

Abstract
tert-Butoxide unlocks new reactivity patterns embedded in nitroarenes. Exposure of nitrostilbenes to sodium tert-butoxide was found to produce N-hydroxyindoles at room temperature without an additive. Changing the counterion to potassium changed the reaction outcome to yield solely oxindoles through an unprecedented dioxygen-transfer reaction followed by a 1,2-phenyl migration. Mechanistic experiments established that these reactions proceed via radical intermediates and suggest that counterion coordination controls whether an oxindole or N-hydroxyindole product is formed.
Keywords: tert-butoxide, single electron transfer, nitroarene
Graphical Abstract

tert-Butoxide unlocks divergent reactivity embedded in nitrostilbenes to construct N-hydroxyindoles or oxindoles depending on whether sodium or potassium is the counterion.
Introduction
The ubiquitous nature of N-heterocycles in pharmaceuticals and organic materials has inspired the discovery of new reactivity patterns to facilitate their construction.[1],[2] The development of reductive methods to access these privileged scaffolds by constructing C–N bonds using nitroarenes has received significant attention because of the ready availability and robust nature of nitroarenes. While traditional C–N bond formation methods were developed using a superstoichiometric quantity of a reductant to deoxygenate the nitro-group,[3] recent efforts have focused on the development of catalytic processes that use the combination of a transition metal-[4] a base-metal-[5] or a phosphine[6] catalyst and a stoichiometric reductant. In contrast to these approaches, 175 years ago,[7] sodium methoxide was reported to reduce nitrobenzene to azoxybenzene in boiling methanol (Scheme 1). Subsequent mechanistic investigations in 1962 by Ogata and Mibae suggested that electron-transfer from alkoxide triggered deoxygenation to produce an ArNO intermediate which reacted with an N-hydroxybenzene to produce the azoxy product.[8] After these mechanistic studies, interest in exploiting electron-transfer from alkoxides waned, until a recent resurgence in the use of potassium tert-butoxide as a single-electron reductant.[9] In 2010, the Hayashi and Shi groups reported that tert-butoxide mediated the transition metal-free couplings of iodoarenes.[9a, 9b] Investigations by Murphy and co-workers established that electron-transfer to the iodoarene occurred from the in situ-generated phenanthroline dianion 1.[9d] Inspired by these reports, we were curious if exposure of a nitrostilbene to an alkoxide would generate a nitrosoarene intermediate that might be intercepted intramolecularly to afford an N-heterocycle (Scheme 1). In contrast to our expectations, we found that nitrostilbene 2 could be converted to N-hydroxyindole 5 using simply NaOt-Bu at room temperature without an additive. Strikingly, changing the counterion to potassium triggered an unprecedented dioxygen transfer and [1,2] aryl migration tandem reaction to afford oxindole 6 as the only N-heterocycle from nitrostilbene 2.
Scheme 1.

tert-Butoxide-mediated N-heterocycle formation from nitroarenes.
Results and Discussion
Investigation of the scope and limitations of N-heterocycle formation.
To test our hypothesis that nitrosoarene intermediates could be accessed using an alkoxide and trapped to afford an N-heterocycle, the reactivity of nitrostilbene 2a was examined (Table 1). While no reaction was observed upon exposure of 2a to sodium methoxide in boiling methanol, changing the solvent to tetrahydrofuran resulted in the formation of N-hydroxyindole 5a at room temperature (entries 1 and 2). The yield of 5a was improved by changing the identity of the alkoxide with the best outcome obtained when tert-butoxide was used (entries 2 – 4). Increasing the temperature or the reaction time, however, did not further increase the yield (entries 5 and 6). The reaction was not light-dependent:[10] performing the reaction in a foil-covered vessel provided N-hydroxyindole 5a in 55% yield (entry 7). A solvent screen revealed that N-hydroxyindole formation occurred smoothly in ethereal solvents, but was attenuated in tert-butanol (entries 7 – 10).[11] To our surprise, the identity of the counterion was critical to the reaction outcome: while no reaction was observed when lithium- or magnesium tert-butoxide was employed, 3-phenyl-3-hydroxy-2-oxindole 6a was obtained as the only product using potassium tert-butoxide irrespective of exposure to light (entries 11 – 13). The formation of 6a, whose structure confirmed by X-ray crystallography,[12] involves not only oxygen transfer to the ortho-alkenyl substituent but also a [1,2]-phenyl shift. While oxygen transfer from nitroarenes to pendant acetylenes has been reported to afford N-heterocycles,[13] this oxidation-migration tandem reaction of ortho-alkenyl substituents is unprecedented.
Table 1.
Development of reductive and divergent N-heterocycle formation.

