

Summary
Recently, allele-specific single-cell RNA-seq analysis has demonstrated widespread dynamic random monoallelic expression of autosomal genes (aRME) in different cell types. However, the prevalence of dynamic aRME during pregastrulation remains unknown. Here, we show that dynamic aRME is widespread in different lineages of pregastrulation embryos. Additionally, the origin of dynamic aRME remains elusive. It is believed that independent transcriptional bursting from each allele leads to dynamic aRME. Here, we show that allelic burst is not perfectly independent; instead it happens in a semicoordinated fashion. Importantly, we show that semicoordinated allelic bursting of genes, particularly with low burst frequency, leads to frequent asynchronous allelic bursting, thereby contributing to dynamic aRME. Furthermore, we found that coordination of allelic bursting is lineage specific and genes regulating the development have a higher degree of coordination. Altogether, our study provides significant insights into the prevalence and origin of dynamic aRME and their developmental relevance during early development.
Subject areas: biological sciences, developmental biology, Bioinformatics
Graphical abstract
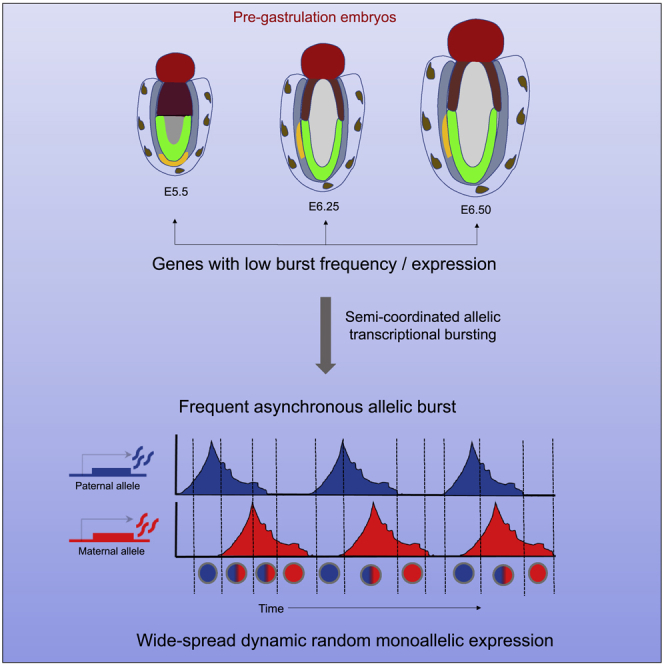
Highlights
-
•
Dynamic aRME is widespread in different lineages of pregastrulation embryos
-
•
Semicoordinated bursting of genes with low burst frequency leads to dynamic aRME
-
•
Degree of coordination of allelic bursting is lineage specific
-
•
Developmental genes have higher degree of coordination of allelic bursting
Biological sciences; Developmental biology; Bioinformatics
Introduction
In a diploid eukaryotic cell, both parental alleles of a gene are usually expressed. However, monoallelic expression of genes is common in phenomena such as genomic imprinting or X chromosome inactivation, where a single allele of a gene is expressed (Bartolomei and Ferguson-Smith, 2011; Gayen et al, 2015, 2016; Harris et al., 2019; Lyon, 1961; Mandal et al., 2020; Saiba et al., 2018; Sarkar et al., 2015). Surprisingly, recent advances on allele-specific single-cell RNA-seq (scRNA-seq) have revealed that many autosomal genes express monoallelically, which is transient in nature (Deng et al., 2014; Gendrel et al., 2016; Gregg, 2017; Reinius et al., 2016; Reinius and Sandberg, 2015; RV et al., 2021). This widespread temporal aRME has been termed as dynamic random monoallelic expression of autosomal genes (aRME). The pioneering study of Deng et al. showed that ∼12–24% of autosomal genes in a mouse blastomere undergo dynamic random monoallelic expression (RME) (Deng et al., 2014). In the same study, analysis of hepatocytes from adult mice and mouse fibroblast cell lines also showed a similar pervasiveness of dynamic aRME (Deng et al., 2014). Subsequently, prevalent dynamic aRME has been reported in various cell types of mice and humans (Borel et al., 2015; Reinius et al., 2016). However, the prevalence of dynamic aRME during pregastrulation development is not known yet. Here, we have profiled the genome-wide pattern of dynamic aRME in different lineages of pregastrulation mouse embryos. It is believed that dynamic aRME creates temporal variation among the cells and therefore can contribute to the cell fate decision and promote cellular plasticity during development. Therefore, profiling the pattern of dynamic aRME during early development is of immense interest.
On the other hand, the origin of dynamic aRME remains poorly understood. It is thought that dynamic aRME is a consequence of stochastic transcriptional burst (Eckersley-Maslin and Spector, 2014; Reinius and Sandberg, 2015). It is known that transcription happens through discrete bursts such that the state of a gene keeps switching randomly from an active to an inactive state, which leads to discontinuous production of mRNA (Raj et al., 2006; Raj and van Oudenaarden, 2008; Suter et al., 2011; Tunnacliffe and Chubb, 2020). The sporadic nature of transcriptional bursting is proposed to be a major driver of spontaneous heterogeneity in gene expression, which in turn drives diversity of cell behavior in differentiation and disease. In general, burst kinetics are characterized through the simple two-state model of transcription (Larson, 2011; Peccoud and Ycart, 1995). This model assumes that promoter activity can switch stochastically from transcriptional “on” to “off” state, with transcripts being produced only in the “on” state. Moreover, the transition between on and off state and the rate of transcription are determined by a single rate-limiting step and often the RNA decay rate is used to normalize these kinetic parameters. Furthermore, kinetics is mainly estimated by the burst size and burst frequency. The burst size is described as the average number of synthesized mRNA while a gene remains in an active state, whereas burst frequency is the rate at which bursts occur per unit time. For a long time, the analysis of transcriptional burst kinetics, including burst size and frequency, was mainly relied on single-molecule RNA-fluroscence in situ hybridization or live-cell imaging and therefore restricted to a few selected loci of the genome (Raj et al., 2006). Recent advancements in allele-specific expression analysis of many genes at a single cell level have made it possible to analyze transcriptional burst kinetics at the allelic level genome wide more extensively (Ochiai et al., 2020; Sun and Zhang, 2020). However, the kinetics of bursting at the allelic level remains poorly understood. Investigation of burst kinetics at the allelic level is important to understand the periodic fluctuation of the abundance of transcripts from each allele and how it can contribute to cellular and phenotypic variability. To explore this, we have profiled genome-wide allele-specific transcriptional burst kinetics in different lineages of pregastrulation mouse embryos through allele-specific scRNA-seq. Moreover, we have extended our analysis to explore the biological relevance of the allelic burst kinetics. Finally, we have investigated the association between allelic bursting and dynamic aRME.
