

SUMMARY
The core cohesin subunit STAG2 is recurrently mutated in Ewing sarcoma but its biological role is less clear. Herein, we demonstrate that cohesin complexes containing STAG2 occupy enhancer and polycomb repressive complex (PRC2) marked regulatory regions. Genetic suppression of STAG2 leads to a compensatory increase in cohesin-STAG1 complexes, but not in enhancer rich regions, and results in reprogramming of cis-chromatin interactions. Strikingly, in STAG2 knockout cells, the oncogenic genetic program driven by the fusion transcription factor EWS/FLI1 was highly perturbed, in part due to altered enhancer-promoter contacts. Moreover, loss of STAG2 also disrupted PRC2-mediated regulation of gene expression. Combined, these transcriptional changes converged to modulate EWS/FLI1, migratory and neurodevelopmental programs. Finally, consistent with clinical observations, functional studies revealed that loss of STAG2 enhances the metastatic potential of Ewing sarcoma xenografts. Our findings demonstrate that STAG2 mutations can alter chromatin architecture and transcriptional programs to promote an aggressive cancer phenotype.
Keywords: Cohesin, Ewing sarcoma, fusion oncoprotein, metastasis, STAG2, STAG1, EWS/FLI1, PRC2, POU3F2
Graphical Abstract
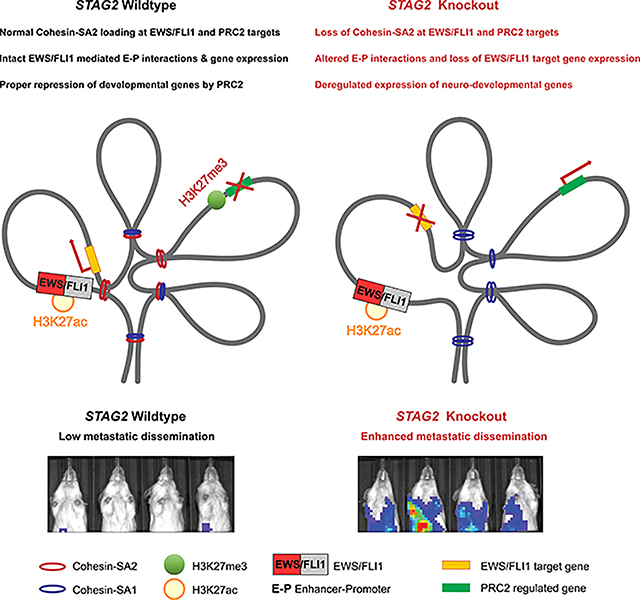
Adane et al. demonstrate that deletion of STAG2 changes the distribution of cohesin complexes and leads to reprograming of cis-chromatin interactions in Ewing sarcoma. STAG2 loss attenuated EWS/FLI1 driven oncogenic programs and disrupted PRC2 regulated developmental processes to enhance the metastatic potential of Ewing sarcoma cells.
INTRODUCTION
Massively parallel sequencing efforts have revealed cancer-associated mutations in the cohesin complex, a multiprotein complex important in sister chromatid cohesion and gene regulation (Romero-Perez et al., 2019). STAG2 is a member of the cohesin complex, composed of SMC1A, SMC3, and RAD21, forming a ringed structure that can surround two strands of DNA. The fourth member of the complex is one of three members of the STAG protein family: STAG1, STAG2, or STAG3 (Haering et al., 2008; Nasmyth, 2002). Studies have demonstrated that cohesin complexes containing either STAG1 (cohesin-SA1) or STAG2 (cohesin-SA2) are expressed concurrently in mitotic cells and bind to overlapping, as well as unique, chromatin locations (Cuadrado et al., 2019; Kojic et al., 2018; Losada et al., 2000). The cohesin complex plays an important role in regulating sister chromatid alignment during cell division. Therefore, when loss of function mutations in STAG2 were first identified in cancer, it was hypothesized that these events would cause cohesin dysfunction and improper chromosomal segregation resulting in aneuploidy (Solomon et al., 2011). However, subsequent studies demonstrated that STAG2 mutations are not significantly associated with aneuploidy in hematopoietic malignancies, Ewing sarcoma, or bladder carcinoma (Balbas-Martinez et al., 2013; Crompton et al., 2014; Kon et al., 2013; Solomon et al., 2013; Tirode et al., 2014). The cohesin complex also plays a critical role in chromatin regulation of gene expression. Cohesin maintains chromatin accessibility at transcription factor binding sites during cell division and promotes DNA-DNA contacts that form the basis of enhancer-promoter interactions and define the boundaries of topologically associated domains at CTCF binding sites (Dowen et al., 2014; Kagey et al., 2010; Wendt et al., 2008). Therefore, we hypothesized that loss of STAG2 alters cohesin function resulting in changes in gene regulation in Ewing sarcoma.
Studies have demonstrated that patients with STAG2-mutated Ewing sarcoma have a higher rate of metastatic disease and worse outcomes (Crompton et al., 2014; Tirode et al., 2014). Therefore, we also hypothesized that altered gene expression associated with STAG2 loss would be associated with an increase in metastatic potential. To test these hypotheses, we chose the pediatric solid tumor Ewing sarcoma as our model system, a malignancy defined by a simple genome and an oncogenic rearrangement between the EWSR1 gene and an ETS-family transcription factor encoding gene, most commonly FLI1, resulting in an EWS/FLI1 fusion. STAG2 mutations are present in 15–20% of tumors and lead to loss of expression of the gene (Brohl et al., 2014; Crompton et al., 2014; Tirode et al., 2014). In this study, we genetically ablated STAG2 in Ewing sarcoma cell lines expressing wild-type STAG2 and examined the phenotypic, transcriptional, and epigenetic effects of STAG2 loss.
Results
Depletion of STAG2 alters the composition of the cohesin complex and is synthetic lethal with STAG1 loss
In order to evaluate the oncogenic role of loss-of-function mutations in STAG2, we initially explored the Cancer Dependency Map (DepMap) database (Tsherniak et al., 2017). Our analysis revealed that, distinct from other core subunits of the cohesin complex, CRISPR/Cas9-based disruption of STAG2 does not affect the proliferation and/or viability of most Ewing sarcoma cell lines (Figures 1A). A subset of cell lines in the database bear STAG2 mutations. Thus, we focused on two cell lines, TC71 and A673, which express wildtype STAG2. These cell lines, however, show relatively contrasting phenotypes with respect to STAG2 deletion, with TC71 showing a modest growth defect and A673 showing a modest growth advantage. STAG2 is an X-linked gene, and our previous study has shown that recurrent mutations of STAG2 in Ewing sarcoma invariably result in lack of expression of the protein product (Crompton et al., 2014). Thus, we generated isogenic clonal STAG2 knockout (KO) cells, along with controls, and subjected them to long-term in vitro proliferation assays (Figure S1A). In liquid culture, STAG2 deleted A673 cells showed a mild, yet statistically significant, growth advantage, consistent with DepMap data (Figure 1B). In contrast, TC71 cells displayed similar rate of growth relative to nontargeting and parental controls in liquid culture and a mild growth defect in semi-solid methylcellulose media (Figures 1B and S1B). Moreover, given the important role of the cohesin complex in maintaining proper cohesion and segregation of sister chromatids, we also evaluated cell cycle progression. Our results showed relatively similar distribution of cells in all phases of the cell cycle in control and STAG2 deleted A673 cells (Figure 1C).
Figure 1. Loss of STAG2 does not consistently alter cell growth but changes the composition of the cohesin complex and renders cells sensitive to STAG1 deletion.

(A) Hockey plots depicting the distribution of STAG2, SMC1A, SMC3 and RAD21 gene effect (CERES) scores across the 789 cell lines in CRISPR (Avana) Depmap v20Q3 data. (B) Line graphs showing mean ± sd of cumulative doublings for parental or clonally selected non-targeting (NT) or STAG2 KO A673 and TC71 cells. Pairwise comparative analysis for exponential growth fitted curves (extra-sum-of-squares F test, ** P < 0.01, ns=not significant). (C) EdU incorporation-based cell cycle profiling and a representative flow cytometry plots (left) are shown. Two independent experiments as mean ± sd barplots (right). 2-way ANOVA ns=not significant. (D-E) Sub-cellular fractionation (D) or Immunoprecipitation with SMC1A, SMC3 or IgG (E) followed by western blot was performed for the indicated proteins. (F) Genome-scale CRISPR/Cas9 screen in isogenic NT and STAG2 KO A673 clonal cells. Shown is the volcano plot for gene effect size vs. −log10(P-value) for the genome-wide differential analysis of isogenic NT vs. STAG2 KO processed reads; dots represent genes (limma eBayes for MAGeCK gene effect scores, effect size ≤ −0.3, adjusted P ≤ 0.10). See also Figure S1.
Because STAG2 is one of the core subunits of the cohesin complex, we next employed biochemical assays and investigated the impact of STAG2 loss on the complex. Purified cytoplasmic, soluble-nuclear and chromatin bound fractions from control and STAG2 KO cells were immunoblotted for several cohesin complex subunits. Strikingly, we observed increases in STAG1 levels with STAG2 KO in the chromatin bound fraction in A673 and soluble nuclear fraction of A673 and TC71 cells (Figures 1D and S1C). In addition, RAD21 levels were reduced in the soluble nuclear fraction. Next, to test whether changes in STAG2 abundance affect the assembly of the complex, we immunoprecipitated cell extracts with antibodies against SMC1A or SMC3 and immunoblotted for all the core subunits of the complex. Consistent with the cell fractionation experiments, we observed increased levels of STAG1 and decreased levels of RAD21 in association with the SMC proteins in STAG2 KO cells (Figures 1E and S1D).
The absence of consistent cell proliferation alterations, and the increased levels of cohesin-SA1, suggest that at least the essential, cell cycle associated function of STAG2 is being compensated by its paralog STAG1. Consistent with this notion and in good agreement with previous findings by other investigators, (Benedetti et al., 2017; Liu et al., 2018; van der Lelij et al., 2017) a genome-scale CRISPR/Cas9 screen identified STAG1 as the top synthetic lethal dependency in isogenic STAG2 KO A673 cells compared to controls (Figure 1F). This finding was also validated in independent experiments comparing the effect of STAG1 deletion in isogenic settings, as well as in WT and STAG2 mutant parental Ewing sarcoma cell lines (Figures S1E-H). Taken together, these results strongly suggest that STAG2 mutations in Ewing sarcoma do not predominantly and universally perturb the canonical cell cycle associated function of the cohesin complex and that the likely contribution of these mutations to Ewing sarcoma pathogenesis is through other mechanisms.
Cohesin-SA1 and cohesin-SA2 bind to overlapping and unique regions in Ewing sarcoma cells
In addition to the canonical role of maintaining sister chromatid cohesion during cell division, the cohesin complex also mediates dynamic, high frequency intrachromosomal interactions including those conjoining enhancers to promoters (Hansen et al., 2018). Therefore, we hypothesized that loss of STAG2 disrupts cis-chromatin interactions in Ewing sarcoma cells. First, to determine the genome-wide distribution and relative abundance of the cohesin complexes in the presence and absence of STAG2, we performed calibrated Chromatin Immunoprecipitation Sequencing (ChIP-Seq) analysis using antibodies against STAG2, STAG1 and SMC1A in isogenic A673 cells. Consistent with our biochemical analyses, genome-wide binding of STAG1 increased significantly in STAG2 KO cells (Figures 2A and 2B). There was a trend toward increased levels of SMC1 although this did not reach statistical significance.
Figure 2. STAG2 occupies PRC2 and enhancer marked regions and its deletion is incompletely compensated by STAG1.
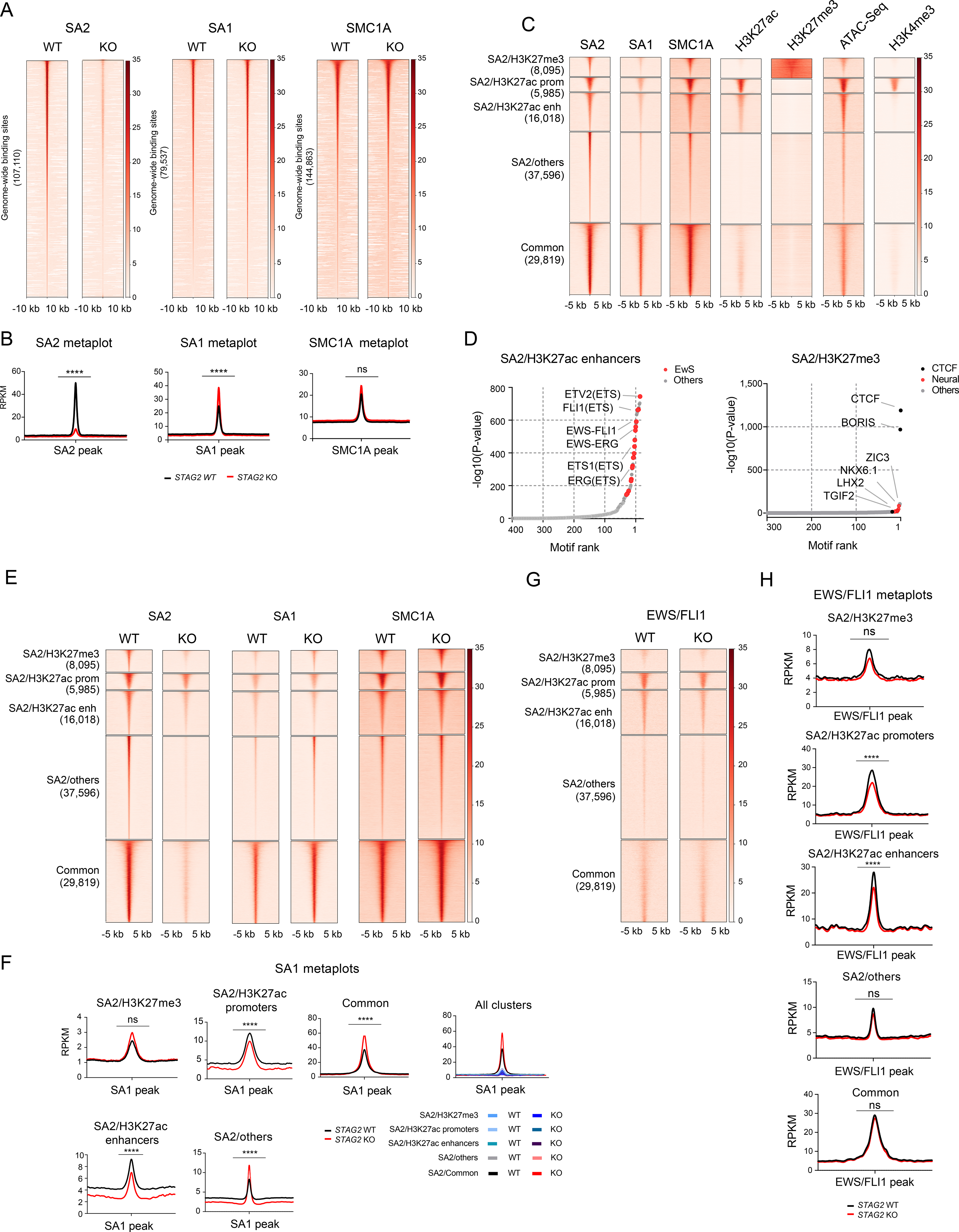
(A) Genome-wide heatmaps of SA2, SA1 and SMC1A ChIP-Seq peak centered signal in A673 cells expressing WT or KO STAG2. Regions are ranked based on WT signal. (B) Read density metaplots showing average RPKM normalized signal for SA2, SA1 and SMC1A in WT (black) and STAG2 KO (red) A673 cells. Differential read density in KO vs. WT conditions (unpaired t-test with Welch’s correction, **** P < 0.0001, ns = not significant). (C) Clustered heatmaps of ChIP-Seq peak centered signal for SA2, SA1, SMC1A, H3K27ac, H3K27me3, chromatin accessibility (ATAC-Seq) and H3K4me3. Cluster regions are ranked by SA2 signal. (D) Hockey plots depicting motifs enriched in SA2/H3K27ac enhancers and SA2/H3K27me3 regions defined in Figure 2C. (E) Clustered heatmaps depicting SA2, SA1 and SMC1A signal in the cohesin regions defined in Figure 2C in control and STAG2 KO A673 cells. (F) Metaplots showing average SA1 signal in the cohesin regions defined in Figure 2C. Differential read density in KO vs. WT conditions (unpaired t-test with Welch’s correction, **** P < 0.0001, ** P < 0.01). (G) Clustered heatmap depicting EWS/FLI1 signal in cohesin regions defined in Figure 2C in control and STAG2 KO A673 cells. (H) Metaplot showing average EWS/FLI1 signal in cohesin regions defined in Figure 2C. Differential read density in KO vs. WT (unpaired t-test with Welch’s correction, **** P < 0.0001, ** P < 0.01). See also Figure S2.
