

Summary:
Meningioma-1 (MN1) overexpression in AML is associated with poor prognosis, and forced expression of MN1 induces leukemia in mice. We sought to determine how MN1 causes AML. We found that overexpression of MN1 can be induced by translocations that result in hijacking of a downstream enhancer. Structure predictions revealed that the entire MN1 coding frame is disordered. We identified the myeloid progenitor-specific BAF complex as the key interaction partner of MN1. MN1 over-stabilizes BAF on enhancer chromatin, a function directly linked to the presence of a long polyQ-stretch within MN1. BAF over-stabilization at binding sites of transcription factors regulating a hematopoietic stem/progenitor program prevents the developmentally appropriate decommissioning of these enhancers and results in impaired myeloid differentiation and leukemia. Beyond AML, our data details how the overexpression of a polyQ protein – in the absence of any coding sequence mutation – can be sufficient to cause malignant transformation.
Keywords: Meningioma-1, BAF, SWI/SNF, leukemia, AML, IDP, polyQ
eTOC blurb
Meningioma-1 (MN1) translocations result in overexpression of MN1 through enhancer hijacking, or expression of an MN1-fusions protein. MN1 is an intrinsically disordered polyQ protein. MN1 overexpression is sufficient to cause malignant transformation via over-stabilization of the BAF complex at critical enhancers.
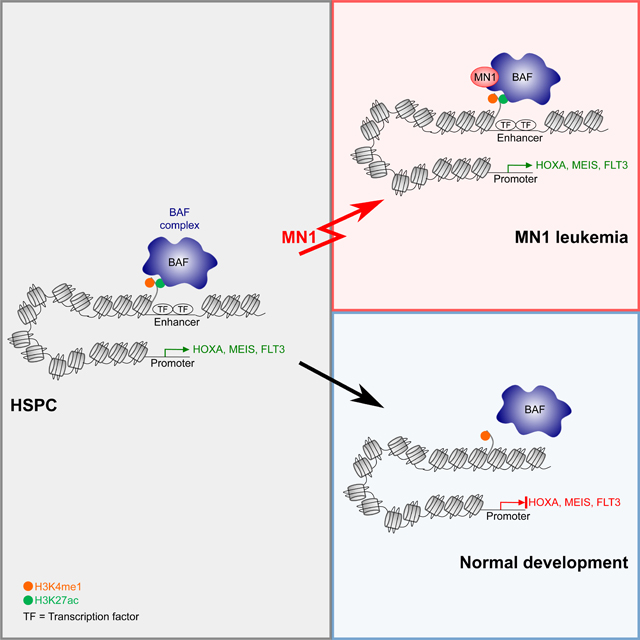
Graphical Abstract
Introduction:
Acute myeloid leukemia (AML) is an aggressive myeloid malignancy (Dombret and Gardin, 2016). Meningioma-1 (MN1) is a transcriptional coactivator that undergoes rearrangements in AML and brain tumors. Most of the coding frame of MN1 is confined to a single large exon, which serves as the N-terminal fusion partner in MN1-ETV6 and MN1-STAT3 fusions in AML, MN1-FLI1 in AMKL (Buijs et al., 1995; Buijs et al., 2000; Dang et al., 2017; Grosveld, 2007; Wang et al., 2020), and MN1-BEND2 or MN1-CXXC5 fusions in brain tumors (Sturm et al., 2016). Notably, no fusion RNA or fusion protein could be identified in the majority of MN1-translocation FISH+ AML and brain tumors (Sturm et al., 2016; Wang et al., 2020). MN1 translocations without the generation of a fusion protein result in high expression of MN1 (Wang et al., 2020).
Overexpression of full length MN1 in mouse bone marrow progenitors induces AML as a single hit (Heuser et al., 2011). Furthermore, side-by-side comparison of full length MN1 and MN1-FLI1 revealed that both are capable of inducing serial replating in methylcellulose and leukemia in mice. The major contribution of the FLI1 fusion partner in this model is a skewing of the phenotype towards the megakaryocytic lineage, resulting in AMKL in animals transplanted with MN1-FLI1 transduced bone marrow progenitors, and myelomonocytic AML in animals transplanted with MN1 only transduced progenitors (Dang et al., 2017; Wenge et al., 2015). This recapitulated the phenotype observed in patients. In leukemias with MN1-ETV6 translocations, the disruption and resulting haploinsufficiency of the ETV6 may contribute to leukemogenesis.
High expression of MN1 occurs across multiple cytogenetic subgroups and has been associated with chemotherapy resistance and poor outcome (Haferlach et al., 2012; Heuser et al., 2006; Langer et al., 2009; Metzeler et al., 2009; Pogosova-Agadjanyan et al., 2020; Thol et al., 2012; Wang et al., 2020; Xiang et al., 2013). How MN1 causes leukemia is poorly understood. MN1 has no sequence homology to any other protein, yet the protein is conserved among species. Relatively little is known about its functions. MN1 binds to chromatin of actively transcribed loci (Heuser et al., 2011) and increases transcription in the presence of sequence specific transcription factors and CBP/p300, but does not have direct transactivating activity itself (Sutton et al., 2005; van Wely et al., 2003). Furthermore, a recent bioinformatics-based study excluded MN1 as a potential DNA-binding protein (Lambert et al., 2018), suggesting that the interaction with DNA is likely indirect. The molecular mechanism of how MN1 modulates transcription is not understood. Furthermore, MN1 has no validated interaction partners. Here, we investigate how high expression of MN1 causes malignant transformation in AML.
Results:
High expression of full length MN1 in t(12;22) AML is the result of enhancer hijacking.
We first sought to further characterize MN1-ETV6 translocations with and without the generation of an MN1-fusion protein in human AML. Specifically, we asked whether MN1 translocations resulted in placing MN1 in the proximity of downstream regulatory regions that could induce the high expression of MN1 or MN1-FP in a panel of cell lines with a t(12;22) translocation between the MN1 and ETV6 loci (Figure 1A): In UCSD-AML1 cells, the MN1 breakpoint is located in intron 1, resulting in an MN1-ETV6 fusion RNA and protein. In Mutz-3 and AMU-AML-1 cells, the breakpoint is located in the 3’UTR in exon 2, therefore no fusion protein can be identified (Gotou et al., 2012) (Figure 1B). We performed ChIP-seq for active chromatin marks (H3K27ac, H3K4me1) and Mediator (MED1) associated with enhancer regions (Figure 1C, 1D top panel, and S1A). ChIP-seq identified a large intragenic and downstream regulatory region in the ETV6 locus in cells with MN1-ETV6 translocations - with and without fusion protein (Figure 1D top panel and S1A, right panel), as well as in non-MN1 translocated control AML cell lines (Molm14, Monomac6). H3K27ac was detected on the MN1 promoter in t(12;22) UCSD-AML1, Mutz-3 and AMU-AML1 cells, regardless of whether a fusion protein was generated, but not in Molm14 or Monomac6 control cells (Figure 1C and S1A, left panel). To investigate putative 3D chromatin looping between the MN1 promoter and ETV6 regulatory sites, we performed circular chromosome conformation capture analysis (4C) in UCSD-AML1 and Mutz-3 cells (both have a t(12;22) translocation, with a fusion protein generated in UCSD-AML1, but no fusion protein in Mutz-3) (Figure 1D, bottom panel,1E and S1B–D). Molm14 and MonoMac6 without translocation were used as control. AMU-AML1 cells were no longer available and therefore could not be included. 4C analysis results clearly demonstrated significant physical contact between the MN1 promoter and the multiple ETV6 downstream regulatory regions including the ETV6 enhancer in UCSD-AML1 and Mutz-3 cells but not the control cell lines as analyzed by PeakC (Geeven et al., 2018) (Figure 1D+E and S1B–D). Furthermore, the ETV6 enhancer demonstrated characteristics of a super enhancer based on the ROSE algorithm (Figure 1F). High expression of MN1 in t(12;22) translocated leukemias without a fusion protein, including AMU-AML1 and Mutz-3, has previously been reported on both a protein and RNA level (Gotou et al., 2012; Riedel et al., 2016; Wang et al., 2020), and is re-confirmed in UCSD-AML1 and Mutz-3 cells using qPCR (Figure 1G, AMU-AML1 cells were no longer available). Taken together, our results demonstrate that the t(12;22) translocation results in hijacking of enhancer regions within and downstream of the ETV6 locus: the MN1 promoter contacts the strong ETV6 enhancer, resulting in high expression of full length MN1 in Mutz-3, and an MN1-ETV6 fusion protein in UCSD-AML1 cells. Interestingly, STAT3 and FLI1 – the two other described fusion partners of MN1 – also are regulated by large downstream enhancers (Figure S1E–G). This suggests that a similar mechanism drives MN1 or MN1-FP expression in these leukemias. Our findings solidify the role of MN1 as a bona fide oncogene in human leukemia.
Figure 1. The MN1 promoter interacts with ETV6 regulatory regions in t(12;22) leukemias with and without MN1 fusion protein.

(A) Schematic of t(12;22) translocations.
(B) Schematic of MN1 and ETV6 loci involved in t(12;22) translocation in UCSD-AML1, Mutz-3 and AMU-AML1 cells. UCSD-AML1 cells express a MN1-ETV6 fusion protein. Translocations in Mutz-3 and AMU-AML1 cells involve the native MN1 stop codon in the fusion. As a result, no fusion protein is generated, and only full-length MN1 is expressed on an RNA and protein level.
(C) ChIP-seq tracks for H3K27ac (top) and H3K4me1 (middle) in t(12;22) cells UCSD-AML1 (red), </p/>Mutz-3 (orange) and AMU-AML1 (yellow), as well as control cells without MN1-translocation Molm14 (dark blue) and Monomac6 (light blue). On the left side the MN1 locus is shown, the right side shows the ETV6 locus. The black boxes marks a region within and downstream of ETV6 identified as interacting with the MN1 promoter by 4C (see (D)).
(D) Schematic of 4C experiment anchored at the MN1 promoter.
(E) Interaction between the MN1 promoter and ETV6 enhancer / regulatory elements in t(12;22) UCSD-AML1 (MN1-ET6 FP) and Mutz-3 (no FP), but not Molm14 or Monomac6 control cells. Blue: 4C sequencing tracks, red: statistically significant interaction acc PeakC (Geeven et al., 2018).
(F): ROSE algorithm identifies the ETV6 enhancer as super-enhancer in UCSD-AML1, Mutz-3 and AMU-AML1 cells.
(G): high expression of MN1 in t(12;22) UCSD-AML1 (as part of a MN1-ET6 FP) and Mutz-3 (no FP), but not Molm14 or Monomac6.
See also Figure S1
MN1 interacts with the myeloid progenitor specific BAF complex
We next set out to determine how high levels of MN1 cause AML. MN1 is not known to play a role in normal hematopoiesis. MN1’s physiological function primarily relates to embryonic craniofacial development. The Mn1 knockout mouse is not viable after birth due to palatal abnormalities (Meester-Smoor et al., 2005), precluding studies of postnatal hematopoiesis. However, this model afforded an opportunity to interrogate fetal liver hematopoiesis. We found no numeric abnormality in fetal liver stem and progenitor populations at day E16.5 (Figure S2). Consistent with our observation, deleting MN1 had no significant effect on the clonogenic potential of human CD34+ stem and progenitor cells (Sharma et al., 2020). The available data on MN1 in normal hematopoiesis thus provides no immediate insight as to MN1’s ability to drive hematologic malignancy. We therefore turned to functional studies using MN1 in leukemia cells. We first determined the MN1 interactome by performing pulldown of the tagged protein in MN1-induced leukemias followed by mass spectrometry (CoIP-MS, Figure 2A, Table S1). For confirmation, we also deployed proximity-dependent biotinylation followed by mass spectrometry in common myeloid progenitors (CMPs, Figure S3A, Table S2) as well as 293T cells (Bio-ID (Roux et al., 2012), Figure 2B, Table S2). Pull downs in leukemia cells were performed in the presence of high concentrations of chymostatin as MN1 is rapidly degraded upon cell lysis by myeloid cell specific proteases (Zhong et al., 2018). CoIP-MS of tagged MN1 in murine MN1-driven leukemia cells identified three major components: the BAF (mSWI/SNF) complex, the Kmt2c/d complex, and, with the lowest abundance the spliceosome (Figure 2A). The members of the spliceosome identified in this experiment were different from spliceosome members identified with the BioID-system in transfected 293T cells (Fig 2B). No spliceosome components were identified in MN1-BioID transduced murine CMPs (Figure S3A). Most likely, the identification of spliceosomal proteins represents a contaminant. The complex with the second highest abundance, the COMPASS-like complex including Kmt2d (Mll2/4), was identified in murine MN1 leukemia cells (Figure 2A) as well as in 293T cells using BioID (Figure 2B). The BAF complex was supported by all three experimental setups (Figure 2A+B, Figure S3A). The highest abundance of detected peptides belonged to members of the BAF complex.
Figure 2. MN1 interacts with the myeloid progenitor specific BAF complex to drive AML.