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | MOR (2 equiv) | solvent | h | T (°C) | yield, %a | 5a:6a |
|
| ||||||
| 1 | NaOMe | MeOH | 16 | 70 | n.r. | ... |
| 2 | NaOMe | THF | 16 | 25 | 14 | >20:1 |
| 3 | NaOEt | THF | 16 | 25 | 26 | >20:1 |
| 4 | NaOt-Bu | THF | 16 | 25 | 55 | >20:1 |
| 5 | NaOt-Bu | THF | 16 | 60 | 57 | >20:1 |
| 6 | NaOt-Bu | THF | 40 | 25 | 60 | >20:1 |
| 7b | NaOt-Bu | THF | 40 | 25 | 55 | >20:1 |
| 8 | NaOt-Bu | dioxane | 40 | 25 | 53 | >20:1 |
| 9 | NaOt-Bu | 2-MeTHF | 40 | 25 | 55 | >20:1 |
| 10 | NaOt-Bu | t-BuOH | 40 | 25 | 38 | >20:1 |
| 11 | LiOt-Bu | THF | 16 | 25 | 0 | ... |
| 12 | Mg(Ot-Bu)2 | THF | 16 | 25 | 0 | ... |
| 13c,d | KOt-Bu | THF | 16 | 25 | 62 | <1:20 |
Isolated after silica gel chromatography.
Reaction performed in a foil-covered vessel.
The outcome of the reaction did not change if light was excluded.
The X-Ray structure of 6a was deposited in the Cambridge Crystallographic Database (CCDC 198003).
THF = tetrahydrofuran.
The scope and limitations of the NaOt-Bu-mediated reductive cyclization was explored by developing conditions to alkylate the N-hydroxyl group as well examining the effect of changing the electronic- and steric environment of the nitrostilbenes (Table 2). The substrates for this study were either commercially available or readily prepared in one-step from 2-bromonitroarenes and styrene using a Heck reaction.[14] Because N-alkoxy- or N-acetoxyheterocycles are more stable,[15] we explored telescoping the reaction by adding an electrophile to determine if the sensitive N-hydroxy functionality could be alkylated or acetylated. We found that a methyl-, benzyl-, or benzoate group could be easily added to the initially formed N-hydroxyindole to improve the reproducibility of the reaction sequence and ease purification. Our investigation of a series of nitrostilbenes that varied their electronic- and steric nature revealed that higher yields were obtained in the presence of electron-donating groups positioned para to the styryl group (entries 2 and 5). In contrast, the presence of electron-withdrawing groups severely attenuated the reaction yield. Gratifyingly, the yield of N-hydroxyindole could be rescued if the reaction was heated to 100 °C (entries 4 and 7). Changing the electronic nature of the β-aryl group had less of an overt effect on the reaction outcome: while higher yields were observed for electron-rich aryl groups, the presence of a fluorine did not diminish the yield as substitution on the nitroarene moiety (entries 8 – 11). While adding a stronger electron-withdrawing Cl did reduce the yield, increasing the temperature of the reaction increased the yield of 5o. The reactivity of naphthalene-derived 2q illustrated that N-hydroxyindole formation was relatively insensitive to steric constraints (entry 12).
Table 2.
Scope of NaOt-Bu-mediated N-hydroxindole formation.

| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | # | nitroarene 2 | N-heterocycle 5 | R3 | yield, %a |
|
| |||||
| 1 | a |

|

|
H | 60 |
| Me | 67 (63)b | ||||
| Bn | 60 | ||||
| Bz | 59 | ||||
| 2 | b |

|

|
H | 58 |
| Me | 62 | ||||
| 3 | c |

|

|
H | 53 |
| Me | 42 | ||||
| 4 | e |

|

|
H | 24 (38)c |
| Me | 22 (44)c | ||||
| 5 | g |

|
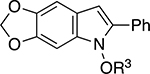
|
Me | 63 |
| 6 | h |

|
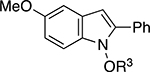
|
Me | 44 |
| 7 | i |

|

|
H | 25 (60)c |
| Me | 17 (63)c | ||||
| 8 | l |

|

|
H | 46 |
| Me | 60 | ||||
| 9 | m |

|

|
H | 68 |
| Me | 75 | ||||
| 10 | n |

|

|
H | 67 |
| Me | 59 | ||||
| 11 | o |

|

|
H | 25 (53)c |
| Me | 28 (55)c | ||||
| 12 | q |

|

|
Me | 52 |
Isolated after silica gel chromatography.
Reaction performed on 2 mmol scale.
The tert-BuO-mediated reduction was performed at 100 °C for 16 h.
After our initial investigation into the scope of using NaOt-Bu as the reductant, we turned our attention to exploring the reactivity of nitrostilbenes towards KOt-Bu (Table 3). Analysis of the reactivity patterns revealed several differences in comparison to N-hydroxyindole formation. First, the success of the oxygen-transfer-migration reaction was less dependent on the electronic nature of the nitrostilbene with chloro-, bromo- and even trifluoromethyl groups tolerated (entries 2 – 10). Further, the opposite electronic trend was observed for β-aryl groups: more electron-deficient groups led to higher oxindole yields (entries 11 – 15). Naphthalene 2q demonstrated that oxindole formation tolerated an increased steric environment around the nitro-group to afford 6q albeit in diminished yield relative to 6a (entry 16). In contrast to N-hydroxyindole formation, the yield was not improved when the reaction temperature was increased.
Table 3.
Scope of KOt-Bu-mediated oxindole formation.

| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | # | R1 | R2 | Ar | yield, %a |
|
| |||||
| 1 | a | H | H | Ph | 62 (55)b |
| 2 | b | MeO | H | Ph | 43 |
| 3 | c | F | H | Ph | 43 |
| 4 | d | Cl | H | Ph | 48 |
| 5 | e | F3CO | H | Ph | 57 |
| 6 | f | F3C | H | Ph | 38 |
| 7 | g | O–CH2–O | Ph | 41 | |
| 8 | h | H | MeO | Ph | 49 |
| 9 | j | H | F | Ph | 74 |
| 10 | k | H | Br | Ph | 49 |
| 11 | l | H | H | 4-MeOC6H4 | 41 |
| 12 | m | H | H | p-Tol | 38 |
| 13 | n | H | H | 4-FC6H4 | 56 |
| 14 | o | H | H | 4-ClC6H4 | 50 |
| 15 | p | H | H | 4-CF3C6H4 | 66 |
| 16 | q |

|

|
41 | |
Isolated after silica gel chromatography.
Reaction performed on 2 mmol scale.
To assess the impact of the ortho-styryl substituent on the reaction outcome, substrates 2s, Z-2a and 2r were investigated (Scheme 2). The position of the phenyl substituent was found to control which N-heterocycle was formed: exposure of α-phenylnitrostyrene 2s to tert-butoxide afforded only N-methoxyindole 5sa using either KOt-Bu or NaOt-Bu after telescoping the reaction with MeI, although an increased reaction temperature was required using NaOt-Bu. The stereochemistry of the 2-nitrostilbene was also critical to the reaction outcome. While E-nitrostilbene formed either N-hydroxyindole 5a or oxindole 6a depending on identity of the tert-butoxide counterion, the reaction of Z-2a afforded only oxindole 6a irrespective of whether sodium- or potassium tert-butoxide was used. Similar reactivity was observed when nitrostilbene 2r was exposed to NaOt-Bu, which produced only oxindole 6r where one of the oxygen atoms was transferred to the ortho-alkenyl substituent.[16] These results suggest that steric interactions between the β-phenyl substituent and a reactive intermediate or stabilization of a radical- or charged intermediate by the α-phenyl substituent can override the counterion effect.
Scheme 2.

Effect of phenyl substituent position on the reaction outcome.
Mechanistic EPR investigations.
To investigate if the tert-butoxide-mediated N-heterocycle formation involved the formation of radical intermediates, the reaction was analyzed using X-band, solution-phase EPR spectroscopy (Figures 1 and 2). At room temperature, reaction mixtures from addition of KOtBu and NaOtBu exhibit EPR spectra near g = 2.00. The additional most-common feature of the spectra is the existence of a three-line pattern, typical for a radical interacting with a 14N (I = 1) nucleus. We attribute these peaks to radical intermediates of an electron transfer from tert-butoxide (Eox = +0.10 V vs SCE)[17] to nitrostilbene (Ered = −1.13 V vs SCE).[4g, 18] The high potential gap of ΔE = 1.03 V between the oxidation peak potential of t-BuO− and the reduction peak potential of nitrostilbene suggests that coordination of the counterion occurs to facilitate the transfer.[19] This hypothesis is supported by the simulation of the X-band EPR spectrum from the NaOt-Bu-mediated reaction, which afforded the best match with experiment when a nitro radical anion was assumed coordinated to the sodium counterion (Figure 1a).[20]
Figure 1.

Experimental (black) and simulated (red) X-band EPR spectra of tert-butoxide-mediated reduction of nitrostilbene 2a (THF, 298 K, 1 h) (a, result of NaOt-Bu reaction, b, result of KOt-Bu reaction) Data collection parameters: Frequency = 9.4306 GHz, Power = 0.2 mW, Modulation = 0.1 G; a. Fitting parameters for 7 are giso = 2.005, Aiso = 29 MHz, and an line width for isotropic broadening, lw =[0.81 0.53] in mT (the first Gaussian and the latter Lorentzian broadening); b. Fitting parameters for 8 are giso = 2.009, A = [22.1 18.2] MHz, lw = 1.09 mT, τcorr = 5 ns, and weight = 0.85; and fitting parameters for 9 are giso = [2.007], A = [21.2 14.7 11.9 8.7], and lw = 0.2 mT, relative contributions to spectral intensity are 93% 8 and 7% 9.
The foregoing outcome suggests that counterion identity is critical to directing the radical formation pathway. For example, the EPR spectrum obtained from the NaOt-Bu-mediated reaction displays only three well-resolved peaks and no further couplings to 1H in Figure 1a. The absence of additional peaks from the latter couplings strongly suggests that the spin density is localized only in the nitro group, not delocalized onto the aromatic ring. We propose that the small size as well as strong Lewis acidity of Na+ allows for effective binding to the nitro group and prevention of spin density delocalizing on the aryl ring. In contrast, a spectrum displaying significantly more complicated hyperfine interactions was observed when KOt-Bu was employed (Figure 1b).[21],[22] Simulation of the spectra with a variety of possible radicals revealed a best match when assuming a mixture of 93% of 8 and 7% of 9. We interpret these data to suggest that the nitro radical is less effective in coordinating the larger potassium counterion. As a result of the weaker coordination, the formation of two spin isomers is allowed, one featuring an NO2 radical and one with spin density delocalized onto the aryl ring. We note that this preliminary interpretation requires that the radical is not delocalized across both moieties. This outcome could occur if interactions with K+ drive the nitro-groups away from planarity with the stilbene.[23] Extension of this interpretation could explain the less complex EPR spectrum for the Na+ system: stronger binding of the nitro radical anion to Na+ produces only one, NO2-localized isomer. We tentatively interpret the difference in coordination in these simulated spectra to account for the difference in reaction outcome: the lack of coordination to the counterion could enable oxygen-atom transfer from 8 to the ortho-styryl substituent to afford oxindole 6a, which would not be possible when it is bound to the sodium ion.[24]
These EPR experiments spurred us to examine the effect of a crown ether on the reaction outcome (Scheme 3). When 15-crown-5 was added in combination with NaOt-Bu, we found that N-hydroxyindole 5a was no longer formed, and oxindole 6a was produced as the only N-heterocyclic product. This phenomenon proved to be general: exposure of 2-nitrostilbenes 2c, 2g, or 2l produced only oxindoles 6c, 6g, or 6l albeit in lower yield than using KOt-Bu.[25] In contrast, the addition of 18-crown-6 to the KOt-Bu-mediated reaction resulted in no change to the outcome—oxindole 6a was the only N-heterocycle formed. Analysis of the reaction using EPR spectroscopy reveals a substantial change in spectral shape when the crown ether is present, potentially suggesting changes in spin identity via crown-ether binding to the counterion.[26] Together, these experiments suggest that coordination of the counterion controls whether N-hydroxyindole 5 or oxindole 6 is produced.
Scheme 3.

