

Abstract
Numerous mass spectrometry-based strategies ranging from hydrogen–deuterium exchange to ion mobility to native mass spectrometry have been developed to advance biophysical and structural characterization of protein conformations and determination of protein–ligand interactions. In this study, we focus on the use of ultraviolet photodissociation (UVPD) to examine the structure of human carbonic anhydrase II (hCAII) and its interactions with arylsulfonamide inhibitors. Carbonic anhydrase, which catalyzes the conversion of carbon dioxide to bicarbonate, has been the target of countless thermodynamic and kinetic studies owing to its well-characterized active site, binding cavity, and mechanism of inhibition by hundreds of ligands. Here, we showcase the application of UVPD for evaluating structural changes of hCAII upon ligand binding on the basis of variations in fragmentation of hCAII versus hCAII–arylsulfonamide complexes, particularly focusing on the hydrophobic pocket. To extend the coverage in the midregion of the protein sequence, a supercharging agent was added to the solutions to increase the charge states of the complexes. The three arylsulfonamides examined in this study largely shift the fragmentation patterns in similar ways, despite their differences in binding affinities.
Graphical Abstract
INTRODUCTION
Owing to their critical roles in many mechanisms and cellular processes related to disease states, proteins remain the most common targets for the development of inhibitors that selectively bind and disrupt protein function, typically via formation of noncovalent complexes.1,2 Numerous methods have been developed to characterize the structures of protein–inhibitor complexes and the conformational reorganization of the proteins as well as determine the binding affinities and elucidate the binding sites.1 Well-established biophysical methods used in this context include surface plasmon resonance (SPR), isothermal titration calorimetry, UV–visible, fluorescence, and circular dichroism spectroscopies, nuclear magnetic resonance (NMR) spectrometry, and X-ray crystal-lography, among others.3–5 A number of mass spectrometry methods have also been used in the context of drug development and the characterization of protein targets, ranging from the vast array of powerful high throughput proteomics methods6,7 to more specialized techniques like hydrogen/deuterium exchange used to probe the impact of mutations and binding events on protein stability and conformation.8 A more newly emerging strategy, termed native mass spectrometry, has gained traction in the field of structural biology owing to the ability to interrogate intact protein complexes in the gas phase with high sensitivity.9–11 From the native mass spectra, stoichiometries of the protein–ligand complexes can be determined and relative binding affinities, estimated.12–17 Moreover, the combination of native MS with tandem mass spectrometry (MS/MS) of intact, noncovalently bound protein–ligand complexes offers the potential for structural characterization and localization of the ligand binding site.12,18
In the context of MS/MS methods for protein complexes, collisional activated dissociation typically causes preferential cleavage of noncovalent bonds, which lead to the ejection of bound ligands but little information regarding binding site or specific structural changes of the protein.19–21 Electron-based activation techniques, including electron-capture dissociation (ECD) and electron-transfer dissociation (ETD), preserve noncovalent interactions and generate sequence ions whose abundances reflect the flexibility of the region of the protein from which the backbone cleavages occur (i.e., correlation with crystallographic B-factors).22,23 Surface-induced dissociation (SID) has provided incredible insight into the quaternary structures of multimeric protein complexes and subunit organization.24 In ultraviolet photodissociation (UVPD), absorption of high-energy photons (typically 193 nm) results in the activation of proteins to excited electronic states in a way that promotes backbone cleavages and retention of non-covalent interactions.25 This process results in holo-type sequence ions in which ligands may remain bound, and the pattern of apo and holo product ions reveal the ligand binding sites.26–28 Furthermore, changes in the fragmentation patterns of the apo-proteins versus protein-ligand complexes suggest conformational re-organization of the proteins upon ligand binding. In essence, regions of the proteins that are more flexible exhibit enhanced backbone cleavages, whereas more highly structured regions stabilized by networks of noncovalent interactions display diminished backbone cleavages.26–28 The latter outcome has been attributed to inhibition of the separation of fragment ions during the activation/dissociation process, thus attenuating the detection of those products. These features make UVPD a versatile tool for the characterization of protein–ligand interactions and have catalyzed a number of recent studies.23,26–36 For example, UVPD has been used to elucidate the binding sites of both a cofactor (NADPH) and an inhibitor (methotrexate) to dihydrofolate reductase (DHFR) and to monitor its conformational changes upon ligand binding.28 The extensive array of conformational changes of adenylate kinase was examined during each step of its catalytic cycle, further demonstrating the sensitivity of UVPD to changes in protein structure.30 In addition, UVPD was used to decipher structural changes promoted by single amino acid mutations in the GTP-ase protein K-Ras31 and DHFR.32 More recently, a multistage UVPD approach was employed to characterize the impact of a single amino acid mutation of the dimeric human mitochondrial enzyme branched-chain amino acid transferase 2 on its conformation and to localize the binding location of its cofactor, pyridoxal phosphate.34 These previous studies have demonstrated the utility of UVPD for mapping binding sites, monitoring conformational changes related to ligand binding, and deciphering the modulation of protein–ligand interactions by single point mutations. The present work aims to evaluate the sensitivity of UVPD to variations in binding affinities and binding geometries of protein–ligand complexes as demonstrated for human carbonic anhydrase II (hCAII) and three arylsulfonamide inhibitors, ethoxzolamide, chlorothiazide, and furosemide.
The well-characterized crystal structure of human carbonic anhydrase II (hCAII) and extensive understanding of its binding interactions with arylsulfonamide inhibitors have made it a target of numerous fundamental biophysical, kinetic, and thermodynamic studies.37–50 Carbonic anhydrase catalyzes the conversion of carbon dioxide to bicarbonate and thus plays an essential role in controlling the pH of cells and CO2 transport. Overexpression of this enzyme is known to cause glaucoma, and mutations can cause many other diseases including osteopetrosis and cerebral calcification.43 Due to these reasons, an array of inhibitors has been developed, and numerous biophysical studies have provided conformational details about the protein.37–50 Several mass spectrometry studies have also evaluated the binding constants of carbonic anhydrase interacting with sulfonamide inhibitors on the basis of ion abundances in ESI mass spectra.15–17 In addition, the relative stabilities of the carbonic anhydrase complexes have been estimated using sustained off-resonance irradiation collision-induced dissociation.51–54
Our work highlights the utility of UVPD for the identification of structural changes of hCAII upon ligand binding, ones primarily observed in the hydrophobic pocket. As shown here, the addition of a supercharging agent to the solutions allowed access to higher charge states of the complexes and offered higher sequence coverage upon UVPD. No significant differences were identified between ligands with different binding affinities, which suggests that the affinity of the ligands does not significantly affect the structure of carbonic anhydrase for the different sulfonamide complexes.
EXPERIMENTAL SECTION
Sample Preparation.
Human carbonic anhydrase II (Sigma-Aldrich, St. Louis, MO), ammonium acetate (Sigma-Aldrich, St. Louis, MO), and HPLC grade water (Millipore, Burlington, MA) were purchased. Ethoxzolamide, chlorothiazide, and furosemide were purchased from Sigma-Aldrich (St. Louis, MO). Equimolar protein/ligand solutions were prepared at a concentration of 10 μM with 100 mM ammonium acetate. A supercharging agent, m-nitrobenzyl alcohol (m-NBA, Sigma-Aldrich, St. Louis, MO), was added at 0.5% to the protein solutions to improve sequence coverage upon UVPD and generate extensive holo-product ions without disrupting the protein–ligand interactions. Solutions were desalted using Micro Biospin P-6 gel columns (Bio-Rad Laboratories Inc., Hercules, CA) for MS analysis.
Mass Spectrometry.
A Thermo Fisher Scientific Orbitrap Elite mass spectrometer coupled to a 193 nm Coherent Excistar XS excimer laser for UVPD in the HCD cell was used. The resolving power was set to 240 K at m/z 400. The solutions were infused via a gold/palladium-coated static tip with applied voltage ranging from 0.9 to 1.1 kV at a capillary temperature of 200 °C. UVPD was performed using a single 1.0–2.0 mJ laser pulse in the HCD cell. The charge states of the protein and protein–ligand complexes were selected using an isolation width of 20–40 m/z to allow sufficient ion abundance for this moderately large protein complex. An AGC target of 5 × 105 with a maximum injection time of 2 s was used. 500 scans were averaged for each spectrum. All results were replicated three times.