(A) MN1 CoIP in MN1-driven murine leukemia cells followed by mass spectrometry
(B) MN1 proximity-dependent biotinylation (BioID) in 293 cells followed by mass spectrometry (A+B) Node color represents the average protein abundance (log2 normalized signal intensity) of two independent experiments. Color scale goes from low abundance (blue) to high abundance (red). The canonical BAF complex shows the highest abundance. Connectivity map is based on the STRING database.
(C) Schematic of the BAF, PBAF and non-canonical (nc) BAF complex based on (Mashtalir et al., 2018). Complex defining subunits are depicted in blue. The Smarcd2 subunit (green) has documented roles in myeloid development. Smarca4 (Brg1, red) is the ATPase subunit in hematopoietic progenitor cells, while in hematopoietic stem cells the ATPase subunit is Smarca2 (Brm).
(D) Functional validation of the BAF core ATPase subunit Smarca4 in MN1 leukemia – experimental schematic. CMP cells were isolated from Smarca4 wt and Smarca4 f/f littermates. Cells were transduced with MN1 (GFP), sorted for GFP, then transduced with cre recombinase (dTomato). Sorted cells were injected into mice and subjected to in vitro assays.
(E and F) Results from replating assays in semisolid medium. Colonies (E) and total cells (F) were counted every 5–6 days for a total of 3 platings. Graph shows mean and SD, n = 3 individual experiments, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (unpaired t-test)
(G) Leukemic burden of Smarca4f/f and Smarca4−/− MN1 AML cells (GFP+) in the peripheral blood of mice 20 days after transplant as detected by flow cytometry., n=5–6 animals per group, ****p<0.0001 (unpaired t-test)
(H) Kaplan Meier analysis of mice transplanted with Smarca4f/f and Smarca4−/− MN1 AML cells. # denotes mice that succumbed to leukemia not fully deleted for Smarca4. Two individual CMP donors per group, n=5–6 animals per group. p<0.05 (Cox-Mantel).
Key functions of the BAF complex include ATP-dependent nucleosome remodeling as well as establishing and maintaining enhancers in an active state (Alver et al., 2017; Mittal and Roberts, 2020; Wang et al., 2017). The exact function and composition of BAF complexes is highly context dependent, and there are numerous tissue- and development-specific sub-complexes (Buscarlet et al., 2014; Krasteva et al., 2012; Krasteva et al., 2017; Mashtalir et al., 2018; Michel and Kadoch, 2017; Priam et al., 2017; Pulice and Kadoch, 2016; Schick et al., 2019; St Pierre and Kadoch, 2017; Wang et al., 2017; Witzel et al., 2017). Our leukemia cell specific CoIP-MS specifically identifies the canonical BAF complex based on the presence of Arid1a, Dpf2 (Baf45d), and no detection of PBRM1/BAF180 (Figure 2C – blue subunits). Furthermore, the predominant Smarcd subunit is Smarcd2, which plays roles in myeloid specification, particularly granulocyte development (Figure 2C-green subunit) (Michel and Kadoch, 2017; Priam et al., 2017; Witzel et al., 2017). In both experimental setups based on murine CMP cells we identified Smarca4 (Brg1) as the ATPase subunit (Figure 2A, Figure S3A, Figure 2C – red subunit). The predominant ATPase subunit in long-term hematopoietic stem cells is Smarca2 (Brm). When cells exit the long term stem cell compartment to become short term hematopoietic stem cells or progenitors, Smarca2 is gradually switched out for Smarca4 (Buscarlet et al., 2014). Smarca4 has also been identified as the predominant subunit in several leukemia models, where it is functionally relevant and represents a non-oncogene dependence being required for leukemia cell growth and survival (Buscarlet et al., 2014; Cruickshank et al., 2015; Shi et al., 2013). The ATPase subunit in the BAF complex identified in our experiments thus is the progenitor/leukemia associated Smarca4.
The core ATPase subunit of the MN1-interacting BAF complex, Smarca4, is required for MN1 leukemogenesis.
In order to establish the functional relevance of the BAF complex in MN1-driven murine leukemias, we transduced CMPs from mice with a floxed allele for the catalytic subunit Smarca4 (f/f) or wild type (wt) littermates with MN1. MN1 transformed cells were then transduced with cre-recombinase (cre) and plated in methylcellulose or transplanted into mice (Figure 2D). Smarca4 deleted (f/f cre) cells showed significantly reduced replating ability compared to Smarca4 wt cre cells (Figure 2E+F). Deletion of Smarca4 also significantly reduced day 20 leukemic burden (Figure 2G) and increased the overall survival compared to Smarca4 wt cre transplanted animals (Figure 2H, Figure S3B - D). Of note, all mice succumbing to leukemia in the Smarca4 f/f cre group showed incomplete deletion of the Smarca4 allele (Figure 2H, marked by #). We thus confirmed the absolute requirement of Smarca4 for MN1-induced leukemias since deletion of Smarca4 abrogated serial replating, the leukemia specific differentiation block, and in vivo leukemogenesis.
MN1 colocalizes with the BAF complex on enhancer chromatin.
We next determined MN1 binding sites on chromatin in relation to Smarca4 using ChIP-Seq. We found that MN1 specifically co-localizes with a subset of Smarca4 peaks at active enhancers as demonstrated by H3K27 acetylation and H3K4 monomethylation at the majority of MN1/Smarca4 dual peaks (Figure 3A–C). Motif analysis of MN1/Smarca4 co-occupied loci showed strong enrichment for Ets factors (Erg, Etv2, Fli1) as well as early hematopoietic transcription factor binding sites such as Runx1, Gata2, Myb, and Meis1 (Figure 3D, Table S3) when compared to Smarca4 only occupied sites. Meis1 was previously shown to be present on sites of MN1 binding and essential to MN1 mediated leukemia (Heuser et al., 2011). Myb is ubiquitously overexpressed in AML and promotes proliferation in numerous different AML models (Jiang et al., 2019; Pattabiraman et al., 2014; Roe et al., 2015). In addition to ChIP, we also demonstrate co-localization of BAF complex members and MN1 using immunofluorescence microscopy, staining for MN1 and the BAF complex member Arid1a in MN1-driven murine leukemia cells (Figure 3E and F). In summary, we identify the BAF complex as a functionally important MN1 interaction partner. MN1 colocalizes with the BAF complex at the binding sites of transcription factors that regulate hematopoietic stem and progenitor specific expression programs.
Figure 3. MN1 colocalizes with the BAF complex on enhancer chromatin.

(A) ChIP-seq tracks of Flt3 and Gata2 loci from MN1 driven murine leukemia cells. MN1 track on top (red), below Smarca4 track (blue), H3K27ac (dark grey), and H3K4me1 (light grey).
(B) Distribution of genomic elements associated with MN1, Smarca4, H3K27ac and H3K4me peaks.
(C) Venn diagram of overlap between MN1, Smarca4, H3K4me1 and H3K27ac.
(D) HOMER motif analysis of MN1 and Smarca4 co-occupied regions compared to Smarca4 –only. Number on the left denotes the rank of the shown motif.
(E) Immunofluorescence microscopy images of fixed and stained cells. Scale bars are 10μm. Representative images of MN1-driven murine leukemia cells co-stained for MN1 (red) and Arid1a (green). DNA was counterstained with DAPI (blue).
(F): Threshold overlap score analysis (TOS, (Stauffer et al., 2018)) of the immunofluorescence images in (E). Shown are three individual fields per cell line (representing nearly all the evaluable cells in each experiment).
A full list of the HOMER analysis is available in Table S3.
MN1 is an intrinsically disordered protein, and overexpression redistributes MN1 within the nucleus.
Upon closer inspection of MN1 fluorescent staining in murine MN1 leukemia cells, we noted a punctate pattern that included perfectly round structures suggestive of speckle formation in the nucleoplasm (Figure 4A). A similar staining pattern was observed when staining for MN1-ETV6 in human AML cells with an MN1-ETV6 translocation using the MN1 antibody (Fig. 4B). We next compared MN1 staining at endogenous expression levels and under conditions of overexpression in 293 cells. Long exposure of cells stained for endogenous MN1 revealed a diffuse pattern that appeared to localize to the nucleolus (Fig 4C top row). In contrast, overexpression of MN1 resulted in redistribution of MN1 into nuclear speckle-like structures (Figure 4C, bottom row, and 4D). Formation of such structures is promoted by a sufficiently high concentration of proteins with intrinsic disorder (Chong et al., 2018; Watson and Stott, 2019). The MN1 protein, although highly conserved (Figure S4), is sparsely annotated in the UniProt database, which is largely explained by the lack of sequence homology of MN1 to any other protein. However, MN1 also scored highly as an intrinsically disordered protein (IDP) using several different prediction algorithms (Figure 4E, Figure S5A–H). Two groups previously identified critical regions within MN1 using a series of deletion mutants (Kandilci et al., 2013; Lai et al., 2014). Surprisingly, large portions of the C-terminus of MN1 are dispensable for leukemogenesis. Two regions are absolutely required for the transforming ability of MN1 (Figure 4F shaded in red/orange). Within the second essential region, we noted a stretch of 28 successive Glutamines (polyQ-stretch 2 or pQ2, Figure 4E blue box and 4F). A second Q-rich region is also present at AA291–309 (pQ1, Figure 4E black box, and 4F). PolyQ-stretches are best known as mediators of neurodegenerative disorders such as Huntington’s disease, spinobulbar muscular atrophy (SBMA) and spinocerebellar ataxias (SCA). Several of these disorders share a common underlying cause, the pathogenic massive elongation of a polyQ-stretch. This results in the formation of protein aggregates, which ultimately lead to neuronal cell death.
Figure 4. MN1 is an intrinsically disordered protein with a long polyQ-stretch.

(A) Representative MN1-driven murine leukemia cells stained for MN1 (red) and DNA counterstained with DAPI (blue).
(B) Representative MN1-ETV6 translocated human UCSD-AML1 leukemia cells stained for MN1 (red) and DNA counterstained with DAPI (blue).
(C) Nuclear distribution of MN1 in untransfected 293 cells (top panels, long exposure to capture low baseline level expression of MN1) and in MN1 transfected 293 cells (bottom panels, short exposure). MN1 detection using a polyclonal MN1 antibody
(D) confirmatory MN1 staining in 293 cells transfected with HA-tagged MN1 using an HA-antibody (top panels) or co-expression of an intracellular single-chain variable fragment binding the linear HA epitope (“Frankenbody”, bottom panels)
(E) IUPRED2 prediction for intrinsic disorder of MN1. The x-axis shows the amino acid position, the y-axis shows the score for the predicted disorder with a score closer to 1 indicating higher disorder. Blue box marks the region of the 28-polyQ-stretch that was subsequently deleted (ΔpQ).
(F) Summary of published MN1 deletion studies (Kandilci et al., 2013; Lai et al., 2014; Wenge et al., 2015). Generally, red/orange areas were previously determined as necessary for MN1 oncogenic function, while green areas are dispensable. Specifically, the most N-terminal fragment AA1–200 was absolutely required for leukemia initiation in all studies. In contrast, deletion of AA201–404 did not affect penetrance or differentiation, and only minimally affected latency. This region encompasses the first Q-rich region, a stretch of several consecutive PQQQ motives. A series of deletions in AA404–570, which encompasses the second polyQ-rich region, abrogated leukemogenesis, with only one mutant (Δ458–560+Δ570–1119 (Kandilci et al., 2013)) reported to induce hematopoietic malignancy with incomplete penetrance, long latency and both T-lymphoid and myeloid differentiation. Remarkably, deletion of the large AA570–1119 region did not affected leukemogenesis. MN1 with a deletion in AA1120 – 1320 induced in a completely penetrant and aggressive myeloid neoplasm but failed to induce the profound differentiation block of full length MN1, when combined with deletions in AA570–1119, latency was also increased. Finally, the very C-terminus encoded by exon 2 (AA1250–1320) is not required (Kandilci et al., 2013).
Overexpression of MN1, but not MN1ΔpQ, stabilizes H3K27ac at MN1-bound loci.
The polyQ-stretch in MN1 is one of the longest in the human proteome outside of pathologic elongation (Sorek et al., 2019). PolyQ-stretches are typically found in regions of intrinsic disorder and are important domains facilitating higher order complexes or self-aggregation, thus playing pivotal roles in cellular processes (Wu and Fuxreiter, 2016). More recently, intrinsically disordered regions (IDRs) and IDPs have been identified to form membrane-less organelles to help compartmentalize the nucleus and supply an environment with a highly concentrated and stable association of the transcription machinery (Chong et al., 2018; Sabari et al., 2018; Watson and Stott, 2019). IDRs have also been identified in oncogenic mutants and fusion proteins, and have been demonstrated to play key roles in the ability of these oncogenes to aberrantly activate transcription (Boulay et al., 2017; Wan et al., 2017). Finally, theoretical modeling suggests that over-stabilization of transcriptional hubs alone could be sufficient to induce malignant transformation, but this has not been shown experimentally (Hnisz et al., 2017). We reasoned that the overexpression of MN1 might do precisely that, and that the polyQ-stretch is central to MN1’s activity at these sites.
