

Abstract
Analysis of catalytic activity of nucleic acid enzymes is crucial for many applications, ranging from biotechnology to the search for antiviral drugs. Commonly used analytical methods for quantifying DNA and RNA reaction products based on slab-gel electrophoresis are limited in throughput, speed, and accuracy. Here we report the optimization of high throughput methods to separate and quantify short nucleic acid reaction products using DNA sequencing instruments based on capillary electrophoresis with fluorescence detection. These methods afford single base resolution without requiring extensive sample preparation. Additionally, we show that the utility of our system extends to quantifying RNA products. The efficiency and reliability of modern instruments offers a large increase in throughput but complications due to variations in migration times between capillaries required us to develop a computer program to normalize the data and quantify the products for automated kinetic analysis. The methods presented here greatly increase sample throughput and accuracy and should be applicable to many nucleic acid enzymes.
Keywords: Capillary electrophoresis, DNA polymerase, DNA and RNA analysis, Quantification and analysis software, Sanger sequencer, High throughput
Subject Category: Enzymatic assays and analysis
Graphical Abstract
1. Introduction:
Nucleic acid enzymes play important roles in biology and biotechnology Understanding the catalytic mechanism, rates of conversion of substrate to product, and substrate selectivity of these enzymes is crucial in understanding their biological function and potential therapeutic benefits. Traditionally, slab-gel polyacrylamide gel electrophoresis (PAGE) has been the method of choice to separate radiolabeled nucleic acids for mechanistic enzyme analysis [1–7]. The method is customizable to separate nucleic acids of almost any size used for in vitro experiments and is relatively easy to set up. While isotope labeling methods have the advantage of being quite sensitive and artifact free, they also have many disadvantages including the exposure to ionizing radiation and the short half-life of commonly used isotopes, necessitating frequent radiolabeling to maintain a strong signal to noise ratio. Besides the use of radioactive isotopes, the major limitations for this method are the low throughput, slow separation, and lack of automation. Quantification of data from slab-gel PAGE also requires manual scanning and analysis which is time consuming and further limits the number of samples that can be analyzed and can introduce errors.
Capillary electrophoresis (CE) was first described in the literature in the late 1970’s [8, 9] and has competed to replace PAGE for nucleic acid analysis. In CE, nucleic acids that have a fluorescent label are separated by size and charge as they migrate through a capillary filled with a pumpable sieving polymer. Modern instruments are fully automated and have a polymer pump that fills the capillaries with the polymer mixture after each run in a matter of minutes. The large surface area in the capillary dissipates heat efficiently, allowing electrophoresis at high-voltage and therefore rapid run times [10]. One paper reports a method to sequence close to 1,000 bases in 40 minutes [11]. Furthermore, CE has the benefit of automated sample loading and fluorescence detection, eliminating the need for radioactive tracers. The greater throughput led CE to become the method employed to sequence the Human Genome [12, 13].
Around 15 years ago, our lab reported methods to adapt the potential advantages of CE to separate single nucleotide incorporation reaction products [14]. At that time however, most instruments were limited to a single capillary which bottlenecked throughput and required ~2.5 hours for separation of a 12-point time course, in part due to the long 57 cm capillary required to get good separation of reaction products. The lack of a capillary oven on the instrument necessitated addition of a large excess of unlabeled DNA complementary to the template strand to prevent re-annealing of the labeled primer and template, despite the presence of 8M urea or 100% formamide in the sample before injection [14]. Finally, the sensitivity of older instruments was reasonable, but did not rival radioactive methods. Ultimately this study was an important proof of concept, however the benefits did not outweigh the limitations relative to slab gel PAGE with the instrumentation available at the time.
Since our last efforts to adapt CE to analyze our samples, there have been significant improvements in the methods by utilizing CE instruments designed for Sanger sequencing that contain multiple capillaries in an array, an autosampler, and built in fluorescence detection [15]. A newer paper utilized an instrument containing 96 capillaries in the capillary array (3730XL Genetic Analyzer), greatly increasing the throughput from the single capillary instruments previously available [15]. This was achieved with a 36 cm capillary and the sensitivity was much better, requiring only a fraction of the material required on previous instruments to get good signal to noise. This study however required longer DNA substrates to achieve single base pair resolution, required an additional sample dilution or cleanup step, relied on commercial LIZ sizing standards as well as custom standards with the same fluorescent label used on the nucleic acid of interest for accurate sizing, and did not provide an automated method for data processing and visualization after integrating the peaks. Here we report our optimization of methods to separate short reaction products by capillary electrophoresis with single base resolution and without any sample cleanup. We also present software to automate analysis of integrated peak data and for use of any custom fluorescently labeled oligo as an internal standard, rather than commercial size standards. Finally, we added electropherogram alignment and visualization features to our software and validated our method with RNA as well as DNA.
2. Results:
2.1. Validation of CE methods against PAGE:
To test the utility of CE against denaturing slab gel PAGE to separate samples from reaction time course experiments, we collected samples at various times by rapid quench methods for a reaction involving the removal of a mismatch buried at the n-2 position in the primer terminus of a double stranded DNA substrate by the 3′—5′ proofreading exonuclease of T7 DNA polymerase (Figure 1). To monitor the progress of the reaction, the primer strand of the DNA substrate was synthesized with a 5′ 6-carboxyfluorescein (6-FAM) label which was shown to give identical rapid quench kinetics as 32P labeled DNA for this enzyme system (data not shown). Based on prior work (14) and the crystal structure of the enzyme, the primer was designed to be long enough so the FAM label did not interfere with DNA binding. The 6-FAM labeled DNA from one syringe was mixed with a large excess of T7 DNA polymerase from the other syringe to start the reaction and time points were quenched with EDTA from the quench syringe and collected in empty 1.5 ml plastic tubes. An aliquot of each sample was removed, mixed with Formamide Loading Dye and separated by denaturing PAGE. The resulting gel was scanned for 6-FAM fluorescence with a laser gel scanner to give the results shown in Figure 1A. The box method in ImageQuaNT was used to manually determine the intensity of each species as a function of time. Concentration of each species was obtained by dividing the intensity of a select band by the sum of the intensities of all bands at that time point and multiplying by the concentration of DNA used in the experiment. The open circles and dashed lines in Figure 1D show the results of the quantification of the experimental data by slab gel PAGE and resulting fits to 2 exponential burst equations for each DNA species.
Figure 1: Exonuclease validation experiment for PAGE versus CE.