Data Analysis.
The UVPD spectra were decharged and deisotoped using the Xtract algorithm (Xcalibur Qual Browser, Thermo Fisher Scientific) with a signal-to-noise ratio of 2, a fit factor of 44%, and remainder of 25%. Sequence coverage maps were generated using ProSight Lite v1.455 with a 10 ppm error tolerance. The searches included all types of fragment ions produced by UVPD (a, a + 1, b, c, x, x + 1, y, y − 1, and z) or HCD (b, y). Fragments (a, a + 1, b, c, x, x + 1, y, y − 1, and z) were also identified using UV-POSIT56 for both UVPD and HCD, and the searches included fragment ions with a designated mass shift corresponding to each ligand mass, both with and without the coordinating Zn2+. Normalized ion abundances were calculated by summing the abundances of all a, b, c, z, y, and z fragment ions associated with a particular backbone position and dividing this by the summed abundance of all identified ions. Data was processed using UVPOSIT, an in-house algorithm, to calculate backbone cleavage propensities. The propensities for backbone cleavages throughout the protein containing R residues were calculated by summing all an, bn, and cn ions originating from cleavage immediately C-terminal to a given residue along with all C-terminal xR−n+1, yR−n+1, and zR−n+1 ions originating from cleavage N-terminal to the same residue. In this way, the degree of backbone cleavages adjacent to each residue are compiled to generate maps of the variation in fragmentation throughout the protein. The images of protein crystal structures (PDB 2ILI, 1Z9Y, 3CAJ) were generated using Pymol (version 1.3).
RESULTS AND DISCUSSION
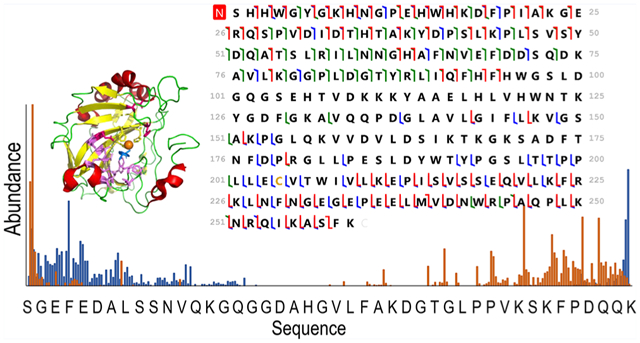
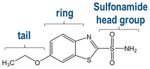
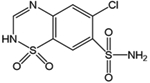
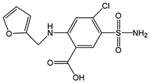
hCAII was showcased for this study because it has been the target of hundreds of biochemical and biophysical studies that have established a vast understanding of its structure/function relationships, thermodynamic and kinetic parameters for binding both substrates and inhibitors (particularly sulfonamides), and mechanistic insight.37 The binding site of hCAII is known to have both hydrophobic and hydrophilic pockets as depicted in Figure S1.37,39 The hydrophobic pocket is formed by the residues Val121, Val143, Leu198, Thr199, His200, and Trp209 that engage in hydrogen bonds with ligands, whereas the hydrophilic pocket is composed of Asn62, His64, Asn67, and Gln92.40–42 The zinc cofactor is tetrahedrally coordinated with His94, His96, and His119. Arylsulfonamides are one of the most extensively studied class of inhibitors, exhibiting high binding affinities (μM to nM) and sharing well-defined binding geometries.37 Sulfonamide drugs bind to the active site of carbonic anhydrase and inhibit its function owing to the similarity of the sulfamido group and the transition state analog of HCO3−, a key step in the reversible hydration of carbon dioxide. The sulfonamide nitrogen atom coordinates to the zinc cofactor, and the NH group engages in hydrogen-bonding with Thr199.37 The aryl ring of the ligand engages in interactions with the hydrophobic portion of the binding pocket. Entry of the substrate (CO2) or an inhibitor (e.g., sulfonamide ligand) into the binding pocket depends on the orientations of loop 2 (composed of residues 230–240) and particularly loop 1 (containing residues 197–206, the hydrophobic side of the binding pocket). The binding of a sulfonamide ligand does not cause major conformational changes to hCAII but rather more modest reorganization of residues lining the active site.37 On the basis of the impressive foundational knowledge of the hCAII structure and binding interactions, for the present study, UVPD was used to characterize the structural changes of hCA II upon binding ethoxzolamide, chlorothiazide, and furosemide (Table 1) with dissociation constants of 0.004 μM (high affinity), 0.4 μM, and 3.1 μM (low affinity), respectively. Differences in the pattern of backbone cleavages between hCAII and hCAII–ligand complexes were mapped onto the crystal structure of the protein to visualize changes in conformation attributed to ligand binding. To further expand the depth of backbone cleavages and the resulting sequence coverage, the addition of a supercharging agent was evaluated to increase the charge states of the complexes prior to UVPD.
Table 1.
Human Carbonic Anhydrase II–Ligand Complexes, Their Binding Affinities, and Ligand Structures
| Complex | MW (Da) | Kd(uM) | Ligand structure |
|---|---|---|---|
| Ethoxzolamide | 258.01 | 0.004 |
|
| Chlorothiazide | 295.72 | 0.4 |
|
| Furosemide | 330.01 | 3.1 |
|
Native MS and Backbone Fragmentation of Carbonic Anhydrase.
ESI of human carbonic anhydrase II resulted in ions in low charge states (9+, 10+, and low abundance 11+) characteristic of native-like proteins (Figure 1A). UVPD of the low charge states provided good sequence coverage ranging from 62% for the 9+ to 66% for 10+ as shown in Figures S2 and 2, but some of the key residues in the midsection of the protein were not mapped, which hampers the structural analysis of the protein–ligand complexes (Figure 2). To extend the coverage, a supercharging agent, m-NBA, was added to the native hCAII solutions in an effort to produce a larger range of charge states. The mechanism of supercharging is still not fully understood with studies supporting that supercharging agents cause a chemical/thermal denaturation57–66 resulting in the unfolding of the protein during ESI as supported by circular dichroism experiments.60,65 However, other studies defend that supercharging does not cause conformational changes as noncovalent ligand binding is not disrupted in protein complexes.67–70 In this work, a low concentration of the supercharging agent was added to slightly increase the charge states while minimizing the effect on the protein conformation. The addition of m-NBA led to the production of charge states between 11+ and 14+ (Figure 1B) without the loss of the zinc cofactor even for the higher charge states. This suggests that the protein structure was maintained in the gas phase and the addition of the supercharging agent at 0.5% did not cause denaturation. Greater sequence coverage was obtained in the middle section of the protein for the 11+ and 12+ charge states as shown in Figure 2. The sequence coverage obtained with UVPD for the different charge states is displayed in Figure S2. An increased coverage is observed from the 9+ and 10+ (62% and 66%) to the 11+ and 12+ (79% and 76%) charge states followed by a decrease in coverage for higher charge states (51% and 50% for 13+ and 14+, respectively). Variations in sequence coverage upon supercharging have also been noted in prior studies, one which focused on collision-induced dissociation (CID) of bovine CA and also used m-NBA to increase the charge states.72 CID of higher charge states did not significantly increase the number of identified fragment ions (28 products for 10+ versus 29 products for 14+), but an increase in the formation of fragment ions retaining zinc was reported.67 In the present study, higher charge states provided higher sequence coverage upon UVPD and also generated a greater number of zinc-containing fragment ions (Figure S3). UVPD is not a process largely mediated by mobile protons like CID, and thus, there is typically not an enhancement of preferential cleavages (occurring adjacent to Asp, Glu, or Pro residues) that is often observed for CID. The drop in sequence coverage for the highest 13+ and 14+ charge states upon UVPD (Figure S3) is attributed to the production of more highly charged fragment ions that fall in regions of the mass spectrum that are more congested and thus less easy to deconvolve. Since the 11+ species of hCAII provided greater sequence coverage and a higher number of fragment ions (Figure 2), it was targeted throughout the rest of the study.
Figure 1.