We thus generated a mutant of MN1 that lacks the polyQ-stretch (MN1-ΔpQ, Figure 4E, blue box, and S6A). We verified that MN1-ΔpQ was expressed at similar levels as wt-MN1, and recognized by a polyclonal antibody (Figure S6B). We then interrogated the effect of MN1 versus MN1ΔpQ on Smarca4 recruitment and the enhancer landscape. MN1 or MN1-ΔpQ and an empty control vector were introduced into HSPCs, and cells were allowed to differentiate for 7 days (Figure 5A and B). At the end of the culture period, Smarca4 binding, H3K27ac and H3K4me1 were determined by ChIP-Seq (Figure 5C). In cells overexpressing MN1 (red tracks), binding sites for MN1 (grey) and Smarca4 overlap with H3K27ac and H3K4me1. This pattern was observed on key transcription factors and signaling molecules in hematopoietic stem and early progenitor cells such as the posterior Hoxa cluster and Flt3, which were previously documented to be critical for MN1 leukemogenesis (Figure 5C). In contrast, Smarca4 binding and H3K27ac at these sites were substantially reduced in untransduced control cells undergoing normal differentiation (Figure 5C light blue tracks and 5E), and in cells overexpressing MN1-ΔpQ (Figure 5C dark blue tracks and 5E). The observed changes in H3K27ac and H3K4me1 in control and MN1-ΔpQ cells were consistent with the published trajectory of these modifications during hematopoietic development (Figure 5C and D) (Lara-Astiaso et al., 2014). We observed a linear correlation between MN1 peak height and the differential in Smarca4 peak height between MN1 and MN1-ΔpQ transduced cells (Figure 5F). On a genome wide level, loci marked with H3K27ac in MN1 but not in MN1-ΔpQ (“dynamic H3K27ac”) were also associated with high MN1 peaks (Figure 5G). Finally, we detected new Smarca4 and H3K27ac peaks emerging in MN1-ΔpQ and control cells at loci associated with differentiation, while ChIP-Seq patterns at these sites in MN1 cells reflected their undifferentiated state (Figure S7A+B). We conclude that the polyQ-stretch is critically important for the recruitment of the BAF complex and sustained H3K27ac at sites of MN1 binding in MN1-transduced cells.
Figure 5. Overexpression of MN1 stabilizes H3K27ac at MN1-bound loci.

(A) Schematic of the experimental setup. Isolated progenitor cells were transduced with either MN1 or MN1-ΔpQ. GFP+ cells and GFP- cells (equals no transduction, noTD) were sorted and maintained in culture for 1 week to allow differentiation, then submitted to ChIP-Seq and RNA-Seq.
(B) Cytospins of cells transduced with the indicated construct at the time point of collection for ChIP-seq analysis (Figure 5) and RNA-Seq (Figure 6) analysis.
(C) ChIP-seq tracks of murine MN1 leukemias and in vitro MN1 transduced or untransduced control HSPCs. The grey MN1 track at the top shows MN1 binding and the dark red track underneath (“MN1-leuk”) shows Smarca4 binging peaks in established leukemias as a reference. The remaining tracks refer to short term cultured cells as shown in A+B. Vertical labeling on the left (“ChIP”) indicates which marks are shown in the respective tracks. From top to bottom, MN1, Smarca4, H3K27ac, H3K4me1. Adjacent horizontal labeling (“TD” = transduction) indicates the construct used to transduce HSPCs or establish leukemias: MN1 (red) or MN1-ΔpQ (dark blue, “ΔpQ”). Untransduced control cells are light blue (“no TD” = no transduction). Shown are two loci with known functions in AML that display high MN1 peaks and aberrantly maintained Smarca4 binding and H3K27 acetylation, the Hoxa-cluster on the left and Flt3 on the right.
(D) ChIP-seq tracks for H3K27ac and H3K4me1 at the Hoxa cluster and Flt3 locus during normal myeloid development. Tracks were obtained from publication Lara-Astiaso et al. Vertical labeling on the left (“ChIP”) indicates which marks are shown in the respective tracks. Adjacent horizontal labeling indicates the cell type. Common myeloid progenitors (CMP, red), granulocyte macrophage progenitors (GMP, dark grey), monocytes (Mono, light grey), granulocytes (Gran, blue).
(E) Tag density plot for the indicated proteins/chromatin marks of Smarca4 bound loci anchored on MN1 in full length MN1 (“F”), MN1-ΔpQ (“Δ”) transformed cells, and untransduced control cells (“N”).
(F) Correlation of MN1 peak height over background (y-axis) with the differential of Smarca4 peak height (x-axis) at loci that loose Smarca4 in MN1-ΔpQ transduced cells (ΔSmarca4, x-axis). R = Pearson correlation.
(G) Violin plot of the genome-wide correlation between dynamic or static H3K27ac peaks (x-axis) and MN1 peak height (y-axis). ChIP-seq identified H3K27ac peaks that are only present in MN1, but not MN1-ΔpQ transformed or untransduced differentiating bone marrow progenitors after 7 days of culture (“dynamic”). In contrast, “static” H3K27ac are present in all the conditions. Dynamic H3K27ac peaks were associated with greater MN1 peak height at that locus (***p=0.0002, Mann Whitney).
See also Figure S7
Overexpression of MN1, but not MN1ΔpQ, stabilizes the binding of the BAF complex to chromatin.
We next performed chromatin fractionation on bone marrow progenitors overexpressing either MN1 or MN1-ΔpQ to directly test whether MN1 mediates tighter binding of the BAF complex to chromatin than MN1-ΔpQ. Western blot analysis as well as quantitative MS show that transduction with MN1 indeed resulted in more stable binding of key BAF complex members to chromatin compared to MN1-ΔpQ controls (Figure 6 A–C, Table S4).
Figure 6. Overexpression of MN1, but not MN1-ΔpQ, aberrantly stabilizes the binding of the BAF complex to chromatin.

(A-C) Chromatin fractionation of bone marrow progenitor cells overexpressing either MN1 or MN1-ΔpQ. Increasing salt concentrations are used to elute proteins bound to chromatin. Proteins with tighter binding need higher salt concentrations to be eluted. Top Panel: Normalized iBAQ quantified peptide abundance in indicated chromatin fractions measured by mass spectrometry. Bottom panel: Western blot images for the respective fractionations. Three key BAF complex members Smarca4 (A), Arid1a (B), and Smarcd2 (C) are shown in the presence of either MN1 or MN1-ΔpQ.
(D) RNA-seq volcano plot showing genes downregulated (left, blue) and genes upregulated (right, orange) in MN1-ΔpQ transformed cells compared to MN1. Labels indicate downregulated MN1 target genes. n=4 for MN1-ΔpQ (includes n=1 noTD), n=3 for MN1.
(E) RT-qPCR confirmation of four key MN1 target loci, Hoxa9, Hoxa10, Meis1, and Flt3, for the three conditions MN1 (red, n=2), MN1-ΔpQ (dark blue, n=2), and noTD (light blue, n=1).
(F) H3K27ac ChIP-Seq signal height and position upstream, downstream and over the coding frame of genes that are part of the leukemogenic MN1 program as defined by Heuser and colleagues (Heuser et al., 2011) and downregulated in MN1-ΔpQ (blue line) compared to MN1 (red line) transformed cells. A set of genes with matched expression levels in MN1 transformed cells that were not part of the MN1-program served as controls.
(G-L) GSEA analysis of differentially regulated genes in MN1-ΔpQ transformed cells compared to MN1. (G) The leukemogenic MN1 program as defined by Heuser and colleagues (Heuser et al., 2011).
(H) Genes downregulated after HOXA9 knock down (KD) (geneset: HOXA9_DN.V1_DOWN). (I) Genes dependent on Myb in the KMT2A/NRASG12D R2 cells (Roe et al., 2015). (J) Gene set associated with myeloid cell development (geneset: BROWN_MYELOID_CELL_DEVELOPMENT_UP). (K) Genes upregulated after HOXA9 knock down (KD) (geneset: HOXA9_DN.V1_UP). (L) Genes regulated by CEBPα. in the KMT2A/NRASG12D R2 cells (Roe et al., 2015)
All genesets can be found in Supplemental Table S5.
We next determined the transcriptional effect of transducing HSPCs with either wild type MN1 or MN1-ΔpQ (Figure 6D). Loss of Smarca4 binding and H3K27ac at MN1 target genes such as Hoxa9, Hoxa10, Meis1 and Flt3 was associated with the downregulation of these genes (Figure 6D+E). On a genome wide level, expression of the “MN1-program” (Heuser et al., 2011) was enriched in wild type MN1 vs MN1-ΔpQ, and downregulation of this program was associated with a decrease in H3K27ac and BRG1 peak height (Figure 6F+G, Figure S7 + 8, Table S5). Furthermore, functional targets of Myb (Roe et al., 2015) and Hoxa9 (Faber et al., 2009) (Figure 6H+I, Figure S8, Table S5) in KMT2A-MLLT3 driven AML were enriched in wild type MN1 vs MN1-ΔpQ. In contrast, genes associated with myeloid development, such as a myeloid development signature described by Brown and colleagues (Brown et al., 2006), and genes upregulated after knockdown of Hoxa9 (Faber et al., 2009) (Figure 6J–K, Figure S8, Table S5) are enriched in MN1-ΔpQ. Work by Kandilci and colleagues suggested that low levels of CEBPα plays a key role in MN1-driven AML, critically contributing to the differentiation block observed in this AML. Overexpression of CEBPα was able to induce differentiation in MN1-AML (Kandilci and Grosveld, 2009). We found strong enrichment of genes regulated by CEBPα in MN1-ΔpQ, confirming the critical role of CEBPα. in driving myeloid differentiation in MN1-AML (Figure 6L, Figure S8, Table S5).
The polyQ-stretch is required for the leukemogenic activity of MN1.
Finally, we investigate the role of the polyQ-stretch in the ability of MN1 to induce serial replating and in vivo leukemogenesis (Figure 7A). Transduction of CMPs with wild type MN1 induced serial replating and a powerful differentiation block as previously reported (Heuser et al., 2011) (Figure 7B+C, Figure S8A). In contrast, MN1-ΔpQ transduced CMPs expanded during the first plating in methylcellulose but did not replate (Figure 7C, Figure S9A). Resultant colonies from the first plating consisted of mostly mature myeloid forms (Figure 7B). Myeloid differentiation in untransduced control cells and MN1-ΔpQ cells was associated with significantly reduced cell cycle (Figure S9B+C). We did not observe a significant increase in cell death/apoptosis in MN1-ΔpQ cells (Figure S9D). In vivo, MN1-ΔpQ cells failed to induce leukemia in recipient mice (Figure 7D+E). This demonstrates the critical requirement of the polyQ-stretch for the leukemogenic activity of MN1.
Figure 7. The MN1 polyQ-stretch is required for leukemogenesis.

(A) Experimental schematic. Isolated CMPs are transduced with either MN1 or MN1-ΔpQ, GFP+ cells are sorted and injected into mice or used for in vitro assays.
(B) Cytospins of cells transduced with the indicated construct after the first methylcellulose plating (shown in C). Representative images of MN1 transduced cells (left) and MN1-ΔpQ transduced cells (right).
(C) Replating assay in semisolid medium. Colonies were counted every 5–6 days for four platings total. MN1 (red triangles) and MN1-ΔpQ (blue circles) represent data from five independent experiments, each plated in duplicate. The empty-GFP control (grey squares) shows data from one experiment plated in duplicates. **p<0.01, ***p<0.001, (unpaired t-test)
(D) Leukemic burden in mice transplanted with MN1 (red triangles) or MN1-ΔpQ-transduced cells (blue circles) in mice 17 days after transplantation. Peripheral blood of transplanted mice was collected and GFP+ cells were detected by flow cytometry. Shown are the mean and SD, n=5–6 animals per group, ****p<0.0001 (t-test)
(E) Kaplan-Meier analysis of mice transplanted with MN1- (red triangles) or MN1-ΔpQ-transduced cells (blue circles). n=4–5 animals per group. p<0.005 (Cox-Mantel).
