

Abstract
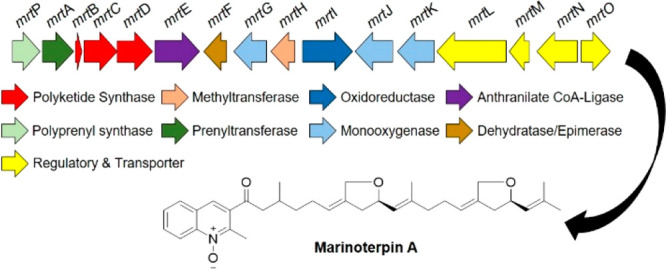
The chemical examination of two undescribed marine actinobacteria has yielded three rare merosesterterpenoids, marinoterpins A–C (1–3, respectively). These compounds were isolated from the culture broth extracts of two marine-derived actinomycetes associated with the family Streptomycetaceae, (our strains were CNQ-253 and AJS-327). The structures of the new compounds were determined by extensive interpretation of 1D and 2D NMR, MS, and combined spectroscopic data. These compounds represent new chemical motifs, combining quinoline-N-oxides with a linear sesterterpenoid side chain. Additionally, consistent in all three metabolites is the rare occurrence of two five-ring ethers, which were derived from an apparent cyclization of methyl group carbons to adjacent hydroxy-bearing methylene groups in the sesterterpenoid side chain. Genome scanning of AJS-327 allowed for the identification of the marinoterpin (mrt) biosynthetic cluster, which consists of 16 open-reading frames that code for a sesterterpene pyrophosphate synthase, prenyltransferase, type II polyketide synthase, anthranilate:CoA-ligase, and several tailoring enzymes apparently responsible for installing the N-oxide and bis-tetrahydrofuran ring motifs.
Introduction
As part of a broad investigation of the novel metabolites from marine-derived actinomycete bacteria,1 we have encountered a series of rare quinoline merosesterterpenoids that possess unusual five-membered ring ethers positioned on a linear sesterterpenoid side chain. Our attention was initially drawn to this linear sesterterpenoid metabolite class as part of an exhaustive chemical examination of our strain AJS-327, a taxonomically unique and distant relative of the genus Streptomyces. Subsequently, three closely related metabolites, marinoterpins A–C (1–3, respectively; Figure 1) were isolated from the culture broths of two marine-derived actinomycetes, our strains CNQ-253 and AJS-327. The two strains, both of which were isolated from the La Jolla, California local marine environment, are related to actinomycetes in the family Streptomycetaceae; however, the weak 16S sequence comparison of strain AJS-327 to type strains shows it is likely a new species or even genus.2
Figure 1.

Structures of marinoterpins A–C (1–3, respectively), illustrating the quinoline and bis-ether-substituted sesterterpenoid side chains. The configurational assignments for 1, which are based on a comparison with model compounds, are tentative.
Results and Discussion
Isolation and Structure Elucidation of Marinoterpins A–C
The EtOAc extracts from the seawater-based culture broths of actinomycete strains AJS-327 and CNQ-253 were fractionated by C-18 reversed-phase column chromatography, followed by C-18 preparative HPLC, to afford marinoterpin A (1) from strain AJS-327 and marinoterpins B (2) and C (3) from strain CNQ-253 (Figure 1).
The molecular formula of marinoterpin A (1) was assigned as C35H45NO4 on the basis of an observed protonated molecular ion at m/z 544.3411 [M + H]+ and a corresponding sodium adduct ion at m/z 566.3231 [M + Na] in the HR-ESI-TOFMS in combination with 13C NMR spectroscopic data. UV absorption at 320 nm coupled with 14 degrees of unsaturation indicated that 1 is composed of an aromatic chromophore with added unsaturation. The IR spectrum of 1 showed characteristic absorption bands at 1180 and 1699 cm–1, indicating the presence of an N-oxide3 and suggesting the presence of a conjugated carbonyl group, respectively. Two substructures, a quinoline-N-oxide moiety and a uniquely modified linear sesterterpenoid chain, were assigned by the analysis of 1D and 2D NMR spectroscopic data (Tables 1 and S1). These data revealed the presence of 5 methyl groups, 9 methylene carbons, 12 methine carbons (9 sp2 carbons), and 9 quaternary carbons, including 1 carbonyl carbon (Table 1 and Figures S1–S7). The characteristic 1H NMR features of the 2-methylquinoline-N-oxide were five aromatic protons at δH 8.66 (1H, d, J = 8.8, H-8), 8.48 (1H, s, H-4), 8.16 (1H, dd, J = 8.2, 1.3, H-5), 7.97 (1H, br t, J = 7.9, H-7), and 7.79 (1H, br t, J = 7.7, H-6) and a methyl group at δH 2.78 (3H, s, H-9). Sequential 1H–1H COSY correlations of H-5 to H-6 to H-7 and to H-8, HMBC NMR correlations of H-4 to C-2 (δC 147.6), C-8a (δC 142.4), and C-5 (δC 130.8), and correlations of H-5 to C-4 (δC 129.5) and C-8a, and of H-8 to C-4a (δC 128.9) and C-8a indicated that the aromatic substructure is a 2,3-disubstituted quinoline ring (Figure 2). HMBC NMR correlations from the methyl group H3-9 to C-2, C-3 (δC 135.5), C-8 (δC 120.0), and C-8a allowed the methyl group to be positioned at the C-2 position of the quinoline ring.4 The reported carbon chemical shifts of the 2-methylquinoline moiety are δC-9 26.1, δC-2 158.0, and δC-8 148.7 in CDCl3. In this case, however, the assignment of an N-oxide is required to explain the upfield carbon chemical shifts (C-9 (δC 16.1), C-2, and C-8).4 These carbon chemical shifts are in good agreement with those previously reported for quinoline-N-oxides.3 The modified sesterterpenoid chain, which features two tetrahydrofuran rings, was defined by comprehensive analysis of the 2D NMR data (Figure 2). 1H NMR data showed that the resonances from the similar tetrahydrofuran components seriously overlapped. The 2D NMR data, however, clearly resolved these resonances. HMBC correlations from the methyl groups H3-20′ (δH 1.73 and δC 25.9) and H3-25′ (δH 1.70 and δC 18.3) to C-19′ (δC 138.0) and C-18′ (δC 125.9), coupled with correlations from the oxygenated methylene protons H2-24′ (δH 4.39 and 4.23) to C-14′ (δC 120.8), C-15′ (δC 140.2), C-16′ (δC 40.2), and C-17′ (δC 77.4), defined the terminus of the sesterterpenoid chain and positioned an ether carbon at C-17′. COSY correlations between H2-13′ (δH 2.02) to H-14′ (δH 5.31), H2-16′ (δH 2.59/2.18) to H-17′ (δH 4.57), and H-18′ (δH 5.19) further established these relationships. HMBC correlations from H2-4′ (δH 1.51/1.37) to C-5′ (δC 27.8) and from H2-2′ (δH 3.12/2.97) to C-1′ (δC 202.9), C-4′ (δC 37.4), and C-21′ (δC 20.1) showed the beginning of the sesterterpenoid chain. Finally, HMBC correlations from H-4 to C-1′ and from H2-2′ to C-1′ demonstrated that the two substructures were connected at position C-1′. The geometry of the C-6′ (δC 121.3) and C-7′ (δC 140.1), C-10′ (δC 126.2) and C-11′ (δC 140.7), and C-14′ and C-15′ double bonds were assigned as Z, E, and Z configurations on the basis of ROESY correlations of H-6′ (δH 5.33) to H-8′ (δH 2.59/2.18), H-10′ (δH 5.19) to H-12′ (δH 2.04), and H-14′ to H-16′, respectively (Figure 3).