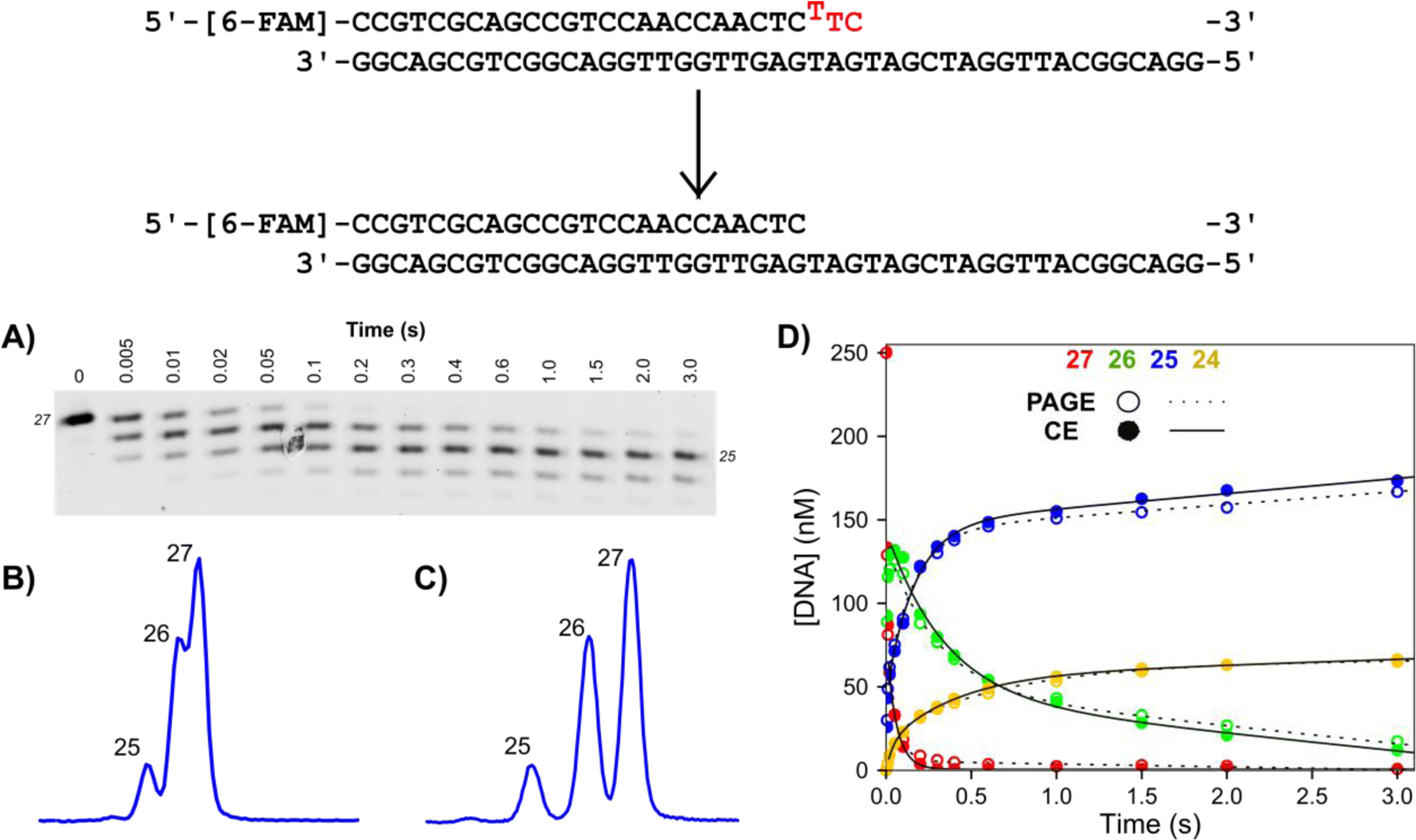
Scheme: The DNA substrate is a 27 nt, 5′-[6-FAM]-labeled primer annealed to the 45 nt template, with a buried T:T mismatch at the n-2 position. Bases to be removed are shown in red. Reaction conditions: A solution of 1.25 μM T7 DNA polymerase, 25 μM thioredoxin, 0.1 mg/ml BSA, and 12.5 mM Mg2+ was mixed with 250 nM FAM-labeled DNA from the other syringe in the quench flow at 20°C to start the reaction. Time points were quenched by mixing with EDTA from the quench syringe to a final concentration of 0.3 M. A) Analysis of time points by denaturing PAGE. Samples were mixed with formamide loading dye, separated by denaturing PAGE with 15% acrylamide/7M urea, and scanned on a Typhoon 9500 scanner with the FAM fluorescence filter. B) Separation of 5 ms time point by CE with POP-7 polymer. The same samples from (A) were injected at 1.6 kV for 15 seconds. Three peaks are visible, however lack of separation between products make quantification difficult. C) Separation of 5 ms time point by CE with POP-6 polymer. The same samples from (A) were injected with the same parameters as in (B). Each product is well resolved to near baseline, facilitating accurate peak integration. D) Overlay of data analyzed by PAGE versus CE. Data for products of varying lengths (27 nt, red; 26 nt, green; 25 nt, blue; 24 nt, yellow) for analysis by PAGE (open circles, dashed line) versus capillary electrophoresis (closed circles, solid line). Net rates and amplitudes obtained by fitting the data to a double exponential burst equation are identical for the two data sets within 2%.
Next, samples from the same experiment were analyzed by CE to compare with data derived from slab gel PAGE. Others have suggested that it is necessary to clean up samples before separation by CE to reduce the salt concentration to get good electrokinetic injection of the nucleic acid [11], however, such a step would greatly increase the cost and time required to analyze our samples by CE. Instead, quenched samples from the experiment described above were diluted ten-fold directly into highly deionized (HiDi) formamide and loaded into a 96 well plate. We used a 3130XL Genetic Analyzer instrument equipped with a 36 cm capillary array (16 capillaries). Injection parameters were optimized to get good injection without performing any further sample preparation until we obtained conditions that gave good injection, as indicated by the large fluorescence signal of the main products. Care was taken to ensure the peak intensities stayed in the linear range for the CCD camera (< 4,000 relative fluorescence units, RFU). The built-in capillary oven provided a constant temperature of 60°C during the separation to prevent reannealing of the primer and template strands. After a 3 minute of pre-run electrophoresis at 15 kV, samples were electrokinetically injected for 12 seconds at 1.6 kV. The run voltage was stepped to 15 kV over 40, 15 second steps then data collection was initiated. Data were collected for 800 seconds before ending the run. The polymer delivery pump replaced the polymer between each run. Our detailed run and injection conditions are listed in Table 2 and in the figure legends. After good injection without sample cleanup was obtained, we worked to optimize the separation capability of the system.