Effect of 15-crown-5 on the reaction outcome.
Reactivity of 18O-labeled reagents.
To determine if the C2- and C3 oxygens in oxindole 6 originated from an intra- or intermolecular reaction, the reactivity of 18O-labeled potassium tert-butoxide and 18O-labeled nitrostilbene 2g was investigated (Scheme 4). When K18Ot-Bu was used, no 18O-incorporation into 6a was observed to suggest that both oxygens were transferred from the nitro-group. To test if oxygen transfer occurred intermolecularly, a mixture of 2a and 2g-18O were submitted to reaction conditions and double crossover of the labeled- and unlabeled oxygens to the oxindole product was observed to afford 16% of 6a and 6a-18O and 64% of 6g-18O2 and 6g-18O. Analysis of the mass spectrum showed an increase of the [M + 2]+ signal for 6a and [M − 2]+ signal for 6g-18O to suggest that only one of the oxygen atoms is transferred, and the parent ion [(M) − OH]+ revealed that intermolecular O-transfer occurred only to the C2-position of the oxindole.
Scheme 4.

Investigation of an intra- or intermolecular mechanism for oxygen transfer.
Potential mechanisms for N-heterocycle formation.
Our data suggests a possible mechanism that accounts for the dependence of the reaction outcome on the identity of the counterion (Scheme 5). Electron transfer from tert-butoxide to nitrostilbene produces radical anion 8 and tert-butoxy radical,[27] which fragments to produce acetone and methyl radical. Mesomer 10 traps sodium ion to produce 7, which accepts a second electron to form 11. Fragmentation of 11 produces nitrosostilbene 12, which undergoes a 6π-electron-five atom electrocyclization to form 13 that isomerizes to produce N-hydroxyindole 5a.[28]
Scheme 5.

Potential mechanism for N-hydroxyindole formation.
Our data suggests that oxindole 6 is formed through an intermolecular mechanism when the counterion is not coordinated to the nitrostilbene radical anion (Scheme 6).[29] The intermolecular oxygen-atom transfer could occur by attack of radical anion 8 onto the nitrostilbene 2 at the β-position to afford nitrite 14,[30] which undergoes a 3-exo-tet radical cyclization to produce epoxide 15.[31],[32],[33] Ring-opening by the proximal nitro group could produce 16.[34],[35],[36] tert-Butoxide-mediated single electron reduction followed by fragmentation produces 18. Hydrogen-atom abstraction by tert-butoxy radical or methyl radical produces ketone 19.[37],[38] Isomerization forms enol 20,[39] which attacks the nitroso group to produce N-heterocycle 21. A subsequent 1,2 phenyl shift affords N-hydroxyoxindole 22,[40] which is reduced to oxindole 6a. To test if oxygen-transfer occurred via an epoxide intermediate, 2-nitrostilbene oxide was examined as a substrate. In line with our mechanistic hypothesis, treatment of 15 with KOt-Bu resulted in the formation of oxindole 6a. Conversion of 2-nitrostilbene oxide to oxindole supports that the intermolecular reaction occurs between a nitroarene radical anion and a neutral nitroarene.
Scheme 6.