Table 1. NMR Spectroscopic Data for Marinoterpins A–C (1–3) (CD3OD, 500 MHz).
| marinoterpin
A (1) |
marinoterpin
B (2) |
marinoterpin
C (3) |
||||
|---|---|---|---|---|---|---|
| no. | δH mult (J, Hz) | δC | δH mult (J, Hz) | δC | δH mult (J, Hz) | δC |
| 2 | 147.6 | 149.6 | 163.1 | |||
| 3 | 135.5 | 134.4 | 138.3 | |||
| 4 | 8.48, s | 129.5 | 7.89, s | 129.2 | 7.67, s | 135.5 |
| 4a | 128.9 | 129.8 | 121.3 | |||
| 5 | 8.16, dd (8.2, 1.3) | 130.8 | 7.97, d (8) | 129.3 | 7.64, d (8) | 128.2 |
| 6 | 7.79, br t (7.7) | 130.2 | 7.69, t (7.5) | 129.5 | 7.29, t (7.5) | 122.6 |
| 7 | 7.97, br t (7.9) | 134.2 | 7.82, t (8) | 131.7 | 7.60, t (8) | 129.8 |
| 8 | 8.66, d (8.8) | 120.0 | 8.60, d (9) | 119.7 | 7.55, d (8) | 114.4 |
| 8a | 142.4 | 138.5 | 138.5 | |||
| 9 | 2.78, s | 16.1 | 2.73, s | 15.0 | 4.55, m | 77.1 |
| 1′ | 202.9 | 3.61, d (7) | 33.1 | 3.33, d (7) | 28.9 | |
| 2′ | 3.12, dd (16.9, 5.8) | 49.8 | 5.33, t (7) | 122.1 | 5.41, t (7) | 121.2 |
| 2.97, dd (16.9, 7.6) | ||||||
| 3′ | 2.17, ma | 30.3 | 139.4 | 138.2 | ||
| 4′ | 1.51, m/1.37, m | 37.4 | 2.18, ma | 40.2 | 2.19, ma | 39.1 |
| 5′ | 1.99 | 27.8 | 2.11, ma | 28.7 | 2.13, ma | 27.7 |
| 6′ | 5.33, ma | 121.3 | 5.32, ma | 121.0 | 5.37, ma | 129.6 |
| 7′ | 140.1 | 140.2 | 138.8 | |||
| 8′ | 2.59, dd (15.2, 5.8)b/2.18, ma | 40.3 | 2.58, dd (15, 5.5)/2.18, ma | 40.3 | 2.58, dd (15, 5.5)/2.18, ma | 39.1 |
| 9′ | 4.57, ma | 77.5 | 4.55, ma | 77.5 | 4.55, ma | 76.3 |
| 10′ | 5.19, d (8.5)b | 126.2 | 5.18, ma | 126.3 | 5.19, ma | 124.8 |
| 11′ | 140.7 | 140.7 | 139.6 | |||
| 12′ | 2.04, ma | 40.0 | 2.06, ma | 40.0 | 2.06, ma | 38.5 |
| 13′ | 2.02, ma | 28.6 | 2.05, ma | 28.6 | 2.05, ma | 28.9 |
| 14′ | 5.31, ma | 120.8 | 5.29, ma | 120.8 | 5.30, ma | 120.1 |
| 15′ | 140.2 | 140.2 | 137.1 | |||
| 16′ | 2.59, dd (15.2,5.8)b/2.18,ma | 40.2 | 2.58, dd (15, 5.5) /2.18, ma | 40.3 | 2.58, dd (15, 5.5) | 39.5 |
| 17′ | 4.57, ma | 77.4 | 4.55, ma | 77.4 | 4.55, ma | 76.1 |
| 18′ | 5.19, d (8.5)b | 125.9 | 5.18, ma | 125.9 | 5.18, ma | 125.2 |
| 19′ | 138.0 | 138.0 | 139.6 | |||
| 20′ | 1.73, s | 25.9 | 1.72, s | 25.9 | 1.73, s | 24.8 |
| 21′ | 1.03, d (6.6) | 20.1 | 1.79, s | 16.4 | 1.61, s | 16.1 |
| 22′ | 4.39, t (14.3)b/4.23, ma | 69.0 | 4.21, d (13)/4.38, t (12.5) | 69.0 | 4.21, d (13)/4.39, t (12.5) | 67.9 |
| 23′ | 1.70, ma | 16.7 | 1.67, s | 16.7 | 1.73, s | 15.1 |
| 24′ | 4.39, t (14.3)b/4.23, ma | 69.0 | 4.21, d (13)/4.38, t (12.5) | 69.0 | 4.21, d (13) /4.39, t (12.5) | 67.9 |
| 25′ | 1.70, ma | 18.3 | 1.69, s | 18.3 | 1.71, s | 15.5 |
Coupling constants were not determined due to overlapping signals.
The coupling constant was derived from 2D homo-J-resolved 1H NMR spectral data.
Figure 2.
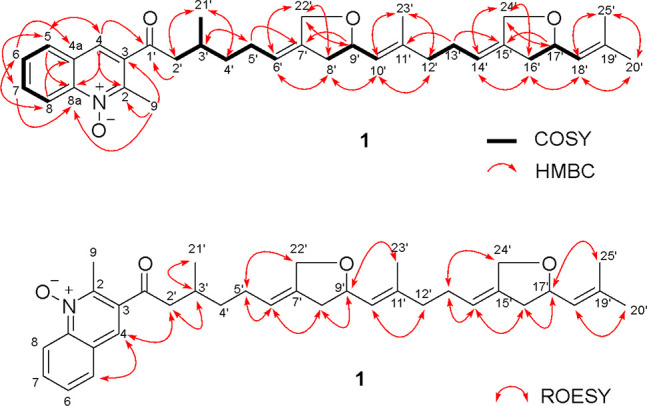
2D COSY, HMBC, and overall ROESY NMR correlations for marinoterpin A (1).
Figure 3.
