

Graphical abstract
First step is the data pre-processing to retrieve the sequence from raw data.The second step is to encode the sequences using on-hot-encoding to make the data readable for the network. The third step is the neural network model construction, and the last step is to classify the sequence as methylated or non-methylated.
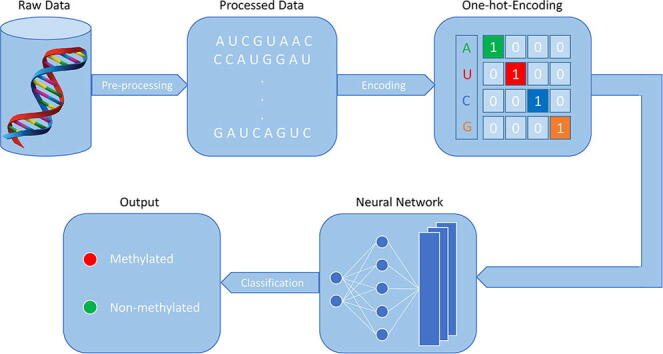
Keywords: Binary-encoding, Deep neural network, N6-methyladenosine (m6A), Motif, Tissue-specific
Abstract
The most communal post-transcriptional modification, N6-methyladenosine (m6A), is associated with a number of crucial biological processes. The precise detection of m6A sites around the genome is critical for revealing its regulatory function and providing new insights into drug design. Although both experimental and computational models for detecting m6A sites have been introduced, but these conventional methods are laborious and expensive. Furthermore, only a handful of these models are capable of detecting m6A sites in various tissues. Therefore, a more generic and optimized computational method for detecting m6A sites in different tissues is required. In this paper, we proposed a universal model using a deep neural network (DNN) and named it TS-m6A-DL, which can classify m6A sites in several tissues of humans (Homo sapiens), mice (Mus musculus), and rats (Rattus norvegicus). To extract RNA sequence features and to convert the input into numerical format for the network, we utilized one-hot-encoding method. The model was tested using fivefold cross-validation and its stability was measured using independent datasets. The proposed model, TS-m6A-DL, achieved accuracies in the range of 75–85% using the fivefold cross-validation method and 72–84% on the independent datasets. Finally, to authenticate the generalization of the model, we performed cross-species testing and proved the generalization ability by achieving state-of-the-art results.
1. Introduction
The post-transcriptional RNA modification is an evolutionarily conserved process of RNAs that exists in all biological organisms [1]. The sophistication of biological knowledge and the evenness of regulation increases as RNA undergoes a post-transcriptional transformation. Over and above 150 different types of RNA post-translational modifications have been discovered, with methylation accounting for two-third of them [2]. N6-methyladenosine (m6A) modification is a typical and generous post-transcriptional RNA modification that affects nearly all cell cycle processes, including translation efficiency, cell growth, and cell viability [3], [4], [5], [6], [7], [8]. This indicates the methylation of the adenosine base at the nitrogen-6 location. Furthermore, m6A modification is a reversible process that can be triggered by methyltransferases and demethylases [9], [10], [11]. Research has shown that m6A is related to the occurrence of diseases including thyroid tumors [12], prostate cancer [13], obesity [14], and acute myelogenous leukemia [15]. m6A is a common transcriptional modification, which may occur in various species such as mammals, plants, and bacteria [16]. Research has shown that m6A acts as a regulator at each stage of mRNA metabolism [17]. This warrants extensive research on m6A modification, however, our current knowledge is still limited regarding m6A modification. Therefore, it is crucial to extensively study m6A, and correctly determine m6A modification sites in the transcriptome.
The m6A sites can be identified using two primary approaches. The first is experimental techniques including Methylated RNA Immunoprecipitation (MeRIP) [18], photo-crossl-inking-assisted (PA)-m6A-seq [19], m6A sequencing (m6A-seq) [20], m6A-crosslinking immunoprecipitation (CLIP) [21], individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) [22], DART-seq [23], MAZTER-Seq [24] and m6A-eCLIP (meCLIP) [25]. These experimental approaches provide a layout for the identification of potential m6A modification sites. Moreover, a few bioinformatics techniques, which are capable of discovering m6A sites directly from the data gathered by experimental techniques have been suggested [26], [27], [28]. However, the sequencing data gathered is already too large to be catered by basic bioinformatics techniques; therefore, there is a requirement for more efficient techniques to identify m6A sites in the transcriptome. The second approach to identify m6A sites is the application of computational techniques to sequencing data. In recent studies using spatial specificity of gene expression, it was discovered that the site of m6A modification varies in different tissues and species. Doa et al., [29] suggested a technique called iRNA-m6A that can detect m6A modification sites in various tissues of human, mouse, and rat by using a support sector machine (SVM), which was applied to the dataset provided by Zhang et al., [30]. This approach significantly enhances the accuracy of identifying m6A modification sites. However, there is still much room for improvement in techniques for detecting m6A sites.