Previous studies have shown that the two cohesin complexes bind to common, as well as non-overlapping regions (Cuadrado et al., 2019; Kojic et al., 2018; Ochi et al., 2020). While cohesin-SA1 often colocalizes with CTCF and cohesin-SA2 at TAD boundaries, cohesin-SA2 occupies regulatory regions replete with cell-type specific enhancers. Therefore, we set out to determine where cohesin-SA2 resides with respect to putative regulatory genomic domains defined by chromatin accessibility and the histone modification marks H3K27ac, H3K27me3 and H3K4me3 associated with enhancers, polycomb repressive complex and active promoters respectively. Our results showed that STAG2 bound regions cluster into five discrete categories. Four of the regions contained high levels of cohesin-SA2 along with either H3K27me3 (SA2/H3K27me), H3K27ac (SA2/H3K27ac-Promoters, SA2/H3K27ac_Enhancers), or without either mark (SA2/Others), whereas the last group has high levels of cohesin-SA1 and cohesin-SA2 (common) (Figure 2C). Motif enrichment analysis revealed that regions bound by cohesin-SA2 and decorated by the H3K27ac mark, both at promoters and enhancers, are enriched with canonical ETS motifs (Figure 2D and S2A). In light of the role that EWS/FLI1 plays in Ewing sarcoma, this data suggests that cohesin-SA2 may partner with the fusion oncogene to hijack and reprogram the gene regulatory landscape. Importantly, while the cohesin-SA2/Others, common, and SA2/H3K27me3 regions were all enriched for the canonical motifs of CTCF and its paralog BORIS (CTCF-like), several neurogenic transcription factors were also enriched in the SA2/H3K27me3 region, suggesting that cohesin-SA2 may contribute to the regulation of neurodevelopmental programs in Ewing sarcoma (Figure 2D and S2A).
Next, we determined how binding of the cohesin subunits changes across these pre-defined regions as a consequence of STAG2 depletion. As expected, the binding signal for STAG2 decreased significantly across all regions (Figure S2B). Strikingly, STAG1 and SMC1A binding increased significantly, or with a trend toward significance, in all regions with the exception of SA2/H3K27ac region, suggesting that cohesin-SA1 does not sufficiently replace cohesin-SA2 around a subset of enhancers and promoters (Figures 2E,F and S2C). Given, the significant enrichment of several ETS motifs in the cohesin-SA2/H3K27ac regions, we also tested whether deletion of STAG2 impacts the distribution of EWS/FLI1 on chromatin. Consistent with the results of the motif enrichment analysis, EWS/FLI1 binding was the strongest at cohesin-SA2 bound enhancer/promoter marked regions and was mostly absent at H3K27me3 marked regions (Figure 2G). Deletion of STAG2 modestly decreased the binding of EWS/FLI1 genome-wide and more prominently at H3K27ac marked regions, without affecting the expression levels of the fusion oncoprotein itself (Figures 2G-H and S2D-F).
Deletion of STAG2 disrupts long-range cis enhancer-promoter interactions, including those anchored by EWS/FLI1
To evaluate how loss of STAG2 impacts cohesin mediated cis-chromatin interactions across the genome, we next performed proximity ligation-based global chromatin conformation profiling by SMC1A HiChIP in control and STAG2 KO A673 cells. Our analysis identified a total of 56,219 high-confidence, cohesin-anchored, chromatin interactions out of which 4500 display significantly altered contact frequency in STAG2 KO cells relative to WT controls (Figure 3A and Table S1). Importantly, more than a third of these altered loops are anchored by a putative enhancer on one end and a gene promoter on the other (Figure 3B and Table S1). The data also revealed that chromatin regions that are furthest from each other are more likely to decrease in contact frequency whereas loops established by closely located interacting pairs are enriched for increase in contact frequency as a result of STAG2 loss (Figure 3C). Notably, roughly 23% of the differential loops involve anchor points that hosted long range contacts that both increase and decrease in frequency of interaction suggesting a role for loop re-organization in a subset of cases.
Figure 3. Loss of STAG2 alters the frequency of cis-chromatin contacts.
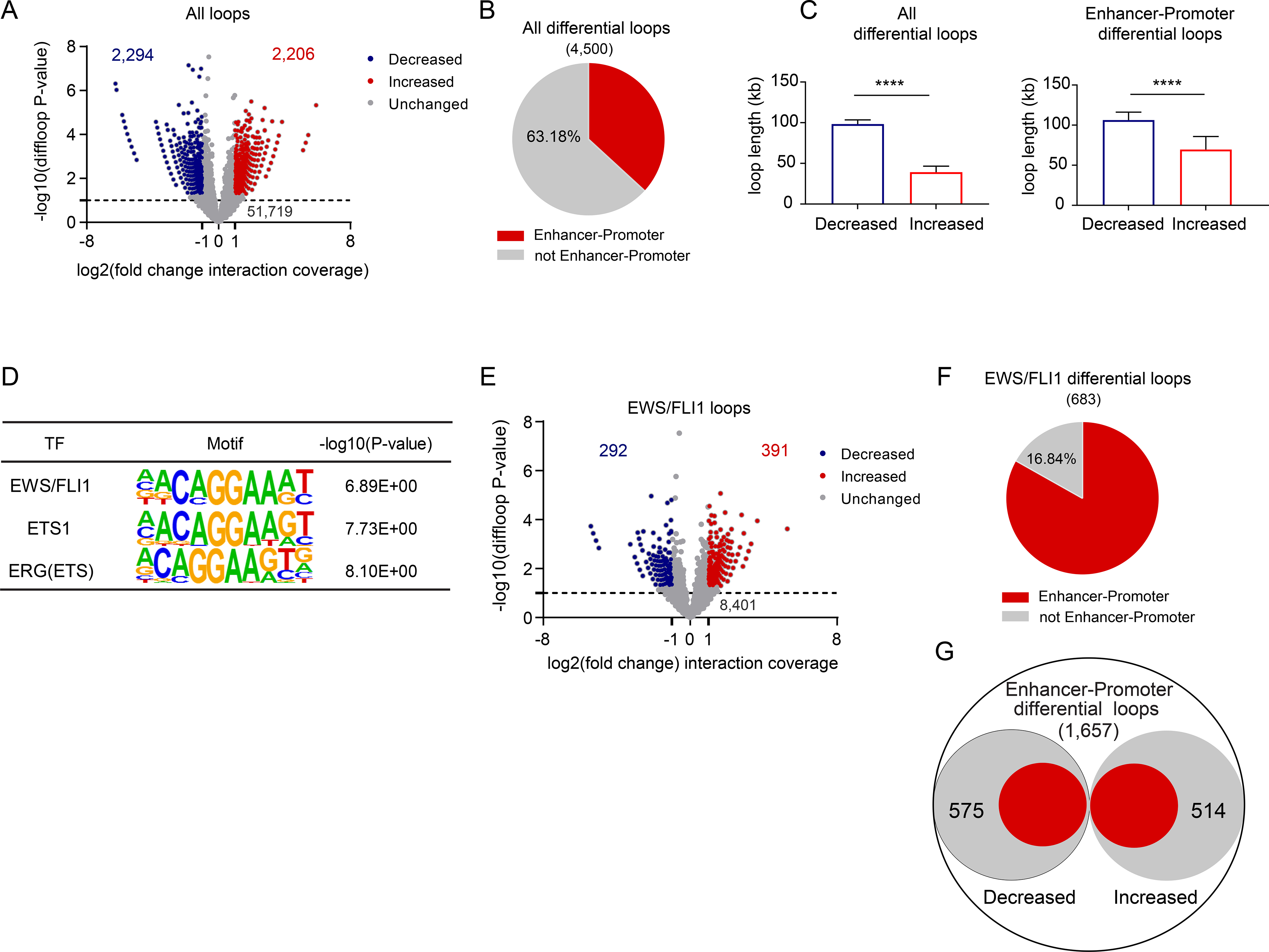
(A) Volcano plot of changes in chromatin loop strength comparing STAG2 WT to KO A673 cells based on SMC1A Hi-ChIP (edgeR over-dispersed Poisson regression, |(FC)|≥2, P≤ 0.05). (B) Pie chart depicting the fraction of differential loops involving enhancer-promoter interactions (two-tailed Fisher exact test, **** P < 0.0001). (C) Median + 95% confidence interval plots for lengths of differential loops (unpaired t-test with Welch correction, **** P < 0.0001). (D) Three of the top 10 enriched motifs for the enhancer regions involved in differential enhancer-promoter loops. (E) Volcano plot depicting the differential status of the 9,084 EWS/FLI1 anchored differential loops (edgeR over-dispersed Poisson regression, |fold change| ≥ 2, P ≤ 0.05). (F) Pie chart depicting the fraction of EWS/FLI1 anchored differential loops involving enhancer-promoter interactions. Two-tailed Fisher exact test, **** P < 0.0001. (G) Diagram depicting the fraction of EWS/FLI1 anchored enhancer-promoter interactions as a subset of all differential enhancer-promoter loops. Two-tailed Fisher exact test **** P < 0.0001. See also Figure S3, Tables S1 and S2.
Given the significant number of altered loops connecting enhancers to promoters, we next asked which sequence-specific transcription factors are likely to preferentially engage these enhancers and mediate long-range chromatin contacts. Motif enrichment analysis identified consensus binding sequences for the dominant oncogenic fusion transcription factor EWS/FLI1, as well as the ETS family members ETS1 and ERG, among the top 10 enriched motifs (Figure 3D and Table S2). Thus, we hypothesized that deletion of STAG2 may also disrupt EWS/FLI1 anchored long-range chromatin contacts.
To test this hypothesis, we performed ChIP-Seq and mapped the genome-wide binding pattern of EWS/FLI1 and overlapped our peaks with two additional studies performed by other investigators in A673 cells (Riggi et al., 2014; Tomazou et al., 2015). We integrated these high-confidence binding sites with H3K27ac ChIP-Seq, chromatin accessibility and GGAA repeat calling from the reference genome to generate a comprehensive view of EWS/FLI1 binding. As expected, EWS/FLI1 bound strongly at promoters, canonical enhancers and enhancers associated with long GGAA microsatellite repeats (Figure S3A). We then asked whether the cohesin complex also binds to these regions to potentially mediate long-range interactions and whether cohesin loading, as well as EWS/FLI1 binding, change in the context of STAG2 loss. Our data revealed that SMC1A and the H3K27ac mark strongly colocalize with EWS/FLI1 at enhancers and promoters (Figure S3B). Importantly, consistent with our previous observations (Figure 2F and 2H), in STAG2 KO cells, EWS/FLI1 binding decreased across multiple regions, with the exception of GGAA repeat marked enhancers, while STAG1 failed to compensate for the loss of STAG2 across all of the EWS/FLI1 bound regions assessed (Figure S3C). Notably, cohesin loading at EWS/FLI1 bound enhancers, particularly at GGAA repeats, is highly selective for Ewing sarcoma cells as we found very low signal for SMC1A or SMC3 binding at these sites in multiple other cell types (Figure S3D). Combined, these results argue that cohesin and EWS/FLI1 cooperatively establish and drive part of the oncogenic enhancer-promoter network in Ewing sarcoma and that the loss of STAG2 may decommission the components necessary for maintaining these interactions.
Next, we interrogated our HiChIP data to identify long-range chromatin contacts that are anchored at least at one edge by EWS/FLI1 and cohesin. Our analysis identified a total of 9084 high confidence loops that satisfy this criterion (Figure 3E and Table S1). In STAG2 depleted cells, there were 683 EWS/FLI1 anchored loops that significantly changed in contact frequency. Strikingly, 83.16% of these loops connected enhancers to promoters with EWS/FLI1 occupying the edge of the loop located at the distal enhancer site (Figure 3F and Table S1). Moreover, as a group, EWS/FLI1 anchored loops accounted for a large subset of all significantly altered enhancer-promoter contacts in STAG2 KO cells (Figure 3G). Similar to all other altered loops, long-range chromatin contacts mediated by EWS/FLI1 also tend to decrease in contact frequency the further apart interacting regions are (Figure S3C). Thus, in aggregate, these findings strongly suggest that loss of STAG2 significantly modifies cis-chromatin interactions and may partially reprogram the oncogenic program driven by EWS/FLI1.
Deletion of STAG2 rewires the transcriptional program of Ewing sarcoma cells
Given the significant changes we observed in the frequency of enhancer-promoter interactions and the important functional role they play in regulating gene expression, we predicted that loss of STAG2 will have a measurable effect on transcriptional output. To test the idea, we performed RNA-seq analysis in two independent Ewing sarcoma cell lines where we deleted STAG2. We found that loss of STAG2 causes a similar pattern of transcriptional changes in both cell lines (Figure 4A and Table S3) with a high degree of overlap between the genes whose expression changed significantly (|log2(FC)| ≥ 1.5, adjusted P ≤ 0.1, Figure S4A). Moreover, gene set enrichment analysis (GSEA) revealed highly concordant transcriptional changes in TC71 and A673 cells (Figures 4B and S4B). Finally, to determine whether the transcriptional changes seen in our cell line models are generalizable and reflective of a conserved biology that emerges as a consequence of STAG2 loss, we performed GSEA using three independent gene expression data sets of primary Ewing sarcoma patient tumors (Crompton et al., 2014; Postel-Vinay et al., 2012; Savola et al., 2011). Our analysis revealed that the differentially expressed genes in each of the two cell lines were significantly enriched in the transcriptomes of patient tumors that carry mutant STAG2 or express low levels (Figures 4C-E and S4C-E). Collectively, these findings indicate that loss of STAG2 produces highly consistent and stable transcriptional changes that may undergo selection to confer a competitive advantage.
Figure 4. Loss of STAG2 alters the EWS/FLI1 driven oncogenic transcriptional program.
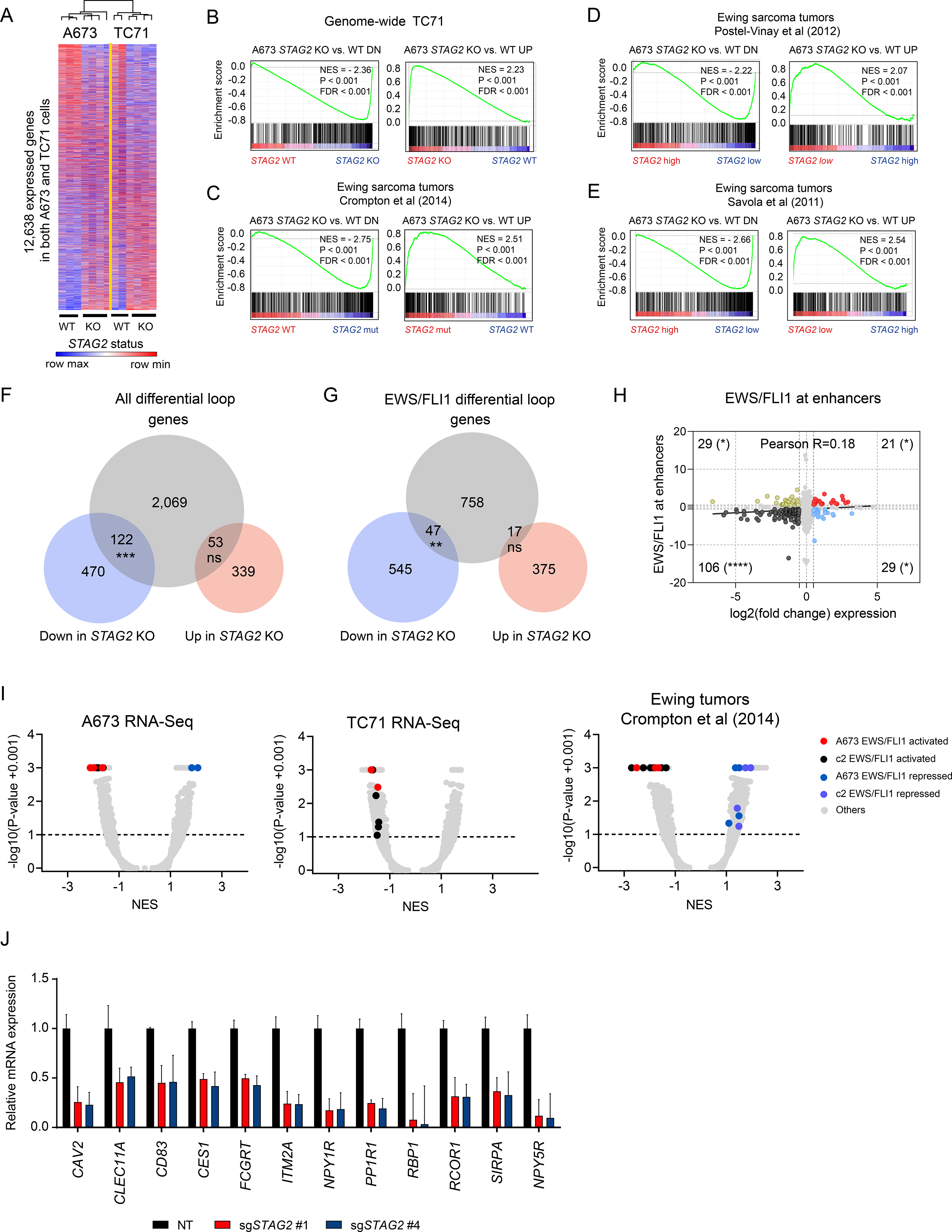
(A) Heatmap and average linkage dendrogram of genome-wide gene expression, log2(normalized counts), in STAG2 KO vs. WT A673 and TC71 cells. (B-E) GSEA plots demonstrating the enrichment of the STAG2 KO vs. WT gene signature for A673 cells in the genome-wide expression changes induced by STAG2 KO in TC71 cells (B) or in three distinct Ewing sarcoma primary patient tumor datasets (C-E). Normalized enrichment score, P-value and FDR are indicated in each plot. (F) Venn diagram depicting the overlap between differentially expressed genes with genes harboring a differential loop within 5kb of their TSS. (Two-tailed Fisher exact test, *** P < 0.001, ns = not significant). (G) Similar analysis as in (F) is shown for EWS/FLI1 anchored differential loops (Two-tailed Fisher exact test, ** P < 0.01, ns = not significant). (H) Scatter plot depicting the overlap between gene expression change and EWS/FLI1 binding at the nearest enhancer assessed by ChIP-seq. Two-tailed Fisher exact test (**** P < 0.0001, * P < 0.05). (I) Volcano plots for GSEA enrichment scores for the genome-wide expression changes induced in A673 and TC71 and by STAG2 mutant vs. STAG2 WT tumor samples from (Crompton et al., 2014) vs. the union of a collection of 12 EWS/FLI1 public gene signatures in A673 cells and MSigDB v7.1 c2 pathways (5,529 gene sets). (J) RT-QPCR based validation of a subset of EWS/FLI1 target genes repressed in STAG2 KO relative to control A673 cells. Data shown as mean ± SD. See also Figure S4 and Table S3.
