

Summary:
TANK binding kinase 1 (TBK1) regulates IFN-I, NF-κB, and TNF-induced RIPK1-dependent cell death (RCD). In mice, biallelic loss of TBK1 is embryonically lethal. We discovered four humans, ages 32, 26, 7, and 8 from three unrelated consanguineous families with homozygous loss-of-function mutations in TBK1. All four patients suffer from chronic and systemic autoinflammation, but not severe viral infections. We demonstrate that TBK1 loss results in hypomorphic but sufficient IFN-I induction via RIG-I/MDA5, while the system retains near intact IL-6 induction through NF-κB. Autoinflammation is driven by TNF-induced RCD as patient-derived fibroblasts experienced higher rates of necroptosis in vitro, and CC3 was elevated in peripheral blood ex vivo. Treatment with anti-TNF dampened the baseline circulating inflammatory profile and ameliorated the clinical condition in vivo. These findings highlight the plasticity of the IFN-I response and underscore a cardinal role for TBK1 in the regulation of RCD.
In Brief:
TBK1 signals activation of antiviral defenses and controls TNF-mediated inflammation. Deletion of TBK1 in mice is embryonically lethal. Humans lacking TBK1 expression survive and have adequate antiviral function. Instead, these individuals suffer from inflammatory pathology driven by sensitivity to TNF-induced cell death that can be effectively treated with anti-TNF therapeutics.
Graphical Abstract
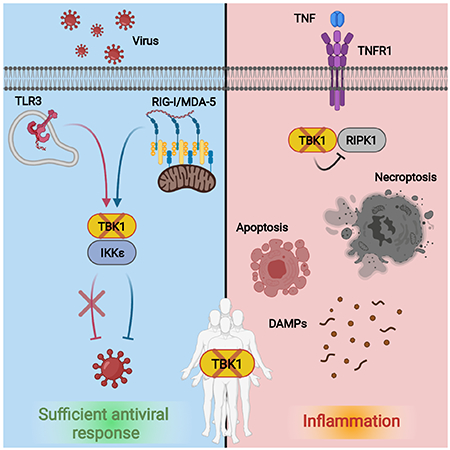
Introduction
Primary immunodeficiencies (PIDs) or Inborn Errors of Immunity (IEIs) are genetic disorders that heighten susceptibility to infection, autoinflammation, severe allergy, autoimmunity, and/or malignancy. Often monogenic, the causes of these clinically-apparent immune dysfunctions have been linked to pathogenic variants in over 400 different genes(Gruber and Bogunovic, 2020; Tangye et al., 2020), but through the increased biochemical assessment of these variants, we are unraveling how genetics influence the functional and clinical presentations of these PIDs and IEIs.
Occasionally, a monogenic IEI will manifest as a combination of weakened pathogen protection and chronic autoinflammation (Bogunovic et al., 2012; Boisson et al., 2012; Cuchet-Lourenço et al., 2018; Zhang et al., 2015). This clinical scenario occurs when the gene in question functions in both promotion and suppression of inflammation. TANK Binding Kinase 1 (TBK1) is one such factor. Classically considered an activator of innate immunity, TBK1 acts as a signaling intermediate upstream of type-I-interferon (IFN-I) and inflammatory cytokine production via NF-κB (Ahmad et al., 2016; Herman et al., 2012). The loss of TBK1 would therefore be expected to produce an immunocompromised state, as was the case in one functional evaluation of two individuals with genetic defects in TBK1 where heterozygous loss-of-function (LoF) mutations conferred susceptibility to Herpes Simplex encephalitis (HSE) (Herman et al., 2012). However, TBK1 has more recently been shown to control early stages of autophagy (Ahmad et al., 2016; Li et al., 2018; Oakes et al., 2017; Pilli et al., 2012; Richter et al., 2016; Thurston et al., 2009) and to negatively regulate inflammatory TNF-mediated cell death via an inactivating interaction with RIPK1 (Lafont et al., 2018; Xu et al., 2018).
In mice, biallelic loss of TBK1 is embryonic lethal, but viability is rescued by TNF, cytokine or receptor, knockout or complementation with kinase-inactive RIPK1 (Bonnard et al., 2000; Hemmi et al., 2004; Perry et al., 2004; Xu et al., 2018). In humans, genetic variants in TBK1 have been linked to familial amyotrophic lateral sclerosis (ALS) (Cirulli et al., 2015; Freischmidt et al., 2015) and frontotemporal dementia (FTD) (Freischmidt et al., 2015; Pottier et al., 2015) with modest associations. Here we report and characterize four patients, two adult siblings, and two children from three unrelated consanguineous families with biallelic LoF mutations in TBK1 who suffer from an early-onset autoinflammatory syndrome not partnered with any overt signs of heightened sensitivity to viral infection.
Results
Four individuals with homozygous LoF variants in TBK1.
A sister (P1: age 32) and brother (P2: age 26) from a consanguineous family of Moroccan descent presented in early childhood with an unknown auto-inflammatory syndrome in the absence of pathogenic autoantibodies (Sup Fig 1A). Both developed polyarthritis in their first year, followed by cutaneous vasculitis, auto-inflammatory features, and mild intellectual disability. P1 developed epileptic and pseudo-epileptic seizures in adolescence, while P2 experienced intermittent periods of motor disturbance starting at age 20 and then generalized seizures from the age of 24 (Sup Table 1 & 2; Data S1). A third patient from a consanguineous Indian family (P3: age 7) suffered from a similar unknown auto-inflammatory syndrome to P1 and P2 that included early-onset arthritis, systemic vasculitis, and auto-inflammatory features. He had one seizure with status epilepticus but has otherwise experienced normal physical and cognitive development up to the time of this report (Sup Table 1 & 2; Data S1). P4 (age 8), who was previously reported as part of a study that correlated autoinflammatory disease with genetic drivers in a cohort of unclassified patients (Kosukcu et al., 2021), experienced recurrent fever, joint pain, erythematous rash, growth failure, and delayed neurocognitive development that was accompanied by enlarged ventricles and brain matter atrophy (Sup Table 1 & 2; Data S1).
Whole exome sequencing (WES) revealed a homozygous nonsense mutation in the helix-loop-helix domain of TBK1 in P1 and P2 (chr12:64890825-G>A; p.Trp619*; p.W619*), a homozygous missense mutation in the kinase domain in P3 (chr12:64868103-T>G; p.Tyr212Asp; p.Y212D), and a homozygous nonsense mutation in the first coiled-coil domain in P4 (chr12:64879775-C>T; p.Arg440Ter; p.R440*) (Kosukcu et al., 2021) (Fig 1A & Sup Fig 1B). All variants are private to the patients, and the lack of any other homozygous predicted loss-of-function (pLoF) variants in the gnomAD database was standout (Karczewski et al., 2020). Further, TBK1 is considered a LoF-intolerant gene based on the ratio of observed and expected pLoF variants reported in gnomAD, which is adjusted for sequence context, coverage, and methylation with a cutoff of < 0.35 (pLoF o/e = 0.254), and in silico damage prediction indicated a probable functional consequence from these variants as they scored above the mutation significance cutoff (MSC) for TBK1. The MSC is a gene-specific threshold of significance for scoring the potential damage of a mutation (CADD: P1 and P2 = 39, P3 = 24.9; P4 = 36; MSC = 10.854) (Itan et al., 2016; Kircher et al., 2014). Combined with a lack of other plausible candidate variants in WES (Sup Table 3 & 4) and the high conservation of the mutated residues (Sup Fig 1C), these genetic lesions stood out as the most likely cause of the patients’ clinical phenotype.
Figure 1. 3 families with homozygous LoF variants in TBK1.

A) Patient family pedigrees and P1, P2, (W619*), P3 (Y212D), and P4 (R440*) variant locations within a schematic of the TBK1 protein.
B – C) Patient-derived hTERTs assessed for baseline TBK1 mRNA expression by qPCR (n = 3) and C) protein expression by western blot.
D) 293Ts 24 hrs after transfection with the TBK1 alleles under study assessed by western blot and E – G) qPCR (n = 3).
To determine if the genetic variants affect mRNA expression and/or protein production, we derived hTERT-immortalized dermal fibroblasts (hTERTs) from P1 and P2 and assessed endogenous baseline TBK1 expression. W619* TBK1 was dramatically less well expressed at the mRNA level compared to WT (Fig 1B) and was undetectable at the protein level (Fig 1C). We also transfected N-terminally FLAG-tagged WT, W619*, and R440* TBK1 into HEK293T cells to check for the possibility of a truncated protein with an N-terminal FLAG but no W619* or R440* TBK1 of any length was visible with anti-FLAG staining (Fig 1D). This absence of protein combined with the lack of mRNA suggested a mechanism of nonsense-mediated mRNA decay and complete loss of expression (LoE). Y212D TBK1 transfected into HEK293Ts, on the other hand, was expressed at comparable levels to WT in HEK293Ts (Fig 1D, E), but, unlike the WT, its overexpression did not trigger downstream signaling, suggesting a severely hypomorphic or loss of function (LoF) variant (Fig 1D – G & Sup Fig 2A). The same was true for G159A, an autosomal dominant (AD) mutation that strangled IFN-I induction and led to HSE susceptibility (Herman et al., 2012), and for S172A, an artificial but established phosphoinactive TBK1 mutant (Fig 1D – G & Sup Fig 2A) (Kishore et al., 2002).
Complete loss of TBK1 is not a complete loss of the IFN-I system.
TBK1 functions downstream of various pattern recognition receptors (PRRs) including TLR3, RIG-I, MDA5, and cGAS/STING to trigger the antiviral IFN-I and IFN-III responses and NF-κB-mediated inflammation (Chu et al., 1999; Fitzgerald et al., 2003; Jønsson et al., 2017; McWhirter et al., 2004; Perry et al., 2004; Sharma et al., 2003; Zhang et al., 2019). Therefore, it is not surprising that Tbk1−/− mouse fibroblasts have a diminished IFN-I response to viral infections (Fitzgerald et al., 2003; Perry et al., 2004; tenOever et al., 2004), and that AD TBK1 deficiency was previously documented in two humans who developed HSE early in life (de Diego and Rodríguez-Gallego, 2014; Herman et al., 2012). We were, therefore, surprised to learn that P1, P2, P3, and P4 all appear to have adequate antiviral protection in the absence of TBK1 based on their clinical history, serology (P1 and P2 were positive for HSV-1 without any HSV-1-related disease) (Sup Table 1), and infection of P1 and P2 hTERTs at baseline with VSV (Sup Fig 1D). Also unexpected was the identification of basal ganglia calcifications in both P1 (Sup Fig 3C, D) and P2 (data not available) since basal ganglia calcifications are tightly associated with type-I interferonopathy, but neither possessed an IFN-I signature in peripheral blood (Sup Fig 1E) (Livingston et al., 2014).
To understand why these individuals do not present clinically with hyper-susceptibility to viral infection, we measured IFN-I activation in P1- and P2-derived hTERTs, Epstein Bar virus transduced B cells (EBV-Bs), P1-hTERTs stably transduced with Y212D TBK1, and CRISPR-generated TBK1 KO A549s. hTERTs and EBV-Bs were stimulated with poly(I:C) (pIC) added directly to the culture media, leading to endosomal uptake and TLR3-mediated detection, or with pIC transfected into the cytoplasm with lipofectamine (L2K) where it engages RIG-I-like receptors. In P1 and P2, endosomal pIC, sensed by TLR3, failed to induce IFN-β and IL-6 (as a measure of NF-κB), but pIC delivered into the cytoplasm produced appreciable, albeit hypomorphic, levels of both cytokines (Fig 2A; Sup Fig 2B, C). When we looked downstream of IFNβ we found that despite reduced IFN-β expression, IFIT1 mRNA levels were comparable to the healthy controls (HCs) (Fig 2A). Ectopic expression of Y212D TBK1 (P3’s allele) in P1 hTERTs appeared severely hypomorphic. It produced far less pIRF3 and pSTAT2 than WT following a 4 hr endosomal pIC stimulation (Fig 2B), and muted IFN-I stimulated gene (ISG) and IL-6 mRNA expression after an overnight endosomal pIC stimulation (Fig 2C; Sup Fig 2D). In response to cytoplasmic pIC, Y212D TBK1 behaved similarly to WT (Fig 2B, C; Sup Fig 2D), which together with the above suggests that Y212D TBK1 is severely hypomorphic in the TLR3 axis but comparably functional to the TBK1-null system downstream of MAVS.
