

Abstract
Fibroblast growth factor receptor (FGFR) is a promising anticancer target. Currently, most FGFR inhibitors lack sufficient selectivity and have nonnegligible activity against kinase insert domain receptor (KDR), limiting their feasibility due to the serious side effects. Notably, compensatory activation occurs among FGFR1–4, suggesting the urgent need to develop selective pan-FGFR1–4 inhibitors. Here, we explored the antitumor activity of DW14383, a novel irreversible FGFR1–4 inhibitor. DW14383 exhibited equivalently high potent inhibition against FGFR1, 2, 3 and 4, with IC50 values of less than 0.3, 1.1, less than 0.3, and 0.5 nmol/L, respectively. It is a selective FGFR inhibitor, exhibiting more than 1100-fold selectivity for FGFR1 over recombinant KDR, making it one of the most selective FGFR inhibitors over KDR described to date. Furthermore, DW14383 significantly inhibited cellular FGFR1–4 signaling, inducing G1/S cell cycle arrest, which in turn antagonized FGFR-dependent tumor cell proliferation. In contrast, DW14383 had no obvious antiproliferative effect against cancer cell lines without FGFR aberration, further confirming its selectivity against FGFR. In representative FGFR-dependent xenograft models, DW14383 oral administration substantially suppressed tumor growth by simultaneously inhibiting tumor proliferation and angiogenesis via inhibiting FGFR signaling. In summary, DW14383 is a promising selective irreversible pan-FGFR inhibitor with pan-tumor spectrum potential in FGFR1–4 aberrant cancers, which has the potential to overcome compensatory activation among FGFR1–4.
Keywords: receptor tyrosine kinase, FGFR inhibitor, DW14383, antitumor activity
Introduction
Fibroblast growth factor receptors (FGFRs) constitute a class of pivotal mediators of a variety of vital physiological processes, including wound repair, embryonic development, wound healing, cell differentiation, proliferation, and migration [1–4]. Alterations in FGFR family signaling have been described in various tumor types [5, 6]. FGFR1 amplification is found in breast tumors, lung cancers, oral squamous carcinoma, rhabdomyosarcoma, and ovarian and bladder cancers [7–13], and FGFR2 alterations have been identified in breast cancers, gastric cancers and endometrial adenocarcinomas [14–16]. Mutations in FGFR3 are known to exist in bladder cancers [17–19], and aberrant expression of FGF19/FGFR4 contributes to hepatocellular carcinoma (HCC) progression [20]. Clinical evidence has indicated that the dysregulation of FGFR is associated with resistance to anticancer agents, such as EGFR inhibitors, angiogenesis inhibitors, and endocrine therapy [3, 21–24]. Therefore, targeting FGFR is a hotspot in the cancer therapy field.
Several FGFR inhibitors have been under development, some of which are currently being tested in clinical trials [25]. The pan-FGFR inhibitor erdafitinib and FGFR1–3 inhibitor pemigatinib were recently approved by the U.S. Food and Drug Administration (FDA) for the treatment of metastatic and unresectable urothelial carcinoma (mUC) in 2019 [25, 26] and unresectable locally advanced or metastatic cholangiocarcinoma with FGFR2 fusion or other rearrangement in 2020 [27], respectively. Notably, most FGFR inhibitors are nonselective inhibitors with multitarget effects and, in particular, simultaneously inhibit kinase insert domain receptor (KDR). The adverse effects, such as serious hypertension induced by systemic KDR inhibition or other nonspecific-targeting-related toxicity, limit the maximum effectiveness of FGFR inhibition treatment for patients with FGFR-driven cancer [2, 3, 28, 29]. Even the two recently launched FGFR inhibitors, erdafitinib and pemigatinib, were reported to exhibit nonnegligible activity against KDR (36.8 nmol/L and 70 nmol/L, respectively) [26, 27]. Moreover, the compensatory activation among FGFR1–4 subtypes should not be overlooked [30]. However, most selective FGFR inhibitors, including the launched pemigatinib, inhibit FGFR1–3 to a similar extent due to the remarkably similar structure in their kinase domains, while the potency of these inhibitors against FGFR4 is comparatively low due to their distinct structure [31, 32]. These findings suggest the urgent need to develop pan-FGFR1–4 inhibitors. A conserved cysteine residue located in the rim of the P-loop can be covalently bound in all four FGFR paralogs [33–37], indicating the possibility of developing an irreversible inhibitor cotargeting FGFR1–4, which might also have the potential to achieve higher selectivity [38]. The irreversible pan-FGFR inhibitors TAS-120 and PRN1371, which selectively inhibit FGFR1–4 with equivalent potency, have been reported to be promising for the treatment of tumors with FGFR alterations in an early trial [39–42]. Moreover, the great success of other irreversible kinase inhibitors, such as the BTK inhibitor ibrutinib and the EGFR inhibitor osimertinib, is well known [43, 44]. These successful applications of covalent kinase inhibitors provide great inspiration for developing therapeutically relevant irreversible pan-FGFR inhibitors.
In this paper, we described a novel selective and irreversible inhibitor of FGFR1–4, DW14383, and focused on evaluating the FGFR-targeting anticancer activity of DW14383 both in vitro and in vivo. DW14383 exhibited equivalently high potent inhibition of FGFR1, 2, 3 and 4, showing more than 1100-fold selectivity for FGFR1 over recombinant KDR, making it one of the most selective FGFR inhibitors over KDR described to date. DW14383 strongly suppressed FGFR1–4 signaling, inducing G1/S cell cycle arrest and antagonizing FGFR-dependent cancer cell proliferation with subnanomolar to nanomolar half maximal inhibitory concentration (IC50) values. Moreover, oral administration of DW14383 substantially suppressed tumor growth in FGFR1-amplified NCI-H1581 and FGF19/FGFR4-driven Hep3B xenograft models by blocking FGFR signaling. These results indicate that DW14383 is a promising FGFR inhibitor for patients with FGFR-aberrant cancer.
