

Abstract
Seventy years ago, we learned from Chris Anfinsen that the stereochemical code necessary to fold a protein is embedded into its amino acid sequence. In water, protein morphogenesis is a spontaneous reversible process leading from an ensemble of disordered structures to the ordered functionally competent protein; conforming to Aristotle's definition of substance, the synolon of matter and form. The overall process of folding is generally consistent with a two state transition between the native and the denatured protein: not only the denatured state is an ensemble of several structures, but also the native protein populates distinct functionally relevant conformational (sub)states. This two‐state view should be revised, given that any globular protein can populate a peculiar third state called amyloid, characterized by an overall architecture that at variance with the native state, is by‐and‐large independent of the primary structure. In a nut shell, we should accept that beside the folded and unfolded states, any protein can populate a third state called amyloid which gained center stage being the hallmark of incurable neurodegenerative disorders, such as Alzheimer's and Parkinson's diseases as well as others. These fatal diseases are characterized by clear‐cut clinical differences, yet display some commonalities such as the presence in the brain of amyloid deposits constituted by one misfolded protein specific for each disease. Some aspects of this complex problem are summarized here as an excursus from the prion's fibrils observed in the brain of aborigines who died of Kuru to the amyloid detectable in the cortex of Alzheimer's patients.
Keywords: aggregation, denaturation, misfolding, protein folding
1. ONCE UPON A TIME …
In the Fore language Kuru means tremor, the shocking symptom of a fatal disease that was endemic among aborigines living in the mountains of Papua‐New Guinea. After WW2 a significant number of Fore suffered from this neurological disorder characterized by ataxia, brain degeneration, progressive dementia and premature death (Figure 1). In 1957, Daniel C. Gajdusek from the NIH moved to New Guinea to work with Michael Alpers, an Australian doctor who discovered that Kuru was endemic in the Fore with a pattern of transmission suggesting some role for a peculiar infectious pathogen.1, 2 The clue was finding that transmission of the pathogen occurred because the Fore were eating the brain of deceased relatives. This endo‐cannibalistic tradition demanded preparation of the brain, a job carried out by women with the help of children, both preferentially infected and thereby ill.
FIGURE 1.

Adapted from Liberski.3 A Fore boy suffering with an advanced kuru
In 1959, a specimen of the brain of a girl who died of Kuru was shipped by Gajdusek to the NIH and injected into the brain of two chimpanzees in an attempt to test whether the presumed pathogen could be transmitted to a primate. Indeed after approximately 2 years one of the two chimpanzees developed the encephalopathy, showing that (a) the mysterious agent could adapt to and grow in a different primate; and (b) the long incubation time and the very small size of the pathogen led to the definition of slow virus.4, 5 A very interesting conclusion of this successful experiment was to unveil that the devastating type of dementia affecting the Fore is a transmissible neurodegenerative disorder caused by a peculiar micro‐organism, an unconventional slow virus. In 1976 Gajdusek was awarded the Noble prize in Medicine or Physiology (shared with BS Blumberg) …“for their discoveries concerning new mechanisms for the origin and dissemination of infectious diseases.”
About 20 years elapsed from the beginning of the campaign in New Guinea and the Nobel to Gajdusek. Over that period the group at the National Institute of Neurological Diseases in Bethesda made a number of interesting discoveries on the generality of the pathogenesis of several neurological disorders of humans and animals. I enjoyed reading an interesting account of that period written by David M. Asher,1 one of the members of that group for 25 years.
I had the fortune to meet Gajdusek when he came to Rome after the Nobel Prize to pay a visit to Jeffries Wyman, formerly professor of Biophysical Chemistry at Harvard but appointed visiting scientist at the Regina Elena and the Biochemistry Institute of the University in Rome; Wyman came in 1962 for a couple of weeks visit that was extended for almost a quarter century.6 When Gajdusek arrived, Wyman was planning to spend 2–3 months in Papua‐New Guinea to live with the aborigines, and Gajdusek was clearly the perfect person to offer first hand advice; I still have the reprint of Gajdusek's paper in Science4 presented to Wyman.
2. TRANSMISSIBLE SPONGIFORM ENCEPHALOPATHIES (TSE)
The understanding that several apparently different fatal neurological diseases, such as Kuru, Creutzfeldt‐Jakob disease (CJD) and the Scrapie of sheep belong to the same family being all transmissible via a mysterious slow virus is recognized as an extremely important achievement of the NIH group led by Gajdusek. Some similarities between Kuru and scrapie were already pointed out in a short communication to The Lancet by Hadlow7; and in 1966 Alpers et al.8 showed that the scrapie agent is an exceptionally small infectious agent, hence a virus. The similarities between Kuru and sporadic CJD were pointed out by Klatzo, Gajdusek and Zigas9 based on plain morphological similarities; these authors emphasized the characteristic spongy look of the brain which explains the name TSE. Figure 2a depicts the image of the brain of a CJD victim showing many holes left by dead cells. Interestingly this peculiar morphology is similar to that of the brain of Augustine D. (Figure 2b), the women studied by Doctor Lois Alzheimer who reported at the beginning of XX century the first case of a devastating dementia. A fundamental experiment made possible by the availability of samples of brain tissue from CJD cadavers, was the transmission of CJD to primates.10
FIGURE 2.
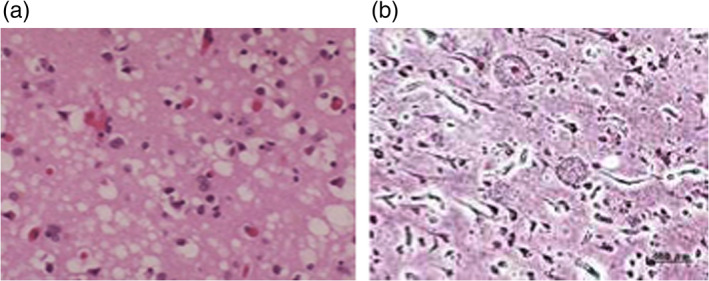
Panel (a): Vacuolation of the cortex of a person who died of Creutzfeldt‐Jacob Disease. Panel (b): Histological preparation of the brain of Mrs Augustine D., the first patient recognized by Doctor Lois Alzheimer to be affected by a devastating dementia, nowadays known as Alzheimer disease. Adapted from Dudhatra et al11
Sporadic CJD is a rare neurodegenerative disease that was reported in the national Press once it was recognized to be the cause of death of the famous Russian choreographer George Balanchine. In the mid‐seventies, new interesting information on CJD was published. Iatrogenic transmission due to cornea grafting from person to person was well documented, the donor being discovered to be a CJD patient after a post mortem examination. A striking case of transmission of CJD to two young people was conclusively attributed to the use of cortical electrodes previously employed for brain surgery of an old patient that eventually died of CJD.12 The conventional sterilization procedure of cortical electrodes was clearly unfit to kill the contaminating pathogen, the slow virus; and years later this path of transmission was proven by implanting a fragment of the very same probe into a chimpanzee who developed a CJD‐like syndrome. Finally, it was discovered that some cases of CJD were familial, a very important result as outlined below.13
A connection between these different neurological disorders was justified by several commonalities, such as transmissibility, extremely small pathogen, very long incubation period, spongy morphology of the brain, lack of inflammatory and immunological response. The transmission of scrapie to mice proved of great practical importance for subsequent laboratory experiments. In summary, the hypothesis that different encephalopathies share a common etiology being all caused by the same type of peculiar infectious agent, gained consensus and paved the way to surprising novelties concerning the nature of the pathogen, an intriguing conundrum. The perseverance of the National Institute of Neurological Diseases in pursuing for many years an expensive and somewhat exotic neuropathology program should be recognized as a virtuoso model.
3. THE SLOW VIRUS IS A … PROTEIN: THE DISCOVERY OF THE PRION
The chemical nature of this slow unconventional virus was for good many years a mystery since numerous attempts to demonstrate the presence of the pathogen's genome, DNA or RNA, failed. These infectious agents were found to lack a number of chemical and physical properties of typical viruses, infectivity surviving treatment with nucleases and being inactivated only by exceptionally high dosage of UV or ionizing radiations. The challenging hypothesis that a protein may be the only component of the infectious particle, the so‐called protein only hypothesis, was proposed but generally viewed with a great deal of skepticism. The challenge stimulated the curiosity of Stanley Prusiner who embarked on the risky project of purifying the pathogen causing scrapie in sheep.
As a student in the Medical school at the University of Penn, Prusiner had the opportunity to meet Britton Chance, a giant of the Biochemistry and Biophysics of last century. Brit helped young Prusiner to obtain a fellowship at NIH (personal communication) to work with Earl Stadman, a classical extremely competent biochemist who gave Prusiner the opportunity to acquire a solid background in protein chemistry that proved essential for the purification of the scrapie agent. After moving to San Francisco, Prusiner started working full time on the problem and did set up a quick reliable biological assay to follow infectivity using golden Syrian hamster. Intracerebral injection of the homogenate of the brain of a scrapie affected sheep induced in the hamster a syndrome with the hallmarks of the spongiform encephalopathies; upon perfecting the procedure, the hamsters developed the neuropathy after an incubation time of approx 60 days. The availability of a reliable biological method to follow infectivity and obtain an answer in a time much shorter than the usual procedure, based on transmission from sheep to sheep, was a winning step.