Results
Dynamic aRME in different lineages of pregastrulation mouse embryos
To investigate the aRME pattern in different lineages of pregastrulation mouse embryos, we performed allele-specific gene expression analysis using the available scRNA-seq data set of E5.5, E6.25, and E6.5 hybrid mouse embryos (Cheng et al., 2019) (Figure 1A). These embryos are derived from two divergent mouse strains (C57Bl/6J and CAST/EiJ). Therefore, they harbor polymorphic sites between the alleles, which allowed us to perform allelic expression profiles of the genes (Figure 1A). We segregated the cells into the three lineages: epiblast (EPI), extraembryonic ectoderm (ExE), and visceral endoderm (VE) based on t-distributed stochastic neighbor embedding (t-SNE) analysis and lineage-specific marker gene expression (Figure S1).
Figure 1.

Genome-wide profiling of aRME in different lineages of pregastrulation embryos
(A) Graphical outline of the workflow: allelic gene expression and burst kinetics analysis in different lineages (EPI, ExE, and VE) of pregastrulation hybrid mouse embryos (E5.5, E6.25, and E6.50) at the single-cell level using published scRNA-seq data set. Hybrid mouse embryos were obtained from crossing between two divergent mouse strains C57 and CAST.
(B) Estimation of the mean percent of autosomal genes showing monoallelic expression per cell of each lineage (EPI, ExE, and VE) at different stages (E5.5, E6.25, and E6.5).
(C) Estimating the mean percent of autosomal genes showing monoallelic expression per cell of each lineage of embryos at different stages.
(D) Estimating the mean percent of autosomal genes exhibiting monoallelic expression per embryo of each lineage at different stages. For this analysis, for each lineage, we considered embryos which had at least 10 cells for that lineage.
First, we quantified the autosomal gene's allelic expression pattern in an individual cell of different lineages. Considering the technical noise such as allelic dropout can lead to a false estimation of monoallelic expression, especially for lowly expressed genes, we removed those genes from our analysis. We considered only those genes, which had at least mean ten reads per cell for each lineage of a specific developmental stage. We considered a gene as monoallelic if at least 95% of the allelic reads was originated from only one allele. We found that an average of ∼15–20% of genes showed monoallelic expression either from CAST or C57 allele per cell, and the pattern was almost similar across the three lineages EPI, ExE, and VE of different developmental stages (Figure 1B). Moreover, each embryo's allelic expression of different developmental stages showed a very similar pattern (Figure 1C). Interestingly, per-embryo estimation of the mean percent of genes with monoallelic expression by pooling an individual embryo's cells resulted in significant reduction in the fraction of genes with monoallelic expression (0.8–2% genes per embryo) (Figure 1D). Based on this, we assumed that the allelic expression pattern of the individual gene might vary from cell to cell in each embryo's lineage at a particular stage. To test our assumption, we investigated the status of the allelic pattern of individual genes across the cells of each lineage of each developmental stage. Indeed, we found a considerable variation of the gene's allelic status across the cells, indicating the presence of cell-to-cell dynamic aRME (Figure 2). We observed four different patterns of allelic expression; Cat1: nonrandom monoallelic (1–2%), Cat 2: random monoallelic with one allele (4–39%), Cat 3: random monoallelic with either allele (30–81%), and Cat 4: biallelic (10–29%) (Figure 2). Altogether, our analysis revealed a high degree of cell-to-cell dynamic aRME (category 2: 4–39% and category 3: 30–81%) in each lineage of pregastrulation embryos, indicating dynamic allelic expression is a general feature of gene expression affecting many genes during development. We validated our allelic expression analysis through profiling the allelic expression of X-linked genes (Figure S2).
Figure 2.

Dynamic aRME in different lineages of pregastrulation embryos
Quantification of the percent of genes showing the different category of allelic expression (Cat1: nonrandom monoallelic, Cat 2: random monoallelic with one allele, Cat 3: random monoallelic with either allele, Cat 4: biallelic) in three different lineages EPI, ExE, and VE across the various developmental stages.