In recent years, deep learning models have insignificantly impacted the field of bioinformatics. Various computational methodologies comprising deep-learning techniques, including Gene2Vec [31], DeepM6ASeq [32], BERMP [33], DNA6mA-MINT [34], pcPromoter-CNN [35], 4mCPred-CNN [36], im6A-TS-CNN [37], and iPseU-CNN [38] have been suggested. Liu et al., proposed a CNN-based solution in im6A-TS-CNN, which utilizes one-hot encoding technique to encode the data samples and then classify the data using CNN architecture using the datasets of Homo sapiens, Rattus norvegicus, and Mus musculus. Dao et al., [29] proposed an SVM-based machine-learning model to classify m6A sites on the same datasets. They used various encoding schemes including mono-nucleotide binary encoding, physical-chemical property matrix, and nucleotide chemical property.
In light of the above studies, the current research is focused on building a computational method that can detect m6A modification sites in human, mouse, and rat tissues. To create a benchmark dataset based on the experimental analysis performed by Zhang et al., [30], we first compiled experimentally validated m6A and non-m6A sequences. To formulate the samples, only a single encoding scheme, one-hot-encoding, was used. Motivated by the increasing implementation of deep-learning algorithms, we proposed a universal CNN-based approach called, TS-m6A-DL. Performing 5-fold cross-validation and independent testing revealed that, TS-m6A-DL, outperforms the current state-of-the-art approaches. Additionally, cross-species validation testing proved the stability of our model achieving state-of-the-art results.
2. Materials and methods
2.1. Benchmark datasets
A major step in training an efficient computational model is the construction of high-quality datasets. Zhang et al., [30] produced an efficient antibody-independent m6A detection tool, m6A-REF-seq, to classify the sites of alteration in various tissues including brain, liver, kidney, heart, and testis of Homo sapiens, Mus musculus, and Rattus norvegicus species. Considering the excellent caliber of these data, benchmark dataset was developed using them. To further enhance the data integrity, we only used sequences with a length of 41 nucleotides (nt) having the m6A site in the middle for positive sequences. Sample sequences exceeding 80% resemblance were eliminated using the CD-HIT software [39] to prevent duplication and eliminate homology bias. The negative sequences, which were experimentally proved as non-methylated, for the abovementioned tissues were extracted by fulfilling the 41nt length requirement containing Adenine in the center. They show m6A consensus motif but were not enriched in the analysis of m6A. Since an unbalanced dataset replicates biasness towards one class [40], random negative sequences equal to the number of positive sequences were retrieved. The datasets are divided into training and independent datasets to critically analyze the developed model’s performance and generalization. There is minimal overfitting when the proposed technique is appropriate for the independent dataset [29]. Table 1 lists the specifics of the positive and negative sequences in each dataset.
Table 1.
Benchmark datasets of different tissues of Human, Mouse, and Rat. Here, the methylated sequences are referred as positive, and non-methylated sequences are referred as negative. The negative sequences show an m6A consensus motif but were not enriched in the analysis for m6A.