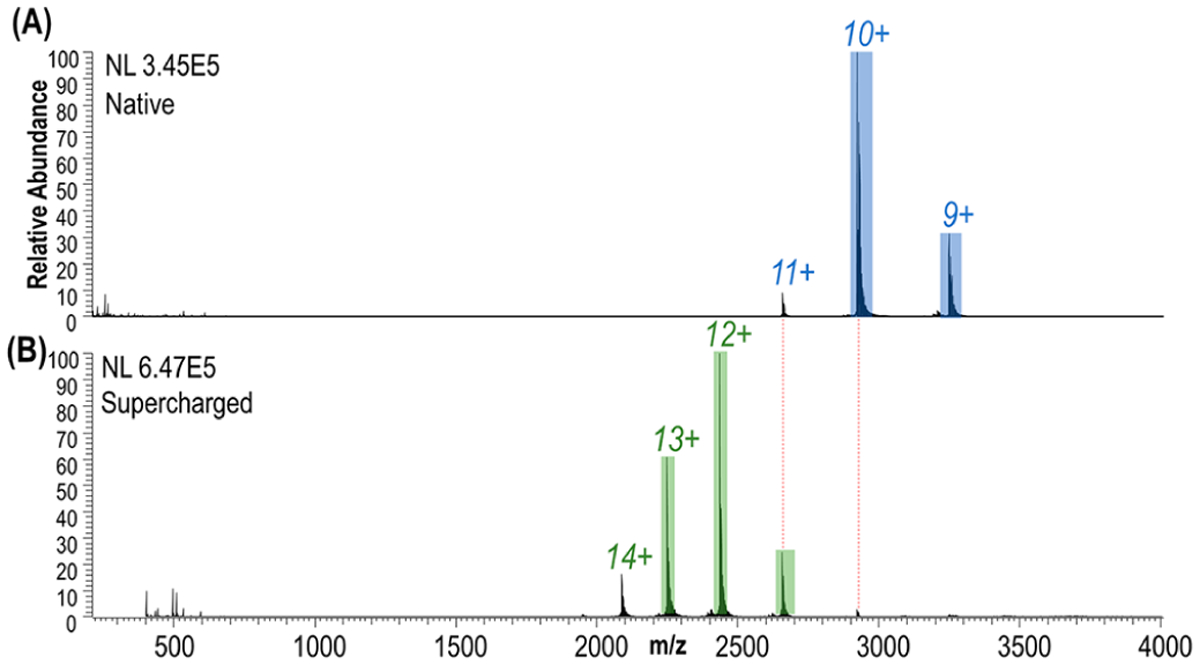
ESI mass spectra of human carbonic anhydrase II in (A) 100 mM ammonium acetate (native) and (B) supercharged.
Figure 2.
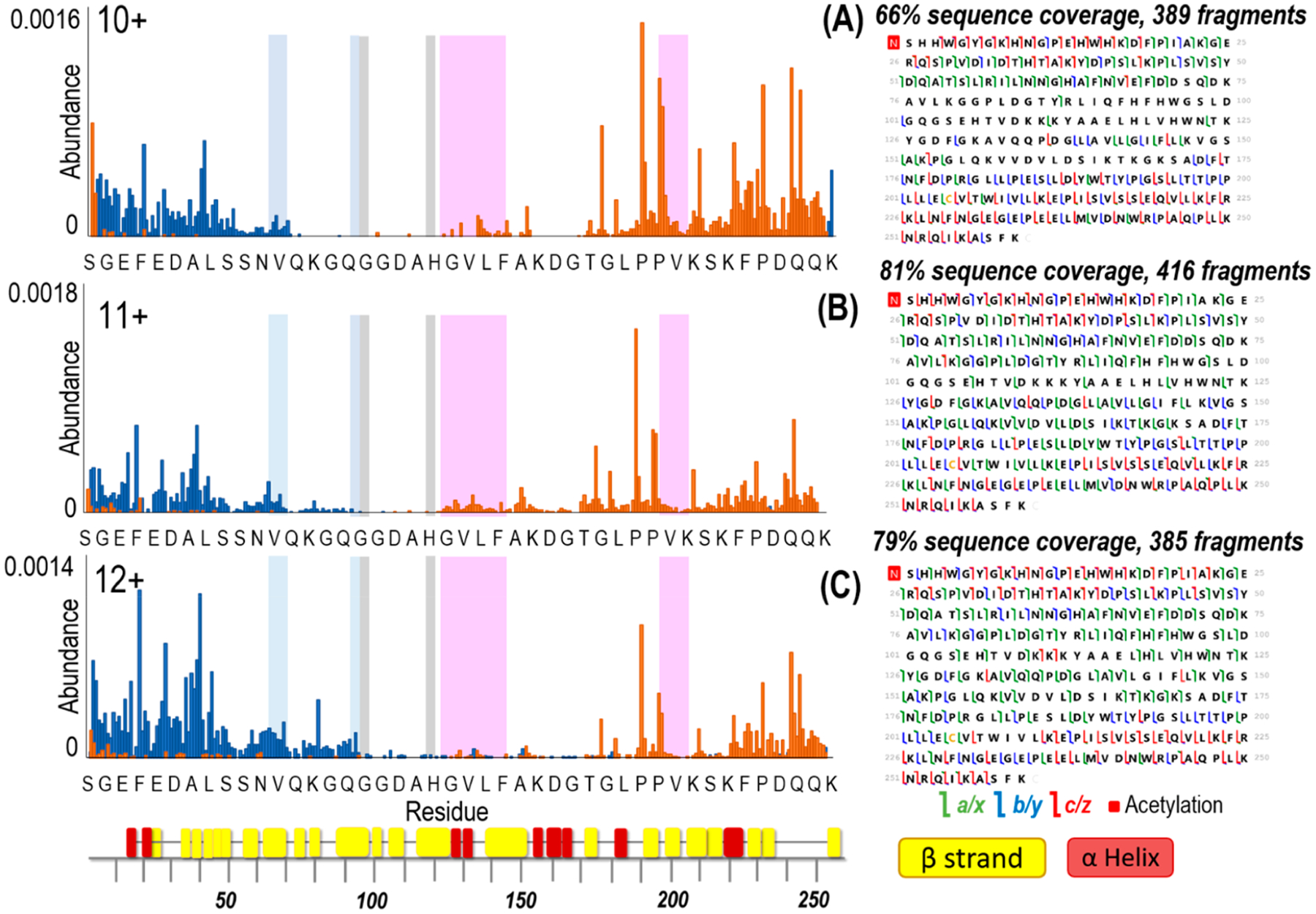
Backbone cleavage propensity maps that correspond to fragment ions produced upon UVPD (1 pulse, 2 mJ) of hCAII for the 10, 11, and 12+ charge states and respective sequence coverage maps. C-terminal ions are depicted in orange and N-terminal ions are depicted in blue. Highlighted are the hydrophobic pocket (pink), the hydrophilic pocket (light blue), and the residues that coordinate zinc (gray). The x-axis shows 1 out of every 6 residues. β-strands and α-helices are labeled underneath the x-axis using colors corresponding to Figure S1.
Analysis of Fragment Ions Generated by UVPD.
The type of ions produced upon UVPD were examined for human carbonic anhydrase II across different charge states. As shown in Figure 3, a diverse array of fragment ions was formed, and the most dominant ions observed were a, x, and y ions, whereas b, c, and z ions as well as a few d and w ions were formed with lower abundances. This is the usual trend observed with UVPD as fragmentation may occur directly from excited states populated upon UV photoabsorption or after internal conversion and intramolecular vibrational energy redistribution (IVR) resulting in CID-like fragmentation. The latter process typically generates b/y-type ions, whereas the former produces a/x and c/z-type ions more unique to UVPD.71 The absence of significant vibrational energy redistribution means that a/x ions frequently retain non-covalent interactions, such as ones stabilizing bound ligands or those associated with secondary structural motifs, and thus, these fragment ions are anticipated to provide the most insight into protein structure and ligand-induced conformational reorganization.
Figure 3.