Allelic bursting is semicoordinated
Next, we explored genome-wide allele-specific transcriptional burst kinetics to investigate the link between dynamic aRME and transcriptional bursting. Based on two-state transcription models, transcription occurs in bursts where the state of a gene keeps switching from ON to OFF state (Figure 3A). Burst kinetics is mainly characterized by burst frequency and burst size. The burst frequency is the rate at which bursts occur per unit time, and burst size is determined by the average number of synthesized mRNA while a gene remains in an active state (Figure 3A). We used single-cell allelic expression (SCALE) to determine the genome-wide burst kinetics of autosomal genes in an allele-specific manner (Jiang et al., 2017). Principally, based on the Empirical Bayes framework, SCALE first categorizes the genes to biallelic, monoallelic, and silent using the allele-specific read counts, and then biallelic genes are further classified as biallelic bursty and biallelic nonbursty. Finally, different burst kinetics parameters are deduced for the biallelic bursty genes. We performed burst kinetics analysis for only E6.5 in EPI (n = 123 cells) and VE (n = 115 cells) cells. For other stages or lineages, there were not a sufficient number of cells for performing SCALE analysis. We considered those autosomal genes (n = 5633 genes for EPI and n = 5791 genes for VE) for SCALE analysis, which had at least mean ten reads per cell in each lineage. In both E6.5 EPI and VE, we found that most of the genes (70–82%) showed bursty expression (Figure 3B; Table S1). Next, we compared the burst kinetics between the alleles of biallelic bursty genes. Interestingly, we found that the alleles of most of the genes showed similar burst kinetics, i.e., they had identical burst frequency and size (Figures 3C and 3D). Only 48 out of 3861 bursty genes (EPI) and 90 out of 4705 bursty genes (VE) showed significantly different allelic burst frequency after false discovery rate correction (Figure 3C). On the other hand, very few genes showed significantly different allelic burst size (Figure 3D). Next, we determined the independence of allelic transcriptional burst. We plotted the percent of cells expressing neither allele (p0) with the percent of cells expressing both alleles (p2), as depicted in Figure 3E. In the perfect independent model, most of the genes (black dots) should lie across the red curve, whereas perfect coordination model genes should lie near the diagonal blue line. Interestingly, we found that most of the genes reside in the middle of between the red and diagonal blue lines, indicating that allelic bursting is neither entirely independent nor perfectly coordinated (Figure 3E). Additionally, the null hypothesis of independence was rejected for most of the genes (Table S1). Altogether these results suggested that alleles of most of the genes have similar burst kinetics; however, allelic bursting was neither entirely independent nor perfectly coordinated.
Figure 3.

Genome-wide profiling of transcriptional burst kinetics
(A) Representation of the two-state model of transcription. kon: the rate at which a gene becomes transcriptionally active (from off to on); koff: the rate at which a gene becomes inactive (from on to off); S is the rate of transcription, while the gene is active; d is the rate of mRNA decay. Burst kinetics is characterized through burst frequency (kon) and the burst size (S/koff)..
(B) Estimating the proportion of autosomal genes with bursty expression in EPI and VE cells of the E6.5 stage.
(C) The burst frequency of the two alleles of most of the genes was highly similar in EPI and VE cells of the E6.5 stage (r = 0.80 and 0.82, respectively). Genes having significantly different allelic burst frequency are marked as a red triangle.
(D) A similar burst size of the two alleles of most of the genes in EPI and VE cells of the E6.5 stage (r = 0.51 and 0.59, respectively). Genes having significantly different allelic burst sizes are marked as a red triangle.
(E) Smooth scatterplot showing independence of allelic transcriptional bursting in EPI and VE cells of E6.5 stage. p0 is the percent of cells expressing neither allele. p2 is the percent of cells expressing both alleles. Black points represent individual genes. The diagonal blue line (p0 + p2 = 1) represents coordinated bursting, whereas the red curve represents perfect independent bursting with shared kinetics.
Next, to get a quantitative assessment of the dependence between the alleles, we constructed a simple two-component stochastic model, inspired from the classic two-state model of transcription (Peccoud and Ycart, 1995) (Figure S4). This model describes two allele's identical kinetic parameters in terms of their rates to switch “on” and “off.” In addition, two parameters stayOn and stayOff have been included that add to “on” and “off” such that an allele that is on would have increased rate (on + stayOn) of remaining on and vice versa, thus leading to bursty transcription. To model the dependency of the transcription of one allele on the other, we assumed that the probability of an allele turning and staying on is multiplied by a parameter lambda, when the other allele is on. Therefore, lambda >1 describes that an allele in “on” state facilitates the other allele also being “on” (the higher the value of lambda, the higher the effect), lambda = 1 describes independent bursting of the two alleles, and lambda <1 describes that an allele being ‘on’ disfavors the other allele staying “on.” In total, for both alleles, we have five parameters in the model. The on and off probabilities are chosen between 0 and 1, and lambda values are chosen between 0.01 and 100. The parameters are converted to probabilities for simulation, and thus, the relative levels of parameters instead of absolute values are more important. To obtain different possible model behaviors, we sampled 35,000 parameter sets chosen uniformly randomly from the aforementioned range. The algorithm used for simulations is given in Figure S4. For each parameter set sampled, we simulate for 10,000 time steps and record the state of the allele at the end. This step is repeated 100 times to calculate the probability of the alleles being on.
First, we validated our model through simulations for independently expressing alleles by testing it against the theoretical relationship between probability of both alleles being off (p0) and both alleles being on (p2). Given the alleles both have the same on probability (p), p0 is given as (1-p)∗(1-p) and p2 is given as p∗p. Therefore, p2 and p0 are related via the following equation:
We found that the values obtained via numerical simulations (Figure 4A) lie very close to this curve and distributed symmetrically along the curve (Figure 4A inset), thus validating the model. Next, to understand how various parameters affect the placement of genes in the p0-p2 plot, we divided the data obtained from simulations into four regions (Figure 4B). The yellow region represents points below the independence curve, orange dots are those lying on the absolute dependence curve, black dots denote the region between the two curves, and red dots are for the region with high p0. Among the four, the experimental data points align best with the black region in the plot. To understand these regions better, we generated the distribution of model parameters corresponding to these regions (Figures 4C and S3).
Figure 4.

Simulation of the dependence between allelic bursting
(A) Comparison of simulation results for independent allele bursting with theoretical curve representing independent bursting on the p0-p2 plot. A distribution of distance of the points from the curve is shown in the inset.
(B) Classification of simulation data based on their position in the p0-p2 plot with respect to the theoretical curves.
(C) Parameter distribution for the genes represented with maroon dots (p0>0.7) in (B). Inset shows a scatterplot between lambda and on probability.
(D) Smooth scatterplot of independence of allelic transcriptional bursting in EPI and VE cells of E6.5 stage including dropout-prone genes.
(E) PCA1 components of the parameter regimes labeled in (B).
(F) Scatterplot between p0 and p2 constructed by using two constraints on the parameter values: on > 0.1 and 10 < lambda<50.