Materials and methods
Compounds
DW14383 (Fig. 1) was synthesized in Professor Wen-hu Duan’s Laboratory at the Shanghai Institute of Materia Medica (chemical formula: C22H20BrN5O2S, Mw: 498.40). TAS-120, AZD4547, and BLU9931 were purchased from Selleck Chemicals (Houston, USA).
Fig. 1.
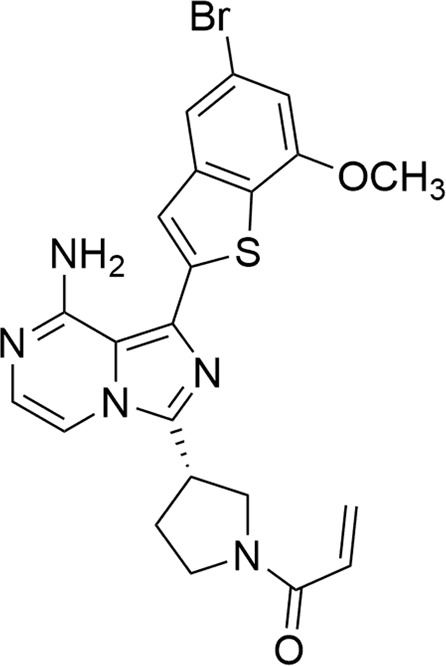
The chemical structure of DW14383.
ELISA kinase assays
Enzyme-linked immunosorbent assays (ELISAs) were carried out to determine the effects of DW14383 on the activities of various tyrosine kinases, as previously described [38]. The inhibition rate (%) was calculated using the following equation: [1 − (A490/A490 control)] × 100%. The IC50 values were calculated from the inhibition curves based on two separate experiments using a modified 4-parameter logistic model. If the inhibitory rate is higher than 50% at the minimum tested drug concentration, the conclusion is that the IC50 is lower than the tested minimum drug concentration. And if the inhibitory rate is lower than 50% at the maximum tested drug concentration, then the IC50 is higher than the tested maximum drug concentration.
Cell culture
All cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured according to the suppliers’ instructions. BaF3 cells were engineered to express human wild-type TEL-FGFR1 or TEL-KDR that stably expressed constitutively active FGFR1 or KDR. All the cell lines were authenticated via short tandem repeat analysis by Genesky Biopharma Technology (last tested in 2018).
Cell proliferation assay
Cell proliferation was determined using a Cell Counting Kit (CCK)-8 assay or sulforhodamine B (SRB) assay. On the same day, the cells were seeded in 96-well cell culture plates, and various concentrations of compounds were added before culturing for 72 h at 37 °C. The IC50 values were calculated by concentration-response curve fitting using the four-parameter method.
Cell cycle analysis
The effects of the added compounds on cell cycle progression and population distribution were determined by flow cytometry. A total of 2 × 105 cells were seeded in 6-well plates and treated with compounds at the indicated concentration or with a vehicle control. After 24 h, the cells were collected, fixed and stained with propidium iodide (10 μg/mL) for 30 min and then analyzed using a flow cytometer (FACSCalibur instrument; Becton, Dickinson & Co.). The data were plotted using CellQuest software (Becton, Dickinson & Co).
Western blot analysis
KG1, SNU16, BaF3/TEL-FGFR1, BaF3/TEL-KDR, UMUC14 and Hep3B cells were treated with the indicated doses of DW14383 for 2 h and then lysed in preheated 2% sodium dodecyl sulfate (SDS). The obtained protein lysates were resolved by SDS-PAGE. Primary antibodies against p-FGFR (Y653/654), FGFR1, FGFR2, FGFR3, FGFR4,p-FRS2α (Y196), FRS2α, p-ERK1/2(T202/Y204), ERK1/2, p-RB(S807/811), RB, PARP and c-Myc were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against GAPDH were purchased from KANGCHEN (Shanghai, China).
Irreversibility assay
The recovery of enzyme activity after DW14383 administration was evaluated by a rapid dilution experiment, as previously described [34]. A mixed solution of FGFR1 kinase (2 nmol/L) and excess DW14383 (100-fold IC50) was preincubated at ambient temperature for 30 min. This enzyme-inhibitor complex was then diluted 1:100 into reaction mixtures containing 262 μmol/L ATP and the substrate peptide (5-FAM-KKSRGDYMTMQIG-CONH2).
In vivo antitumor activity assay
For the NCI-H1581 and Hep3B xenograft model experiments, the indicated tumor cells (5 × 106 cells per mouse) were injected subcutaneously (s.c.) into the right flank of nude mice. After growing to 700–800 mm3, the well-developed tumors were cut into 1-mm3 fragments and transplanted s.c. into the right flank of female nude mice (4–6 weeks old) using a trocar. The mice were randomized when the tumor reached a mean volume of ~100–150 mm3 and were treated with DW14383 at the indicated doses via gavage once daily for 14 or 21 days. Tumor volume was measured twice per week with a microcaliper and quantified using the modified ellipsoidal formula (tumor volume = 1/2 (length × width2)). Tumor growth inhibition (TGI) values were determined on the final day of the study as 100 × {1 − [(VTreated Final day − VTreated Day 0)/(VControl Final day − VControl Day 0)]}. The data are presented as the means ± standard error of the mean (SEM), while statistical significance was calculated by unpaired Student’s t test (NS, P > 0.05; P < 0.05; P < 0.001). Animal procedures were approved by the Institutional Animal Care and Use Committee of the Shanghai Institute of Materia Medica.
Pharmacodynamics studies
To assess the pharmacodynamics of DW14383 in tumors, mice bearing established xenograft tumors were treated with the compound at 25 or 5 mg/kg for 3 days. Two hours following the last administration, the mice were humanely euthanized, and their tumors were resected. The tumors were snap-frozen in liquid nitrogen and then homogenized in 500 μL of protein extraction solution (radioimmunoprecipitation assay, RIPA). The tumor extracts were then subjected to Western blot analysis. The individual bands of p-FGFR1 and p-ERK were scanned and quantified using Gel Pro Analyzer software (Media Cybernetics, Inc.). The relative tyrosine phosphorylation of each sample at the indicated time points was then calculated, with the average value of vehicle-treated sample used set at 100%.