Table 2:
Capillary electrophoresis separation parameters
| Parameter | Value |
|---|---|
| Oven_Temp | 60–65°C |
| Poly_Fill_Vol | 6500 steps |
| Current_Stability | 5 μAmps |
| Pre_Run_Voltage | 15 kV |
| Pre_Run_Time | 180 seconds |
| Injection_Voltage | Variable |
| Injection_Time | Variable |
| Voltage_Number_of_Steps | 40 steps |
| Voltage_Step_Interval | 15 seconds |
| Data_Delay_Time | 1 second |
| Run_Voltage | 15 kV |
| Run_Time | 800 seconds |
Initially, samples were separated with Performance Optimized Polymer 7 (POP-7) which is the standard self-coating, pumpable polymer for long read sequencing runs traditionally performed on the ABI 3130XL Genetic Analyzer instrument. With POP-7 polymer installed on the instrument, three peaks were detected for the 0.005 second sample time point, as expected based on the PAGE analysis (Figure 1B). However, the separation between peaks was not sufficient for accurate peak integration. POP-6 is the most viscous of the commercial polymers designed for use on the Genetic Analyzer series of instruments and is commonly used to separate small nucleic acid fragments [10] so we chose to test the separation performance of this polymer on our samples. Samples were injected with the same parameters, but with POP-6 polymer filling the capillaries. This resulted in near baseline separation of the three reaction products (Figure 1C). The greater separation between peaks allowed accurate peak integration with GeneMapper analysis software. Peak integration was performed with the Microsatellite Default preset and a data set of 16 time points could be analyzed in a matter of seconds. A table of peak data (migration time, peak height, peak area, etc.) was exported from GeneMapper. The table was imported into Excel to manually calculate the fractional peak area for each peak at each time point. Since the identity of each peak in the electropherograms is known from PAGE, the size of each peak at each time point could be manually determined. The results of the quantification and fitting the resulting data to a 2 exponential burst equation are shown as the closed data points and solid lines respectively in Figure 1D. Importantly, only a very slight difference (less than 2% of amplitudes) between the data points for CE versus PAGE was detected, indicating the same information can be extracted from the samples regardless of whether they are analyzed by PAGE or CE. Additionally, single base resolution was achieved in a run cycle of only 25 minutes after the oven reaches temperature, including: filling the array with polymer, pre-run electrophoresis, sample injection, and separation. Analysis of a time course by PAGE in comparison took almost 6 hours to: cast the gel, preheat the gel, load samples, run the gel and scan, then integrate individual bands. For PAGE, analysis of the same samples on different gels gave a variation of 5% at most at 6-FAM-DNA concentrations of 200 nM in the reaction. Lower 6-FAM labeled DNA concentrations could be detected but produced significantly more fluorescence artifacts due to the higher gain on the PMT required to visualize the bands, resulting in higher errors. For CE the same samples were separated in different injections at reaction DNA concentrations down to 1 nM and the variation between the calculated product concentration was less than 3%. We have not quantified the variation at lower DNA concentrations however the limit of detection is at least 0.1 nM 6-FAM-DNA in the reaction, rivaling the sensitivity of experiments with 32P labeled DNA substrates.
2.2. Normalized migration time with Cy3 internal standard:
While the experiment above could be analyzed manually in Excel, careful attention to detail was required as migration times for identical products varies significantly between capillaries in the array (Figure S1) which greatly complicated the analysis. This method became quite time consuming with many samples with many peaks and intermediates that rise and fall. Typically, a set of internal standards labeled with a unique dye is used to determine the size of each peak relative to the standard. We found that use of commercial size standards was expensive and inaccurate due to the large effect of dye identity on the migration of short oligonucleotides [10]. Therefore, we opted for a single sulfindocyanine (Cy3) labeled oligo as an internal standard, with a much lower cost than commercial size standards and customizable to the size range for each experiment. The Cy3 fluorophore is compatible with the G5 dye filter set pre-programmed in the data collection software (fluorescence emission on the yellow channel) and the modification is easily added during commercial oligonucleotide synthesis.
We optimized the concentration of Cy3 internal standard added using a few principles. The concentration of internal standard was optimized to give a maximum of ~ 500 RFU while the FAM peaks were around 3,000 RFU. Using a low concentration of internal standard prevented any artifacts from preferential injection. We found that a final concentration of Cy3 internal standard between 0.8 and 2 nM in HiDi formamide gave the desired results when 10 μl of the formamide/internal standard solution was mixed with 1 μl of quenched reaction samples at concentrations between 35 and 125 nM. For a simple test of our method to normalize the migration time, we performed a single nucleotide incorporation experiment where dATP was added to a solution of T7 DNA polymerase and FAM labeled DNA and the amount of product formed as a function of time was monitored (Figure 2). The Cy3 internal standard oligo was added to the HiDi formamide to a concentration of 1.5 nM, before dispensing 10 μl into each well on the plate and adding 1 μl of quenched sample to each well. To correct for varying migration times between capillaries, the following formula was used: Xn,Corrected = 100 × (Xn/XIS), where Xn,Corrected is the corrected migration time of a peak of n nucleotides in length, XIS is the migration time of the Cy3 internal standard peak, and Xn is the migration time of the peak of n nucleotides in length (Figure 2A). We found that after this basic arithmetic, the corrected migration times for each product fell within relatively narrow ranges across all capillaries which greatly facilitated accurate sizing. While this method of standardization of peak migration time was effective, it was still time consuming for large data sets so further automation was warranted.
Figure 2: Single nucleotide incorporation experiment and internal standard normalization.
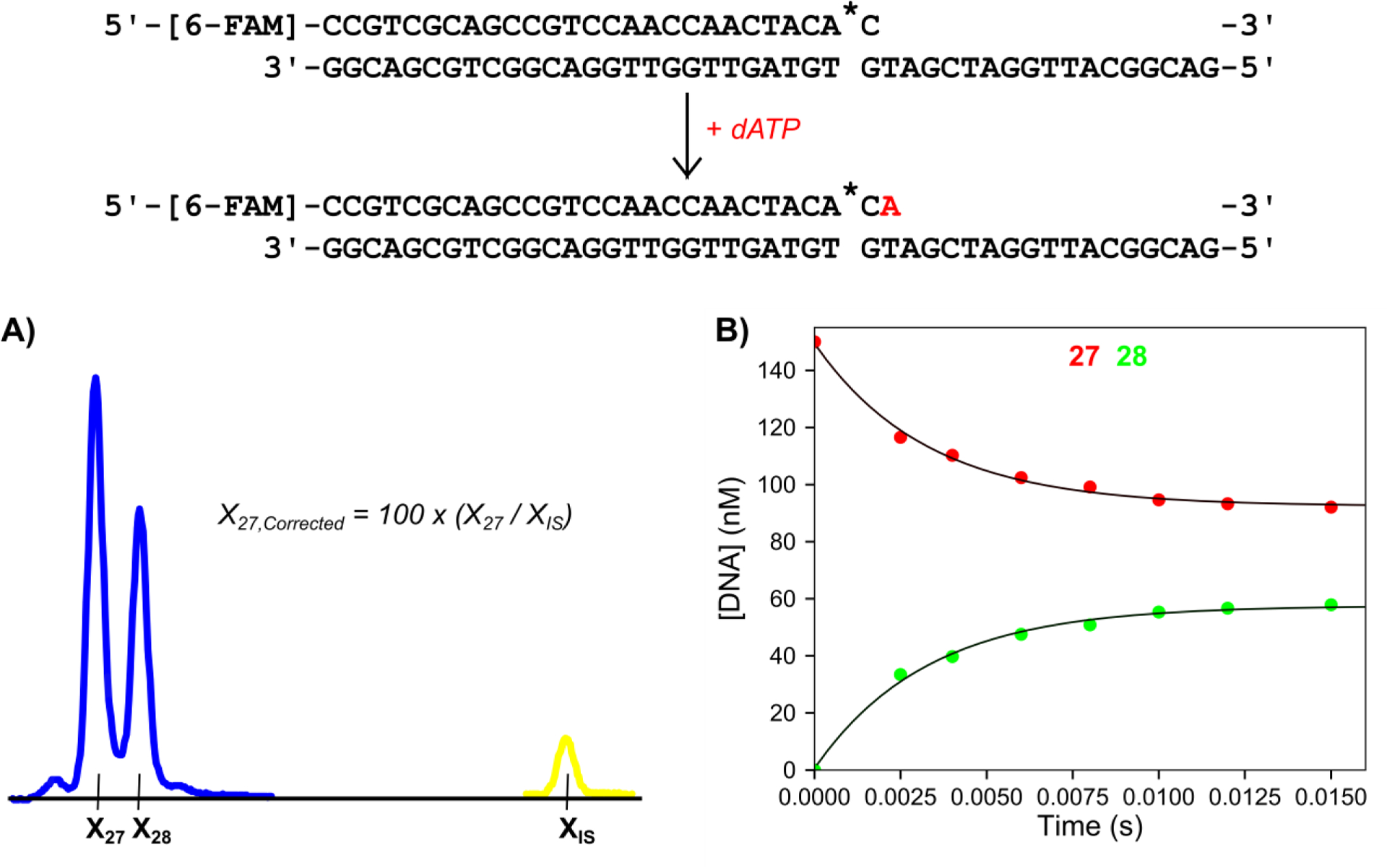