| Species | Tissues | Positive |
Negative |
||
|---|---|---|---|---|---|
| Training | Testing | Training | Testing | ||
| Human | Brain | 4605 | 4604 | 4605 | 4604 |
| Liver | 2634 | 2634 | 2634 | 2634 | |
| Kidney | 4574 | 4573 | 4574 | 4573 | |
| Mouse | Brain | 8025 | 8025 | 8025 | 8025 |
| Liver | 4133 | 4133 | 4133 | 4133 | |
| Kidney | 3953 | 3952 | 3953 | 3952 | |
| Heart | 2201 | 2201 | 2200 | 2200 | |
| Testis | 4707 | 4706 | 4707 | 4706 | |
| Rat | Brain | 2352 | 2351 | 2352 | 2351 |
| Liver | 1762 | 1762 | 1762 | 1762 | |
| Kidney | 3433 | 3432 | 3433 | 3432 | |
2.2. Sequence encoding
In neural networks, making the sequence readable for the network using an efficient encoding scheme is the foremost prerequisite that directly affects the model’s performance. One-hot-encoding is a widely used encoding scheme that can accurately represent the nucleotides as a four-dimensional binary vector. We can represent the nucleotides as follows:
2.3. Network architecture
An input layer, several intermediary hidden layers, and an output layer comprise a neural network. After converting the sequence into a numerical format to make it readable for the network, the input layer receives the 41 x 4 matrix as an input. The proposed network architecture is illustrated in Fig. 1. The input matrix is fed to the first convolution layer with 64 filters with a kernel size of 3 and a stride of 1. The number of feature maps in DNNs multiplies with the network’s depth, resulting in a drastic increase in the number of parameters and computational requirements when larger kernel sizes are used. Therefore, this convolution layer is followed by another convolution layer with 16 filters having unit kernel size and the same stride. The concept was first introduced by Lin et al., in their Network in Network study [41]. This particular strategy has been used in this study, to reduce dimensionality by reducing the number of feature maps while keeping the most important features and neglecting the features which are not contributing. The convolution layer can be represented mathematically as follows:
| (1) |
where ‘X’ represents the input, ‘i’ represents the output position index, and ‘k’ determine the kernel index. is a weight matrix, where ‘M’ is the window size and ‘N’ is the number of input channels.
Fig. 1.

Architecture of the proposed network, TS-m6A-DL. First step is to encode the sequence data to make it readable for the network. Then the encoded data is fed to the convolution layer; Conv1D(f,k,s), where f is the number of filters, k is the kernel size, and s is the stride. The convolution layer is followed by the pooling layer MaxPool(2), where 2 is the pooling size; which is then followed by the dropout layer Dropout(0.6), where 0.6 is the dropout rate.
This layer is then followed by a max-pooling layer with a pool size of 2, to downsample the content of feature maps, shrinking their height and width whilst preserving their salient features. It can be calculated mathematically by the following equation:
| (2) |
where ‘X’ represents for the input feature map, ‘i’ represents the output position index, ‘k’ denotes the kernel index, and ‘M’ denotes the pool window size.
To avoid the overfitting bias, we used a dropout function with a rate of 0.6. The output after the dropout was flattened and simultaneously fed as input to another convolution layer, repeating the same procedure as above, two more times. The three flattened outputs after every block were concatenated and fed as input to the dense layer containing 16 nodes, followed by the output layer, which determines whether the sequence is methylated or non-methylated. Each convolution layer including the dense layer uses a rectified linear function (ReLU) as the activation function, except for the output layer which uses the sigmoid function for classification. ReLU and sigmoid can be expressed mathematically as follows:
| (3) |
| (4) |
We further used the L2 regularization approach for the kernel and bias within the convolution layer to prevent overfitting, setting the rate to 0.001. The Nadam (Nesterov accelerated Adaptive Moment Estimation) optimizer with a learning rate of 0.0021 was used to construct the model accompanying binary cross-entropy as the loss function. The maximum training epoch was set to 50, and the training batch size was set to 32. Furthermore, during the training phase, we also used the early stop strategy through which the training process would halt if the prediction accuracy on the validation set stopped improving for 10 epochs.
3. Evaluation metrics
K-fold cross-validation and independent testing are widely used to assess the performance of a model. To analyze the effectiveness and robustness of the model, we performed both of the above-mentioned tests on our model, defining K equals to 5 in our model. To objectively assess the results from the analysis and for an unbiased comparison with the previous state-of-the-art methods, we used the same widely used metrics including sensitivity (Sn), specificity (Sp), accuracy (ACC), and Matthews correlation coefficient (MCC).
| (5) |
| (6) |
| (7) |
| (8) |
where TP, TN, FP, and FN represent the number of true positives, true negatives, false positives, and false negatives, respectively. Furthermore, we also used the area under the curve (AUC) which is a useful metric for assessing predictive performance of a model.
4. Results and discussion
We developed a generalized model for the classification of m6A sites based on the data defined in the Materials and methods. To prove the model’s stability in order to classify tissue-specific m6A modifications in humans, mice, and rats; we performed both 5-fold cross-validation and independent testing, and formulated the results in Table 2. The proposed approach was found to be robust when the findings of the 5-fold cross-validation and independent tests were compared with existing state-of-the-art models.
Table 2.