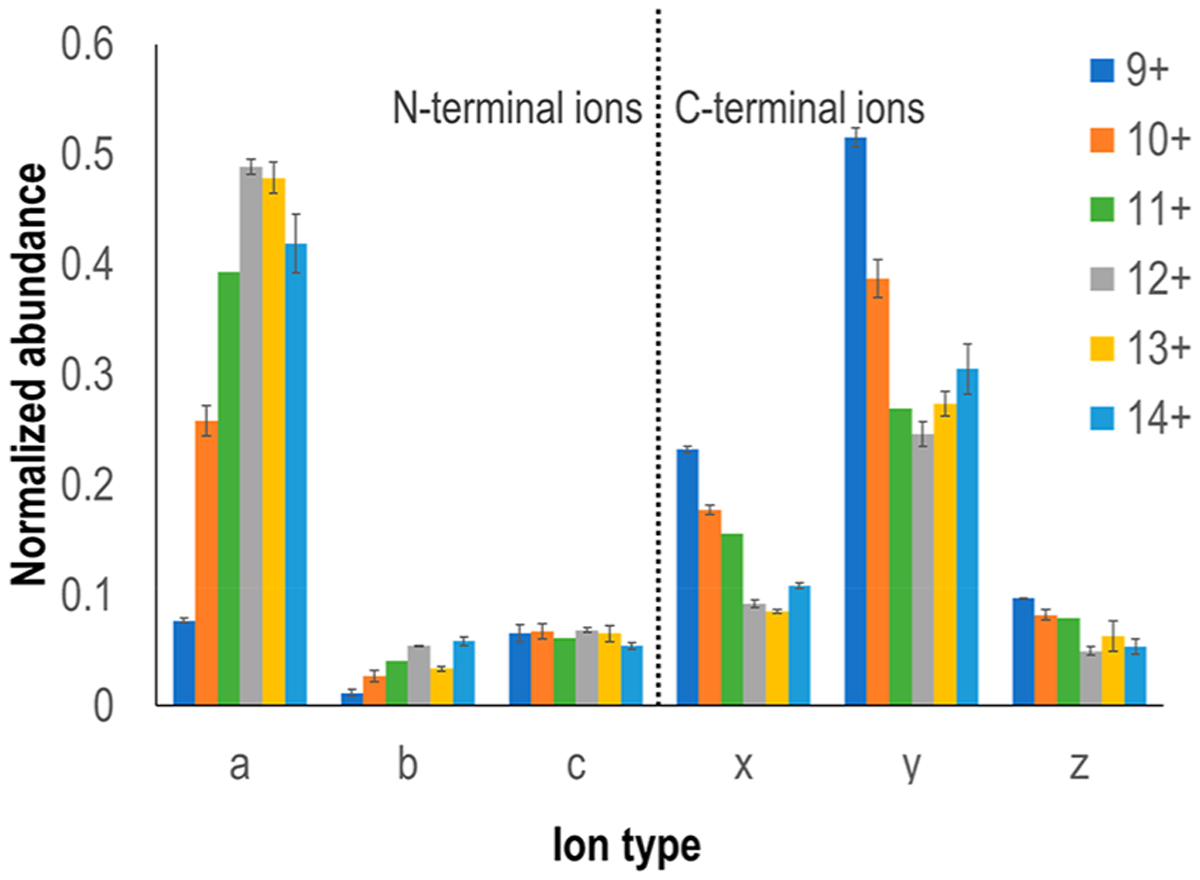
Summed abundance of product ion type observed as a function of precursor ion charge state for human carbonic anhydrase II.
Interestingly, fragment ions containing the C-terminus (x, y, z) were more abundant upon UVPD of the protein in lower charge states, and their abundance decreased with increasing charge of the protein. The trend reversed for the fragment ions containing the N-terminus (a, b, c); the abundances of these ions increased with the charge state of the protein. The variations in the preferential formation of N-terminal versus C-terminal fragment ions likely arise from a combination of several factors: conformational changes owing to increases in the charge state, adjustment of the initial positions of protons as more protons are added,72 location of secondary structural motifs that favor direct dissociation from excited states versus internal conversion, intramolecular vibrational energy redistribution, and preferential fragmentation via CAD-like path-ways, and shifts in salt bridges and other charge-stabilizing interactions.73 Nevertheless, our data suggests that the protein structure is largely maintained in the gas phase and the addition of the supercharging agent does not cause significant conformational changes since all the precursor charge states interrogated (9+ to 14+) showed that the zinc cofactor remained bound to the protein. Variations in the propensities for the formation of N-terminal versus C-terminal fragment ions as a function of precursor charge state is not uncommon in the activation and dissociation of intact proteins, and deciphering the underlying reasons via a more elaborate data-mining investigation of multiple proteins may facilitate optimization of experimental parameters to maximize sequence coverage in top-down proteomics.
Backbone Fragmentation of Carbonic Anhydrase–Ligand Complexes.
Electrospray ionization of solutions containing human carbonic anhydrase II and one ligand (ethoxzolamide, chlorothiazide, or furosemide) with 0.5% NBA resulted in the formation of abundant 1:1 complexes in charge states ranging from 10+ to 14+ (Figures S4–S6). Without 0.5% NBA, the charge states range from 9+ to 11+ (Figure S7). For each complex examined, the 11+ charge state was isolated and subjected to 193 nm UVPD (1 pulse at 2 mJ per pulse). Figures S8–S10 display the UVPD mass spectra of hCAII and hCAII–ligand complexes (all 11+ charge state), demonstrating high sequence coverage ranging from 63% to 66% for the complexes.
Backbone cleavage propensity maps throughout the entire protein sequence were generated by summing the abundances of all a, b, c, z, y, and z fragment ions originating from cleavages C- and N-terminal to each residue and dividing by the summed abundance of all identified ions. The backbone cleavage propensity maps for hCAII and hCAII–ethoxzolamide are depicted in Figure 4. The corresponding maps for the complexes containing the other two inhibitors (chlorothiazide and furosemide) are displayed in Figures S11 and S12. It is evident that in certain regions backbone fragmentation is suppressed upon ligand binding. On the basis of previous studies,26–28,30–33,35 conformational changes of a protein that are related to ligand binding may be reflected in the variations of the backbone cleavage efficiencies upon UVPD. In essence, regions of a protein that engage in intramolecular interactions with a ligand may exhibit suppressed fragmentation (compared to the ligand-free protein) owing to a reduced ability of the fragments to separate from each other. Similarly, other regions of the protein that undergo conformational reorganization as a result of ligand binding may display enhanced fragmentation upon UVPD. In this context, the various N-terminal and C-terminal fragment ions serve as the reporters of the backbone cleavage propensities throughout the protein, and the comparison of the abundances of the fragment ions for the apoprotein (no ligand) versus the corresponding protein–ligand complex may be used to map the variations in backbone cleavage propensities that arise from ligand binding. To better visualize the changes in fragmentation between the ligand-free protein and each protein–ligand complex, difference plots were constructed by subtraction of the abundances of fragment ions generated from carbonic anhydrase from the abundances of fragment ions of the carbonic anhydrase–ligand complexes (Figure 5). The corresponding histograms of the p-values calculated from the t test for each difference plot are shown in Figure S13, thus allowing the application of a confidence level of 99% to filter the most meaningful differences in backbone fragmentation. In the resulting series of difference plots in Figure 5, negative values signify a decrease in UVPD fragmentation efficiency of the carbonic anhydrase–ligand complex relative to the apoprotein, whereas positive values indicate an increase in UVPD fragmentation efficiency of the carbonic anhydrase–ligand complex relative to the apoprotein.
Figure 4.