Figure 2. Complete loss of TBK1 is not a complete loss of the IFN system, and inactive TBK1 interferes with IKKε-mediated IFN-I activation.
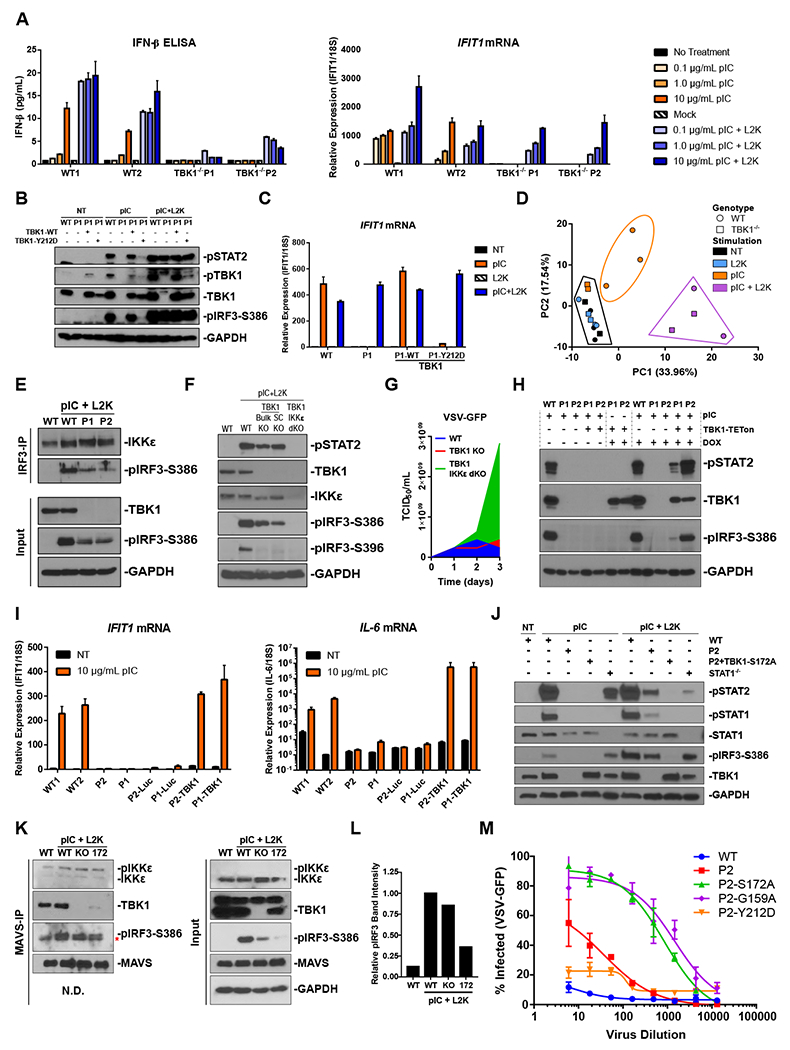
A) hTERTs stimulated with pIC overnight either in the endosome (TLR3) or transfected into the cytoplasm (RIG-I/MDA5) with L2K (n = 3).
B) P1 hTERTs stably expressing either DOX-inducible WT or Y212D TBK1 4 hps C) or after an overnight stimulation (n = 3).
D) PCA of sample variance determined by RNAseq on P1 and P2 hTERTs compared to 3 HC-derived lines following overnight stimulation with endosomal (10 μg/mL) or cytoplasmic (transfected) (0.1 μg/mL) pIC.
E) Immunoprecipitation of IRF3 from hTERTs stimulated with cytoplasmic pIC (1 μg/mL) for 4 hrs.
F) CRISPR generated TBK1 and TBK1/IKKε dKO A549s stimulated with cytoplasmic pIC (1 μg/mL) for 4 hrs.
G) VSV-GFP titrated on A549s. To calculate the TCID50, infected wells were considered GFP+ if they displayed a mean intensity above the mean of the uninfected wells.
H) hTERTs transduced with DOX-inducible TBK1 exposed to endosomal pIC (10 μg/mL) for 4 hrs following a 2-day DOX pre-treatment. IFN-I signaling was assessed by western blot.
I) hTERTs complemented with WT TBK1 were stimulated with endosomal pIC (10 μg/mL) for 4 hrs and assessed by qPCR (n = 3).
J) P2-derived hTERTs complemented with phospho-inactive S172A TBK1 were stimulated with endosomal (10 μg/mL) or cytoplasmic pIC (0.1 μg/mL) for 4 hrs.
K) Immunoprecipitation of MAVS from TBK1 KO A549s complimented with phospho-inactive S172A TBK1. Cells were transfected with 1 μg/mL pIC for 4 hrs. The asterisk indicates a non-specific band in the pIRF3 blot of the IP.
L) Band intensity quantification of pIRF3 that co-IPed with MAVS in B (left panel, upper band). Values are relative to WT pIC+L2K.
M) hTERTs transfected with pIC (0.1 μg/mL) 6 hrs before an overnight VSV-GFP infection (n = 3).
The residual ability of TBK1-null cells to react to cytoplasmic, but not endosomal pIC, was noted globally in RNAseq on hTERTs from P1 and P2 24 hrs after endosomal or cytoplasmic pIC stimulation. In principal component analysis, P1 and P2 transfected with pIC clustered together with similarly treated HCs, while P1 and P2 hTERTs that received endosomal pIC clustered with untreated and mock-transfected samples (Fig 2D). At the same time, pIC-transfected P1 and P2 hTERTs produced a muted transcriptional profile to HC cells (Sup Fig 4A). We also assessed cGAS/STING functionality in P1 and P2 and found a hamstrung IFN-I and NF-κB response to transfected DNA (Sup Fig 2E), in agreement with previous reports that cGAS signaling is markedly diminished in the absence of TBK1 (Fang et al., 2017). This demonstrated that a TBK1-null system can transduce signals through RIG-I-like PRRs and MAVS but not TLR3- and TRIF-dependent pathways. Meanwhile, similar induction of ISGs between the patients and HCs highlights the plasticity of the IFN-I response since it appears that antiviral immunity is largely adequate despite stunted cytokine production.
We next aimed to define the mechanism of TBK1-independent IFN-I induction. The presence of phosphorylated and nuclear IRF3, the principal IFN-I transcription factor, in P1 and P2 (Fig 2E, Sup Fig 4B, C) suggested that IFN-I production in a TBK1-null system involves a compensatory kinase, such as the other non-canonical IKK-related kinase, IKKε. Known to have overlapping function with TBK1, IKKε is capable of phosphorylating IRF3 (and IRF7) to initiate IFN-I production (Fitzgerald et al., 2003; Perry et al., 2004; Sharma et al., 2003; tenOever et al., 2004) and was the most likely compensatory factor. We first established binding between IKKε and IRF3 in TBK1-null hTERTs (Fig 2E), and then assessed IRF3 and STAT2 phosphorylation as measures of signaling up and downstream of IFN-I cytokine production following cytoplasmic pIC stimulation. Unlike in intracellular-pIC stimulated WT and TBK1-null hTERTs (Fig 2E), EBV-Bs (Sup Fig 2C), and A549s (Fig 2F), pIRF3 and pSTAT2 were absent in A549s doubly knocked out for TBK1 and IKKε (dKOs) (Fig 2F) and TBK1-null cells pretreated with the TBK1/IKKε inhibitor, MRT67307 (MRT) (Sup Fig 2F). Moreover, the dKO A549s were susceptible at baseline to VSV infection whereas TBK1 KOs were not (Fig 2G; Sup Fig 1D). Finally, to confirm that the lost TLR3 function was the result of absent TBK1, we complemented P1 and P2 hTERTs with lentiviruses driving TBK1 expression from either a CMV or TETon promoter. In both contexts, TBK1 restoration rescued TLR3 signaling (Fig 2H, I). These results confirm that IKKε activates IFN-I production in the absence of TBK1 via MAVS-associated PRRs and provide a plausible mechanism for the lack of overt susceptibility to viral infection in human TBK1 deficiency.
Phospho-inactive TBK1 is more disruptive to IFN-I induction than complete loss of TBK1 expression.
It is important to note that the above results do not reconcile the different disease manifestations between the patients reported here and the individuals with heterozygous mutations in TBK1 that suffered from HSE (Herman et al., 2012). To do that, we complemented P2 hTERTs with a phospho-inactive form of TBK1 (S172A) (Kishore et al., 2002) and the previously characterized kinase-dead allele from one of the patients who developed HSE (G159A) (Herman et al., 2012). S172A did not rescue TLR3 function. Instead, it disrupted IRF3 and STAT2 phosphorylation more than the TBK1-null system (Fig 2J). To understand how S172A antagonizes IKKε’s ability to phosphorylate IRF3, we assessed the impact of S172A on the interaction between IKKε and MAVS in A549s but found no difference between S172A, WT or TBK1-null cells (Fig 2K, L). This suggests that S172A sterically blocks IKKε’s ability to phosphorylate IRF3 at MAVS, not the initial arrival of IKKε to the scaffold, though the marked decrease of MAVS-bound S172A remains curious. Functionally, this antagonized IFN-I response beyond complete LoE manifested in a heightened susceptibility to VSV 6 hrs after an intracellular pIC stimulation for both G159A and S172A (Fig 2M). In contrast to the S172A and G159A mutations, P3’s allele (Y212D) was not overtly susceptible to infection (Fig 2M), likely a result of the hypomorphicity—as opposed to complete LoF—of the Y212D variant, which has a similar, if not slightly better, signaling capacity in MAVS-mediated pathways to the TBK1-null system (Fig 2B, C, Sup Fig 2D). Thus, IKKε interference enhances IFN-I antagonism and likely contributes to the discordant anti-viral phenotype between these TBK1-null patients and the heterozygous individuals described previously (Herman et al., 2012).
TNF-driven RIPK1-mediated cell death is a source of autoinflammation in TBK1 deficiency
TBK1 participates in multiple other immunoregulatory networks beyond IFN-I and NF-κB activation. In addition to a well-established role in autophagy, which appeared unaffected in P1 and P2 hTERTs (Sup Fig 4D, E), and only slightly slowed in TBK1/IKKε dKO A549s (Sup Fig 4F), TBK1 also acts as an important secondary brake on tumor necrosis factor α (TNF)-induced cell death.
The different avenues on which the TNF receptor signaling complex (TNFR1-SC) can transmit depend on the downstream components that are recruited following binding of the cytokine to the receptor. Under most conditions, TNF initiates a pro-inflammatory and pro-survival signal via K63 and M1 linear ubiquitination of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) by two E3 ubiquitin (Ub) ligases: the cellular inhibitor of apoptosis proteins (cIAP) and the linear Ub chain assembly complex (LUBAC) (Fig 3A). These Ub chains tether the TNFR1-SC, RIPK1 included, to NEMO and its associated kinases to turn on the NF-κB and MAPK pathways (Peltzer and Walczak, 2019; Ting and Bertrand, 2016; Yuan et al., 2019). However, the balance of adapter and secondary messenger proteins downstream of the receptor can tip converting TNF signaling into a cell death-inducing event (Fig 3A). If that happens, cell death occurs via either apoptosis or necroptosis. In this context, RIPK1, activated by TNFR, dissociates from the TNFR1-SC and recruits death domain proteins in a complex that cleaves caspase-8 (CASP8) to initiate apoptosis (Feoktistova et al., 2016; Micheau and Tschopp, 2003).
Figure 3. TNF-driven RIPK1-mediated cell death is a source of autoinflammation in TBK1 deficiency.

A) Cartoon depicting the pro-survival and pro-death arms of the TNF signaling pathway.
B) FLAG IP of the TNFR-SC in hTERTs following a 15 min stimulation with 1 μg/mL FLAG-TNF.
C) hTERTs stimulated with TNF over 24 hrs (n = 3).
D) hTERTs stimulated with TNF assessed for loss of pCYLD. Phosphorylation at this site decreases deubiquitinase activity.
E) hTERTs pretreated for 30 min with either birinapant and zVAD, or birinapant, zVAD, and Nec-1 were exposed to TNF for 24 hrs. Relative viability was measured by resazurin reduction (n = 3).
F – G) hTERTs complemented with DOX-inducible TBK1 were pretreated for 30 min as indicated and then stimulated with TNF (5 ng/mL) for 24 hrs. Relative viability was measured by resazurin reduction (F n = 3, G n = 6).
