rottapel R, and Moffat J. (2014). Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol. Syst. Biol. 10, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, and Crabtree GR (2012). Dynamics and memory of heterochromatin in living cells. Cell 149, 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker D, Broche J, Laistner L, Adam S, Bashtrykov P, and Jeltsch A. (2020). Engineering of Effector Domains for Targeted DNA Methylation with Reduced Off-Target Effects. Int. J. Mol. Sci. 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman L, and Gersbach CA (2018). Editing the Epigenome: Reshaping the Genomic Landscape. Annu. Rev. Genomics Hum. Genet. 19, 43–71. [DOI] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016a). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. ELife 5, e19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Witkowsky LB, Guglielmi B, Replogle JM, Gilbert LA, Villalta JE, Torigoe SE, Tjian R, and Weissman JS (2016b). Nucleosomes impede Cas9 access to DNA in vivo and in vitro. ELife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovestadt V, Jones DTW, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz H-J, et al. (2014). Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–541. [DOI] [PubMed] [Google Scholar]
- Iglesias N, Currie MA, Jih G, Paulo JA, Siuti N, Kalocsay M, Gygi SP, and Moazed D. (2018). Automethylation-induced conformational switch in Clr4 (Suv39h) maintains epigenetic stability. Nature 560, 504–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, et al. (2018). p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, and Gong C-X (2016). Tau and neurodegenerative disease: the story so far. Nat. Rev. Neurol. 12, 15–27. [DOI] [PubMed] [Google Scholar]
- Isaac RS, Jiang F, Doudna JA, Lim WA, Narlikar GJ, and Almeida R. (2016). Nucleosome breathing and remodeling constrain CRISPR-Cas9 function. ELife 5, e13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M, Santos DA, Saunders RA, Horlbeck MA, Hawkins JS, Scaria SM, Norman TM, Hussmann JA, Liem CR, Gross CA, et al. (2020). Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat. Biotechnol. 38, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GJ, and Doudna JA (2018). CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, and Huang B. (2016a). A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. U. S. A. 113, E3501–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, and Huang B. (2016b). A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. 113, E3501–E3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-L, Li G-H, Fu J, Fu Y-W, Zhang L, Chen W, Arakaki C, Zhang J-P, Wen W, Zhao M, et al. (2018). Highly efficient genome editing via CRISPR–Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 46, 10195–10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, and Jaenisch R. (2016). Editing DNA Methylation in the Mammalian Genome. Cell 167, 233–247.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Angstman JF, Richardson ME, Linder SJ, Cascio VM, Tsai SQ, Ho QH, Sander JD, Reyon D, Bernstein BE, et al. (2013). Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 31, 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlambo T, Nitsch S, Hildenbeutel M, Romito M, Müller M, Bossen C, Diederichs S, Cornu TI, Cathomen T, and Mussolino C. (2018). Designer epigenome modifiers enable robust and sustained gene silencing in clinically relevant human cells. Nucleic Acids Res. 46, 4456–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Geen H, Bates SL, Carter SS, Nisson KA, Halmai J, Fink KD, Rhie SK, Farnham PJ, and Segal DJ (2019). Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin 12, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, and Wu H. (2016). Differential methylation analysis for BS-seq data under general experimental design. Bioinforma. Oxf. Engl. 32, 1446–1453. [DOI] [PubMed] [Google Scholar]
- Park M, Patel N, Keung AJ, and Khalil AS (2019). Engineering Epigenetic Regulation Using Synthetic Read-Write Modules. Cell 176, 227–238.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Jih G, and Moazed D. (2015). Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348, 1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T. (2016). deepTools2: a next generation web server for deepsequencing data analysis. Nucleic Acids Res. 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Replogle JM, Norman TM, Xu A, Hussmann JA, Chen J, Cogan JZ, Meer EJ, Terry JM, Riordan DP, Srinivas N, et al. (2020). Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat. Biotechnol. 38, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger V, Wang C-W, Geething NC, Spink BJ, Campbell A, To W, Scholle MD, Yin Y, Yao Y, Bogin O, et al. (2009). A recombinant polypeptide extends the in vivo half-life of peptides and proteins in a tunable manner. Nat. Biotechnol. 27, 1186–1190. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, and Zhang F. (2015). High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 16, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer Y, Shivalila CS, Soldner F, Markoulaki S, and Jaenisch R. (2015). Tracing Dynamic Changes of DNA Methylation at Single-Cell Resolution. Cell 163, 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, and Jurkowski TP (2017). Efficient targeted DNA methylation with chimeric dCas9–Dnmt3a–Dnmt3L methyltransferase. Nucleic Acids Res. 45, 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak YE, Kleinstiver BP, Nuñez JK, Hsu JY, Horng JE, Gong J, Weissman JS, and Joung JK. (2017). Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors. Nat. Methods 14, 1163–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P. (2015). Sambamba: fast processing of NGS alignment formats. Bioinforma. Oxf. Engl. 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarjan DR, Flavahan WA, and Bernstein BE (2019). Epigenome editing strategies for the functional annotation of CTCF insulators. Nat. Commun. 10, 4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, Prabhu AV, Fernandopulle MS, Patel R, Abshari M, et al. (2019). CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 104, 239–255.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van MV, Fujimori T, and Bintu L. (2021). Nanobody-mediated control of gene expression and epigenetic memory. Nat. Commun. 12, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, and Qi LS (2019). A CRISPR–dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 431, 34–47. [DOI] [PubMed] [Google Scholar]
- Xu X, Tao Y, Gao X, Zhang L, Li X, Zou W, Ruan K, Wang F, Xu G, and Hu R. (2016). A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CD, Richardson CD, and Corn JE (2019). Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 21, 1468–1478. [DOI] [PubMed] [Google Scholar]
- Yu R, Wang X, and Moazed D. (2018). Epigenetic inheritance mediated by coupling of RNAi and histone H3K9 methylation. Nature 558, 615–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z-M, Lu R, Wang P, Yu Y, Chen D, Gao L, Liu S, Ji D, Rothbart SB, Wang Y, et al. (2018). Structural basis for DNMT3A-mediated de novo DNA methylation. Nature 554, 387–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) A schematic of the CRISPRoff-V1 construct and various linker sequences used to generate protein variants.
(B) A time course of CLTA-GFP silencing after transfection of CRISPRoff-V1 variants or controls dCas9-KRAB (gray) and dCas9-D3A-D3L (orange).
(C) A crystal structure of DNMT3A (orange) and DNMT3L (yellow) in complex with CpG-containing DNA (PDB 5YX2). The arrows point to the dCas9 attachment positions for CRISPRoff-V1 and CRISPRoff-V2.
(D) A schematic of four CRISPRoff-V2 constructs that varies BFP as a linker between dCas9 and KRAB or separated from CRISPRoff by a P2A sequence. The V2.3 and V2.4 constructs encode NLS sequences at the amino and carboxyl termini of dCas9.
(E) A western blot of dCas9, dCas9-KRAB, CRISPRoff construct protein expression.
(F) A time course of CLTA-GFP silencing after transfection of the V1 and V2.1, V2.3, V2.4 constructs, along with dCas9-KRAB and dCas9 only controls.
(G) A time course of Snrpn-GFP silencing after transfection of dCas9-D3A-3L (orange), dCas-9KRAB (gray), or CRISPRoff-V1 (black) and V2 (blue).
(H) A representative flow cytometry plot of HEK293T CLTA-GFP cells 6 days after transfection of mRNA encoding CRISPRoff.
(I) Representative flow cytometry plots of multiplexing gene targeting of two genes simultaneously, measured at 30 days post-transfection.
(J) Quantification of gene silencing measured at 31 days post-transfection of CRISPRoff with four simultaneous sgRNAs targeting ITGB1, CLTA, CD81, and CD151.
(K) Quantification of cells with CLTA, CD81, and CD151 silenced in cells that have ITGB1 either silenced (blue) or unsilenced (gray) in the four gene knockdown experiment.
The error bars in B, F, G, J, and K represent SD from three independent experiments.
(A) Flow cytometry histograms of CRISPRoff expression (BFP) before and after doxycycline (dox) treatment. After 24 hours of dox administration, the media was replaced with media without doxycycline (1d and 2d post dox-wash) to turn off CRISPRoff expression.
(B) Quantification of K562 cells with CD81 silenced 10 days after initial dox-induction of CRISPRoff expression. Doxycycline was included in the media for either 3 days (middle) or 4 days (right) prior to washing cells to remove doxycycline.
(C) Quantification of cells with ITGB1, CD81, or CD151 silenced in HeLa and U2OS measured at 18 days post-CRISPRoff-V1 transfection with sgRNAs targeting the indicated genes.
(D) A representative flow cytometry plot of CD81 expression in iPS cells after transfection of CRISPRoff with either non-targeting sgRNAs or sgRNAs targeting CD81.
(E, F) A comparison of cells with CLTA (E) and H2B (F) silenced 10 days after transfection of CRISPRoff with dCas9 from S. pyogenes (dSpyCas9) or S. aureus (dSauCas9) or dCas12a from Lachnospiraceae bacterium (dLbCas12a).
The error bars in B, C, E, and F are SD from three independent experiments.