First, we looked at the region with high p0. The distribution of parameters revealed that these points had very low “on” probability and high values of lambda (Figure 4C). A scatterplot between these two parameters further shows that for high values of lambda, the probability to switch to an “on” state must be very small (Figure 4C inset). In the experimental data, none of the points lie in this region, possibly because of the elimination of very low expressed dropout prone genes with “on” probabilities lower than 0.1. To test this hypothesis, we included low expressed dropout prone genes in the p0-p2 plot (Figure 4D) and observed the region with high p0 populated, thereby validating that high p0 region of the graph corresponds to dropout-prone alleles.
We next wanted to understand which parameters contribute most to the regional separation on the p0-p2 plot. To do so, we performed principal component analysis (PCA) on the parameter sets corresponding to each region. For all regions, the first PCA component (PC1) explained >90% variance, and major contributor to PC1 is the dependence parameter lambda (Figure 4E). Looking at the range of lambdas for each region of the p0-p2 plot, we found that the region between the two theoretical curves (complete dependence, complete independence) corresponds to moderate values of lambda (between 10 and 50). Simulating parameter sets in this range of lambda values gave us a close similarity (Figure 4F) to the experimental plots (Figure 3E). Together, this quantitative analysis highlights the mechanistic underpinnings underlying observed experimental data, endorsing a semicoordinated allelic bursting for most genes.
Dynamic aRME is linked to allele-specific transcriptional burst kinetics
Next, we delineated the correlation between allelic transcriptional burst kinetics and dynamic aRME. First, we wanted to see if there is any correlation between bursty gene expression and dynamic aRME. Interestingly, we found that most dynamic aRME genes (Cat 2 and Cat 3) showed bursty expression (Figure 5A). Especially for Cat 3 aRME genes, more than 92% of genes showed bursty expression (Figure 5A). On the other hand, most biallelic genes (Cat 4) for EPI cells showed nonbursty expression (Figure 5A). Altogether, these results suggested that dynamic aRME is generally linked with bursty expression. Next, we examined if there is any correlation between the allelic expressions of genes with the allelic burst kinetics. To test this, we performed a pairwise correlation test between different burst kinetics parameters and the sum of allelic read counts for each gene across the cells (Figure 5B). We found that the total expression of alleles is positively correlated (r = 0.65–0.77) with allelic burst frequency. On the other hand, although allelic expression was positively correlated with the burst size (r = 0.12–0.18) and the proportion of unit time the allele remains active (r = 0.23–0.34), the correlation value was much lower than the burst frequency. To get more insights into this aspect, we compared the burst frequency and burst size of alleles with the percent of cells expressing that corresponding allele (Figure 5C) or the mean expression of alleles (Figure 5D). Interestingly, we found that the proportion of cells express one allele of genes/mean expression of alleles is substantially dependent on the burst frequency of that allele rather than burst size (Figures 5C and 5D). Overall, allelic expression was directly proportional to the allelic burst frequency such that alleles expressing high showed high allelic burst frequency and those expressing low had low allelic burst frequency. Altogether, these analyses suggested that burst frequency among the different kinetics parameters is crucial for monoallelic gene expression. Next, we delineated if dynamic aRME is dependent on the overall expression level. Interestingly, comparison of expression levels between bursty vs. nonbursty genes revealed that nonbursty genes always have significantly higher expression than the bursty genes (Figure 6A). Next, we hypothesized that the proportion of cells with the monoallelic expression might depend on the gene's expression level. We analyzed the correlation between gene expression level and percent of cells showing the monoallelic expression for that gene to test our hypothesis. As expected, we found a high negative correlation (r = −0.58 to −0.61) (Figure 6B). Altogether, these results indicated that the extent of a gene's monoallelic expression depends on its expression level and allelic burst frequency. Based on our observation and analysis, we proposed a model highlighting how transcriptional burst kinetics can contribute to the dynamic aRME (Figure 6C). We propose that bursty genes with asynchronous allelic burst kinetics build up the dynamic aRME landscape. Genes with lower expression and/or lower burst frequency frequently undergo monoallelic expression (Figure 6C). On the other hand, genes with high expression and/or high allelic burst frequency express most of the time biallelically (Figure 6C).
Figure 5.

Association between burst kinetics and dynamic aRME
(A) Quantification of the proportion of dynamic aRME genes (Cat 2 & Cat 3) with bursty expression and proportion of biallelic genes (Cat 4) having bursty expression in EPI and VE cells of E6.5 stage.
(B) Pairwise correlation between different allelic bursting kinetics parameters (burst frequency: konCAST and konC57; Proportion of unit time that the gene stays in active form: pCAST = konCAST/(konCAST + koffCAST) ; pC57 = konC57/(konC57 + koff57); burst size: SCAST/koffCAST and SC57/koffC57) and expression level (sum of normalized allelic read counts (log)) of the alleles in EPI and VE cells of E6.5 stage.
(C) Scatterplot representing an estimate of burst size and a burst frequency of the CAST and C57 allele of autosomal genes. The gene's color is profiled based on the percent of cells expressing the CAST or C57 allele.
(D) Scatterplot representing an estimate of burst size and burst frequency of the CAST and C57 allele of autosomal genes. The color of the gene is depicted based on the mean allelic expression.
Figure 6.

Relation of bursting and dynamic aRME with the gene expression level
(A) Comparison of the expression level of bursty vs. nonbursty genes in EPI and VE cells of the E6.5 stage. ∗∗∗p value <0.001 (Mann–Whitney two-sided U test).
(B) Correlation plot between the mean expression of gene and percent of cells showing monoallelic expression for that gene (r = −0.58 in EPI and −0.61 in VE).
(C) Model representing how transcriptional burst kinetics can lead to dynamic aRME.