See also Figure S9
Discussion
70–80 percent of patients with AML with translocations or high expression of MN1 die within 2 years of diagnosis (Haferlach et al., 2012; Heuser et al., 2006; Langer et al., 2009; Metzeler et al., 2009; Thol et al., 2012; Wang et al., 2020; Xiang et al., 2013). How MN1 causes AML had so far been a mystery, as its structure is unresolved, and no interaction partners were known. Using a proteomics approach, we identified the canonical BAF complex as the main interaction partner of oncogenic MN1. Specifically, our MN1 pulldown in leukemia cells identified SmarcD2, a subunit with functional importance in myeloid differentiation (Michel and Kadoch, 2017; Priam et al., 2017; Witzel et al., 2017). We also identified Smarca4 as the predominant ATPase subunit. Smarca4 is a known essential gene in several other AML models (Buscarlet et al., 2014; Cruickshank et al., 2015; Shi et al., 2013). In vivo Smarca4 deletion has only moderate effects on hematopoietic stem and progenitor (HSPC) compartments, while more mature myeloid compartments and T-cell maturation are severely affected. Nevertheless, the published differential sensitivity between normal HSPCs and AML on Smarca4 suggests a potential therapeutic window for Smarca4 inhibition in AML (Buscarlet et al., 2014). We show that overexpressed MN1 aberrantly stabilizes this hematopoietic progenitor BAF complex at transcription factor binding sites on enhancers that regulate hematopoietic stem and progenitor specific expression programs. We propose a model where these sites are bound cooperatively by a set of hematopoiesis specific transcription factors, including Meis1, Myb, Runx1, and Gata2, (as has been previously shown in the context of normal hematopoiesis (Wilson et al., 2010) and KMT2A-rearranged leukemia (Roe et al., 2015)). Decommissioning of these enhancers is required for the differentiation stage appropriate downregulation of key loci such as the later Hoxa cluster, Meis1 or Flt3. Specifically, studies by Heuser and colleagues as well as our group have documented the absolute functional requirement of Hoxa9 and Meis1 expression for the initiation and maintenance of MN1-AML (Heuser et al., 2011; Riedel et al., 2016). In our model, MN1 mediated stabilization of the BAF complex at these critical loci is associated with aberrant maintenance of an immature progenitor program resulting in differentiation block, proliferation and leukemia.
The ability of MN1 to stabilize the BAF complex on chromatin is strictly dependent on the presence of the polyQ-stretch in MN1. Deletion of the polyQ-stretch impairs MN1’s ability to over-stabilize the BAF complex binding to chromatin, thus releasing the differentiation block. PolyQ-stretches are best known for their role in neurodegenerative diseases – the so-called polyQ diseases, which includes Huntington’s disease as the most prominent example. In the polyQ diseases, elongation of the polyQ-stretch causes protein aggregates that result in neuronal death and neurodegeneration. Interestingly, overexpression of proteins such as Ataxin-1 or AR with a non-expanded polyQ-stretch in murine models of SCA-1 and SBMA phenocopied clinical and histologic features of polyQ elongation, albeit less severe and with a longer latency (Duvick et al., 2010; Fernandez-Funez et al., 2000; Monks et al., 2007; Nedelsky et al., 2010). This suggests that neurodegenerative pathology in these disorders is caused at least in part by increasing polyQ dosage, which can be achieved either by elongation of the polyQ-stretch, or overexpression of the unexpanded protein. The overexpression of MN1 may thus result in increased polyQ dosage similar to the elongation of the polyQ-stretch in neurodegenerative disease. In contrast to neurodegenerative diseases, the consequence of increased poly-Q dosage is not cell death, but uncontrolled growth and malignant transformation. The unstructured nature of intrinsically disordered regions (IDRs) poses formidable challenges for drug development. Nevertheless, there is interest in developing polyQ disruptors for neurodegenerative diseases, and it is conceivable that such an approach could also have activity in MN1-AML.
IDRs have been identified in several oncogenes, and were critical for the transforming activity of oncogenes such as mutant ENL (Wan et al., 2017) or the EWS-FLI fusion (Boulay et al., 2017). However, both an oncogenic mutation (point mutation or fusion) and the IDR were required. In contrast, overexpression of wild type MN1 is sufficient to induce a rapidly fatal AML. Although some MN1 translocation events do result in the generation of an MN1 fusion protein (MN1-ETV6, MN1-STAT3 and MN1-FLI1), the fusion partner is not required for leukemic transformation. The MN1 leukemia model thus presents strong evidence that the mere overexpression of an intrinsically disordered protein is sufficient to cause leukemic transformation – without the need for any coding sequence mutation.
Our study has several limitations. Most importantly, we have not identified the specific member of the BAF complex that interacts with MN1, or the respective domains within both proteins. Future studies will need to clarify the precise interaction domains. The disruption of protein-protein interactions is emerging as an interesting strategy for targeted leukemia therapy as exemplified by a series of compounds currently in early clinical trials which disrupt the interaction between KMT2A-fusions and their binding partner Menin (Grembecka et al., 2012; Krivtsov et al., 2019). It will also be important to clarify whether MN1 at physiologic expression levels also binds to the BAF complex or whether this is only observed at pathologic expression levels, and what determines which specific subset of SMARCA4 containing BAF complexes that are co-occupied by MN1 on chromatin. Similarly, we were not able to determine whether MNΔpQ still interacts with chromatin. We were unable to ChIP MN1ΔpQ despite similar expression levels to full length MN1 and preserved interaction with the ChIP antibody on IP-Western. However, cell numbers were also limiting due to poor expansion of these cells. Since we cannot exclude a technical failure as an alternative explanation, further studies are required to clarify this point. Another limitation relates to the relatively long timeframe between transduction of progenitor cells with MN1 or MN1ΔpQ and the epigenomic/transcriptomic analysis. When correlating MN1 peak height with changes in Smarca4 and H3K27ac, we observed a linear correlation between MN1 peak height and Smarca4 changes, but not the more indirect H3K27ac changes. H3K27ac loss was still associated with higher MN1 peaks, but the correlation was not linear. This may in part be due to nonuniform effects of BAF complex member alterations on the chromatin landscape of associated loci even when measuring on an extremely short time scale (Schick et al., 2021). In addition, the fact that our analysis also read out large scale secondary changes in chromatin modifications and gene expression made it difficult to distinguish and quantify primary changes induced by MN1 vs MNΔpQ.
Finally, future studies are required to mechanistically evaluate other domains that were previously shown to contribute to, and be required for, MN1’s oncogenic function. Of particular interest are the first 200 amino acids, which were found to be universally required. Another area of potential interest is a second Q-rich region, which by itself has minimal impact on leukemogenesis as reported by Lai and colleagues (Lai et al., 2014). However, it would be interesting to explore whether this region contributes to the hematopoietic malignancies with long latency and incomplete penetrance induced by the Δ458–560+Δ570–1119 variant reported by Kandilci and colleagues (Kandilci et al., 2013).
Our findings have relevance beyond hematopoietic cancers. MN1 translocations are rare in AML (Wang et al., 2020), but MN1 overexpression is common, and may be driven by several other alterations of the non-coding genome. Furthermore, translocations without fusion protein generation are common in pediatric brain tumors (Sturm et al., 2016). It is conceivable that MN1, or other intrinsically disordered proteins, undergo similar events in multiple additional cancers. Enhancer hijacking is not well captured with current clinical diagnostic techniques, thus, these events are easily missed. MN1 translocation events resulting in high MN1 expression exceed the frequency of fusion proteins detectable by RNA-Seq, or translocations visible by cytogenetics. The comparatively high frequency of these translocations particularly in primitive neuroectodermal tumors (PNETs, 17%) was only revealed after FISH probes were specifically developed to search for disruption of the MN1 locus. Future systematic studies of the non-coding genome are warranted and may uncover similar oncogenic events in multiple other cancers.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kathrin M. Bernt, MD (berntk@email.chop.edu).
Materials availability:
All model constructs made for this manuscript will be made available with a simple MTA.
Data and code availability:
4C, ChIP-Seq and RNA-Seq are available through GEO (https://www.ncbi.nlm.nih.gov/geo/): ChIP-Seq of murine leukemias: GSE154602, RNA-Seq: GSE154997, ChIP-Seq of human leukemia cell lines: GSE154985 and GSE171394, 4C: GSE171526. Uncut blots and gels are available at Mendeley: Mendeley Data, V1, doi: 10.17632/8k9shrb22x.1 (https://data.mendeley.com/datasets/8k9shrb22x/1)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals:
Mice for transplants were maintained at the Animal Research Facility at the Children’s Hospital of Philadelphia. Fully backcrossed 6–8 week old female recipient C57BL/6 mice were obtained from Jackson laboratory and allowed to acclimatize for one week prior to transplantation. Mn1+/− mice were obtained from Trevor Williams at the University of Colorado and maintained as heterozygotes. Mice were housed in a ASBL2 SPF barrier facility in group housing (5 animals/cage). 3–6 month old fully backcrossed C57BL/6 male and female donors were used for leukemia generation.
Fully backcrossed C57BL/6 Smarca4f/f were maintained at the Animal Research Facility at the Université de Montréal, Canada. Thigh and hip bones of two 3–6 month old fully backcrossed C57BL/6 donors ( male and female) were used for leukemia generation. Whole bone marrow cells were isolated from donor animals and frozen in 10% DMSO after red cell lysis. Bone marrow cells were shipped to the University of Pennsylvania on dry ice. All animal breeding and experiments were approved by the Internal Animal Care and Use Committees of the respective institutions.
Cell Lines:
Human cell lines were purchased from the American Type Culture Collection (ATCC) and the German Collection of Microorganisms and Cell Cultures (DSMZ). UCSD-AML1 were grown in RPMI 1640 medium supplemented with 10% heat inactivated FBS, 1% Penicillin/Streptomycin, and human GM-CSF (10ng/mL). Mutz-3 were grown in alpha-MEM (with ribo- and deoxyribonucleosides) + 20% heat inactivated FBS, 1% Penicillin/Streptomycin + 20% vol conditioned medium of cell line 5637 (HTB-9). 5637, Molm14 and Monomac6 were grown in RPMI 1640 medium supplemented with 10% heat inactivated FBS, and 1% Penicillin/Streptomycin. Human 293 cells were grown in DMEM medium supplemented with 10% heat inactivated FBS, and 1% Penicillin/Streptomycin. Murine derived leukemia cells were grown in IMDM medium supplemented with 15% heat in activated FBS, 1% Penicillin/Streptomycin, 1% L-Glutamine, murine IL-3, murine IL-6 (10ng/mL), and murine SCF (20ng/mL). All cells were grown in a humidified incubator at 37°C and 5% CO2. Cell lines were authenticated and tested for mycoplasma every 6 months in culture.
AMU-AML1 cell line:
AMU-AML1 cells were originally established from a patient with acute myeloid leukemia with multilineage dysplasia as published by Gotou and colleagues (Gotou et al., 2012). Karyotyping of AMU-revealed a t(12;22) translocation as the only cytogenetic abnormality (46, XY, t(12;22)(p13;q11.2) The breakpoints were mapped to the 5’ untranslated region to intron 1 of TEL and the 3’ untranslated region of MN1. The chimeric transcript and protein of MN1-TEL could not be detected by reverse-transcriptase polymerase chain reaction or Western blot analysis. However, the expression levels of the MN1 transcript and protein were high in AMU-AML1 cells when compared with other cell lines with t(12;22)(p13;q11–12). In the original publication, sustained in vitro proliferation of AMU-AML1 cells is described. However, neither our laboratory nor Dr. Hanamura’s laboratory succeeded in expanding these cells in in vitro culture.
METHOD DETAILS
Chromatin Immunoprecipitation (ChIP):
For histone marks, cells were crosslinked at room temperature for 10 min in 1% formaldehyde in PBS. Crosslinking reaction was stopped by adding Glycine to a final concentration of 0.125 M. All following buffers were supplemented with protease inhibitors (Complete Mini, Roche or Halt, Thermo Fisher Scientific) and 200μM Chymostatin (Cayman) (Zhong et al., 2018), steps were performed at 4°C. Cells were washed twice in ice cold PBS and lysed in ChIP lysis buffer (1% SDS, 10mM EDTA, 50mM Tris-HCl pH8.1). Cells were sonicated with a qSonica sonicator for 25 min. After spinning for 10 min at full speed, supernatant was removed and diluted 1:10 in ChIP dilution buffer (0.01% SDS, 1.1% Triton-X100, 1.2mM EDTA, 16.7mM Tris-HCl pH8.1, 167 mM NaCl). Lysates were incubated with target antibodies overnight at 4°C with rotation. Protein A+G coated magnetic beads were added for 3h, then washed with Low Salt Buffer (0.8% SDS, 1% Triton-X100, 2mM EDTA, 20mM Tris-HCl pH8.1, 150mM NaCl), High Salt Buffer (0.8% SDS, 1% Triton-X100, 2mM EDTA, 20mM Tris-HCl pH8.1, 500mM NaCl), LiCl Buffer (0.25 M LiCl, 1% IGEPAL-CH 630, 1% deoxycholic acid sodium salt, 1mM EDTA, 10mM Tris-HCl pH8.1), and TE Buffer (1mM EDTA, 10mM Tris-HCl pH8.1). Crosslinking was reversed in Elution Buffer (1% SDS, 0.1 M NaHCO3, 200mM NaCl) at 65°C overnight. Beads were eluted adding RNase A (20μg/mL) at 37°C and Proteinase K at 55°C (100μg/mL) for 1h each. DNA was purified using a PCR purification kit according to the manufacturer’s instructions. DNA was quantified using the Qubit assay.