Infectivity proved to survive harsh experimental conditions such as exposure to detergents, high temperatures and treatment with micrococcal nuclease or proteinase K. These peculiar biochemical stability helped finding conditions to purify the protein carrying infectivity, that Prusiner called Prion for Proteinacious Infectious Particle.14, 15 The purified Prion had a molecular mass of 27–30 kD and a propensity to aggregate forming long polymers called “rods,” similar to the so called scrapie‐associated fibrils (Figure 3). The unusual resistance of the Prion to heat and proteolytic attack was correlated to its aggregated state that is more stable than the corresponding native state.16, 17
FIGURE 3.
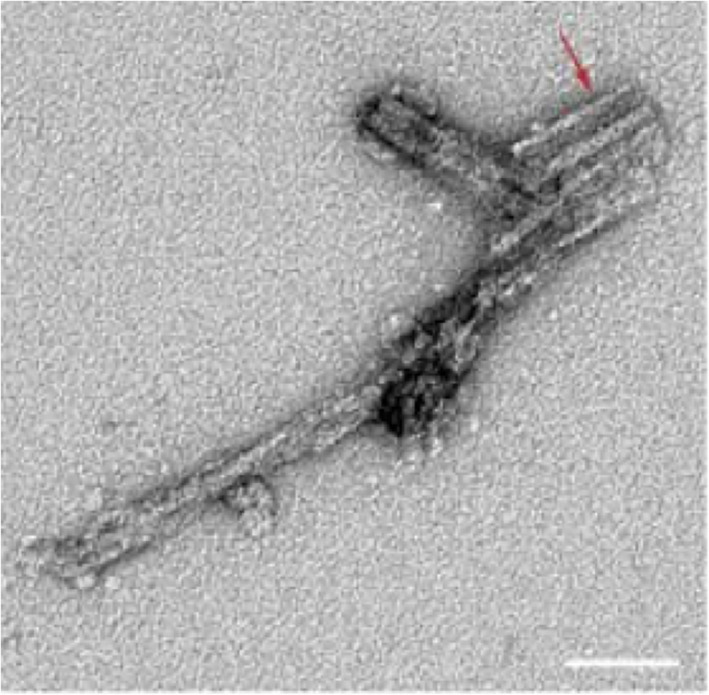
Two‐dimensional negative stain image of PrP rod isolated from prion‐infected hamster brain. Paired PrP fibers with a double helical substructure, separated by an 8–10 nm gap. Adapted from Terry et al21
Next fundamental hurdle was the mechanism underlying replication in the hamster's brain of an infectious agent devoid of DNA/RNA. As outlined above, an additional peculiar feature of these encephalopathies is the absence of inflammatory and immunological responses which led Brunori and Talbot18 to suggest that the Prion protein is coded in the human genome and expressed in the normal brain; but modified after biosynthesis to acquire a pathological state. The critical step in the epiphany of pathogenicity was suggested to be a proteolytic activation, which actually proved incorrect. In April 1985, the expression of PrP in the normal brain was indeed demonstrated in an seminal paper published by Weissman, Prusiner, Hood et al.19 Starting from the sequence of a seven amino acid fragment of PrP27‐30, these authors isolated from hamster's brain one clone containing a 2 kb DNA insert that was sequenced and allowed to fish out a cDNA clone. Almost simultaneously Chesebro et al.20 also reported a cDNA clone obtained from mouse brain, an important confirmation that PrP is normally expressed in the brain. In essence these pioneering results provided intriguing insight into the properties of the mysterious infectious agent, starting with the determination of its amino acid sequence. Moreover, it was proven that PrP is encoded in the host genome (called PrPcell) and expressed in normal and infected brain, as well as in other tissues. The function of PrPcell in normal individuals is still quite uncertain except for an intriguing role in regulating sleep, as discovered by studying a disease called (familial or sporadic) fatal insomnia shown to be caused by a mutation in the PrPcell gene.22, 23
In 1986, the Prion moved center stage in the media and public opinion because of the breakout of the so called Mad Cow Disease, an epidemic that originated in the UK but soon became a general health problem in many European countries. Around the end of 1987, the British Authorities officially recognized that people were victims of what was called Bovine Spongifom Encephalopathy (BSE), a variant of Creutzfeld‐Jacob disease (vCJD) that proved to be a fatal neurological disorder with aggressive clinical features.24 This syndrome was shown to be an epizoosis accidentally transmitted to humans eating meat from Prion infected cattle, which were made ill by being fed with Prion contaminated meal containing the remains of calf or sheep infected by scrapie. Once the chain of transmission of mad cow disease was clarified, the European Health Authorities imposed a drastic eradication program demanding the incineration of millions of cattle (mostly in the UK) and a ban on selling cuts of beef and sheep considered at risk. By 2014, the total number people victims of the vCJD amounted to approx. 200 people. The drastic measures imposed by the Health Authorities stopped the epidemic, and today the fear that exploded after the transmission mechanism was made public has vanished.
A fundamental conundrum to be tackled was the molecular mechanism underlying the transformation of PrPcell into PrPscrapie to account for the multiplication of the slow virus and thereby the long incubation time. The brilliant theory proposed by C. Weissman and S.B. Prusiner25, 26 envisaged that (a) the 3D structure of PrPscrapie is different from that of PrPcell, and (b) the former binds to PrPcell inducing a change in conformation of the normal protein which acquires “pathogenicity.” But how to explain these far reaching statements and support the hypothesis?
The determination of the 3D structure of the Prion by NMR due to Wüthrich and coworkers27, 28 provided some important information to unveil the molecular mechanism of induction of pathogenicity. As depicted in Figure 4, PrPcell is a globular protein with a two‐over‐two helical bundle and a long intrinsically disordered tail. PrPscrapie on the other hand is a conformational variant with reorganization of two of the four helices into β‐strands. A transition from fully helical to partially β‐strand had been detected also by far UV‐CD. Could PrPscrapie be a misfolded variant of the protein? As discussed below, exposure of the two β‐strands in PrPscrapie increases the propensity to aggregate and in fact induces a kernel for the onset of the amyloid fibril growth.
FIGURE 4.
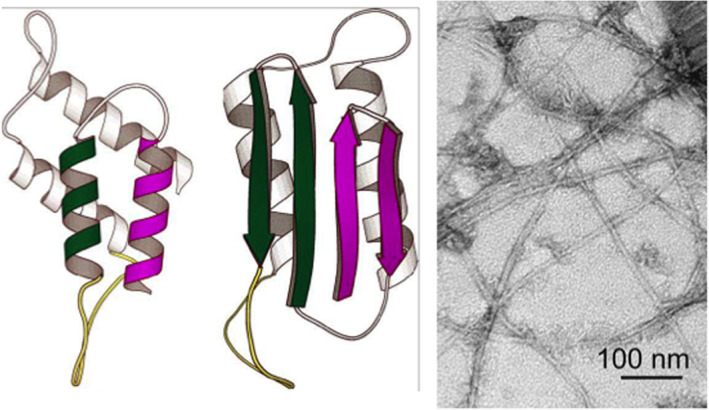
Protein misfolding and the amyloid state. On the left the models for the two conformational states of the Prion protein (PrPcell and PrPscrapie), having the same amino‐acid sequence.27, 28 PrPcell (on the left) is all α‐helical: two of the four helices (shown in color) are converted into β‐sheet in the PrPscrapie state (on the right); adapted from Prusiner.25 The right panel shows an electron microscopy image of the amyloid state of Aβ, adapted from Tycko and Wyckner.57 Regardless of the amino acid sequence or the native 3D structure of the protein involved in the specific neurological disorder, the aggregate consists of thread‐like fibrils of about 7–10 nm diameter which is rich in β‐sheet and thermodynamically very stable
In a nutshell, this misfolding transition may represent the mechanistic commonality among all the Prion diseases (Kuru/scrapie/CJD/BSE) and may also be crucial in several devastating neurological disorders caused by aggregation of misfolded proteins. These discoveries paved the way to the conceptual connection between prions and the amyloid state, which nowadays is considered of fundamental importance in the pathogenesis of several neurodegenerative diseases.
4. PROTEIN FOLDING AND MISFOLDING
The recognized pioneer of the protein folding field is Chris Anfinsen29 who demonstrated working with bovine ribonuclease that protein (un)folding is a reversible chemical reaction, and information to acquire the native folded state is embedded in the amino acid sequence. It is generally accepted that functional specificity and efficiency of proteins is strictly dependent on their native 3D structure (the Form) defined by the atomic coordinates. The morphogenetic event leading from a disordered random coil polypeptide with a genetically defined amino acid sequence (the Matter) to the functionally competent ordered structure has defined “the protein folding problem.” The transition to the native state demands the formation of very many weak bonds and a hydrophobic collapse to compensate for the huge decrease in entropy.30 In water, this spontaneous process is dictated by the primary structure of the protein, the amino acid sequence conforming to the general Aristotelian definition of substance as the synolon of matter and form (substantia causa sui).
Protein folding is an extremely complex chemical reaction even for a relatively small globular protein (<100 amino acids). According to Michael Levitt,31 in 1975 Francis Crick stated: “it is very difficult to conceive of a scientific problem that would not be solved in the coming twenty years…except for a model of brain function and protein folding.” Indeed since Anfinsen's seminal experiment, the protein folding problem has attracted and obsessed many outstanding scientists who published fundamental contributions concerning (a) the forces involved in the stabilization of the folded state, (b) the role of the hydrophobic effect and the hydrophobic collapse, (c) the thermodynamics underlying the stability of a native protein, (d) the analysis of the unfolding equilibrium isotherm, (e) the kinetics to detect folding intermediates and define a mechanism, and so on.