The observation that enhancer-promoter interactions and transcriptional programs are concurrently altered in STAG2 KO cells prompted us to investigate whether the two are directly linked. To this end, we selected all the differential loops that are anchored within 5 kb of a transcriptional start site and annotated the nearest expressed gene. We identified 2000 loops and the associated genes (differential loop genes) that satisfied the criteria. Using this list, we interrogated our transcriptional data generated in A673 cells focusing only on genes that undergo significant change in expression (|log2(FC)| ≥ 1.5, adjusted P ≤ 0.1) in the context of STAG2 loss. This integrative analysis of HiChIP and RNA-seq data identified a significant association between loss of gene expression and altered cohesin mediated cis-chromatin interaction (Fig 4F and Table S3). Indeed, nearly 20% of genes repressed in STAG2 depleted cells also display differential chromatin loops proximal to their transcriptional start sites. Notably, genes upregulated upon STAG2 deletion did not have a statistically significant association with the presence of an altered loop near their promoters. Given the substantial number of altered chromatin contacts we identified that are anchored by EWS/FLI1, we expanded our analysis for the subset of genes that are associated with these loops. We identified a total of 822 genes that are associated with differential EWS/FLI1 anchored loops. Importantly, the results similarly showed that there is a significant association with the presence of an altered loop near the transcriptional start site (TSS) of downregulated but not upregulated genes in STAG2 KO cells (Figures 4G, S4F and Table S3). Taken together, these results show that the cohesin complex anchors critical chromatin loops near gene promoters and that the loss of STAG2 disrupts these structures to produce consistent and stable changes to genetic programs that may drive tumorigenesis.
STAG2 loss attenuates the EWS/FLI dependent oncogenic program
EWS/FLI1 is a dominant oncogenic fusion transcription factor and, as such, drives a defined genetic program in Ewing sarcoma cells. Given the alterations we observed in its binding at enhancer marked regions, chromatin contacts involving enhancers and promoters anchored by EWS/FLI1, and their significant association with at least downregulated genes, we sought to determine the extent to which STAG2 loss specifically perturbs the EWS/FLI1 dependent oncogenic program. To directly test this idea, we initially evaluated the relationship between the change in EWS/FLI1 binding as assessed by ChIP-Seq and the level of change in expression of the nearest gene. Our analysis revealed that on average more genes lose EWS/FLI1 binding and get repressed in STAG2 KO cells relative to controls (Figure S4G). Importantly, EWS/FLI1 binding loss at enhancers, but not at promoters, significantly correlated with downregulation of gene expression (Figure 4H and S4H).
Next, we generated a robust list of EWS/FLI1 dependent gene expression signatures by utilizing several previously published studies (Table S4). Using this refined compendium of EWS/FLI1 signatures, we performed GSEA on the transcriptome of STAG2 KO cells. Intriguingly, in both A673 and TC71 cells, loss of STAG2 markedly repressed the EWS/FLI1 dependent oncogenic program (Figure 4I & S4I). Similar analysis performed in three independent Ewing tumor data sets also showed attenuation of the oncogenic program as a consequence of STAG2 mutation (Figures 4I and S4I, Table S4). Further, we took advantage of a recently generated, single cell RNA-seq based, transcriptomic signature of EWS-FLI1 dubbed IC-EwS (Aynaud et al., 2020) and performed GSEA on our STAG2 KO cell lines and STAG2 mutant primary tumors. Consistent with the results generated using our refined compendium of signatures, the IC-EwS signature was significantly repressed in STAG2 KO and STAG2 mutant cells, further confirming that loss of STAG2 dominantly limits part of the oncogenic program (Figure S4J). Finally, we performed RT-QPCR and validated the suppression of a number of EWS/FLI1 target genes in STAG2 KO cells relative to controls (Figure 4J). Combined, the results of these studies strongly argue that STAG2 loss modulates the EWS/FLI1 oncogenic program in part through disruption of long-range chromatin contacts anchored by EWS/FLI1.
Polycomb repressive complex-mediated gene regulation is disrupted in STAG2 KO cells
In order to gain a broader insight into the transcriptional changes induced by loss of STAG2, we next performed a comprehensive enrichment analysis using gene sets available through the Molecular Signatures Database (MSigDB) collection. Our results further confirmed that Ewing sarcoma and EWS/FLI1 specific programs are altered in STAG2 KO cells as we found 4 Ewing sarcoma-related signatures out of the top 50 gene sets (Figure 5A and Table S5). Strikingly, we also found several PRC2-associated gene signatures as significantly enriched in our transcriptional data, suggesting that deletion of STAG2 likely disrupts PRC2-mediated regulation of gene expression (Figure 5A and Table S5). To directly assess the involvement of PRC2-dependent epigenetic dysregulation, we first performed calibrated ChIP-Seq for the PRC2 modification mark H3K27me3 in control and STAG2 KO cells. We found that the level of H3K27me3 mark is decreased in STAG2 KO cells genome-wide (Figures 5B and 5C). Importantly, the PRC2 mark is enriched at a subset of genomic regions preferentially occupied by cohesin-SA2, and these regions also lose H3K27me3 significantly in STAG2 KO cells (Figure S5A & B).
Figure 5. Loss of STAG2 perturbs PRC2-mediated regulation of gene expression.
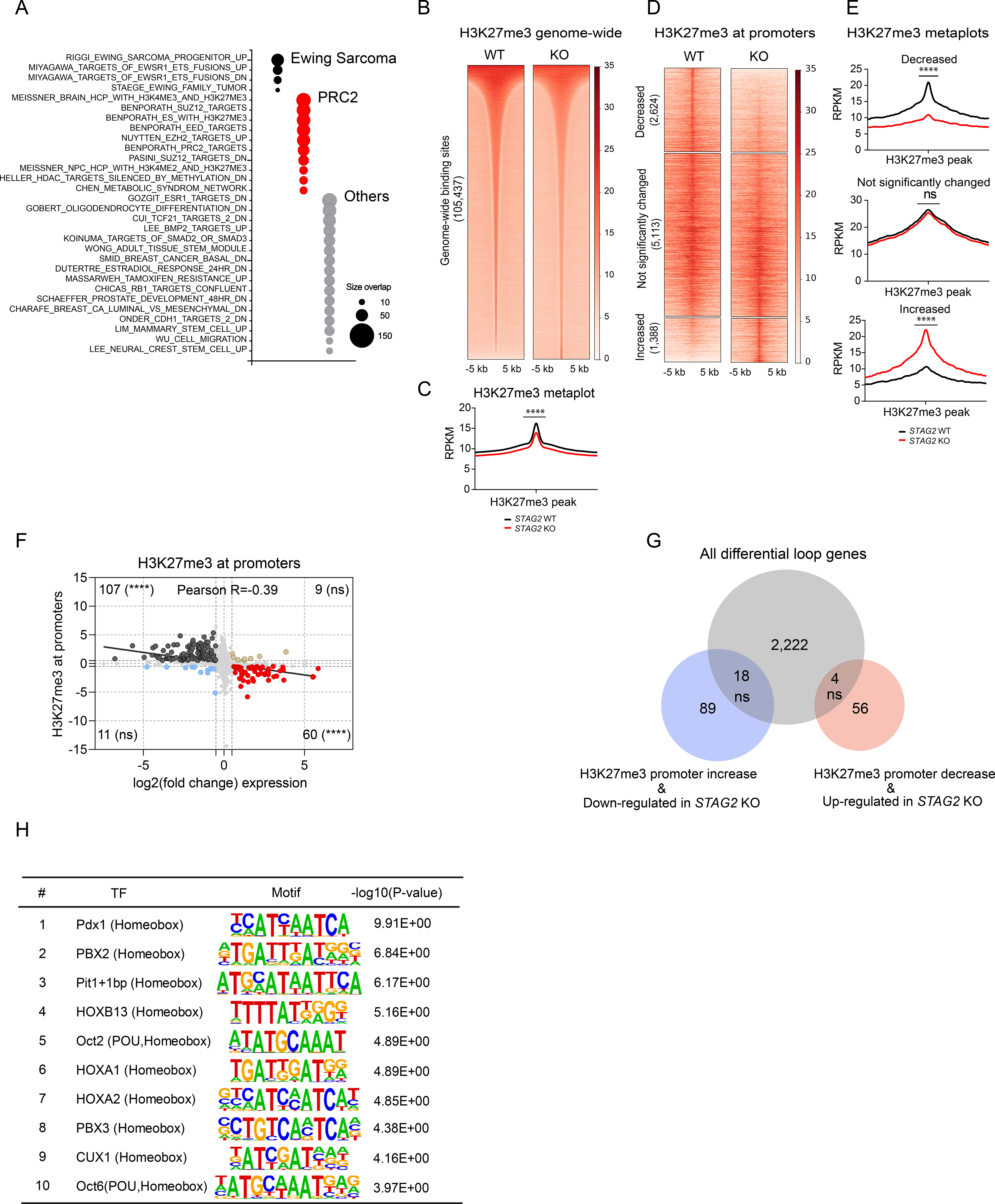
(A) Bubble plot summarizing top 50 significant enrichments (size overlap≥25, P≤0.05, FDR≤0.05) for the MSigDB v7.1 c2 collection. Enriched gene sets are clustered in representative functional categories. (B) Genome-wide heatmaps of H3K27me3 ChIP-Seq signal in A673 cells expressing WT or STAG2 KO centered on significant peaks identified in either or both conditions. (C) Metaplots showing average genome-wide H3K27me3 signal in STAG2 KO and WT A673 cells. (unpaired t-test with Welch’s correction, **** P < 0.0001). (D) Clustered heatmaps depicting TSS +/− 5kb promoter regions with decreased, not significantly changed, or increased H3K27me3 ChIP-Seq binding in STAG2 KO vs. WT A673 cells (|Delta(area under curve signal)| ≥ 1.5). (E) Metaplots showing average H3K27me3 signal in the promoter regions defined in Figure 5C. Differential read density for STAG2 KO vs. WT A673 within clusters (unpaired t-test with Welch’s correction, **** P< 0.0001, ns = not significant). (F) Scatter plot depicting the overlap between the genes with significant change for H3K27me3 ChIP-Seq binding at promoter regions with differentially expressed genes. Two-tailed Fisher exact test, **** P < 0.0001, ns = not significant. (G) Venn diagram showing the overlap between the subset of genes with concurrent changes in expression and H3K27me3 levels at the promoter (shown in Figure 5F) with the total list of genes harboring a differential loop within 5kb of the TSS. Two-tailed Fisher exact test, ns = not significant. (H) List of top 10 enriched motifs at the promoters of genes with altered H3K27me3 levels in STAG2 KO cells. See also Figure S5, Tables S5 and S6.
The PRC2 complex often localizes near gene promoters to regulate transcription. Thus, we evaluated how the H3K27me3 mark changes, specifically within 3 kilobases of TSS in the context of STAG2 loss. We identified 2624 sites with decreased and 1388 sites with increased levels of H3K27me3 (Figures 5D and 5E). To determine whether these changes are associated with, and thus likely to regulate, gene expression we assessed the degree of overlap between differentially expressed genes (|log2(FC)| ≥ 1.5, adjusted P ≤ 0.1) and genes with altered levels of H3K27me3 at the promoter. Our analysis revealed a strong correlation between gene expression and levels of H3K27me3 at the promoter (Figure 5F). Indeed, we identified 107 genes that were repressed with significant promoter proximal deposition of H3K27me3 and 60 genes that were induced in STAG2 KO cells that concurrently lost the PRC2 mark (Figure 5F). Next, we asked whether changes to cis-chromatin contacts in the proximity of gene promoters are associated with altered levels of H3K27me3 in the subset of genes that were differentially expressed. Our analysis revealed a lack of any statistically significant correlation, suggesting that the change in PRC2-mediated regulation of gene expression in the wake of STAG2 loss is likely independent of alterations to long-range enhancer-promoter interactions (Figure 5G). Notably, although we did not find changes to the levels of H3K27ac genome-wide in STAG2 KO cells, we found a very strong inverse correlation between the levels of H3K27me3 and H3K27ac levels at the promoters of differentially expressed genes (Figures S5C-G), suggesting that active epigenetic modification is taking place in the vicinity of the TSS. We thus decided to check whether this is mediated by a few select transcription factors. To test the idea, we performed motif enrichment analysis using the genomic sequences that were differentially marked by H3K27me3 near the promoter for all genes. Interestingly, our analysis revealed the enrichment of a number of Homebox and POU family pioneer neurodevelopmental transcription factors (Figure 5H and Table S6). These results suggest that loss of STAG2 may perturb PRC2 mediated regulation of developmental programs.
STAG2 loss reprograms migratory and neurodevelopmental transcriptional programs
Our data thus far highlights changes in the enhancer-promoter interaction network, a partial dampening of the EWS/FLI1 dependent oncogenic transcriptional program and disruption of PRC2-mediated regulation of gene expression as key emergent molecular manifestations of STAG2 loss. To gain insight into the potential functional outcomes of these changes, we performed gene ontology analysis which leverages transcriptional signatures to infer cellular processes that uniquely define the network of differentially expressed genes. Our data revealed the striking enrichment of three functional categories in the top 50 gene ontology signatures (Figure 6A and Table S7). First, we identified several processes indicative of an altered invasive and migratory behavior as exemplified by GO terms such as locomotion, motility, taxis and biological adhesion. Second, we observed a strong enrichment of early developmental and morphogenetic processes. Third and likely a subcategory of the second feature, we identified a robust enrichment of neurodevelopmental processes including the GO terms Neurogenesis, Neuron differentiation, Neuron projection and Axon development. Thus, at the cellular level, depletion of STAG2 appears to alter the migratory potential of Ewing sarcoma, along with the hijacking and reprogramming of early developmental programs that may reinforce these properties.
Figure 6. Depletion of STAG2 enhances the migration and metastatic potential of Ewing sarcoma cells.
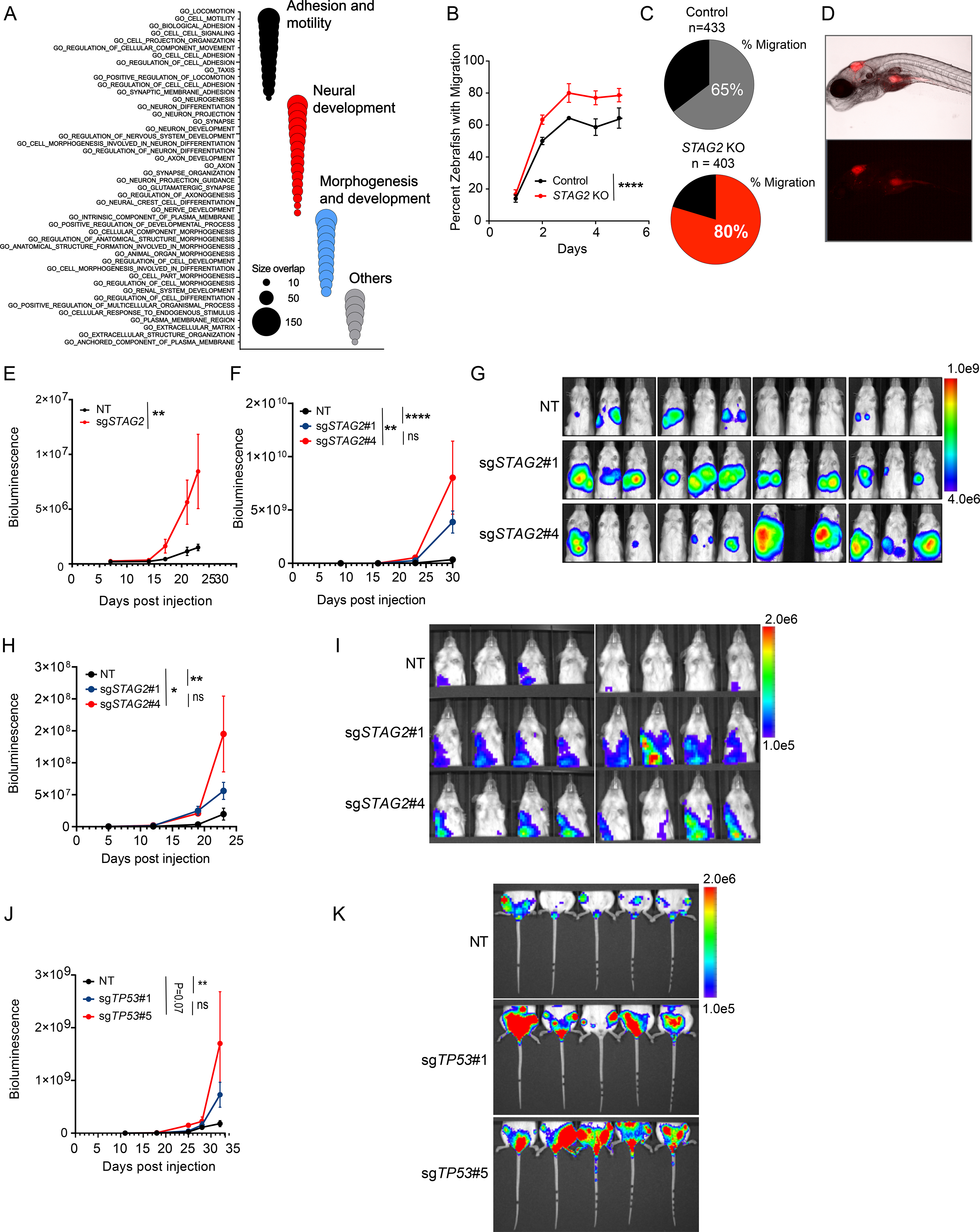
(A) Bubble plot summarizing top 50 significant enrichments (size overlap≥10, P≤0.05, FDR≤0.05) for the MSigDB v7.1 c5 collection. Enriched gene sets are clustered in representative functional categories. (B) Line graph represents mean ± sd of the percentage of zebrafish that displayed migration to at least one of the three regions of interest as a function of the days post injection. (2-way ANOVA, **** P < 0.0001). (C) Pie chart showing the percentage of zebrafish with migration of Ewing cells to at least one of the three sites examined three days post injection. (D) Representative brightfield (top) and fluorescence (541/565 nm bottom) images displaying migration of TC71 cells to the yolk sack. (E) Quantification of bioluminescence signal collected for lower extremities after blocking upper abdominal cavity. Line graph represents mean ± SEM, N = 8 per group. (2-way ANOVA, ** P<0.001). (F) Quantification of bioluminescence signal collected for upper thoracic cavity after blocking lower abdomen. Line graph represents mean ± SEM, N = 12 per group (2-way ANOVA, **** P < 0.0001,** P < 0.01, ns = not significant). (G) Bioluminescence images of mice described in Figure 6F. (H) Quantification of bioluminescence signal collected for upper thoracic cavity while blocking lower abdomen. Line graph represents mean ± SEM, N = 8 per group. (2-way ANOVA, **** P 0.0001,** P < 0.01, ns=not significant). (I) Bioluminescence images of mice described in Figure 6H. (J) Quantification of bioluminescence signal collected for lower extremities after blocking upper abdominal cavity. Line graph represents mean ± SEM, N = 5 per group. (2-way ANOVA, ** P < 0.01, ns = not significant). (K) Bioluminescence images of mice described in Figure 6J. See also Figure S6 and Table S7.