If CASP8 is inactive, RIPK1 will phosphorylate RIPK3 (Cho et al., 2009; Mompeán et al., 2018; Newton et al., 2014; Polykratis et al., 2014; Wu et al., 2013; Zhang et al., 2009), which in turn triggers the mixed lineage kinase domain-like protein (MLKL) to oligomerize and form pores in the membrane that drive necroptosis (Cai et al., 2014; Dondelinger et al., 2014; Huang et al., 2017; Wang et al., 2014; Wu et al., 2013, 2011). TBK1 can suppress these cell death processes by placing an inactivating phosphate on RIPK1 at T189 (Fig 3A) (Xu et al., 2018). It is because of this mechanism that TBK1-null murine cells undergo RIPK1-mediated cell death (RCD) in response to TNF more readily than WT (Lafont et al., 2018; Xu et al., 2018).
Suspecting elevated rates of cell death might feed into these patient’s chronically inflamed state, we measured the impact of TBK1 deficiency on TNF signaling in the patient-derived hTERTs. An IP for FLAG-TNF revealed that TNFR-SC assembled all components involved in NF-κB activation, while a TNF stimulation time course showed normal target gene (IL-6) induction in the absence of TBK1 (Fig 3B, C), demonstrating proper function of the proinflammatory arm of the TNF signaling pathway in the absence of TBK1.
However, the impact of TBK1 deficiency on TNF-induced RCD was substantial. CYLD is a deubiquitinase that dismantles the poly-Ub link between RIPK1 and NEMO to potentiate RCD (Draber et al., 2015; Kovalenko et al., 2003; Ting and Bertrand, 2016; Trompouki et al., 2003; Wright et al., 2007). We found that CYLD loses its inactivating phosphorylation mark faster in TBK1-null hTERTs than WT (Fig 3D). We also found that complementation with WT TBK1 completely rescued pCYLD stability, but complementation with G159A, the kinase-dead allele from the patient who developed HSE without baseline autoinflammation, did so only partially (Sup Fig 5A). This difference in pCYLD stability may indicate the minimum level of inactivated (phosphorylated) CYLD required to prevent RIPK1 deubiquitination and may help explain why the patient with the G159A allele did not suffer from autoinflammation. Thus, in addition to its already described function as a direct inhibitor of cell death via RIPK1 phosphorylation (Lafont et al., 2018; Xu et al., 2018), TBK1 also likely plays a role in regulating the ubiquitination status of RIPK1 by controlling CYLD.
All this disrupted regulation culminated in a higher rate of necroptosis in response to TNF in P1 and P2 hTERTs compared to WT following inhibition of the cIAPs (birinapant) and CASP8 (zVAD) (Fig 3E). Importantly, this effect was rescued by the RIPK1-inhibitor, Necrostatin-1 (Nec-1) (Fig 3E), and by genetic complementation with WT TBK1 (Fig 3F). Complementation with either Y212D TBK1 (P3’s mutation) or S172A TBK1 produced a partial rescue of RCD (Fig 3G, Sup Fig 5B), which means that even fully inactive TBK1, if expressed, can provide some anti-RCD function. A complete role reversal compared to the effect of LoF TBK1 in IFN-I induction, where the IKKε-mediated anti-viral response was weaker with inactive TBK1 than in the TBK1-null system (Fig 2J–M). At the same time, TBK1/IKKε dKO A549s, but not TBK1 single KOs, died under necroptosis-inducing conditions, which highlights the importance of IKKε in preventing TNF-induced cell death and demonstrates that its anti-death capacity varies across cell types (Sup Fig 5C).
When we parsed this death response to confirm the specific mechanism, we found clear evidence for necroptosis and signs of mild apoptosis in TBK1-null cells. Under necroptotic conditions with zVAD and birinapant, we observed more pRIPK1 and pMLKL in TBK1-null cells than WT in response to TNF stimulation (Sup Fig 5D, E). Under apoptotic conditions in the absence of zVAD, we observed small amounts of cleaved CASP3 (CC3) with TNF and birinapant, indicating low-level apoptotic activity in both genotypes (Sup Fig 5F). However, this level of apoptosis was not enough to produce appreciable killing in our viability assay (Sup Fig 5G). On the other hand, pyroptosis, an inflammasome-driven mechanism of cell death precipitated by CASP1 cleavage (Chan and Schroder, 2020), did not appear to participate in the TNF-induced cell death response, at least not in hTERT fibroblasts following TNF stimulation or plasmid DNA transfection (Sup Fig 5H, I).
Another factor we considered was the contribution of IFNγ to the inflammatory phenotype. IFNγ was recently shown to synergize with TNF to kill mice across the range of regulated cell death mechanisms (Karki et al., 2021), and we were curious to see if a combinatorial effect would manifest in TBK1 deficient cells. We carried out stimulations in various death-predisposing conditions and found that IFNγ not only can enhance the cell-killing capacity of TNF, but it can also kill on its own under the right conditions (Sup Fig 5J). In conjunction with zVAD and birinapant, IFNγ killed both WT and TBK1-null cells, though WT was not as sensitive as P1 (Sup Fig 5J). Further, we found that this mode of cell death is RIPK1-mediated when pretreatment with Nec-1 rescued viability in at least WT TBK1-expressing hTERTs (Sup Fig 5J).
TBK1 and IKKε regulate cell death via CYLD.
Intrigued by the faster inactivation of CYLD in P1 and P2 (Fig 3D), we probed the possibility that TBK1 and IKKε phosphorylate CYLD (Hutti et al., 2009; Xu et al., 2020), and assessed the contribution of that interaction to RCD. Mechanistically, this interaction could impact the prodeath propensity of RIPK1 as CYLD functions to remove poly-ub chains that prevent RIPK1 from joining the death-signaling complex (Moquin et al., 2013; Xu et al., 2020). We found that transfected TBK1 and IKKε phosphorylate transfected CYLD in a 293T overexpression system, whereas the kinase-dead TBK1-K38A did not (Fig 4A, B). Chemical inhibition of TBK1/IKKε with MRT also abolished CYLD phosphorylation in response to TNF in WT and NEMO-null Jurkats (Fig 4C) and increased cell death beyond the impact of NEMO deficiency (Fig 4D, E). This suggested that in addition to directly phosphorylating RIPK1, TBK1 and IKKε restrain CYLD-mediated deubiquitination of RIPK1 to further curb an RCD response to TNF. To define CYLD’s contribution to RCD regulation, we showed that shRNA KD of CYLD in Jurkats and CYLD KO in mouse ear fibroblasts reduced the rate of TNF-induced necroptosis in NEMO-null cells, even in the presence of MRT (Fig 4F, G). Finally, we completely rescued the necroptotic effect of TNF in NEMO-null, MRT-treated, CYLD KD Jurkats with the RIPK1 inhibitor, Nec-1 to confirm that this is all in the context of RCD (Fig 4H). Combined, these data support the notion that TBK1 exerts partial regulation of TNF-mediated cell death via phosphorylation of CYLD.
Figure 4. TBK1 and IKKε regulate cell death via CYLD.

A) CYLD overexpressed with WT or kinase-dead K38A TBK1 in 293EBNAs.
B) CYLD overexpressed with TBK1 or IKKε in 293EBNAs.
C – E) WT or NEMO-null Jurkats pretreated for 30 min with MRT and then stimulated with TNF for C) 15 min, D) 0-4 hrs E) 24 hrs (n = 3).
F) CYLD knocked down in WT or NEMO-null Jurkats by shRNAs. Cells were pretreated with zVAD and MRT for 30 min before stimulation with TNF for 24 hrs (n = 3).
G) Murine ear fibroblasts were treated with zVAD and MRT for 30 min before a 24 hr TNF stimulation (n = 3).
H) CYLD knocked down in NEMO-null Jurkats by shRNAs. Cells were pretreated for 30 min with zVAD, MRT, and Nec-1 before TNF stimulation for 24 hrs (n = 3).
Significance was evaluated by T-tests, **p≤0.01, ***p≤0.001.
Ex vivo inflammation in TBK1 deficiency.
The high rate of RCD in TBK1 deficiency represents a plausible source for the autoinflammation in these patients. We suspected that a predisposition towards TNF-induced cell death might create a feedforward loop of immunostimulatory DAMP release that fuels continued inflammation. Consistent with this hypothesis, we found apoptotic activity, indexed by CC3, globally in P2 and specifically in the T cells, pDCs, and non-canonical monocytes of P1 (Fig 5A) that tracked with a nonspecific inflammatory profile across multiple cell types in whole blood immunophenotyping via CyTOF. The canonical immune activation marker, CD69, was most conspicuous, elevated in all immune cell subsets in P1 and P2 (Fig 5A), while other inflammatory markers appeared to be restricted to specific compartments. PD-1 and CD57 were high in CD4 T cells indicating a shift towards an active, and potentially exhausted, effector profile (Fig 5A, B). CD8 T cells also appeared to be in an active state, though this was better represented by an increase in CD38, while the less mature, and uncommitted, CD4−/CD8− and CD4+/CD8+ T cells were high in PD-1 (Fig 5A). Finally, monocytes, mDCs, and NK cells displayed augmented signaling compared to HC, indicated by elevated STAT1, STAT3, MK2, and ErK phosphorylation. (Fig 5A).
Figure 5. Baseline immunophenotype of blood from TBK1-deficient individuals.
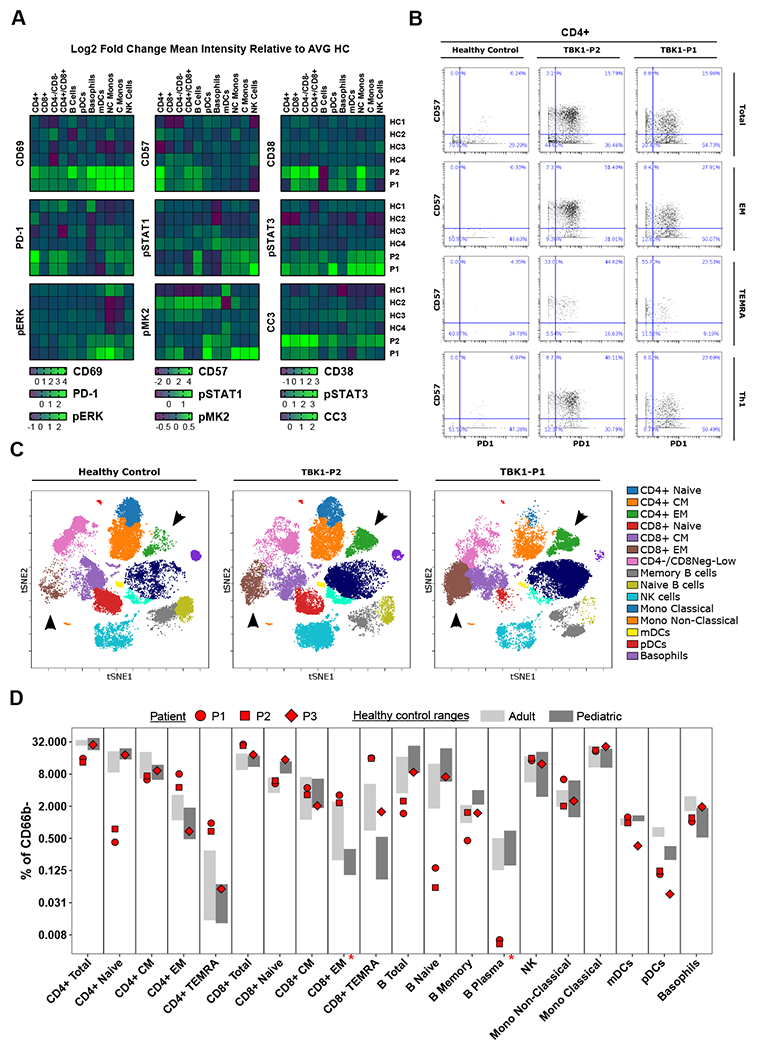
CyTOF immunophenotyping of whole blood from TBK1-deficient patients and HCs. The data in A and D is from a separate experiment from the data in B and C. The experiment that generated B and C had 1 HC.
A) Mean intensity of inflammatory markers relative to the average of 4 HCs.
B) Manually gated CD4 T cells and subsets from P1 and P2 compared to HC.
C) Unsupervised clustering of agranulocytes (CD66b-) displayed on a multidimensional tSNE plot.