Scheme: The DNA substrate is a 27 nt, 5′-[6-FAM]-labeled primer containing a phosphorothioate linkage at the 3′ end (denoted by *), annealed to a 45 nt template strand. The added nucleotide is shown in red. Reaction conditions: A solution of 75 nM T7 DNA polymerase (exo−), 1.5 μM Thioredoxin, 0.1 mg/ml BSA, and 150 nM FAM-DNA was mixed with 100 μM dATP and 12.5 mM Mg2+ to start the reaction in the quench flow instrument at 20°C. Time points were quenched by mixing with EDTA from the quench syringe (0.3 M final concentration). A) Electropherogram showing the 15 ms time point with the Cy3 internal standard. Samples were injected for 15 seconds at 2.6 kV. Blue peaks correspond to FAM-labeled reaction products and the yellow peak corresponds to the Cy3-labeled internal standard. Xn is the migration time of the peak for DNA n nucleotides in length. XIS is the migration time of the Cy3 labeled internal standard oligo. The formula for the corrected migration time for the 27 nt peak is shown in the panel and is used to normalize the variable migration times between capillaries. B) Plot of DNA concentration versus time for varying length products derived from the burst experiment. The 27 nt primer is shown in red and the 28 nt primer is shown in green as a function of time. This representative data set is part of the included example data provided with the software. Both sets of data are shown fit to single exponential functions.
2.3. Analysis software:
To automate data processing following peak integration, we created analysis software that takes the output from peak identification and integration in GeneMapper and provides simple, reliable automation of data processing with an easy-to-use graphical user interface. Details about the packages used in programming the software are given in the methods section and a detailed instruction manual for the software can be found bundled with the software on the source code page. Here we will outline the principles and main features of the software. A link to the installer bundle can be found in the data availability statement at the end of the paper and contains everything needed to install the software on a computer with a Windows operating system, as well as example data files and an instruction manual. Most features are compatible with Macintosh systems however there are still some bugs. The software is provided as open-source so users can easily optimize it for their use.
Formatting the data for compatibility use with our analysis software begins at the data collection phase. First, a properly formatted results group must be created with the Results Group Editor in the 3130XL Data Collection Software. Recommended settings for this tab are shown in Figure S2 but most importantly must include an underscore as the delimiter in the file name alone with and the sample name. Next the samples are entered into the Plate Manager. In the Plate Editor, the important columns and information to enter are as follows: for Sample Name, include experiment time point; for Analysis Method, select “Microsatellite Default;” for Results Group, choose the results group made above; for Instrument Protocol, choose the instrument protocol created based on the parameters in Table 2. The plate can then be analyzed. After the data are collected, peak detection and integration are performed in GeneMapper software. First the samples are added to the project and analyzed to detect peaks and perform peak integration. The data for the blue and yellow dye channels are then exported and saved as a tab delimited text file containing for each peak: dye channel, peak area, peak height, and data point (migration time). The exported .txt file can then be analyzed in our software to normalize the migration time relative to the standard and calculate the fraction of material in each peak.
The first panel of the software calculates the fractional area and size for each peak. The time point corresponding to each set of peaks is determined by entering the number of underscores (previously entered in the results group) before the time point data, as explained in the instruction manual. Noise which was identified as a peak can then be filtered out for the FAM peaks of interest and/or for the internal standard peaks by selecting a minimum height filter threshold for the peaks. Next, normalized migration times for each FAM peak for each time point are calculated to begin sizing and to minimize issues of varying migration times between capillaries. Our internal standard elutes much later than the FAM peaks so using the equation in Figure 2A, larger oligonucleotides have larger corrected migration times than shorter oligos. The software provides a list of all corrected difference ranges which is then used to locate the difference range corresponding to each polymer. Difference ranges for each polymer are then either entered manually into the software or ranges can be imported from an Excel file to save time. Difference ranges for specific oligos can be determined by injection of unreacted oligos of known length, or a time point can be analyzed by PAGE with standards and then peaks can be assigned to difference ranges. Care must be taken in this step but once these ranges are determined one time, they can be used over and over again for experiments with the same starting oligo. Similar steps are required to ensure accurate sizing using the LIZ standard so this is not a disadvantage of our method. Data are then processed in the software to return the fractional area and size of each polymer at each time point. Finally, the concentration of nucleic acid in the experiment is entered to scale the fractional area to give concentrations of each species. The software can then export a text file containing concentration and size versus time data that can be directly used in various data fitting software packages or output as a publication-quality figure directly from the software.
2.4. Electropherogram alignment / figure preparation with analysis software.
Raw electropherograms can be viewed easily in Gene Mapper however the .fsa file format of the electropherograms makes manual electropherogram manipulation and alignment difficult. Raw electropherograms can also be viewed with our software without performing the fractional area versus size calculations described above, however peaks cannot be aligned nor intensity normalized without first performing fractional area and size analysis. If fractional area and sizing is performed first, the raw electropherograms can be corrected with similar principles as were used for sizing and fractional area determination. The software reads the raw .fsa files and aligns the internal standard peaks to have the same migration time for each time point. Since injection efficiency is variable between capillaries, we programmed a feature to modify the raw RFU intensity to fractional area which makes visualization much easier. Finally, we added a feature to display the electropherograms in a 3D plot, containing the time point of the electropherogram on the X-axis, the normalized migration time on the Y-axis, and the fractional area on the Z-axis (Figure 3). Additionally, time on the X-axis can be shown on a logarithmic scale to better visualize experiments where time points are skewed towards shorter reaction times (not shown).
Figure 3: 3-D plot of aligned and normalized single nucleotide incorporation experiment electropherograms.