Results
DW14383 is a potent pan-FGFR inhibitor with high selectivity over KDR
DW14383 exhibited equipotent activity against FGFR1, 2, 3 and 4, with IC50 values of less than 0.3, 1.1, less than 0.3, and 0.5 nmol/L, respectively (Table 1). The widely validated selective FGFR1–3 inhibitor AZD4547 [45, 46] and irreversible FGFR1–4 inhibitor TAS-120 [47–49] were also tested as positive controls. DW14383 was more potent than TAS-120 and comparable to AZD4547 (Table 1). Testing against a small panel of representative human kinases, including the highly homologous kinase KDR, was also conducted. No obvious inhibitory effects were observed against other tested kinases, including the typical angiogenesis-regulating kinases (VEGFR1, KDR, PDGFRα, and PDGFRβ) (Table 1). Notably, DW14383 displayed more than 1100-fold selectivity for FGFR1 over recombinant KDR, making it one of the most selective FGFR inhibitors over KDR described to date. These data suggested that DW14383 was a selective pan-FGFR kinase inhibitor.
Table 1.
Kinase selectivity profile of compounds.
| Kinase | IC50 (nmol/L) | ||
|---|---|---|---|
| DW14383 | TAS-120 | AZD4547 | |
| FGFR1 | <0.3 | 3.9 ± 0.8 | 0.4 ± 0.1 |
| FGFR2 | 1.1 ± 0.3 | 4.2 ± 0.0 | 1.0 ± 0.5 |
| FGFR3 | <0.3 | 8.1 ± 0.9 | 2.3 ± 0.2 |
| FGFR4 | 0.5 ± 0.1 | 4.1 ± 1.2 | 49.8 ± 5.4 |
| KDR | 335.8 ± 25.2 | >1000 | 57.9 ± 22.1 |
| VEGFR-1 | >100 | >100 | >100 |
| PDGFR-α | >1000 | >100 | >1000 |
| PDGFR-β | >100 | >100 | >1000 |
| C-Kit | >100 | >100 | >100 |
| Flt-3 | >1000 | >100 | >1000 |
| EGFR | >1000 | >1000 | >1000 |
| ErbB2 | >1000 | >100 | >1000 |
| ErbB4 | >100 | >100 | >1000 |
| c-Src | >1000 | >1000 | >1000 |
| ABL | >1000 | >1000 | >1000 |
| EPH-A2 | >1000 | >1000 | >1000 |
| IGFIR | >100 | >100 | >1000 |
aValues are the mean ± SD or estimated values of two or more independent assays.
DW14383 is an irreversible FGFR inhibitor
A dilution method [50] was used to evaluate the irreversible kinase inhibition caused by DW14383 treatment. The known irreversible FGFR inhibitor TAS-120 was used as a positive control. FGFR1 kinase was used as a representative kinase. In this assay, FGFR1 kinase and excess DW14383 or TAS-120 were preincubated for 30 min. The enzyme-inhibitor mixture was then diluted 100-fold into a reaction buffer containing a high concentration of ATP and the indicated substrate peptide. Under this condition, the enzyme activity was continuously measured by evaluating the percentage of phosphorylated substrate. Similar to TAS-120, a significantly lower conversion was observed over 60 min when DW14383 was added to FGFR1, and the recovery curve of DW14383 was similar to that of the background group (Fig. 2). These results suggested that DW14383 covalently reacted with FGFR1 and irreversibly suppressed kinase activity.
Fig. 2. FGFR1 kinase activity is irreversibly inhibited by DW14383 and TAS-120.

The conversion rate describes the percentage of phosphorylated substrate peptide. The results of the peptide phosphorylation by FGFR after preincubation without enzyme (background), without inhibitor (control) or with the indicated inhibitor are shown.
DW14383 inhibits cellular FGFR signaling
To further examine the efficacy of DW14383 on FGFR signaling, we selected four typical human cancer cell lines harboring FGFR aberrations and analyzed the changes in the phosphorylation levels of FGFR and ERK after treatment with DW14383. The Western blot results indicated that in the FGFR-translocated KG1 cell line, DW14383 substantially inhibited p-FGFR1 and p-ERK, the major downstream signaling molecules (Fig. 3a). Similar results were found in SNU16 cells with FGFR2 amplification (Fig. 3b) and UMUC14 cells with FGFR3 mutation (Fig. 3c). Based on the significant FGFR4 inhibition activity of DW14383, we also investigated Hep3B cells with abnormal activation of the FGF19/FGFR4 signaling axis [50]. With FGF19 stimulation, the levels of p-FGFR4 and p-ERK in the Hep3B cells were increased. DW14383 treatment at 20 nmol/L completely inhibited FGFR4 phosphorylation and ERK phosphorylation (Fig. 3d).
Fig. 3. DW14383 inhibits FGFR signaling.

Phosphorylation of FGFRs and ERK after treatment with DW14383 for 2 h was evaluated by Western blotting of KG1 (a), SNU16 (b), UMUC14 (c) and Hep3B (d) cells.
These results indicated that DW14383 significantly inhibited FGFR signaling.
In vitro antitumor activity of compound DW14383 with selectivity
We then chose different FGFR-addicted cancer cell lines to further test the antiproliferative effect of DW14383 in vitro (Table 2). DW14383 treatment for 72 h dose-dependently blocked the proliferation of FGFR1-, FGFR2-(IC50 < 0.3 nmol/L) and FGFR3-dependent cell lines (IC50 = 20.1 ± 12.7 nmol/L). DW14383 also exhibited potent inhibition of FGF19/FGFR4-mediated Hep3B cell proliferation (Table 2). This high potency further supported the potential use of DW14383 for treating HCC cell lines harboring FGF19/FGFR4 aberrations. The positive controls AZD4547 and TAS-120 also showed a significant antiproliferation effect against FGFRs-driven cell lines.