After a period of relative stagnation, in the nineties we witnessed major progress in the field with the introduction of new concepts and novel experimental approaches to unveil the mechanism and explore early folding events. Extensive experimental work led to the hypothesis that folding may proceed via parallel pathways which would account for observations obtained by catching intermediates or by investigating the structural properties using kinetic analysis, site directed mutagenesis or high resolution proton NMR.32, 33, 34, 35
In 1995, Wolyness, Onuchich and collaborators36 presented the funnel theory to describe in general terms the molecular events leading from the unfolded ensemble to the “unique” native state, as depicted in Figure 5. The protein's energy landscape is represented as a funnel to highlight the entropy/enthalpy compensation in going downhill. The width of the funnel is a representation of entropy: at the top, the extremely large number of structures (>1030) depicts the ensemble of disordered states when the polypeptide is unfolded (random coil). The formation of intramolecular short range and long range weak bonds is associated with a steep reduction in the width of the funnel and progressive decrease in the number of states, going through a productive but still expanded on‐pathway intermediate(s). At the bottom of the funnel, the minimal entropy native protein is the dominant state.
FIGURE 5.
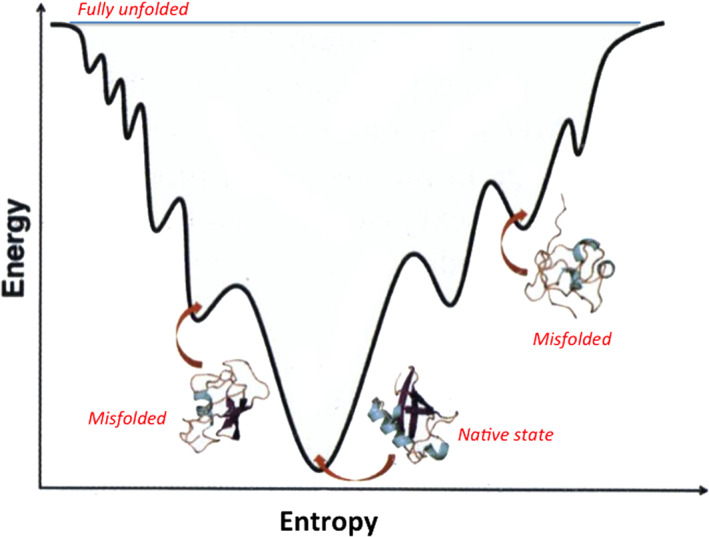
Schematic representation of the folding funnel, with sketches of the structure(s) of a globular protein. The width of the funnel is proportional to entropy; and the ordinate to enthalpic contribution increasing up to down. At the top, a myriad of random coil disordered states of the denatured system (not shown). At the bottom, the native folded state with correct topology. In the process, the formation of weak bonds is associated to the progressive stabilization of structural elements and a decrease in entropy
As shown in Figure 5, the surface of the folding funnel is complex with bumps and minima, implying that the transition to the native state may proceed via parallel pathways36 accounting for several experimental findings. The population of off‐pathway misfolded states is crucial to the onset of amyloid formation and pathophysiology, as commented below. Given the underlying complexity, it was surprising to find37 that (a) the equilibrium (un)folding very often conforms to a two state behavior, suggesting the absence of a significant population of equilibrium intermediates; (b) the time course of (un)folding is consistent with a single exponential behavior implying that folding involves crossing a barrier characterized by an activation energy and a maximum along the reaction coordinate; thus obtaining information on the structure of the transition state (TS) proved most valuable.
This formidable problem was tackled by Alan Fersht38, 39 by introducing the so‐called Φ‐value analysis, based on measuring the effect on the folding rate constant of specific single site mutation(s). Since for small globular proteins the kinetics of folding ranges from microsec to sec, the rate must be estimated by rapid reaction methods (mostly stopped flow and temperature jump).
The principle of the procedure is illustrated in Figure 6. To obtain reliable information on the folding transition state, a large number of single site (conservative) mutants must be expressed and characterized by kinetics and computer simulation. The structural constrains due to interactions between the amino acid side chains making contacts in the TS were exploited by Vendruscolo et al.40 to introduce restrains in the simulation of molecular dynamics trajectories, and thereby to acquire structural information on the folding TS. The Φ‐value analysis has been an important breakthrough providing information on the folding mechanism of a protein, given the crucial role of the transition state in unveiling the folding pathway. Naturally, the intrinsic complexity of the folding process implies that the TS structure indeed describes an ensemble of states. Nevertheless, the model paved the way to a number of predictions that have been tested by ad hoc experiments on folded globular proteins but also on intrinsically disordered proteins.41
FIGURE 6.
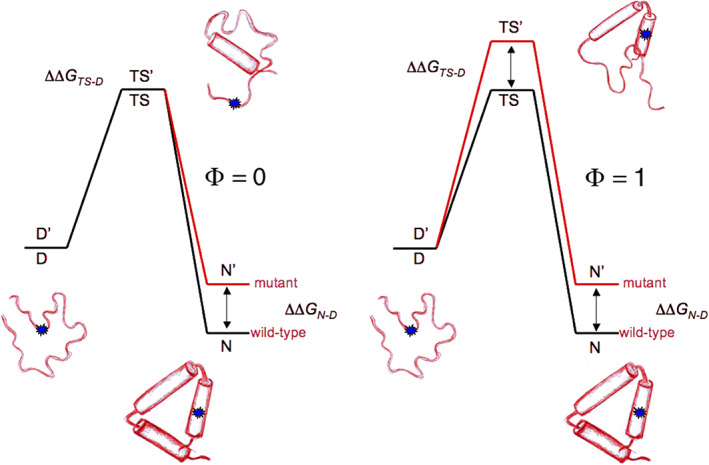
The Φ‐value analysis introduced by Alan Fersht et al38, 39 to characterize the folding/unfolding transition state (TS). A conservative mutation introduced in the middle of one of the 3 helices (small blue circle), is associated—in this hypothetical case—with a destabilization of the native state (from N to N′). The rate constant for the transition Denatured‐to‐Native (D to N) provides an estimate of the transition state barrier height. On the left: if the mutation has no effect on the barrier height, Φ is set to zero to indicate that the amino acid that has been changed makes no contacts with other side chains in the TS. On the right: in this case the mutation has an effect on the barrier height with an increase (TS to TS′) equal to the perturbation of the ground state (N to N′), and Φ is set to one; therefore the amino acid under scrutiny must be in contact(s) with other amino acids in the TS (drawing by S. Gianni, with permission)
Very many proteins conforming either to a simple two‐state/one barrier process or sometimes a three‐state/two barrier process39 have been investigated by kinetics and mutagenesis (Figure 7). In some case, the Φ‐value analysis highlighted the structure of a misfolded intermediate that may be characterized by minor structural differences in the topology of just one β‐sheet.42 This may acquire general significance given that a misfolded intermediate is often assumed to be crucial to trigger the nucleation of an amyloid.
FIGURE 7.
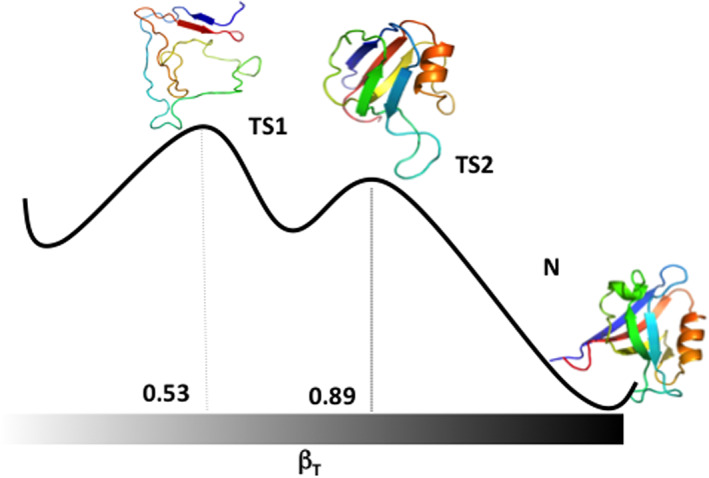
The folding pathway of PDZ2 from experiment and simulation.42 The transition Denatured‐to‐Native (left‐to‐right) proceeds through two barriers defining the relevant transition states (TS1 and TS2) that are located at different relative values of the reaction coordinate expressed in terms of the Tanford parameter βT
5. THE AMYLOID FOLD AND NEURODEGENERATIVE DISEASES
The term amyloid was introduced in 1853 by R. Virchow to describe waxy tissue deposits thought to be composed of carbohydrates that were detected by optical microscopy in the brain of people who died of fatal neurological disorders. These aggregates were visualized through the absorption of Congo red, a dye employed in histology which was known to bind to starch, a similarity than accounts for the introduction of the term amyloid from the greek ἄμυλον.
A broad range of fatal human neurodegenerative diseases such as Alzheimer's and Parkinson's, are characterized by the presence of amyloid fibrils in the brain of the cadavers.43, 44, 45 These fibrils were shown to arise and grow because of (a) misfolding of peptides or proteins that fail to mature into a stable native state, and/or (b) failure of the housekeeping biochemical machinery geared to clear cellular garbage. The crucial role of misfolding in the onset of these disorders of the brain (and other organs) led to identify this new class of devastating diseases as “misfolding disorders,” which prevail in older people. These disorders have some commonalities being fatal, characterized by a long incubation time and lack of specific effective treatment. Thus the hash tag unfortunately is: No cure only care.