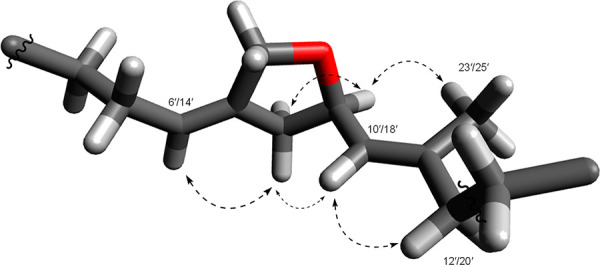
3D molecular model of one five-ring ether in marinoterpin A (1), showing the identical NOE correlations for both ethers using Avogadro molecular modeling software. (NOE correlations are the dashed arrows).
We attempted to evaluate the relative configurations at the C-3′, C-9′ (δC 77.5), and C-17′ position of 1 on the basis of ROESY NMR data. Unfortunately, ROESY NMR correlations did not provide convincing information. However, we could clearly see the relationship of the adjacent five-membered ring ether protons by the ROESY analysis (Figure 3), and correlations between H-4 and H2-2′ established the solution conformation of the quinoline ring (Figure 2.). 2D homo-J-resolved 1H NMR data showed the large coupling constants (8.5 Hz each) of H-10′ and H-18′ with no NOE correlations between H-9′ (δH 4.57) and H-10′ and H-17′ and H-18′, indicating that H-9′ and H-10′ and H-17′ and H-18′ were in anti-configurations. The specific rotation of 1 ([α]D25 + 5.91, c = 0.05, CH3OH) suggested, based on model studies with chiral synthetic methylene tetrahydrofurans, that C-9′ and C-17′ were both in R*, R* configurations.5 We also attempted to define the absolute configuration of 1 by X-ray methods. Although 1 is a solid, we were unable to obtain a crystal of an appropriate size and structure. As a result, we could not assign the configuration at C-3′.
Marinoterpin B (2) was isolated as an optically active colorless oil ([α]D25 + 5.00, c = 0.08, CH3OH). The molecular mass of 2 was obtained from the HR-ESI-TOFMS spectrum, which showed a protonated molecular ion at m/z 528.3469 [M + H]+, (calcd for C35H46NO3, 528.3478). On this basis, and considering the NMR data, the molecular formula was assigned as C35H45NO3, which indicated that marinoterpin B (2) was composed of 14 double-bond equivalents analogous to 1. The UV spectrum of 2 displayed three absorption bands at λmax 204, 237, and 318 nm, suggesting the presence of the quinoline aromatic nucleus without additional conjugation. An initial analysis of 2 by 1H, 13C and 2D NMR methods clearly illustrated that 2 is an analogous 2-methylquinoline-N-oxide-based merosesterterpenoid. As in 1, the 2-methylquinoline-N-oxide ring in 2 was assembled by the interpretation of COSY and HMBC spectral data (Table S2). COSY correlations of four methine signals from H-5 to H-8 (δH 7.97, 7.69, 7.82, and 8.60) established four consecutive aromatic protons. HMBC correlations from H-6 to C-4a and C-8, H-7 to C-5 and C-8a, H-8 to C-4a and C-6, and H-5 to C-4, C-7, and C-8a confirmed the presence of the quinoline ring system (C-4a to C-8a). The H-4 aromatic proton signal, which was observed as a singlet at δH 7.89, showed HMBC correlations to C-2, C-3, C-8a, and C-1′, thus confirming the presence of the quinoline ring. The aromatic methyl group, H3-9 (δH 2.73), was positioned at C-2 on the basis of HMBC correlations from H3-9 to C-2 and C-3 and the chemical shift observed for 1. Based upon the NMR data and the comparison with 1, marinoterpin B (2) was also assigned as a 2-methylquinoline-N-oxide (Figure 1).
Detailed 1D and 2D NMR data for 2 allowed the sesterterpenoid side chain from C-6′ onward to be assigned as identical to that of 1 (Tables 1 and S2). On the basis of the above observations and analyses, the structure of 2 was assigned for marinoterpin B. While it is tempting to assign the relative configurations in 2 as those in 1 based upon 2D NMR and optical rotation data, we can only suggest analogous configurations for marinoterpin B (2).
Marinoterpin C (3), a minor metabolite, was obtained as a colorless oil that showed a protonated molecular ion at m/z 528.3468 [M + H]+, which is consistent with a molecular formula of C35H46NO3 that indicated 3 contained the same number of degrees of unsaturation as in 1 and 2. An initial analysis of the 1H NMR spectral data for 3 (Table 1) provided obvious evidence that this compound had an oxygenated sesterterpenoid side chain identical to that in 1 and 2. The only difference between marinoterpin B and C was observed in the aromatic chromophore. The chemical shifts of five aromatic protons were shifted slightly upfield (δH 7.67, 7.64, 7.60, 7.55, and 7.29). Additionally, the absence of a signal for the aromatic methyl in the NMR spectrum of 3 and the presence of a new oxygenated methylene carbon signal at δ 77.1 suggested the absence of the N-oxide and a replacement of the methyl at C-2 by a hydroxymethyl group. Consequently, the analysis of the 2D COSY, HMQC and HMBC NMR data for 3 led to the confident assignment shown in Figure 1.
Marinoterpin A Biosynthetic Gene Cluster
Sesterterpenes are a small class of terpene natural products that are not commonly isolated from bacteria.6 As a result, only a few bacterial geranylfarnesyl polyprenyl synthases have been biochemically characterized, such as those involved in the biosynthesis of somaliensenes A and B,7 atolypenes,8 and the sestermobaraenes.9 Marinoterpins 1–3 are structurally related to the aurachins, a small family of natural products bearing prenylated quinoline, 4-quinolone, or quinoline N-oxide cores. While the marinoterpins are composed of previously unknown sesterterpenoid side chains with two unusual five-membered ethers, the aurachins more commonly possess sesquiterpenoid and monoterpenoid side chains (Figure 4). The basic aurachins A–D (4–7, respectively) were isolated from the myxobacterium Stigmatella aurantiaca strain Sg a15,10 aurachin RE (8) was isolated from Rhodococcus erythropolis JCM 6824,11 and aurachin SS (9) with a monoterpenoid side chain was isolated from a culture of Streptomyces sp. NA04227.12 Stable isotope enrichment studies with 13C- and 18O-labeled precursors revealed that the quinoline and quinolone cores in 4–7 were synthesized from anthranilic acid and acetate,10 suggesting the involvement of a polyketide synthase.
Figure 4.

Select aurachin prenylated natural products.