Table 2.
Antiproliferative activity of compounds on FGFR-addicted cell lines.
| Cell line | Gene aberration | IC50 (nmol/L)a | ||
|---|---|---|---|---|
| DW14383 | TAS-120 | AZD4547 | ||
| KG1 | FGFROP2-FGFR1 | <0.3 | <0.3 | 7.2 ± 0.1 |
| NCI-H1581 | FGFR1 amplification | <0.3 | <0.3 | 11.6 ± 2.2 |
| SNU16 | FGFR2 amplification | <0.3 | <0.3 | 2.0 ± 0.5 |
| NCI-H716 | FGFR2 amplification | <0.3 | <0.3 | 1.7 ± 0.7 |
| UMUC14 | FGFR3 mutation | 20.1 ± 12.7 | / | 83.0 ± 35.7 |
| Hep3B | FGF19/FGFR4 amplification | 27.0 ± 1.0 | 125.2 ± 26.4 | / |
| BaF3/TEL-FGFR1 | TEL-FGFR1 fusion | <0.2 | / | 0.8 ± 0.2 |
| BaF3/TEL-KDR | TEL-KDR fusion | 481.4 ± 131.1 | / | / |
aValues are the mean ± SD or estimated values of two or more independent assays.
Our previous study demonstrated that FGFR inhibition exerts antiproliferative effects by arresting cells in the G1/S phase [51]. Thus, we investigated the influence of DW14383 on cell cycle arrest. The NCI-H1581 and SNU16 cell lines were used as representative FGFR cell lines. Consistently, DW14383 treatment-induced G1/S cell cycle arrest in both cell lines (Fig. 4a). We also found that the downregulation of the cell cycle regulator c-Myc is essential for the suppression of cell proliferation mediated by FGFR inhibition [51]. Consistently, DW14383 treatment dramatically downregulated c-Myc levels in representative FGFR-addicted H1581 cells (Fig. 4b). In addition, the phosphorylation of RB, the critical molecule for late G1 cell cycle progression [52], was also inhibited upon DW14383 treatment. However, no sub-G1 cell population or obvious PARP cleavage were observed (Fig. 4a, b), largely excluding the possibility that DW14383 induced apoptosis. These results suggested that DW14383 inhibited the proliferation of FGFR-addicted cells by arresting cells at G1/S phase rather than apoptosis.
Fig. 4. In vitro antitumor activity of compound DW14383 with selectivity.

a The influence of DW14383 on cell cycle arrest was evaluated in NCI-H1581 and SNU16 cells by FACS. The data are presented as the means ± SD. ***P < 0.001, using Student’s t test. b NCI-H1581 cells were treated with DW14383 at 0.4, 2 or 10 nmol for the indicated time (24 or 48 h) followed by immunoblot analysis. c The antiproliferative effect of DW14383 was measured by CCK-8 or SRB in a panel of cancer cell lines. d The antiproliferative effect of DW14383 was measured by CCK-8 assay in BaF3/TEL-FGFR1 and BaF3/TEL-KDR cells. e Phosphorylation of FGFR or KDR and indicated downstream signaling proteins after treatment with DW14383 for 2 h, was evaluated by Western blotting of BaF3/TEL-FGFR1 and BaF3/TEL-KDR cells.
Next, we performed cell proliferation assays using 12 other cell lines with low FGFR expression or low FGFR activation. We found that DW14383 exhibited almost 3000-fold less potency in these cell lines (Fig. 4c).
Next, to validate the cellular selectivity of DW14383 for FGFR over KDR, we used BaF3/TEL-FGFR1 and BaF3/TEL-KDR, which represent FGFR1- and KDR-driven cells, respectively. DW14383 significantly inhibited the BaF3/TEL-FGFR1 cell proliferation (IC50 < 0.2 nmol/L), while it had no obvious inhibitory effect on the proliferation of the BaF3/TEL-KDR cells (IC50 = 481.4 ± 131.1 nmol/L) (Table 2 and Fig. 4d). Consistently, FGFR1 signaling in the BaF3/TEL-FGFR1 cells was blocked at a lower concentration (1 nmol/L). However, even at 500 nmol/L, DW14383 had no obvious inhibitory effect against KDR signaling in the BaF3/TEL-KDR cells (Fig. 4e). These results further confirmed the selectivity of DW14383 for FGFR over KDR.
All these data strongly demonstrated that DW14383 dramatically and selectively inhibited the proliferation of FGFR-addicted cancer cell lines.
Antitumor effect of compound DW14383 in vivo
We then investigated the in vivo antitumor effect of DW14383 in FGFR-aberrant cell xenograft models. The indicated dose of DW14383 was administered to mice with FGFR1-amplified NCI-H1581-derived tumors or FGF19/FGFR4-driven Hep3B-derived tumors for 14 or 21 consecutive days. The irreversible pan-FGFR inhibitor TAS-120 was used as control. The well-validated FGFR1–3 inhibitor AZD4547 and FGFR4 inhibitor BLU9931 were used as positive system controls. Consistent with the reported data [45, 53, 54], a significant antitumor effect was observed (Fig. 5). In the NCI-H1581 model, DW14383 significantly suppressed tumor growth at a dose of 5 mg/kg (TGI = 62.6%) or 25 mg/kg (TGI = 84.6%). In addition, the efficacy of DW14383 at 25 mg/kg was more potent than TAS-120 at 50 mg/kg (Fig. 5a–c). No significant differences in body weights between the four groups were observed throughout the treatment period (Fig. 5d). We further tested the anticancer activity of DW14383 at a lower dose in a human HCC Hep3B xenograft model. DW14383 had a significant dose-dependent inhibitory effect on Hep3B transplantation tumor growth, resulting in a TGI of 97.8% at a dose of 5 mg/kg (Fig. 6a–c), suggesting that DW14383 exhibits promising potential for antitumor therapy to treat liver cancer. The efficacy of DW14383 at 5 mg/kg was more potent than that of TAS-120 at 5 mg/kg (Fig. 5a–c). In addition, the tumor-bearing mice did not exhibit significant weight loss during the administration of DW14383, indicating good tolerance (Fig. 6d). These results revealed that DW14383 effectively inhibited tumor growth in NCI-H1581 and Hep3B subcutaneous xenograft models.