Relevance of allelic burst kinetics to development
Next, we extended our analysis to explore the biological perspectives of allelic transcriptional burst kinetics. Since it is believed that stochastic allelic bursting/dynamic aRME provides developmental plasticity, we investigated the correlation between the degree of coordination of allelic bursting and development. To test this, we categorized genes into four different major classes based on their allelic coordination: highly coordinated, semicoordinated, independent, and genes with low p0 and high p2 (Figure 7A). Next, we performed gene ontology (GO) biological process analysis of these different categories of genes in EPI and VE cells of E6.5 (Table S2). Interestingly, we found that in EPI E6.5 cells, the highly coordinated genes showed significant enrichment to different developmental processes, including gastrulation, mesoderm development, and embryonic development (Figures 7B, Table S3). We did not find any such development-related enrichment in case of independent genes. Additionally, genes with low p0 and high p2 did not show enrichment for developmental genes (Figure 7A). However, in the case of VE cells, we did not find significant enrichment of the developmental genes in neither highly coordinated nor independent classes, suggesting that they are primarily in a semicoordinated state. Altogether, this analysis indicated that many genes regulating the development have higher degree of coordination of allelic bursting. Next, we performed a cross-comparison of allelic coordination of the genes between EPI and VE cells. To do this, we selected the common genes between VE and EPI cells from the SCALE output (Figure 7C). We found that while majority of genes was intersected between EPI and VE cells, many genes were not (Figure 7C). Certainly, many genes related to each cell state are excluded from this intersection because they are only expressed in one lineage and therefore did not pass the quality control in SCALE analysis in other lineage specially owing to their low expression. Next, we categorized those common/intersecting genes into four different major classes based on allelic coordination: highly coordinated, semicoordinated, independent, and genes with low p0 and high p2 (Figure 7D). Cross-comparison of these four classes of genes between EPI and VE cells showed the degree of coordination of allelic bursting changes between these two lineages for many genes, emphasizing the biological significance of allelic burst kinetics (Figure 7E). Interestingly, GO biological process analysis of unique genes of each category related to EPI and VE revealed a distinct pattern of biological functions (Table S4).
Figure 7.

Genes with higher allelic bursting coordination enriched for gene sets with developmental functions
(A) Scatterplot representing different categories of the genes based on allelic transcriptional bursting in EPI and VE cells of E6.5: low p0 high p2 (green asterisk), highly coordinated (genes with p0+p2 ≥ 0.90: slate gray square within blue dotted lines), independent (genes near the red curve line; with a threshold ±0.05 [upper and lower red curve line] from the main red curve lines: rosewood triangle), and semicoordinated (Persian green dots).
(B) Gene ontology biological process representing enrichment of highly coordinated genes (EPI, E6.5) in different developmental processes.
(C) Venn diagram representing the common genes between EPI E6.5 and VE E6.5 cells.
(D) Scatterplot representing different categories of the genes (low p0 high p2, highly coordinated, independent, and semicoordinated) based on allelic transcriptional bursting in EPI and VE cells of E6.5.
(E) Cross-comparison of the degree of coordination between EPI E6.5 and VE E6.5 cells based on different categories (genes with low p0 high p2, highly coordinated, independent, and semicoordinated) of allelic burst kinetics.
Discussion
It is believed that dynamic aRME creates temporal variation among the cells and thereby can contribute to the cell fate decision and promote cellular plasticity during development (Gregg, 2017; Huang et al., 2018; Montag et al., 2018; Ng et al., 2018). Therefore, investigating dynamic aRME during early development is crucial. In the present study, we show widespread dynamic aRME in different lineages of pregastrulation mouse embryos. Notably, dynamic aRME is more prevalent (∼69–88% genes) in pregastrulation embryos than in the blastomeres (∼12–24%) reported by Deng et al. (Deng et al., 2014) (Figure 2). This robust increase in the fraction of dynamic aRME in pregastrulation embryos indicates that dynamic allelic expression is a general feature of gene expression affecting many genes during development. However, our analyses have one caveat that is worth discussing. Our estimation of dynamic aRME might be erroneous to some extent owing to the allelic dropout effect of scRNA-seq. Although we believe that through eliminating the low expressed genes and Spike-in normalization of allelic read counts, we have significantly reduced the chances of false estimation of aRME owing to the allelic dropout. Indeed, previous reports have shown that low expressed genes are highly prone to undergo allelic dropout (Kim et al., 2015; Santoni et al., 2017; Wainer-Katsir and Linial, 2020; Zhao et al., 2017). Nevertheless, we believe that using a split-cell strategy as used in blastomere analysis by Deng et al., (2014) would have reduced the allelic dropout level more precisely. In split-cell experiments, RNA lysate from individual cells is split into two equal volume and then processed for sequencing. Using the allelic call from the split pair, stochastic dropout can be estimated and thereby false positive in monoallelic expression estimation can be eliminated. However, we could not perform this experiment as the scRNA-seq data set used for this study lacked split-cell experiment data. Separately, among these aRME genes, some gene's allelic expression pattern might be mitotically heritable, as reported earlier (Eckersley-Maslin et al., 2014; Gendrel et al., 2014; Gimelbrant et al., 2007; Jeffries et al., 2016; Zwemer et al., 2012). In the future, investigation on the clonal cell population can disentangle the mitotically stable aRME from the dynamic aRME.
On the other hand, we have profiled genome-wide allele-specific burst kinetics of autosomal genes to understand the implication of allelic bursting on the dynamic aRME. We found that majority of the autosomal genes have bursty expression, and alleles of most of the genes have similar burst kinetics, which is consistent with previous reports in other cell types (Figures 3B, 3C, and 3D) (Jiang et al., 2017). However, we found that allelic bursting is not perfectly independent; instead, it happens in a semicoordinated fashion (Figures 3E and 4). Finally, we demonstrate that dynamic aRME is linked to semicoordinated allelic bursting. We show that majority of dynamic aRME genes have bursty expression, whereas most of the biallelic genes were found to be nonbursty. Moreover, we found that the extent of dynamic aRME is determined by burst frequency rather than burst size or how long an allele remains active. Notably, we found that dynamic aRME was highly dependent on the overall expression level of a gene. Altogether, we propose that semicoordinated allelic bursting for the genes with lower burst frequency leads to frequent asynchronous allelic bursting, thereby creating widespread dynamic aRME (Figure 6C). On the other hand, nonbursty genes or bursty genes with high allelic burst frequency and/or high expression levels exhibit frequent biallelic expression (Figure 6C).