D) Immune cell subsets at baseline represented as percent of agranulocytes (CD66b-). HC range n = 4, Pediatric HC range n = 5. An asterisk indicates the cell type was not identified in P3.
TBK1 deficiency also had an impact on immune cell proportions. In the T cell compartment, we found activated and expanded CD4 and CD8 effector populations in P1 and P2 (Fig 5B – D), specifically effector memory (EM), CD45RA re-expressing EMs (TEMRAs), and cells fitting the Th1 classification (Fig 5B). This signature resembled the splenocyte phenotype of mice with a conditional TBK1 KO in DCs (Xiao et al., 2017) as well as mice with a catalytically-inactive truncated TBK1 (Marchlik et al., 2010). Naïve CD4 cells were depleted in both P1 and P2 (Fig 5D), potentially reflecting effector cell expansion, while B cells exhibited a diminished naïve population alongside fewer plasma cells (Fig 5D). All of this was accompanied by a nonspecific inflammatory cytokine signature in Luminex on patient serum (Sup Fig 6).
Like P1 and P2, the immune profile in P3 contained expanded CD8 TEMRAs (Fig 5D) and elevated expression of CD69 in CD4 and CD8 TEMRAs as well as CD8 CM cells (Sup Fig 7A). Consistent with P1 and P2, P3 also exhibited fewer pDCs relative to age-matched HCs (Fig 5D). Blood was not available for P4.
Single-cell RNA sequencing of patient peripheral blood mononuclear cells supports cell death and hints at an anti-apoptotic signaling.
To get a better understanding of the cell death predisposition in TBK1 deficiency, we performed single-cell RNA sequencing on peripheral blood mononuclear cells (PBMCs) from P1 and P2 and an age-matched HC. Unsupervised graph-based clustering of 9833 cells integrated from patients and control resolved 19 clusters annotated in a semi-supervised fashion based on marker gene expression profiles (Fig 6A, B). Differential gene expression analysis between the patients and HC (Fig 6C) found that, in addition to several nuclear genes (MTRNR2L1, MTRNR2L10, MTRNR2L12), the patients express high levels of mitochondria-related genes, evidence of the putative cell death signature (Fig 6D). Closer examination of mitochondrial gene expression at cell-type-specific resolution revealed that myeloid cells (monocytes: cluster 1,4,11 and mDCs: cluster 13) have the greatest elevations in mitochondrial gene expression between the patients and HC (Fig 6E).
Figure 6. Single-cell RNA sequencing of patient PBMCs supports cellular death and hints at anti-apoptotic cell death signaling pathways.

A) UMAP of the integrated dataset of 9833 PBMCs from the two TBKT−/− patients and the HC. Points are colored by the results of unsupervised graph-based clustering.
B) Major immune cell populations and their subsets annotated according to marker gene expression patterns.
C) UMAP from A colored by patient identity.
D) Global PBMC differential gene expression between TBK1−/− patients and the HC reveals elevated levels of mitochondrial-related genes. Labeled points and flagged genes represent mitochondrial genes with adjusted p-value < 10−100.
E) Clusters in the myeloid cell compartment exhibit increased levels of mitochondrial gene expression in TBK1−/− patients.
F) Expression feature plot of the top DEG, MTRNR2L1, expressed exclusively in TBK1−/− myeloid cells.
Of particular note, the top differentially expressed gene (DEG) within the myeloid cells was the nuclear-encoded MTRNR2L1 (Fig 6F) (Yong and Tang, 2018). Also known as humanin-like 1 or HN1, MTRNR2L1 encodes a secreted 21 amino acid peptide that reportedly protects epithelial cells from TNF-induced apoptosis by allosterically inhibiting the function of pro-apoptotic proteins BAX, tBID, and BimEL (Gottardo et al., 2014; Morris et al., 2020). We suspect that HN1, along with other Humanin-like genes in the DEG list, plays an important role in dictating which cell death pathways are utilized in TBK1 deficiency.
Anti-TNF therapy quells inflammation in TBK1 deficiency.
Informed by the collective data, we implemented an anti-TNF regimen to block the source of inflammation in TBK1 deficiency. After approximately 4 months of anti-TNF treatment, P1’s clinical condition began to improve. She was cautiously tapered off steroids and has not had any significant neurological sequelae, cutaneous vasculitis, or arthritis since. P3 was also initiated on anti-TNF and responded positively. Unfortunately, P2 developed an allergic reaction to two different forms of anti-TNF treatment (adalimumab and infliximab) and was placed on anti-IL-6R, but to no clinical effect (Sup Table 1). Results from P4’s anti-TNF treatment are pending.
Analyses of post-treatment blood from P1 also demonstrated improvement of her condition, with most of the aberrant immune cell populations at, or returning to, the HC range (Fig 7A). Specifically, the low naïve CD4 T cell levels rose, while effector CD4 and CD8 T cells, naïve and plasma B cells, and eosinophils all returned to the HC range (Fig 7A, B). pDCs, on the other hand, remained low and unchanged from baseline (Fig 7A). Furthermore, treatment correlated with a quieting of various dysregulated inflammatory markers and a drop in apoptotic activity across the immune landscape (Fig 7C). CD69 and pSTAT3 levels were universally affected but fell most prominently in the myeloid compartment, while a decrease in ErK activation was limited to non-classical monocytes, mDCs, and basophils. In lymphocytes, NK cells experienced substantial drops in pSTAT1, pSTAT3, and CD69, and inflammation generally resolved across various T and B cell subsets in unique patterns for each marker (Fig 7C). These changes in inflammatory activity occurred as CC3 levels resolved from a mild but appreciably elevated state in most lymphocytes (Fig 7C & Sup Fig 7B). P1 and P3’s prognosis solidifies that the clinical manifestation of TBK1 deficiency is driven by TNF-mediated cell death.
Figure 7. Anti-TNF therapy quells inflammation in TBK1 deficiency.

CyTOF immunophenotyping of whole blood from P1 collected pre (day 0) and post (day 131) administration of anti-TNF treatment (Infliximab) compared to 4 HCs.
A) Immune cell subsets represented as percent of agranulocytes (CD66b-).
B) Eosinophils as a percent of granulocytes (CD66b+).
C) Mean intensity of inflammatory markers relative to the average of 4 HCs.
Discussion
Human TBK1 deficiency manifests in a somewhat unexpected phenotype. Loss of a gene traditionally thought of as crucial for innate immune activation produces a clinical syndrome defined primarily by systemic autoinflammation. While PID and IEIs often manifest as inflammatory conditions alongside, or independent of, an elevated risk of infection, complete TBK1 deficiency does not appear to render humans especially vulnerable to infectious disease, though the possibility exists that these individuals may be more sensitive to highly pathogenic viruses like SARA-CoV-2 given their hypomorphic ability to induce IFN-I cytokine (Gottardo et al., 2014). Instead, the salient consequence for human health is chronic and systemic autoinflammation precipitated by an imbalance in TNF signaling that favors higher rates of immunostimulatory forms of cell death, which may contribute to the lack of infectious disease susceptibility.
As a centrally positioned intermediate in TLR3-, STING-, and MAVS-mediated immune activation, expectations were that humans lacking TBK1 function would be at a disadvantage when it came to their antiviral immunity. It has been shown that TBK1-null mouse fibroblasts have a diminished IFN-I response to viral infections (Fitzgerald et al., 2003; Perry et al., 2004; tenOever et al., 2004), and humans harboring heterozygous LoF mutations in TBK1 are susceptible to HSE (Herman et al., 2012). Yet, the TBK1-deficient patients described here have shown no signs of overt sensitivity to viral infection over their nearly thirty-year life history. The capability of their antiviral immunity then has to come from the compensatory function of IKKε, which can phosphorylate IRF3 at MAVS in the absence of TBK1 in mouse and human cells (Fig 2) (Fitzgerald et al., 2003; Perry et al., 2004; Sharma et al., 2003; tenOever et al., 2004). However, it appears that the quality of TBK1 impacts the competency of IKKε’s compensatory function. In the IFN-I context, IKKε-mediated activation of IRF3 was attenuated by expression of phospho-inactive TBK1 to levels below the protein-null system (Fig 2J–M), but in response to TNF, phospho-inactive TBK1 had the opposite effect rendering cells less susceptible to TNF-induced RCD than the null system (Fig 3G, Sup Fig 5B). This biochemical distinction between LoF and LoE argues that AD LoF variants can have epistatic effects that are pathway-specific and more profound than complete LoE. It also suggests that different TBK1 variants may lead to autoinflammation or susceptibility to viral disease, or potentially both depending on the affected pathways.
It is important to note, however, that this IFN-I defect and its potential consequence for human health is a secondary component of this disease, a notion that simultaneously challenges the necessity of TBK1 in innate immunity and contends that the prepotent role for TBK1 is in RIPK1 negative regulation. The higher propensity for RCD in response to TNF in TBK1 deficiency predisposes TBK1-null individuals to a vicious inflammatory feedforward loop, as any minor stimulus that triggers the release of TNF can cause cells to die. Unlike traditional apoptosis, which is immune-silent, RCD, in the form of either apoptosis via cleaved CASP8 or necroptosis via oligomerization of MLKL, produces DAMPs that present a potent immune stimulus. Most of the time in healthy individuals, TNF engagement initiates a pro-inflammatory and pro-survival signal because RIPK1 is tethered to NEMO by K63 and M1 Ub chains maintained by the E3 ligases cIAP1/2/TRAF2 and the LUBAC complex (SHARPIN, HOIP, HOIL). This tether ensures that TNF-mediated RIPK1 activation will drive the NEMO-associated kinases involved in the NF-κB and MAPK pathways (Peltzer and Walczak, 2019; Ting and Bertrand, 2016; Yuan et al., 2019).
Should the Ub tether fail, RIPK1 gains the potential to dissociate from TNFR1, form a complex with CASP8 and RIPK3/MLKL, and initiate RCD. However, there is backup regulation as RIPK1 can be inactivated by the canonical NF-κB inhibiting kinases (IKKα/β), the noncanonical IKKs, TBK1 and IKKε, and, to a lesser extent, the TNFR-SC-independent MAP kinase-activated protein kinase 2 (MK2) (Amin et al., 2018; Annibaldi et al., 2018; Peltzer and Walczak, 2019; Ting and Bertrand, 2016). Interestingly, it has been suggested that none of these RIPK1 checkpoint kinases can compensate for each other, meaning that if one functions incorrectly TNF becomes cytotoxic (Peltzer and Walczak, 2019), but our data in TBK1/IKKε dKO A549s seems to indicate some cell-type specificity to that point. In contrast to murine cells, which are more susceptible to RCD without TBK1 (Lafont et al., 2018; Xu et al., 2018), TBK1 KO A549s could not be killed by TNF while the TBK1/IKKε dKOs could (Sup Fig 5C), suggesting that IKKε can compensate, at least in part, for TBK1 in RCD prevention in a human cell line and that the relative IKKε expression across tissues may contribute to varying RCD susceptibility. These data also provide a hint as to why humans can survive TBK1 deficiency, but mice cannot: perhaps human IKKε is better suited to pick up the slack than its murine ortholog.
We also show that direct phosphorylation of RIPK1 is not the only way that TBK1 can steer the outcome of TNF receptor engagement. TBK1 can influence the deconstruction of the Ub tether and deubiquitination of RIPK1 after it has dissociated from TNFR1 by inactivating CYLD (Fig 4). Deubiquitinases such as CYLD counter the function of cIAP1/2 and LUBAC to dampen the NF-κB response downstream of the TNFR-SC (Brummelkamp et al., 2003; Kovalenko et al., 2003; Lork et al., 2017; Trompouki et al., 2003). Without TBK1, CYLD has more leeway to dismantle the poly-Ub links that keep RIPK1 away from MLKL and CASP8 and deubiquitinate TNFR1-independent RIPK1 in a process that further encourages RCD (Lork et al., 2017). Therefore, negative regulation of CYLD by TBK1 potentially contributes to these patients’ autoinflammatory condition by enhancing necroptosis.
Ultimately, the source of morbidity in TBK1 deficiency is RCD-triggered DAMP detection, which leads to generalized autoinflammation likely fed, at least in part, by IL-1s. Specifically, IL-1α was elevated in P1 and P2 (Sup Fig 6A) and might, if IL-1s are released in response to DAMPs, explain why P2 and P4 experienced partial improvement on anakinra (Sup Table 1; Data S1). Also, the specifics of this mechanism of autoinflammation distinguish this disease from one caused by a constitutively active RIPK1 that resists cleavage by CASP8 (Lalaoui et al., 2020; Tao et al., 2020). While both conditions stem from dysregulated RIPK1 that leads to unchecked RCD, the cleavage-resistant RIPK1 patients also suffer from proinflammatory signaling that is RIPK1-dependent, an absent feature in TBK1 deficiency.