For Smarca4 and HA ChIP, the protocol was the same as described for histone marks with the following differences: cells were crosslinked at room temperature for 50 min in PBS with DSG (Thermo Fisher Scientific) at 2mM final concentration, then formaldehyde was added. Beads were washed twice in Low Salt Buffer (0.1% SDS, 1% Triton-X100, 2mM EDTA, 20mM Tris-HCl pH8.1, 150mM NaCl), twice in High Salt Buffer (0.1% SDS, 1% Triton-X100, 2mM EDTA, 20mM Tris-HCl pH8.1, 500mM NaCl) and once in TE Buffer (1mM EDTA, 10mM Tris-HCl pH8.1).
Western Blotting:
Samples were loaded on 4–12% 10 well precast Bis-Tris gels and run in MOPS running buffer (NuPage). Proteins were wet transferred in 1x transfer buffer (10x stock: 144g Glycine, 30.2g Tris Base, fill to 1L with MilliQ H2O) with 10% methanol and 0.01% SDS using a 0.45μm nitrocellulose membrane (Bio-Rad) and the XCell II Blot Module (Invitrogen) at 16V for 2h on ice. Membranes were blocked in 5% milk in 0.05% TBST for 1h, then antibody at indicated concentrations in 5% milk in 0.05% TBST was added. Membranes were incubated overnight at 4°C while rocking. Antibody was removed and membranes washed three times in 0.05% TBST, secondary antibody coupled to HRP was diluted 1:1000 in 5% milk in 0.05% TBST and added at room temperature for 1h. After three more washes with 0.05% TBST, membranes were incubated with Western Lighting RTM Plus-ECL reagent for 1min and exposed on HyBlot CL Autoradiography Film (Denville Scientific) in the darkroom.
Virus production:
Retroviral supernatants were produced by cotransfecting ΔΨ and the respective MSCV plasmid into 293 cells grown on poly-L-lysine using FuGene6 according to the manufacturer’s instructions. Viral supernatant was harvested, concentrated using PEG, and stored at −80°C.
Protein Immunoprecipitation (IP):
For the BioID system, the protocol was adapted from Roux and colleagues (Roux et al., 2012). All buffers were supplemented with protease inhibitors (Complete mini, Roche) and all IP steps were performed at 4°C. Two 10-cm dishes of 293T cells were transfected with 5μg BioID-MN1, or 5μg BioID-empty and 5μg HA-MN1 using FuGene6 according to manufacturer’s instructions. 24h after transfection, 50μM Biotin was added, IP was started 24h later. Whole cells or nuclei were lysed in 300μL BioID-IP lysis+wash buffer (50mM Tris-HCl pH7.4, 150mM NaCl, 2.5mM MgCl2, 1% Triton-X 100, fresh 1mM DTT). Lysates were sonicated for 40s (Qsonica sonicator) and spun at full speed for 7min in a tabletop centrifuge. The supernatant was filled to 1mL with BioID-IP lysis+wash buffer and 60μL Streptavidin coated beads were added for incubation overnight while rotating. Beads were washed 3 times with BioID-IP lysis+wash buffer and twice with BioID wash buffer (50mM Tris-HCl pH7.4, 50mM NaCl). Beads were eluted by adding a 1:1 mixture of 4x LDS Buffer (NuPage) and 10mM Biotin + Reducing agent (1x final, NuPage) followed by 10min at 95°C. For Co-IPs murine leukemia cells established using Ty1-tagged MN1 (test) or HA tagged MN1 (control) were used. All buffers were supplemented with protease inhibitor (Complete Mini, Roche) and 200μM Chymostatin (Cayman), all steps were performed at 4°C. 4–5 million cells were lysed in 300 μL BioID-IP lysis+wash buffer (described above). 250U Benzonase (Sigma) was added and lysates were incubated for 1h on ice. After spinning at full speed for 8min in a tabletop centrifuge supernatant was removed and filled to 600μL total volume with BioID-IP lysis+wash buffer. 5μg Ty1 antibody and 35μL protein A+G coated beads were added and incubated for 3–4h rotating. Beads were washed twice with BioID-IP lysis+wash buffer and twice with BioID high salt wash buffer (50mM Tris-HCl pH7.4, 500mM NaCl). Beads were eluted by adding 2% SDS in PBS followed by 10 min at 90°C, or room temperature.
Isolation and transduction of bone marrow progenitors:
Freshly isolated or previously frozen murine bone marrow cells were lineage depleted by staining cells with biotinylated lineage antibodies (CD3, CD4, CD8, Gr1, B220, CD19 all 1:100, IL7R 1:30, Ter119 1:400) for 30 min on ice. After washing the cells, streptavidin coated magnetic Dynabeads were added and incubated for 1h. After removal of the beads, the remaining cells were stained with FCɣ-PE, ckit-A647, Sca1-PE-Cy7, Streptavidin-APC-Cy7 (all 1:100), and CD34-FITC (1:30) for 1h. Cells were sorted on an Aria Fusion at the CHOP flow core. CMPs were defined as Lin- ckit+ sca1- CD34 intermediate FCɣ intermediate. LSKs were defined as Lin- ckit+ sca1+. Most experiments were conducted using CMPs as the cell of origin. ChIP experiments in Figure 5 used CMPs and LSKs (“HSPCs”). After sort, cells were prestimulated for 24h in murine bone marrow medium (see cell culture) supplemented with murine Flt3 ligand (20ng/mL) and murine TPO (20ng/mL). Cells were transduced on retronectin coated wells by adding the respective virus. 48 to 72 hours later cells were sorted for fluorescent marker expression on an Aria Fusion (BD) or Astrios EQ (Beckman Coulter).
Timed matings and analysis of fetal liver hematopoiesis:
For fetal liver experiments, timed matings between two MN1+/− animals were set up, females were checked daily for vaginal plugs and separated after mating. Due to hypospermia pairs were monitors for several months before pregnancies were identified. Pups were obtained on day E16.5 and fetal livers were isolated by dissection. In parallel, non-hematopoietic tissue was obtained for genotyping. Fetal liver hematopoietic stem and progenitor cells were isolated by lineage depleting cells with biotinylated lineage antibodies (CD3, CD4, CD8, Gr1, B220, CD19 all 1:100, IL7R 1:30, Ter119 1:400) as detailed above. Lin- cells were stained with CD150-PE, ckit-A647, Sca1-PE-Cy7, Streptavidin-APC-Cy7 (all 1:100), and CD48-Pacific Blue (1:30) for 1h and analyzed by flowcytometry.
Transplantation:
For MN1 or MN1-ΔpQ, bone marrow cells were transduced and sorted as above, and 10,000 cells/mouse were transplanted into 650cGy irradiated mice via tail vein injection. For Smarca4f/f and wt littermate control cell primary transplants, CMPs were sorted as described above. Cells were sequentially transduced with MSCV-MN1-GFP and MSCV-cre-dTomato. Double positive cells were sorted and 70,000 cells/mouse transplanted as described above.
RNA extraction:
RNA was extracted using the Qiagen micro or mini kit according to the manufacturer’s instructions. Resulting RNA was stored at −80°C until further use.
Flow cytometry:
To measure engraftment of transplanted cells, mice were bled 17–22 days after transplant and blood was analyzed for GFP expression.
Cell cycle was determined using the Click-iT EdU A647 flow cytometry assay kit according to the manufacturer’s instructions. DAPI was added to visualize DNA content.
Cell death/apoptosis was determined by staining with Annexin V APC and DAPI.
Differentiation status of cells was determined by staining cells with Ly-6G/Ly-6C PE-Cy7 (Gr1) and CD11b APC (Mac-1) for 30min in PBS with 2% FBS and 1% P/S on ice.
For all experiments data was acquired on a Cytoflex S flow cytometer (Beckman Coulter) and analyzed using FlowJo and Graphpad Prism software.
Replating assay:
Murine cells were plated in M3234 Methylcellulose supplemented with 1% P/S, murine IL-3 (10ng/mL), murine IL-6 (10ng/mL), and murine SCF (20ng/mL), and 18mL IMDM medium. Cells were replated every 5–6 days.
Immunofluorescence:
Murine leukemia cells:
Cells were washed twice in PBS then resuspended in PBS and pipetted into 16-well glass slides (LabTek chamber slides). After letting the cells settle for 30 min at RT cells were fixed with 2–4% PFA in PBS for 10 min at RT. Cells were washed 3 times with PBS, and blocked and permeabilized for 2h at 4°C in Fix+Perm buffer (1% BSA + 0.2% Triton X-100 in PBS). Primary antibodies were diluted in Fix+Perm buffer 1:50 and slides were incubated with antibody O/N at 4°C. Cells were washed 3 times with PBS. Secondary antibodies were diluted 1:500 in Fix+Perm buffer and incubated 2h at 4°C. Cells were washed once in Fix+Perm buffer and twice in PBS. DNA was stained with 280nM DAPI in PBS for 4min at RT followed by 3 washes in PBS. Slides were mounted using Vectashield Vibrance Antifade Mounting Medium and imaged the same day. Images were taken on a Leica DM5000B equipped with a DFC365FX camera using an HCX PL APO 100x/1.40 – 0.70 OIL objective.
Adherent HEK293 cells:
Cells were grown on 16-well glass slides (LabTek chamber slides). Transfection with the respective plasmids was done using FuGENE6 (Promega). After 48h cells were fixed in 2% PFA in PBS for 10 min at RT. Staining was done as described above. For “Frankenbody”-experiments, live cells were incubated with Hoechst dye before fixing in 2% PFA in PBS for 7min at RT. After 3 washes with PBS slides were mounted and imaged as described above.
Overlap between MN1 and ARID1A was quantified using Threshold overlap score analysis (Stauffer et al., 2018).
Chromatin Fractionation Assay:
The protocol described by and Porter and colleagues (Porter et al., 2017) was used. Cells were lysed on ice for 5 min in 100μL HC buffer (20mM Tris-HCl pH7.4, 10mM NaCl, 3mM MgCl2). NP-40 was added to a final concentration of 0.1% and spun for 5 min at 6000g. Pelleted nuclei were resuspended in 100μL RIPA 0 buffer (50mM Tris pH8, 1% NP-40, 0.25% Sodium deoxycholate, 0 mM NaCl) by pipetting up and down 15 times, incubated on ice for 5 min, then spun for 3 min at 6500g. Supernatant was collected and labelled as fraction 0. This was repeated using RIPA buffers with increasing NaCl concentrations. All buffers were ice cold and supplemented with Halt (Thermo Scientific) and 0.2mM Chymostatin (Cayman). All centrifugation steps were carried out at 4C. Equal amounts of each collected fraction were used for analysis by Western Blot.
RT-qPCR:
Extracted RNA was quantified using a Nanodrop 8000 Spectrophotometer. MN1 was quantified using the MN1 Ipsogen kit for clinical absolute quantification of MN1 according to the manufacturer’s instructions. For all other qPCR analyses 1μg RNA was used to generate cDNA using the qScript cDNA Supermix (Quanta Biosciences) according to the manufacturer’s instructions. Reactions were run in technical duplicates using the PowerUP SYBR green MasterMix (Applied Biosciences) and indicated primer pairs on a QuantStudio5 qPCR machine (Applied Biosystems). RT-qPCR primers (mouse):
| Name | Sequence 5′ – 3′ | |
| Actin | F: TATGAGCTGCCTGACGGC; | R: CAGCAATGCCTGGGTACA |
| Hprt: | F: GCAGCGTTTCTGAGCCAT; | R: CATCGCTAATCACGACGC |
| Hoxa9 | F: AGGCAAGGCCAGATTTGA; | R: AGGCAAGGCCAGATTTGA |
| Hoxa10 | F: ATCAGCCGTAGCGTCCAC; | R: AGTTCTGGGGCAGAGGCT |
| Meis1: | F: CACAGGAGACCCGACAATG; | R: CATGTCCCCCGAGTTGAC |
| Flt3: | F: TTCCTCAAAGCACCCCAG; | R: TGTACCCATCCTCCAGGC |
Library Preparation and sequencing of DNA and RNA:
Purified ChIP-DNA and isolated RNA was sent to Genewiz for library preparation and Illumina sequencing. For RNA-Seq, library prep was performed on PolyA selected mRNA species (eukaryotic). Multiplexed sequencing was conducted on Illumina HiSeq Sequencers to a read depth of 20–30 million per sample.
RNA-Seq and GSEA analysis:
Raw short read files were quantified by using Kallisto [version 0.45.0, (Bray et al., 2016)] against reference genome (Human GRCh37 or Mouse GRCm38). Output from Kallisto was then directly imported into DESeq2 (Release 3.1) (Love et al., 2014) in order to identify differentially expressed genes (DEG). DEGs were defined as genes with False Discovery Rate (FDR) less than 0.05, adjusted p-value of ≤ 0.01, and fold change ≥ 1.2. The volcano plot was made using R package “EnhancedVolcano” (Version 1.6, Blighe K, Rana S, Lewis M (2020). EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. R package version 1.6.0, https://github.com/kevinblighe/EnhancedVolcano). All analysis was carried out using R, version 3.6.3. GSEA analysis, plots, and heatmaps were carried out using GSEA (Subramanian et al., 2005) (version 4.0.) downloaded directly from Broad Institute. In order to use GSEA tool evaluating our RNA-Seq outcome, we selected ‘Standard GSEA’ as recommended, using normalized read counts obtained from DEseq2. All default parameters were applied.