What is the mechanism whereby a misfolded state triggers the formation of an amyloid fibril? Long ago Linus Pauling, Nobel laureate in Chemistry, made the blunt statement that proteins are sticky (personal communication). It has been proposed that the molecular mechanism describing the link between misfolding and disease is, by‐and‐large, associated to the propensity of peptides or proteins to stick together largely via their hydrophobic core, a fundamental step in the onset of the pathology.44, 45, 46, 47, 48, 49 Amyloid is a rather generic name to indicate a peculiar state. The long fibrils containing a single type of protein or peptide specific for a given disease, are thread like structures of typically approx 7–13 nm diameter, constituted by several protofilaments (2–7 nm diameter) that associate laterally. The fibrils are stabilized by a canonical cross‐β amyloid fold with the β‐strands orthogonal to the fibril axis (Figure 8); the interacting fibrils have the same orientation of the N‐ and C‐terminals, which maximizes the number of H‐bonds and hydrophobic interactions along the main axis. The considerable mechanical strength of the amyloid is attributed to the extended set of hydrogen bonds between the β‐sheets and to the lateral association of protofilaments producing more complex structures (48, 49 and references therein).
FIGURE 8.
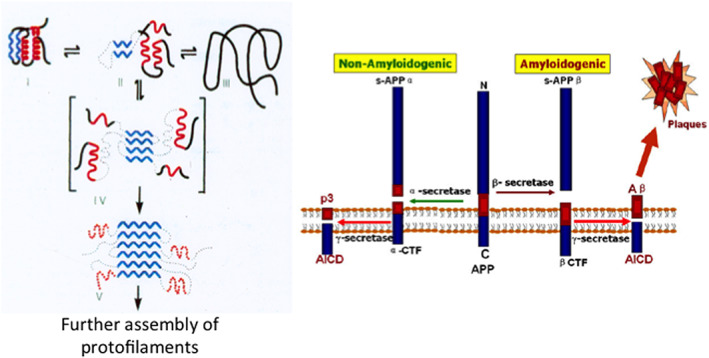
On the left, schematic representation of the folding of a small globular protein, and the formation of amyloid. The native folded state (on the top left) is assembled with two α‐helices (in red) and two β‐sheets (in blue). In such a case, the onset of initial aggregation and formation of the amyloid depends on the population of a misfolded intermediate (in square parenthesis). The exposed β‐sheet initiates intermolecular interactions within a kernel which is followed by elongation and formation of protofilaments. On the right, the biochemistry and aggregation of Aβ. The transmembrane Amyloid Precursor Protein67 is cleaved by two secretases that can produce either amyloidogenic Aβ peptides yielding amyloid plaques; or NON‐amyloidogenic peptides that are cleared out. Inspired by a scheme presented by Prof. G. Petsko (New York)
A remarkable feature of the amyloid is the monotony of the basic architecture, a finding that initially impressed people and led to believe that in all cases the structure was indeed the same. Despite the recurrent canonical features of the amyloid fibers, it is worth noticing how the work of several groups has contributed in defining the finest details of the 3D structure of several types of amyloids extracted from the brain of cadavers (ex vivo), or assembled in vitro from synthetic polypeptides. Polymorphs have been identified more often when amyloids were assembled from short peptides but also detected with whole proteins such as Tau50 or α‐synuclein.51 In some cases, polymorphism has been correlated to the binding of different cofactors or truncation of a domain or interactions with cellular components, as deduced from intriguing structures solved using mostly solid state NMR or cryo electron microscopy.52, 53, 54
Notwithstanding the variety and complexities of the amyloid fold, the basic morphology of the core populated in vitro and in vivo seems, by‐and‐large, independent of the specific amino acid sequence of the protein or polypeptide involved. The huge number of folds characteristic of native globular proteins, each dictated by its specific sequence, underlies the selection process driven by evolutionary pressure; a mechanism that at present is not obvious for the amyloid fold. In addition it is unknown if the polymorphs of the same protein may or may not have different roles in the onset of cellular dysfunctions or in the time course of the disease. This intriguing problem may be considered vis‐à‐vis Chris Dobson's statement made at the beginning of this century that “in principle, any protein can assemble into an amyloid state” (personal communication).
An important property of amyloid is the remarkable resistance to heat and to proteolytic attack; after all, proteinase K was employed by Prusiner15 in the purification procedure of the prion obtaining a fragment called PrP27‐30. There is convincing evidence that the overall stability of the fibril exceeds that of the native state; and the rate of depolymerization is an uphill very slow event, making the process essentially irreversible. It may be said that amyloid is a stable almost imperishable state conferring immortality to a protein, which thereby eludes the physiological cellular homeostasis. This almost perennial deposit of fibrils has been initially considered the major reason for the pathology, a viewpoint that has been recently revised.
When and how does a protein commit to a misfolded rather than a native state? It has been reported that aggregation leading to formation of an amyloid can be initiated not only from a misfolded intermediate but also from the native state. A schematic representation of a possible molecular path critical in the onset of amyloid formation is depicted in Figure 8, where a folding intermediate exposes a β‐sheet acting as a kernel for aggregation. We mentioned above how the pathogenetic prion PrPscrapie is generated by the conversion of two of the four α‐helices of PrPCell into a doublet of exposed β‐sheet (see Figure 4), a rather dramatic conformational change.27, 28 In other cases however, the structural difference between a misfolded and a productive intermediate can be rather subtle; as shown for example with the circularly permuted variant of a PDZ domain, a globular protein of about 100 amino acids that mediates protein–protein interaction.42
6. THE PROPENSITY TO FORM AN AMYLOID IS BAD NEWS
In spite of marked clinical peculiarities, different neurodegenerative disorders display some commonalities, such as transmissibility with a long incubation time, lack of immunological/inflammatory response, no effective therapy and presence of amyloid deposits in the nervous tissue, the hallmark of a neurodegenerative disease. Chiti and Dobson47 published a fairly complete list of misfolding diseases of humans; a limited subset is presented in Table 1. These neurodegenerative disorders are characterized by different physiopathological and clinical features; yet from a biochemical viewpoint each one is associated to the folding defect of one specific protein with propensity to aggregate into an amyloid. As discussed above, although the overall architecture of the amyloid is quite uniform, fibrils originating from the same misfolded protein display a significant degree of polymorphism that should not be overlooked.55 In addition it is obvious that the chemistry of the different specific proteins must impose some stereochemical discrimination to explain why an amyloid fibril containing a mixture of different misfolded proteins has not been discovered as yet. This seems consistent with the finding by Silvestrini et al.56 that the amyloid from CJD individuals that are heterozygous for the mutation VAL210ILE of PrP contains the wild‐type as well as the mutant.
TABLE 1.
Selected human neurodegenerative diseases associated with protein misfolding and formation of extracellular or intracellular amyloid
| Disease | Protein or peptide | Length/aa | Structure |
|---|---|---|---|
| Alzheimer's | APP β40‐42 | 37–48 | ID |
| + Tau protein | 352–441 | ID | |
| Parkinson's | α‐Synuclein | 140 | ID |
| TSE | Prion/fragments | 230 | α‐Helix/ID |
| ALS | Superoxide dismutase | 154 | β‐Sheet/Ig‐like |
Abbreviations: ALS, amyotrophic lateral sclerosis; APP, amyloid precursor protein; ID, intrinsically disordered; TSE, transmissible spongiform encephalopathies.
How much does the aggregation propensity of a polypeptide depend on its amino acid sequence? In most cases the proteins involved display some regions having a high level of hydrophobicity, high propensity to form β‐structures, the presence of an alternate set of hydrophobic and hydrophilic residues, and a limited number of charged residues.47, 53, 57 Is there solid evidence in favor of the statement one disease‐one misfolded protein? A clue to this question emerged already at the beginning of this story from interesting clinical and genetic studies of Creutzfeld‐Jacob Disease patients displaying familiarity that was shown to be associated to mutations of the PrPcell gene.13, 56, 58, 59 This trend was eventually confirmed once it was reported that some of the natural mutants of APP/Amyloid Precursor Protein, of α‐synuclein, and of superoxide dismutase, are characterized by a propensity to aggregate which is enhanced relative to the corresponding wt. These mutants are associated to an early onset of the corresponding disorder that is, Alzheimer's disease (AD), Parkinson's disease (PD) and Lou Gerhig's disease (ALS), respectively.
Among the age related neurodegenerative disorders, ALZHEIMER DISEASE (AD) is the most frequent in the older group of people. The Alzheimer's Association (USA) claims that as much as one half of people over 85 years of age may be affected by AD and will slowly but irreversibly loose independence and precipitate into fully fledged dementia. Discovered in 1905 by Dr Lois Alzheimer (Figure 2), this serious fatal disorder has been considered for decades almost a curiosity of interest only to clinical neurologists. However over the last 3 decades, with the progressive expansion in the number of cases linked to increase in life expectancy, Alzheimer disease has become a problem of general concern.