In its entirety, TBK1 deficiency is a complex and unexpected IEI that produces chronic and systemic autoinflammation driven by a heightened sensitivity to RCD. TNF-targeting therapies are a viable treatment option that, if administered early, might stave off the detrimental consequences of chronic inflammation.
Limitations of the Study
We acknowledge that important additional information surrounding TBK1 deficiency could be gleaned from tissue biopsies, such as the liver, that were not available. There also remains an important association between heterozygous mutations in TBK1 and ALS/FTD (Cirulli et al., 2015; Freischmidt et al., 2015). P1 and P2 have intellectual disabilities, but none of the four patients described currently experience related signs or symptoms of ALS/FTD. However, follow-up evaluations of all patients and their heterozygous parents could provide new details surrounding this CNS connection. Detailed genetic and biochemical evaluations are required to further delineate each variation’s ability to regulate IFN-I induction and contain IFNγ- and TNF-mediated cell death pathways. Finally, certain experiments used only one patient cell line due to different growth rates of available material.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Requests for resources, reagents, and additional information should be directed to the lead contact, Dusan Bogunovic (dusan.bogunovic@mssm.edu).
Materials availability
All plasmids and cell lines used in this study are available from the lead contact.
Data and code availability
Counts matrices for bulk and single-cell RNAseq experiments are available on Mendeley Data (bulk RNAseq DOI: 10.17632/648zntv5dx.1; single-cell RNAseq DOI: 10.17632/96hx5z4wv8.1). This study did not generate any unique code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
The patients or their parents provided informed consent granting the use of their samples and associated data and images in this publication according to the requirements set forth by the Institutional Review Board at Erasmus University Medical Center (protocol METC-2012387), the National Institute of Diabetes and Digestive and Kidney Diseases/National Institute of Arthritis, Musculoskeletal and Skin Diseases Institutional Review Board, or the Hacettepe University Ethics Committee (GO 15/744). Please refer to Data S1 (clinical history) and Supplemental Table 1 for information about the patients’ age, sex, and disease details.
Cell lines
Patient- and HC-derived dermal fibroblasts, immortalized with human telomerase (hTERT), HEK293Ts, and A549 cells were cultured in DMEM containing 10% fetal bovine serum (FBS) (Invitrogen), 1% GlutaMAX (GIBCO), and 1% penicillin/streptomycin (GIBCO). All cell lines reconstituted with TBK1 were transduced with lentivirus and either selected with puromycin or sorted by RFP or BFP expressed as a separate protein from the same plasmid. EBV-transformed lymphoblastoid cell lines (EBV-B cells) were generated by infecting PBMCs from the patients or HCs with EBV supernatants and were cultured in RPMI with 10% FBS, 1% GlutaMAX (GIBCO), and 1% penicillin/streptomycin (GIBCO). Single-cell clones from CRISPR-generated KOs in A549s were isolated via limiting dilution and screened by Western Blot and qPCR (data not shown) for the absence of TBK1 and IKKε, and competence of signaling in response to transfected pIC. CRISPR editing was performed with Alt-R® S.p. Cas9 Nuclease V3 (IDT 1081058) and Alt-R crRNA guide RNAs (IDT) transfected into A549s with a 4D-Nucleofector (Lonza) using the manufacturer’s optimized protocol. 3T8 are CD3+ Jurkat T-cells, and 8321 are a NEMO-null derivative of 3T8 produced by ICR191 mutagenesis and selection for cells that do not respond to phorbol myristate acetate (PMA) (He and Ting, 2002).
METHOD DETAILS
Genetic analyses: TBK1 variant detection
P1 and P2 genomic DNA was extracted from peripheral blood samples using standard procedures. Diagnostic chromosomal investigation in P1 and P2 using high-resolution SNP-array (Illumina Infinium® CytoSNP-850K v1.2) excluded copy number variants but identified shared regions of homozygosity in the siblings fitting with the consanguinity between their parents.
P1’s DNA was enriched for diagnostic whole exome sequencing using the Sureselect Clinical Research Exome (CRE) Capture Enrichment kit (Agilent Technologies) and paired-end sequenced on the Illumina Hiseq platform. Data were demultiplexed by Illumina software bclfastq, sequence reads were mapped to the genome with the BWA algorithm (http://bio-bwa.sourceforge.net/) and variant calling was performed by the Genome Analysis Toolkit (http://www.broadinstitute.org/gatk/). With this sequencing strategy, the average coverage of the exome is aimed at 50X, and within P1 more than 91% of the RefSeq sequences were covered at least 20X. Variants were filtered using the Cartagenia software package on minor allele frequency (MAF<1% in population databases), location (within an exon or exon-intron boundaries) in genes included in the Intellectual Disability gene panel (version 4; X no. of genes), and homozygous state. No candidate was identified and expanded variant filtering was based on homozygous variants located in the shared regions of homozygosity between P1 and P2. This revealed a homozygous nonsense variant in TBK1 (chr12:64890825G>A; p.(W619*)), which was confirmed by Sanger sequencing. In P2, the homozygous presence of TBK1 p.(W619*) was confirmed by Sanger sequencing.
For P3, genomic DNA was extracted from peripheral blood using the Maxwell® 16 Blood DNA Purification Kit (Promega). Exome sequence libraries from P3, the parents, and a healthy sibling were prepared using the Twist Custom Panel EF Workflow (Twist Bioscience) and paired-end sequencing was performed on an Illumina HiSeq2000 instrument. The GATK base quality score recalibration, indel realignment, duplicate removal was applied for data analysis. SNP/INDEL discovery and genotyping were conducted using standard hard filtering parameters according to GATK Best Practices recommendations (Van der Auwera et al., 2013; DePristo et al., 2011). Gemini and Exomiser software packages were used to further prioritize the variants (Paila et al., 2013; Robinson et al., 2014). Variants with a total minor allele frequency < 0.001 in the Genome Aggregation Database (gnomAD) and a read depth > 20 were considered for the analysis and a consanguineous inheritance model was assumed (Sup Table 2).
Autoantibody Array
The presence of autoantibodies in circulation was detected with an IgG autoantibody array (RayBiotech PAH-AIDG-G1-16). Plasma from five healthy donors, two donors with systemic lupus erythematosus (SLE), and two with Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) were included as controls. All plasma was clarified and diluted 1:200 before testing. Slides were scanned at 10 μm resolution using an Axon GenePix 4300 microarray scanner (Molecular Devices) using a laser at 532 nm with 100% power/PMT Gain 560. A raw 16-bit .tiff format image file was generated for each slide and analyzed with Genepix Pro 7.0 software (Molecular Devices).
Cloning and mutagenesis
The TBK1 CDS was cloned from a HC fibroblast cell line and ligated into the Gateway (Thermo Fisher) compatible pDONR223 and subsequently flipped into pTRIP-X-IRES-PuroR-2A-RFP or subcloned with In-Fusion (Takara) into a lentiviral compatible vector with a TETon 3G (TRE3G) promoter (Takara) and BFP expressed from a separate constitutive promoter. The TETon system was combined with a separate lentiviral compatible vector containing the reverse tetracycline-controlled transactivator (rtTA) (Addgene: 66810). Site-directed mutagenesis (SDM) was performed with QuickChange II (Agilent) in the pDONR223 backbone. The mutations were then flipped with Gateway or In-Fused into the desired expression construct. Lentivirus for stable gene expression was produced in HEK293T cells transfected according to the protocol for lipofectamine 2000 (Thermo Fisher) with pCAGGS-VSV-G, pCMV-Gag/Pol, and genes of interest at a 1:4:5 ratio. Media was changed the next morning and virus was collected at 48 and 72 hpt.
Immunoblots
Cells were lysed in RIPA buffer (Thermo Fisher 89900) with protease/phosphatase inhibitor cocktail (Cell Signaling #5872) for at least 10 min on ice. Samples were centrifuged to remove insoluble complexes and then boiled with NuPage sample buffer (Thermo Fisher NP0007) containing 50 mM DTT. The samples were subjected to gel electrophoresis and semi-dry transfer. Membranes were blocked in 5% BSA, then incubated overnight with primary antibody followed by HRP-conjugated secondary antibodies and visualized on film. Fig 1C, 1D, 2B, 2J, 2K, 4A, 4B, 4C, 4D, Sup Fig 2F, and Sup Fig 5I are representative of at least three replicate experiments. 2F, 2H, 3D, Sup Fig 2C, Sup Fig 5D, Sup Fig 5E, Sup Fig 5F, and Sup Fig 5H are representative of two replicate experiments. Fig 2E, Fig 3B, Sup Fig 4D and 4F, and Sup Fig 5A are single experiments.
Immunoprecipitation
Cells were lysed in 50 mM Tris, pH 6.8, 0.5% NP-40, 200 mM NaCl, 10% glycerol, 1 mM EDTA, plus 1X Protease/Phosphatase inhibitor cocktail (Cell Signaling Technology). Lysates were sonicated and then incubated with primary Ab at the dilution recommended by the manufacturer or with anti-FLAG conjugated protein G dynabeads overnight at 4C. If the Ab was not preconjugated to the beads, the immune complex was then conjugated to magnetic protein G dynabeads (Thermo Fisher) for 20 min at RT in wash buffer (0.02% TWEEN-20 in PBS) the next day. Immunoprecipitates were eluted with 20 - 30 uL 50 mM Glycine HCl, pH 2.75 for 2 min and analyzed on an immunoblot as described above.
qPCR
Total RNA was isolated from cells in culture with RNeasy spin columns (QIAGEN 74104). cDNA was synthesized with the High-Capacity RT Kit (Applied Biosystems 4368814) and measured in qPCR with the TaqMan Master Mix II with UNG (Thermo Fisher 4440038) on a LightCycler 480 (Roche) with the following primers/probes: 18S (4318839), GAPDH (4453320), IFIT1 (Hs01911452), TBK1 (HS00179410_m1), IL-6 (Hs00985639_m1), MX1 (Hs200895608_m1), ISG15 (Hs00192713_m1), IFI44L (Hs00915292_m1), RSAD2 (Hs00369813_m1). Expression was normalized to GAPDH or 18S and then made relative to an untreated calibrator (NT HC) via the ΔΔCt method.
RNAseq
hTERTs derived from P1, P2, and three HCs were stimulated with 10 μg/mL pIC, 10 μg/mL pIC + Lipofectamine 2000 (Thermo Fisher), Lipofectamine 2000 alone, or left untreated (NT) for 24 hrs. Total RNA was isolated with RNeasy spin columns (QIAGEN 74104), treated with RNase-Free DNase (QIAGEN 79254), and prepped for RNAseq with the TruSeq RNA Sample Prep Kit v2 (Illumina). 150 bp unpaired reads were generated on a NextSeq 550 (Illumina). Results were analyzed in BaseSpace (Illumina) using the RNA-Seq Alignment (STAR) and RNA-Seq Differential Expression (DESeq2) modules.
Single-cell RNA sequencing sample processing
Within 16 hours of isolation, fresh PBMCs were processed for 3’-end droplet-based scRNA-seq on the 10 Genomics Chromium (version 2) platform. PBMC collection and scRNA-seq from P1, P2 and an age-matched HC were performed in a single experiment to mitigate batch effects. For each sample, 12,000 cells were loaded onto a single lane of the 10X Genomics Chromium chip (version 2) and scRNA-seq libraries were prepared according to the manufacturer’s instructions. Libraries from the 3 donors were pooled and sequenced on the Illumina HiSeq 4000 in paired-end configuration (Read 1: 26nt; Read 2: 98nt).
Cell Death Assay
Cells were plated at ~80-90% confluency in a 96-well plate the day before. Different concentrations of the inhibitors were required to induce RCD in different cell lines. hTERTs and A549s were pretreated with 10 μM of the SMAC-mimetic, Birinapant (ApexBio: A4219), 100 μM of the pan-caspase inhibitor, Z-Val-Ala-DL-Asp(OMe)-fluoromethylketone (zVAD) (Bachem 4027403), and/or 10 μM of the RIPK1 inhibitor, Necrostatin-1 (Nec-1) (Selleck Chemicals S8037) for 30 min before a 24 hr stimulation with human or murine TNFα. Jurkats and mouse-ear fibroblasts were treated with 1 μM Birinapant, 25 μM zVAD, and/or 5 μM MRT67307. Viability was measured with the Deep Blue Cell Viability Kit (BioLegend 424702) or CellTiter-Glo 2.0 (Promega G9241) on a Synergy H1 Hybrid Reader (BioTEK).