ChIP-Seq analysis:
Raw short read files were aligned against reference genome (Human GRCh37 or Mouse GRCm38) using BWA [version 1.19.0, (Li and Durbin, 2009)]. We used the reads aligned to the reference genome to detemine transcription factor binding sites and histone modification levels. We first removed duplicate reads and reads aligned to human mitochondrial DNA in order to improve peak identification accuracy. We then applied Model-based Analysis of ChIP-Seq (MACS2) (Zhang et al., 2008) to the bam files to predict histone marker binding peaks (broad peaks option) or transcription factor binding peaks (narrow peaks option). Macs2 output of fold enrichment for peak’s summit against random Poisson distribution with local lambda was used to represent peak relative height. We removed any peaks identified overlapping ENCODE blacklist for human [http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg19-human] or for mouse [http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse] (Amemiya et al., 2019). To identify and rank super-enhancer regions we employed the ROSE tool with all default parameters (http://younglab.wi.mit.edu/super_enhancer_code.html) (Whyte et al., 2013). Homer (annotatePeaks.pl) was used to associate each identified super-enhancer region with gene annotation. The figures were generated using in house R code.
To assess the degree of correlation between MN1 peak height and loss of Smarc4 we measured peak height difference between MN1_Smarca4 and MN1pQdel_Smarca4 (Diff_Smarca4) at peaks identified by MACS2. We determined the degree of correlation between peak height of MN1 (over input) and Diff_Smarca4 using R function “cor”. The signficance of correlation is estimated using R function “cor.test”. We employ R package ggplot2 to plot the figure.
Homer (Heinz et al., 2010) “Annotating Regions in the Genome program” (annotatePeaks.pl) was used to annotated identified ChiP-Seq peak regions using MACS2. Ngs.plot (Shen et al., 2014) was applied to plot ChIP-Seq data average profiles and heatmaps. Motif analysis was conducted using the Homer tool ‘findMotifsGenome’. We used all Samrca4 detected peaks as our background in ‘findMotifsGenome’ while searching MN1 and Smarca4 co-occupied peak motifs. Motif logo plots were generated by Homer. Identified motifs were inspected carefully. Any dubious outputs were removed as suggested by Homer program manual.
4C Experimental Work Flow and Analysis:
4C was performed as described in (Krijger et al., 2020; van de Werken et al., 2012a; van de Werken et al., 2012b). In short, 10 million cells were crosslinked for 10 minutes at room temperature in a 2% formaldehyde buffer. Fixation was quenched by adding glycine (0.13M final concentration) and cells were spun down at 4 degrees at 500g for 3 minutes. Cells were washed in cold PBS twice and subsequently lysed for 10 minutes on ice. Nuclei were washed in cold PBS, flash frozen and stored at −80 degrees. After sample collection for all four cell lines, nuclei pellets were thawed on ice and resuspended in 1x restriction buffer. For the first restriction, DpnII enzyme was used. The first ligation was performed in 1 ml total volume overnight, followed by purification of the 3C template using phenol/chloroform extraction and ethanol precipitation. Second restriction digestion was done using Csp6I enzyme followed by a second ligation in 7 ml total volume. 4C template was purified using phenol/chloroform extraction and ethanol precipitation. DNA pellet was resuspended in 500ul mQ and purified once more using Ampure beads to a 1:1 ratio. Four first round PCR reactions with 200ng 4C template each were performed as described in (Krijger et al., 2020) the MN1 promoter viewpoint with the following primer pairs:
TACACGACGCTCTTCCGATCTGTTGGCCTCCTTAAGAGATC (reading 1) plus
ACTGGAGTTCAGACGTGTGCTCTTCCGATCGAGCCGAAGTGCAGTAGA (nonreading 1) and
TACACGACGCTCTTCCGATCTGGATATATACTCTTAAGACACAAAGGATC (reading 2) plus
ACTGGAGTTCAGACGTGTGCTCTTCCGATCGGAGGGGTCTTAGTGTGAAA (nonreading 2).
First round PCR reactions were pooled (4× 50ul) and 50/200 ul was purified using 50ul Ampure beads. Elution was done in 50 ul mQ and 10 ul of the first PCR product was used for the second round PCR using the Truseq universal primer AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT and a barcoded reverse RPI primer CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCT. PCR products were purified using Roche high pure PCR product purification kit (11732676001) according to manufacturer’s protocol, pooled and sequenced 1× 75 basepairs on a Illumina Nextseq 550. 4C reads were filtered and processed using the 4C pipeline (de Wit lab; https://github.com/deWitLab/4C_mapping) and mapped to the Etv6 locus. The peakC R package (de Wit lab; https://github.com/deWitLab/peakC) (Geeven et al., 2018) was used to call peaks in the 4C data with the following parameters: wsize=41, qWd=3.5, qWr=4. Significantly called peaks for UCSD-AML1 and Mutz-3 are indicated in Supplementary Figure 1 in red and 4C peaks called in both samples are highlighted as red boxes in Figure 1.
Alignment File Visualization:
ChiP-Seq alignment files were visualized using Integrative Genomics Viewer (IGV) (Robinson et al., 2011).
Mass Spectrometry (MS)-based proteomics analysis:
Sample preparation for bottom-up proteomics analysis was performed using S-trap (Protifi, Huntington, NY) following the manufacturer’s protocol with slight modifications. Protein was firstly reduced with 10 mM DTT for 30 min at 45 °C, and then alkylated with 20 mM IAA for 30 min at room temperature in the dark. Additional SDS was added to make the final concentration as 5% before proteins were transferred to the S-trap. Trypsin in 50 mM TEAB solution was used to digest 1.5 hour at 47°C. Peptides were eluted using 50% acetonitrile/0.2% formic acid, vacuum centrifuged to dryness, and kept at −20 °C before further analysis.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed using either Thermo-Fisher Orbitrap Fusion equipped with nano-EasyLC 1000 (Co-IP samples) or Q Exactive HF-X coupled with Ultimate 3000 nano-LC (Chromatin fractionation samples). Tryptic peptides were desalted using C18 stage tip, and separated on a home-packed capillary column (75 μm * 20 cm, 3 μm C18-AQ resin) using a 100 min gradient (2–5% B for 0–5 min, 5–35% B from 5 to 95 min, 35–95% B from 95 to 100 min and wash for 5 min at 95% B before return to 2% B for re-equilibration) at a flow rate of 400 nl/min. Mobile phase A consisted of 0.1% formic acid in water, and mobile phase B consisted of 0.1% formic acid in 80% acetonitrile. Full scan mass range of m/z 300–1500 was analyzed in the Orbitrap at 60,000 FWHM (200 m/z) resolution and 5.0e5 AGC target value with maximum injection time to 50 ms. Determined charge states between 2 and 5 were required, and 60 s dynamic exclusion window was used with isotopes excluded. For Fusion, MS/MS was performed in normal mode ion trap with the TopSpeed (3 seconds) using data-dependent acquisition. HCD collision energy was set to 27% with stepped collision energy 3%, AGC target to 1.0e4 and maximum injection time to 70 ms. For HF-X, MS/MS was performed in Orbitrap at 15,000 with TopN (top 15) using data-dependent acquisition. HCD collision energy was set to 28%, AGC target to 1.0e5 and maximum injection time to 50 ms.
Raw files were analyzed by MaxQuant (v1.6.0.16) against Uniprot mouse proteome (download on 20180703) plus human MN1 and common contaminant database. The search included fixed modification of carbamidomethyl cysteine and variable modifications of methionine oxidation, and N-terminal acetylation. All other values used default settings for label-free quantification with match between enabled. Proteins labeled as ‘only identified by site’, or ‘reverse’ or ‘potential contaminant’ were excluded from further analysis. Protein intensities are iBAQ log2 transformed and normalized by subtracting the average.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Graphpad Prism. Statistical details of standard molecular and cell biology experiments as well as in vivo experiments (number of experiments, replicates, definition of center, and dispersion and precision measures, p-value definition, statistical test used) are detailed in the respective figure legends.
Supplementary Material
CoIP of TY1-tagged MN1 in murine leukemia cells. Related toFigure 2. Direct CoIP of TY1-tagged MN1 in murine MN1-driven leukemia cells mass spectrometry data. Empty cells indicate no detection.
Bio-ID of MN1 in human 293 and murine CMPs. Related toFigure 2. BioID-IP of MN1 in human 293 cells and murine CMP cells mass spectrometry data.
TF motive enrichment of Smarca4/MN1 dual peaks. Related toFigure 3. HOMER motif enrichment analysis of Smarca4 and MN1 co-occupied loci in murine MN1- driven leukemia cells compared to Smarca4 only peaks.
Smarca4, Arida1a and Smarcd2 chromatin fractionation. Related toFigure 6. Mass spectrometry data and normalization of chromatin fractionation experiment.
Gene sets. Related toFigure 6. Gene sets used for GSEA and ChIP-Seq analysis.
Key Resources Table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Western blot, immunoprecipitation and immunofluorescence antibodies and reagents | ||
| Smarca4 | Abcam | Cat # ab110641, RRID:AB_10861578 |
| Arid1a | Santa Cruz | Cat # sc-32761, clone PSG3, RRID:AB_673396 |
| Smarcd2 | Bethyl Labs | Cat # A301–596A, RRID:AB_1078812 |
| HA | Abcam | Cat # ab9110, RRID:AB_307019 |
| Ty1 | LifeTech | Cat # MA5–23513, clone BB2, RRID:AB_2610644 |
| MN1 | Lifespan Biosciences | Cat # LS-C416119–100 |
| H3K4me1 | Abcam | Cat # ab8895, RRID:AB_306847 |
| H3K27ac | Abcam | Cat # ab4729, RRID:AB_2118291 |
| Goat anti rabbit secondary A555 conjugated | LifeTech | Cat # A21428, RRID:AB_2535849 |
| Mouse polyclonal anti-actin | Milipore Sigma | Cat # MAB1501, RRID:AB_2223041 |