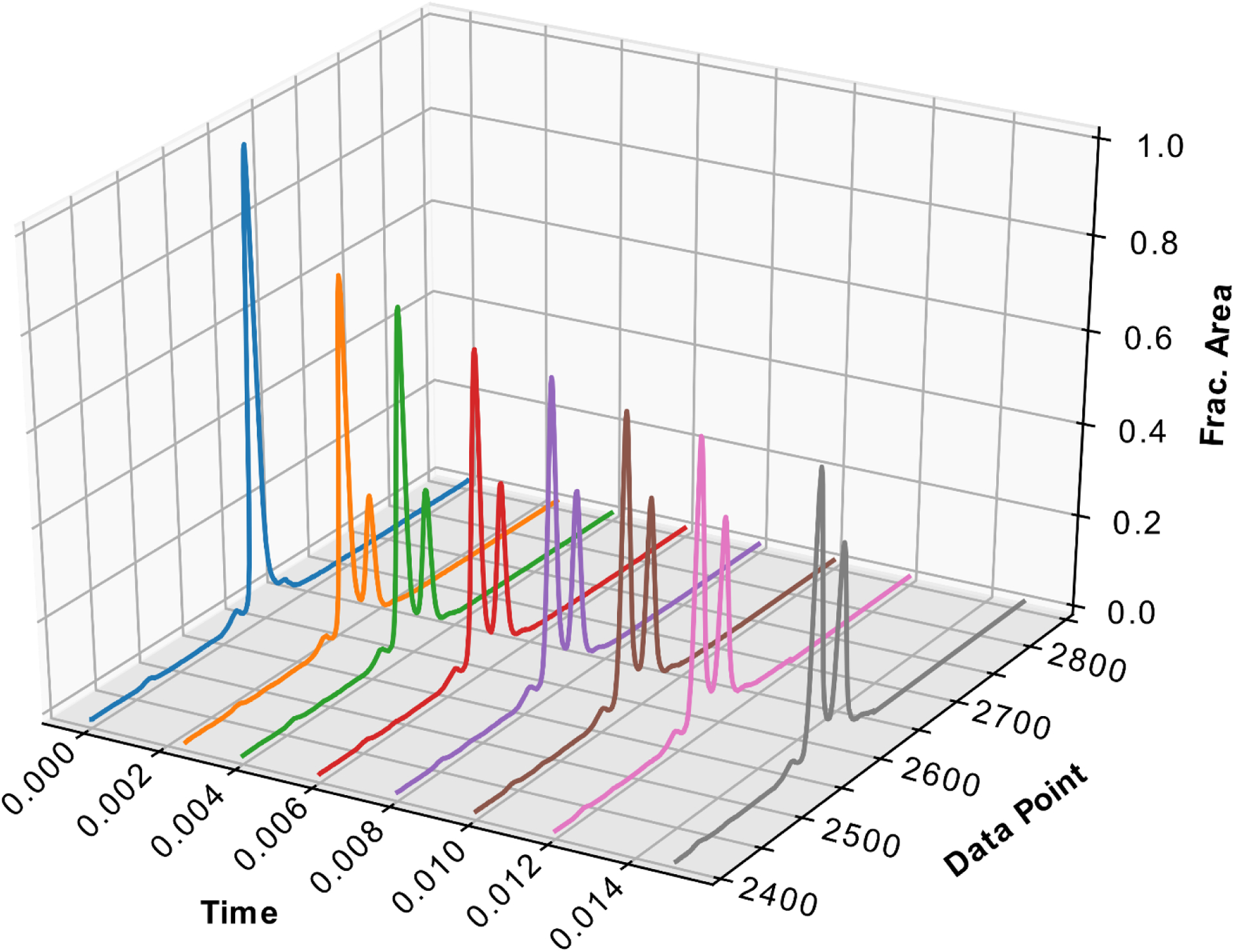
Data is from the experiment described in Figure 2. Time is shown on the X axis, “Data Point” shown on the Y-axis represents the normalized migration time (relative to the internal standard), and the fractional area normalized to peak height is shown on the Z-axis. The normalized migration time was calculated by the analysis software to correct for the differences in migration time between capillaries. Fractional area was calculated in the analysis software to determine a scaling factor for each electropherogram.
2.5: Separation of RNA by capillary electrophoresis:
To test the utility of our method for RNA dependent enzymes, we performed experiments with the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) RNA dependent RNA polymerase (RdRp) complex. RNA is prone to reannealing during electrophoresis which can complicate analysis. We wanted to see if we could analyze short RNA substrates by CE without artifacts. As a test experiment, a solution of SARS-CoV-2 RdRp complex, and a 5′-[6-FAM] labeled 20 nt RNA primer annealed to a 40 nt RNA template was mixed with UTP and the triphosphate active form of the antiviral drug Remdesivir (RTP) which acts as an ATP analog, extending the primer by 5 bases (Figure 4). Samples were prepared as above by dilution into formamide/Cy3 internal standard and analyzed with similar instrument parameters but with the oven temperature set to a slightly higher 65°C. Figure 4A shows the separation of the products of the short (20 – 27 nt) RNA primer and the separation between the Cy3 internal standard and the FAM RNA peaks. We selected this particular electropherogram as it has the most peaks and is therefore the most difficult to resolve. Since the amplitude of the 21 nt product is low, it appears to blend with the 20 nt peak. Peak integration / quantification however is minimally affected by this lack of resolution since the 21 nt peak accounts for only a small fraction of the total peak area for this time point. No additional peaks corresponding to dsRNA were observed indicating that the run conditions were sufficient to completely denature the RNA. The analysis software was used as above to provide the RNA species concentration versus time graph in Figure 4B.
Figure 4: Validation of RNA separation and analysis by CE.