Viral challenges
Cells were exposed to VSV-GFP at indicated concentrations or dilutions from stock for the indicated amount of time. Following the incubation period, cells were fixed in 4% PFA for 20 min at 4C and stained with DAPI. GFP signal from infected cells was acquired on a Celigo Imaging Cytometer (Nexcelom) and quantified in CellProfiler (McQuin et al., 2018).
ELISA and Luminex
Plasma collected by Ficoll isolation from heparinized whole blood was clarified by centrifugation. Circulating cytokine levels were determined in magnetic Luminex assays with the Bio-Plex Pro Human Inflammation (BioRad 171al001m) and the MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel (Millipore Sigma HCYTMAG-60K-PX30) according to the manufacturer’s protocol.
ELISAs were performed according to the manufacturer’s protocols. Human IFN-β ELISA Kit (PBL Assay Science 41410-2); Human IL-6 DuoSet ELISA (R&D Systems DY206-05); IL-18 Human ELISA Kit (Thermo Fisher BMS267-2).
Immunofluorescent Microscopy
hTERTs from patients and HCs were plated on μ-Slide chamber slides, transfected with 10 μg/mL pIC+L2K for 4 hrs. Staining was carried out with the Cytofix/Cytoperm kit (BD 554714). Primary for IRF3 (Cell Signaling Technology 11904) and Actin (Cell Signaling Technology 3700) were incubated overnight at 4C, and subsequently labeled with fluorescent secondary for 45 min at RT. Chamber wells were filled with 300 uL PBS, and images were acquired on a DMi8 widefield microscope (Leica) with a 40X oil-immersion objective.
For autophagy experiments, cells were grown to 50% confluency on 13-mm-diameter coverslips and fixed with 100% methanol on ice before washing with 1x PBS. Blocking was done in 10% FBS-PBS, and cells permeabilized with 0.1% Triton X-100 in PBS. Staining was performed in a moist chamber using the following antibodies: LC3B (1:500 dilution; Abcam), and Alexa Fluor 488 (1:750 dilution; Thermo Fisher Scientific). Images were taken with a 63x oil-immersion lens on an Axio Observer widefield fluorescence microscope (Zeiss).
Mass cytometry (CyTOF)
Whole blood collected by venipuncture into sodium heparin vacutainer tubes was shipped overnight from Rotterdam to New York before being stained and processed for mass cytometry immediately upon arrival. For intracellular staining, whole blood was stabilized immediately at the time of the draw in Proteomic Stabilizer PROT1 (SmartTube) and frozen at −80°C.
Frozen samples were thawed according to the manufacturer’s protocol. Once thawed, samples were washed with barcode permeabilization buffer (Fluidigm) and barcoded with Fluidigm’s Cell-ID 20-Plex Pd Barcoding Kit, and then washed and pooled into a single tube, Fc-blocked (FcX, Biolegend) and heparin-blocked to prevent non-specific binding. Cells were then stained with a cocktail of markers to identify major immune populations. All antibodies in the panel were either conjugated in-house with X8 MaxPar conjugation kits (Fluidigm) or purchased from Fluidigm. The antibody cocktail was filtered through an Amicon filter with 0.1 μm pores before staining.
After surface staining, the samples were permeabilized with methanol and stored for at least 12 hours in methanol at −80°C. Samples were washed, heparin-blocked, and stained with a cocktail of phosphorylation and signaling antibodies. The stained samples were washed and incubated in freshly diluted 2.4% formaldehyde containing 125nM Ir Intercalator (Fluidigm), 0.02% saponin and 30 nM OsO4 (ACROS Organics) for 30 minutes at room temperature. Samples were washed and acquired immediately after staining. Samples were washed once with PBS+0.2% BSA, once in PBS, and once in CAS buffer (Fluidigm). They were next resuspended at a concentration of 1 million cells per mL in CAS buffer containing a 1/20 dilution of EQ beads (Fluidigm). After instrument tuning and optimization, samples were run at an acquisition rate of < 300 events per second on a Helios mass cytometer (Fluidigm) with a modified wide-bore injector (Fluidigm).
This study contains data pertaining to P1 and P2 from two separate rounds of CyTOF. The first employed a panel of surface markers on whole blood samples shipped overnight from Rotterdam to New York. This data is found in Fig 5B – C. The second run involved blood collected from P1 and P2 on the day treatment was initiated, and from blood collected from P1 after 131 days of treatment. For this pre-treatment/post-treatment evaluation, all blood was fixed immediately upon collection with Proteomic Stabilizer PROT1 (SmartTube) and frozen at −80°C where it was stored until it was shipped to New York for staining and data collection. All CyTOF data pertaining to P3 was acquired in a third experiment in which blood was fixed immediately upon collection with Proteomic Stabilizer PROT1 (SmartTube) and then frozen on dry ice and shipped via overnight delivery the same day to New York for staining and data collection.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as the mean ± standard deviation where relevant. Exact values of n are reported in the figure legends.
Autoantibody Array
Genepix Pro 7.0 software (Molecular Devices) was used for analysis of 16-bit .tiff image file for each slide. Local backgrounds were subtracted, and the median net intensity in relative fluorescence units was determined for each autoantigen. All plasma was run in triplicate. The data is presented as the ratio of patient average/HC average+2SDs.
Image Quantification
Immunofluorescence microscopy was used to evaluate, percent VSV-GFP infection, nuclear or cytoplasmic IRF3 location, and LC3 puncta formation as measures of IFN-I signaling and autophagy, respectively. CellProfiler was used in all cases for quantification (McQuin et al., 2018). To measure percent infection, DAPI was segmented (global, Minimum Cross-Entropy) to provide a total cell count. GFP was segmented (global, Minimum Cross-Entropy) to define the total infected area. Nuclei that overlapped with the infected area were considered infected. To determine IRF3 localization image illumination was corrected, intensities were log2 transformed, and then DAPI (adaptive, Otsu, two classes) and Actin (propagation, global, Otsu, two classes) were segmented to determine cell, cytoplasm, and nuclear compartments. Mean IRF3 intensity was measured in each compartment of each cell and displayed as the nuclear/cytoplasmic ratio. To quantify LC3 puncta, DAPI (global, Minimum Cross-Entropy) and LC3 (Watershed - image, adaptive, Otsu, three classes - foreground) signals were used to segment cellular compartments. A Gaussian filter was applied (sigma = 1) and puncta (speckles) were enhanced using the EnhanceOrSuppressFeatures module. The enhanced image was segmented for puncta (global, Otus, two classes), which were related to their respective parental cell.
Viral Challenges
Percent infection was quantified in CellProfiler using the method described in the Image Quantification section of the Methods. A one-way ANOVA was used to test for significant differences between the infected samples in Sup Fig 1D.
Cell Death Assay
Percent cell death was calculated relative to the untreated or DMSO-treated condition for each cell line. The raw readout was either fluorescence intensity of resazurin reduction proportional to the number of metabolically active cells if the Deep Blue Cell Viability (BioLegend 424702) reagent was used or luminescence proportional to the amount of ATP for CellTiter-Glo (Promega G9241). Specifically, background was subtracted from blank wells containing media and either Deep Blue or CellTiter-Glo reagent without cells. The reading from each well was then normalized to the mean intensity of the NT or DMSO-treated condition (e.g., one replicate of P1 zVAD+Bir+TNF/mean of the P1 untreated wells). This produced a relative percent viability with the inverse the relative percent cell death. Each experiment used at least 3 replicates per sample. T-tests were used to calculate significance in Fig 5 and Sup Fig 5B, and multiple T-tests were used to calculate significance in Sup Fig 5B (**p≤0.01, ***p≤0.001, ****p≤0.0001).
Luminex
Samples, run in duplicate, were quantified on a MAGPIX xMAP Instrument (Luminex). Cytokine concentrations were quantified on standard curves and relative expression was plotted relative to the HC mean.
RNAseq
hTERT fibroblast from P1 and P2 were compared to cell lines from 3 HC in RNAseq. FASTQ files were uploaded to BaseSpace (Illumina), aligned to hg19 with the RNA-Seq Alignment (STAR) module, and differential gene expression was calculated with the RNA-Seq Differential Expression (DESeq2) module. All genes displayed in Sup Fig 4 had a q < 0.05 when the average of HCs treated with pIC+L2K were compared to the average of NT HCs.
Mass Cytometry (CyTOF)
Samples from P1, P2, and P3 were evaluated with CyTOF. An immunophenotyping panel of baseline markers was used to evaluate P1 and P2 compared to a single HC (Fig 5B – C). Pre/post treatment samples from P1 were compared to 4 adult HCs (Fig 5A, D; Fig 7; Sup Fig 7B). A baseline sample from P3 was measured against samples from his mother sister. To contrast immune cell subsets against a larger pool of age-relevant HCs, immune cells as a percent of CD66- cells were compared to 5 pediatric HCs from a separate batch. FCS files were normalized and concatenated with Fluidigm acquisition software, and the barcoded samples were deconvoluted with a MATLAB-based debarcoding application (“Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm”). Analysis was done in Cytobank. Cells were identified as Ir191/193-positive and Ce140-negative events. Doublets were excluded based on Mahalanobis distance and barcode separation and the Gaussian parameters acquired by Helios CyTOF software. tSNE analysis, biaxial gating, and unsupervised clustering of immune populations were also performed in Cytobank. Mean signal intensities were calculated, and relative expression/activation was determined by normalization relative to the mean for HCs.
Single-cell RNA sequencing data processing and analysis
Single-cell RNAseq was performed on PBMCs from P1, P2, and an adult HC. Base call sequencing image files were extracted off the sequencing, demultiplexed per sample and converted into paired FASTQ reads using the mkfastq command in the 10X Genomics CellRanger suite. FASTQ reads were aligned to the human CGRh38 genome reference with the CellRanger count function using the default parameters. Gene x cell matrix outputs for each donor were further processed using DoubletDetector to identify putative doublets.
Both gene x cell matrices and DoubletDetector output were read into the R statistical framework and analyzed with the single-cell RNA-seq analysis package, Seurat (v4.0.1). Cells with detectable expression of fewer than 500 genes, greater than 10% UMIs from mitochondrial gene transcripts, or identified as “doublet” by DoubletDetector were removed from further analysis. Gene expression data were independently normalized using SCTransform with the parameter ‘vars.to.regress’ set to regress out effects primarily due to mitochondrial gene expression. The top 3000 shared highly variable genes were then selected to proceed with integration analyses (immunoglobulin genes excluded). To minimize sample-specific effects, all datasets were integrated on the first 30 canonical correlation components using the SCTransform integration workflow described by the developer. Next, dimensional reduction was performed by principal component analysis (PCA) on the integrated dataset. Unsupervised graph-based clustering was performed on the first 30 principal components using the smart local moving algorithm at a resolution of 0.8 and clustering results were visualized by the Uniform manifold approximation and projection (UMAP) method. Cluster marker genes lists were generated using Seurat’s FindAllMarkers command. Gene lists were manually inspected clusters were annotated based on literature search and our expertise in immunology in a supervised fashion. Transcriptional differences between TBK1−/− patients and the HC were conducted using Seurat’s FindMarkers command. Statistical cutoffs are noted in the legend of Fig 6.
Supplementary Material
Sup Fig 1. No autoimmunity, baseline VSV susceptibility, or type-I IFNopathy associated with TBK1 mutations at conservation residues, Related toFigures 1 and 2. A) Serological screen of P1 and P2 serum for autoreactive antibodies against known epitopes linked to common autoreactive targets. B) Sanger sequencing of genomic DNA confirming the mutation status of each patient. C) Evolutionary conservation of the TBK1 residues mutated in each patient. Multiple protein sequence alignment of TBK1 was generated with the COBALT tool from NCBI. Red indicates that an amino acid differs from the human ortholog. The locations of the examined variants are indicated by the red arrow. D) hTERTs were infected with VSV-GFP for 1 hr at an MOI of 0.1, 1, and 10, washed three times with PBS and incubated overnight (n=3). In a separate experiment, VSV-GFP was titrated on A549s overnight. A one-way ANOVA was used to test for differences in infection between cell lines (n=3). E) Expression of six ISGs (IFI27, IFI44L, IFIT1, ISG15, RSAD2, SIGLEC1) measured in peripheral blood by qPCR. The median relative expression in the patients was compared to 10 HCs to calculate an IFN-score (Livingston et al., 2014).