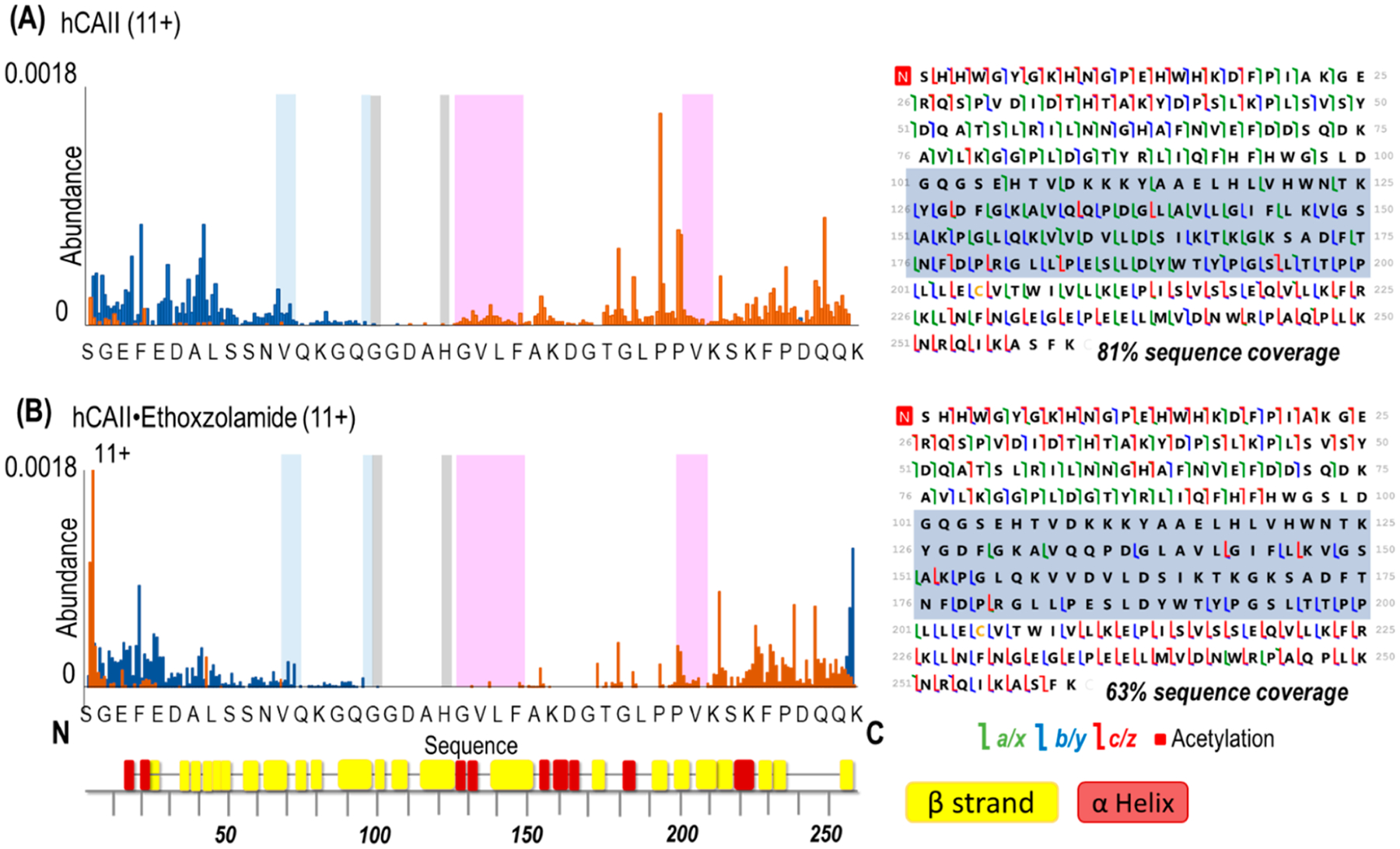
Backbone cleavage propensity maps (including both apo and holo ions) produced upon UVPD (1 pulse, 2 mJ) of (A) hCAII and (B) hCAII bound to ethoxzolamide and respective sequence coverage maps (left). Backbone cleavages producing C-terminal ions are depicted in orange, and those producing N-terminal ions are depicted in blue. Highlighted are the hydrophobic pocket (pink), the hydrophilic pocket (light blue), and the residues that coordinate zinc (gray). The x-axis shows 1 out of every 6 residues. Shaded regions in the sequence maps highlight decreased sequence coverage for the hCAII complex. β-strands and α-helices are labeled underneath the x-axis using colors corresponding to Figure S1.
Figure 5.
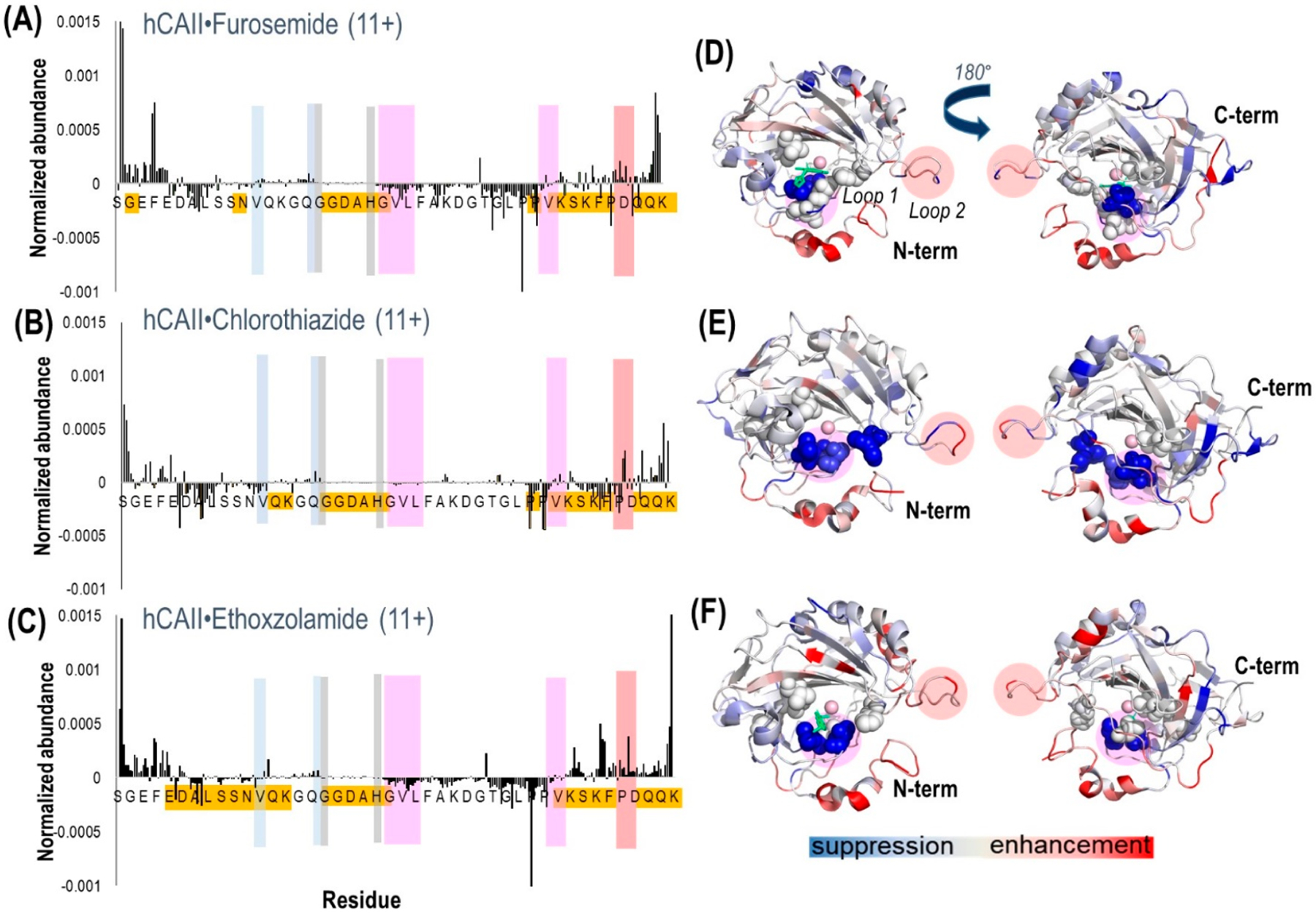
Plots of the differences in abundances of fragment ions (both apo and holo ions) obtained from hCAII complexed with (A) furosemide, (B) chlorothiazide, and (C) ethoxzolamide. Highlighted are the hydrophobic pocket/loop 1 (pink), the hydrophilic pocket (light blue), the residues that coordinate zinc (gray), and loop 2 (peach). Highlighted in gold are the residues where the most meaningful differences in backbone fragmentation occurred. The crystal structure of hCAII shows the suppression (blue-shaded residues) or enhancement (red-shaded residues) of backbone cleavages upon UVPD for the complexes containing (D) furosemide (PDB 1Z9Y), (E) chlorothiazide (PDB 2ILI), and (F) ethoxzolamide (PDB 3CAJ). For the structures, the sulfonamide ligand is depicted in green. There is no reported crystal structure of hCAII–chlorothiazide, so the structure is shown for hCAII. Regions shaded in a pink circle (residues 197–206) indicate loop 1, and those shaded in a peach circle (residues 230–240) indicate loop 2. The residues corresponding to the hydrophobic pocket (V121, V143, L198, T199, H200, W209) are represented as spheres.