Interestingly, our analysis revealed that the degree of coordination of allelic bursting for many genes varied between developmental lineages EPI and VE (Figures 7D and 7E). Moreover, we found that unique genes of each coordination category between VE and EPI have distinct biological functions. Importantly, we found that genes involved in development have a higher degree of coordination of allelic bursting in EPI E6.5 cells (Figure 7B). Notably, we found key genes including Brachyury T, Eomes, Pou5f1, Fgfr1, etc. involved in gastrulation/germ layer formation showed higher coordination (Table S3). Beyond plasticity, the high degree of allelic coordination of developmental genes in EPI E6.5 could also reflect latent/structural heterogeneity within the epiblast at the onset of gastrulation as the initial germ layers begin to be specified. Therefore, in the future, it will be worth to investigate further the potential link between high coordination and lineage commitment during pregastrulation. Altogether, these results indicate the biological significance of allelic burst kinetics and related dynamic aRME. In the future, more extensive investigations are necessary to understand further the biological implications of allelic bursting/dynamic aRME in a wide range of biological processes and diseases.
Together, our study shed light on the kinetics of transcriptional bursting at allelic level and the biological relevance of it. In the future, extensive studies are necessary to understand the regulatory network behind semicoordinated allelic bursting. In a perfect independent model, regulation of allelic expression should be autonomous, whereas, in an alternative model of perfect dependence, there can be shared allelic expression regulation. We believe that autonomous as well as shared regulation of the alleles result in semicoordinated transcriptional bursting. Interestingly, a recent study has shown that chromatin conformations are variable between alleles, and each allele can behave independently, indicating that regulation of allelic expression can be self-governing (Finn et al., 2019). Moreover, it has been shown that while allelic burst frequency is regulated through an enhancer, burst size is controlled by the core promoter (Larsson et al., 2019). Moreover, a recent report suggests stochastic switching between methylated and unmethylated states at many regulatory loci occurs in a sequence-dependent manner, which can be another mechanism behind stochastic allelic transcriptional bursting (Onuchic et al., 2018).
Limitation of the study
In this study, we have provided significant insights into the prevalence and origin of dynamic aRME and their developmental relevance during early mammalian development. One potential caveat of our study is that our estimation of dynamic aRME might be erroneous to some extent owing to the allelic dropout effect of scRNA-seq. Although we believe that through eliminating the low expressed genes in our analysis, we have significantly reduced the chances of false estimation of aRME owing to the allelic dropout.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| Seurat (version 3.1.5) | (Butler et al., 2018; Stuart et al., 2019) | https://satijalab.org/seurat/ |
| VCF tools | (Danecek et al., 2011) | https://github.com/vcftools/vcftools |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| SCALE | (Jiang et al., 2017) | https://github.com/yuchaojiang/SCALE |
| Julia 1.5.1. | (Bezanson et al., 2017) | https://epubs.siam.org/doi/pdf/10.1137/141000671 |
| gProfiler | (Raudvere et al., 2019) | https://biit.cs.ut.ee/gprofiler_archive3/e102_eg49_p15/gost |
| ggplot2 | (Wickham, 2016) | https://ggplot2.tidyverse.org/ |
| R | R core team | https://www.R-project.org/ |
| Samtools | (Li et al., 2009) | http://www.htslib.org/ |
| BEDTools | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| Code for simulation | This paper | https://github.com/csbBSSE/aRME |
Resource availability
Lead contact
Further information on resources and reagents should be directed to lead contact, Srimonta Gayen (srimonta@iisc.ac.in)
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
No experimental model system used for this study.
Method details
Data acquisition
Single-cell transcriptome datasets used for this study were acquired from Gene Expression Omnibus (GEO) under the accession number “GEO:GSE109071” (Cheng et al., 2019). For our research, we analyzed a single-cell dataset generated from E5.5, E6.25, and E6.50 hybrid mouse embryos (C57BL/6J × CAST/EiJ). E5.5 and E6.25 embryos were derived from the following cross: C57(F) × CAST(M), whereas E6.5 were derived from CAST(F) × C57(M).
Lineage identification
All the single cells (510 cells) of different stages were subjected to a dimension reduction algorithm using t-distributed stochastic neighbor embedding (t-SNE) to identify lineages. Three thousand most variable genes were used for the analysis. t-SNE was performed using Seurat (version 3.1.5) (Butler et al., 2018; Stuart et al., 2019). The allocation of each cluster to cell lineages to EPI, ExE, and VE lineages was based on the expression of bona fide marker genes: Oct4 for EPI, Bmp4 for ExE, and Amn for VE.
Allele-specific expression and burst kinetics analysis
For allelic expression analysis of genes, first, we constructed in silico CAST specific parental genome by incorporating CAST/EiJ specific SNPs into the GRCm38 (mm10) reference genome using VCF tools (Danecek et al., 2011). CAST specific SNPs were obtained from the Mouse Genomes Project (https://www.sanger.ac.uk/science/data/mouse-genomes-project). Reads were mapped onto both C57BL/6J (mm10) reference genome and CAST/EiJ in silico parental genome using STAR with no multi-mapped reads. To exclude any false positive, we only considered those genes with at least 1 informative SNPs (at least 3 reads per SNP site). In genes having more than 1 SNP, we took an average of SNP-wise reads to have the allelic read counts. We normalized allelic read counts using Spike-in control as described in Sun et al. 2020 (Sun and Zhang, 2020). First, we calculated the sum of reads mapping to all Spike-in molecule in each cell of 510 cells. Next, we divided each cell’s Spike-in reads with the highest Spike in value from the array of 510 cells to obtain the normalization factor for each cell that are all between 0 and 1. Finally, we normalized the allelic read counts in each cell by dividing the original read count by the corresponding normalization factor. We considered those genes which had at least mean 10 reads per cell for each lineage of a specific developmental stage. Allelic expression was calculated individually for each gene using formula = (Maternal/Paternal reads) ÷ (Maternal reads + Paternal reads). A gene was considered monoallelic if at least 95% of the allelic reads came from only one allele. We performed genome-wide allele-specific burst kinetics analysis using SCALE (Jiang et al., 2017).