| Goat anti mouse secondary H+L-HPR conjugated | BioRad | Cat# 170–6516, RRID:AB_11125547 |
| Goat anti rabbit secondary H+L-HPR conjugated | BioRad | Cat# 170–6515, RRID:AB_11125142 |
| 10% BisTris Gel | Invitrogen | Cat# NP0301 |
| Pierce™ Protein A/G Magnetic Beads (IP) | Pierce | Cat # 88802 |
| Protein A+G coated beads (ChIP) | Magna ChIP, Millipore | Cat # 16–663 |
| Western Lighting RTM Plus-ECL | Perkin-Elmer | Cat# NEL104001EA |
| Flow cytometry antibodies | ||
| CD3, Biotin, anti-mouse | Biolegend | Cat# 100304, RRID:AB_312669 |
| CD4, Biotin, anti-mouse | Biolegend | Cat# 100404, RRID:AB_312689 |
| CD8α, Biotin, anti-mouse | Biolegend | Clone 53–6.7, Cat# 100704, RRID:AB_312743 |
| Gr1 (Ly6-G/Ly6-C), Biotin, anti-mouse | Biolegend | Clone RB6–8C5y, Cat# 108404, RRID:AB_313369 |
| Gr1 (Ly6-G/Ly6-C), PeCy7, anti-mouse | Biolegend | Cat# 108416, RRID:AB_313381 |
| MAC1, APC, anti-mouse | Biolegend | Cat# 101212, RRID:AB_312795 |
| B220, Biotin, anti-mouse/human | Biolegend | Clone RA3–6B2, Cat# 103204, RRID:AB_312989 |
| CD19, Biotin, anti-mouse | Biolegend | Clone 6D5, Cat# 115504, RRID:AB_313369 |
| IL-7Rα (CD127), Biotin, anti-mouse | Biolegend | Clone A7R34, Cat# 135006, RRID:AB_2126118 |
| Ter-119, Biotin, anti-mouse | Biolegend | Clone TER-119, Cat# 116204, RRID:AB_313705 |
| Streptavadin, APC-Cy7 | Biolegend | Cat# 405208 |
| ckit (CD117), Alexa Fluro 647, anti-mouse | Biolegend | Clone 2B8, Cat# 105818, RRID:AB_493474 |
| Sca-1 (Ly6A), Pe-Cy7, anti-mouse | Invitrogen | Clone D7, Ref# 25–5981-82, RRID:AB_469669 |
| FCγR, PE, anti-mouse | Biolegend | Cat# 101307, RRID:AB_312806 |
| CD34, FITC, anti-mouse | Biolegend | Cat# 553733 |
| CD150, PE, anti-mouse | Biolegend | Clone TC15–12F12.2, Cat # 115903, RRID:AB_313682 |
| CD48, Pac Blue, anti-mouse | Biolegend | Clone HM48–1, Cat # 103417, RRID:AB_756139 |
| Dyanbeads M-280, Streptavidin | Invitrogen | Ref# 11206D |
| Annexin-APC | BD Biosciences | 550474 |
| Recombinant DNA | ||
| MSCV-MN1-IRES-GFP | Bernt Lab, cDNA from Ellen Zwaarthof | N/A |
| MSCV-Ty1-MN1-IRES-GFP | Tanja Gruber | N/A |
| MSCV-HA-MN1-IRES-GFP | Bernt Lab – newly generated | N/A |
| HA-MN1ΔpQ-HA-IRES-GFP | Bernt Lab – newly generated | N/A |
| MSCV-IRES-dTomato | Addgene | Plasmid #107229 |
| MSCV-Cre-IRES-dTomato | Armstrong Lab | N/A |
| MSCV-IRES-GFP | Armstrong Lab | N/A |
| Mouse strains | ||
| C57BL/6NJ | Jackson laboratories | Strain # 005304 |
| C57BL/6 Smarca4f/f | Julie Lessard | N/A |
| Mn1+/− | Trevor Williams | N/A |
| Cytokines | ||
| Recombinant murine IL-3 | PeproTech | Cat# 213–13 |
| Recombinant murine IL-6 | PeproTech | Cat# 216–16 |
| Recombinant murine SCF | PeproTech | Cat# 250–03 |
| Recombinant murine TPO | PeproTech | Cat# 315–14 |
| Recombinant murine FLT3-L | PeproTech | Cat# 250–31L |
| Recombinant human GM-CSF | PeproTech | Cat# 315–03 |
| Cell lines | ||
| UCSD-AML1 (female) | DSMZ | Cat# ACC691, RRID:CVCL_1853 |
| AMU-AML1 (male) | Ichiro Hanamura | N/A |
| Mutz-3 (male) | DSMZ | ACC-295, RRID:CVCL_1433 |
| Monomac6 (male) | DSMZ | ACC124, RRID:CVCL_1426 |
| 5637 (male) | DSMZ | ACC-35, RRID:CVCL_0126 |
| Molm14 (male) | DSMZ | ACC-777, RRID:CVCL_7916 |
| HEK293 (“293”) (female) | ATCC | CRL-1573 RRID:CVCL_0045 |
| Cell Culture Reagents | ||
| RPMI-1640 Media | VWR | Cat# 10–040-CV |
| IMDM (Iscove’s modif of DMEM) | VWR | Cat# 45000–366 |
| DMEM | VWR | Cat# 45000–312 |
| Alpha MEM | HyClone | Cat# SH30265.01 |
| Methylcellulose M3234 | Sigma | Cat# M7140 |
| Fetal Bovine Serum | Life Technologies | Cat# 10438026 |
| L-Glutamine | Life Technologies | Cat# 25030081 |
| Penicillin-Streptomycin (10,000 U/mL) | Invitrogen | Cat# 15140122 |
| Fugene 6 Transfection Reagent | VWR | Cat# PAE2692 |
| OptiMEM | Thermo | Cat# 31985–062 |
| Retronectin | Clontech Laboratories | Cat# T100B |
| Polyethylene glycol | Sigma | Cat# P4338 |
| Trypan Blue Solution | Mediatech | Cat# MT25–900-CI |
| Phosphate Buffered Saline | Mediatech | Cat# MT21–031-CV |
| Lookout mycoplasma PCR detection kit | Sigma | Cat # MP0035–1KT |
| Other Reagents | ||
| BD Pharmlyse | Fisher BD | Cat# 555899 |
| Chemicals | ||
| TritonX-100 | VWR | Cat# 9002–93-1 |
| Hydrochloric Acid | Fisher Scientific | Cat# A144 |
| Sodium Chloride (NaCl) | Sigma Aldrich | Cat# S9888 |
| NP-40 (IGEPAL) | Alfas Aesar | Cat# J61055 |
| Sodium dodecyl sulfate (SDS) | Teknova | Cat# S0288 |
| Sodium Deoxycholate | Alfas Aesar | Cat# J62288 |
| Tris-HCl | Roche | Cat # 10812846001 |
| Lithium Chloride (LiCl) | Alfas Aesar | Cat# 36217 |
| Ethylenediaminetetraacetic acid (EDTA) | VWR | Cat# E1777 |
| Sodium Bicarbonate (NaHCO3) | Fisher Scientific | Cat# S233 |
| Halt protease and phosphatase inhibitor coctail | Thermo Scientific | Cat# 1861281 |
| Kits | ||
| QiAquick PCR Purification Kit | Qiagen | Cat# 28106 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74136 |
| RNeasy Plus Micro Kit | Qiagen | Cat# 74034 |
| ZymoPURE II Plasmid Maxi Prep Kit | Zymo | Cat# 11–555B |
| Click-iT EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Invitrogen | Cat# C10419 |
| Qubit Protein Assay Kit |
LifeTechnologies | Cat# Q33211 |
| Ipsogen MN1 ProfileQuant | Qiagen | Cat# 676813 |
| Oligonucleotides | ||
| For primer sequences (qRT-PCR and 4C) please refer to Table S6 | ||
| Software and Algorithms | ||
| FlowJo (Flow cytometry) | FlowJo software | Version 10.5.3 |
| Graphpad Prism | Graphpad software | Prism 8 |
| MaxQuant | MaxQuant software | Version 1.6.0.16 http://www.maxquant.org |
| Kallisto (RNA-Seq alignment) | (Bray et al., 2016) | Version 0.45.0 |
| DESeq2 (DEG) | (Love et al., 2014) | Release 3.1 |
| EnhancedVolcano (volcano plots) | (Blighe K, Rana S, Lewis M, 2020) | R package version 1.6.0, https://github.com/kevinblighe/EnhancedVolcano |
| GSEA | (Subramanian et al., 2005) | Version 4.0 |
| BWA (ChIP-Seq alignment) | (Li and Durbin, 2009) | Version 1.19.0 |
| MACS2 (ChIP-Seq peak finding) | (Zhng et al., 2008) | N/A |
| Homer (Motif analysis) | (Heinz et al., 2010) | annotatePeaks.pl |
| ROSE (super enhancer identification) | (Whyte et al., 2013) | Young lab: http://younglab.wi.mit.edu/super_enhancer_code.html |
| Image J | National Institute of Health |
https://imagej.nih.gov/ij/
|
| TOS (Threshold overlap score analysis of IF) – ImageJ plugin | (Stauffer et al., 2018) |
http://sites.imagej.net/EzColocalization/plugins/
|
| 4C read mapping | (Geeven et al., 2018) | de Wit lab: https://github.com/deWitLab/4C_mapping |
| peakC (4C peak calling) | (Geeven et al., 2018) | de Wit lab; https://github.com/deWitLab/peakC |
| Ngs.plot (Visualization) | (Shen et al., 2014) | annotatePeaks.pl |
| Integrative Genomics Viewer (IGV)(Visualization) | (Robinson et al., 2011) | Version 2.7 https://software.broadinstitute.org/software/igv/ |
| Encode Peak Blacklist - human | (Amemiya et al., 2019) | http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg19-human |
| Encode Peak Blacklist - mouse | (Amemiya et al., 2019) | http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/mm10-mouse |
| Deposited Data | ||
| Human 4C data | This paper | https://www.ncbi.nlm.nih.gov/geo/: GSE171526 |
| Human ChIP-Seq data – UCSD-AML1, AMU-AML1 and controls | This paper | https://www.ncbi.nlm.nih.gov/geo/: GSE154985 |
| Human ChIP-Seq data – Mutz3 | This paper | https://www.ncbi.nlm.nih.gov/geo/: GSE171394 |
| Murine ChIP-Seq data | This paper | https://www.ncbi.nlm.nih.gov/geo/: GSE154602 |
| Murine RNA-Seq data | This paper | https://www.ncbi.nlm.nih.gov/geo/: GSE154997 |
| Uncut blots and gels | This paper | Mendeley Data, V1, doi: 10.17632/8k9shrb22x.1 https://data.mendeley.com/datasets/8k9shrb22x/1 |
Highlights:
MN1 translocations in AML result in MN1 overexpression due to enhancer hijacking.
MN1 interacts with the myeloid progenitor BAF complex.
MN1 over-stabilizes the BAF complex at critical enhancers.
Overexpression of the polyQ protein MN1 is sufficient to cause AML.
Acknowledgments:
We thank Ivan Maillard, Vikram Paralkar, Patricia Ernst, Stuart Orkin, Scott Armstrong, Leonard Zon and Martin Carroll for helpful discussions. We thank Ellen Zwaarthoff for providing the MN1 cDNA, Michael Heuser for providing HA-MN1, and Tanja Gruber for providing TY1-MN1. We thank Trevor Williams for providing MN1+/− mice, and Laura Bennett and Nancy Speck for help with timed matings and the isolation of fetal liver cells. We thank the animal and flow cores at the Children’s Hospital of Philadelphia and the University of Pennsylvania, as well as the Mass Spec Core at the University of Colorado and the University of Pennsylvania. Funding: This work was supported by start-up funds from the Division of Pediatric Oncology and the Abramson Cancer Research Center at the Children’s Hospital of Philadelphia, funding from the Doris Duke Foundation [2014104 to KMB], Hyundai Hope on Wheels [to KMB], the Emerson Collective [to KMB], NIH P01CA196539 [to BAG], a Robert Arceci Award from Leukemia and Lymphoma Society [to BAG], the CHOP Foerderer Award [to SSR] and the American Society of Hematology (ASH) [to SSR].
Footnotes
Declaration of interests: KMB hold a patent on the use of DOT1L inhibitors for MN1 leukemia. KMB has received research funding from Syndax and has previously consulted for Agios. IH received research funding from Bristol-Myers Squibb (BMS), Celgene, MSD, Astellas, Otsuka, Ono, Kyowa Kirin, Sanofi, Shionogi, Zenyaku, Daiichi Sankyo, Taiho, Takeda, Chugai, Eli Lilly, Nihon Shinyaku, Novartis, Pfizer, Fujimoto, Tanabe-Mitsubishi, Fukuyu Hospital, Yamada Yohojo, and received honoraria, or holds membership on an entity’s board of directors, speaker’s bureau, or its advisory committees for Celgene, Janssen, Takeda, Ono, BMS, Novartis, Daiichi Sankyo, Kyowa Kirin, Eisai, Nihon-Shinyaku, Pfizer, AbbVie, Otsuka, Shionogi, Mundi, CSL, MSD. All other authors have no competing interests to declare.