Fig. 5. DW14383 significantly suppressed tumor growth in the NCI-H1581 xenografts.

a, b Tumor growth inhibition in the NCI-H1581 model treated with DW14383 at the indicated doses. c Weights of tumor tissues from the three groups on day 14 after treatment was initiated. d Body weight change during the treatment period. The data in panel a and c are presented as the means ±SEM. *P < 0.05, **P < 0.01, ***P < 0.001, using Student’s t test.
Fig. 6. DW14383 significantly suppressed tumor growth in the Hep3B xenografts.

a, b Inhibition of tumor growth in the Hep3B model treated with DW14383 at the indicated doses. c Weights of the tumor tissues from all three groups on day 14 at the end of treatment. d Body weight change in all three groups throughout the experiment. The data in panel a and c are presented as the means ±SEM. ***P<0.001, using Student’s t test.
DW14383 significantly suppressed FGFR signaling and inhibited tumor cell proliferation and angiogenesis in FGFR-dependent transplanted tumor tissues
Furthermore, the expression of tumor mitotic index (Ki67) and platelet endothelial cell adhesion molecules (CD31) after treatment was also measured. As shown in Fig. 7a, Ki67 levels and CD31-positive endothelial cells were reduced in a dose-dependent manner upon DW14383 administration in the Hep3B model, indicating that DW14383 significantly inhibited the growth of FGFR-dependent xenografts by exerting simultaneous effects on tumor cell growth and angiogenesis.
Fig. 7. Functional effects of DW14383 in vivo.
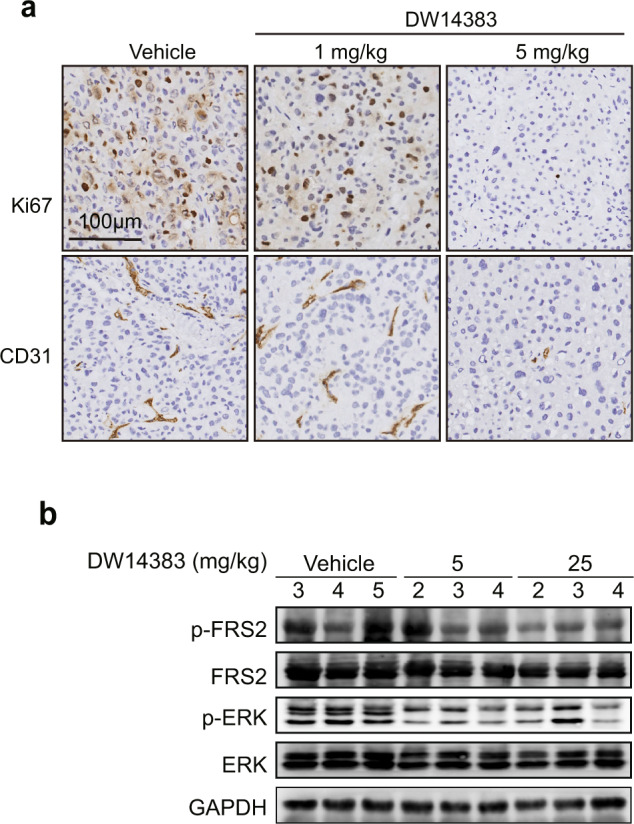
a Ki67 (above) and CD31 (bottom) levels in the Hep3B xenografts were evaluated by IHC. b The NCI-H1581 model was treated with DW14383 for 3 days, and the phosphorylation of FRS2 and ERK in the tumor tissue was evaluated by Western blotting.
To evaluate the in vivo FGFR inhibition by DW14383, NCI-H1581 tumors were harvested 2 h after 3 days administration of 25 or 5 mg/kg DW14383, and FGFR signaling in the tumors was examined. Activation of FGFR substrate 2α (FRS2α), a key adaptor protein largely specific to FGFR, was examined as a surrogate for p-FGFR in the FGFR1-amplified scenario [51]. In agreement with the suppressed tumor growth, marked reduction in the intratumoral phosphorylation of FRS2α and downstream ERK were observed upon oral administration of DW14383 (Fig. 7b).
Discussion
FGFR aberration has been shown to contribute to carcinogenesis, metastasis and the resistance to anticancer agents [55–57], making FGFR an attractive target. To date, most FGFR inhibitors have lacked sufficient selectivity, limiting their feasible use due to serious side effects associated with inhibiting activity against other kinases, especially VEGFR. In addition, it should not be neglected that compensatory activation occurs among FGFR1–4 subtypes [30], and the activity of most known FGFR inhibitors towards FGFR4 is relatively weak, suggesting the urgent need to develop selective pan-FGFR1–4 inhibitors.
Herein, we described an irreversible pan-FGFR inhibitor, DW14383, which potently inhibited FGFR1–4 kinase activities at subnanomolar to nanomolar IC50 levels. DW14383 blocked the phosphorylation of corresponding FGFR kinases and key downstream MEK/ERK activation [58] in the DW14383-responsive scenarios. In turn, in representative FGFR1-, FGFR2-, FGFR3-, and FGFR4-addicted cancer cell lines, DW14383 displayed superior antiproliferative activity in vitro via G1/S cell cycle arrest. Furthermore, the oral administration of DW14383 at the optimal dose suppressed tumor growth in FGFR1-amplified NCI-H1581 and FGF19/FGFR4-amplified Hep3B xenograft mice by suppressing FGFR signaling. Moreover, the in vitro and in vivo efficacy of DW14383 was more potent than that of TAS-120, an irreversible FGFR1–4 inhibitor, in clinical trials. Furthermore, IHC staining of Ki67 and CD31 suggested that DW14383 antagonizes FGFR-mediated tumor growth by inhibiting tumor proliferation and angiogenesis.