In AD patients, the amyloid includes extracellular plaques made of Aβ (a fragment of APP), and intracellular neurofibrillary tangles made of hyperphosphorylated Tau protein. Tau is a microtubule‐associated protein that regulates the assembly and structural stability of microtubules in neurons and in axons. From the biochemical standpoint, Tau is characterized as an intrinsically disordered protein. The abnormal hyperphosphorylation of Tau is cause of detrimental pathological functions inducing microtubules to disassemble, free Tau to aggregate into paired helical filaments, and ultimately neurodegeneration.60, 61 Evidence suggests that hyperphosphorylation is linked to an imbalance between the activities of cellular protein kinases and phosphatases. There is evidence that in AD, the β‐amyloidogenic peptides play in vivo a significant role in favoring the imbalance between kinases and phosphatases. Therefore targeting the pathology of Tau is an intriguing strategy to search for Tau‐based therapeutics to control Alzheimer's disease.
Besides AD, there are several other neurological disorders related to the intracellular formation of Tau‐neurofibrillary tangles. Given the importance of tauopathies, several scientists have investigated the problem of unveiling the high resolution structure of the neurofibrillary tangles containing hyperphosphorylated Tau. This project has been successfully tackled using mainly cryo EM.54, 62, 63, 64 It may be of interest to mention that structural data are available also for the chronic traumatic encephalopathy (CTE), a syndrome induced by repetitive mild traumatic brain injury.
The primary amyloid in AD is represented by extracellular accumulation of the so called plaques, aggregates made of the Aβ fragments from the APP. A simplified scheme of the biochemistry involved is outlined in Figure 8. APP can be cleaved by proteolytic enzymes called secretases; the successive cuts by β‐secretase and γ‐secretase produce two peptides (Aβ40 and Aβ42) that have a significant propensity to polymerize and yield amyloid plaques.65, 66, 67 As outlined above, these events are paralleled by the accumulation of hyperphosphorylated Tau with formation of intracellular neurofibrillary tangles.68 Attempts to inhibit the secretases have unfortunately failed to improve the clinical setting69; this stimulated studies to unveil the details of the molecular mechanism underlying Aβ40/Aβ42 aggregation process.
It is accepted that a classical nucleation‐propagation mechanism is inadequate to describe the complexities of the time course of fibrillation, and the crucial role of secondary nucleation has been advocated. Therefore to discover drugs that may interfere with polymerization it is necessary to identify reaction intermediates and their kinetics of formation and interconversion. Determination of the rate constants for the various microscopic steps and their dependence on experimental conditions, is very complex. Breaking down the significant parameters demands (a) extensive experiments to unveil the role of protein concentration and seeding on the time course of aggregation, and (b) very sophisticated mathematical analysis.70, 71
Under physiological conditions these amyloidogenic peptides are processed via the normal degradation paths. However with aging, homeostasis begins to decline and normal housekeeping mechanisms become defective because of cumulative damage or shortage of sufficient energy supply. Baranczak and Kelly72 have reviewed available strategies to interfere with the progression of protein aggregation involved in several degenerative diseases including transthyretin amyloidosis. The survey of currently used drugs to modify the time course of the disease is compared with novel approaches to combat the decline of protein homeostasis linked to stress and aging (see below).
Following a therapeutic strategy based on fishing out small molecules interfering with the primary nucleation of Aβ40/42, animal models proved useful. Given that the propensity to aggregate has been correlated to charge and hydrophobicity,73 several mutants Aβ peptides with increased or decreased aggregation propensity have been expressed in the brain of the fruit fly Drosophila.74 Remarkably, the level of damage to the physical performance and life span of these mutant flies increases with the decrease in solubility of the mutant peptides. Likewise employing the worm C. elegans as an additional model system, Habchi et al.75 discovered that bexarotene, an approved anticancer drug, seems (a) to control primary nucleation by delaying the formation in vitro of larger aggregates and toxic species, and (b) to rescue locomotors properties of the worm C. elegans expressing the mutant peptides.
PARKINSON'S DISEASE (PD) is the second most prevailing among the neurodegenerative diseases of humans, with 7 to 10 million Parkinson patients worldwide; the prevalence, which is 40 per 10,000 in the fourth decade of life, raises 50 fold above 80 years. Goedert, Jakes and Spillantini76 published a fascinating account of the history of the discovery of α‐synuclein, its location in the Lewy's bodies and its role in PD; the paper was published in the Journal of Parkinson's Disease to celebrate the 200th anniversary of the first description of the disease by Dr. James Parkinson. The authors recall the identification of three types of synuclein, the demonstration that only α‐synuclein (140 aa) is pathogenic; and the following identification of the first mutants involved in the early onset of the clinical syndrome. The story unfolded over 20 years and was marked by several breakthroughs, starting from the identification of several mutations causing familial PD. The discovery of several mutants of α‐synuclein that affected the clinical course of the disease was strong evidence that indeed aggregation of this protein is a crucial component of the molecular mechanism; a special case is that of individuals carrying the mutation A53T who develop a severe form of PD often associated with dementia.
As depicted in Figure 9, the unfolded state of α‐synuclein monomer, crucial to the molecular pathogenesis of PD, tends to aggregate forming oligomers and protofibrils that coalesce into bigger aggregates and Lewy's bodies (which are not observed in any other neurodegenerative disease). α‐Synuclein aggregation propensity seems to be linked to the extent of exposure of the N‐terminus of the protein.77
FIGURE 9.
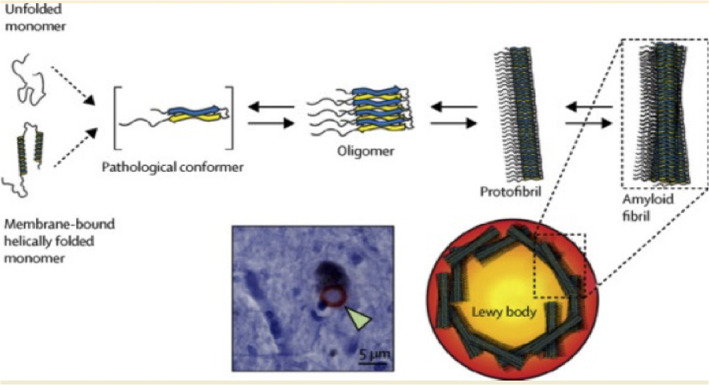
The pathway of aggregation of α‐synuclein in going from unfolded monomers (on the left) to amyloid fibrils and to Lewy's bodies, the latter seen by microscopy in the brain of people who died of Parkinson disease
Structural studies of the α‐synuclein amyloid provided interesting structural information on the polymorphism of the fibrils. David Eisenberg and coworkers55 have determined the cryo‐EM structures of full‐length recombinant α‐synuclein organized in two distinct polymorphs composed of protofilaments with highly conserved kernel structure. The structures of the two polymorphs unveil the atomic interactions of the steric zippers, with differences that should be taken into account when designing specific drugs targeting the fibrils. Using CryoEM, more and more groups78, 79 have tackled the 3D structure of α‐synuclein from different sources to unveil the extent of polymorphism and its significance.
AMYOTROPHIC LATERAL SCLEROSIS (ALS), the most common motor neuron disease in adults, is caused by selective killing of these neurons and thereby progressive paralysis; its prevalence is 2–3 cases per 100,000 people, the risk increasing by a factor of approx. 10 after 60 years of age.80 The syndrome was first described in 1869 by the French neurobiologist and physician Dr. Jean‐Martin Charcot. In the US and Canada, for good many years it has been referred to as Lou Gerhig's disease from the name of a famous baseball player who in the 1930s had to abandon the sport because of progressive paralysis and subsequent death. The story was made popular by a movie in which the role of Lou was played by the famous actor Gary Cooper.
This fatal disease appears at present correlated to misfolding of the Cu,Zn‐enzyme superoxide dismutase or its apo form, at least for those patients (approx. 10%) found to be carriers of mutations in SOD1, defining a group called familial ASL. The finding that SOD activity of the mutant(s) is normal excluded the hypothesis that the neuron killing mechanism is due to oxidative damage.80 ALS is associated with many mutations in the gene encoding for superoxide dismutase, mutations associated to variable survival times of the carrier. Oliveberg and collaborators81 have demonstrated a linear correlation between the survival time of the carrier and the folding pattern of the different SOD1 mutants, a remarkable finding. The mutations are found to perturb the protein by destabilizing either the monomer (eventually loosing a metal), or the dimer interface. Given the general rule that aggregation propensity is maximal when the overall charge of the protein is close to neutrality, it is not surprising that two mutants with the highest net charge are outliers in the linear plot of survival versus protein stability. This rather simple but fascinating picture may need confirmation given the complexities of the in vivo situation. Nevertheless, it is implicit that misfolded SOD1 mutants will tend to aggregate in the motor neurons forming an amyloid and producing toxic effects.82 The significance for the pathology of monomer–monomer assembly and formation of mixed hybrids in mutants of SOD1 has been highlighted by Horovitz and collaborators83 using a mass spectrometry approach.
The sporadic ALS group which includes approx. 90% of cases, seems to have no apparent genetic linkage. Nevertheless Rotunno and Bosco84 argued that the propensity to misfold characteristic of the SOD1 mutants may emerge also in the wild type enzyme either by metal depletion or by destabilization of the dimeric assembly.85 Considering clinical similarities between familial and sporadic ALS, and the similar age of onset for both groups (between 50 and 60 years),80 it is possible that both converge on a common pathway. To sum up, while the molecular mechanism underlying SOD1 toxicity is still under discussion, the detection of misfolded wild‐type SOD1 in human post‐mortem samples of sporadic ALS supports the hypothesis that aggregation of misfolded wild type SOD1 may be involved also in the pathogenesis of the sporadic ALS. It should not forgotten however that other hypotheses for the molecular mechanism of damage of motor neurons were proposed and discussed, such as: abnormal accumulation of glutamate and of neurofilaments, defects in the induction of vascular endothelial cell growth factor, and mutations of a second gene termed ALS2. In a nutshell, the molecular cause of sporadic ALS is still an open problem.