A number of notable differences are observed upon inspection of the fragmentation trends for hCAII relative to its complexes: cleavages at some backbone sites were enhanced; others were significantly suppressed upon ligand binding. Most of the significant variations in backbone cleavage efficiency upon UVPD occurred near the putative binding region (His-64, His-94, His-96, His-119, Val-121, Val-143, Thr-199, and Thr-200).37–44 To help visualize these differences, the residues for which backbone fragmentation is enhanced or suppressed are highlighted in the carbonic anhydrase structures in Figure 5D–F. The red shading represents locations of enhanced UVPD fragmentation, and the blue shading indicates positions of suppressed UVPD fragmentation for the sulfonamide complexes. The presence of the ligand generally suppressed fragmentation in the active site loop composed of residues 197–206 (loop 1) where Leu 198, Thr 199, and His 200 are known to participate in ligand binding by electrostatic contacts (gas phase) or hydrophobic interactions (solution),37,74 implying the establishment of noncovalent interactions of hCAII with the ligand. All three ligands interact with carbonic anhydrase through the sulfonamide headgroup (SO2NH), the aryl ring, and the tail region (see the structures in Table 1). The ionized sulfonamide interacts with the zinc cofactor, and the sulfonamide headgroup interacts with residues that are hydrogen-bonding acceptors or donors such as Thr199. The aryl group interacts with the hydrophobic pocket of CA, thus explaining why all inhibitors suppress UVPD fragmentation of the hydrophobic pocket.
Most of the enhanced fragmentation of the carbonic anhydrase–ligand complexes occurred in the N- and C-terminus regions. An increased degree of backbone fragmentation of the N- and C-terminus regions has been commonly observed upon UVPD of other protein–ligand complexes,75 suggesting that the terminal ends cleave more readily, perhaps owing to preferential stabilization of other regions of the protein upon ligand binding. In general, enhanced fragmentation near the N- and C-termini has been observed in many top-down studies of both denatured and native-like proteins owing to their greater exposure on the surfaces, and thus, these terminal regions are in many ways the least diagnostic of specific conformational changes.
Another region of the carbonic anhydrase–ligand complexes that exhibits notable variations in fragmentation encompasses loop 2 (residues 230–240), a structural element that modulates the entrance to the binding pocket and is composed of a very flexible loop motif that is prone to significant conformational rearrangement. The variation in fragmentation near the loop 2 region is rationalized by the conformational flexibility of loop 2 in the presence of the ligand. These observations are generally in agreement with the findings of an extensive modeling study of hCAII (apo CA and CA with CO2)74 that has shown that carbonic anhydrase has multiple conformations of loops 1 and 2, including the open, semiopen, and closed conformations. Once a ligand binds, loop 1 is expected to close and compress the pocket in order to bury the ligand in the active site and move it closer to the residues involved in hydrogen bonding. Loop 2, on the other hand, is a surface loop and can only adopt open or superopen conformations to allow the entrance of the ligand into the binding pocket.
The region where zinc binds (His 94, His 96, and His 119) and consequently where the ligand interacts via the Zn–N bond did not show significant differences in backbone cleavage propensity for the ligand-bound versus ligand-free forms of hCAII. This lack of change in the backbone cleavage propensity is rationalized on the basis of the significant stability of the zinc binding site, thus mitigating its structural reorganization during ligand binding.
Suppressed fragmentation was uniquely observed for the carbonic anhydrase–chlorothiazide complexes in the hydrophilic portion of the binding pocket (Asn62, His64, Asn67, and Gln92). This interesting difference might be related to the fact that chlorothiazide is significantly more hydrophilic (XLogP3 = −0.2) than ethoxzolamide and furosemide (XLogP3 = 2), meaning that chlorothiazide might preferentially participate in hydrophilic contacts that stabilize this region of the protein.76
There was no clear correlation between the binding affinities of the three sulfonamide ligands and the backbone cleavage patterns (i.e., the abundances of fragment ions or the overall degree of enhancement or suppression of fragmentation) upon UVPD. Although a much more extensive study with numerous ligands is warranted, this outcome is consistent with previous evidence that inhibitors containing aromatic functional groups all orient into the hydrophobic binding pocket of the binding site in a similar manner.
CONCLUSION
Here, we used UVPD to study conformational changes of human carbonic anhydrase II upon binding of sulfonamide ligands with different affinities. A supercharging agent was used to improve coverage in the middle of the protein sequence as well as to increase the number of holo ions. The apo and holo ions produced after UVPD of the human carbonic anhydrase II–ligand complexes provided insight into some structural changes of the protein after ligand binding. Most of the significant variations in backbone cleavage efficiency upon UVPD occurred near the binding region. The presence of the ligand generally suppressed fragmentation in the hydrophobic pocket possibly due to noncovalent interactions between hCAII and the ligand and enhanced fragmentation in the loop 2 and N- and C-terminus regions, suggesting greater conformational flexibility of these regions in the presence of the ligand. The three arylsulfonamides examined in this study cause similar structural changes despite their differences in binding affinities. Nevertheless, UVPD allowed the characterization of general conformational changes upon ligand binding, and this outcome is consistent with previous evidence that inhibitors containing aromatic functional groups all orient into the hydrophobic binding pocket of the binding site in a similar fashion. A more extensive study is necessary in the future to continue to assess conformational changes of human carbonic anhydrase II upon binding of different types of ligands with different affinities.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the Robert A. Welch Foundation (F-1155) and National Institute Of General Medical Sciences of the National Institutes of Health under awards R01GM121714 and R35GM139658. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Robert A. Welch Foundation or the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.1c00004.
Supporting figures include MS1 spectra, examples of MS/MS spectra, a sequence map, backbone cleavage maps, statistical analysis, and distributions of different fragment ion types (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jasms.1c00004
The authors declare no competing financial interest.
REFERENCES
- (1).Du X; Li Y; Xia Y-L; Ai S-M; Liang J; Sang P; Ji X-L; Liu SQ Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci 2016, 17, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fry DC Protein–Protein Interactions as Targets for Small Molecule Drug Discovery. Biopolymers 2006, 84, 535–552. [DOI] [PubMed] [Google Scholar]
- (3).Acharya KR; Lloyd MD The Advantages and Limitations of Protein Crystal Structures. Trends Pharmacol. Sci 2005, 26, 10–14. [DOI] [PubMed] [Google Scholar]
- (4).Yu H Extending the Size Limit of Protein Nuclear Magnetic Resonance. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Loo JA Studying Noncovalent Protein Complexes by Electrospray Ionization Mass Spectrometry. Mass Spectrom. Rev 1997, 16, 1–23. [DOI] [PubMed] [Google Scholar]
- (6).Zhang Z; Wu S; Stenoien DL; Pasa-Tolic L High Throughput Proteomics. Annu. Rev. Anal. Chem 2014, 7, 427–454. [DOI] [PubMed] [Google Scholar]
- (7).Gillet LC; Leitner A; Aebersold R Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annu. Rev. Anal. Chem 2016, 9, 449–472. [DOI] [PubMed] [Google Scholar]