Performance of TS-m6A-DL; Before the underscore, characters h, m, and r stand for human, mouse, and rat, respectively; and the b,h,k,l, and t after underscore denotes brain, heart, kidney, liver, and testis, respectively.
| Species | 5-fold Cross Validation |
Independent Testing |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ACC | Sn | Sp | MCC | AUC | ACC | Sn | Sp | MCC | AUC | |
| h_b | 0.7507 | 0.8191 | 0.6823 | 0.5068 | 0.8262 | 0.7384 | 0.8123 | 0.6646 | 0.4822 | 0.8097 |
| h_k | 0.8099 | 0.8393 | 0.7804 | 0.6211 | 0.8904 | 0.8020 | 0.8045 | 0.7996 | 0.6042 | 0.8802 |
| h_l | 0.8335 | 0.8595 | 0.8075 | 0.6684 | 0.9135 | 0.8056 | 0.8204 | 0.7908 | 0.6115 | 0.8784 |
| m_b | 0.7985 | 0.8134 | 0.7836 | 0.5974 | 0.8831 | 0.7877 | 0.8291 | 0.7462 | 0.5774 | 0.8725 |
| m_h | 0.7823 | 0.8150 | 0.7496 | 0.5664 | 0.8504 | 0.7502 | 0.7931 | 0.7072 | 0.5023 | 0.8234 |
| m_k | 0.8222 | 0.8259 | 0.8186 | 0.6451 | 0.9079 | 0.8076 | 0.8421 | 0.7732 | 0.6168 | 0.8892 |
| m_l | 0.7506 | 0.7953 | 0.7060 | 0.5044 | 0.8288 | 0.7203 | 0.7805 | 0.6600 | 0.4438 | 0.7913 |
| m_t | 0.7756 | 0.8179 | 0.7333 | 0.5544 | 0.8630 | 0.7644 | 0.8425 | 0.6863 | 0.5354 | 0.8432 |
| r_b | 0.7955 | 0.8227 | 0.7682 | 0.5922 | 0.8758 | 0.7728 | 0.8132 | 0.7324 | 0.5475 | 0.8543 |
| r_k | 0.8341 | 0.8400 | 0.8281 | 0.6683 | 0.9066 | 0.8327 | 0.8522 | 0.8132 | 0.6660 | 0.9083 |
| r_l | 0.8348 | 0.8347 | 0.8348 | 0.6706 | 0.9025 | 0.8101 | 0.8547 | 0.7656 | 0.6227 | 0.8853 |
4.1. Comparison with existing methods
To validate our model, we used the same validation approach as that used for the validation of iRNA-m6A [29] and m6A-TS-CNN [37], retaining the same number of folds, to obtain a better comparative study. Fig. 2, Fig. 2b show the comparison results in terms of accuracy, as determined by 5-fold cross-validation and independent testing. High accuracy on independent dataset implies that the model has the ability to identify m6A sites for an unknown sequence. A complete comparison using all five evaluation matrices; ACC, Sn, Sp, MCC, and AUC is shown in Table S1 and S2 for the 5-fold cross-validation and independent testing, respectively.
Fig. 2.

Comparison of the models; iRNA-m6A, m6A-TS-CNN, and TS-m6A-DNN in term of accuracy.
4.2. Cross-species testing
Since the datasets come from various organisms and tissues, it is useful to see how a model trained on samples from a particular tissue of one species can recognize m6A in the same tissues of another species. Therefore, for the same tissues in different species, we applied cross-species testing and provided the accuracies in Fig. 3, Fig. 3, Fig. 3v for the brain, kidney, and liver, respectively. The x-axis shows the datasets on which we trained our model and the y-axis shows the datasets on which we tested our model.
Fig. 3.

Cross-species testing. Here, h,m, and r on x and y-axis represents the species human, mouse, and rat.
The results indicate that the proposed tool, TS-m6A-DL, is efficient for cross-species testing for the classification of m6A sites, proving its universality. The complete results using the same five evaluation matrices are illustrated in Table S3.
4.3. Motif analysis
Motifs were calculated from the first activation layer [42]. Each input sequence produced a corresponding activation map in the first layer from which we selected the maximum activation. This maximum activation was mapped back to the input sequence to select a subsequence with the filter-size length. The selected subsequences from each filter were aligned and used to find the motif using position weight matrix (PWM) technique [43]. Multiple motifs were generated using 64 filters in the first layer. Then, we compared these motifs with biologically reported motifs [30]. We found a strong match of ACA motif detected by our model with that reported by Zhang et al., [30]. Fig. 4 shows the motif detected by our model in which the first A is the putative m6A.