Sup Fig 2. MAVS-mediated PRRs have hypomorphic signaling capacity in TBK1 deficient cells, Related toFigure 2. A) ISG mRNA expression in HEK293Ts following transfection with the various TBK1 alleles under study (n=3). B) hTERTs stimulated with pIC overnight either in the endosome (TLR3) or transfected into the cytoplasm (RIG-I/MDA5) with L2K. IL-6 was measured by ELISA (n=3). C) P1- and P2-derived EBV-Bs respond to cytoplasmic pIC. EBV-Bs were stimulated with pIC for 4 hrs either in the endosome (TLR3) or transfected into the cytoplasm (RIG-I/MDA5) with L2K. EBV-Bs do not express TLR3 and so these data were not a factor in assessing TLR3 function in a TBK1-null system. D) P1 hTERTs stably transduced with Y212D TBK1 (P3’s allele) stimulated with endosomal or transfected pIC overnight (n=3). E) hTERTs were transfected with plasmid DNA overnight to assess cGAS/STING function. IFIT1 mRNA was measured by qPCR and IL-6 was measured by ELISA (n=3). F) hTERTs were pretreated with 5 μM MRT for 30 min before transfection with pIC. Lysates were collected 4 hpt.
Sup Fig 3. Chest and brain imaging of P1 and P2, Related toFigure 1and the Human Subjects section of the STAR Methods. Chest CT scans of (A) P1 at age 28 and (B) P2 at age 13. C – D) CT of P1 showing basal ganglia calcifications (white arrows). E – G) MRI of P1 (ADC, DWI, T2w sequences, respectively) showing ischaemia in the right-sided frontotemporal region and basal ganglia. H) Magnetic Resonance Venography (MRV) of P1 showing stenosis and partial occlusion of the carotic arteries and middle cerebral artery (black arrows). I – J) MRI FLAIR in P2 showing multiple white matter lesions (indicated by white arrows) and a region with atrophy and gliosis (black arrow in I). K – L) MRI of P2 (T1 with gadolinium contrast and T2w sequences, respectively) showing infratentorial gadolinium-enhancing white matter lesion and non-enhancing lesion (white arrows).
Sup Fig 4. Severely muted transcriptomic response to TLR3-detected pIC in P1, a comparable transcriptomic response to pIC detected by RIG-I/MDA5 that involves IRF3 nuclear translocation, and normal baseline autophagy in TBK1-deficient hTERTs and A549s, Related toFigure 2. For RNAseq, hTERTs were stimulated overnight with 10 ug/mL pIC alone or transfected with L2K. The average of the unstimulated HCs served as the calibrator for differential expression. A) All differentially expressed genes with q < 0.05 in the average of HC pIC+L2K when compared to the average NT HC. B) hTERTs from P1, P2, and two HCs were stimulated with transfected pIC (10 μg/mL) for 2 and 4 hrs and then stained for total IRF3. Cellular localization of IRF3 was determined via widefield microscopy. C) The ratio of nuclear to cytoplasmic IRF3 mean intensity was determined in Cellprofiler. Each dot represents an individual cell. D) hTERTs were starved in EBSS media and autophagy markers were measured by western blot. E) The rate of baseline autophagy measured by the number of LC3 puncta/cell quantified in Cellprofiler. Images are representative of two different fields of view for each sample that were combined in the quantification. F) A549s were stimulated with 5 mM metformin (MET) and autophagy markers were measured by western blot.
Sup Fig 5. TNF- and IFNγ-induced cell death, Related toFigures 3 and 4. A) hTERTs complemented with the indicated TBK1 allele were pretreated with DOX for 48 hrs before stimulation with 100 ng/mL TNF. B) Phospho-inactive S172A TBK1 partially rescues the predisposition towards RCD in TBK1-null cells. P2 hTERTs were transduced with lentivirus to stably express phospho-inactive S172A TBK1. Cells were pretreated for 30 min as indicated and then exposed to TNF for 24 hrs. Relative viability was measured by resazurin reduction (n=4). C) A549s were treated as indicated for 30 min before an overnight stimulation with TNF and assessed for relative viability. Significance was evaluated with multiple T-tests, ****p≤0.0001 (n=4). D – F) Western blots assessing markers of necroptotic and apoptotic mechanisms of cell death. G) hTERTs were pretreated with birinapant or DMSO for 30 min before an overnight stimulation with TNF (n=3). H) Western blot assessing markers of pyroptosis. I) hTERTs transfected with 1 ug/mL of non-expressing plasmid DNA were assessed for markers of inflammasome activation and pyroptosis via western blot of whole cell lysate and culture supernatant. THP1s differentiated to macrophage with 50 ng/mL PMA for 24 hrs, and then stimulated with 200 ng/mL LPS for 4 hrs and then 200 ng/mL LPS + 50 mM ATP for 2 hrs in opti-MEM served as a positive control for inflammasome activation. J) 10 ng/mL IFNγ was given to hTERTs 24 hrs before exposure to birinapant, zVAD, Nec-1, and TNF. Nec-1 was added 1 hr before birinapant and zVAD, which were given 1 hr before TNF. Viability was measured by resazurin reduction 24 hrs after exposure to TNF (n=4).
Sup Fig 6. Elevated cytokines detected in Luminex, Related toFigure 5. A) Differentially regulated cytokines in P1 and P2 across B – C) 2 different Luminex panels covering 66 different analytes. The number of HCs in A for a particular cytokine equals the number of HCs on the panel that cytokine was measured. D) IL-18 ELISA on baseline plasma from P1 and P2. The dotted line indicates the assay’s limit of detection.
Sup Fig 7. Baseline immunophenotyping and apoptosis in immune cell populations detected by CyTOF, Related toFigures 5 and 7. A) CD69 expression on T cell populations in Family 2. B) CC3 levels from immune cell populations in whole blood analyzed by CyTOF from before and after P1 received anti-TNF treatment. HC n = 4. Pre = treatment day 0. Post = day 131 after treatment initiation.
Highlights.
Homozygous LoF TBK1 produces systemic autoinflammation, not overt viral disease.
Expressed but inactive TBK1 inhibits IFN-I induction more than no TBK1.
Autoinflammation is driven by TNF-induced cell death caused by dysregulated RIPK1.
Treatment with anti-TNF resolves clinical disease.
Acknowledgments
We thank Adeeb Rahman, Brian Lee, Daniel Geanon, Geoffrey Kelly, Kevin Tuballes, and Laura Walker from the Mount Sinai Human Immune Monitoring Center for their help designing and performing the CyTOF and scRNAseq experiments. We thank Dr. Frans Verheijen of the Dept. of Clinical Genetics at Erasmus MC for his assistance. The graphical abstract was created with BioRender. This research was supported by NIAID grants R01 AI127372, R01 AI148963, and R01AI151029, as well as the Hirsch Scholar Award to D.B. J.T. was funded by NIAID F31 AH 38363.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
D.B. is the founder of Lab11 Therapeutics.
References
- Ahmad L, Zhang SY, Casanova JL, and Sancho-Shimizu V (2016). Human TBK1: A Gatekeeper of Neuroinflammation. Trends Mol. Med 22, 511–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin P, Florez M, Najafov A, Pan H, Geng J, Ofengeim D, Dziedzic SA, Wang H, Barrett VJ, Ito Y, et al. (2018). Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl. Acad. Sci. U. S. A 115, E5944–E5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annibaldi A, Wicky John S, Vanden Berghe T, Swatek KN, Ruan J, Liccardi G, Bianchi K, Elliott PR, Choi SM, Van Coillie S, et al. (2018). Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol. Cell 69, 566–580.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D, Salem S, Radovanovic I, Grant AV, Adimi P, et al. (2012). Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science 337, 1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, Abhyankar A, Israël L, Trevejo-Nunez G, Bogunovic D, et al. (2012). Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol 13, 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, Itié A, Wakeham A, Shahinian A, Henzel WJ, et al. (2000). Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19, 4976–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Nijman SMB, Dirac AMG, and Bernards R (2003). Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424, 797–801. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang H-C, Choksi S, Liu J, Ward Y, Wu L, and Liu Z-G (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AH, and Schroder K (2020). Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med 217, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, and Chan FK-M (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu WM, Ostertag D, Li ZW, Chang L, Chen Y, Hu Y, Williams B, Perrault J, and Karin M (1999). JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity 11, 721–731. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu Y, Wang Q, Krueger BJ, et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchet-Lourenço D, Eletto D, Wu C, Plagnol V, Papapietro O, Curtis J, Ceron-Gutierrez L, Bacon CM, Hackett S, Alsaleem B, et al. (2018). Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Diego RP, and Rodríguez-Gallego C (2014). Other TLR Pathway Defects. In Stiehm’s Immune Deficiencies, (Elsevier; ), pp. 687–710. [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. (2014). MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 7, 971–981. [DOI] [PubMed] [Google Scholar]
- Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, Spilgies L, Surinova S, Taraborrelli L, Hartwig T, et al. (2015). LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 13, 2258–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Wang C, Jiang Q, Lv M, Gao P, Yu X, Mu P, Zhang R, Bi S, Feng J-M, et al. (2017). NEMO-IKKβ Are Essential for IRF3 and NF-κB Activation in the cGAS-STING Pathway. J. Immunol 199, 3222–3233. [DOI] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, and Leverkus M (2016). Ripoptosome Analysis by Caspase-8 Coimmunoprecipitation. Cold Spring Harb. Protoc 2016, pdb.prot087403. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao S-M, and Maniatis T (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol 4, 491–496. [DOI] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci 18, 631–636. [DOI] [PubMed] [Google Scholar]
- Gottardo MF, Jaita G, Magri ML, Zárate S, Moreno Ayala M, Ferraris J, Eijo G, Pisera D, Candolfi M, and Seilicovich A (2014). Antiapoptotic factor humanin is expressed in normal and tumoral pituitary cells and protects them from TNF-α-induced apoptosis. PLoS One 9, e111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber C, and Bogunovic D (2020). Incomplete penetrance in primary immunodeficiency: a skeleton in the closet. Hum. Genet 139, 745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K-L, and Ting AT (2002). A20 inhibits tumor necrosis factor (TNF) alpha-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol. Cell. Biol 22, 6034–6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, and Akira S (2004). The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med 199, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman M, Ciancanelli M, Ou Y-H, Lorenzo L, Klaudel-Dreszler M, Pauwels E, Sancho-Shimizu V, Pérez de Diego R, Abhyankar A, Israelsson E, et al. (2012). Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med 209, 1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Zheng X, Wang Z, Chen X, He W, Zhang Y, Xu J-G, Zhao H, Shi W, Wang X, et al. (2017). The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol. Cell. Biol 37, e00497–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM, Hahn WC, and Cantley LC (2009). Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol. Cell 34, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, Scott E, Shah I, Stenson PD, Gleeson J, et al. (2016). The mutation significance cutoff: gene-level thresholds for variant predictions. Nat. Methods 13, 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jønsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, Egedal JH, Kjolby M, Mohammadi P, Prabakaran T, et al. (2017). IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun 8, 14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, Zheng M, Sundaram B, Banoth B, Malireddi RKS, et al. (2021). Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 184, 149–168.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, Lange G, Caroll J, Reitz B, Donnelly A, et al. (2002). IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem 277, 13840–13847. [DOI] [PubMed] [Google Scholar]
- Kosukcu C, Taskiran EZ, Batu ED, Sag E, Bilginer Y, Alikasifoglu M, and Ozen S (2021). Whole exome sequencing in unclassified autoinflammatory diseases: more monogenic diseases in the pipeline? Rheumatology (Oxford). 60, 607–616. [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, and Courtois G (2003). The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424, 801–805. [DOI] [PubMed] [Google Scholar]
- Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, Draberova H, von Mässenhausen A, Bhamra A, Henderson S, et al. (2018). TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol 20, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaoui N, Boyden SE, Oda H, Wood GM, Stone DL, Chau D, Liu L, Stoffels M, Kratina T, Lawlor KE, et al. (2020). Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Xu D, Wang Y, Zhou Z, Liu J, Hu S, Gong Y, Yuan J, and Pan L (2018). Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 8627, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Lin J-P, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CGEL, Falconer A, et al. (2014). A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J. Med. Genet 51, 76–82. [DOI] [PubMed] [Google Scholar]