- (8).Konermann L; Pan J; Liu Y-H Hydrogen Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. Chem. Soc. Rev 2011, 40, 1224–1234. [DOI] [PubMed] [Google Scholar]
- (9).Pacholarz KJ; Garlish RA; Taylor RJ; Barran PE Mass Spectrometry Based Tools to Investigate Protein–Ligand Interactions for Drug Discovery. Chem. Soc. Rev 2012, 41, 4335–4355. [DOI] [PubMed] [Google Scholar]
- (10).Eschweiler JD; Kerr R; Rabuck-Gibbons J; Ruotolo BT Sizing Up Protein-Ligand Complexes: The Rise of Structural Mass Spectrometry Approaches in the Pharmaceutical Sciences. Annu. Rev. Anal. Chem 2017, 10, 25–44. [DOI] [PubMed] [Google Scholar]
- (11).Boeri Erba E; Petosa C The Emerging Role of Native Mass Spectrometry in Characterizing the Structure and Dynamics of Macromolecular Complexes. Protein Sci. 2015, 24, 1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ishii K; Noda M; Uchiyama S Mass Spectrometric Analysis of Protein-Ligand Interactions. Biophysics and Physicobiology 2016, 13, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sharon M; Robinson CV The Role of Mass Spectrometry in Structure Elucidation of Dynamic Protein Complexes. Annu. Rev. Biochem 2007, 76, 167–193. [DOI] [PubMed] [Google Scholar]
- (14).Heck AJR Native Mass Spectrometry: A Bridge Between Interactomics and Structural Biology. Nat. Methods 2008, 5, 927–933. [DOI] [PubMed] [Google Scholar]
- (15).Daniel JM; McCombie G; Wendt S; Zenobi R Mass Spectrometric Determination of Association Constants of Adenylate Kinase with Two Noncovalent Inhibitors. J. Am. Soc. Mass Spectrom 2003, 14, 442–448. [DOI] [PubMed] [Google Scholar]
- (16).Jecklin MC; Schauer S; Dumelin CE; Zenobi R Label-Free Determination of Protein–Ligand Binding Constants Using Mass Spectrometry and Validation Using Surface Plasmon Resonance and Isothermal Titration Calorimetry. J. Mol. Recognit 2009, 22, 319–329. [DOI] [PubMed] [Google Scholar]
- (17).Erba EB; Zenobi R Mass Spectrometric Studies of Dissociation Constants of Noncovalent Complexes. Annu. Rep. Prog. Chem., Sect. C: Phys. Chem 2011, 107, 199–228. [Google Scholar]
- (18).Macias LA; Santos IC; Brodbelt JS Ion Activation Methods for Peptides and Proteins. Anal. Chem 2020, 92 (1), 227–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).McCammon MG; Hernández H; Sobott F; Robinson CV Tandem Mass Spectrometry Defines the Stoichiometry and Quaternary Structural Arrangement of Tryptophan Molecules in the Multiprotein Complex TRAP. J. Am. Chem. Soc 2004, 126, 5950–5951. [DOI] [PubMed] [Google Scholar]
- (20).Benesch JL; Aquilina JA; Ruotolo BT; Sobott F; Robinson CV Tandem Mass Spectrometry Reveals the Quaternary Organization of Macromolecular Assemblies. Chem. Biol 2006, 13, 597–605. [DOI] [PubMed] [Google Scholar]
- (21).Hall Z; Hernandez H; Marsh JA; Teichmann SA; Robinson CV The Role of Salt Bridges, Charge Density, and Subunit Flexibility in Determining Disassembly Routes of Protein Complexes. Structure 2013, 21, 1325–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lermyte F; Sobott F Electron Transfer Dissociation Provides Higher-Order Structural Information of Native and Partially Unfolded Protein Complexes. Proteomics 2015, 15, 2813–2822. [DOI] [PubMed] [Google Scholar]
- (23).Li H; Wongkongkathep P; Van Orden SL; Ogorzalek Loo RR; Loo JA Revealing Ligand Binding Sites and Quantifying Subunit Variants of Non-Covalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment. J. Am. Soc. Mass Spectrom 2014, 25, 2060–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhou M; Wysocki VH Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas Phase. Acc. Chem. Res 2014, 47, 1010–1018. [DOI] [PubMed] [Google Scholar]
- (25).Brodbelt JS; Morrison LJ; Santos I Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules. Chem. Rev 2020, 120, 3328–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).O’Brien JP; Li W; Zhang Y; Brodbelt JS Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2014, 136, 12920–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cammarata M; Brodbelt JS Structural Characterization of Apo-Myoglobin by Ultraviolet Photodissociation. Chem. Sci 2015, 6, 1324–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cammarata M; Thyer R; Rosenberg J; Ellington A; Brodbelt JS Structural Characterization of Dihydrofolate Reductase Complexes by Top-down Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2015, 137, 9128–9135. [DOI] [PubMed] [Google Scholar]
- (29).Morrison LJ; Brodbelt JS 193 nm Ultraviolet Photodissociation Mass Spectrometry of Tetrameric Protein Complexes Reflects Quaternary and Secondary Protein Topology. J. Am. Chem. Soc 2016, 138, 10849–10859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mehaffey MR; Cammarata MB; Brodbelt JS Tracking the Catalytic Cycle of Adenylate Kinase by Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem 2018, 90, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cammarata M; Shardon C; Rosenberg J; Singleton J; Fast W; Brodbelt JS Impact of G12C Mutation on Conformation of GTPase K-Ras Probed by Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2016, 138, 13187–13196. [DOI] [PubMed] [Google Scholar]
- (32).Cammarata M; Thyer R; Lombardo M; Anderson A; Wright D; Ellington A; Brodbelt JS Characterization of Trimethoprim Resistant E. Coli Dihydrofolate Reductase Mutants by Mass Spectrometry and P21L Inhibition by Propargyl-Linked Antifolates. Chem. Sci 2017, 8, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mehaffey MR; Schardon CL; Novelli ET; Cammarata MB; Webb LJ; Fast W; Brodbelt JS Investigation of GTP-Dependent Dimerization of G12X K-Ras Variants Using Ultraviolet Photodissociation Mass Spectrometry. Chem. Sci 2019, 10, 8025–8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sipe SN; Brodbelt JS Impact of Charge State on 193 nm Ultraviolet Photodissociation of Protein Complexes. Phys. Chem. Chem. Phys 2019, 21, 9265–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mehaffey MR; Sanders JD; Holden DD; Nilsson CL; Brodbelt JS Multi-Stage Ultraviolet Photodissociation Mass Spectrometry to Characterize Single Amino Acid Variants of Human Mitochondrial BCAT2. Anal. Chem 2018, 90, 9904–9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sanders JD; Mullen C; Watts E; Holden DD; Syka JEP; Schwartz JC; Brodbelt JS Enhanced Sequence Coverage of Large Proteins by Combining Ultraviolet Photodissociation with Proton Transfer Reactions. Anal. Chem 2020, 92, 1041–1049. [DOI] [PubMed] [Google Scholar]
- (37).Krishnamurthy VM; Kaufman GK; Urbach AR; Gitlin I; Gudiksen KL; Weibel DB; Whitesides GM Carbonic Anhydrase as a Model for Biophysical and Physical-Organic Studies of Proteins and Protein–Ligand Binding. Chem. Rev 2008, 108, 946–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Iyer R; Barrese AA; Parakh S; Parker CN; Tripp BC Inhibition Profiling of Human Carbonic Anhydrase II by High-Throughput Screening of Structurally Diverse, Biologically Active Compounds. J. Biomol. Screening 2006, 11 (7), 782–791. [DOI] [PubMed] [Google Scholar]
- (39).Tripp BC; Smith K; Ferry JG Carbonic Anhydrase: New Insights for an Ancient Enzyme. J. Biol. Chem 2001, 276, 48615–48618. [DOI] [PubMed] [Google Scholar]
- (40).Alterio V; Di Fiore A; D’Ambrosio K; Supuran CT; De Simone G Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev 2012, 112 (8), 4421–4468. [DOI] [PubMed] [Google Scholar]
- (41).Avvaru BS; Busby SA; Chalmers MJ; Griffin PR; Venkatakrishnan B; Agbandje-McKenna M; Silverman DN; McKenna R Apo-Human Carbonic Anhydrase II Revisited: Implications of the Loss of a Metal in Protein Structure, Stability, and Solvent Network. Biochemistry 2009, 48 (31), 7365–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ma H; Li A; Gao K Network of Conformational Transitions Revealed by Molecular Dynamics Simulations of the Carbonic Anhydrase II Apo-Enzyme. ACS Omega 2017, 2, 8414–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wambo TO; Chen LY; McHardy SF; Tsin AT Molecular Dynamics Study of Human Carbonic Anhydrase II in Complex with Zn2+ and Acetazolamide on the Basis of All-Atom Force Field Simulations. Biophys. Chem 2016, 214–215, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gaspari R; Rechlin C; Heine A; Bottegoni G; Rocchia W; Schwarz D; Bomke J; Gerber H-D; Klebe G; Cavalli A Kinetic and Structural Insights into the Mechanism of Binding of Sulfonamides to Human Carbonic Anhydrase by Computational and Experimental Studies. J. Med. Chem 2016, 59, 4245–4256. [DOI] [PubMed] [Google Scholar]