Fig. 4.

The ACA-motif detected by our model, TS-m6A-DL.
5. Conclusion
Since m6A is involved in a variety of biological processes, precise detection of m6A sites is critical for scientific investigations to understand its regulatory function and to obtain various insights into drug design. In this study, we present TS-m6A-DL, a deep-learning-based universal model for detecting m6A sites in various tissues of humans, mice, and rats. To validate our model, we performed 5-fold cross-validation and independent testing, and the achieved results exemplified that the TS-m6A-DL tool performs better than the previous state-of-the-art tools. A web-based server was developed and made available at http://nsclbio.jbnu.ac.kr/tools/TS-m6A-DL/ for the benefit of the research community. We expect that the new framework will be useful for detecting m6A-sites and facilitate drug development.
CRediT authorship contribution statement
Zeeshan Abbas: Conceptualization, Methodology, Software, Writing - original draft, Writing - review & editing. Hilal Tayara: Conceptualization, Methodology, Software, Writing - original draft, Writing - review & editing. Quan Zou: Conceptualization, Validation, Supervision, Writing - review & editing. Kil To Chong: Conceptualization, Validation, Supervision, Writing - review & editing, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported in part by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2020R1A2C2005612) and in part by the Brain Research Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No. NRF-2017M3C7A1044816).
Footnotes
Supplementary data associated with this article can be found, in the online version, athttps://doi.org/10.1016/j.csbj.2021.08.014.
Contributor Information
Quan Zou, Email: zouquan@nclab.net.
Kil To Chong, Email: kitchong@jbnu.ac.kr.
Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sun Xi Hong, Zhao Ling Ping, Zou Quan, Wang Zhan Bin. Identification of microrna genes and their mrna targets in festuca arundinacea. Appl Biochem Biotechnol. 2014;172(8):3875–3887. doi: 10.1007/s12010-014-0805-6. [DOI] [PubMed] [Google Scholar]
- 2.Toone Eric J. Advances in enzymology and related areas of molecular biology, volume 240. John Wiley & Sons. 2011 [PubMed] [Google Scholar]
- 3.Wang Xiao, Zhao Boxuan Simen, Roundtree Ian A., Lu Zhike, Han Dali, Ma Honghui, Weng Xiaocheng, Chen Kai, Shi Hailing, He Chuan. N6-methyladenosine modulates messenger rna translation efficiency. Cell. 2015;161(6):1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Yang, Li Yue, Toth Julia I., Petroski Matthew D., Zhang Zhaolei, Zhao Jing Crystal. N 6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16(2):191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bokar Joseph A. Springer; 2005. The biosynthesis and functional roles of methylated nucleosides in eukaryotic mrna. In Fine-tuning of RNA functions by modification and editing; pp. 141–177. [Google Scholar]
- 6.Rehman Mobeen Ur, Hong Kim Jee, Tayara Hilal, Chong Kil to. m6a-neuraltool: convolution neural tool for rna n6-methyladenosine site identification in different species. IEEE Access. 2021;9:17779–17786. [Google Scholar]
- 7.Alam Waleed, Ali Syed Danish, Tayara Hilal, Chong Kil to. A cnn-based rna n6-methyladenosine site predictor for multiple species using heterogeneous features representation. IEEE Access. 2020;8:138203–138209. [Google Scholar]
- 8.Ali Syed Danish, Alam Waleed, Tayara Hilal, Chong Kil. Identification of functional pirnas using a convolutional neural network. IEEE/ACM Trans Comput Biol Bioinf. 2020 doi: 10.1109/TCBB.2020.3034313. [DOI] [PubMed] [Google Scholar]