Given the enrichment of cell motility-associated signatures, we next sought to evaluate the migratory potential of Ewing sarcoma cells. The zebrafish provides a robust model system to accurately track fluorescently labeled cells in vivo (El-Naggar et al., 2015; Olson and Nechiporuk, 2018). Thus, we injected control and STAG2 KO cells into the hindbrain of zebrafish larvae (control n=433, STAG2 KO n=403) two days post fertilization and monitored the movement of cells daily for 5 additional days via microscopy (Figure S6A). For controls, we injected fluorescently labeled microspheres and confirmed that there is no passive diffusion up to 5 days post injection (Figure S6B). At each time point, we scored colonization of the dorsal surface, yolk sack, and tail. We observed enhanced migration of STAG2 KO cells, which migrated to at least one distal site in 85% of the zebrafish injected, whereas STAG2 WT cells only do so in 65% of the cases (Figures 6B-D).
STAG2 loss enhances the metastatic potential of Ewing sarcoma xenografts
Based on the transcriptional signature we identified, the results of the migration assay in zebrafish, and the reported clinical association between loss-of-function mutations in STAG2 and the incidence of metastasis (Crompton et al., 2014), we hypothesized that deletion of STAG2 will enhance the metastatic potential of Ewing sarcoma cells. To test the hypothesis, we performed extensive xenograft studies by transplanting control and STAG2 KO Ewing sarcoma cells into immunocompromised NSG mice. First, we generated polyclonal STAG2 KO TC71 cells and injected them via tail vein into recipient mice (Figure S6C). While the overall disease burden was similar, STAG2 KO cells metastasized to the hindlegs and marrow space significantly more relative to controls (Figures 6E and S6D-H). Next, we used clonally selected control and STAG2 KO A673 cells and transplanted them via tail vein into recipient mice. In this model we measured the extent of metastasis to visceral organs. We observed significantly more infiltration of lungs and liver tissue in STAG2 KO recipient mice compared to controls (Figures 6F and 6G). Finally, we employed a semi-orthotopic xenograft model by transplanting cells into the hindleg intramuscular space. The data revealed that STAG2 KO A673 cells grow more aggressively at the primary site (Figures S6I and S6J) and also metastasized to distal organs better than their WT counterparts (Figures 6H and 6I). We further confirmed the finding by ex-vivo imaging of lungs recovered from recipient mice at the study end point (Figures S6K and S6L).
STAG2 and TP53 mutations are often concurrent and carry dismal clinical outcome (Tirode et al., 2014). Both A673 and TC71 cells, which we have used for most of our studies, are mutants for TP53. Thus, we asked whether deletion of TP53 in the background of STAG2 mutation can further modulate disease course in vivo. To this end, we utilized TC32 cells, which are mutant for STAG2 but carry a WT copy of TP53. We transplanted control and TP53 deleted cells via tail vein injection into recipient mice and measured disease course by bioluminescence imaging. We observed that TP53 KO cells generate a significantly more aggressive disease (Figures S6M and S6N) associated with higher infiltration of hind legs and lungs (Figures 6J,K and S6O, P). Taken together, the results of our studies strongly argue for a role for STAG2 loss in promoting metastatic behavior in vivo and also provide experimental evidence for the clinical associations observed previously.
The neurodevelopmental transcription factor POU3F2 modulates the metastatic potential of STAG2 deleted Ewing sarcoma cells
To identify critical mediators of metastatic behavior downstream of STAG2, we focused on genes that were induced in the wake of STAG2 depletion. Two neurodevelopmental transcription factors, POU3F2 and NR2F1, were consistently upregulated in STAG2 KO cells relative to controls (Figures 7A and 7B). POU3F2 is a pioneer transcription factor critical for early neuroepithelial progenitor specification and linked to metastatic properties in multiple tumor types (Bishop et al., 2017; Fane et al., 2019; Urban et al., 2015; Yao et al., 2017; Zeng et al., 2018). Likewise, NR2F1 is an orphan nuclear receptor important for many aspects of neurogenesis and implicated in tumor cell dormancy, invasion and metastasis (Armentano et al., 2007; Armentano et al., 2006; Gao et al., 2019; Sosa et al., 2015; Zhou et al., 1999). Therefore, we decided to investigate the potential role of these two transcription factors in modulating the metastatic capacity of Ewing sarcoma cells.
Figure 7. The neurodevelopmental transcription factor POU3F2 modulates the metastatic potential of STAG2 KO Ewing sarcoma cells.
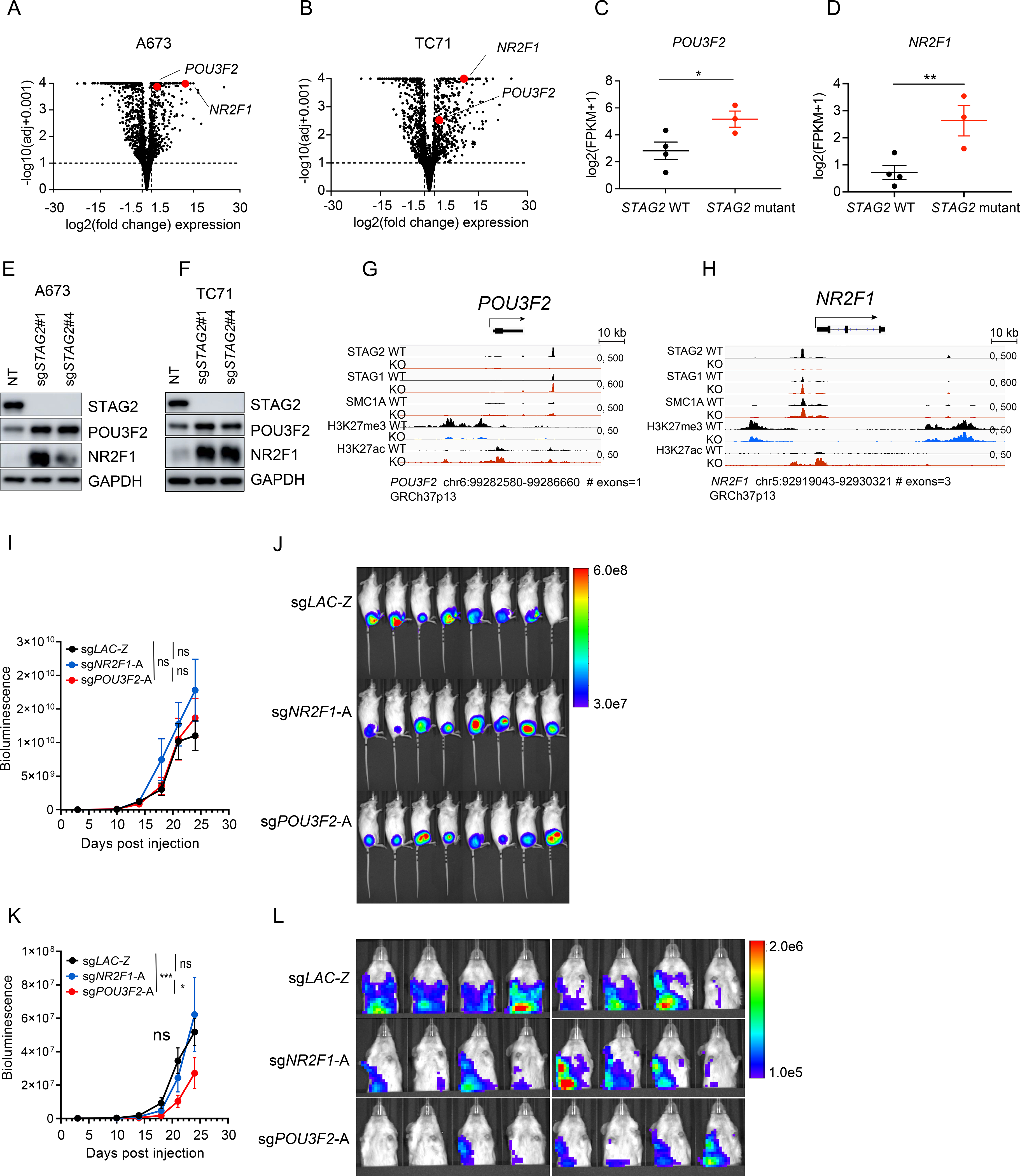
(A-B) Volcano plot depicting the transcriptional changes induced by STAG2 KO vs. control in (A) A673 and (B) TC71 cells. POU3F2 and NR2F1 are highlighted in red. Significance cutoffs: |log2(FC)| ≥ 1.5, adjusted P ≤ 0.10). (C-D) Scatter dot plot depicting the mean ± SEM mRNA log2(FPKM+1) expression of (C) POU3F2 and (D) NR2F1 in three STAG2 mutant tumors vs. the top four STAG2 WT tumors with highest STAG2 expression from (Crompton et al., 2014) data. Mann-Whitney non-parametric t-test (** P < 0.001, * P < 0.05). (E-F) Validation of an increase in POU3F2 and NR2F1 protein levels in (E) A673 and (F) TC71 cells clonally selected for STAG2 loss. (G-H) Integrated genomics viewer track showing SA2, SA1, SMC1, H3K27me3 and H3K27ac signals at the (G) POU3F2 and (H) NR2F1 locus in control and STAG2 KO A673 cells. (I) Quantification of bioluminescence signal for whole body. Line graph represents mean ± SEM, N = 8 per group (2-way ANOVA, ns = not significant). (J) Bioluminescence images of mice described in Figure 7I. (K) Quantification of bioluminescence signal for upper thoracic cavity after blocking the primary tumor at the lower abdomen. Line graph represents mean SEM, N = 8 per group (2-way ANOVA, *** P < 0.001, * P < 0.05, ns = not significant). (L) Bioluminescence images of mice described in Figure 7K. See also Figure S7.
Consistent with our findings in cell line models, STAG2 mutant Ewing tumors express elevated levels of POU3F2 and NR2F1 relative to controls (Figures 7C, D and S7A-D). We further confirmed these results by western blot analysis in both clonally selected and polyclonal STAG2 KO A673 and TC71 cells (Figures 7E,F and S7E,F). The expression of lineage specifying transcription factors is often regulated by the polycomb repressive complex. In agreement with this notion, we observed marked loss of H3K27me3 near the promoters of both genes and a concurrent increase in the active transcription mark H3K27ac in STAG2 KO cells (Figures 7G and 7H). Moreover, a prominent STAG2 peak is lost at an upstream enhancer region of POU3F2 and near the TSS of NR2F1, suggesting that in the absence of STAG2, PRC2-mediated suppression of these transcription factors is compromised.
To directly test the functional role of POU3F2 and NR2F1 in the metastatic process, we generated POU3F2 and NR2F1 KO cells in the background of clonally selected STAG2 KO A673 cells (Figures S7G and S7H). Transplantation of POU3F2 and NR2F1 KO cells resulted in comparable levels of tumor at the primary site (Figures 7I and 7J). However, POU3F2 KO, but not NR2F1 KO, cells displayed reduced infiltration of visceral organs, suggesting that in STAG2 KO cells the increase in POU3F2 expression is partially responsible for the enhanced metastatic potential of Ewing sarcoma cells (Figure 7K, L).
DISCUSSION
Emerging data demonstrates that cohesin plays a significant role in transcriptional regulation by securing DNA-DNA contacts between enhancers and promoters. By broadly surveying the epigenetic and transcriptional landscape coupled with chromatin interactions, we demonstrate a unique functional role of STAG2 in Ewing sarcoma pathogenesis. Specifically, our studies revealed a critical role for STAG2 in governing the establishment of gene-regulatory architecture underlying oncogenic and developmental programs that undergo reprogramming upon loss of STAG2 to promote disease progression.
While STAG2 and its paralog STAG1 appear to have redundant roles in chromatin cohesion and segregation, STAG2 localized to genomic loci marked by epigenetic modifications associated with enhancers in Ewing sarcoma cells. Importantly, depletion of STAG2 resulted in a compensatory increase in STAG1 protein levels. However, examination of the relative genomic distribution of the increased STAG1 revealed that enhancer marked regions were insufficiently bound by additional cohesin-SA1. Intriguingly, these enhancer rich domains were enriched for ETS binding motifs and were also strongly occupied by EWS/FLI1. Consistent with this observation, depletion of STAG2 resulted in reconfiguration of a subset of enhancer-promoter interactions including those associated with EWS/FLI1. Moreover, genes regulated by EWS/FLI1 were among the most enriched for alterations in expression as a consequence of STAG2 loss. While one obvious prediction would be that loss of STAG2 reinforces the EWS/FLI oncogenic program, surprisingly, STAG2 deletion repressed a subset of EWS/FLI regulated genes. In this way, STAG2 loss appears to modulate the expression of a subset of genes directly regulated by EWS/FLI, effectively dampening the activity of the oncogene and promoting an EWS/FLI “low” state.
Our data further showed that STAG2 also occupied a subset of PRC2-marked regions, which tend to contain binding motifs for transcription factors involved in neurodevelopmental programs. Notably, these regions were devoid of ETS binding motifs and were poorly occupied by EWS/FLI1, suggesting that cohesin-SA2 may function independently of the EWS/FLI oncogenic program to regulate a neurogenic program. Indeed, STAG2 loss altered H3K27me3 levels at the promoters of many genes and also resulted in the deregulation of neurodevelopmental programs.
The exact mechanism by which loss of STAG2 changes the nature of long-range cis-chromatin interactions or the regulation of developmental genes by PRC2 is currently unknown. Given the lack of compensation by cohesin-SA1 at enhancer rich regions, it is tempting to speculate that the absence of a productive cohesin complex at these sites may drive the collapse of enhancer-promoter contacts. However, our data showed that a number of enhancer-promoter interactions in these regions were preserved, and at times were even strengthened in STAG2 KO cells arguing against a simple, all or none mode of regulation. In addition, at cohesin-SA2 and PRC2 enriched regions, we observed marked deregulation of gene expression without pervasive changes to cis-chromatin interactions. Chromatin interactions are highly dynamic, and a static snapshot garnered from HiChIP is likely to miss a subset of interactions potentially explaining some of the gaps in our understanding of the underlying process. Additionally, recent findings have revealed that cis-chromatin interactions involving polycomb-marked regions may occur at distances bridging several mega-bases, which may put them outside of the purview of our analysis (Kraft et al., 2020; Rhodes et al., 2020; Zhang et al., 2020). Therefore, additional studies are needed to further delineate the exact molecular mechanism by which loss of STAG2 brings about fundamental changes to transcriptional programs.