- (45).Chakravarty S; Kannan KK Refined Structures of Three Sulfonamide Drug Complexes of Human Carbonic Anhydrase I Enzyme. J. Mol. Biol 1994, 243, 298–309. [DOI] [PubMed] [Google Scholar]
- (46).Alterio V; Vitale RM; Monti SM; Pedone C; Scozzafava A; Cecchi A; De Simone G; Supuran CT Carbonic Anhydrase Inhibitors: X-ray and Molecular Modeling Study for the Interaction of a Fluorescent Antitumor Sulfonamide with Isozyme II and IX. J. Am. Chem. Soc 2006, 128, 8329–8335. [DOI] [PubMed] [Google Scholar]
- (47).Fisher SZ; Maupin CM; Budayova-Spano M; Govindasamy L; Tu C; Agbadje-McKenna M; Silverman DN; Voth GA; McKenna R Atomic Crystal and Molecular Dynamics Simulation Structures of Human Carbonic Anhydrase II: Insights into the Proton Transfer Mechanism. Biochemistry 2007, 46, 2930–2937. [DOI] [PubMed] [Google Scholar]
- (48).Di Fiore A; Pedone C; Antel J; Waldeck H; Witte A; Wurl M; Scozzafava A; Supuran CT; De Simone G Carbonic Anhydrase Inhbitors: The X-Ray Crystal Structure of Ethoxzolamide Complexes to Human Isoform II Reveals the Importance of Thr200 and Gln92 for Obtaining Tight-Binding Inhibitors. Bioorg. Med. Chem. Lett 2008, 18, 2669–2674. [DOI] [PubMed] [Google Scholar]
- (49).Temperini C; Cecchi A; Scozzafava A; Supuran CT Carbonic Anhydrase Inhibitors. Comparison of Chlorthalidone, Indapamide, Trichloromethiazide, and Furosemide X-Ray Crystal Structures in Adducts with Isozyme II, When Several Water Molecules Make the Difference. Bioorg. Med. Chem 2009, 17, 1214–1221. [DOI] [PubMed] [Google Scholar]
- (50).Avvaru BS; Kim CU; Sippel KH; Gruner SJ; Agbandje-McKenna M; Silverman DN; McKenna R A Short Strong Hydrogen Bond in the Active Site of Human Carbonic Anhydrase II. Biochemistry 2010, 49, 249–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cheng X; Chen R; Bruce JE; Schwartz BL; Anderson GA; Hofstadler SA; Gale DC; Smith RD; Gao J; Sigal GB; Mammen M; Whitesides GM Using Electrospray Ionization FTICR Mass Spectrometry to Study Competitive Binding of Inhibitors to Carbonic Anhydrase. J. Am. Chem. Soc 1995, 117, 8859–8860. [Google Scholar]
- (52).Gao J; Cheng X; Chen R; Sigal GB; Bruce JE; Schwartz BL; Hofstadler SA; Anderson GA; Smith RD; Whitesides GM Screening Derivatized Peptide Libraries for Tight Binding Inhibitors to Carbonic Anhydrase II by Electrospray Ionization-Mass Spectrometry. J. Med. Chem 1996, 39, 1949–1955. [DOI] [PubMed] [Google Scholar]
- (53).Wu Q; Gao J; Joseph-McCarthy D; Sigal GB; Bruce JE; Whitesides GM; Smith RD Carbonic Anhydrase-Inhibitor Binding: From Solution to the Gas Phase. J. Am. Chem. Soc 1997, 119, 1157–1158. [Google Scholar]
- (54).Gao J; Wu Q; Carbeck J; Lei QP; Smith RD; Whitesides GM Probing the Energetics of Dissociation of Carbonic Anhydrase-Ligand Complexes in the Gas Phase. Biophys. J 1999, 76, 3253–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fellers RT; Greer JB; Early BP; Yu X; LeDuc RD; Kelleher NL; Thomas PM ProSight Lite: Graphical Software to Analyze Top-down Mass Spectrometry Data. Proteomics 2015, 15, 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Rosenberg J; Parker WR; Cammarata MB; Brodbelt JS UV-POSIT: Web-Based Tools for Rapid and Facile Structural Interpretation of Ultraviolet Photodissociation (UVPD) Mass Spectra. J. Am. Soc. Mass Spectrom 2018, 29 (6), 1323–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sterling HJ; Cassou CA; Trnka MJ; Burlingame AL; Krantz BA; Williams ER The Role of Conformational Flexibility on Protein Supercharging in Native Electrospray Ionization. Phys. Chem. Chem. Phys 2011, 13, 18288–18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sterling HJ; Kintzer AF; Feld GK; Cassou CA; Krantz BA; Williams ER Supercharging Protein Complexes from Aqueous Solution Disrupts their Native Conformations. J. Am. Soc. Mass Spectrom 2012, 23, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sterling HJ; Williams ER Origin of Supercharging in Electrospray Ionization of Noncovalent Complexes from Aqueous Solution. J. Am. Soc. Mass Spectrom 2009, 20, 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Sterling HJ; Prell JS; Cassou CA; Williams ER Protein Conformation and Supercharging with DMSO from Aqueous Solution. J. Am. Soc. Mass Spectrom 2011, 22, 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Iavarone AT; Jurchen JC; Williams ER Supercharged Protein and Peptide Ions Formed by Electrospray Ionization. Anal. Chem 2001, 73, 1455–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Iavarone AT; Williams ER Supercharging in Electrospray Ionization: Effects on Signal and Charge. Int. J. Mass Spectrom 2002, 219, 63–72. [Google Scholar]
- (63).Iavarone AT; Williams ER Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J. Am. Chem. Soc 2003, 125, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Going CC; Williams ER Supercharging with m-Nitrobenzyl Alcohol and Propylene Carbonate: Forming Highly Charged Ions with Extended, Near-Linear Conformations. Anal. Chem 2015, 87 (7), 3973–3980. [DOI] [PubMed] [Google Scholar]
- (65).Sterling HJ; Daly MP; Feld GK; Thoren KL; Kintzer AF; Krantz BA; Williams ER Effects of Supercharging Reagents on Noncovalent Complex Structure in Electrospray Ionization from Aqueous Solutions. J. Am. Soc. Mass Spectrom 2010, 21, 1762–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Going CC; Xia Z; Williams ER New Supercharging Reagents Produce Highly Charged Protein Ions in Native Mass Spectrometry. Analyst 2015, 140, 7184–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Yin S; Loo JA Top-Down Mass Spectrometry of Supercharged Native Protein-Ligand Complexes. Int. J. Mass Spectrom 2011, 300 (2–3), 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Lomeli SH; Peng IX; Yin S; Loo RRO; Loo JA New Reagents for Increasing ESI Multiple Charging of Proteins and Protein Complexes. J. Am. Soc. Mass Spectrom 2010, 21, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Lomeli SH; Yin S; Loo RRO; Loo JA Increasing Charge While Preserving Noncovalent Protein Complexes for ESI-MS. J. Am. Soc. Mass Spectrom 2009, 20, 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Ogorzalek Loo RR; Lakshmanan R; Loo JA What Protein Charging (and Supercharging) Reveal about the Mechanism of Electrospray Ionization. J. Am. Soc. Mass Spectrom 2014, 25, 1675–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Julian R The Mechanism behind Top-Down UVPD Experiments: Making Sense of Apparent Contradictions. J. Am. Soc. Mass Spectrom 2017, 28 (9), 1823–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Morrison LJ; Brodbelt JS Charge Site Assignment in Native Proteins by Ultraviolet Photodissociation (UVPD) Mass Spectrometry. Analyst 2016, 141, 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Loo RRO; Loo JA Salt Bridge Rearrangement (SaBRe) Explains the Dissociation Behavior of Noncovalent Complexes. J. Am. Soc. Mass Spectrom 2016, 27 (6), 975–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Ma H; Li A; Gao K Network of Conformational Transitions Revealed by Molecular Dynamics Simulations of the Carbonic Anhydrase II Apo-Enzyme. ACS Omega 2017, 2, 8414–8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Crittenden CM; Novelli ET; Xu GN; Giles DH; Fies WA; Mehaffey MR; Dalby KN; Webb LJ; Brodbelt JS Structural Evaluation of Protein/Metal Complexes via Native Electrospray Ultraviolet Photodissociation Mass Spectrometry. J. Am. Soc. Mass Spectrom 2020, 31, 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Cheng T; Zhao Y; Li X; Lin F; Xu Y; Zhang X; Li Y; Wang R; Lai L Computation of Octanol-Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model 2007, 47, 2140–2148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.