- 9.Jia Guifang, Ye Fu., Zhao Xu., Dai Qing, Zheng Guanqun, Yang Ying, Yi Chengqi, Lindahl Tomas, Pan Tao, Yang Yun-Gui. N 6-methyladenosine in nuclear rna is a major substrate of the obesity-associated fto. Nat Chem Biol. 2011;7(12):885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jia Guifang, Ye Fu., He Chuan. Reversible rna adenosine methylation in biological regulation. Trends Genet. 2013;29(2):108–115. doi: 10.1016/j.tig.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Yamei Niu, Zhao Yong-Sheng Wu, Li Ming-Ming, Wang Xiu-Jie, Yang Yun-Gui. N6-methyl-adenosine (m6a) in rna: an old modification with a novel epigenetic function. Genomics Proteomics Bioinf. 2013;11(1):8–17. doi: 10.1016/j.gpb.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Zhi-Hao, Feng Shao-Ting, Zhang Di, Cao Xu-Chen, Yu Yue, Wang Xin. The functions and prognostic values of m6a rna methylation regulators in thyroid carcinoma. Cancer Cell Int. 2021;21(1):1–15. doi: 10.1186/s12935-021-02090-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai Jiarong, Yang Fei, Zhan Hailun, Situ Jie, Li Wenbiao, Mao Yunhua, Luo Yun. Rna m6a methyltransferase mettl3 promotes the growth of prostate cancer by regulating hedgehog pathway. OncoTargets Ther. 2019;12:9143. doi: 10.2147/OTT.S226796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Machiela Mitchell J., Lindström Sara, Allen Naomi E., Haiman Christopher A., Albanes Demetrius, Barricarte Aurelio, Berndt Sonja I., Bas Bueno-de Mesquita H., Chanock Stephen, Michael Gaziano J. Association of type 2 diabetes susceptibility variants with advanced prostate cancer risk in the breast and prostate cancer cohort consortium. Am J Epidemiol. 2012;176(12):1121–1129. doi: 10.1093/aje/kws191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bansal H., Yihua Q., Iyer Swaminathan Padmanabhan, Ganapathy S., Proia D., Penalva L.O., Uren P.J., Suresh U., Carew J.S., Karnad A.B. Wtap is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28(5):1171–1174. doi: 10.1038/leu.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodi Zsuzsanna, Button James D., Grierson Donald, Fray Rupert G. Yeast targets for mrna methylation. Nucl Acids Res. 2010;38(16):5327–5335. doi: 10.1093/nar/gkq266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Boxuan Simen, Roundtree Ian A., He Chuan. Post-transcriptional gene regulation by mrna modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer Kate D., Saletore Yogesh, Zumbo Paul, Elemento Olivier, Mason Christopher E., Jaffrey Samie R. Comprehensive analysis of mrna methylation reveals enrichment in 3 utrs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Kai, Zhike Lu., Wang Xiao, Fu Ye, Luo Guan-Zheng, Liu Nian, Han Dali, Dominissini Dan, Dai Qing, Pan Tao. High-resolution n6-methyladenosine (m6a) map using photo-crosslinking-assisted m6a sequencing. Angew Chem. 2015;127(5):1607–1610. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominissini Dan, Moshitch-Moshkovitz Sharon, Schwartz Schraga, Salmon-Divon Mali, Ungar Lior, Osenberg Sivan, Cesarkas Karen, Jacob-Hirsch Jasmine, Amariglio Ninette, Kupiec Martin. Topology of the human and mouse m 6 a rna methylomes revealed by m 6 a-seq. Nature. 2012;485(7397):201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 21.Ke Shengdong, Alemu Endalkachew A., Mertens Claudia, Gantman Emily Conn, Fak John J., Mele Aldo, Haripal Bhagwattie, Zucker-Scharff Ilana, Moore Michael J., Park Christopher Y. allowing the potential for 3 utr regulation. Genes Develop. 2015;29(19):2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linder Bastian, Grozhik Anya V., Olarerin-George Anthony O., Meydan Cem, Mason Christopher E., Jaffrey Samie R. Single-nucleotide-resolution mapping of m6a and m6am throughout the transcriptome. Nat Methods. 2015;12(8):767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer Kate D. Dart-seq: an antibody-free method for global m 6 a detection. Nat Methods. 2019;16(12):1275–1280. doi: 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandey Radha Raman, Pillai Ramesh S. Counting the cuts: Mazter-seq quantifies m6a levels using a methylation-sensitive ribonuclease. Cell. 2019;178(3):515–517. doi: 10.1016/j.cell.2019.07.006. [DOI] [PubMed] [Google Scholar]