Potential intermolecular mechanism for oxindole formation.
Conclusion
In conclusion, we have discovered a novel tert-butoxide-mediated reaction of 2-nitrostilbenes that produces either a N-hydroxyindole or an oxindole depending on the identity of the counterion. The reactivity patterns exhibited by the 2-nitrostilbenes suggest that the N-heterocyclic products are formed by either an intra- or intermolecular reaction, where the degree of coordination of the counterion to the radical anion dictate which mechanism occurs. Our findings illustrate that nitrosoarene reactive intermediates can not only be generated at room temperature from nitroarenes but that unprecedented reactivity patterns can be triggered divergently to produce functionalized N-heterocycles. In addition to leveraging this novel reactivity to construct different sized N-heterocycles, future studies are also aimed at obtaining a deeper understanding of the spin dynamics and spin Hamiltonian parameters of the observed radical intermediates to conclusively assign their identity.
Supplementary Material
Acknowledgments
TGD, YZ, and HZ are grateful to the National Science Foundation CHE-1564959 and the National Institutes of Health (NIGMS) R01GM138388 for their generous financial support. We thank Professor Leslie Fung and Dr. Daniel McElheny for assistance with the EPR spectrometer and Mr. Furong Sun (UIUC) for high resolution mass spectrometry data.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Edwankar CR, Edwankar RV, Namjoshi OA, Rallapappi SK, Yang J, Cook JM, Curr. Opin. Drug Discovery Dev 2009, 12, 752–771; [PubMed] [Google Scholar]; b) McGrath NA, Brichacek M, Njardarson JT, J. Chem. Educ 2010, 87, 1348–1349; [Google Scholar]; c) Kochanowska-Karamyan AJ, Hamann MT, Chem. Rev 2010, 110, 4489–4497; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Baumann M, Baxendale IR, Ley SV, Nikbin N, Beilstein J Org. Chem 2011, 7, 442–495; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Vitaku E, Smith DT, Njardarson JT, J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- [2].a) Liu D, Zhao G, Xiang L, Eur. J. Org. Chem 2010, 2010, 3975–3984; [Google Scholar]; b) Stokes BJ, Driver TG, Eur. J. Org. Chem 2011, 2011, 4071–4088; [Google Scholar]; c) Wu X-F, Neumann H, Beller M, Chem. Rev 2012, 113, 1–35; [DOI] [PubMed] [Google Scholar]; d) Gulevich AV, Dudnik AS, Chernyak N, Gevorgyan V, Chem. Rev 2013, 113, 3084–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Cadogan JIG, Cameron-Wood M, Mackie RK, Searle RJG, J. Chem. Soc 1965, 4831–4837; [Google Scholar]; b) Sundberg RJ, Yamazaki T, J. Org. Chem 1967, 32, 290–294; [Google Scholar]; c) Sundberg RJ, Kotchmar GS, J. Org. Chem 1969, 34, 2285–2288; [Google Scholar]; d) Cadogan JIG, Acc. Chem. Res 1972, 5, 303–310; [Google Scholar]; e) Bartoli G, Palmieri G, Bosco M, Dalpozzo R, Tetrahedron Lett 1989, 30, 2129–2132; [Google Scholar]; f) Bartoli G, Bosco M, Dalpozzo R, Palmieri G, Marcantoni E, J. Chem. Soc., Perkin Trans 11991, 2757–2761; [Google Scholar]; g) Sapountzis I, Knochel P, J. Am. Chem. Soc 2002, 124, 9390–9391; [DOI] [PubMed] [Google Scholar]; h) Dohle W, Staubitz A, Knochel P, Chem. Eur. J 2003, 9, 5323–5331; [DOI] [PubMed] [Google Scholar]; i) Dalpozzo R, Bartoli G, Curr. Org. Chem 2005, 9, 163–178; [Google Scholar]; j) Gao H, Xu Q-L, Yousufuddin M, Ess DH, Kürti L, Angew. Chem. Int. Ed 2014, 53, 2701–2705. [DOI] [PubMed] [Google Scholar]
- [4].a) Akazome M, Kondo T, Watanabe Y, J. Org. Chem 1993, 58, 310–312; [Google Scholar]; b) Akazome M, Kondo T, Watanabe Y, J. Org. Chem 1994, 59, 3375–3380; [Google Scholar]; c) Tollari S, Cenini S, Crotti C, Gianella E, J. Mol. Catal 1994, 87, 203–214; [Google Scholar]; d) Söderberg BC, Shriver JA, J. Org. Chem 1997, 62, 5838–5845; [Google Scholar]; e) Ragaini F, Sportiello P, Cenini S, J. Organomet. Chem 1999, 577, 283–291; [Google Scholar]; f) Smitrovich JH, Davies IW, Org. Lett 2004, 6, 533–535; [DOI] [PubMed] [Google Scholar]; g) Davies IW, Smitrovich JH, Sidler R, Qu C, Gresham V, Bazaral C, Tetrahedron 2005, 61, 6425–6437; [Google Scholar]; h) Ragaini F, Rapetti A, Visentin E, Monzani M, Caselli A, Cenini S, J. Org. Chem 2006, 71, 