7. OPEN QUESTIONS ON THE MOLECULAR PATHOPHYSIOLOGY OF AMYLOIDOSIS
Is the mechanism of cellular toxicity understood? At the beginning people thought that the extensive amyloid plaques detected in the brain of an Alzheimer patient where the sole or the primary cause of damage. It seems that this hypothesis may have to be abandoned; after all, pharmacological trials employing drugs to disassemble or dissociate the amyloid plaques were not encouraging.86 Nowadays emphasis has shifted, the species believed to be toxic to cells being preferentially the oligomeric states (in parenthesis in Figure 10), transients on the way to grow into larger aggregates and fibrils.70, 87 Their capacity to spread by diffusion and their higher surface‐to‐volume ratio are presumed to increase the level of cellular damage. Recent investigation on α‐synuclein oligomers by Fusco et al.88 unveiled some of the basic structural features responsible for neuronal toxicity. α‐synuclein oligomers display a highly lipophilic subdomain promoting interaction with the cell membrane and a structured region disrupting membrane integrity by inserting into the lipid bilayer; site directed mutations in the membrane binding region of the protein reduce toxicity in neuroblastoma cells, confirming this model.
FIGURE 10.
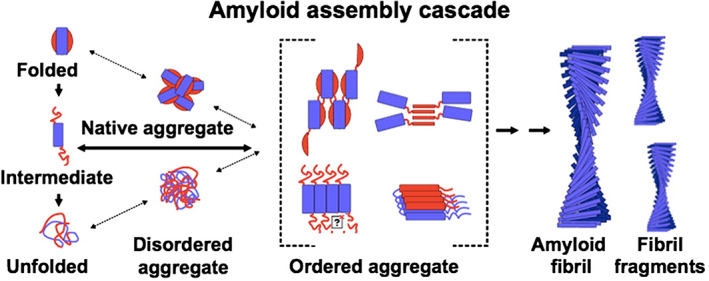
Cartoon with an outlook of the overall population of states in going from biosynthesis on the left to the amyloid fibril on the right. The set of ordered aggregates is crucial in the amyloid assembly cascade, from Radford and Weissmann45 modified
The strategy of Dobson and collaborators70 has been to crack the molecular mechanism by which aggregation occurs and thereby attempt control. Unveiling the number of species involved and their molecular features may pave the way to drug design in order to block the expansion of the amyloid and to quench cell toxicity. Although the microscopic mechanism of aggregation of misfolded monomers is extremely complex, considerable progress has been made by investigating the polymerization time course by experiments and sophisticated data analysis.49, 71 The first order conclusion confirms that indeed smaller oligomers are likely to be the toxic state(s) and therefore should be considered primary pharmacological targets. Moreover these Authors discovered a group of small molecules that are effective in reducing the rates of primary or secondary nucleation; and also engaged in trials using single‐domain antibodies designed to bind specifically to short stretches of amino acid residues of the target.70
Why is there a specific pattern of tissue or organ specificity? Most often amyloidosis is not systemic, and indeed in some cases, such as Parkinson's disease, there is a prevailing localization of the α‐synuclein fibrils deposition in the area of the substantia nigra. A reasonable explanation for this setting of the tissue target is lacking, as far as I know. One may be led to believe that this is linked to a localized overproduction of the relevant protein; this hypothesis however seems somewhat difficult to generalize. The only clear cut case of APP overproduction was reported in the Down's syndrome carriers, but this is not clarifying the matter. This question is still a conundrum.
The mechanism whereby longevity affects the onset of disease is still somewhat of a mystery, although it seems likely that failure of the cellular defense machineries may be probably involved. Indeed, it should not be forgotten that in the case of mutant proteins characterized by an increased propensity to aggregate, deposition of the fibrils almost always begins late in life although the mutant protein is synthesized from the time of conception.
Protein homeostasis in healthy eukaryotic cell is a complex network which is very effective in clearing the cell from molecular garbage thereby maintaining the health of the organism. It is well established that molecular chaperones fulfill a crucial job by binding to oligomeric or misfolded species that need a second chance to achieve the native state. A decline in activity of the proteostasis network that may emerge under stress or aging leads to accumulation of misfolded proteins, thereby triggering the beginning of aggregation.89, 90 At present the biochemical processes that jeopardize the efficiency of the homeostasis network are not quite clear. Aging is so complex that modifications of the different components of the network or lack of energy supply may all be reasonably involved. To combat the decline of proteostasis may avoid imbalance of the network and allow to rescue full activity; with decrease in the concentration of toxic oligomeric species and thereby reduction in the rate of fibrillation, which is the beginning of the collapse and cell death (70 and references therein).
As discussed by Seaman91, 92 and by Small, Petsko and collaborators93, 94 an alternative possibility to account for a failure of cellular homeostasis is the progressive disfunction of the Retromer. A handicap of this multi protein intracellular carrier may enhance the production of APP‐derived amyloidogenic peptides, feeding the growth of amyloid plaques. Stabilization of the Retromer multiprotein assembly has been attempted by Young et al.95 and by Mecozzi et al.96; the latter have discovered small molecular weight pharmacological chaperones that may have beneficial effects on the stability of the Retromer. Clearly more original work is desirable.
In the case of degeneration of the substantia nigra in idiopathic Parkinson disease, McNaught and Jenner97 reported that inhibition of the ubiquitin‐proteasome pathway may be responsible for altered α‐synuclein processing and Lewy's body formation.
An autocatalytic time course of these incurable diseases is another intriguing problem, considering that in general a long incubation time is followed by an accelerated deterioration of the clinical picture towards the end. Why is there a sort of accelerated time course in the accumulation of the clinical deficit? There is more than one intriguing hypothesis based on the growth and stability of the patient's own amyloid aggregates.82 On one hand, we cannot ignore that accumulation of amyloid by exponential growth will, at one point, exceed the capabilities of the innate housekeeping safeguards based on proteasome control. A possible intervention would be to reduce the supply of new monomers quenching over expression and blocking the feeding of new units fueling uncontrolled exponential growth of aggregates.98, 99 Another lead is suggested by the dependence of fibrillation kinetics on the square root of the concentration of the aggregation competent denatured/misfolded state, consistent with a fragmentation assisted growth that will generate new oligomers enhancing the rate of formation of new fibrils and “catalyzing”—so to speak—the speed of growth of the amyloid. If confirmed, the latter mechanism may indicate new interesting targets for pharmacological attack of the growth of the amyloid, in an attempt to slow down the progression of brain damage.100
8. THE SCIENTIFIC AND SOCIO‐ECONOMIC CHALLENGE IN AN AGING POPULATION*
World population growth has been accompanied by a progressive increase in the number of older people, life expectancy rising above 80 years in many developed countries. Alzheimer's and Parkinson's diseases, Amyotrophic Lateral Sclerosis and other neurodegenerative disorders are known to be strongly age‐related. Alzheimer's disease alone will affect between one‐third to one‐half of people above 85 years of age; thus the number of Alzheimer's patients, nowadays estimated at 40 million worldwide, is anticipated to increase to around 140 million by 2050. Given that no therapy is at present available for these fatal neurodegenerative disorders, a forthcoming neurological tsunami is likely to have a devastating impact on individuals, families, caregivers and societies at large.
When cognitive problems first begin and before they are sufficiently severe to impair markedly a subject's ability to carry out daily activities, the pathology results in mild cognitive impairment that however will progress to a full‐fledged dementia. With time, disabilities impair normal autonomous life, and eventually these patients require total assistance: considerable successes in terms of caring and improvements in quality of life have been achieved, even though such services are often overburdened. Moreover, education, diet, physical exercise, cognitive stimulation, and treatment of diabetes, hypertension, obesity, might improve cognitive status; these effects, however, are small and have to be confirmed with more extensive clinical studies that are in progress.
The dimension of this global neurological challenge and the ensuing socio‐economic scenario is daunting and devastating. The individual, social and financial burden of assisting these disabled patients surely will grow. By 2050 the economic toll is expected to rise to about one trillion US$ per year in the USA alone. The magnitude of the problem calls for a broad responsible initiative and a strong global commitment to fundamental research in order to discover an original and effective therapy.
As outlined above, in spite of the evident clinical differences among them, neurodegenerative diseases have some fundamental commonalities. Today the primary goal should be understanding the fundamental cause, mechanism and progression of these disabling diseases; the discovery that protein misfolding and amyloid formation is likely to be a unifying molecular mechanism shared by these different disorders has been an important step forward. As discussed above, a sensible strategy is to discover methodologies and drugs that either prevent or interfere with the formation and growth of amyloid. Because of the heavy personal and economic impact of neurodegenerative diseases, and since pharmaceutical companies alone are unlikely to invest in the kind of fundamental research necessary to crack the problem, large‐scale public funding is urgently needed and should be considered by Society an investment and not an expense.
(*): This issue was discussed by the international scientific Committee appointed on the occasion of the G7 for SCIENCE meeting, held at the Accademia Nazionale dei Lincei in Rome, May 2017.
9. CODA: EX UNO … PLURA
I guess that at least a billion people around the globe must have handled, at least once, a one dollar bill. They may have noticed the Great Seal of the United States of America (see Figure 11, left) which was officially adopted by an act of the US Congress in 1782. The American Eagle depicted behind the shield, is below a crown containing 13 stars representing the original colonies, and holds the symbols of peace and war, an olive twig and many arrows. The motto E PLURIBUS … UNUM, inscribed in the golden ribbon, is symbolic and prophetic of the US Union.