At the cellular level, STAG2 loss enhanced the migratory and metastatic potential of Ewing sarcoma cells. These finding are in good agreement with clinical observations which showed strong correlation between loss-of-function STAG2 mutations alone or in combination with TP53 loss and the incidence of metastasis in patients with Ewing sarcoma (Crompton et al., 2014; Tirode et al., 2014). Given the pleotropic consequences of STAG2 loss, the relative contribution of each towards the metastatic phenotype is currently unclear. Recent data demonstrates that titration of EWS/FLI1 transcriptional activity promotes cellular plasticity, which may be necessary for Ewing sarcoma cells facing changes in environmental pressures, such as cells undergoing metastasis (Franzetti et al., 2017). Although we didn’t find a strong overlap between our own data set and the proteomics-based characterization by Franzetti and colleagues, likely due to differences in the model systems utilized, each of our findings converge on the idea that an EWS/FLI “low” state may enable metastases (Franzetti et al., 2017). On the other hand, there is a growing body of evidence highlighting the role of latent developmental program hijacking in metastatic disease (Gupta et al., 2005; Pomerantz et al., 2020). In our study, we observed a dominant representation of neurodevelopmental programs as the transcriptional fingerprints of STAG2 loss. In addition, the expression of a number of neurodevelopmental transcription factors were altered in the wake of STAG2 depletion, including the neural crest specifiers FOXD3 and SOX9, as well as the master regulator of neural progenitor cell differentiation POU3F2 (Cheung and Briscoe, 2003; Lukoseviciute et al., 2018). Interestingly, POU3F2 was one of the most significantly induced genes in STAG2 KO cells. Moreover, recent studies in melanoma and prostate cancer have revealed a critical role for POU3F2 in driving the metastatic process (Bishop et al., 2017; Fane et al., 2019). Thus, we used POU3F2 as a linchpin to assess the potential contribution of the altered neurodevelopmental programs to the metastatic process in Ewing sarcoma. Importantly, our studies revealed that POU3F2 may indeed contribute to the enhanced metastatic potential observed in STAG2 KO cells as its repression partially attenuated their metastatic invasion. Therefore, taken together, our results provide a strong direct evidence for the involvement of altered neurogenic programs in driving the metastatic process downstream of STAG2 loss. Furthermore, our results also implicate STAG2 loss-mediated modulation of the EWS/FLI1 dependent oncogenic program as an additional candidate mediator of metastasis in Ewing sarcoma, a finding which warrants validation in future studies. Finally, because the cohesin complex plays several important roles in the cell, other functions not explored in this work, such as its role in DNA damage and repair (Meisenberg et al., 2019; Mondal et al., 2019), RNA processing (Kim et al., 2019), immune signaling (Ding et al., 2018), and telomere maintenance (Daniloski and Smith, 2017) may be altered in the context of STAG2 mutations, topics also worthy of exploration in future studies.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kimberly Stegmaier (Kimberly.stegmaier@dfci.harvard.edu).
MATERIALS AVAILABILITY
This study did not generate new unique reagents. Any additional material or reagent described in this manuscript will be made available with a simple MTA.
DATA AND CODE AVAILABILITY
The data from this study including all ChIP-Seq, RNA-Seq, ATAC-Seq and HiChIP was uploaded at GEO under the accession number GSE116495. The proteomics data from this study is available at the following link http://massive.ucsd.eduMSV000082954. Ewing sarcoma tumor RNA-Seq data previously reported by (Crompton et al., 2014) is available at dbGaP with the following accession number dbGaP: phs000804.v1.p1. Ewing sarcoma ChIP-Seq previously reported by (Riggi et al., 2014) is available at GEO with the following accession number GEO: GSE61953. Ewing sarcoma ChIP-Seq and RNA-seq data previously reported by (Tomazou et al., 2015) is available at the following link http://tomazou2015.computational-epigenetics.org. Ewing sarcoma ChIP-Seq data previously reported by (Kojic et al., 2018) is available at GEO with the following accession number GEO: GSE101921. Ewing sarcoma ChIP-Seq data previously reported at ENCODE: ENCSR000DZP is available at GEO under the accession number GEO: GSM935376. Ewing sarcoma tumor Affymetrix arrays data previously reported by (Postel-Vinay et al., 2012) available at GEO with the following accession number GEO: GSE34620. Ewing sarcoma tumor Affymetrix arrays data previously reported by (Savola et al., 2011) is available at GEO with the following accession number GEO: GSE17618. Ewing sarcoma RNA-Seq data previously reported by (Smith et al., 2006) is available at GEO with the following accession number GEO: GSE53066. Ewing sarcoma RNA-Seq data previously reported by (Sankar et al., 2014) is available at NCBI with the following accession number NCBI: PRJNA176544. Ewing sarcoma Affymetrix arrays data previously reported by (Smith et al., 2006) is available at Array express with the following accession number Array Express: E-GEOD-4560. Ewing sarcoma RNA-Seq data previously reported by (He et al., 2017) is available at GEO with the following accession number GEO: GSE73092. Ewing sarcoma IC-EwS gene signature previously reported by (Aynaud, 2020) is available at the following link https://www.sciencedirect.com/science/article/pii/S2211124720300747?via=ihub.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
All mice were housed at Dana-Farber Cancer institute’s animal facility. All mouse procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Dana-Farber Cancer Institute. For all experiments, female 6–8 weeks old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were obtained from Jackson laboratory and allowed to acclimatize for one week prior to transplantation. The details of individual xenograft studies including the method of transplantation, the cell dose used, and nature of data acquisition is described in the Methods Details section.
Zebrafish
Zebrafish (Danio rerio) were raised and maintained according to standard protocol (Westerfield, M., 2000). Tg(nacre−/−; roy−/−) zebrafish, commonly called caspers, were used for all in vivo experiments (White et al., 2008). Adult fish were maintained in a recirculating commercial housing system at 28.5°C under a 14:10 light: dark condition schedule. The optimal temperature for zebrafish growth occurs at 28°C; however, to accommodate the injected cancer cell lines, zebrafish larvae used in xenotransplantation (XT) experiments were incubated at 35°C to allow for normal growth and development of both zebrafish and injected human cell lines (Haldi et al., 2006; Lee et al., 2005). All zebrafish larvae were maintained in E3 embryo medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) in 10 cm petri dishes. Larvae were cleaned and provided with new media daily. Larvae were euthanized by tricaine overdose (1mg/ml) at seven days post fertilization (dpf), followed by 6.15% bleach solution to ensure complete mortality. Use of zebrafish in this study was approved by the Dalhousie University Committee on Laboratory Animals (protocol # 17–005)
Cell Lines
All cell lines used in this study were previously genotyped and confirmed to express the appropriate EWS-ETS rearrangement using either a combination of whole-exome sequencing and transcriptome sequencing or a combination of STR profiling and RT-PCR (Crompton et al., 2014). A673 (female), EW8 (male), and SKPNDW (male) cells were grown in Dulbecco’s modified Eagle’s media (Mediatech) with 10% FBS (Sigma-Aldrich) and 1% penicillin streptomycin glutamine (Gibco). A673 medium was supplemented with 1 mmol/L sodium pyruvate (Gibco). TC71 (male) and TC32 (female) cells were grown in RPMI (Mediatech) with 10% FBS and 1% penicillin streptomycin glutamine (Gibco). A673, TC71, TC32 and EW8 were provided by Dr. Todd Golub (Broad Institute, Cambridge, MA) and SKPNDW by Dr. Alejandro Sweet-Cordero (Stanford University, Stanford, CA).
METHOD DETAILS
CRISPR/Cas9 genome editing
LentiCRISPRv2 plasmid backbone encoding Cas9 nuclease and puromycin resistance gene was digested with the restriction endonuclease BsmbI, gel extracted and used in a ligation reaction with synthetic oligonucleotide fragments for guide RNA sequences (gRNA) targeting a gene of interest. Oligos were purchased from Integrated DNA Technologies (IDT), annealed and end phosphorylated via T4 polynucleotide kinase before using in the ligation reaction. All gRNA sequences used in this manuscript are provided in the Key Resources Table under the oligonucleotide heading. Lentiviral particles were generated by co-transfecting cells with 2 μgs of lentiCRISPRv2-gRNA constructs along with viral packaging plasmids pVSVg (Addgene 8454) and pPAX2 (Addgene 19319) into HEK293T cells using TransIT-VirusGEN transfection reagent (Mirus). Ewing sarcoma cells plated 2–3 days prior were transduced with fresh viral particles, cultured for 48 hours and subjected to puromycin selection at 1 μg/ml for 72 additional hours. Gene KO was confirmed by immunoblotting for the respective targets.
KEY RESOURCES TABLE.
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-STAG2 (Western blot) | Santa Cruz | Cat#sc-81852; RRID: AB_2199948 |
| Rabbit anti-STAG2 (ChIP-seq) | Cell Signaling | Cat#5882; RRID:AB_10834529 |
| Rabbit anti-STAG1(Western blot) | Bethyl | Cat# A302-579A;RRID:AB_2034857 |
| Rabbit anti-STAG1(ChIP-seq) | Bethyl | Cat# A302-578A; RRID:AB_2034858 |
| Rabbit anti-SMC1A | Bethyl | Cat# A300-055A; RRID:AB_2192467 |
| Rabbit anti-RAD21 | Bethyl | Cat# A300-080A; RRID:AB_2176615 |
| Rabbit anti-SMC3 | Bethyl | Cat# A300-060A, RRID:AB_67579 |
| Rabbit anti-H3K27me3 | Cell Signaling | Cat# 4395, RRID:AB_11220433 |
| Rabbit anti-EWS/FLI1 | Abcam | Cat# ab15289; RRID:AB_301825 |
| Rabbit anti -alpha TUBULIN | Cell Signaling | Cat# 2144; RRID:AB_2210548 |
| Mouse anti-GAPDH | Santa Cruz | Cat# sc-47724; RRID:AB_627678 |
| Rabbit anti-H3K27ac3 | Abcam | Cat# ab4729, RRID:AB_2118291 |
| Rabbit anti-POU3F2 | Cell Signaling | Cat# 12137; RRID:AB_2797827 |
| Rabbit anti-NR2F1 | Cell Signaling | Cat# 6364; RRID:AB_11220432 |
| Normal Rabbit IgG-R | Cell Signaling | Cat# 2729; RRID:AB_1031062 |
| Drosophila H2Az(spike-in Antibody) | Active Motif | Cat#61686; RRID: AB_2737370 |
| Rat anti-MouseIgG2a HRP conjugated secondary | GenWay | Cat# 25-783-70745;RRID:AB_1028627 |
| Sheep Anti-Mouse IgG ECL Antibody, HRP Conjugated | GE Healthcare | Cat# NA9310-1ml; RRID:AB_772193 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bovine serum albumin | Sigma-Aldrich | Cat#F2442 |
| Phosphate Buffer Saline | DFCI supply center | NA |
| RPMI medium | DFCI supply center | NA |
| DMEM medium | DFCI supply center | NA |
| Penicillin-Streptomycin | Gibco | Cat#15070063 |
| Sodium Pyruvate | ThermoFisher | Cat#21051040 |
| Puromycin | Gibco | Cat#A1113802 |
| Polybrene | American Bio | Cat#AB01643 |
| TransIT-VirusGEN transfection reagent | Mirus | Cat#MIR 6704 |
| X-tremeGENE™ HP DNA Transfection Reagent | Sigma | Cat#6366236001 |
| ClonaCell-TCS medium | Stem Cells | Cat#03814 |
| MTT labeling reagent | Sigma | Cat#11 465 007 001 |
| Trypan Blue | Gibco | Cat#15250061 |
| Cell titer Glo | Promega | Cat#G7570 |
| 4′,6-diamidino-2-phenylindole (DAPI) | Sigma | Cat#D9542 |
| Pierce IP Lysis Buffer | Thermofisher | Cat#87788 |
| 16% methanol-free Formaldehyde solution (w/v) | Thermofisher | Cat#28908 |
| 5M NaCl | Thermofisher | Cat#AM9760G |
| Tris-EDTA pH 8.0 | Sigma-Aldrich | Cat#93283 |
| Ultrapure 0.5M EDTA | Invitrogen | Cat#15575-038 |
| Ultrapure 1M Tris-HCl pH 8.0 | Invitrogen | Cat#15568-025 |
| Ultrapure 10% SDS | Invitrogen | Cat#15553-035 |
| 2.5M Glycine solution | BostonBioProducts | Cat#C-4375 |
| Halt protease inhibitors | Thermofisher | Cat#78429 |
| phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat#P7626 |
| Transposase,TAGMENT DNA Enzyme | Illumina | Cat#15027865 |
| Dynabeads Protein A | Invitrogen | Cat#10004D |
| Lithium Chloride | Sigma | Cat#L9650 |
| NP40 | Fisher | Cat#FNN021 |
| Deoxycholic acid | Fisher | Cat#BP349-100 |
| NaHCO3 | Sigma | Cat#S5761 |
| NaN3 | USB | Cat#21610 |
| Triton X-100 | Sigma | Cat#0992-93-1 |
| Igepal CA-630 | Sigma | Cat#I8896 |
| Agencourt AMPure XP | Beckman Coulter | Cat#A63881 |
| Critical Commercial Assays | ||
| qScript cDNA synthesis Kit | QuantaBio | Cat#95047-025 |
| SYBR Green FastMix | QuantaBio | Cat#95072-250 |
| Click-iT™ Plus EdU Flow Cytometry Assay Kit | Thermofisher | Cat#C10634 |
| Subcellular Protein Fractionation Kit | Thermofisher | Cat#87787 |
| Deposited Data | ||
| ChIP-Seq, RNA-Seq, ATAC-Seq, Hi-ChIP | This study | GEO:GSE116495 |
| Ewing sarcoma Proteome | This study | http://massive.ucsd.eduMSV000082954 |
| Ewing sarcoma RNA-Seq | (Crompton et al., 2014) | dbGaP: phs000804.v1.p1 |
| Ewing sarcoma ChIP-Seq | (Riggi et al., 2014) | GEO: GSE61953 |
| Ewing sarcoma ChIP-Seq, RNA-seq | (Tomazou et al., 2015) | http://tomazou2015.computational-epigenetics.org |
| Ewing sarcoma ChIP-Seq | (Kojic et al., 2018) | GEO: GSE101921 |
| Ewing sarcoma ChIP-Seq | ENCODE: ENCSR000DZP | GEO: GSM935376 |
| Ewing sarcoma Affymetrix arrays | (Postel-Vinay et al., 2012) | GEO: GSE34620 |
| Ewing sarcoma Affymetrix arrays | (Savola et al., 2011) | GEO: GSE17618 |
| Ewing sarcoma RNA-Seq | (Smith et al., 2006) | GEO: GSE53066 |
| Ewing sarcoma RNA-Seq | (Sankar et al., 2014) | NCBI: PRJNA176544 |
| Ewing sarcoma Affymetrix arrays | (Smith et al., 2006) | Array Express: E-GEOD-4560 |
| Ewing sarcoma RNA-Seq | (He et al., 2017) | GEO: GSE73092 |
| Ewing sarcoma IC-EwS gene signature | (Aynaud, 2020) | https://www.sciencedirect.com/science/article/pii/S2211124720300747?via=ihub |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) | The Jackson Laboratory | Cat# JAX:005557;RRID:IMSR_JAX:005557 |
| Zebrafish: Tg(nacre−/−; roy−/−)(Caspers) | Dr. Leonard Zon Lab, DFCI | NA |
| Experimental Models: Cell Lines | ||
| Human: A673 (Female) | Dr. Todd Golub Lab, Broad Institute | NA |
| Human: TC71 (Male) | Dr. Todd Golub Lab, Broad Institute | NA |
| Human: TC32 (Female) | Dr. Todd Golub Lab, Broad Institute | NA |
| Human: EW8 (Male) | Dr. Todd Golub Lab, Broad Institute | NA |
| Human: SKPNDW (Male) | DR. Alejandro Sweet-Cordero Lab, Stanford University | NA |
| Oligonucleotides | ||
| sgSTAG2#1 gRNA AATGTCTTACTGCTCTACAA | IDT | NA |
| sgSTAG2#4 gRNA GCTGAATGTCATCCTCCCGT | IDT | NA |
| sgSTAG1#2 gRNA GGAATTAGAGGAGCAGGCCG | IDT | NA |
| sgSTAG2#3 gRNA CAGCGAGCTTGAAGAAACAG | IDT | NA |
| sgNT1 gRNA GTAGCGAACGTGTCCGGCGT | IDT | NA |
| sgNT2 gRNA GACCGGAACGATCTCGCGTA | IDT | NA |
| sgPOU3F2-A gRNA GCTGTAGTGGTTAGACGCTG | IDT | NA |
| sgNR2F1-A gRNA GTACTGGCCTGGATTGGGCT | IDT | NA |
| sgLACZ gRNA AACGGCGGATTGACCGTAAT | IDT | NA |
| Recombinant DNA | ||
| pVSVg (addGene 8454) | addGene | RRID:Addgene_8454 |
| pPAX2 (addGene 19319) | addGene | RRID:Addgene_1226 |
| lentiCRISPR v2 | addGene | RRID:Addgene_52961 |
| pSpCas9BB-2A-GFP(PX458) | addGene | RRID:Addgene_48138 |
| CMV-GFP-T2A-Luciferase plasmid | SBI | Cat#BLIV101PA-1 |
| Software and Algorithms | ||
| Prism 8 | Graphpad | http://www.graphpad.com |
| FlowJo V9.9.6 | Tree Star | http://www.flowjo.com/ |
| ImageQuant TL 8.2 | Cytiva | https://www.cytivalifesciences.com |
Generation of clonal STAG2 KO Ewing sarcoma cells
For TC71 cells, the protocol outlined above was used with the exception of the use of X-tremeGENE™ HP DNA Transfection Reagent (Roche). Puromycin selected cells were seeded in semi-solid methylcellulose-based medium (ClonaCell-TCS Medium, Stemcell Technologies) at clonal density and individual colonies were picked, expanded in liquid culture and gene editing was confirmed by western blotting for STAG2. For A673 cells, pSpCas9BB-2A-GFP(PX458) (Addgene#48138) expressing Cas9 nuclease and EGFP plasmid were used to clone gRNA sequences targeting STAG2. A673 cells were transiently transfected with 10 μg of pSpCas9BB-2A-GFP-gRNA using X-tremeGENE™ HP DNA Transfection Reagent. Seventy-two hours later, GFP+ cells were FACS sorted, allowed to expand in liquid culture and re-seeded in ClonaCell-TCS Medium (Stemcell Technologies) at clonal density. Single colonies were picked and expanded in culture and eventually validated for deletion of STAG2 by western blots.