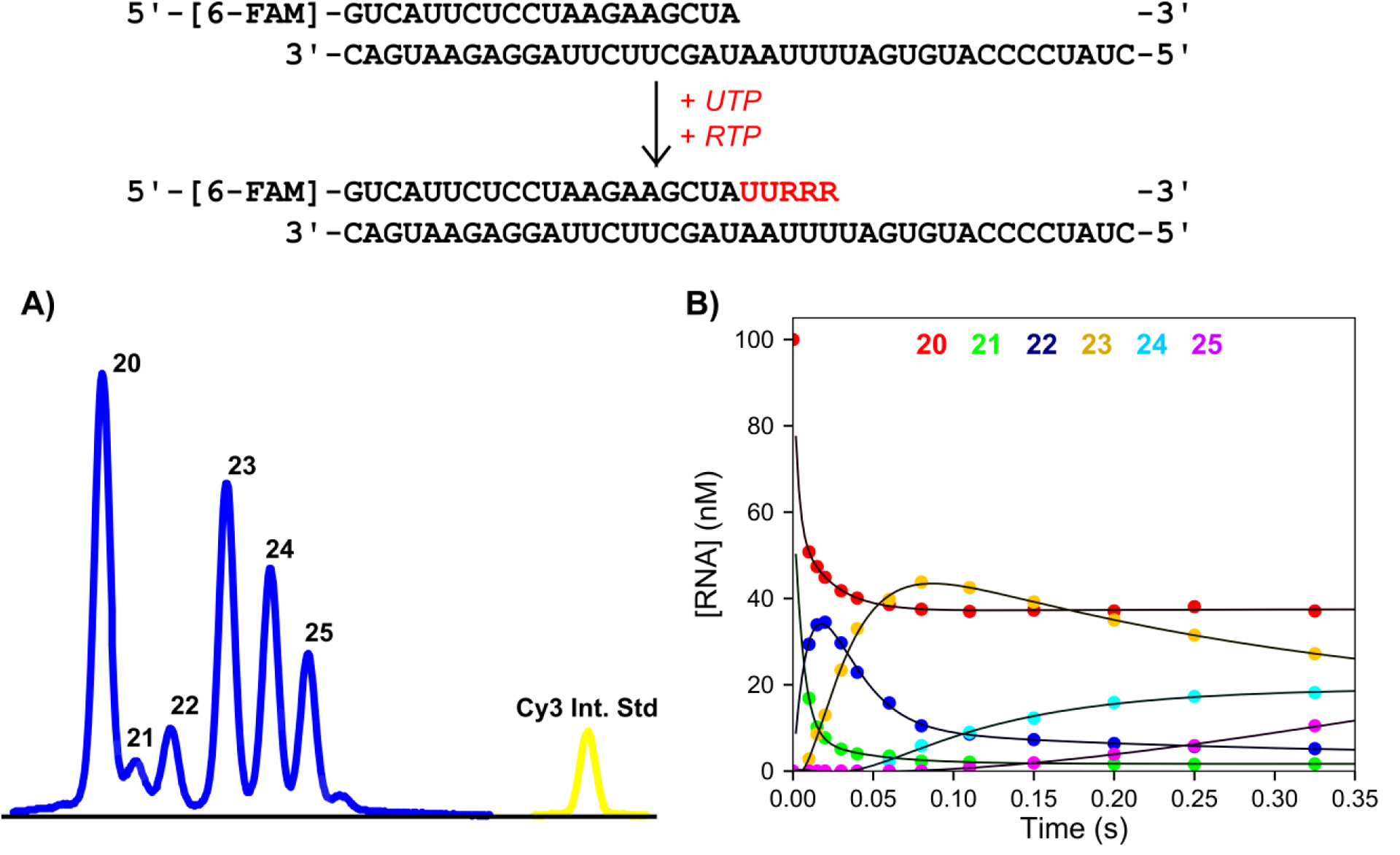
Scheme: The RNA substrate used in the kinetics assay is shown, consisting of a 20 nt, 5′-[6-FAM]-labeled primer annealed to a 40 nt template. UTP and Remdesivir triphosphate were added and the extended RNA bases are shown in red. Experimental conditions: A solution of 1.5 μM SARS CoV-2 RdRp (NSP12/7/8 plus 6 μM NSP8), 100 nM FAM-RNA, and 5 mM Mg2+ was mixed with 150 μM UTP and 40 μM Remdesivir-triphosphate to start the reaction in the quench flow at 37°C. Time points were quenched by mixing with EDTA from the quench syringe to 0.3 M. A) Sample electropherogram for a 0.325 second time point. Samples were injected for 6 seconds at 3.6 kV. Blue peaks correspond to FAM-labeled RNA products, while the yellow peak corresponds to the Cy3 internal standard. B) Concentration of RNA versus time. Data for different species were determined using the analysis software and are colored as listed in the top of the panel. Data are shown fit to a double exponentials function.
3. Discussion:
For our kinetic analysis of DNA polymerases, we have traditionally used radiolabeled nucleic acid substrates and relied on tedious and time-consuming slab gel PAGE methods to resolve and quantify products. Therefore, we sought to find new methods for analysis of our samples. Since our previous attempt to use CE, instrumentation has improved significantly warranting further investigation into this method. In our previous studies [14], we were greatly limited by the instrumentation available to us at the time. CE technology has greatly improved since those efforts and now instruments optimized for Sanger sequencing are highly sensitive to multiple dyes, contain a capillary oven to improve sample denaturation, and contain up to 96 capillaries to greatly improve throughput. Despite significant advances, most studies on these instruments are geared toward sequencing longer DNA fragments and few report quantitative results. One study, however, showed good quantitative single base pair separation of slightly longer DNA substrates (50 nt) on an instrument containing 96 capillaries [15]. Our aim was to build on these results by optimizing the method to achieve single base pair resolution of short nucleic acid substrates commonly used in enzymology experiments, and automate peak sizing and processing following peak detection and integration.
We show that using POP-6 instead of POP-7 allows single base resolution of short nucleic acid substrates commonly used in our experiments. As with many other systems that go from low throughput to high throughput, manual analysis methods were no longer a reasonable option due to the large quantities of data produced. Automating the process with our analysis software was key to enjoying the benefits of the increased throughput. Our analysis software circumvents problems with peak sizing caused by variable migration times between capillaries by using a Cy3 internal standard in each sample to normalize migration time. This greatly facilitates speed and accuracy of sizing and analysis. Our software also aligns raw electropherograms from the sequencer and allows peak height to be normalized to fractional area. Electropherograms can then be displayed in multiple 2D plots or a single 3D plot containing multiple electropherograms (Figure 3).
We were able to purchase a refurbished 16 capillary instrument for a fraction of the cost of a new 96 capillary instrument. The 16 capillary instrument was more than adequate for our studies, although these methods will likely work just as well on newer instruments with more capillaries as the raw data and peak integration files are similar. For the reactions shown here, we used the blue channel to monitor 6-FAM fluorescence and the yellow channel to monitor Cy3 fluorescence for the internal standard. The most recent version of our software however allows use of any dye for both the oligo of interest and the internal standard oligonucleotide. Additional modifications to the software could also be included in the future to analyze reactions where dual labeled substrates are analyzed relative to an internal standard. Furthermore, coupling the techniques presented here with mass spec to analyze oligos as they come out of the capillary could allow oligo sizing without the use of an internal standard. The open-source software we provide can be easily adapted to new protocols.