Characterization of the aua,13rau,11 and sau(12) clusters responsible for the biosynthesis of 4–7, 8, and 9, respectively, revealed the following set of core genes shared between the three clusters: a prenyltransferase, an acyl carrier protein (ACP), two β-ketoacyl-ACP synthases, and a benzoate:CoA ligase (Figure 5). The presence of two β-ketoacyl-ACP synthases and an ACP in all three clusters further supported that a type II polyketide synthase (PKS) was involved in the assembly of the aurachin core. Unlike modular type I PKS systems where catalytic domains are arranged linearly across multimodular megasynthases, type II PKS systems contain dissociated domains that are used iteratively during the synthesis of a polyketide. A minimal type II PKS cluster typically includes a ketosynthase (KS or KSα) that forms C–C bonds through Claisen-like condensation reactions with activated acyl and malonyl extender units, a KSβ or chain-length factor (CLF) that determines the length of the carbon chain, and an ACP containing a phosphopantetheine arm that tethers the growing acyl chain.14−16
Figure 5.

Prenylated quinoline biosynthetic clusters. Organization of the marinoterpin biosynthetic gene cluster (mrt) from Streptomyces sp. AJS-327 (GenBank accession o. MW452943), the aurachin biosynthetic gene cluster (aua) from Stigmatella aurantiaca Sg a15 (GenBank accession nos. AM404078 for locus I, HE580420 for locus II, and HE580421 for locus III), the aurachin RE biosynthetic cluster (rau) from Rhodococcus erythropolis JCM 6824 (GenBank accession no. AB694012), and the aurachin SS biosynthetic gene cluster (sau) from Streptomyces sp. NA04227 (GenBank accession no. KY810820). The arrows, which are color coded based on the putative function and point in the direction of transcription, represent open reading frames. MEP is the methylerythritol 4-phosphate pathway.
As the genome of Streptomyces sp. AJS-327 was recently sequenced (GenBank accession number SKBR00000000), the 6.5 Mbp genome was scanned for putative aurachin-like biosynthetic clusters. Initial screening using AntiSMASH17 failed to identify any type II PKS clusters that would be responsible for synthesizing marinoterpin A. With access to the aua, sau, and rau clusters, the anthranilate:CoA ligase, KSα, and KSβ homologues, respectively, were used to screen the genome of AJS-327 using the BLAST+ package.18 A 22 kbp region on scaffold one contained “hits” to the biosynthetic hooks, and an annotation of the corresponding region (the mrt cluster) revealed 16 open-reading frames that encode a sesterterpene diphosphate synthase (mrtP), a prenyltransferase (mrtA), two β-ketoacyl-ACP synthases (mrtC and D), an ACP (mrtB), and a benzoate:CoA ligase (mrtD). In addition to the genes required to synthesize the prenylated quinoline core, the mrt cluster also codes for a variety of tailoring enzymes to further functionalize the disubstituted quinoline ring, including two cytochrome P450s (mrtJ and K), a flavin monooxygenase (mrtG), an FAD-dependent oxidoreductase (mrtI), an NAD-dependent epimerase or dehydratase (mrtF), and a methyltransferase (mrtH) (Figure 5 and Table S4). Because only marinoterpin A has been isolated from AJS-327, it is unclear what role the methyltransferase MrtH plays in its biosynthesis, if any. A detailed investigation of the mass spectrometry data using the Global Natural Product Social Molecular Networking (GNPS) platform19 did not identify any methylated marinoterpin derivatives.
Based on the similarities between the aua, rau, sau, and mrt gene clusters, a biosynthetic route for marinoterpin A (1) was proposed (Figure 6). It must be noted that the biosynthetic cluster responsible for assembling marinoterpins B (2) and C (3) is currently unknown because the compounds were isolated from Streptomyces. sp. CNQ-253 and its genome has not yet been sequenced. However, due to structural similarities to 1, especially the presence of two tetrahydrofuran rings on the sesterterpenoid side chain, the biosyntheses of 2 and 3 are also proposed.
Figure 6.

Proposed biosynthesis of marinoterpin A (1) from strain AJS-327 and those of marinoterpins B (2) and C (3) from strain CNQ-253. Polyketide synthase abbreviations are ketosynthase (KSα), chain length factor (CLF), and acyl carrier protein (ACP).
The biosynthesis of 1–3 starts with the activation and priming of anthranilate. This priming mechanism was established for the biosynthesis of 4–7, where two aryl-CoA ligases, namely, AuaEII and AuaE, are required for the activation of anthranilate and ACP loading, respectively.20 Interestingly, while the aua cluster contains two anthranilate-CoA ligase homologues, the sau, rau, and mrt clusters only contain one copy. A sequence comparison with various aryl-CoA ligases and anthranilate-specific adenylation domains (Figure S21) suggests that the priming mechanism catalyzed by MrtE is analogous to the EncN-mediated priming of benzoate in enterocin biosynthesis.21 The first half-reaction adenylates anthranilate to form the anthraniloyl-AMP intermediate, while the second half-reaction uses transthioesterification with either CoA or the phosphopantetheine prosthetic group of MrtB to eliminate AMP and form either anthraniloyl-CoA or anthraniloyl-MrtB, respectively.
As observed with the biosynthesis of 4–713 and supported with 13C feeding studies,10 KSα (MrtC) and KSβ (MrtD) catalyze two extensions with malonyl-CoA after anthranilate is transferred to the ACP (MrtB) to yield the intermediate 10. The release of 10 and the decarboxylation of the second malonyl-CoA extender unit affords the 4-hydroxy-2-methylquinoline intermediate 11. The formation of the C25 geranylfarensyl diphosphate (GFPP) side chain proceeds through the condensation of hemiterpenes dimethylallyl diphosphate and isopentenyl diphosphate using the polyprenyl diphosphate synthase MrtP; in a similar reaction catalyzed by AuaA,22 the isoprenoid side chain is attached to the 4-hydroxy-2-methylquinoline intermediate at position C-3 using the prenyltransferase MrtA to yield the 3-geranylfarnesyl-2-methyl-4-quinolone intermediate 12. A phylogenetic comparison of the characterized polyprenyl diphosphate synthases (Figure S22) and prenyltransferases (Figure S23) shows that the geranylfarnesyl diphosphate synthases are more closely related to each other than to shorter chain polyprenyl synthases, and the prenyltransferases clade together based on the type of intermediate the isoprenoid unit is attached to.
Following the formation of the aurachin-related intermediate 12, a series of reactions, including N-oxidation and bis-ether ring formation on the sesterterpenoid side chain, are required for its conversion to 1. While the furanosesterterpenoids isolated from the sponge Luffariella variabilis also contain a five-membered ring ether on their sesterterpene chains,23 the corresponding biosynthetic cluster is not known. Given the number of tailoring enzymes present in the mrt cluster, it’s unclear what enzymes are ultimately involved in forming the tetrahydrofuran rings, as well as the order of events. Therefore, in an attempt to tease apart the enzymes involved in converting 12 to 1, we implemented a bioinformatic investigation that focused on the monooxygenase MrtG and cytochrome P450s MrtJ and MrtK.