FIGURE 11.

On the left, the Great Seal of the USA printed in the one dollar bill; on the right the seal of protein structure complexity. Prepared with the help of Dr Giorgio Giardina, Rome, IT)
Considering the complexity and multiplicity of states available to proteins, I felt inspired by the Great Seal and conceived a variant shown in Figure 11 right where the 3D protein models around the Eagle convey a representation of multiplicity. Inscribed in the shield, the unique (=EX UNO …) amino acid sequence of the protein is the basic chemistry dictating in water the 3D structure of a native protein; whose model depicted below shows several α‐helices and one β‐sheet. The denatured protein at the top is supposed to represent the huge myriad of disordered states corresponding to a maximum entropy landscape. Since every protein under appropriate conditions may aggregate into an amyloid, the third state with its basic 3D architecture is depicted on the right.
At this point, the classical motto may be rephrased into EX UNO … PLURA, if we believe that a protein demands the neutral (as suggested by Prof. Luca Serianni, Linceo and renowned Latinist).
AUTHOR CONTRIBUTIONS
Maurizio Brunori: Conceptualization; writing‐original draft; writing‐review & editing.
ACKNOWLEDGMENTS
I like to express my deep appreciation to Prof Stefano Gianni (Rome) for invaluable scientific collaboration and generous help; and sincere thanks to Prof Andrea Bellelli (Rome) and Prof Michael Wilson (Colchester, UK) for reading and commenting this manuscript.
Brunori M. From Kuru to Alzheimer: A personal outlook. Protein Science. 2021;30:1776–1792. 10.1002/pro.4145
Dedicated to the memory of Professor Christopher M. Dobson (Sept 8, 2019).
REFERENCES
- 1.Asher DM. Kuru: Memories of the NIH years. Philos Trans R Soc B. 2008;4002:3618–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajdusek DC, Zigas V. Degenerative disease of the central nervous system in New Guinea: The endemic occurrence of ‘kuru’ in the native population. N Engl J Med. 1957;257:974–978. [DOI] [PubMed] [Google Scholar]
- 3.Liberski PP. Kuru: A journey back in yime from Papua New Guinea to the Neanderthals' extinction. Pathogens. 2013;2:472–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gajdusek DC. Unconventional viruses and the origin and disappearance of kuru. Science. 1977;197:943–960. [DOI] [PubMed] [Google Scholar]
- 5.Gajdusek DC, Gibbs CJ Jr, Alpers MP. Experimental transmission of a kuru‐like syndrome to chimpanzees. Nature. 1966;209:794–796. [DOI] [PubMed] [Google Scholar]
- 6.Brunori M. Hemoglobin is an honorary enzyme. Trends Biochem Sci. 1999;24:158–161. [DOI] [PubMed] [Google Scholar]
- 7.Hadlow WJ. Scrapie and kuru. Lancet. 1959;2:289–290. [Google Scholar]
- 8.Alpers T, Haig DA, Clarke MC. The exceptionally small size of the scrapie agent. Biochem Biophys Res Commun. 1966;22:278–284. [DOI] [PubMed] [Google Scholar]
- 9.Klazlo I, Gajdusek DC, Zigas V. Pathology of kuru. Lab Invest. 1959;8:799–847. [PubMed] [Google Scholar]
- 10.Gibbs CJ Jr, Gajdusek DC, Asher DM, et al. Creutzfeldt–Jakob disease (spongiform encephalopathy): Transmission to the chimpanzee. Science. 1968;161:388–389. [DOI] [PubMed] [Google Scholar]
- 11.Dudhatra GB, Avinash K, Modi CM, et al. Transmissible spongiform encephalopathies affecting humans. ISRN Infect Dis. 2013;2013:387925. [Google Scholar]
- 12.Bernoulli C, Siegfried J, Baumgartner G, et al. Danger of accidental person‐to‐person transmission of Creutzfeldt–Jakob disease by surgery. Lancet. 1977;1:478–479. [DOI] [PubMed] [Google Scholar]
- 13.Bertoni JM, Brown P, Goldfarb LG, Rubenstein R, Gajdusek DC. Familial Creutzfeldt–Jakob disease (codon 200 mutation) with supranuclear palsy. JAMA. 1992;268:2413–2415. [PubMed] [Google Scholar]
- 14.Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. [DOI] [PubMed] [Google Scholar]
- 15.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. [DOI] [PubMed] [Google Scholar]
- 16.Prusiner SB. Some speculations about prions, amyloid, and Alzheimer's disease. N Engl J Med. 1984;310:661–663. [DOI] [PubMed] [Google Scholar]
- 17.Prusiner SB. Scrapie prions, brain amyloid, and senile dementia. Curr Top Cell Regul. 1985;26:80–91. [DOI] [PubMed] [Google Scholar]
- 18.Brunori M, Talbot B. A mechanism for prion replication. Nature. 1985;314:676. [DOI] [PubMed] [Google Scholar]
- 19.Oesch B, Westaway D, Walchli M, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell. 1985;40:735–746. [DOI] [PubMed] [Google Scholar]
- 20.Chesebro B, Race R, Wehrly K, et al. Identification of scrapie prion protein‐specific mRNA in scrapie‐infected and uninfected brain. Nature. 1985;315:331–333. [DOI] [PubMed] [Google Scholar]
- 21.Terry C, Wenborn A, Gros N, et al. Ex vivo mammalian prions are formed of paired double helical prion protein fibrils. Open Biol. 2016;6:160035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunori M, Silvestrini MC, Pocchiari M. The scrapie agent and the prion hypothesis. Trends Biochem Sci. 1988;13:309–313. [DOI] [PubMed] [Google Scholar]
- 23.Cortelli P, Gambetti P, Montagna P, Lugaresi E. Fatal familial insomnia: Clinical features and molecular genetics. J Sleep Res. 1999;8:23–29. [DOI] [PubMed] [Google Scholar]
- 24.Prusiner SB. Prion diseases and the BSE crisis. Science. 1997;278:245–251. [DOI] [PubMed] [Google Scholar]
- 25.Prusiner SB. Molecular biology and pathogenesis of prion diseases. Trends Biochem Sci. 1996;21:482–487. [DOI] [PubMed] [Google Scholar]
- 26.Weissmann C. A theory for prion propagation. Nature. 1991;352:679–680. [DOI] [PubMed] [Google Scholar]
- 27.Riek R, Hornemann S, Wider G, et al. NMR structure of the mouse prion protein domain PrP(121−231). Nature. 1996;382:180–182. [DOI] [PubMed] [Google Scholar]
- 28.Riek R, Wider G, Billeter M, et al. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci U S A. 1998;95:11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anfinsen CB, Haber E, Sela M, White FHJ. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc Natl Acad Sci U S A. 1961;47:1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tanford C. Protein denaturation. Adv Protein Chem. 1968;24:1–95. [PubMed] [Google Scholar]
- 31.Levitt M. Though the breach. Curr Opin Struct Biol. 1996;6:193–194. [DOI] [PubMed] [Google Scholar]
- 32.Dobson CM, Radford SE. Understanding how proteins fold: The lysozyme story so far. Trends Biochem Sci. 1994;19:31–39. [DOI] [PubMed] [Google Scholar]
- 33.Fersht AR, Itzhaki LS, Elmasry NF, Matthews JM, Otzen DE. Single versus parallel pathways of protein folding and fractional formation of structure in the transition state. Proc Natl Acad Sci U S A. 1994;91:426–10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mallela K, Englander SW. A unified mechanism for protein folding: Predetermined pathways with optional errors. Protein Sci. 2007;16:449–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Creighton TE. Protein folding: An unfolding story. Curr Biol. 1995;5:353–356. [DOI] [PubMed] [Google Scholar]
- 36.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins. 1995;21:167–195. [DOI] [PubMed] [Google Scholar]
- 37.Jackson SE, Fersht AR. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two‐state transition. Biochemistry. 1991;30:10428–10435. [DOI] [PubMed] [Google Scholar]
- 38.Fersht AR. Optimization of rates of protein folding: The nucleation‐condensation mechanism and its implications. Proc Natl Acad Sci U S A. 1995;21:10869–10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. [DOI] [PubMed] [Google Scholar]
- 40.Vendruscolo M, Pace E, Dobson CM, Karplus M. Three key residues form a critical contact network in a protein folding transition state. Nature. 2001;409:641–645. [DOI] [PubMed] [Google Scholar]
- 41.Toto A, Malagrinò F, Visconti L, et al. Templated folding of intrinsically disordered proteins. J Biol Chem. 2020;295:6586–6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gianni S, Ivarsson Y, De Simone A, et al. Structural characterization of a misfolded intermediate populated during the folding process of a PDZ domain. Nat Struct Mol Biol. 2010;17:1431–1437. [DOI] [PubMed] [Google Scholar]
- 43.Buxbaum JN, Linke RP. A molecular history of amyloidosis. J Mol Biol. 2012;41:142–159. [DOI] [PubMed] [Google Scholar]
- 44.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. [DOI] [PubMed] [Google Scholar]
- 45.Radford SE, Weissmann JS. The molecular and cellular mechanisms of amyloidosis. J Mol Biol. 2012;421:139–141. [DOI] [PubMed] [Google Scholar]
- 46.Buell AK, Dobson CM, Knowles TP. The physical chemistry of the amyloid phenomenon: Thermodynamics and kinetics of filamentous protein aggregation. Essays Biochem. 2014;56:11–39. [DOI] [PubMed] [Google Scholar]
- 47.Chiti F, Dobson CM. Protein misfolding, amyloid formation and human disease: A summary of progress over the last decade. Annu Rev Biochem. 2017;86:27–68. [DOI] [PubMed] [Google Scholar]
- 48.Eisenberg D, Juncker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:348–396. [DOI] [PubMed] [Google Scholar]
- 50.Scheres SHW, Zhang W, Falcon B, Goedert M. Cryo‐EM structures of tau filaments. Curr Opin Struct Biol. 2020;64:17–25. [DOI] [PubMed] [Google Scholar]