Cumulative cell growth
Parental, non-targeting controls, and STAG2 KO TC71 and A673 cells were plated in replicates at equal low densities and grown for 3 to 4 days. Live cell counts were assessed by trypan blue exclusion staining and cells were re-plated at the same original density. This was repeated every 3 to 4 days for approximately 2 weeks. At each interval, cell counts were used to determine the number of cell doublings using the formula LOG((x/y),2), where x=total cells per flask, and y=total number of cells seeded per flask. Cell doublings per interval were added to produce a cumulative cell doubling value.
Cell viability
To determine the effects of STAG1 deletion on cell viability in STAG2 mutant or STAG2 depleted Ewing sarcoma cells, cells were plated in 384-well plates at a concentration of 1000 cells per well in 50 μL of medium. Cell viability was measured by adding 10 μL of CellTiter-Glo ATP-based assay (Promega). Luminescence was read using the FLUOstar Omega microplate reader (BMG LabTech).
Analysis of growth in semi-solid methylcellulose media
Parental, non-targeting control and STAG2 KO TC71 cells were plated in semi-solid methylcellulose-based medium (ClonaCell-TCS Medium, Stemcell Technologies) at a density of 2500 cells/ml in 6 well plates. Fourteen days later, wells containing colonies propagated in culture were stained with 700 μl of a 1:1 mixture of PBS and MTT labeling reagent (Roche) by incubating for 60 minutes in an incubator at 37°C, >95% humidity and 5% carbon dioxide. Plates were imaged using ImageQuant LAS 4000 imager (GE healthcare) and pictures of individual wells were taken. Colony number in each well and volume per individual colony were determined from the images using the ImageQuant TL 8.2 image analysis software (Cytiva).
Cell cycle profile analysis by EDU incorporation
For cell cycle profile analysis, the Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit (ThermoFisher Scientific) was used. Log phase growing non-targeting control and STAG2 KO A673 cells were pulsed with 10 μM of the modified nucleotide analogue EdU (5-ethynyl-2’ -deoxyuridine) for 90 minutes in an incubator at 37°C, >95% humidity and 5% carbon dioxide. Cells were harvested, washed, fixed, permeabilized and treated with a reaction cocktail containing Alexa flour 647 conjugated picolyl azide to label incorporated EdU. Cells were treated with RNase for 15 minutes, stained with 4′,6-diamidino-2-phenylindole (DAPI) and subsequently analyzed by flow cytometry.
Sub-cellular protein fractionation
Cells were grown to 80% confluence and a total of five million cells were harvested. The subcellular Protein Fractionation Kit for Cultured Cells (ThermoFisher Scientific) was used to isolate proteins in the soluble cytoplasmic fraction, nuclear soluble fraction and chromatin-bound fraction according to the manufacturer’s protocol. Harvested cells were treated with a plasma membrane permeabilization reagent to release soluble cytoplasmic proteins followed by treatment with a reagent to dissolve plasma, mitochondrial and ER/Golgi membranes while maintaining nuclear membrane integrity. Intact nuclei were isolated by centrifugation and their protein content was extracted. Chromatin bound nuclear proteins were released by treating with micrococcal nuclease for 15 minutes at room temperature.
Co-immunoprecipitation
Cells were grown to 80% confluence and a total of five million cells were harvested, washed with ice cold phosphate buffer saline (PBS) and lysed with lysis buffer (pH 7.4, 0.025M Tris, 0.15M NaCl, 0.001M EDTA, 1% NP40, 5% glycerol) supplemented with protease inhibitor cocktail. Five μgs of antibodies were conjugated with 25 μl of magnetic beads. Protein lysates were quantified and resuspended at a concentration of 1 mg/ml in lysis buffer. 250 μls of lysates (per IP) were incubated with the antibody coupled magnetic beads for two hours at room temperature with gentle rotation, washed and eluted to release target antigens.
Chromatin Immunoprecipitation-Sequencing
Cells (20 million per ChIP reaction for all but EWS/FLI1, which required 40 million cells) were crosslinked with warm 1% methanol-free formaldehyde (ThermoFisher) for 10 minutes at room temperature rotating at 12 RPM. The reaction was quenched by adding glycine to a final concentration of 0.125M and incubating for an additional 5 minutes at room temperature rotating at 12 RPM. Cell pellets were washed three times with ice cold PBS and resuspended in 1 ml of SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl pH8) supplemented with protease inhibitor cocktail including phenylmethylsulfonyl fluoride (PMSF) and incubated at RT for 2 minutes with gentle rotation. Lysates were centrifuges at 15,000 G for 10 minutes at 4°C and the pellet was re-suspended in 900 μl of ChIP IP buffer (2:1 SDS lysis buffer : triton dilution buffer), transferred to milliTUBE (covaris). Sonication was performed on a E220 Focus Ultra Sonicator (Covaris) using the setting (duty cycle 5%, peak power 140W, cycles per burst 200, Temperature 4°C, time 30 minutes/millitube). ChIP inputs from sheared chromatin were de-crosslinked by adding de-crosslinking buffer (NaHCO3, NaCl, RNase A, Proteinase K) and incubating for two hours at 65°C in a thermal cycler. The remaining sheared chromatin was incubated with primary antibody coupled to Protein A DynaBead (Beckman Coulter, antibody bead conjugation was performed for 16 hours) overnight, rotating at 4°C. As a calibration control, antibody against a drosophila specific histone variant H2Av and drosophila chromatin (active motif) were used as per the recommendation of the manufacturer. The next day, ChIP product was eluted from the Dynabeads in 100 μl of elusion buffer and de-crosslinked for 12 hours at 65°C. For both Input and chipped material, AMPure XP beads (Beckman coulter) were used to purify DNA. Information regarding antibodies used is presented in key resources table under the heading of Antibodies.
ChIP-Seq Library Preparation & Sequencing
ChIP-Seq libraries were prepared using Swift S2 Acel reagents on a Beckman Coulter Biomek i7 liquid handling platform from approximately 1 ng of DNA according to the manufacturer’s protocol and 14 cycles of PCR amplification. Finished sequencing libraries were quantified by a Qubit fluorometer and samples were QC’D using a Bioanalyzer Tapestation (Agilent Technologies 2200) to determine fragment size. Library pooling and indexing was evaluated with shallow sequencing on an Illumina MiSeq. Subsequently, libraries were sequenced on a NovaSeq targeting 40 million 100bp read pairs by the Molecular Biology Core facilities at Dana-Farber Cancer Institute.
HiChIP
HiChIP was performed on the A673 clonal lines sgNT-1c4 and sgSTAG2–1c6 in duplicate based on a previously published protocol (Mumbach et al., 2016) with a few adaptations which were performed as previously described (Weintraub et al., 2017). The A673 clonal lines sgNT-1c4 and sgSTAG2–1c6 were tested with duplicates for each line crosslinked on separate occasions. The SMC1A antibody used in the ChIP experiments was by Bethyl A300–055A. Libraries were sequenced 100×100 on an Illumina Hi-Seq 2500 platform.
ATAC-Sequencing
ATAC-Seq was performed on A673 clones (sgNT-1c4, sgNT-2c3, sgSTAG2–1c6, and sgSTAG2–4c5). (Buenrostro et al., 2013) Cells were collected, and apoptotic cells removed with the Annexin V MicroBead Kit (Miltenyi Biotec) according to the manufacturer’s protocol. To prepare nuclei, we spun 50,000 cells at 500 × g for 5 minutes. Cells were lysed using cold lysis buffer (10 mM Tris-Cl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL CA-630). Immediately after lysis, nuclei were spun at 500 × g for 10 minutes using a refrigerated centrifuge. Immediately following the nuclei prep, the pellet was resuspended in the transposase reaction mix (25 μL 2x TD buffer, 2.5 μL Transposase (Illumina) and 22.5 μL of nuclease free water). The transposition reaction was carried out for 30 minutes at 37 °C. Directly following transposition the sample was purified using a Qiagen Minelute kit. Transposed DNA was eluted in 10 μL Elution Buffer (10mM Tris buffer, pH 8). We then performed size selection to target fragments of 115–600 bp by Pippin Prep according to manufacturer’s instructions. Next, we amplified 20 μL of library fragments after adding 25 μL of NEBnext PCR master mix and 2.5 μL of custom Nextera PCR primers 1 and 2. PCR was performed using the following conditions: 72°C for 5 minutes, 98°C for 30 seconds, followed by thermocycling at 98°C for 10 seconds, 63°C for 30 seconds and 72°C for 1 minute for 12 cycles. We then performed a SPRI PCR cleanup (AMPure XP Beads; Beckman Coulter/Agencourt) and confirmed that the final library fragments were 100–800 bp by Tapestation.
Genome-wide expression profiling
Total RNA was extracted with an RNeasy Kit (Qiagen) from STAG2 wild-type and STAG2 knockout clones (A673.sgNT-1c4, A673.sgNT-2c3, A673.sgLacZ-2c1, A673.sgSTAG2–1c4, A673.sgSTAG2–1c6, A673.sgSTAG2–4c3, A673.sgSTAG2–4c5, TC71.sgNT-1c6, TC71.sgNT-2c5, TC71.sgSTAG2–1c6, TC71.sgSTAG2–4c15, TC71.sgSTAG2–4c18, TC71.sgSTAG2–1c3). For the A673 clones, Poly(A) RNA was isolated using the NEBNext mRNA magnetic isolated module (New England Biolabs) and paired-end libraries were prepared using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs) according to the manufacturer’s protocols with the following modifications: the PCR library enrichment conditions were adjusted to 12 cycles and the PCR library reaction was purified twice by size selection with Agencourt AMPure XP beads (Beckman Coulter). Libraries were subjected to 50 base paired-end sequencing (Illumina HiSeq 2000). For the TC71 clones, Total RNA was extracted with the RNeasy Kit and on-column DNA digestion (Qiagen). Poly(A) mRNA was isolated, and libraries were prepared using the TruSeq Stranded mRNA Kit (Illumina) according to the manufacturer’s protocol. Strand-specific mRNA sequencing libraries were pooled and sequenced on a NextSeq500 instrument with single-end 75bp reads to a depth of 30–40M reads/sample.
Quantitative PCR
RNA was extracted from cells using the RNeasy Mini kit and on-column DNA digestion (Qiagen) and quantified using Qubit RNA HS Assay (ThermoFisher Scientific). cDNA was prepared using qScript cDNA synthesis Kit (QuantaBio) and a C1000 Touch Thermal Cycler (Bio-Rad). Data were collected in technical triplicates and biological duplicate using SYBR Green FastMix (QuantaBio) on QuantStudio™ 6 -Flex Real-Time PCR Systems (Life technologies) and analyzed using the ΔΔCT method (Livak and Schmittgen, 2001).
Zebrafish model of migration
Fluorescently labeled TC71 cells clonally selected for STAG2 KO (sgSTAG2–1c6, sgSTAG2–4c15, and sgSTAG2–4c18) or WT STAG2 expression (sgNT-1c6 and sgNT-2c5) were injected into the hindbrain ventricle (HBV) of zebrafish larvae 2 days post-fertilization (dpf). The needle tip was positioned perpendicular to the otolith of the zebrafish larvae and inserted directly into the HBV for the transplantation of 50–100 cells. After injection, zebrafish were monitored with brightfield and fluorescent microscopy and scored for presence of migrated TC71 cells to the dorsal surface, yolk sac and tail. To demonstrate that migration does not occur passively, larvae with fluorescent microspheres (~10 microns) were also injected into the HBV at 2 dpf. Larvae were monitored to confirm that there was no microsphere migration into the dorsal surface, yolk sac and tail. Each TC71 clone was injected into a minimum of 120 zebrafish. For analysis, all STAG2 KO clones were combined (n = 403) and all WT STAG2 clones were combined (n = 433). The percentage of fish in the STAG2 KO group and Control group were calculated and a 95% confidence interval was estimated with the modified Wilson method. Fisher’s exact test was used to determine whether a difference in the rate of migration was significant at day 3 post-injection, the day where the maximum migration rate was reached in this experiment.
Murine xenograft studies
Two tail vein injection mouse xenograft experiments were conducted to test the effect of STAG2 knockout on metastasis. In the first experiment, A673 cells clonally selected for STAG2 KO or WT STAG2 expression (sgNT-1c4, sgNT-2c3, sgSTAG2–1c4, sgSTAG2–1c6, sgSTAG2–4c5, and sgSTAG2–4c5) were transduced with the lentiviral plasmid pFUW-Luc-Neo to constitutively express luciferase. One million cells were injected into the tail vein of each NSG mouse such that 6 mice were injected with each clone. Mice were then monitored for the presence of metastasis by serial bioluminescence imaging. For analysis, data from mice injected with STAG2 KO clones were combined and mice with WT STAG2 clones were combined. The two groups were compared by mixed-effects model with multiple comparisons. One mouse treated with sgSTAG2–4c5 clones died from disease prior to day 30.
In the second experiment, TC71 cells were transduced with the lentiviral plasmid encoding GFP-T2A-Luciferase and selected by fluorescence-activating cell sorting (FACS) for GFP expression. Cells were then transduced with lentiviral CRISPR Cas9 STAG2-targeting guides (sgSTAG2–1 and sgSTAG2–4) or a non-targeting guide (sgNT-1). Cells were selected for transduction by puromycin for five days and knockout of STAG2 was confirmed by western immunoblot before 500,000 cells of each condition were injected into the tail vein of NSG mice. Mice were then monitored for the presence of metastasis by serial bioluminescence imaging with the abdomen blocked to measure metastatic sites (lower extremities) distant from the initial target organs of the injection (liver and lungs). Five mice were injected with each sgSTAG2 treated condition and 10 mice were injected with sgNT-1 treated cells. One mouse injected with sgNT-1 and one mouse injected with sgSTAG2–4 treated cells died shortly after tail vein injection and was excluded from bioluminescence analysis. For analysis, data from mice injected with STAG2 KO clones were combined. The two groups were compared by mixed-effects model with multiple comparisons.
A third semi-orthotopic intramuscular injection mouse xenograft experiment was conducted to test the effect of STAG2 knockout on metastasis. Clonally selected A673 cells (NT1c4, sgSTAG2#1-c6, sgSTAG2#4-c5) were transduced with the lentiviral plasmid encoding GFP-T2A-Luciferase and selected by fluorescence-activating cell sorting (FACS) for GFP expression. Cells were washed and resuspended in 1:1 mixture of Matrigel/PBS and injected directly into the hind limb cranial thigh muscle away from sciatic nerve at a concentration of 50,000 cells per mouse in 50 μl Matrigel solution. We used 8 NSG mice per group and monitored for disease progression and the presence of metastasis by serial bioluminescence imaging of whole body as well as the upper thoracic cavity after blocking the primary tumor site. At the conclusion of the experiment, lungs from all recipient mice were extracted and BLI imaging was performed ex vivo tissue culture plates.
In a separate experiment, TC32 cells were transduced with the lentiviral plasmid encoding GFP-T2A-Luciferase and selected by fluorescence-activating cell sorting (FACS) for GFP expression. Cells were then transduced with lentiviral CRISPR Cas9 TP53-targeting guides (sgTP53–1 and sgTP53–5) or a non-targeting guide (sgNT-1). These polyclonal cells were selected for transduction by puromycin for five days and knockout of TP53 was confirmed by western immunoblot before 250,000 cells of each condition were injected into the tail vein of NSG mice (n=5 per group). Mice were then monitored for overall disease progression and the presence of metastasis by serial bioluminescence imaging of whole body and with the abdomen blocked to measure metastatic colonization of the lower extremities.
To assess the effect of POU3F2 and NR2F1 on the metastatic potential of STAG2 KO cells, we used one of the clonally selected STAG2 KO cells (sgSTAG2#1-c6) and transduced it with the lentiviral plasmid encoding GFP-T2A-Luciferase and selected by fluorescence-activating cell sorting (FACS) for GFP expression. Next, we transduced these cells with lentiviral CRISPR Cas9 targeting guides against (sgPOU3F2-A, sgNR2F1-A or LACZ). Successfully transduced cells were selected with 1 μ/ml of puromycin for 3 days and knockout of each gene validated by western blot. Cells were injected via the intramuscular route as described above at a concentration of 50,000 cells per mouse (n=8 per group) and disease progression was monitored by serial bioluminescence imaging as described above.
DepMap data analysis
CERES Gene effect scores for cohesin genes STAG2, RAD21, SMC1A and SMC3 were downloaded from the CRISPR (Avana) Public Depmap v20Q3 portal https://depmap.org/portal/download/ for the 16 Ewing sarcoma and 773 other lineage cell lines in the database. A lower CERES score indicates a higher likelihood that the gene is essential for the cell line. A CERES score of 0 indicates not essentiality, while a score of −1 is comparable with the median of all pan-essential genes, i.e., the genes which are essential for every cell line. A CERES score below −0.5 is estimated as indicative of gene essentiality for the cell line, while a score above 0.5 is estimated as indicative of tumor suppressor effect. For each cohesin gene, the essentiality effect on Ewing sarcoma cells vs. all other cell lines as background, was visualized on hockey plots.
Genome scale CRISPR/Cas9 screen
The genome-scale CRISPR-Cas9 screening was performed using the Broad Institute’s Avana library. The isogenic A673 cell lines with either STAG2 knockout or non-targeting controls were screened with the Avana library, containing 73,372 guides and an average of 4 guides per gene.(Doench et al., 2016; Meyers et al., 2017) The screens were conducted in a pooled experiment as previously described.(Aguirre et al., 2016; Meyers et al., 2017).