During aurachin biosynthesis in S. aurantiaca Sg a15, the Rieske oxygenase AuaF installs the N-hydroxylase on aurachin D (7) to form aurachin C (6).24 In the rau cluster, the cytochrome P450 RauA catalyzes N-hydroxylation.11 While the mrt cluster does not contain a Rieske oxygenase homologue, it does contain two cytochrome P450s and a flavin monooxygenase; both enzyme classes are known to catalyze N-oxidation. A comparative analysis of the two cytochrome P450s from the marinoterpin cluster with the N-hydroxylase-catalyzing P450 from the rau cluster showed that RauA shares only a 20% amino acid identity with MrtJ and a 21% identity with MrtK. Although the two P450s from the mrt cluster have low similarities to RauA, it cannot be ruled out that one is responsible for installing the N-hydroxyl moiety in 1. Interestingly, an unknown monooxygenase was proposed to install the secondary alcohol on the C-9′ position of the farnesyl side chain in aruachin RE (8),11 and a similar enzyme may be responsible for oxidizing the linear sesterterpene chain in coleifolide B.25 It is thus speculated that one of the cytochrome P450s (MrtJ or K) could oxidize the C-9′ and C-17′ methylene groups on the sesterterpene side chain to secondary alcohols in the marinoterpin biosynthesis to form intermediate 13, and radical abstraction of the methyl moieties at C-22′ and C-24′ could lead to tetrahydrofuran ring formation. Alternatively, the oxidation of C-22′ and C-24′ in 13, followed by intramolecular cyclization and dehydration, could afford the five-membered ring ethers.
To evaluate whether the flavin monooxygenase MrtG could be involved in N-oxidation, a phylogenetic analysis with characterized flavin monooxygenases was carried out, which suggested that MrtG is more closely related to AuaG than other N-oxidizing flavin monooxygenases, such as those involved in valanimycin biosynthesis26−28 (Figure S24). During a detailed biosynthetic investigation of aurachins A (4) and B (5), AuaG was shown to catalyze the migration of the farnesyl group from the C-3 position to the C-4 position on the quinoline core through a semipinacol rearrangement,29 thus converting aurachin C (6) to 5. Based on their similarity, we propose that MrtG is a unique flavin monooxygenase that may be involved with the bis-ether ring formation observed in 1–3.
Conclusions
While linear sesterterpenoids are rare, sesterterpenoids and their cyclized analogs in particular are common metabolites, especially from sponges.30−32 The isoquinoline N-oxides are common components of plants, whereas the quinoline N-oxides are commonly produced by bacteria.33−44 However, to the best of our knowledge, this is the first time linear quinoline merosesterterpenoids containing the replicate tetrahydrofuran functionalities observed in 1–3 have been described. The identification of the marinoterpin (mrt) biosynthetic cluster from Streptomyces sp. AJS-327 and a comparison to other aurachin gene clusters allowed us to propose a biosynthetic route for the 3-geranylfarnesyl-2-methylquinoline core shared among marinoterpins A–C. Future biochemical characterization experiments with the mrt tailoring enzymes will ultimately shed light on the reactions needed to furnish the two tetrahydrofuran rings on the linear sesterterpenoid side chain as well as the N-oxidation observed in marinoterpins A and B.
Experimental Section
General Experimental Procedures (Strain AJS-327)
Optical rotations were measured on a JASCO P-2000 polarimeter, and UV spectra were recorded with a Varian Cary UV–visible spectrophotometer. Fourier transform infrared (FT-IR) spectra were obtained using a PerkinElmer 1600 FT-IR spectrophotometer. 1D and 2D NMR spectral data were obtained on a JEOL 500 MHz NMR spectrometer. The NMR chemical shifts were referenced to the residual solvent peaks (δH 3.31 and δC 49.0 for CD3OD). The chemical shift values are reported in parts per million, and the coupling constants are reported in Hertz. High-resolution ESI-TOF mass spectra were provided by the mass spectrometry facility at the Department of Chemistry and Biochemistry at the University of California, San Diego, La Jolla, CA on an Agilent 6530 HR-TOF LCMS system. Preparative HPLC separations were performed using a Shimadzu SCL-10AVP instrument with a Shimadzu SPD-M10AVP diode array detector system, and a Luna 10 μm C18 (2) column (10 × 250 mm, 10 μm, Phenomenex) was used at a flow rate of 3 mL/min for preparative HPLC.
Sample Collection, Bacterial Isolation, and Identification (Strain AJS-327)
Strain AJS-327 was isolated from an unidentified detached sponge fragment collected on the beach 200 m south of Scripps Pier in La Jolla, CA. The sponge fragment was collected in a sterile 50 mL tube and transported to the laboratory, where it was cut into small fragments with sterile scissors within 1 h and used for isolation with A1 medium-based agar plates (10 g of soluble potato starch, 4 g of yeast extract, 2 g of peptone, 750 mL of natural seawater, 250 mL of distilled water, and 18 g of agar). Strain AJS-327 was identified by partial (1380 bp) 16S rDNA sequence analysis using a NCBI BLASTn search. The closest matching type strains were the Streptomyces cacoi strain NBRC 12748 (96% identity; accession no. NR_041061.1), the Streptomyces oryzae strain NBRC 109761 (96% identity; accession no. NR_146025.1), the Streptomyces artemisiae strain YIM 63135 (96% identity; accession no. NR_116242.1), and the Streptomyces armeniacus strain NBRC 12555 (96% identity; accession no. NR_112247.1). The very poor matches strongly indicate that our strain represents a novel lineage within the actinomycete family Streptomycetaceae.
Cultivation and Extraction of Strain AJS-327
Strain AJS-327 was cultured in 12 × 1 L volumes of a seawater-based A1 medium (10 g of starch, 4 g of yeast extract, and 2 g of peptone) while shaking at 200 rpm for 15 days at 27 °C. The whole culture broth was extracted with ethyl acetate (2 × 12 L), and the ethyl acetate extract was concentrated to yield 2.0 g of the organic extract.
General Experimental Procedures (Strain CNQ-253)
Optical rotations were measured using a JASCO P-2000 polarimeter with a 10 cm cell. UV spectra were obtained using Varian Cary 50 Bio UV–visible spectrophotometer. IR spectra were acquired on a PerkinElmer 1600 series FTIR spectrometer. 1H, 13C, and 2D NMR spectral data were obtained on a Varian Inova 500 MHz NMR spectrometer. High-resolution mass spectra were recorded on a ThermoFinnigan MAT900XL instrument with an Agilent ESI-TOF detector at The Scripps Research Institute, La Jolla, CA. Low-resolution LC/MS spectra were obtained on a Hewlett-Packard HP1100 integrated LC-MS system with a reversed-phase C18 column (Agilent, 4.6 mm × 100 mm, 5 μm) at a flow rate of 0.7 mL/min. Reversed-phase HPLC separations were performed using a semipreparative C18 Phenomenex Luna (2) 5 μm (10 mm × 250 mm) column with a CH3CN/H2O gradient solvent system. Preparative HPLC was performed using a Waters model 4000 system with a UV variable-wavelength detector monitoring at 210 nm using a C18 Nova-Pak 6 μm 60 Å, (40 mm × 300 mm) column.