(A) An RNA-sequencing TPM (transcripts per kilobase million) plot for HEK293T cells transfected with CRISPRoff and NT (non-targeting) sgRNAs compared to untransfected HEK293T cells.
(B, C, D) RNA-sequencing TPM (transcripts per kilobase million) are plotted for HEK293T cells transfected with CRISPRoff and either NT sgRNAs compared to sgRNAs targeting CLTA (B), HIST2H2BE (C), or RAB11A (D). The data are representative of the average of two independent replicates.
(E, F) Representation of gene expression changes +/– 1000 kb from the annotated targeted gene for CD81, CD151, ITGB1 (E) and RAB11, HIST2H2BE, CLTA (F). Each box represents a gene.
(G) A barplot of genome-wide average CpG coverage obtained from whole genome bisulfite sequencing. WT indicates untransfected cells; NT indicates CRISPRoff delivered with non-targeting sgRNAs; T indicates CRISPRoff delivered with sgRNAs targeting CLTA or DYNC2LI1. The experiments were performed in two replicates. CLTA experiments are presented in Figure 2, DYNC2LI1 experiments are presented in Figure 5.
(H) A barplot of genome-wide average CpG methylation (beta-values) in the samples described in Figure S3G.
(I) Heatmap plots comparing average CpG methylation (beta-values) of 20-CpG sliding windows between untransfected (WT) replicates (left), WT and NT (middle), and WT and targeting sgRNAs (right) for CLTA (top row) and DYNC2LI1 (bottom row) experiments. Red color indicates highest density and blue color indicates lowest density. White areas indicate the absence of windows with the respective average methylation levels.
(J) A heat map showing pairwise correlations between genome-wide CpG methylation profiles of the samples described in Figure S3G, including replicates. Samples are sorted by unsupervised hierarchical clustering. Dark brown color indicates highest correlation, light yellow color indicates lowest correlation. This analysis highlights the variations in global CpG methylation between samples for CLTA and DYNC2LI1 experiments.
(K) A close up of a 17 kb genomic region containing the ZSCAN16 gene. DNA methylation profiles for untransfected, NT, and targeting sgRNA cells from the CLTA experiment are shown. The box highlights a differentially methylated region at the gene promoter, indicating a gain of CpG methylation in the targeting sgRNA cells compared to the control cells.
(L) A close up of a 30 kb genomic region containing the RPS6KA6 gene. The box highlights a gain of CpG methylation at the promoter in the targeting sgRNA cells. A CpG island located in the promoter of the gene is indicated in green.
(A) A bar plot of HEK293T cells reactivating CRISPRoff-silenced H2B (black), Snrpn-GFP (gray), or CLTA (blue) 9 days after Cas9-mediated knockout of DNMT1.
(B) Time course measurements of cells with reactivated CLTA expression after increasing doses of 5-aza-dC in HEK293T cells with CLTA silenced by CRISPRoff.
(C) A comparison of CLTA-GFP expression 28 days post transfection of the four TET fusions in Figure 3A co-transfected with one sgRNA sequence or a pool of three sgRNAs. Error bars represent the range of two technical replicates.
(D) A schematic of the CLTA CGI (green) with sgRNA binding sites annotated (a, b, c). The lollipop plot shading represents the percent of CpG methylation, as measured by bisulfite-PCR of CRISPRoff-silenced cells. Gene annotations were obtained from UCSC Genome Browser.
(E) A schematic of vectors that express a CLTA-targeting sgRNA and MS2 coat protein (MCP) fusion to various transcriptional activators.
(F) Violin plots that represent median CLTA-GFP fluorescence 2 days post-transfection of sgRNAs targeting CLTA and either dCas9 or dCas9 and MCP-fused transactivators into cells with endogenously expressed CLTA-GFP.
(G) A time course of HEK293T cells with CLTA-GFP reactivation after transfection of sgRNAs targeting CLTA and either TETv4 only, or TETv4 along with various MCP-fused transactivator domains into cells with CRISPRoff-silenced CLTA. Untreated cells are represented in white circles.
(H) A time course of HEK293T cells with CLTA-GFP reactivation after transfection of sgRNAs targeting CLTA and either dCas9-VPR or dCas9 along with various MCP-fused transactivator domains, or untransfected cells. The transfections were performed in the absence of TETv4 to measure persistent gene activation in the absence of DNA demethylation.
The error bars in G and H are SD from three independent experiments.
(A) A plot comparing the phenotype score (γ) of genes between technical replicates of the CRISPRoff (left) and CRISPRoff D3A methyltransferase mutant (right) genome-wide screens. The negative control sgRNAs are highlighted in blue.
(B) Violin plots of the phenotype score (γ) of all genes from each screen.