The isogenic A673 cell lines with either STAG2 knockout or non-targeting controls were screened in replicate (n=4) with the Avana CRISPR library after pooled transduction into a population of cells. The essential genes in the isogenic lines were identified using MAGeCK.(Li et al., 2014) The Avana guides were filtered to include guides that only map to one location in the genome as MAGeCK does not model the cutting effect of guides that map to multiple locations in the reference genome. Next, the guides were filtered to only include guides that had predicted efficacy scores of >0.95 as modeled by CERES on a large collection of cancer cell lines in order to minimize off-target effects.(Meyers et al., 2017) Thus, the maximum-likelihood analysis of gene essentialities function from MAGeCK was run with 4 replicates of each isogenic line (A673.sgSTAG2–1c6, A673.sgSTAG2–4c5, A673.sgNT-1c4, A673.sgNT-2c3) and approximately 41,978 guides mapping to 17,146 genes. The MAGeCK maximum-likelihood analysis assigned a gene essentiality score for each isogenic line collapsing the replicates.
RNA-Seq analysis
The human reads were mapped to the GRCh37/hg19 human genome using STAR v2.7.3 with standard parameters --outSAMtype BAM SortedByCoordinate --outSAMunmapped Within --outSAMattributes NH HI NM MD AS XS --outReadsUnmapped Fastx --outSAMstrandField intronMotif --quantMode TranscriptomeSAM GeneCounts --quantTranscriptomeBan IndelSoftclipSingleend. Quality control for the mapped reads and for replicate reproducibility was performed using SARTools v1.7.3 (Varet et al., 2016). Gene level reads were summarized by counting the reads that overlapped the GRCh37/hg19 annotated gene exons, by using the featureCounts v1.6.3 method implemented in the Subread v2.0.0 package (http://subread.sourceforge.net)(Liao et al., 2014). Gene counts were normalized and used to quantify differential genes between the experimental and control conditions using DESeq2 v1.24.0. Genes with ≥10 reads across at least 3 samples (A673 data) and across at least 2 samples (TC71 data) were annotated as “expressed”. Differentiability for the expressed genes was assessed with DESeq2 based on the robust shrunken log2 fold change scores and the approximate posterior estimation for GLM coefficients (apeglm v1.6, Zhu et al., (2019)) method for effect size. The cutoffs for differentially expressed genes were |log2(fold change)|≥1.5 and adjusted P-value≤0.10. Heatmap for transcriptional data visualization was created by using the Morpheus software platform (https://software.broadinstitute.org/morpheus/) based on the log2(1+ DESeq2 normalized counts) data. The RNA-Seq data for this study are available for download from the Gene Expression Omnibus (GEO) repository (GSE 116495) upon manuscript publication.
ChIP-Seq data analysis
Quality control tests for unmapped sequences were performed based on the FastQC v.0.11.5 software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). All of the ChIP-Seq data sets were aligned to the GRCh37/hg19 human genes and Drosophila Melanogaster dm6 genes using bowtie2–2.3.5 with the standard options. PCR duplicates were removed with the Picard v2.18.2 MarkDuplicates tool (Li and Durbin, 2010). The Active Motif Spike-in Normalization protocol was then applied to each hg19 sample by multiplying the human tag counts with the normalization factors derived from the uniquely mapped Drosophila reads as ratios between the sample with the lowest dm6 counts vs. the dm6 counts for that sample.
For each mark the reads mapped on the STAG2 wild-type clones A673.sgNT-1c4 and A673.sgNT-2c3 were merged and labeled as “STAG2 WT”. Similarly, for each mark the reads mapped on the STAG2 knockout clones A673.sgSTAG2–1c6, A673.sgSTAG2–4c5 were merged and labeled as “STAG2 KO” with the exception of H3K27me3 for which only the A673.sgSTAG2–1c6 clone is available and labeled as “STAG2 KO”.
The dm6 normalization factors for individual clones and for the merged clones were very close to 1. The mapped reads for individual and for merged clones were normalized in units of Reads Per Kilobase per Million (RPKM or rpm/bp) and coverage tracks for the RPKM signal were created as bigwig files for bins of size 20 base pairs by using the bamCoverage tool available in deepTools v2.5.3. (Ramirez et al., 2016).
Peak calling was performed against input controls using the model-based MACS2 v2.1.1.20160309 software (Zhang et al., 2008), with the cut-off FDR ≤ 0.01. Narrow peaks were identified for all marks except H3K27me3 for which MACS2 was called with the broad peak option. Area under Curve (AUC) RPKM normalized signal across genomic regions was computed with the bwtool software (Pohl and Beato, 2014). Peaks with low area under curve coverage (< 300 RPKM) were disregarded and the ENCODE black-listed regions for hg19 (available at https://www.encodeproject.org/annotations/ENCSR636HFF/) were removed from each set of peak regions. Quality control tests for the mapped reads were performed by using the ChIPQC library available from Bioconductor v3.6 (Carroll et al., 2014). The distances between replicates for STAG2 WT and STAG2 KO clones were estimated based on the multiBamSummary function available from the deepTools v2.5.3. (Ramirez et al., 2016) and visualized on correlation heatmaps and PCA plots. The peaks were annotated with the closest hg19 genes by using the annotatePeaks function available in the Homer v4.11 package (Heinz et al., 2010) and the GREAT annotation platform (McLean et al., 2010).
ATAC-Seq
ATAC-Seq data were collected using paired-end 50 bp reads from HiSeq, Illumina at the Center for Cancer Genome Discovery at the Dana-Farber Cancer Institute (2015). Quality control tests for unmapped reads were performed based on the FastQC v.0.11.5 software (Babraham Bioinformatics, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). All of the ATAC-Seq data sets were aligned to the GRCh37/hg19 human genes using bwa v0.7.17 (https://github.com/lh3/bwa) with the mem -M options (Li and Durbin, 2010). The mitochondrial reads (chr M) were removed with samtools after alignment. PCR duplicates were removed with Picard v2.18.2 MarkDuplicates tool. The reads mapped on the STAG2 wild-type clones were merged and labeled “STAG2 WT”, and similarly, the reads mapped on the STAG2 knockout clones were merged and labeled “STAG2 KO”. The mapped reads were normalized in units of Reads Per Kilobase per Million (RPKM or rpm/bp) and coverage tracks for the RPKM signal were created as bigwig files for bins of size 20 base pairs by using the bamCoverage tool available in deepTools v2.5.3 (Ramirez et al., 2016). Model-based peaks were identified using model-based MACS2 v2.1.1.20160309 software (Zhang et al., 2008), with the cut-off FDR ≤ 0.01. The ATAC-Seq data for this study is available for download from the Gene Expression Omnibus (GEO) repository (GSE 116495) upon manuscript publication.
ChIP-Seq and ATAC-Seq visualization and analysis
ChIP-Seq binding peaks and normalized binding signal were visualized on the Integrative Genomic Viewer (IGV) v2.4.0 platform.(Thorvaldsdottir et al., 2013) Gene promoter regions were defined as the +/− 3 kb intervals around the hg19 gene transcription start site (TSS). Enhancer regions were required to exist outside of the promoter regions defined as +/− 3 kb from a TSS. The BEDTools v2.27 (Quinlan and Hall, 2010) was used to perform various genomic region analyses (sorting, intersection, merging). Heatmaps of AUC ChIP-Seq normalized signal occupancy on genomic regions were created using the computeMatrix and the plotHeatmap tools available in the deepTools v2.5.3 suite. The plotProfile tool from deepTools v2.5.3 was used to create metaplots based on the average normalized scores across genomic regions. Differential genome-wide mark binding in STAG2 KO vs. STAG2 WT clones was quantified based on the unpaired t-test with Welch correction (cutoff P-value ≤ 0.05) for the area under curve (AUC) RPKM normalized signal across the genome-wide regions in the two conditions. For a specific genomic region, the STAG2 KO vs. STAG2 WT changes in signal occupancy were classified as “increase”, “decrease” or “not significant” based on the absolute cut-off 1.5 for the delta area under curve scores.
Defining Cohesin clusters
SA2 binding peak regions not overlapping with SA1 peaks were labeled “SA2-only”. Overlapping peak regions for both SA1 and SA2 were labeled “Common”. SA2-only regions bound by H3K27me3 broad peaks were labeled “SA2/H3K27me3”. SA2-only regions bound by H3K27ac peaks (promoter or enhancer sites) were labeled “SA2/H3K27ac-Promoters” and “SA2/H3K27ac-Enhancers”, respectively. SA2-only regions without H3K27me3 or H3K27ac overlapping peaks were labeled “SA2/others”.
Motif enrichment analysis
The motif enrichment analysis was performed for genomic regions and gene promoters using HOMER v4.11 platform (Heinz et al., 2010) with the significance cutoff P < 0.01. For motif discovery in input genomic regions, HOMER v4.11 used a background control of 100,000 random sequences that matched the GC-content of the queried input sequences. The random sequences were selected from HOMER’s own pre-built internal repository of the collection of human (hg19) sequences of predefined lengths (8,10,12,..) that were extracted from the TSS (+/−50 kb) regions. HOMER removes the bias introduced by lower-order oligo sequences through an additional auto-normalization step, by correcting any imbalances between the input target regions and background sequences in 1-mers, 2-mers and 3-mers. This auto-normalization procedure attempts to remove some of the sequence bias associated with certain genomic regions and the noise that may have been introduced by experimental sequencing.
Generation of EWS/FLI1 gene signatures
A compendium of 33 gene sets: 15 EWS/FLI1 gene signatures in A673 cells and 18 EWS/FLI1 signatures in various other contexts was created based on the following resources:
The A673 IC-EwS on single cell transcriptomics data (Aynaud et al., 2020).
Five A673 shEWS/FLI1 transcriptomic signatures: RNA-Seq Lessnick-Pais (2016) GSE53066, Lessnick-Sarkar(2012) PRJNA176544, Tomazou(2015) RNA-Seq http://www.medical-epigenomics.org/papers/tomazou2015, Delattre-Kovar(2017) GSE73092) and Affymetrix Lessnick-Smith(2006) E-GEOD-4560. Each of the RNA-Seq data sets was processed separately based on the RNA-Seq pipeline employed for our study, and the Affy expression data was analyzed by using the limma eBayes package with standard cutoffs (|log2(fold change)|≥1.5, adjusted P≤0.1).
Consensus A673 RNA of 590 EWS/FLI1 activated, respectively 1,392 repressed gene signatures, based on the rule “3 out of 5” A673 shEWS/FLI1 transcriptomic signatures.
A DNA A673 consensus EWS/FLI1 gene target signature based on the gene targets for the EWS/FLI1 bound regions in our “high coverage” A673 EWS/FLI1 ChIP-Seq data which overlap with A673 FLI1 peaks identified in two external studies (Riggi et al., 2014; Tomazou et al., 2015).
Consensus A673 RNA/DNA EWS/FLI1 of 325 activated and 537 repressed by intersecting the Consensus A673 RNA and Consensus A673 ChIP-Seq EWS/FLI1 gene targets.
the collection of 16 EWS/FLI1 and Ewing sarcoma signatures in various contexts (not A673 cells) available from the MSigDB v7.1 c2 database.
two core MSigDB Ewing sarcoma Hancock-Lessnick (2008) signatures.
GSEA analysis of transcriptome and proteomic data
GSEA v4.1 software (Subramanian et al., 2005) was used to identify functional associations of the molecular phenotypes induced by STAG2 loss with the compendia of EWS/FLI1 activated and repressed signatures on A673 cells merged with the MSigDB v7.1 collection c2 of curated pathways and experimental gene sets. The molecular phenotypes induced by STAG2 KO vs. STAG2 wild-type were measured by (i) RNA-Seq expression of the A673 and TC71 cells, (ii) proteome mass spectrometry of A673 cells, (iii) RNA-Seq expression of Ewing sarcoma tumors with loss-of -unction mutations in STAG2 compared to tumors expressing wild type STAG2.(Crompton et al., 2014) (iv) RNA-Seq expression of Ewing sarcoma tumors with low STAG2 expression vs. high STAG2 expression identified in ~ top 12% tumors in two transcriptome data sets with no information about STAG2 mutation status: (Savola et al., 2011) GSE17679, 42 Ewing sarcoma tumors, and Postel-Vinay (2012) GSE34620, 117 Ewing sarcoma tumors.
For each of these datasets, the hg19 genes were ranked based on the expression fold change in STAG2 KO (low) vs. STAG2 wild type (high) phenotypes. The goal of GSEA was to identify the gene sets that are distributed at the top or at the bottom of the ranked list of genes based on the Kolmogorov-Smirnov enrichment test. Gene sets with absolute Normalized Enrichment Score ≥1.3, a nominal P ≤ 0.05 and an FDR ≤ 0.25 for the Kolmogorov-Smirnov test were considered significant hits. The results were visualized on volcano plots for the Normalized Enrichment Score (NES) vs. −log10(P) and on GSEA plots.
Enricher and GO analysis
Overlapping enrichment analysis for the genes with significantly altered expression induced by STAG2 KO in A673 cells (|log2(fold change)| ≥1.5, adjusted P < 0.10), was performed against the MSigDB v7.1 collections c2 (canonical pathways and experimental gene sets) and c5 (Gene Ontology). Significant enrichments were quantified based on hypergeometric test (P-value ≤0.05, FDR ≤0.05) and size overlap ≥10.
HiChIP data analysis
HiChIP raw reads were aligned to hg19 human reference genome using HiC-Pro v2.10 (Servant et al., 2015). Each of the four replicate samples was sequenced to a depth > 150M reads. Moreover, each sample passed stringent quality control with a minimum of 23% of all reads mapping to intrachromosomal loci. High-confidence loop calls were inferred using hichipper (Lareau and Aryee, 2018) using the merged union of 368,178 SMC1A ChIP-Seq peaks from both the WT and STAG2 KO cell lines in our study into 144,863 genomic regions. Long range interactions spanning two anchor regions, termed DNA loops, were derived from linked paired-end reads that overlapped restriction fragments containing this consensus peak of possible loop anchors. In total, 1,836,186 interactions spanning pairs of 102,398 genomic loci were observed among the four samples. As a majority of these interactions represent background proximity ligation, we performed stringent filtering to identify putative biologically functional DNA loops.
We called a set of 56,219 loops naive to any additional genetic or epigenetic annotation that met stringent criteria. These loops contained at least four reads with paired-end tags (PETs) in two or more replicates and were statistically-significant at a distance-dependent P-value of 1% and FDR of 1% based on the per-loop measures from the loop proximity bias correction algorithm originally implemented in Mango (Phanstiel et al., 2015). The set of 56,219 stringently filtered loops served as a basis for differential loop calling between the wild-type and STAG2 KO mutant cell lines. To identify loops with significant changes in contact coverage (either decrease or increase) in A673 with STAG2 KO compared to STAG2 WT cells, differential loops were called at using the diffloop (Lareau et al., 2018) package with the cutoffs |abs(fold change contact coverage)|≥2 and P-value ≤0.05. Loop edges were annotated for their overlap with gene promoter regions and with ChIP-Seq binding sites for H3K27ac enhancers and EWS/FLI1. Loop were annotated for TSS+/−5kb promoter region landing for hg19 genes, and separately for expressed hg19 genes, in accordance with the 5kb resolution of our HiChIP data.
Enhancer-Promoter loops were defined as the significant loops connecting a H3K27ac enhancer binding site on one anchor and a landing gene promoter (TSS+/−5kb region) on the opposite anchor. Enhancers were identified in the H3K27ac A673 ChIP-Seq data in this study as the H3K27ac binding sites outside the TSS+/−3kb regions. Loop anchors were fully annotated with the hosted enhancer binding sites and gene targets (Table S1).
EWS/FLI1 status was assigned to the significant loops that host a EWS/FLI1 binding site (ChIP-Seq peak) on one anchor and a gene promoter (TSS+/−5kb) region on the opposite anchor. Thus, an EWS/FLI1 loop intermediates the “long interactions” between any of the EWS/FLI1 binding sites on one anchor to any of the gene promoter regions on the opposite anchor.
For the purpose of the EWS/FLI1 loop annotation, we defined a consensus of 11,519 ChIP-Seq EWS/FLI1 binding sites on A673 cells by intersecting the peaks identified in the A673 EWS/FLI1 ChIP-Seq high coverage data in this study (MACS2, FDR ≤0.01) with the union of the EWS/FLI1 peaks identified in two published A673 EWS/FLI1 ChIP-Seq studies (Riggi et al., 2014; Tomazou et al., 2015). Based on the consensus EWS/FLI1 peaks we identified 8,957 EWS/FLI1 loops. The anchors of the EWS/FLI1 loops were annotated with the EWS/FLI1 ChIP-Seq binding sites and gene promoter landing regions and with the STAG2 KO vs. WT differential status information for the host loop and for the gene. The fully annotated collection of all 23,580 EWS/FLI1 binding site – promoter long interactions harbored by the 8,957 EWS/FLI1 loops is available (Table S6) as a resource for exploratory data analyses. The median loop length was examined for association with the differential loop status (increase or decrease) induced by STAG2 KO for all loops, and separately for enhancer-promoter and EWS/FLI1 loops (unpaired t-test with Welch correction, P-value ≤0.10).
QUANTIFICATION AND STATISTICAL ANLAYSIS
Data are expressed as mean ± SEM or mean ± SD as indicated in the figure legends. Group size was determined on the basis of the results of preliminary experiments and no statistical method was used to predetermine sample size. The indicated sample size (n) represents biological replicates. Group allocation and outcome assessment were not performed in a blinded manner. All samples that met proper experimental conditions were included in the analysis. Details of statistical analysis used to determine significance are presented in the respective figure legends.
Supplementary Material
Table S1. Results of the SMC1A HiChIP data analysis, related to Figure 3.