Bacterial Isolation and Identification (Strain CNQ-253)
The actinomycete strain CNQ-253 was obtained from a marine sediment sample collected at a depth of −46 m off San Diego, CA in 2001. A partial 16S rDNA sequence analysis (991 bps) indicated that the closest formally described species is S. cacaoi (accession no. AB184183, 97.6%); however, the relatively low level of identity suggests that strain CNQ-253 belongs to a new Streptomyces species.
Fermentation and Extraction (Strain CNQ-253)
Strain CNQ-253 was cultured at 27 °C for six days while shaking at 215 rpm in ten × 1 L volumes of the liquid medium A1Bfe+C (composed of 10 g of starch, 4 g of yeast extract, 2 g of peptone, 1 g of CaCO3, 40 mg of Fe2(SO4)3·4H2O, and 100 mg of KBr per 1 L of seawater). Amberlite XAD-7 resin (20 g/L) was added at the end of the fermentation period to adsorb extracellular secondary metabolites, and the culture and resin were shaken at a low speed for two additional hours. The resin and cell mass were collected by filtering them through cheesecloth and washed with DI water to remove salts. The resin, cell mass, and cheesecloth were then soaked for 2 h in acetone, after which the acetone extract was filtered and reduced to dryness, and the residue was taken up in ethyl acetate to give 1.95 g of a brown oily organic extract from a 10 L culture.
Isolation of Marinoterpin A (1)
The organic broth extract of strain AJS-327 (2.0 g) was fractionated by C-18 reversed-phase vacuum flash chromatography using a step-gradient solvent system consisting of MeOH and H2O. The MeOH/H2O (100:0) fraction contained compound 1. The fraction was subjected to reversed-phase HPLC separation (Phenomenex Luna 10 μm, C18 (2) 250 × 10 mm column; 4 mL/min) using a gradient elution from 70% to 100% aqueous MeOH for 30 min to obtain compound 1. Marinoterpin A was further purified by reversed-phase HPLC (Phenomenex Luna 10 μm, 250 × 10 mm column; 3 mL/min; UV detection at 230 nm) using isocratic elution (85% aqueous MeOH) to obtain 1 (3 mg, tR = 23.0 min).
Isolation of Marinoterpin B (2) and C (3)
The organic broth extract of strain CNQ-253 was fractionated by C-18 reversed-phase vacuum liquid chromatography (H2O/CH3OH, gradient 90:10 to 0:100%) to give six fractions. Fraction four was then further purified by preparative RP HPLC (Prep Nova-Pak HRC 18, 6 μm, 300 mm × 40 mm) with CH3CN/H2O as eluent, followed by semipreparative isocratic HPLC to give marinoterpin B (2, 18.5 mg). Fraction five was further purified by isocratic RP HPLC to yield marinoterpin B (2, 12.2 mg), and marinoterpin C (3, 1.5 mg).
Marinoterpin A (1)
Amorphous solid; [α]D25 + 5.91 (c = 0.05, CH3OH); UV (CH3OH) λmax (log ε) 202 (4.34), 234 (4.21), 320 (3.37) nm; IR (film) νmax 3404, 1699, 1404, 1325, 1181, 1075 cm–1; 1H and 13C NMR data, see Table 1; HRMS (ESI/Q-TOF) m/z [M + H]+ Calcd for C35H46NO4 534.3427, found 544.3411; [M + Na]+ Calcd for C35H46NO4Na 566.3246, found 566.3231.
Marinoterpin B (2)
Colorless viscous oil; [α]D25 + 5.00 (c = 0.08, CH3OH); UV (CH3OH) λmax (log ε) 204 (3.80), 237 (3.72), 318 (2.89); IR (KBr) νmax 3317, 2924, 2851, 1750, 1677, 1500, 1442, 1378, 1310, 1226, 1099, 750 cm–1; 1H and 13C NMR data, see Table 1; HRMS (ESI/Q-TOF) m/z [M + H]+ Calcd for C35H46NO3 528.3478, found 528.3469.
Marinoterpin C (3)
Colorless viscous oil; [α]D25 + 4.62 (c = 0.106, CH3OH); UV (CH3OH) λmax (log) 230 (3.74), 236 (2.89), 276 (2.72); IR (KBr) νmax 3320, 2924, 2854, 1715, 1643, 1589, 1456, 1378, 1093, 756 cm–1; 1H and 13C NMR data, see Table 1; HRMS (ESI/Q-TOF) m/z [M + H]+ Calcd for C35H46NO3 528.3478, found 528.3468.
Acknowledgments
This research is a result of the financial support, in part, from the National Cancer Institute under award CA044848 to W.F. and from the Moore Foundation GBMF7621 to J.M.W. We thank Ted Molinski, UCSD, for insightful discussions on approaches to the absolute configurations of the marinoterpins and C. Kauffman, SIO, for assistance with bacterial cultivations (strain CNQ-253).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00262.
Full NMR spectral data and high resolution mass spectrometry data for marinoterpins A–C and phylogenetic analyses of the marinoterpin flavin monooxygenase, polyprenyl diphosphate synthase, prenyltransferase, and aryl-CoA ligase (PDF)
Accession Codes
Marinoterpin GenBank accession no. MW452943. The AJS-327 genome has been deposited at DDBJ/ENA/GenBank under accession no. SKBR00000000.