- 25.Roberts Justin T., Porman Allison M., Johnson Aaron M. Identification of m6a residues at single-nucleotide resolution using eclip and an accessible custom analysis pipeline. RNA. 2021;27(4):527–541. doi: 10.1261/rna.078543.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng Jia, Lu Zhiliang, Liu Hui, Zhang Lin, Zhang Shaowu, Chen Yidong, Rao Manjeet K., Huang Yufei. A protocol for rna methylation differential analysis with merip-seq data and exomepeak r/bioconductor package. Methods. 2014;69(3):274–281. doi: 10.1016/j.ymeth.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Zhen, Zhao Pei, Li Fuyi, Wang Yanan, Ian Smith A., Webb Geoffrey I., Akutsu Tatsuya, Baggag Abdelkader, Bensmail Halima, Song Jiangning. Comprehensive review and assessment of computational methods for predicting rna post-transcriptional modification sites from rna sequences. Brief Bioinf. 2020;21(5):1676–1696. doi: 10.1093/bib/bbz112. [DOI] [PubMed] [Google Scholar]
- 28.Lorenz Daniel A., Sathe Shashank, Einstein Jaclyn M., Yeo Gene W. Direct rna sequencing enables m6a detection in endogenous transcript isoforms at base-specific resolution. Rna. 2020;26(1):19–28. doi: 10.1261/rna.072785.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dao Fu-Ying, Lv Hao, Yang Yu-He, Zulfiqar Hasan, Gao Hui, Lin Hao. Computational identification of n6-methyladenosine sites in multiple tissues of mammals. Comput Struct Biotechnol J. 2020;18:1084–1091. doi: 10.1016/j.csbj.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Zhang, Chen Li-Qian, Zhao Yu-Li, Yang Cai-Guang, Roundtree Ian A., Zhang Zijie, Ren Jian, Xie Wei, He Chuan, Luo Guan-Zheng. Single-base mapping of m6a by an antibody-independent method. Sci Adv. 2019;5(7):eaax0250. doi: 10.1126/sciadv.aax0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou Quan, Xing Pengwei, Wei Leyi, Liu Bin. Gene2vec: gene subsequence embedding for prediction of mammalian n6-methyladenosine sites from mrna. Rna. 2019;25(2):205–218. doi: 10.1261/rna.069112.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Yiqian, Hamada Michiaki. Deepm6aseq: prediction and characterization of m6a-containing sequences using deep learning. BMC Bioinf. 2018;19(19):1–11. doi: 10.1186/s12859-018-2516-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Huang, Yu Ningning He, Chen Zhen Chen, Li Lei. Bermp: a cross-species classifier for predicting m6a sites by integrating a deep learning algorithm and a random forest approach. Int J Biol Sci. 2018;14(12):1669. doi: 10.7150/ijbs.27819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rehman Mobeen Ur, Chong Kil To. Dna6ma-mint: Dna-6ma modification identification neural tool. Genes. 2020;11(8):898. doi: 10.3390/genes11080898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shujaat Muhammad, Wahab Abdul, Tayara Hilal, Chong Kil To. pcpromoter-cnn: a cnn-based prediction and classification of promoters. Genes. 2020;11(12):1529. doi: 10.3390/genes11121529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abbas Zeeshan, Tayara Hilal, Chong Kil To. 4mcpred-cnn-prediction of dna n4-methylcytosine in the mouse genome using a convolutional neural network. Genes. 2021;12(2):296. doi: 10.3390/genes12020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Kewei, Cao Lei, Du Pufeng, Chen Wei. im6a-ts-cnn: identifying the n6-methyladenine site in multiple tissues by using the convolutional neural network. Mol Ther-Nucl Acids. 2020;21:1044–1049. doi: 10.1016/j.omtn.2020.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tahir Muhammad, Tayara Hilal, Chong Kil To. ipseu-cnn: identifying rna pseudouridine sites using convolutional neural networks. Mol Ther-Nucl Acids. 2019;16:463–470. doi: 10.1016/j.omtn.2019.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Weizhong, Godzik Adam. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 40.Wang Ruohan, Wang Zishuai, Wang Jianping, Li Shuaicheng. Splicefinder: ab initio prediction of splice sites using convolutional neural network. BMC Bioinf. 2019;20(23):1–13. doi: 10.1186/s12859-019-3306-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin Min, Chen Qiang, Yan Shuicheng. Network in network. arXiv preprint arXiv:1312.4400; 2013.
- 42.Quang Daniel, Xie Xiaohui. Factornet: a deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods. 2019;166:40–47. doi: 10.1016/j.ymeth.2019.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xia Xuhua. Position weight matrix, gibbs sampler, and the associated significance tests in motif characterization and prediction. Scientifica. 2012;2012 doi: 10.6064/2012/917540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.