Notably, DW14383 can irreversibly bind to FGFR; thus, it exhibits excellent equivalent potent inhibition against FGFR1–4. Aberrances in FGFR family members (FGFR1–4) are observed in multiple types of human malignancies, particularly those with high frequency or lacking effective treatments [1, 2]. For example, FGFR1 was reported in 17% of squamous non-small cell lung carcinomas [59] and ~20% of breast tumors [7]. FGFR2 aberration was described in ~15% of intrahepatic cholangiocarcinomas [60], 4% of triple-negative breast cancer [61], and 5%–10% of gastric cancer cases [62]. FGFR3 mutations are known to occur in nearly 75% of nonmuscle invasive urothelial cell carcinomas [2, 63]. FGF19/FGFR4 aberration contributes to HCC progression [54]. Thus, the characteristics of DW14383 against FGFR1–4 with equivalent potency suggest its pan-tumor spectrum potential in these FGFR1–4 aberrant cancers and its consequent potential to overcome compensatory activation among FGFR1–4. In addition, impressive clinical data of the recently approved pan-FGFR inhibitor erdafitinib in FGFR3-aberrant urothelial carcinoma (mUC) [25, 26] and the FGFR1–3 inhibitor pemigatinib in FGFR2-aberrant cholangiocarcinoma [27] further promote pan-FGFR selective inhibitors development and inspire the recruitment of patients harboring other FGFR aberrant tumor types for clinical trials.
Moreover, DW14383 showed selectivity against FGFR. In particular, it displayed more than 1100-fold selectivity for FGFR1 over recombinant KDR, making it one of the inhibitors most selective for FGFR over KDR described to date. Furthermore, cell proliferation and cellular signaling tests in BaF3/TEL-FGFR1 and BaF3/TEL-KDR cells confirmed the selectivity of DW14383 against FGFR over KDR. These results suggest that DW14383 does not induce the side effects of multitarget inhibitors, particularly by targeting KDR-related angiogenesis kinase, and maximize the efficacy of FGFR inhibition in FGFR-aberrant patients.
In summary, DW14383 is an irreversible, selective pan-FGFR inhibitor that coinhibits the kinase activities of FGFR1–4. DW14383 is a promising inhibitor deserving further investigation in patients with FGFR-aberrant cancers.
Acknowledgements
This work was supported by grants from the “Personalized Medicines --Molecular Signature-based Drug Discovery and Development” Strategic Priority Research Program of the Chinese Academy of Sciences (Nos. XDA12020000 and XDA12020103), the National Natural Science Foundation of China for Innovation Research Group (No. 81821005), the National Natural Science Foundation of China (No. 81773762), the Collaborative Innovation Cluster Project of Shanghai Municipal Commission of Health and Family Planning (2020CXJQ02).
Author contributions
JD and JA conceived the project; JA supervised the project; MDD, YLW, YD, YMS and YCJ performed the research; MDD, YLW, XP, LLL and JA analyzed the data; JF, YMW and WHD contributed to DW14383 design and synthesis; MDD, XP and JA wrote the paper.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Meng-di Dai, Yue-liang Wang, Jun Fan.
References
- 1.Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Disco. 2009;8:235–53.. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17:318–32.. doi: 10.1038/nrc.2017.8. [DOI] [PubMed] [Google Scholar]
- 3.Touat M, Ileana E, Postel-Vinay S, Andre F, Soria JC. Targeting FGFR signaling in cancer. Clin Cancer Res. 2015;21:2684–94.. doi: 10.1158/1078-0432.CCR-14-2329. [DOI] [PubMed] [Google Scholar]
- 4.Hallinan N, Finn S, Cuffe S, Rafee S, O’Byrne K, Gately K. Targeting the fibroblast growth factor receptor family in cancer. Cancer Treat Rev. 2016;46:51–62. doi: 10.1016/j.ctrv.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 5.Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25:552–63.. doi: 10.1093/annonc/mdt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giacomini A, Chiodelli P, Matarazzo S, Rusnati M, Presta M, Ronca R. Blocking the FGF/FGFR system as a “two-compartment” antiangiogenic/antitumor approach in cancer therapy. Pharmacol Res. 2016;107:172–85.. doi: 10.1016/j.phrs.2016.03.024. [DOI] [PubMed] [Google Scholar]
- 7.Courjal F, Cuny M, SimonyLafontaine J, Louason G, Speiser P, Zeillinger R, et al. Mapping of DNA amplifications at 15 chromosomal localizations in 1875 breast tumors: definition of phenotypic groups. Cancer Res. 1997;57:4360–7. [PubMed] [Google Scholar]
- 8.Fischbach A, Rogler A, Erber R, Stoehr R, Poulsom R, Heidenreich A, et al. Fibroblast growth factor receptor (FGFR) gene amplifications are rare events in bladder cancer. Histopathology. 2015;66:639–49.. doi: 10.1111/his.12473. [DOI] [PubMed] [Google Scholar]
- 9.Freier K, Schwaenen C, Sticht C, Flechtenmacher C, Muhling J, Hofele C, et al. Recurrent FGFR 1 amplification and high FGFR1 protein expression in oral squamous cell carcinoma (OSCC) Oral Oncol. 2007;43:60–6. doi: 10.1016/j.oraloncology.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Gorringe KL, Jacobs S, Thompson ER, Sridhar A, Qiu W, Choong DYH, et al. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin Cancer Res. 2007;13:4731–9. doi: 10.1158/1078-0432.CCR-07-0502. [DOI] [PubMed] [Google Scholar]