Author Contributions
W.F., M.C.K., and J.M.W. conceived of and organized the project; M.C.K., R.N.A., and C.B. isolated, purified, and identified the marinoterpins; J.M.W. identified the marinoterpin cluster, carried out phylogenetic and bioinformatic analyses of the marinoterpin enzymes, and proposed the biosynthesis of 1–3; R.C. undertook the bacterial fermentation and culture extractions; and all authors contributed to writing the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Fenical W. Marine Microbial Natural Products: The Evolution of a New Field of Science. J. Antibiot. 2020, 73 (8), 481–487. 10.1038/s41429-020-0331-4. [DOI] [PubMed] [Google Scholar]
- Kim M. C.; Cullum R.; Machado H.; Smith A. J.; Yang I.; Rodvold J. J.; Fenical W. Photopiperazines A-D, Photosensitive Interconverting Diketopiperazines with Significant and Selective Activity against U87 Glioblastoma Cells, from a Rare, Marine-Derived Actinomycete of the Family Streptomycetaceae. J. Nat. Prod. 2019, 82 (8), 2262–2267. 10.1021/acs.jnatprod.9b00429. [DOI] [PubMed] [Google Scholar]
- Kunze B.; Hofle G.; Reichenbach H. The Aurachins, New Quinoline Antibiotics from Myxobacteria: Production, Physico-Chemical and Biological Properties. J. Antibiot. 1987, 40 (3), 258–265. 10.7164/antibiotics.40.258. [DOI] [PubMed] [Google Scholar]
- Sridharan V.; Ribelles P.; Ramos M. T.; Menendez J. C. Cerium(IV) Ammonium Nitrate is an Excellent, General Catalyst for the Friedlander and Friedlander-Borsche Quinoline Syntheses: Very Efficient Access to the Antitumor Alkaloid Luotonin A. J. Org. Chem. 2009, 74 (15), 5715–5718. 10.1021/jo900965f. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Bringley D. A.; Silverman S. M. Asymmetric Synthesis of Methylenetetrahydrofurans by Palladium-Catalyzed [3 + 2] Cycloaddition of Trimethylenemethane with Aldehydes-a Novel Ligand Design. J. Am. Chem. Soc. 2011, 133 (20), 7664–7667. 10.1021/ja201181g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Gustafson K. R.. Sesterterpenoids: Chemistry, Biology, and Biosynthesis. Nat. Prod. Rep. 2020. Dec. 22, 10.1039/D0NP00070A. Epub ahead of print. PMID: 33350420. [DOI] [PubMed]
- Yang Y.; Zhang Y.; Zhang S.; Chen Q.; Ma K.; Bao L.; Tao Y.; Yin W.; Wang G.; Liu H. Identification and Characterization of a Membrane-Bound Sesterterpene Cyclase from Streptomyces somaliensis. J. Nat. Prod. 2018, 81 (4), 1089–1092. 10.1021/acs.jnatprod.7b01033. [DOI] [PubMed] [Google Scholar]
- Kim S.-H.; Lu W.; Ahmadi M. K.; Montiel D.; Ternei M. A.; Brady S. F. Atolypenes, Tricyclic Bacterial Sesterterpenes Discovered using a Multiplexed In Vitro Cas9-TAR Gene Cluster Refactoring Approach. ACS Synth. Biol. 2019, 8 (1), 109–118. 10.1021/acssynbio.8b00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou A.; Dickschat J. S. The Biosynthetic Gene Cluster for Sestermobaraenes- Discovery of a Geranylfarnesyl Diphosphate Synthase and a Multliproduct Sesterterpene Synthase from Streptomyces mobaraensis. (2020). Angew. Chem., Int. Ed. 2020, 59 (45), 19961–19965. 10.1002/anie.202010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofle G.; Kunze B. Biosynthesis of Aurachins A-L in Stigmatella aurantiaca: A Feeding Study. J. Nat. Prod. 2008, 71 (11), 1843–1849. 10.1021/np8003084. [DOI] [PubMed] [Google Scholar]
- Kitagawa W.; Ozaki T.; Nishioka T.; Yasutake Y.; Hata M.; Nishiyama M.; Kuzuyama T.; Tamura T. Cloning and Heterologous Expression of the Aurachin RE Biosynthesis Gene Cluster Afford a New Cytochrome P450 for Quinoline N-Hydroxylation. ChemBioChem 2013, 14 (9), 1085–1093. 10.1002/cbic.201300167. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Yang C. L.; Xiao Y. S.; Zhang B.; Deng X. Z.; Yang L.; Shi J.; Wang Y. S.; Li W.; Jiao R. H.; Tan R. X.; Ge H. M. Aurachin SS, a New Antibiotic from Streptomyces sp. NA04227. J. Antibiot. 2017, 70 (7), 853–855. 10.1038/ja.2017.50. [DOI] [PubMed] [Google Scholar]
- Sandmann A.; Dickschat J.; Jenke-Kodama H.; Kunze B.; Dittmann E.; Müller R. A Type II Polyketide Synthase from the Gram-Negative Bacterium Stigmatella aurantiaca is Involved in Aurachin Alkaloid Biosynthesis. Angew. Chem., Int. Ed. 2007, 46 (15), 2712–2716. 10.1002/anie.200603513. [DOI] [PubMed] [Google Scholar]
- Chen A.; Re R. N.; Burkart M. D. Type II Fatty Acid and Polyketide Synthases: Deciphering Protein-Protein and Protein-Substrate Interactions. Nat. Prod. Rep. 2018, 35 (10), 1029–1045. 10.1039/C8NP00040A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertweck C.; Luzhetskyy A.; Rebets Y.; Bechthold A. Type II Polyketide Synthases: Gaining a Deeper Insight into Enzymatic Teamwork. Nat. Prod. Rep. 2007, 24 (1), 162–190. 10.1039/B507395M. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Tang Y. In Vitro Analysis of Type II Polyketide Synthase. Methods Enzymol. 2009, 459, 367–393. 10.1016/S0076-6879(09)04616-3. [DOI] [PubMed] [Google Scholar]
- Blin K.; Shaw S.; Steinke K.; Villebro R.; Ziemert N.; Lee S. Y.; Medema M. H.; Weber T. Antismash 5.0: Updates to the Secondary Metabolite Genome Mining Pipeline. Nucleic Acids Res. 2019, 47 (W1), W81–W87. 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C.; Coulouris G.; Avagyan V.; Ma N.; Papadopoulos J.; Bealer K.; Madden T. L. Blast+: Architecture and Applications. BMC Bioinf. 2009, 10, 421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Carver J.; Phelan V.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34 (8), 828–837. 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistorius D.; Li Y.; Mann S.; Müller R. Unprecedented Anthranilate Priming Involving Two Enzymes of the Acyl Adenylating Superfamily in Aurachin Biosynthesis. J. Am. Chem. Soc. 2011, 133 (32), 12362–12365. 10.1021/ja203653w. [DOI] [PubMed] [Google Scholar]