Inclusion and diversity: One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, and Roberts CW (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun 8, 14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, et al. (2017). Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178 e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brown AL, Wilkinson CR, Waterman SR, Kok CH, Salerno DG, Diakiw SM, Reynolds B, Scott HS, Tsykin A, Glonek GF, et al. (2006). Genetic regulators of myelopoiesis and leukemic signaling identified by gene profiling and linear modeling. J Leukoc Biol 80, 433–447. [DOI] [PubMed] [Google Scholar]
- Buijs A, Sherr S, van Baal S, van Bezouw S, van der Plas D, Geurts van Kessel A, Riegman P, Lekanne Deprez R, Zwarthoff E, Hagemeijer A, and et al. (1995). Translocation (12;22) (p13;q11) in myeloproliferative disorders results in fusion of the ETS-like TEL gene on 12p13 to the MN1 gene on 22q11. Oncogene 10, 1511–1519. [PubMed] [Google Scholar]
- Buijs A, van Rompaey L, Molijn AC, Davis JN, Vertegaal AC, Potter MD, Adams C, van Baal S, Zwarthoff EC, Roussel MF, and Grosveld GC (2000). The MN1-TEL fusion protein, encoded by the translocation (12;22)(p13;q11) in myeloid leukemia, is a transcription factor with transforming activity. Mol Cell Biol 20, 9281–9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscarlet M, Krasteva V, Ho L, Simon C, Hebert J, Wilhelm B, Crabtree GR, Sauvageau G, Thibault P, and Lessard JA (2014). Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood 123, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, and Tjian R. (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank VA, Sroczynska P, Sankar A, Miyagi S, Rundsten CF, Johansen JV, and Helin K. (2015). SWI/SNF Subunits SMARCA4, SMARCD2 and DPF2 Collaborate in MLL-Rearranged Leukaemia Maintenance. PLoS One 10, e0142806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang J, Nance S, Ma J, Cheng J, Walsh MP, Vogel P, Easton J, Song G, Rusch M, Gedman AL, et al. (2017). AMKL chimeric transcription factors are potent inducers of leukemia. Leukemia 31, 2228–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombret H, and Gardin C. (2016). An update of current treatments for adult acute myeloid leukemia. Blood 127, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, Giesler GJ, Zoghbi HY, and Orr HT (2010). SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron 67, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber J, Krivtsov AV, Stubbs MC, Wright R, Davis TN, van den Heuvel-Eibrink M, Zwaan CM, Kung AL, and Armstrong SA (2009). HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 113, 2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, et al. (2000). Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 408, 101–106. [DOI] [PubMed] [Google Scholar]
- Geeven G, Teunissen H, de Laat W, and de Wit E. (2018). peakC: a flexible, non-parametric peak calling package for 4C and Capture-C data. Nucleic Acids Res 46, e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotou M, Hanamura I, Nagoshi H, Wakabayashi M, Sakamoto N, Tsunekawa N, Horio T, Goto M, Mizuno S, Takahashi M, et al. (2012). Establishment of a novel human myeloid leukemia cell line, AMU-AML1, carrying t(12;22)(p13;q11) without chimeric MN1-TEL and with high expression of MN1. Genes Chromosomes Cancer 51, 42–53. [DOI] [PubMed] [Google Scholar]
- Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, et al. (2012). Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 8, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosveld GC (2007). MN1, a novel player in human AML. Blood Cells Mol Dis 39, 336–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach C, Kern W, Schindela S, Kohlmann A, Alpermann T, Schnittger S, and Haferlach T. (2012). Gene expression of BAALC, CDKN1B, ERG, and MN1 adds independent prognostic information to cytogenetics and molecular mutations in adult acute myeloid leukemia. Genes Chromosomes Cancer 51, 257–265. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser M, Beutel G, Krauter J, Dohner K, von Neuhoff N, Schlegelberger B, and Ganser A. (2006). High meningioma 1 (MN1) expression as a predictor for poor outcome in acute myeloid leukemia with normal cytogenetics. Blood 108, 3898–3905. [DOI] [PubMed] [Google Scholar]
- Heuser M, Yun H, Berg T, Yung E, Argiropoulos B, Kuchenbauer F, Park G, Hamwi I, Palmqvist L, Lai CK, et al. (2011). Cell of origin in AML: susceptibility to MN1-induced transformation is regulated by the MEIS1/AbdB-like HOX protein complex. Cancer Cell 20, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Gao R, Cao C, Forbes L, Li J, Freeberg S, Fredenburg KM, Justice JM, Silver NL, Wu L, et al. (2019). MYB-activated models for testing therapeutic agents in adenoid cystic carcinoma. Oral Oncol 98, 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandilci A, and Grosveld GC (2009). Reintroduction of CEBPA in MN1-overexpressing hematopoietic cells prevents their hyperproliferation and restores myeloid differentiation. Blood 114, 1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandilci A, Surtel J, Janke L, Neale G, Terranova S, and Grosveld GC (2013). Mapping of MN1 sequences necessary for myeloid transformation. PLoS One 8, e61706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasteva V, Buscarlet M, Diaz-Tellez A, Bernard MA, Crabtree GR, and Lessard JA (2012). The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood 120, 4720–4732. [DOI] [PubMed] [Google Scholar]
- Krasteva V, Crabtree GR, and Lessard JA (2017). The BAF45a/PHF10 subunit of SWI/SNF-like chromatin remodeling complexes is essential for hematopoietic stem cell maintenance. Exp Hematol 48, 58–71 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PHL, Geeven G, Bianchi V, Hilvering CRE, and de Laat W. (2020). 4C-seq from beginning to end: A detailed protocol for sample preparation and data analysis. Methods 170, 17–32. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, Ross KN, Perner F, Olsen SN, Pritchard T, et al. (2019). A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 36, 660–673 e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CK, Moon Y, Kuchenbauer F, Starzcynowski DT, Argiropoulos B, Yung E, Beer P, Schwarzer A, Sharma A, Park G, et al. (2014). Cell fate decisions in malignant hematopoiesis: leukemia phenotype is determined by distinct functional domains of the MN1 oncogene. PLoS One 9, e112671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 172, 650–665. [DOI] [PubMed] [Google Scholar]
- Langer C, Marcucci G, Holland KB, Radmacher MD, Maharry K, Paschka P, Whitman SP, Mrozek K, Baldus CD, Vij R, et al. (2009). Prognostic importance of MN1 transcript levels, and biologic insights from MN1-associated gene and microRNA expression signatures in cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 27, 3198–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. (2014). Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashtalir N, D’Avino AR, Michel BC, Luo J, Pan J, Otto JE, Zullow HJ, McKenzie ZM, Kubiak RL, St Pierre R, et al. (2018). Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288 e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meester-Smoor MA, Vermeij M, van Helmond MJ, Molijn AC, van Wely KH, Hekman AC, Vermey-Keers C, Riegman PH, and Zwarthoff EC (2005). Targeted disruption of the Mn1 oncogene results in severe defects in development of membranous bones of the cranial skeleton. Mol Cell Biol 25, 4229–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler KH, Dufour A, Benthaus T, Hummel M, Sauerland MC, Heinecke A, Berdel WE, Buchner T, Wormann B, Mansmann U, et al. (2009). ERG expression is an independent prognostic factor and allows refined risk stratification in cytogenetically normal acute myeloid leukemia: a comprehensive analysis of ERG, MN1, and BAALC transcript levels using oligonucleotide microarrays. J Clin Oncol 27, 5031–5038. [DOI] [PubMed] [Google Scholar]
- Michel BC, and Kadoch C. (2017). A SMARCD2-containing mSWI/SNF complex is required for granulopoiesis. Nat Genet 49, 655–657. [DOI] [PubMed] [Google Scholar]
- Mittal P, and Roberts CWM (2020). The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol 17, 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks DA, Johansen JA, Mo K, Rao P, Eagleson B, Yu Z, Lieberman AP, Breedlove SM, and Jordan CL (2007). Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc Natl Acad Sci U S A 104, 18259–18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, Nie Z, Neale G, and Taylor JP (2010). Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron 67, 936–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman DR, McGirr C, Shakhbazov K, Barbier V, Krishnan K, Mukhopadhyay P, Hawthorne P, Trezise A, Ding J, Grimmond SM, et al. (2014). Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 123, 2682–2690. [DOI] [PubMed] [Google Scholar]
- Pogosova-Agadjanyan EL, Moseley A, Othus M, Appelbaum FR, Chauncey TR, Chen IL, Erba HP, Godwin JE, Jenkins IC, Fang M, et al. (2020). AML risk stratification models utilizing ELN-2017 guidelines and additional prognostic factors: a SWOG report. Biomark Res 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter EG, Connelly KE, and Dykhuizen EC (2017). Sequential Salt Extractions for the Analysis of Bulk Chromatin Binding Properties of Chromatin Modifying Complexes. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priam P, Krasteva V, Rousseau P, D’Angelo G, Gaboury L, Sauvageau G, and Lessard JA (2017). SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPvarepsilon dependent mechanism. Nat Genet 49, 753–764. [DOI] [PubMed] [Google Scholar]
- Pulice JL, and Kadoch C. (2016). Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb Symp Quant Biol 81, 53–60. [DOI] [PubMed] [Google Scholar]
- Riedel SS, Haladyna JN, Bezzant M, Stevens B, Pollyea DA, Sinha AU, Armstrong SA, Wei Q, Pollock RM, Daigle SR, et al. (2016). MLL1 and DOT1L cooperate with meningioma-1 to induce acute myeloid leukemia. J Clin Invest 126, 1438–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe JS, Mercan F, Rivera K, Pappin DJ, and Vakoc CR (2015). BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell 58, 1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick S, Grosche S, Kohl KE, Drpic D, Jaeger MG, Marella NC, Imrichova H, Lin JG, Hofstatter G, Schuster M, et al. (2021). Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat Genet 53, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick S, Rendeiro AF, Runggatscher K, Ringler A, Boidol B, Hinkel M, Majek P, Vulliard L, Penz T, Parapatics K, et al. (2019). Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat Genet 51, 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Jyotsana N, Gabdoulline R, Heckl D, Kuchenbauer F, Slany RK, Ganser A, and Heuser M. (2020). Meningioma 1 is indispensable for mixed lineage leukemia-rearranged acute myeloid leukemia. Haematologica 105, 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E. (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, et al. (2013). Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev 27, 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorek M, Cohen LRZ, and Meshorer E. (2019). Open chromatin structure in PolyQ disease-related genes: a potential mechanism for CAG repeat expansion in the normal human population. NAR Genomics and Bioinformatics 1, e3–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Pierre R, and Kadoch C. (2017). Mammalian SWI/SNF complexes in cancer: emerging therapeutic opportunities. Curr Opin Genet Dev 42, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer W, Sheng H, and Lim HN (2018). EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci Rep 8, 15764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I, et al. (2016). New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 164, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton AL, Zhang X, Ellison TI, and Macdonald PN (2005). The 1,25(OH)2D3-regulated transcription factor MN1 stimulates vitamin D receptor-mediated transcription and inhibits osteoblastic cell proliferation. Mol Endocrinol 19, 2234–2244. [DOI] [PubMed] [Google Scholar]
- Thol F, Yun H, Sonntag AK, Damm F, Weissinger EM, Krauter J, Wagner K, Morgan M, Wichmann M, Gohring G, et al. (2012). Prognostic significance of combined MN1, ERG, BAALC, and EVI1 (MEBE) expression in patients with myelodysplastic syndromes. Ann Hematol 91, 1221–1233. [DOI] [PubMed] [Google Scholar]
- van de Werken HJ, de Vree PJ, Splinter E, Holwerda SJ, Klous P, de Wit E, and de Laat W. (2012a). 4C technology: protocols and data analysis. Methods Enzymol 513, 89–112. [DOI] [PubMed] [Google Scholar]
- van de Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Oz Y, Bouwman BA, et al. (2012b). Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods 9, 969–972. [DOI] [PubMed] [Google Scholar]
- van Wely KH, Molijn AC, Buijs A, Meester-Smoor MA, Aarnoudse AJ, Hellemons A, den Besten P, Grosveld GC, and Zwarthoff EC (2003). The MN1 oncoprotein synergizes with coactivators RAC3 and p300 in RAR-RXR-mediated transcription. Oncogene 22, 699–709. [DOI] [PubMed] [Google Scholar]
- Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA, et al. (2017). ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Chen X, Hui S, Ni J, Yin Y, Cao W, Zhang Y, Wang X, Ma X, Cao P, et al. (2020). Ectopia associated MN1 fusions and aberrant activation in myeloid neoplasms with t(12;22)(p13;q12). Cancer Gene Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lee RS, Alver BH, Haswell JR, Wang S, Mieczkowski J, Drier Y, Gillespie SM, Archer TC, Wu JN, et al. (2017). SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet 49, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M, and Stott K. (2019). Disordered domains in chromatin-binding proteins. Essays Biochem 63, 147–156. [DOI] [PubMed] [Google Scholar]
- Wenge DV, Felipe-Fumero E, Angenendt L, Schliemann C, Schmidt E, Schmidt LH, Thiede C, Ehninger G, Berdel WE, Arteaga MF, and Mikesch JH (2015). MN1-Fli1 oncofusion transforms murine hematopoietic progenitor cells into acute megakaryoblastic leukemia cells. Oncogenesis 4, e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. (2010). Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7, 532–544. [DOI] [PubMed] [Google Scholar]
- Witzel M, Petersheim D, Fan Y, Bahrami E, Racek T, Rohlfs M, Puchalka J, Mertes C, Gagneur J, Ziegenhain C, et al. (2017). Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat Genet 49, 742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, and Fuxreiter M. (2016). The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 165, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L, Li M, Liu Y, Cen J, Chen Z, Zhen X, Xie X, Cao X, and Gu W. (2013). The clinical characteristics and prognostic significance of MN1 gene and MN1-associated microRNA expression in adult patients with de novo acute myeloid leukemia. Ann Hematol 92, 1063–1069. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Prinz A, Steger J, Garcia-Cuellar MP, Radsak M, Bentaher A, and Slany RK (2018). HoxA9 transforms murine myeloid cells by a feedback loop driving expression of key oncogenes and cell cycle control genes. Blood Adv 2, 3137–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CoIP of TY1-tagged MN1 in murine leukemia cells. Related toFigure 2. Direct CoIP of TY1-tagged MN1 in murine MN1-driven leukemia cells mass spectrometry data. Empty cells indicate no detection.
Bio-ID of MN1 in human 293 and murine CMPs. Related toFigure 2. BioID-IP of MN1 in human 293 cells and murine CMP cells mass spectrometry data.
TF motive enrichment of Smarca4/MN1 dual peaks. Related toFigure 3. HOMER motif enrichment analysis of Smarca4 and MN1 co-occupied loci in murine MN1- driven leukemia cells compared to Smarca4 only peaks.
Smarca4, Arida1a and Smarcd2 chromatin fractionation. Related toFigure 6. Mass spectrometry data and normalization of chromatin fractionation experiment.
Gene sets. Related toFigure 6. Gene sets used for GSEA and ChIP-Seq analysis.
Data Availability Statement
4C, ChIP-Seq and RNA-Seq are available through GEO (https://www.ncbi.nlm.nih.gov/geo/): ChIP-Seq of murine leukemias: GSE154602, RNA-Seq: GSE154997, ChIP-Seq of human leukemia cell lines: GSE154985 and GSE171394, 4C: GSE171526. Uncut blots and gels are available at Mendeley: Mendeley Data, V1, doi: 10.17632/8k9shrb22x.1 (https://data.mendeley.com/datasets/8k9shrb22x/1)