In silico model
The model was simulated using Julia 1.5.1(Bezanson et al., 2017). The code is available at https://github.com/csbBSSE/aRME. The plots were made using the ggplot2 package from R 4.0. PCA of the parameter sets was done using prcomp function from R 4.0.
Gene ontology
Gene ontology analysis was performed using g:GOSt from gProfiler (https://biit.cs.ut.ee/gprofiler_archive3/e102_eg49_p15/gost) with g:SCS multiple testing correction method and selected the functional terms which are passing FDR < 0.05 from GO:BP (Raudvere et al., 2019).
Quantification and statistical analysis
All statistical analysis was performed usin the R software (https://www.R-project.org/). Mann–Whitney two-sided U test was used for statistical significance analysis and p values < 0.05 was considered as significant. For correlation analysis, Pearson test was used.
Acknowledgments
This study is supported by Department of Biotechnology (DBT), India (BT/PR30399/BRB/10/1746/2018), Department of Science and technology (DST-SERB), India (CRG/2019/003067), DBT-Ramalingaswamy fellowship (BT/RLF/Re-entry/05/2016) and Infosys Young Investigator award to SG. We also thank DST-FIST [SR/FST/LS11-036/2014(C)], UGC-SAP [F.4.13/2018/DRS-III (SAP-II)] and DBT-IISc Partnership Program Phase-II (BT/PR27952-INF/22/212/2018) for infrastructure and financial support.
Author contributions
SG conceptualized and supervised the study. Bioinformatic analyses were performed by HCN. DC and SM helped with the analysis. MKJ and KH performed simulation. SG, HCN, MKJ, and KH wrote the manuscript. The final manuscript was approved by all the authors.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102954.
Supplemental information
Data and code availability
This study did not generate any unique datasets. The code used for simulation is available at https://github.com/csbBSSE/aRME. For questions regarding the raw data from the current study, please contact the lead contact. All software's used in this study are commercially available.
References
- Bartolomei M.S., Ferguson-Smith A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011;3:1–17. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanson J., Edelman A., Karpinski S., Shah V.B. Julia: a fresh approach to numerical computing. SIAM Rev. 2017;59:65–98. doi: 10.1137/141000671. [DOI] [Google Scholar]
- Borel C., Ferreira P.G., Santoni F., Delaneau O., Fort A., Popadin K.Y., Garieri M., Falconnet E., Ribaux P., Guipponi M. Biased allelic expression in human primary fibroblast single cells. Am. J. Hum. Genet. 2015;96:70–80. doi: 10.1016/j.ajhg.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Pei Y., He L., Peng G., Reinius B., Tam P.P.L., Jing N., Deng Q. Single-cell RNA-seq reveals cellular heterogeneity of pluripotency transition and X chromosome dynamics during early mouse development. Cell Rep. 2019;26:2593–2607.e3. doi: 10.1016/j.celrep.2019.02.031. [DOI] [PubMed] [Google Scholar]
- Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q., Ramsköld D., Reinius B., Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley-Maslin M.A., Spector D.L. Random monoallelic expression: regulating gene expression one allele at a time. Trends Genet. 2014;30:237–244. doi: 10.1016/j.tig.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley-Maslin M.A., Thybert D., Bergmann J.H., Marioni J.C., Flicek P., Spector D.L. Random monoallelic gene expression increases upon embryonic stem cell differentiation. Dev. Cell. 2014;28:351–365. doi: 10.1016/j.devcel.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn E.H., Pegoraro G., Brandão H.B., Valton A.L., Oomen M.E., Dekker J., Mirny L., Misteli T. Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell. 2019;176:1502–1515.e10. doi: 10.1016/j.cell.2019.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayen S., Maclary E., Buttigieg E., Hinten M., Kalantry S. A primary role for the tsix lncRNA in maintaining random X-chromosome inactivation. Cell Rep. 2015;11:1251–1265. doi: 10.1016/j.celrep.2015.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayen S., Maclary E., Hinten M., Kalantry S. Sex-specific silencing of X-linked genes by Xist RNA. Proc. Natl. Acad. Sci. U S A. 2016;113:E309–E318. doi: 10.1073/pnas.1515971113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendrel A.V., Attia M., Chen C.J., Diabangouaya P., Servant N., Barillot E., Heard E. Developmental dynamics and disease potential of random monoallelic gene expression. Dev. Cell. 2014;28:366–380. doi: 10.1016/j.devcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Gendrel A.V., Marion-Poll L., Katoh K., Heard E. Random monoallelic expression of genes on autosomes: parallels with X-chromosome inactivation. Semin. Cell Dev. Biol. 2016;56:100–110. doi: 10.1016/j.semcdb.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Gimelbrant A., Hutchinson J.N., Thompson B.R., Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318:1136–1140. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- Gregg C. The emerging landscape of in vitro and in vivo epigenetic allelic effects. F1000Res. 2017;6:2108. doi: 10.12688/f1000research.11491.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C., Cloutier M., Trotter M., Hinten M., Gayen S., Du Z., Xie W., Kalantry S. Conversion of random X-inactivation to imprinted X-inactivation by maternal PRC2. Elife. 2019;8 doi: 10.7554/elife.44258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.C., Bennett K., Gregg C. Epigenetic and cellular diversity in the brain through allele-specific effects. Trends Neurosci. 2018;41:925–937. doi: 10.1016/j.tins.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries A.R., Uwanogho D.A., Cocks G., Perfect L.W., Dempster E., Mill J., Price J. Erasure and reestablishment of random allelic expression imbalance after epigenetic reprogramming. RNA. 2016;22:1620–1630. doi: 10.1261/rna.058347.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y., Zhang N.R., Li M. SCALE: modeling allele-specific gene expression by single-cell RNA sequencing. Genome Biol. 2017;18 doi: 10.1186/s13059-017-1200-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.K., Kolodziejczyk A.A., Illicic T., Teichmann S.A., Marioni J.C. Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat. Commun. 2015;6 doi: 10.1038/ncomms9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson D.R. What do expression dynamics tell us about the mechanism of transcription? Curr. Opin. Genet. Dev. 2011;21:591–599. doi: 10.1016/j.gde.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A.J.M., Johnsson P., Hagemann-Jensen M., Hartmanis L., Faridani O.R., Reinius B., Segerstolpe Å., Rivera C.M., Ren B., Sandberg R. Genomic encoding of transcriptional burst kinetics. Nature. 2019;565:251–254. doi: 10.1038/s41586-018-0836-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon M.F. Gene action in the X-chromosom of the mouse. Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Mandal S., Chandel D., Kaur H., Majumdar S., Arava M., Gayen S. Single-cell analysis reveals partial reactivation of X chromosome instead of chromosome-wide dampening in naive human pluripotent stem cells. Stem Cell Rep. 2020;14:745–754. doi: 10.1016/j.stemcr.2020.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag J., Kowalski K., Makul M., Ernstberger P., Radocaj A., Beck J., Becker E., Tripathi S., Keyser B., Mühlfeld C. Burst-like transcription of mutant and wildtype MYH7-alleles as possible origin of cell-to-cell contractile imbalance in Hypertrophic Cardiomyopathy. Front. Physiol. 2018;9 doi: 10.3389/fphys.2018.00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng K.K.H., Yui M.A., Mehta A., Siu S., Irwin B., Pease S., Hirose S., Elowitz M.B., Rothenberg E.V., Kueh H.Y. A stochastic epigenetic switch controls the dynamics of T-cell lineage commitment. Elife. 2018;7 doi: 10.7554/eLife.37851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai H., Hayashi T., Umeda M., Yoshimura M., Harada A., Shimizu Y., Nakano K., Saitoh N., Liu Z., Yamamoto T. Genome-wide kinetic properties of transcriptional bursting in mouse embryonic stem cells. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aaz6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuchic V., Lurie E., Carrero I., Pawliczek P., Patel R.Y., Rozowsky J., Galeev T., Huang Z., Altshuler R.C., Zhang Z. Allele-specific epigenome maps reveal sequence-dependent stochastic switching at regulatory loci. Science. 2018;361 doi: 10.1126/science.aar3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peccoud J., Ycart B. Markovian modeling of gene-product synthesis. Theor. Popul. Biol. 1995;48:222–234. doi: 10.1006/tpbi.1995.1027. [DOI] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., Peskin C.S., Tranchina D., Vargas D.Y., Tyagi S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006;4:1707–1719. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A., van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008;135:216–226. doi: 10.1016/j.cell.2008.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. G:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B., Mold J.E., Ramsköld D., Deng Q., Johnsson P., Michaëlsson J., Frisén J., Sandberg R. Analysis of allelic expression patterns in clonal somatic cells by single-cell RNA-seq. Nat. Genet. 2016;48:1430–1435. doi: 10.1038/ng.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinius B., Sandberg R. Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat. Rev. Genet. 2015;16:653–664. doi: 10.1038/nrg3888. [DOI] [PubMed] [Google Scholar]
- RV P., Sundaresh A., Karunyaa M., Arun A., Gayen S. Autosomal clonal monoallelic expression: natural or artifactual? Trends Genet. 2021;37:206–211. doi: 10.1016/j.tig.2020.10.011. [DOI] [PubMed] [Google Scholar]
- Saiba R., Arava M., Gayen S. Dosage compensation in human pre-implantation embryos: X-chromosome inactivation or dampening? EMBO Rep. 2018;19:e46294. doi: 10.15252/embr.201846294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni F.A., Stamoulis G., Garieri M., Falconnet E., Ribaux P., Borel C., Antonarakis S.E. Detection of imprinted genes by single-cell allele-specific gene expression. Am. J. Hum. Genet. 2017;100:444–453. doi: 10.1016/j.ajhg.2017.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar M.K., Gayen S., Kumar S., Maclary E., Buttigieg E., Hinten M., Kumari A., Harris C., Sado T., Kalantry S. An Xist-activating antisense RNA required for X-chromosome inactivation. Nat. Commun. 2015;6 doi: 10.1038/ncomms9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M., Zhang J. Allele-specific single-cell RNA sequencing reveals different architectures of intrinsic and extrinsic gene expression noises. Nucleic Acids Res. 2020;48:533–547. doi: 10.1093/nar/gkz1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter D.M., Molina N., Gatfield D., Schneider K., Schibler U., Naef F. Mammalian genes are transcribed with widely different bursting kinetics. Science. 2011;332:472–474. doi: 10.1126/science.1198817. [DOI] [PubMed] [Google Scholar]
- Tunnacliffe E., Chubb J.R. What is a transcriptional burst? Trends Genet. 2020;36:288–297. doi: 10.1016/j.tig.2020.01.003. [DOI] [PubMed] [Google Scholar]
- Wainer-Katsir K., Linial M. BIRD: identifying cell doublets via biallelic expression from single cells. Bioinformatics. 2020;36:i251–i257. doi: 10.1093/bioinformatics/btaa474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Use R! Springer International Publishing; 2016. ggplot2:Elegant Graphics for Data Analysis. [DOI] [Google Scholar]
- Zhao D., Lin M., Pedrosa E., Lachman H.M., Zheng D. Characteristics of allelic gene expression in human brain cells from single-cell RNA-seq data analysis. BMC Genomics. 2017;18:860. doi: 10.1186/s12864-017-4261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwemer L.M., Zak A., Thompson B.R., Kirby A., Daly M.J., Chess A., Gimelbrant A.A. Autosomal monoallelic expression in the mouse. Genome Biol. 2012;13 doi: 10.1186/gb-2012-13-2-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets. The code used for simulation is available at https://github.com/csbBSSE/aRME. For questions regarding the raw data from the current study, please contact the lead contact. All software's used in this study are commercially available.