3748–3753; [DOI] [PubMed] [Google Scholar]; i) Ragaini F, Ventriglia F, Hagar M, Fantauzzi S, Cenini S, Eur. J. Org. Chem 2009, 2185–2189; [Google Scholar]; j) Okuro K, Gurnham J, Alper H, J. Org. Chem 2011, 76, 4715–4720; [DOI] [PubMed] [Google Scholar]; k) Okuro K, Gurnham J, Alper H, Tetrahedron Lett 2012, 53, 620–622; [Google Scholar]; l) Jana N, Zhou F, Driver TG, J. Am. Chem. Soc 2015, 137, 6738–6741; [DOI] [PubMed] [Google Scholar]; m) Ansari NH, Dacko CA, Akhmedov NG, Söderberg BCG, J. Org. Chem 2016, 81, 9337–9349; [DOI] [PubMed] [Google Scholar]; n) Zhou F, Wang D-S, Guan X, Driver TG, Angew. Chem. Int. Ed 2017, 56, 4530–4534; [DOI] [PubMed] [Google Scholar]; o) Ford RL, Alt I, Jana N, Driver TG, Org. Lett 2019, 21, 8827–8831. [DOI] [PubMed] [Google Scholar]
- [5].a) Srivastava RS, Nicholas KM, Chem. Commun 1998, 2705–2706; [Google Scholar]; b) Srivastava RS, Nicholas KM, Organometallics 2005, 24, 1563–1568; [Google Scholar]; c) Gui J, Pan C-M, Jin Y, Qin T, Lo JC, Lee BJ, Spergel SH, Mertzman ME, Pitts WJ, La Cruz TE, Schmidt MA, Darvatkar N, Natarajan SR, Baran PS, Science 2015, 348, 886–891; [DOI] [PubMed] [Google Scholar]; d) Cheung CW, Ploeger ML, Hu X, Nat. Commun 2017, 8, 14878; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shevlin M, Guan X, Driver TG, ACS Catal 2017, 5518–5522; [Google Scholar]; f) Cheung CW, Ploeger ML, Hu X, ACS Catal 2017, 7, 7092–7096; [Google Scholar]; g) Rauser M, Ascheberg C, Niggemann M, Angew. Chem. Int. Ed 2017, 56, 11570–11574. [DOI] [PubMed] [Google Scholar]
- [6].a) Nykaza TV, Ramirez A, Harrison TS, Luzung MR, Radosevich AT, J. Am. Chem. Soc 2018, 140, 3103–3113; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nykaza TV, Cooper JC, Li G, Mahieu N, Ramirez A, Luzung MR, Radosevich AT, J. Am. Chem. Soc 2018, 140, 15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Zinin N, J. Prakt. Chem 1845, 36, 93–107; [Google Scholar]; b) Klinger H, Ber. Dtsch. Chem. Ges 1882, 15, 865–867; [Google Scholar]; c) Fry HS, Cameron JL, J. Am. Chem. Soc 1927, 49, 864–873. [Google Scholar]
- [8].Ogata Y, Mibae J, J. Org. Chem 1962, 27, 2048–2052. [Google Scholar]
- [9].a) Sun C-L, Li H, Yu D-G, Yu M, Zhou X, Lu X-Y, Huang K, Zheng S-F, Li B-J, Shi Z-J, Nat. Chem 2010, 2, 1044; [DOI] [PubMed] [Google Scholar]; b) Shirakawa E, Itoh K.-i., Higashino T, Hayashi T, J. Am. Chem. Soc 2010, 132, 15537–15539; [DOI] [PubMed] [Google Scholar]; c) Chang W-W, Li Z-J, Yang W-W, Gao X, Org. Lett 2012, 14, 2386–2389; [DOI] [PubMed] [Google Scholar]; d) Zhou S, Anderson GM, Mondal B, Doni E, Ironmonger V, Kranz M, Tuttle T, Murphy JA, Chem. Sci 2014, 5, 476–482; [Google Scholar]; e) Toutov AA, Liu W-B, Betz KN, Fedorov A, Stoltz BM, Grubbs RH, Nature 2015, 518, 80–84; [DOI] [PubMed] [Google Scholar]; f) Barham JP, Coulthard G, Kane RG, Delgado N, John MP, Murphy JA, Angew. Chem. Int. Ed 2016, 55, 4492–4496; [DOI] [PubMed] [Google Scholar]; g) Barham JP, Coulthard G, Emery KJ, Doni E, Cumine F, Nocera G, John MP, Berlouis LEA, McGuire T, Tuttle T, Murphy JA, J. Am. Chem. Soc 2016, 138, 7402–7410; [DOI] [PubMed] [Google Scholar]; h) Smith AJ, Young A, Rohrbach S, O’Connor EF, Allison M, Wang H-S, Poole DL, Tuttle T, Murphy JA, Angew. Chem. Int. Ed 2017, 56, 13747–13751; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Bugaenko DI, Dubrovina AA, Yurovskaya MA, Karchava AV, Org. Lett 2018, 20, 7358–7362; [DOI] [PubMed] [Google Scholar]; j) Cumine F, Palumbo F, Murphy JA, Tetrahedron 2018, 74, 5539–5545; [Google Scholar]; k) Wu L, Annibale VT, Jiao H, Brookfield A, Collison D, Manners I, Nat. Commun 2019, 10, 2786; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Xu Y, Shi X, Wu L, RCS Adv 2019, 9, 24025–24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].For reports on the light-dependency of electron transfer from tert-butoxide, see: Xu Z, Gao L, Wang L, Gong M, Wang W, Yuan R, ACS Catal 2015, 5, 45–50;Nocera G, Young A, Palumbo F, Emery KJ, Coulthard G, McGuire T, Tuttle T, Murphy JA, J. Am. Chem. Soc 2018, 140, 9751–9757.
- [11].Refer to the Supporting Information for more details.
- [12].The crystal structure of 6a was deposited in the Cambridge Crystallographic Database (CCDC 1980039).