4. Materials and methods
4.1. Enzymes, reagents, and oligonucleotides:
The wild-type exo+ and exo− (D5A E7A) T7 DNA polymerase were expressed in E. coli and purified with a polyethyleneimine precipitation step followed by three chromatography steps, as described previously [16]. Thioredoxin was expressed in E. coli and purified with a heat denaturation step followed by two chromatography steps, as described previously [16]. SARS-CoV-2 RNA dependent RNA polymerase complex proteins were expressed in E. coli and purified with multiple chromatography steps, as previously described [17]. Bovine serum albumin (BSA), dNTPs and NTPs were purchased from New England Biolabs. GS-443902 (Remdesivir triphosphate, RTP) was provided by Gilead Sciences Inc. and the concentration of RTP was determined by absorbance at 245 nm using the extinction coefficient 24,100 M−1 cm−1, determined by NMR with formamide as an internal standard. Buffer components and Ethylenediaminetetraacetic acid (EDTA) were from Fisher Scientific. Boric acid, acrylamide, and N,N′-methylenebisacrylamide were from Sigma Aldrich. Highly deionized (HiDi) Formamide, performance optimized polymer (POP)-6, and POP-7 were purchased from ThermoFisher Scientific. HiDi Formamide was aliquoted and stored at −80°C until use. nanoPOP Buffer (10×) with EDTA was purchased from Molecular Cloning Laboratories. Diethyl pyrocarbonate treated water was used in all experiments with RNA. Oligonucleotides used in this study are listed in Table 1, along with their respective extinction coefficients at 260 nm, provided by the manufacturer. All oligos were synthesized by Integrated DNA Technologies. The Cy3 internal standard oligo was purchased with HPLC purification and the RNA oligos were purchased with RNase-free HPLC purification. All other oligonucleotides were purchased with standard desalting then purified in house by denaturing PAGE to greater than 98% full length oligo. The phosphorothioate oligo was purchased as a racemic mixture of the two diastereomers. All DNA oligos were resuspended in 66.2 Buffer (6 mM Tris-HCl pH 7.5, 6 mM NaCl, 0.2 mM EDTA) before storing at −20°C. Double stranded DNA substrates were prepared by mixing primer and template strands at a 1:1.05 molar ratio in DNA Annealing Buffer (10 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA) before heating to 95°C for 3 minutes and cooling slowly to room temperature over 1 hour. RNA oligo stocks were resuspended in RNA annealing buffer (10 mM Tris-HCl pH 7, 50 mM NaCl, 0.1 mM EDTA) and stored at −20°C. Double stranded RNA substrates were prepared by mixing the primer and template strands at a 1:1 molar ratio in RNA annealing buffer, heating to 75°C for 3 minutes, then cooling to room temperature over approximately 1 hour.
Table 1:
Oligonucleotides used in this study
| Oligo Name | DNA / RNA | Sequence 5’ -> 3’ | Extinction Coefficient, 260 nm (M−1 cm−1) |
|---|---|---|---|
| Cy3 Int. Std. | DNA | [Cy3]-CCGTGAGTTGGTTGGACGGCTGCGAGGC | 266,800 |
| FAM-27 | DNA | [6-FAM]-CCGTCGCAGCCGTCCAACCAACTCAAC | 266,660 |
| FAM-27-PThio | DNA | [6-FAM[-CCGTCGCAGCCGTCCAACCAACTACA*C | 267,660 |
| 45-Buried MM | DNA | GGACGGCATTGGATCGATGTAGAGTTGGTTGGACGGCTGCGACGG | 439,600 |
| 45mer | DNA | GGACGGCATTGGATCGATGTGTAGTTGGTTGGACGGCTGCGACGG | 435,100 |
| FAM-20 | RNA | [6-FAM]-GUCAUUCUCCUAAGAAGCUA | 222,360 |
| 40mer | RNA | CUAUCCCCAUGUGAUUUUAAUAGCUUCUUAGGAGAAUGAC | 403,100 |
phosphorothioate linkage
4.2. Kinetics experiments:
Experiments with T7 DNA polymerase were performed in T7 Reaction Buffer (40 mM Tris-HCl pH 7.5, 1 mM EDTA, 12.5 mM MgCl2, 1 mM DTT, 50 mM NaCl) [18] at 20 °C. Experiments with SARS-CoV-2 RNA dependent RNA polymerase complex were performed in SARS-CoV-2 Reaction Buffer (40 mM Tris-HCl pH 7, 5 mM MgCl2, 1 mM DTT, 50 mM NaCl) [17] at 37°C. Quench flow experiments were performed on a KinTek RQF-3 rapid quench instrument with a circulating water bath set to the appropriate reaction temperature. Reaction buffer without magnesium was loaded into the two drive syringes and 0.6 M EDTA was loaded into the quench syringe. All components were allowed to equilibrate for at least 30 minutes on ice before performing the experiment. All experiments were repeated to ensure reproducibility and a single representative data set is shown in each figure.
4.3. PAGE analysis of samples:
Samples for PAGE were diluted with Formamide Loading Dye (5% sucrose, 90% formamide, 10 mM EDTA, 0.025 % [w/v] bromophenol blue, 0.025 % [w/v] xylene cyanol) at a ratio of 1:2 loading dye to sample. The samples were then heat-denatured for 3 min at 95 °C and loaded onto a 15% denaturing polyacrylamide gel (7 M urea), preheated to 50°C using the Bio-Rad Sequi-Gen GT gel apparatus. Electrophoresis was carried out at 100 watts at a constant temperature of 50°C for approximately 3 hours. Gels were scanned on a Typhoon FLA 9500 laser scanner (GE Healthcare) with the FAM fluorescence filter. Products at each time point were quantified with ImageQuant software (GE Healthcare).
4.4. Capillary gel electrophoresis:
Capillary electrophoresis was performed on an Applied Biosystems 3130XL Genetic Analyzer instrument (equipped with a 36 cm capillary array with 16 capillaries), purchased as a refurbished instrument from ThermoFisher Scientific. Data collection was managed with 3130XL Series Data Collection Software 4. A custom run module was created from the template RapidSeq36_POP6 using the run parameters in Table 2. A custom instrument protocol was then created with the custom run module and the G5 dye set. Run parameters were the same for each experiment, with the exception of the oven temperature which was set to 60°C for all DNA experiments and 65°C for RNA experiments. Injection parameters varied based on the concentration of nucleic acid in each experiment and injection parameters for each experiment are given in the figure legends. The Cy3 internal standard oligo was diluted from a 200 nM stock to a final concentration between 0.8 and 2 nM in HiDi Formamide, depending on the concentration of nucleic acid in the experiment. Ten μl of the formamide/internal standard mixture was added to each well. Samples were diluted by adding 1 μl of quenched sample to the 10 μl of formamide/internal standard mixture. Heat denaturation of the samples before injection was not necessary to completely denature the nucleic acids.
4.5. Analysis software:
The analysis software was written in Python 3 using packages tailored for bioinformatics, including matplotlib v3.3.3 [19], Pandas v1.15 [20], and Biopython v1.78 [21]. The .txt files exported from GeneMapper contained the peak areas, peak heights, and data points (migration times) needed for plotting and analysis. Peak data was extracted and manipulated using the Pandas package. Given that the exported text files from Gene Mapper contained only peak areas and not raw data, Biopython was used to extract x and y values for each point in the electropherogram from the .fsa files. Specifically, the Bio.SeqIO module gave all the raw values needed for plotting. Peak alignment was done by first assigning a reference internal standard peak. Each non-internal standard product peak could then be vectorized to shift chromatographs in alignment relative to the reference internal standard peak. After alignment, the processed raw data was imported into matplotlib to prepare 2D and 3D plots.
4.6. Data analysis:
Data fitting was performed with the simulation software, KinTek Explorer [22, 23] v9 (www.kintekexplorer.com). This software was also used in preparing figures for kinetic data. Quench flow data were fit in the software using the afit function to either a single exponential, double exponential, or double exponential burst equation. The equation for a single exponential used is , where A0 is the y-intercept, A1 is the amplitude, b1 is the decay rate and t is time. The equation for a double exponential used is , where A0 is the y-intercept, A1, A2, b1, b2 are the amplitudes and decay rates for the first and second phases, respectively. The equation for a double exponential burst used is , where A0 is the y-intercept, A1, A2, b1, b2 are the amplitudes and decay rates for the first and second phases, respectively, and b3 is the rate of the linear phase.