Table S3. Transcriptional changes and chromatin modifications induced by STAG2 loss, related to Figure 4.
Table S4. EWS/FLI1 gene set enrichments associated with transcriptional changes induced by STAG2 loss, related to Figure 4.
Table S5. Top 100 MSigDB c2 gene sets enriched for the genes with altered expression (592 decreased, 392 increased) induced by STAG2 KO vs. WT in A673 cells, related to Figure 5.
Table S7. Top 100 MSigDB c5 gene sets enriched for the genes with altered expression (592 decreased, 392 increased) induced by STAG2 KO vs. WT in A673 cells, related to Figure 6.
Highlights.
Cohesin-SA2 occupies enhancer and PRC2 marked regulatory regions in Ewing
sarcoma
STAG2 depletion alters a subset of EWS/FLI1 anchored enhancer-promoter interactions
Loss of STAG2 modifies oncogenic and developmental transcriptional programs
STAG2 loss enhances the migratory and metastatic potential of Ewing sarcoma cells
ACKNOWLEDGMENTS
This work was funded by the NCI R01 CA204915, RO1 supplement 3R01 CA204915-03S1, St Jude’s Collaborative Research Consortium, Curing Kids’ Cancer, St. Baldrick’s Foundation (BDC,NVD), the NCI K08 CA188073 (BDC), Ewing Cancer Foundation of Canada (JNB), the C17 Childhood Cancer Canada Foundation (JNB). BKAS is supported by Department of Defense PRCRP Horizon Award (CA181249). SL is a Leukemia Lymphoma Society (LLS) fellow. DL is supported by TC32GM007753 from NIGMS. We thank Nick Barrowman at the Children’s Hospital of Eastern Ontario and Clement Ma at Dana-Farber Cancer Institute for their guidance on statistical analyses.
DECLARATION OF INTERESTS
None of the authors declare a conflict of interest for this project but report the following relationships:
NVD is a current employee of Genentech, Inc., a member of the Roche Group.
RAY is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics and Dewpoint Therapeutics.
MJA has financial interests in Monitor Biotechnologies (formerly known as Beacon Genomics)
KS receives grant funding as part of the DFCI/Novartis Drug Discovery Program, consults for and has stock options in Auron Therapeutics, consulted for AstraZeneca and Kronos Bio.
BDC receives research funding from Gradalis for an unrelated project and spouse was previous employed by Shire and Mersana and currently works for Acceleron.
Footnotes
Inclusion and diversity
One or more of the authors of this manuscript self-identifies as an underrepresented ethnic minority in science, self-identifies as a member of the LGBTQ+ community, and received support from a program designed to increase minority representation in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. (2016). Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer discovery 6, 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armentano M, Chou SJ, Tomassy GS, Leingartner A, O’Leary DD, and Studer M (2007). COUP-TFI regulates the balance of cortical patterning between frontal/motor and sensory areas. Nat Neurosci 10, 1277–1286. [DOI] [PubMed] [Google Scholar]
- Armentano M, Filosa A, Andolfi G, and Studer M (2006). COUP-TFI is required for the formation of commissural projections in the forebrain by regulating axonal growth. Development 133, 4151–4162. [DOI] [PubMed] [Google Scholar]
- Aynaud MM, Mirabeau O, Gruel N, Grossetete S, Boeva V, Durand S, Surdez D, Saulnier O, Zaidi S, Gribkova S, et al. (2020). Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep 30, 1767–1779 e1766. [DOI] [PubMed] [Google Scholar]
- Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, Lapi E, Castro-Giner F, Beltran S, Bayes M, et al. (2013). Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet 45, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti L, Cereda M, Monteverde L, Desai N, and Ciccarelli FD (2017). Synthetic lethal interaction between the tumour suppressor STAG2 and its paralog STAG1. Oncotarget 8, 37619–37632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, Jama R, Nip KM, Angeles A, Johnson F, et al. (2017). The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov 7, 54–71. [DOI] [PubMed] [Google Scholar]
- Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, et al. (2014). The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10, e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TS, Liang Z, Salama R, Stark R, and de Santiago I (2014). Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M, and Briscoe J (2003). Neural crest development is regulated by the transcription factor Sox9. Development 130, 5681–5693. [DOI] [PubMed] [Google Scholar]
- Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS, et al. (2014). The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 4, 1326–1341. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Gimenez-Llorente D, Kojic A, Rodriguez-Corsino M, Cuartero Y, Martin-Serrano G, Gomez-Lopez G, Marti-Renom MA, and Losada A (2019). Specific Contributions of Cohesin-SA1 and Cohesin-SA2 to TADs and Polycomb Domains in Embryonic Stem Cells. Cell Rep 27, 3500–3510 e3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniloski Z, and Smith S (2017). Loss of Tumor Suppressor STAG2 Promotes Telomere Recombination and Extends the Replicative Lifespan of Normal Human Cells. Cancer Res 77, 5530–5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Diep J, Feng N, Ren L, Li B, Ooi YS, Wang X, Brulois KF, Yasukawa LL, Li X, et al. (2018). STAG2 deficiency induces interferon responses via cGAS-STING pathway and restricts virus infection. Nat Commun 9, 1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schujiers J, Lee TI, Zhao K, and Young RA (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Naggar AM, Veinotte CJ, Cheng H, Grunewald TG, Negri GL, Somasekharan SP, Corkery DP, Tirode F, Mathers J, Khan D, et al. (2015). Translational Activation of HIF1alpha by YB-1 Promotes Sarcoma Metastasis. Cancer Cell 27, 682–697. [DOI] [PubMed] [Google Scholar]
- Fane ME, Chhabra Y, Smith AG, and Sturm RA (2019). BRN2, a POUerful driver of melanoma phenotype switching and metastasis. Pigment Cell Melanoma Res 32, 9–24. [DOI] [PubMed] [Google Scholar]
- Franzetti GA, Laud-Duval K, van der Ent W, Brisac A, Irondelle M, Aubert S, Dirksen U, Bouvier C, de Pinieux G, Snaar-Jagalska E, et al. (2017). Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 36, 3505–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XL, Zheng M, Wang HF, Dai LL, Yu XH, Yang X, Pang X, Li L, Zhang M, Wang SS, et al. (2019). NR2F1 contributes to cancer cell dormancy, invasion and metastasis of salivary adenoid cystic carcinoma by activating CXCL12/CXCR4 pathway. BMC Cancer 19, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, Naber SP, and Weinberg RA (2005). The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet 37, 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haering CH, Farcas AM, Arumugam P, Metson J, and Nasmyth K (2008). The cohesin ring concatenates sister DNA molecules. Nature 454, 297–301. [DOI] [PubMed] [Google Scholar]
- Haldi M, Ton C, Seng WL, and McGrath P (2006). Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis 9, 139–151. [DOI] [PubMed] [Google Scholar]
- Hansen AS, Cattoglio C, Darzacq X, and Tjian R (2018). Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T, Surdez D, Rantala JK, Haapa-Paananen S, Ban J, Kauer M, Tomazou E, Fey V, Alonso J, Kovar H, et al. (2017). High-throughput RNAi screen in Ewing sarcoma cells identifies leucine rich repeats and WD repeat domain containing 1 (LRWD1) as a regulator of EWS-FLI1 driven cell viability. Gene 596, 137–146. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, He X, Liu J, Duan Z, Kim T, Gerard J, Kim B, Pillai MM, Lane WS, Noble WS, et al. (2019). Systematic proteomics of endogenous human cohesin reveals an interaction with diverse splicing factors and RNA-binding proteins required for mitotic progression. J Biol Chem 294, 8760–8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic A, Cuadrado A, De Koninck M, Gimenez-Llorente D, Rodriguez-Corsino M, Gomez-Lopez G, Le Dily F, Marti-Renom MA, and Losada A (2018). Distinct roles of cohesin-SA1 and cohesin-SA2 in 3D chromosome organization. Nat Struct Mol Biol 25, 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, et al. (2013). Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 45, 1232–1237. [DOI] [PubMed] [Google Scholar]
- Kraft K, Yost KE, Murphy S, Magg A, Long Y, Corces MR, Granja JM, Mundlos S, Cech TR, Boettiger A, and Chang HY (2020). Polycomb-mediated Genome Architecture Enables Long-range Spreading of H3K27 methylation. bioRxiv, 2020.2007.2027.223438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, and Crews CM (2017). Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 16, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau CA, and Aryee MJ (2018). hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat Methods 15, 155–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau CA, Aryee MJ, and Berger B (2018). diffloop: a computational framework for identifying and analyzing differential DNA loops from sequencing data. Bioinformatics 34, 672–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LM, Seftor EA, Bonde G, Cornell RA, and Hendrix MJ (2005). The fate of human malignant melanoma cells transplanted into zebrafish embryos: assessment of migration and cell division in the absence of tumor formation. Dev Dyn 233, 1560–1570. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu Y, Xu H, Van der Jeught K, Li Y, Liu S, Zhang L, Fang Y, Zhang X, Radovich M, Schneider BP, et al. (2018). Somatic mutation of the cohesin complex subunit confers therapeutic vulnerabilities in cancer. J Clin Invest 128, 2951–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Losada A, Yokochi T, Kobayashi R, and Hirano T (2000). Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J Cell Biol 150, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoseviciute M, Gavriouchkina D, Williams RM, Hochgreb-Hagele T, Senanayake U, Chong-Morrison V, Thongjuea S, Repapi E, Mead A, and Sauka-Spengler T (2018). From Pioneer to Repressor: Bimodal foxd3 Activity Dynamically Remodels Neural Crest Regulatory Landscape In Vivo. Dev Cell 47, 608–628 e606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisenberg C, Pinder SI, Hopkins SR, Wooller SK, Benstead-Hume G, Pearl FMG, Jeggo PA, and Downs JA (2019). Repression of Transcription at DNA Breaks Requires Cohesin throughout Interphase and Prevents Genome Instability. Mol Cell 73, 212–223 e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal G, Stevers M, Goode B, Ashworth A, and Solomon DA (2019). A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin-mutant cancers. Nat Commun 10, 1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, and Chang HY (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 13, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K (2002). Segregating sister genomes: the molecular biology of chromosome separation. Science 297, 559–565. [DOI] [PubMed] [Google Scholar]
- Ochi Y, Kon A, Sakata T, Nakagawa MM, Nakazawa N, Kakuta M, Kataoka K, Koseki H, Nakayama M, Morishita D, et al. (2020). Combined Cohesin-RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov 10, 836–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson HM, and Nechiporuk AV (2018). Using Zebrafish to Study Collective Cell Migration in Development and Disease. Front Cell Dev Biol 6, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Heidari N, and Snyder MP (2015). Mango: a bias-correcting ChIA-PET analysis pipeline. Bioinformatics 31, 3092–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl A, and Beato M (2014). bwtool: a tool for bigWig files. Bioinformatics 30, 1618–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, Gusev A, Korthauer KD, Severson TM, Ha G, et al. (2020). Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet 52, 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postel-Vinay S, Veron AS, Tirode F, Pierron G, Reynaud S, Kovar H, Oberlin O, Lapouble E, Ballet S, Lucchesi C, et al. (2012). Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet 44, 323–327. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JDP, Feldmann A, Hernandez-Rodriguez B, Diaz N, Brown JM, Fursova NA, Blackledge NP, Prathapan P, Dobrinic P, Huseyin MK, et al. (2020). Cohesin Disrupts Polycomb-Dependent Chromosome Interactions in Embryonic Stem Cells. Cell Rep 30, 820–835 e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, et al. (2014). EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 26, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Perez L, Surdez D, Brunet E, Delattre O, and Grunewald TGP (2019). STAG Mutations in Cancer. Trends Cancer 5, 506–520. [DOI] [PubMed] [Google Scholar]
- Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, Beckerle MC, Sharma S, and Lessnick SL (2014). Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res 20, 4584–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savola S, Klami A, Myllykangas S, Manara C, Scotlandi K, Picci P, Knuutila S, and Vakkila J (2011). High Expression of Complement Component 5 (C5) at Tumor Site Associates with Superior Survival in Ewing’s Sarcoma Family of Tumour Patients. ISRN Oncol 2011, 168712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, and Lessnick SL (2006). Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 9, 405–416. [DOI] [PubMed] [Google Scholar]
- Solomon DA, Kim JS, Bondaruk J, Shariat SF, Wang ZF, Elkahloun AG, Ozawa T, Gerard J, Zhuang D, Zhang S, et al. (2013). Frequent truncating mutations of STAG2 in bladder cancer. Nat Genet 45, 1428–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, et al. (2011). Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333, 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, Ekpin E, George A, Zheng Y, Lam HM, et al. (2015). NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat Commun 6, 6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete-Lalami S, Rusch M, et al. (2014). Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 4, 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomazou EM, Sheffield NC, Schmidl C, Schuster M, Schonegger A, Datlinger P, Kubicek S, Bock C, and Kovar H (2015). Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep 10, 1082–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Kobi D, Ennen M, Langer D, Le Gras S, Ye T, and Davidson I (2015). A Brn2-Zic1 axis specifies the neuronal fate of retinoic-acid-treated embryonic stem cells. J Cell Sci 128, 2303–2318. [DOI] [PubMed] [Google Scholar]
- van der Lelij P, Lieb S, Jude J, Wutz G, Santos CP, Falkenberg K, Schlattl A, Ban J, Schwentner R, Hoffmann T, et al. (2017). Synthetic lethality between the cohesin subunits STAG1 and STAG2 in diverse cancer contexts. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varet H, Brillet-Gueguen L, Coppee JY, and Dillies MA (2016). SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS One 11, e0157022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL, et al. (2017). YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588 e1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. (2008). Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801. [DOI] [PubMed] [Google Scholar]
- Westerfield M (2000). The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). 4th ed., Univ. of Oregon Press, Eugene. [Google Scholar]
- Yao Z, Mich JK, Ku S, Menon V, Krostag AR, Martinez RA, Furchtgott L, Mulholland H, Bort S, Fuqua MA, et al. (2017). A Single-Cell Roadmap of Lineage Bifurcation in Human ESC Models of Embryonic Brain Development. Cell Stem Cell 20, 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Jorapur A, Shain AH, Lang UE, Torres R, Zhang Y, McNeal AS, Botton T, Lin J, Donne M, et al. (2018). Bi-allelic Loss of CDKN2A Initiates Melanoma Invasion via BRN2 Activation. Cancer Cell 34, 56–68 e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jeong M, Huang X, Wang XQ, Wang X, Zhou W, Shamim MS, Gore H, Himadewi P, Liu Y, et al. (2020). Large DNA Methylation Nadirs Anchor Chromatin Loops Maintaining Hematopoietic Stem Cell Identity. Mol Cell 78, 506–521 e506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Qiu Y, Pereira FA, Crair MC, Tsai SY, and Tsai MJ (1999). The nuclear orphan receptor COUP-TFI is required for differentiation of subplate neurons and guidance of thalamocortical axons. Neuron 24, 847–859. [DOI] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Results of the SMC1A HiChIP data analysis, related to Figure 3.
Table S3. Transcriptional changes and chromatin modifications induced by STAG2 loss, related to Figure 4.
Table S4. EWS/FLI1 gene set enrichments associated with transcriptional changes induced by STAG2 loss, related to Figure 4.
Table S5. Top 100 MSigDB c2 gene sets enriched for the genes with altered expression (592 decreased, 392 increased) induced by STAG2 KO vs. WT in A673 cells, related to Figure 5.
Table S7. Top 100 MSigDB c5 gene sets enriched for the genes with altered expression (592 decreased, 392 increased) induced by STAG2 KO vs. WT in A673 cells, related to Figure 6.
Data Availability Statement
The data from this study including all ChIP-Seq, RNA-Seq, ATAC-Seq and HiChIP was uploaded at GEO under the accession number GSE116495. The proteomics data from this study is available at the following link http://massive.ucsd.eduMSV000082954. Ewing sarcoma tumor RNA-Seq data previously reported by (Crompton et al., 2014) is available at dbGaP with the following accession number dbGaP: phs000804.v1.p1. Ewing sarcoma ChIP-Seq previously reported by (Riggi et al., 2014) is available at GEO with the following accession number GEO: GSE61953. Ewing sarcoma ChIP-Seq and RNA-seq data previously reported by (Tomazou et al., 2015) is available at the following link http://tomazou2015.computational-epigenetics.org. Ewing sarcoma ChIP-Seq data previously reported by (Kojic et al., 2018) is available at GEO with the following accession number GEO: GSE101921. Ewing sarcoma ChIP-Seq data previously reported at ENCODE: ENCSR000DZP is available at GEO under the accession number GEO: GSM935376. Ewing sarcoma tumor Affymetrix arrays data previously reported by (Postel-Vinay et al., 2012) available at GEO with the following accession number GEO: GSE34620. Ewing sarcoma tumor Affymetrix arrays data previously reported by (Savola et al., 2011) is available at GEO with the following accession number GEO: GSE17618. Ewing sarcoma RNA-Seq data previously reported by (Smith et al., 2006) is available at GEO with the following accession number GEO: GSE53066. Ewing sarcoma RNA-Seq data previously reported by (Sankar et al., 2014) is available at NCBI with the following accession number NCBI: PRJNA176544. Ewing sarcoma Affymetrix arrays data previously reported by (Smith et al., 2006) is available at Array express with the following accession number Array Express: E-GEOD-4560. Ewing sarcoma RNA-Seq data previously reported by (He et al., 2017) is available at GEO with the following accession number GEO: GSE73092. Ewing sarcoma IC-EwS gene signature previously reported by (Aynaud, 2020) is available at the following link https://www.sciencedirect.com/science/article/pii/S2211124720300747?via=ihub.