- [13].a) Asao N, Sato K, Yamamoto Y, Tetrahedron Lett 2003, 44, 5675–5677; [Google Scholar]; b) Söderberg BCG, Gorugantula SP, Howerton CR, Petersen JL, Dantale SW, Tetrahedron 2009, 65, 7357–7363; [Google Scholar]; c) Patel P, Ramana CV, Org. Biomol. Chem 2011, 9, 7327–7334; [DOI] [PubMed] [Google Scholar]; d) Kumar CVS, Puranik VG, Ramana CV, Chem. Eur. J 2012, 18, 9601–9611; [DOI] [PubMed] [Google Scholar]; e) Patel P, Ramana CV, J. Org. Chem 2012, 77, 10509–10515; [DOI] [PubMed] [Google Scholar]; f) Narendraprasad Reddy B, Ramana CV, Chem. Commun 2013, 49, 9767–9769; [DOI] [PubMed] [Google Scholar]; g) Suneel Kumar CV, Ramana CV, Org. Lett 2014, 16, 4766–4769; [DOI] [PubMed] [Google Scholar]; h) Liu R-R, Ye S-C, Lu C-J, Zhuang G-L, Gao J-R, Jia Y-X, Angew. Chem. Int. Ed 2015, 54, 11205–11208; [DOI] [PubMed] [Google Scholar]; i) Maduli EJM, Edeson SJ, Swanson S, Procopiou PA, Harrity JPA, Org. Lett 2015, 17, 390–392; [DOI] [PubMed] [Google Scholar]; j) Marien N, Brigou B, Pinter B, De Proft F, Verniest G, Org. Lett 2015, 17, 270–273; [DOI] [PubMed] [Google Scholar]; k) Marien N, Luo T, Verniest G, Synlett 2017, 28, 934–938; [Google Scholar]; l) Fu W, Song Q, Org. Lett 2018, 20, 393–396; [DOI] [PubMed] [Google Scholar]; m) Marien N, Reddy BN, De Vleeschouwer F, Goderis S, Van Hecke K, Verniest G, Angew. Chem. Int. Ed 2018, 57, 5660–5664. [DOI] [PubMed] [Google Scholar]
- [14].Bumagin NA, Bykov VV, Sukhomlinova LI, Tolstaya TP, Beletskaya IP, J. Organomet. Chem 1995, 486, 259–262. [Google Scholar]
- [15].For a discussion of the stability and potential decomposition products of N-hydroxyindoles, see: Somei M, Heterocycles 1999, 50, 1157–1211;Penoni A, Palmisano G, Broggini G, Kadowaki A, Nicholas KM, J. Org. Chem 2006, 71, 823–825.
- [16].Exposure of nitrostilbene 2r to KOt-Bu produced multiple decomposition products with only trace oxindole 6r observed.
- [17].Yi H, Jutand A, Lei A, Chem. Commun 2015, 51, 545–548. [DOI] [PubMed] [Google Scholar]
- [18].a) Du P, Brosmer JL, Peters DG, Org. Lett 2011, 13, 4072–4075; [DOI] [PubMed] [Google Scholar]; b) Russell GA, Janzen EG, J. Am. Chem. Soc 1962, 84, 4153–4154. [Google Scholar]
- [19].Enemærke RJ, Christensen TB, Jensen H, Daasbjerg K, J. Chem. Soc., Perkin Trans 22001, 1620–1630. [Google Scholar]
- [20].The observed correlation times are in line with other organic radical systems of this approximate size, modeled with similar spin dynamics. For example, see: Bogdanov AV, Mladenova Kattnig BY, Vorobiev AK, Grampp G, Kokorin AI, J. Phys. Chem. B 2020, 124, 11007–11014;Poncelet M, Driesschaert B, Angew. Chem. Int. Ed 2020, 59, 16451–16454; Angew. Chem. 2020, 132, 16593–16593.
- [21].For a discussion of the EPR spectra potassium derivatives of nitrostilbenes, see: Todres ZV, J. Organomet. Chem 1992, 441, 349–354.
- [22].While both Na+ and K+ possess high natural abundance of nuclear spin, simulations including these hyperfine interactions did not produce a better fit to the data. Refer to the Supporting Information for more details.
- [23].Huang L, Massa L, Karle J, Proc. Nat. Acad. Sci. U.S.A 2008, 105, 13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Analysis of the reaction mixture after 20 h using EPR spectroscopy that radical species were still present. We were not able to identify their structures using simulation, and investigations are on-going to their role in the reaction.
- [25].Oxindole formation was accompanied by azoxy by-product.
- [26].The effect of the addition of 15-crown-5 to NaOt-Bu-mediated reduction of 4-nitrostilbene was also investigated using EPR spectroscopy. Simulations were not able to definitively assign a structure to the radical or radicals responsible for the signals. However, we did notice significant changes in the EPR spectral shape, which is likely attributable to interaction with the crown ether. For more details, refer to the Supporting Information.
- [27].For related studies of alkoxide-mediated nitrosoarene reduction, see: Buck P, Angew. Chem., Int. Ed. Engl 1969, 8, 120–131;Bellobono IR, Gamba A, Sala G, Tampieri M, J. Am. Chem. Soc 1972, 94, 5781–5786;Prato M, Quintily U, Scorrano G, J. Chem. Soc., Perkin Trans 21986, 1419–1424.