- Izumikawa M.; Cheng Q.; Moore B. S. Priming Type II Polyketide Synthases Via a Type II Nonribosomal Peptide Synthetase Mechanism. J. Am. Chem. Soc. 2006, 128 (5), 1428–1429. 10.1021/ja0559707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec E.; Pistorius D.; Müller R.; Li S. M. AuaA, a Membrane-Bound Farnesyltransferase from Stigmatella aurantiaca Catalyzes the Prenylation of 2-Methyl-4-Hydroxyquinoline in the Biosynthesis of Aurachins. ChemBioChem 2011, 12 (11), 1724–1730. 10.1002/cbic.201100188. [DOI] [PubMed] [Google Scholar]
- Ahmadi P.; Higashi M.; de Voogd N. J.; Tanaka J. Two Furanosesterterpenoids from the Sponge Luffariella variabilis. Mar. Drugs 2017, 15 (8), 249–256. 10.3390/md15080249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistorius D.; Li Y.; Sandmann A.; Müller R. Completing the Puzzle of Aurachin Biosynthesis in Stigmatella aurantiaca Sg A15. Mol. BioSyst. 2011, 7 (12), 3308–3315. 10.1039/c1mb05328k. [DOI] [PubMed] [Google Scholar]
- Kurimoto S.-I.; Pu J.-X.; Sun H.-D.; Takaishi Y.; Kashiwada Y. Coleifolides A and B, Two New Sesterterpenoids from the Aerial Parts of Scutellaria coleifolia H. Lév. Chem. Biodiversity 2015, 12 (8), 1200–1207. 10.1002/cbdv.201400248. [DOI] [PubMed] [Google Scholar]
- Parry R. J.; Li W.; Cooper H. N. Cloning, Analysis, and Overexpression of the Gene Encoding Isobutylamine N-Hydroxylase from the Valanimycin Producer, Streptomyces viridifaciens. J. Bacteriol. 1997, 179 (2), 409–416. 10.1128/JB.179.2.409-416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry R. J.; Li W. Purification and Characterization of Isobutylamine N-Hydroxylase from the Valanimycin Producer Streptomyces viridifaciens MG456-hF10. Arch. Biochem. Biophys. 1997, 339 (1), 47–54. 10.1006/abbi.1996.9857. [DOI] [PubMed] [Google Scholar]
- Garg R. P.; Ma Y.; Hoyt J. C.; Parry R. J. Molecular Characterization and Analysis of the Biosynthetic Gene cluster for the Azoxy Antibiotic Valanimycin. Mol. Microbiol. 2002, 46 (2), 505–517. 10.1046/j.1365-2958.2002.03169.x. [DOI] [PubMed] [Google Scholar]
- Katsuyama Y.; Harmrolfs K.; Pistorius D.; Li Y.; Müller R. A Semipinacol Rearrangement Directed by an Enzymatic System Featuring Dual-Function FAD-Dependent Monooxygenase. Angew. Chem., Int. Ed. 2012, 51 (37), 9437–9440. 10.1002/anie.201204138. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wang L.; Jung J. H.; Zhang S. Sesterterpenoids. Nat. Prod. Rep. 2007, 24 (6), 1401–1429. 10.1039/b617259h. [DOI] [PubMed] [Google Scholar]
- Cimino G.; Destefano S.; Minale L.; Riccio R.; Hirtosu K.; Clardy J. 2 Novel Sesterterpene Hydroxyquinols from the Sponge Microciona toxistyla. Tetrahedron Lett. 1979, 20 (38), 3619–3622. 10.1016/S0040-4039(01)95479-6. [DOI] [Google Scholar]
- West L. M.; Faulkner D. J. Acanthosulfate, A Sulfated Hydroxyhydroquinone Sesterterpenoid from the Sponge Acanthodendrilla sp. J. Nat. Prod. 2008, 71 (2), 269–271. 10.1021/np070443h. [DOI] [PubMed] [Google Scholar]
- Debitus C.; Guella G.; Mancini I. I.; Waikedre J.; Guemas J. P.; Nicolas J. L.; Pietra F. Quinolones from a Bacterium and Tyrosine Metabolites from its Host Sponge, Suberea creba from the Coral Sea. J. Mar Biotechnol 1998, 6 (3), 136–141. [PubMed] [Google Scholar]
- Dehmlow E. V.; Schulz H. J. Synthesis of Orellanine, the Lethal Poison of a Toadstool. Tetrahedron Lett. 1985, 26 (40), 4903–4906. 10.1016/S0040-4039(00)94981-5. [DOI] [Google Scholar]
- Edrada R. A.; Proksch P.; Wray V.; Witte L.; Müller W. E. G.; VanSoest R. W. M. Four New Bioactive Manzamine-Type Alkaloids from the Philippine Marine Sponge Xestospongia ashüorica. J. Nat. Prod. 1996, 59 (11), 1056–1060. 10.1021/np9604083. [DOI] [PubMed] [Google Scholar]
- Gill M. Pigments of Fungi (Macromycetes). Nat. Prod. Rep. 1999, 16 (3), 301–317. 10.1039/a705730j. [DOI] [PubMed] [Google Scholar]
- Kitamura S.; Hashizume K.; Iida T.; Miyashita E.; Shirahata K.; Kase H. Studies on Lipoxygenase Inhibitors. II. KF8940 (2-n-Heptyl-4-Hydroxyquinoline-N-Oxide), a Potent and Selective Inhibitor of 5-Lipoxygenase, Produced by Pseudomonas methanica. J. Antibiot. 1986, 39 (8), 1160–1166. 10.7164/antibiotics.39.1160. [DOI] [PubMed] [Google Scholar]
- Kugelman M.; Liu W. C.; Axelrod M.; McBride T. J.; Rao K. V. Indicine-N-Oxide - Antitumor Principle of Heliotropium indicum. Lloydia 1976, 39 (2–3), 125–128. [PubMed] [Google Scholar]
- Machan Z. A.; Taylor G. W.; Pitt T. L.; Cole P. J.; Wilson R. 2-Heptyl-4-Hydroxyquinoline N-Oxide, an Antistaphylococcal Agent Produced by Pseudomonas aeruginosa. J. Antimicrob. Chemother. 1992, 30 (5), 615–623. 10.1093/jac/30.5.615. [DOI] [PubMed] [Google Scholar]
- Molyneux R. J.; James L. F. Loco Intoxication - Indolizidine Alkaloids of Spotted Locoweed (Astragalus lentiginosus). Science 1982, 216 (4542), 190–191. 10.1126/science.6801763. [DOI] [PubMed] [Google Scholar]
- Nicholas G. M.; Blunt J. W.; Munro M. H. Cortamidine Oxide, a Novel Disulfide Metabolite from the New Zealand Basidiomycete (Mushroom) Cortinarius Species. J. Nat. Prod. 2001, 64 (3), 341–344. 10.1021/np000408+. [DOI] [PubMed] [Google Scholar]
- Nishioka H.; Imoto M.; Imaoka T.; Sawa T.; Takeuchi T.; Umezawa K. Antitumor Effect of Piericidin B1 N-Oxide. J. Antibiot. 1994, 47 (4), 447–452. 10.7164/antibiotics.47.447. [DOI] [PubMed] [Google Scholar]
- Nishioka H.; Sawa T.; Isshiki K.; Takahashi Y.; Naganawa H.; Matsuda N.; Hattori S.; Hamada M.; Takeuchi T.; Umezawa K. Isolation and Structure Determination of a Novel Phosphatidylinositol Turnover Inhibitor, Piericidin B1 N-Oxide. J. Antibiot. 1991, 44 (11), 1283–1285. 10.7164/antibiotics.44.1283. [DOI] [PubMed] [Google Scholar]
- Rickards R. W.; Rothschild J. M.; Willis A. C.; de Chazal N. M.; Kirk J.; Kirk K.; Saliba K. J.; Smith G. D. Calothrixins A and B, Novel Pentacyclic Metabolites from Calothrix Cyanobacteria with Potent Activity against Malaria Parasites and Human Cancer Cells. Tetrahedron 1999, 55 (47), 13513–13520. 10.1016/S0040-4020(99)00833-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.