- 11.Heist RS, Mino-Kenudson M, Sequist LV, Tammireddy S, Morrissey L, Christiani DC, et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol. 2012;7:1775–80.. doi: 10.1097/JTO.0b013e31826aed28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang T, Gao GH, Fan GX, Li M, Zhou CC. FGFR1 amplification in lung squamous cell carcinoma: a systematic review with meta-analysis. Lung Cancer. 2015;87:1–7. doi: 10.1016/j.lungcan.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Missiaglia E, Selfe J, Hamdi M, Williamson D, Schaaf G, Fang C, et al. Genomic imbalances in rhabdomyosarcoma cell lines affect expression of genes frequently altered in primary tumors: an approach to identify candidate genes involved in tumor development. Genes Chromosomes Cancer. 2009;48:455–67.. doi: 10.1002/gcc.20655. [DOI] [PubMed] [Google Scholar]
- 14.Dutt A, Salvesen HB, Chent TH, Ramos AH, Onofrio RC, Hatton C, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105:8713–7. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai AL, Meetze K, Vo NY, Kollipara S, Mazsa EK, Winston WM, et al. GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res. 2010;70:7630–9. doi: 10.1158/0008-5472.CAN-10-1489. [DOI] [PubMed] [Google Scholar]
- 16.Kunii K, Davis L, Gorenstein J, Hatch H, Yashiro M, Di Bacco A, et al. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008;68:2340–8. doi: 10.1158/0008-5472.CAN-07-5229. [DOI] [PubMed] [Google Scholar]
- 17.Presta M, Chiodelli P, Giacomini A, Rusnati M, Ronca R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol Ther. 2017;179:171–87.. doi: 10.1016/j.pharmthera.2017.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Kim JY, Al-Hilal TA, Chung SW, Kim SY, Ryu GH, Son WC, et al. Antiangiogenic and anticancer effect of an orally active low molecular weight heparin conjugates and its application to lung cancer chemoprevention. J Control Release. 2015;199:122–31.. doi: 10.1016/j.jconrel.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 19.Kim PH, Cha EK, Sfakianos JP, Iyer G, Zabor EC, Scott SN, et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. Eur Urol. 2015;67:198–201. doi: 10.1016/j.eururo.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raja A, Park I, Haq F, Ahn SM. FGF19-FGFR4 signaling in hepatocellular carcinoma. Cells. Cells. 2019;8:536. doi: 10.3390/cells8060536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–94.. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho HK, Yeo AHL, Kang TS, Chua BT. Current strategies for inhibiting FGFR activities in clinical applications: opportunities, challenges and toxicological considerations. Drug Discov Today. 2014;19:51–62. doi: 10.1016/j.drudis.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 23.Kim B, Wang S, Lee JM, Jeong Y, Ahn T, Son DS, et al. Synthetic lethal screening reveals FGFR as one of the combinatorial targets to overcome resistance to Met-targeted therapy. Oncogene. 2015;34:1083–93.. doi: 10.1038/onc.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saito S, Morishima K, Ui T, Hoshino H, Matsubara D, Ishikawa S, et al. The role of HGF/MET and FGF/FGFR in fibroblast-derived growth stimulation and lapatinib-resistance of esophageal squamous cell carcinoma. BMC Cancer. 2015;15:82. doi: 10.1186/s12885-015-1065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dai SY, Zhou Z, Chen ZC, Xu GY, Chen YH, et al. Fibroblast growth factor receptors (FGFRs): structures and small molecule inhibitors. Cells. 2019;8:614. doi: 10.3390/cells8060614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perera TPS, Jovcheva E, Mevellec L, Vialard J, De Lange D, Verhulst T, et al. Discovery and pharmacological characterization of JNJ-42756493 (Erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol Cancer Ther. 2017;16:1010–20.. doi: 10.1158/1535-7163.MCT-16-0589. [DOI] [PubMed] [Google Scholar]
- 27.Liu PCC, Koblish H, Wu L, Bowman K, Diamond S, DiMatteo D, et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS One. 2020;15:e0231877. doi: 10.1371/journal.pone.0231877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soria JC, DeBraud F, Bahleda R, Adamo B, Andre F, Dientsmann R, et al. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann Oncol. 2014;25:2244–51.. doi: 10.1093/annonc/mdu390. [DOI] [PubMed] [Google Scholar]
- 29.Ghedini GC, Ronca R, Presta M, Giacomini A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev Anticancer Ther. 2018;18:861–72.. doi: 10.1080/14737140.2018.1491795. [DOI] [PubMed] [Google Scholar]
- 30.Gutin G, Fernandes M, Palazzolo L, Paek H, Yu K, Ornitz DM, et al. FGF signalling generates ventral telencephalic cells independently of SHH. Development. 2006;133:2937–46.. doi: 10.1242/dev.02465. [DOI] [PubMed] [Google Scholar]
- 31.Johnson DE, Williams LT. Structural and functional diversity in the Fgf receptor multigene family. Adv Cancer Res. 1993;60:1–41. doi: 10.1016/s0065-230x(08)60821-0. [DOI] [PubMed] [Google Scholar]
- 32.Lesca E, Lammens A, Huber R, Augustin M. Structural analysis of the human fibroblast growth factor receptor 4 kinase. J Mol Biol. 2014;426:3744–56.. doi: 10.1016/j.jmb.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Kalyukina M, Yosaatmadja Y, Middleditch MJ, Patterson AV, Smaill JB, Squire CJ. TAS-120 cancer target binding: defining reactivity and revealing the first fibroblast growth factor receptor 1 (FGFR1) irreversible structure. Chemmedchem. 2019;14:494–500. doi: 10.1002/cmdc.201800719. [DOI] [PubMed] [Google Scholar]
- 34.Wang YM, Li LJ, Fan J, Dai Y, Jiang A, Geng MY, et al. Discovery of potent irreversible pan-fibroblast growth factor receptor (FGFR) inhibitors. J Med Chem. 2018;61:9085–104.. doi: 10.1021/acs.jmedchem.7b01843. [DOI] [PubMed] [Google Scholar]