- [28].For a theoretical discussion of 5-center versus 6-center cyclization of nitrosostryrenes, see: Davies IW, Guner VA, Houk KN, Org. Lett 2004, 6, 743–746;Leach AG, Houk KN, Davies IW, Synthesis 2005, 3463–3467.
- [29].Control experiments established that tert-butoxide did not convert N-hydroxyindole to oxindole. For reports of acid-mediated isomerization, see: Somei M, Noguchi K, Yamagami R, Kawada Y, Yamada K, Yamada F, Heterocycles 2000, 53, 7–10;Somei M, Noguchi K, Yamada F, Hetercycles 2001, 55, 1237–1240.
- [30].The reaction of the oxygen of the nitro group with an olefin has precedence in the [2,3] sigmatropic rearrangement of allylic nitro compounds to allylic alcohols, which occurs via a nitrite intermediate. For leading references, see: Boivin J, El Kaim L, Kervagoret J, Zard SZ, J. Chem. Soc., Chem. Commun 1989, 1006–1008;Dumez E, Rodriguez J, Dulcère J-P, Chem. Commun 1999, 2009–2010;Alameda-Angulo C, Quiclet-Sire B, Schmidt E, Zard SZ, Org. Lett 2005, 7, 3489–3492.
- [31].For examples of 3-exo-tet radical cyclizations, see: Ohkita T, Tsuchiya Y, Togo H, Tetrahedron 2008, 64, 7247–7251;Tsuchiya Y, Izumisawa Y, Togo H, Tetrahedron 2009, 65, 7533–7537.
- [32].Nitrite esters are prone to fragmentation of the N–O bond, see: Barton DHR, Beaton JM, Geller LE, Pechet MM, J. Am. Chem. Soc 1960, 82, 2640–2641;Barton DHR, Pure & Appl. Chem 1968, 16, 1–15.
- [33].In order to obtain a yield greater than 50% of oxindole 6, the nitrosoradical arene must be intercepted by a nitroarene or nitroarene radical anion, or that there is a competing intra- or intermolecular mechanism. Investigations are ongoing to disentangle these potential competive mechanisms
- [34].Cyclic nitrites related to 16 have been postulated as reactive intermediates and observed spectroscopically. For leading references, see: Walker JW, Reid GP, McCray JA, Trentham DR, J. Am. Chem. Soc 1988, 110, 7170–7177;Ohwada T, Kasuga M, Shudo K, J. Org. Chem 1990, 55, 2717–2719;Corrie JET, Gilbert BC, Munasinghe VRN, Whitwood AC, J. Chem. Soc., Perkin Trans 22000, 2483–2491;Abbruzzetti S, Sottini S, Viappiani C, Corrie JET, J. Am. Chem. Soc 2005, 127, 9865–9874.
- [35].For related ring-opening of epoxides by nitrates, see: Golding P, W Millar R, C Paul N, H Richards D, Tetrahedron Lett 1988, 29, 2731–2734;Golding P, Millar RW, Paul NC, Richards DH, Tetrahedron 1993, 49, 7051–7062;Iranpoor N, Salehi P, Tetrahedron 1995, 51, 909–912;Volkova YA, Ivanova OA, Budynina EM, Averina EB, Kuznetsova TS, Zefirov NS, Tetrahedron Lett 2008, 49, 3935–3938.
- [36].For related intramolecular ring-opening of ortho-nitrophenylcyclopropane to form 1-oxo-1,2-benzoxazolium- or 1-oxo-1,2-benzoxazinium ions, see: Fedotov AN, Trofimova EV, Mochalov SS, Shabarov YS, Zh. Org. Khim 1988, 24, 2403–2408;Trofimova EV, Fedotov AN, Mochalov SS, Shabarov YS, Zh. Org. Khim 1991, 27, 1193–1198;Fedotov AN, Trofimova EV, Gulov TE, Bandaev SG, Mochalov SS, Zefirov NS, Russ. J. Org. Chem 2013, 49, 1534–1541.
- [37].α-H-atom abstraction of a cage alkoxy radical was proposed by Barton and co-workers to account for ketone formation from a nitrite ester, see: Barton DHR, Hesse RH, Pechet MM, Smith LC, J. Chem. Soc., Perkin Trans 11979, 1159–1165;Ishmuratov GY, Kharisov RY, Shayakhmetova AK, Botsman LP, Shitikova OV, Tolstikov GA, Chem. Nat. Compd 2005, 41, 643–649.
- [38].Alternatively, ketone 19 could be formed from 17 through an H-atom-abstraction fragmentation sequence, which would avoid the formation of alkoxy radical 18.
- [39].While formation of a ketyl radical could start a chain process, this appears unlikely because an excess of the tert-butoxide reductant is required for a positive reaction outcome.
- [40].For reports of base-mediated [1,2] migrations to form 3-hydroxy-2-indolinones from 2-hyhydroxy-3-indolinones, see: Acheson RM, Booth SRG, J. Chem. Soc C1968, 30–32;Kafka S, Klásek A, Košmrlj J, J. Org. Chem 2001, 66, 6394–6399;Coldham I, Adams H, Ashweek NJ, Barker TA, Reeder AT, Skilbeck MC, Tetrahedron Lett 2010, 51, 2457–2460;Liu M, Zhang C, Ding M, Tang B, Zhang F, Green Chem 2017, 19, 4509–4514
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.