Supplementary Material
Improved separation of short FAM labeled oligonucleotides by CE.
Customizable Cy3 internal standard for more accurate sizing of short nucleic acids
New open source software presented to automate data analysis and visualization
Validation experiments show method is compatible with both DNA and RNA
Acknowledgements:
This work was supported by grants from NIGMS (R01GM114223 and R01AI110577 to KAJ) and the Welch Foundation (F-1604 to KAJ)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement:
The Sequencer analysis software has been licensed with GNU General Public Licensing v3.0 and is freely available at https://github.com/nafanh/DNA-Sequencer-Kinetic-Analysis/releases/tag/v2.0
Conflict of interest:
KAJ is president of KinTek Corporation, which provided the RQF-3 rapid quench-flow instrument, and KinTek Explorer software used in this study.
References
- [1].Donlin MJ, Patel SS, Johnson KA, Kinetic partitioning between the exonuclease and polymerase sites in DNA error correction, Biochemistry, 30 (1991) 538–546. [DOI] [PubMed] [Google Scholar]
- [2].Capson TL, Peliska JA, Kaboord BF, Frey MW, Lively C, Dahlberg M, Benkovic SJ, Kinetic characterization of the polymerase and exonuclease activities of the gene 43 protein of bacteriophage T4, Biochemistry, 31 (1992) 10984–10994. [DOI] [PubMed] [Google Scholar]
- [3].Kati WM, Johnson KA, Jerva LF, Anderson KS, Mechanism and fidelity of HIV reverse transcriptase, Journal of Biological Chemistry, 267 (1992) 25988–25997. [PubMed] [Google Scholar]
- [4].Reddy MK, Weitzel SE, von Hippel PH, Processive proofreading is intrinsic to T4 DNA polymerase, Journal of Biological Chemistry, 267 (1992) 14157–14166. [PubMed] [Google Scholar]
- [5].Jia Y, Patel SS, Kinetic Mechanism of Transcription Initiation by Bacteriophage T7 RNA Polymerase, Biochemistry, 36 (1997) 4223–4232. [DOI] [PubMed] [Google Scholar]
- [6].Anand VS, Patel SS, Transient State Kinetics of Transcription Elongation by T7 RNA Polymerase, Journal of Biological Chemistry, 281 (2006) 35677–35685. [DOI] [PubMed] [Google Scholar]
- [7].te Velthuis AJW, Arnold JJ, Cameron CE, van den Worm SHE, Snijder EJ, The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent, Nucleic acids research, 38 (2010) 203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mikkers FEP, Everaerts FM, Verheggen TPEM, High-performance zone electrophoresis, Journal of Chromatography A, 169 (1979) 11–20. [Google Scholar]
- [9].Jorgenson JW, Lukacs KD, Zone electrophoresis in open-tubular glass capillaries, Analytical Chemistry, 53 (1981) 1298–1302. [PubMed] [Google Scholar]
- [10].Wenz H, Robertson JM, Menchen S, Oaks F, Demorest DM, Scheibler D, Rosenblum BB, Wike C, Gilbert DA, Efcavitch JW, High-precision genotyping by denaturing capillary electrophoresis, Genome research, 8 (1998) 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kotler L, He H, Miller AW, Karger BL, DNA sequencing of close to 1000 bases in 40 minutes by capillary electrophoresis using dimethyl sulfoxide and urea as denaturants in replaceable linear polyacrylamide solutions, ELECTROPHORESIS, 23 (2002) 3062–3070. [DOI] [PubMed] [Google Scholar]
- [12].Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng J-F, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen H-C, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JGR, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AFA, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang S-P, Yeh R-F, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Patrinos A, Morgan MJ, C. International Human Genome Sequencing, C.f.G.R. Whitehead Institute for Biomedical Research, C. The Sanger, C. Washington University Genome Sequencing, U.D.J.G. Institute, C. Baylor College of Medicine Human Genome Sequencing, R.G.S. Center, Genoscope, U.M.R. Cnrs, I.o.M.B. Department of Genome Analysis, G.T.C.S. Center, C. Beijing Genomics Institute/Human Genome, T.I.f.S.B. Multimegabase Sequencing Center, C. Stanford Genome Technology, T. University of Oklahoma’s Advanced Center for Genome, G. Max Planck Institute for Molecular, L.A.H.G.C. Cold Spring Harbor Laboratory, G.B.G.R.C.f. Biotechnology, G. *Genome Analysis, U.S.N.I.o.H. Scientific management: National Human Genome Research Institute, C. Stanford Human Genome, C. University of Washington Genome, K.U.S.o.M. Department of Molecular Biology, D. University of Texas Southwestern Medical Center at, U.S.D.o.E. Office of Science, T. The Wellcome, Initial sequencing and analysis of the human genome, Nature, 409 (2001) 860–921.11237011 [Google Scholar]
- [13].Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X, The sequence of the human genome, Science, 291 (2001) 1304–1351. [DOI] [PubMed] [Google Scholar]
- [14].Hanes JW, Johnson KA, Analysis of single nucleotide incorporation reactions by capillary electrophoresis, Analytical Biochemistry, 340 (2005) 35–40. [DOI] [PubMed] [Google Scholar]
- [15].Greenough L, Schermerhorn KM, Mazzola L, Bybee J, Rivizzigno D, Cantin E, Slatko BE, Gardner AF, Adapting capillary gel electrophoresis as a sensitive, high-throughput method to accelerate characterization of nucleic acid metabolic enzymes, Nucleic Acids Res, 44 (2016) e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dangerfield TL, Johnson KA, Optimized incorporation of an unnatural fluorescent amino acid affords measurement of conformational dynamics governinghigh-fidelity DNA replication, Journal of Biological Chemistry, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dangerfield TL, Huang NZ, Johnson KA, Remdesivir Is Effective in Combating COVID-19 because It Is a Better Substrate than ATP for the Viral RNA-Dependent RNA Polymerase, iScience, 23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Patel SS, Wong I, Johnson KA, Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant, Biochemistry, 30 (1991) 511–525. [DOI] [PubMed] [Google Scholar]
- [19].Hunter JD, Matplotlib: A 2D Graphics Environment, Computing in Science & Engineering, 9 (2007) 90–95. [Google Scholar]
- [20].McKinney W, Data Structures for Statistical Computing in Python, Proceedings of the 9th Python in Science Conference 2010, pp. 51–56. [Google Scholar]
- [21].Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL, Biopython: freely available Python tools for computational molecular biology and bioinformatics, Bioinformatics, 25 (2009) 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Johnson KA, Fitting enzyme kinetic data with KinTek global kinetic explorer, Methods in enzymology, 467 (2009) 601–626. [DOI] [PubMed] [Google Scholar]
- [23].Johnson KA, Simpson ZB, Blom T, Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data, Analytical biochemistry, 387 (2009) 20–29. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.