- 51.Ni X, McGlinchey RP, Jiang J, Lee JC. Structural insights into α‐synuclein fibril polymorphism: Effects of Parkinson's disease‐related C‐terminal truncations. J Mol Biol. 2019;431:3913–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balbach JJ, Ishii Y, Antzutkin ON, Leapman RD, Rizzo NW. Amyloid fibril formation by A beta 16‐22, e seven residue fragment of the Alzheimer's beta amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 2000;39:13748–13759. [DOI] [PubMed] [Google Scholar]
- 53.Gallardo R, Ranson NA, Radford SE. Amyloid structures: Much more than just a cross‐β fold. Curr Opin Struct Biol. 2020;60:7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fitzpatrick AWP, Falcon B, He S, et al. Cryo‐EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li B, Ge P, Murray KA, et al. Cryo‐EM of full‐length α‐synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. 2018;9:3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silvestrini MC, Cardone F, Maras B, et al. Identification of the prion protein allotypes which accumulate in the brain of sporadic and familial Creutzfeldt‐Jakob disease patients. Nat Med. 1997;3:521–525. [DOI] [PubMed] [Google Scholar]
- 57.Tycko R, Wyckner RB. Molecular structures of amyloid and prion fibrils: Consensus versus controversy. Acc Chem Res. 2013;46:1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldfarb LG, Brown P, McCombie WR, Goldgaber D, Swergold GD. Transmissible familial Creutzfeldt–Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the Prnp gene. Proc Natl Acad Sci U S A. 1991;88:10926–10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pocchiari M, Salvatore M, Cutruzzolá F, et al. A new point mutation of the prion protein gene in Creutzfeldt–Jakob disease. Ann Neurol. 1993;34:802–807. [DOI] [PubMed] [Google Scholar]
- 60.Alonso AD, Grundke‐Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule‐associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997;94:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muralidar S, Visaga S, Saravanan A, Diraviyam S, Thirumalai D. Role of tau protein in Alzheimer's disease: The prime pathological player. Int J Biol Macromol. 2020;163:1599–1617. [DOI] [PubMed] [Google Scholar]
- 62.Falcon B, Zhang W, Schweighauser M, et al. Tau filaments from multiple cases of sporadic and inherited Alzheimer's disease adopt a common fold. Acta Neuropathol. 2018;136:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goedert M, Spillantini MG, Falcon B, et al. Tau protein and frontotemporal dementias. Adv Exp Med Biol. 2021;281:177–199. [DOI] [PubMed] [Google Scholar]
- 64.Goedert M, Falcon B, Zhang M, Ghetti B, Scheres SHW. Distinct conformers of assembled Tau in Alzheimer's and Pick's diseases. Cold Spring Harb Symp Quant Biol. 2018;83:163–171. [DOI] [PubMed] [Google Scholar]
- 65.Selkoe PD. Cell biology of the amyloid beta‐protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Biol. 1994;10:373–403. [DOI] [PubMed] [Google Scholar]
- 66.Sisodia SS, Price DL. Role of the beta‐amyloid protein in Alzheimer's disease. FASEB J. 1995;9:366–370. [DOI] [PubMed] [Google Scholar]
- 67.Storey E, Cappai R. The amyloid precursor protein of Alzheimer's disease and the Abeta peptide. Neuropathol Appl Neurobiol. 1999;25:81–97. [DOI] [PubMed] [Google Scholar]
- 68.Bancher C, Brunner C, Lassmann H, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989;477:90–99. [DOI] [PubMed] [Google Scholar]
- 69.De Strooper B, Chávez GL. Learning by failing: Ideas and concepts to tackle γ‐secretases in Alzheimer's disease and beyond. Annu Rev Pharmacol Toxicol. 2015;55:419–437. [DOI] [PubMed] [Google Scholar]
- 70.Dobson CM, Knowles TP, Vendruscolo M. The amyloid phenomenon and its significance in biology and medicine. Cold Spring Harb Perspect Biol. 2020;12:a033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Knowles TPJ, Waudby CA, Devlin GL, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. [DOI] [PubMed] [Google Scholar]
- 72.Baranczak A, Kelly JW. A current pharmacologic agent versus the promise of next generation therapeutics to ameliorate protein misfolding and/or aggregation diseases. Curr Opin Chem Biol. 2016;32:10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. [DOI] [PubMed] [Google Scholar]
- 74.Luheshi LM, Tartaglia GG, Brorsson AC, et al. Systematic in vivo analysis of the intrinsic determinants of amyloid beta pathogenicity. PLoS Biol. 2007;5:e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Habchi J, Arosio P, Perni M, et al. An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Aβ42 aggregates linked with Alzheimer's disease. Sci Adv. 2016;2:e1501244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goedert M, Jakes R, Spillantini MG. The synucleinopathies: Twenty years on. J Parkinsons Dis. 2017;7:S51–S69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stephens AD, Zacharopoulou M, Moons R, et al. Extent of N‐terminus exposure of monomeric alpha‐synuclein determines its aggregation propensity. Nat Commun. 2020;11:2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE. A new era for understanding amyloid structures and disease. Nat Rev Mol Cel Biol. 2018;19:755–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG. Structures of α‐synuclein filaments from multiple system atrophy. Nature. 2020;585:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Celeveland DW, Rothstein D. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. [DOI] [PubMed] [Google Scholar]
- 81.Lindberg MJ, Byström R, Boknäs N, Andersen PM, Oliveberg M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)‐associated SOD1 mutants. Proc Natl Acad Sci U S A. 2005;102:9754–9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lang L, Zetterstrom P, Brannstrom T, et al. SOD1 aggregation in ALS mice shows simplistic test tube behavior. Proc Natl Acad Sci U S A. 2015;112:9878–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cveticanin J, Mondal T, Meiering EM, Sharon M, Horovitz A. Insight into autosomal‐dominant inheritance pattern of SOD!‐associated ALS from native mass spectrometry. J Mol Biol. 2020;432:5995–6002. [DOI] [PubMed] [Google Scholar]
- 84.Rotunno MS, Bosco DA. An emerging role for misfolded wild‐type SOD1 in sporadic ALS pathogenesis. Front Cell Neurosci. 2013;7:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Banci L, Bertini I, Durazo A, Girotto S, Gralla EB. Metal free superoxide dismutase forms soluble oligomero under physiological conditions: A possible general mechanism for familial. Proc Natl Acad Sci U S A. 2007;104:11263–11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: Few candidates, frequent failures. Alzheimer Res Ther. 2014;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bucciantini M, Giannoni E, Chiti F, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. [DOI] [PubMed] [Google Scholar]
- 88.Fusco G, Chen SW, Williamson PTF, et al. Structural basis of membrane disruption and cellular toxicity by α‐synuclein oligomers. Science. 2017;358:1440–1443. [DOI] [PubMed] [Google Scholar]
- 89.Balch WE, Morimoto RI, Andrew Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. [DOI] [PubMed] [Google Scholar]
- 90.Hipp MS, Park S‐H, Hartl FU. Proteostasis impairment in protein‐misfolding and ‐aggregation diseases. Trends Cell Biol. 2014;24:506–514. [DOI] [PubMed] [Google Scholar]
- 91.Seaman MN. The retromer complex – Endosomal protein recycling and beyond. J Cell Sci. 2012;125:4693–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seaman MN. Endosome sorting: GSE complex minds the gap. Nat Cell Biol. 2006;8:648–649. [DOI] [PubMed] [Google Scholar]
- 93.Small SA, Petsko GA. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat Rev Neurosci. 2015;16:126–132. [DOI] [PubMed] [Google Scholar]
- 94.Small SA, Simoes‐Spassov S, Mayeux R, Petsko GA. Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer's disease. Trends Neurosci. 2017;40:592–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Young JE, Fong LK, Frankowski H, et al. Stabilizing the retromer complex in a human stem cell model of Alzheimer's disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Rep. 2018;10:1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mecozzi VJ, Berman DE, Simoes S, et al. Pharmacological chaperones stabilize retromer to limit APP processing. Nat Chem Biol. 2014;10:443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McNaugh KSP, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson's disease. Neurosci Lett. 2001;297:191–194. [DOI] [PubMed] [Google Scholar]
- 98.Ralph GS, Radcliffe PA, Day DM, Carthy JM, Leroux MA. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat Med. 2005;11:429–433. [DOI] [PubMed] [Google Scholar]
- 99.Raoul C, Abbas‐Terki T, Bensadoun JC, et al. Lentiviral‐mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11:423–428. [DOI] [PubMed] [Google Scholar]
- 100.Cyriam P, Tartaglia GG, Morimoto RI, Dobson CM, Vendruscolo M. Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Rep. 2013;5:781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]