- Lork M, Verhelst K, and Beyaert R (2017). CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different. Cell Death Differ. 24, 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchlik E, Thakker P, Carlson T, Jiang Z, Ryan M, Marusic S, Goutagny N, Kuang W, Askew GR, Roberts V, et al. (2010). Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J. Leukoc. Biol 88, 1171–1180. [DOI] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, and Maniatis T (2004). IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc. Natl. Acad. Sci. U. S. A 101, 233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, and Tschopp J (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. [DOI] [PubMed] [Google Scholar]
- Mompeán M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, Wu H, and McDermott AE (2018). The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 173, 1244–1253.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moquin DM, McQuade T, and Chan FK-M (2013). CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8, e76841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DL, Johnson S, Bleck CKE, Lee D-Y, and Tjandra N (2020). Humanin selectively prevents the activation of pro-apoptotic protein BID by sequestering it into fibers. J. Biol. Chem 295, 18226–18238. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al. (2014). Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360. [DOI] [PubMed] [Google Scholar]
- Oakes JA, Davies MC, and Collins MO (2017). TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paila U, Chapman BA, Kirchner R, and Quinlan AR (2013). GEMINI: Integrative Exploration of Genetic Variation and Genome Annotations. PLoS Comput. Biol 9, e1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltzer N, and Walczak H (2019). Cell Death and Inflammation - A Vital but Dangerous Liaison. Trends Immunol. 40, 387–402. [DOI] [PubMed] [Google Scholar]
- Perry AK, Chow EK, Goodnough JB, Yeh W-CC, and Cheng G (2004). Differential Requirement for TANK-binding Kinase-1 in Type I Interferon Responses to Toll-like Receptor Activation and Viral Infection. J. Exp. Med 199, 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, et al. (2012). TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity 37, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van T-M, Lee TH, Chan FKM, Pasparakis M, and Kelliher MA (2014). Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol 193, 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, Brown P, Ravenscroft T, van Blitterswijk M, Nicholson AM, et al. (2015). Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U. S. A 113, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Kohler S, Oellrich A, Wang K, Mungall CJ, Lewis SE, Washington N, Bauer S, Seelow D, Krawitz P, et al. (2014). Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 24, 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, TenOever BR, Grandvaux N, Zhou GP, Lin R, and Hiscott J (2003). Triggering the interferon antiviral response through an IKK-related pathway. Science (80-.). 300, 1148–1151. [DOI] [PubMed] [Google Scholar]
- Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, et al. (2020). Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol 40, 24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao P, Sun J, Wu Z, Wang S, Wang J, Li W, Pan H, Bai R, Zhang J, Wang Y, et al. (2020). A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 577, 109–114. [DOI] [PubMed] [Google Scholar]
- tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh W-C, et al. (2004). Activation of TBK1 and IKKepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol 78, 10636–10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston TLM, Ryzhakov G, Bloor S, von Muhlinen N, and Randow F (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol 10, 1215–1221. [DOI] [PubMed] [Google Scholar]
- Ting AT, and Bertrand MJM (2016). More to Life than NF-κB in TNFR1 Signaling. Trends Immunol. 37, 535–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G, Hatzivassillou E, Tsichritzis T, Farmer H, Ashworth A, et al. (2003). CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 424, 793–796. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang L-F, Wang F-S, and Wang X (2014). Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M, and Sun S-C (2007). Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev. Cell 13, 705–716. [DOI] [PubMed] [Google Scholar]
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. (2013). Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 23, 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, Chen X, Longo NS, Louder M, McKee K, et al. (2011). Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333, 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Zou Q, Xie X, Liu T, Li HS, Jie Z, Jin J, Hu H, Manyam G, Zhang L, et al. (2017). The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J. Exp. Med 214, 1493–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, Mifflin L, Pan L, Amin P, Li W, et al. (2018). TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 174, 1477–1491.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Kalac M, Markson M, Chan M, Brody JD, Bhagat G, Ang RL, Legarda D, Justus SJ, Liu F, et al. (2020). Reversal of CYLD phosphorylation as a novel therapeutic approach for adult T-cell leukemia/lymphoma (ATLL). Cell Death Dis. 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong CQY, and Tang BL (2018). A Mitochondrial Encoded Messenger at the Nucleus. Cells 7, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Amin P, and Ofengeim D (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci 20, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D-W, Shao J, Lin J, Zhang N, Lu B-J, Lin S-C, Dong M-Q, and Han J (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. [DOI] [PubMed] [Google Scholar]
- Zhang H, Han C, Li T, Li N, and Cao X (2019). The methyltransferase PRMT6 attenuates antiviral innate immunity by blocking TBK1-IRF3 signaling. Cell. Mol. Immunol 16, 800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Chao Yuan SV, Li Z, Sanal O, Mansouri D, Tezcan I, et al. (2015). Human intracellular ISG15 prevents interferon-a/b over-amplification and auto-inflammation. Nature 517, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sup Fig 1. No autoimmunity, baseline VSV susceptibility, or type-I IFNopathy associated with TBK1 mutations at conservation residues, Related toFigures 1 and 2. A) Serological screen of P1 and P2 serum for autoreactive antibodies against known epitopes linked to common autoreactive targets. B) Sanger sequencing of genomic DNA confirming the mutation status of each patient. C) Evolutionary conservation of the TBK1 residues mutated in each patient. Multiple protein sequence alignment of TBK1 was generated with the COBALT tool from NCBI. Red indicates that an amino acid differs from the human ortholog. The locations of the examined variants are indicated by the red arrow. D) hTERTs were infected with VSV-GFP for 1 hr at an MOI of 0.1, 1, and 10, washed three times with PBS and incubated overnight (n=3). In a separate experiment, VSV-GFP was titrated on A549s overnight. A one-way ANOVA was used to test for differences in infection between cell lines (n=3). E) Expression of six ISGs (IFI27, IFI44L, IFIT1, ISG15, RSAD2, SIGLEC1) measured in peripheral blood by qPCR. The median relative expression in the patients was compared to 10 HCs to calculate an IFN-score (Livingston et al., 2014).
Sup Fig 2. MAVS-mediated PRRs have hypomorphic signaling capacity in TBK1 deficient cells, Related toFigure 2. A) ISG mRNA expression in HEK293Ts following transfection with the various TBK1 alleles under study (n=3). B) hTERTs stimulated with pIC overnight either in the endosome (TLR3) or transfected into the cytoplasm (RIG-I/MDA5) with L2K. IL-6 was measured by ELISA (n=3). C) P1- and P2-derived EBV-Bs respond to cytoplasmic pIC. EBV-Bs were stimulated with pIC for 4 hrs either in the endosome (TLR3) or transfected into the cytoplasm (RIG-I/MDA5) with L2K. EBV-Bs do not express TLR3 and so these data were not a factor in assessing TLR3 function in a TBK1-null system. D) P1 hTERTs stably transduced with Y212D TBK1 (P3’s allele) stimulated with endosomal or transfected pIC overnight (n=3). E) hTERTs were transfected with plasmid DNA overnight to assess cGAS/STING function. IFIT1 mRNA was measured by qPCR and IL-6 was measured by ELISA (n=3). F) hTERTs were pretreated with 5 μM MRT for 30 min before transfection with pIC. Lysates were collected 4 hpt.
Sup Fig 3. Chest and brain imaging of P1 and P2, Related toFigure 1and the Human Subjects section of the STAR Methods. Chest CT scans of (A) P1 at age 28 and (B) P2 at age 13. C – D) CT of P1 showing basal ganglia calcifications (white arrows). E – G) MRI of P1 (ADC, DWI, T2w sequences, respectively) showing ischaemia in the right-sided frontotemporal region and basal ganglia. H) Magnetic Resonance Venography (MRV) of P1 showing stenosis and partial occlusion of the carotic arteries and middle cerebral artery (black arrows). I – J) MRI FLAIR in P2 showing multiple white matter lesions (indicated by white arrows) and a region with atrophy and gliosis (black arrow in I). K – L) MRI of P2 (T1 with gadolinium contrast and T2w sequences, respectively) showing infratentorial gadolinium-enhancing white matter lesion and non-enhancing lesion (white arrows).
Sup Fig 4. Severely muted transcriptomic response to TLR3-detected pIC in P1, a comparable transcriptomic response to pIC detected by RIG-I/MDA5 that involves IRF3 nuclear translocation, and normal baseline autophagy in TBK1-deficient hTERTs and A549s, Related toFigure 2. For RNAseq, hTERTs were stimulated overnight with 10 ug/mL pIC alone or transfected with L2K. The average of the unstimulated HCs served as the calibrator for differential expression. A) All differentially expressed genes with q < 0.05 in the average of HC pIC+L2K when compared to the average NT HC. B) hTERTs from P1, P2, and two HCs were stimulated with transfected pIC (10 μg/mL) for 2 and 4 hrs and then stained for total IRF3. Cellular localization of IRF3 was determined via widefield microscopy. C) The ratio of nuclear to cytoplasmic IRF3 mean intensity was determined in Cellprofiler. Each dot represents an individual cell. D) hTERTs were starved in EBSS media and autophagy markers were measured by western blot. E) The rate of baseline autophagy measured by the number of LC3 puncta/cell quantified in Cellprofiler. Images are representative of two different fields of view for each sample that were combined in the quantification. F) A549s were stimulated with 5 mM metformin (MET) and autophagy markers were measured by western blot.
Sup Fig 5. TNF- and IFNγ-induced cell death, Related toFigures 3 and 4. A) hTERTs complemented with the indicated TBK1 allele were pretreated with DOX for 48 hrs before stimulation with 100 ng/mL TNF. B) Phospho-inactive S172A TBK1 partially rescues the predisposition towards RCD in TBK1-null cells. P2 hTERTs were transduced with lentivirus to stably express phospho-inactive S172A TBK1. Cells were pretreated for 30 min as indicated and then exposed to TNF for 24 hrs. Relative viability was measured by resazurin reduction (n=4). C) A549s were treated as indicated for 30 min before an overnight stimulation with TNF and assessed for relative viability. Significance was evaluated with multiple T-tests, ****p≤0.0001 (n=4). D – F) Western blots assessing markers of necroptotic and apoptotic mechanisms of cell death. G) hTERTs were pretreated with birinapant or DMSO for 30 min before an overnight stimulation with TNF (n=3). H) Western blot assessing markers of pyroptosis. I) hTERTs transfected with 1 ug/mL of non-expressing plasmid DNA were assessed for markers of inflammasome activation and pyroptosis via western blot of whole cell lysate and culture supernatant. THP1s differentiated to macrophage with 50 ng/mL PMA for 24 hrs, and then stimulated with 200 ng/mL LPS for 4 hrs and then 200 ng/mL LPS + 50 mM ATP for 2 hrs in opti-MEM served as a positive control for inflammasome activation. J) 10 ng/mL IFNγ was given to hTERTs 24 hrs before exposure to birinapant, zVAD, Nec-1, and TNF. Nec-1 was added 1 hr before birinapant and zVAD, which were given 1 hr before TNF. Viability was measured by resazurin reduction 24 hrs after exposure to TNF (n=4).
Sup Fig 6. Elevated cytokines detected in Luminex, Related toFigure 5. A) Differentially regulated cytokines in P1 and P2 across B – C) 2 different Luminex panels covering 66 different analytes. The number of HCs in A for a particular cytokine equals the number of HCs on the panel that cytokine was measured. D) IL-18 ELISA on baseline plasma from P1 and P2. The dotted line indicates the assay’s limit of detection.
Sup Fig 7. Baseline immunophenotyping and apoptosis in immune cell populations detected by CyTOF, Related toFigures 5 and 7. A) CD69 expression on T cell populations in Family 2. B) CC3 levels from immune cell populations in whole blood analyzed by CyTOF from before and after P1 received anti-TNF treatment. HC n = 4. Pre = treatment day 0. Post = day 131 after treatment initiation.
Data Availability Statement
Counts matrices for bulk and single-cell RNAseq experiments are available on Mendeley Data (bulk RNAseq DOI: 10.17632/648zntv5dx.1; single-cell RNAseq DOI: 10.17632/96hx5z4wv8.1). This study did not generate any unique code.