(C) A histogram of the number of genes with the indicated phenotype score (γ) from the CRISPRoff and CRISPRoff mutant screens. The light green and light orange lines correspond to the phenotype scores of negative control sgRNAs.
(D, E) Gene set enrichment analysis (GSEA) for genes associated with DNA replication and ribosome, confirming enrichment of expected essential genes. Genes are ranked from lowest (red) to highest (blue) phenotype scores.
(A) The genes chosen for the sgRNA tiling screens are highlighted on a volcano plot depicting gene phenotype scores from previous genome-wide CRISPRi screens in K562 cells (Horlbeck et al. 2016). The colors represent genes with one (orange), multiple (purple), or no annotated CGI (green).
(B) An aggregate plot comparing the normalized phenotype score for each sgRNA for genes with multiple CGIs. The green line represents screen data from CRISPRoff transfection into HEK293T cells, orange from CRISPRoff mutant into HEK293T, and purple from K562 CRISPRi.
(C) Representative sgRNA activity score profile for TBCD from the three screens. The green bar depicts the annotated CGIs obtained from UCSC Genome Browser.
(D, E) Plots overlaying the sgRNA phenotype scores and MNase signals for GFER and IMMT.
(F) An experimental workflow of tiling sgRNA screens to determine optimal sgRNAs for four endogenously GFP-tagged genes: CLTA, H2B, RAB11A, and VIM. The indicated population in the histograms represent the population of cells that have maintained gene silencing 4 weeks after CRISPRoff transfection.
(G, H, I) Overlay plots of sgRNA activity and MNase signal for CLTA, RAB11A, and VIM from the sgRNA tiling screen.
(A) A schematic of the H2B promoter with two sgRNA sites annotated: sg-A at the TSS and sg-B located ~2 kb upstream of the TSS. The CpG island spans 1.4 kb. Sites 1, 2, and 3 represent regions probed for CpG methylation by bisulfite PCR as described in Figures S7E–S7I.
(B) A time course of H2B silencing after transfection of CRISPRoff with sg-A or sg-B.
(C) A comparison of H3K9me3 profiles at the H2B (HIST2H2BE, colored red) promoter at day 5 (green tracks) and day 30 (purple tracks) post-transfection of CRISPRoff with sg-A, sg-B, or non-targeting (NT) sgRNA. The sgRNA binding sites are labeled, along with CpG island annotations (green), and the basal unmethylated CpG region of the H2B promoter prior to transfection obtained from WGBS of WT untransfected cells in Figure 2.
(D) A comparison of H3K9me3 profiles at the H2B promoter, as described in C, except in experiments using CRISPRoff with a D3A methyltransferase mutation.
(E, F) Quantification of CpG methylation at site 1 and site 3 of the H2B promoter (labeled in S7A) in cells transfected with CRISPRoff and either sg-A (blue), sg-B (orange), or non-targeting (gray) sgRNA. The cells were harvested for bisulfite PCR at day 5 post transfection.
(G, H, I) Quantification of CpG methylation at sites 1, 2, and 3 of the H2B promoter, obtained at 30 days post transfection.
Table S1. A list of statistics from RNA-seq experiments, related to Figure 2. The first tab contains differentially expressed genes in the ITGB1, CD81, and CD151 RNA-seq datasets. The second tab contains RNA-seq statistics for HIST2H2BE and the neighboring genes.
Table S2. A list of differentially methylated CpG regions from whole genome bisulfite sequencing, related to Figure 2. The list consists of data comparing CRISPRoff delivered with CLTA-targeting or non-targeting sgRNA. The table also contains statistics from the CLTA RNA-seq data that pertain to differentially methylated regions.
Table S3. A list of sgRNA protospacer sequences and calculated phenotype scores from the genome-wide CRISPRoff screens, related to Figure 4.
Table S4. A list of sgRNA protospacer sequences, deep sequencing counts, and calculated phenotype scores from the genome scale sgRNA tiling screens, related to Figure 5. Each tab contains the counts from the three genome scale sgRNA tiling screens: HEK293T CRISPRoff, HEK293T CRISPRoff mutant, and K562 CRISPRi.
Table S5. A list of sgRNA protospacer sequences, deep sequencing counts, and calculated phenotype scores from the tiling screens across the promoters of GFP-tagged CLTA, HIST2H2BE (H2B), RAB11A, and VIM, related to Figure 6.
Table S6. A list of sgRNA sequences used for CRISPRoff and CRISPRon experiments, related to Figures 1–7.
Data Availability Statement
The RNA-seq, ChIP-seq, and whole genome bisulfite sequencing data are available on GEO (GSE168012).