- 35.Baillie TA. Targeted covalent inhibitors for drug design. Angew Chem Int Ed. 2016;55:13408–21.. doi: 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- 36.Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–21.. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang G, Liu ZG, Wu JZ, Cai Y, Li XK. Anticancer molecules targeting fibroblast growth factor receptors. Trends Pharmacol Sci. 2012;33:531–41.. doi: 10.1016/j.tips.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 38.Jiang XF, Dai Y, Peng X, Shen YY, Su Y, Wei MM, et al. SOMCL-085, a novel multi-targeted FGFR inhibitor, displays potent anticancer activity in FGFR-addicted human cancer models. Acta Pharmacol Sin. 2018;39:243–50.. doi: 10.1038/aps.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou WJ, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, et al. A structure-guided approach to creating covalent FGFR inhibitors. Chem Biol. 2010;17:285–95.. doi: 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan L, Wang J, Tanizaki J, Huang ZF, Aref AR, Rusan M, et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc Natl Acad Sci U S A. 2014;111:E4869–E77.. doi: 10.1073/pnas.1403438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown WS, Tan L, Smith A, Gray NS, Wendt MK. Covalent targeting of fibroblast growth factor receptor inhibits metastatic breast cancer. Mol Cancer Ther. 2016;15:2096–106.. doi: 10.1158/1535-7163.MCT-16-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brameld KA, Owens TD, Verner E, Venetsanakos E, Bradshaw JM, Phan VT, et al. Discovery of the irreversible covalent FGFR inhibitor 8-(3-(4-Acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (PRN1371) for the treatment of solid tumors. J Med Chem. 2017;60:6516–27.. doi: 10.1021/acs.jmedchem.7b00360. [DOI] [PubMed] [Google Scholar]
- 43.Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 2014;10:957–67.. doi: 10.2217/fon.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finlay MRV, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J Med Chem. 2014;57:8249–67.. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 45.Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, et al. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res. 2012;72:2045–56.. doi: 10.1158/0008-5472.CAN-11-3034. [DOI] [PubMed] [Google Scholar]
- 46.Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18:1855–62.. doi: 10.1158/1078-0432.CCR-11-0699. [DOI] [PubMed] [Google Scholar]
- 47.Sootome H, Fujioka Y, Miura A, Fujita H, Hirai H, Utsugi T. TAS-120, irreversible FGFR inhibitor, was effective tumors harboring FGFR Mutat, refractory or resistant ATP competitive inhibitors. Mol Cancer Ther. 2013;12:A271. [Google Scholar]
- 48.Ochiiwa H, Fujita H, Itoh K, Sootome H, Hashimoto A, Fujioka Y, et al. TAS-120, A highly potent selective irreversible FGFR inhibitor, is effective tumors harboring Var FGFR gene abnormalities. Mol Cancer Ther. 2013;12:A270. [Google Scholar]
- 49.Bahleda R, Meric-Bernstam F, Goyal L, Tran B, He Y, Yamamiya I, et al. Phase 1, first-in-human study of futibatinib, a highly selective, irreversible FGFR1-4 inhibitor in patients with advanced solid tumors. Ann Oncol. 2020;31:1405–12. doi: 10.1016/j.annonc.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joshi JJ, Coffey H, Corcoran E, Tsai J, Huang CL, Ichikawa K, et al. H3B-6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res. 2017;77:6999–7013. doi: 10.1158/0008-5472.CAN-17-1865. [DOI] [PubMed] [Google Scholar]
- 51.Liu H, Ai J, Shen A, Chen Y, Wang X, Peng X, et al. c-Myc alteration determines the therapeutic response to FGFR inhibitors. Clin Cancer Res. 2017;23:974–84.. doi: 10.1158/1078-0432.CCR-15-2448. [DOI] [PubMed] [Google Scholar]
- 52.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 53.Xie L, Su X, Zhang L, Yin X, Tang L, Zhang X, et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Cell Cancer Res. 2013;19:2572–83. doi: 10.1158/1078-0432.CCR-12-3898. [DOI] [PubMed] [Google Scholar]
- 54.Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N, et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 2015;5:424–37.. doi: 10.1158/2159-8290.CD-14-1029. [DOI] [PubMed] [Google Scholar]
- 55.Desai A, Adjei AA. FGFR signaling as a target for lung cancer therapy. J Thorac Oncol. 2016;11:9–20. doi: 10.1016/j.jtho.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 56.Zecchini S, Bombardelli L, Decio A, Bianchi M, Mazzarol G, Sanguineti F, et al. The adhesion molecule NCAM promotes ovarian cancer progression via FGFR signalling. EMBO Mol Med. 2011;3:480–94.. doi: 10.1002/emmm.201100152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. 2019;10:1373. doi: 10.1038/s41467-019-09068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hasse C, Holz O, Lange E, Pisowodzki L, Rebscher N, Christin Eder M, et al. FGFR-ERK signaling is an essential component of tissue separation. Dev Biol. 2014;395:154–66.. doi: 10.1016/j.ydbio.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 59.Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med. 2010;2:62ra93. doi: 10.1126/scitranslmed.3001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun. 2015;6:6087. doi: 10.1038/ncomms7087. [DOI] [PubMed] [Google Scholar]
- 61.Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013–23.. doi: 10.1038/onc.2009.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsumoto K, Arao T, Hamaguchi T, Shimada Y, Kato K, Oda I, et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer. 2012;106:727–32.. doi: 10.1038/bjc.2011.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cappellen D, De Oliveira C, Ricol D, de Medina S, Bourdin J, Sastre-Garau X, et al. Frequent activating mutations of FGFR3 in human bladder and cervix carcinomas. Nat Genet. 1999;23:18–20. doi: 10.1038/12615. [DOI] [PubMed] [Google Scholar]