

Summary
LPIN1 mutations are responsible for inherited recurrent rhabdomyolysis, a life-threatening condition with no efficient therapeutic intervention. Here, we conduct a bedside-to-bench-and-back investigation to study the pathophysiology of lipin1 deficiency. We find that lipin1-deficient myoblasts exhibit a reduction in phosphatidylinositol-3-phosphate close to autophagosomes and late endosomes that prevents the recruitment of the GTPase Armus, locks Rab7 in the active state, inhibits vesicle clearance by fusion with lysosomes, and alters their positioning and function. Oxidized mitochondrial DNA accumulates in late endosomes, where it activates Toll-like receptor 9 (TLR9) and triggers inflammatory signaling and caspase-dependent myolysis. Hydroxychloroquine blocks TLR9 activation by mitochondrial DNA in vitro and may attenuate flares of rhabdomyolysis in 6 patients treated. We suggest a critical role for defective clearance of oxidized mitochondrial DNA that activates TLR9-restricted inflammation in lipin1-related rhabdomyolysis. Interventions blocking TLR9 activation or inflammation can improve patient care in vivo.
Keywords: rhabdomyolysis, inflammation, Toll-like receptor 9, autophagosome, late endosome, mitochondrial quality control, hydroxychloroquine, lipin1
Graphical abstract
Highlights
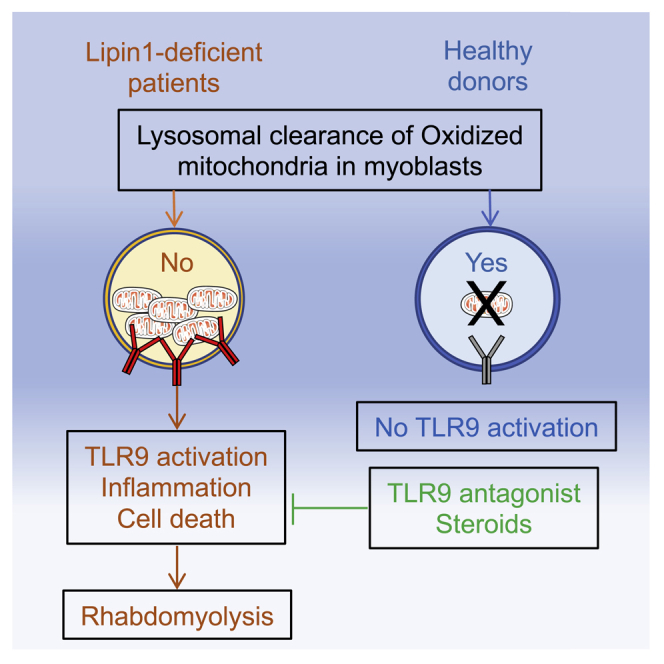
Lipin1 deficiency causes mitophagosome and late endosome dysfunction in human myoblasts
Damaged mitochondria trigger TLR9-mediated inflammation and myoblast death
Mitochondrial DNA elimination or blockade of TLR9 activation prevent myoblast death
Hydroxychloroquine sulfate treatment may prevent rhabdomyolysis episodes in patients
Hamel et al. investigate the mechanism underlying life-threatening rhabdomyolysis in human lipin1 deficiency. They find that endosomal accumulation of mitochondrial DNA in myoblasts induces inflammation and cell death mediated by TLR9 activation in vitro. Treatment of 6 patients with hydroxychloroquine sulfate suggests benefits in preventing rhabdomyolysis episodes.
Introduction
LPIN1 mutations are a common cause of severe and recurrent inherited rhabdomyolysis,1 a disease with dramatic outcome2,3 for which no treatment is available. LPIN1 encodes lipin1, an enzyme expressed in metabolically active tissues that regulates key metabolic pathways, such as adipocyte differentiation and lipid metabolism, nuclear envelope and mitochondrial dynamics, and vacuole fusion.4, 5, 6, 7
Although mouse models in which lipin1 is knocked out selectively in the skeletal muscle have been generated,8,9 important gaps remain in our understanding of the mechanisms linking human LPIN1 mutations to acute rhabdomyolysis. These rodent models exhibit chronic myopathy mainly resulting from the loss of phosphatidate phosphatase-1 (PAP-1) activity and the subsequent remodeling in lipid composition and a defect in fatty acid oxidation, which cause endoplasmic reticulum (ER) stress triggered by lipostasis and the activation of the unfolded protein response. Nonetheless, we found minor alterations in lipid composition and in the expression of key genes regulated by lipin1 in white adipose tissue and muscle from patients, despite a significant decrease in PAP-1 activity,10,11 suggesting that the lack of lipin1 co-transcriptional activity for genes involved in the regulation of adipogenesis and mitochondrial fatty acid oxidation12,13 plays a minor role in human disease.
Fasting and febrile illness are common triggers, suggesting that lipin1 may be involved in the regulation of autophagy and inflammation in humans. In line with this, mouse lipin1 deficiency leads to a reduction in diacylglycerol (DAG) levels and impaired activation of the protein kinase D (PKD)-Vps34 phosphatidylinositol-3 kinase (PI3K) signaling cascade, perturbing the normal maturation of autolysosomes and degradation of damaged mitochondria.14 Contradictory information is available connecting lipin1 with inflammation. Silencing lipin1 expression in human macrophages attenuates the response to endotoxin.15 Moreover, inflammatory infiltrates are absent from muscle biopsies of patients.16 However, lipin1 represses NFATc4 transcriptional activity in adipocytes, thus inhibiting the secretion of inflammatory factors.17
Here, we show that lipin1 synthesizes phosphatidylinositol-3 phosphate (PI3P) selectively at the membrane of autophagosomes and late endosomes, promoting their fusion with hydrolase-containing lysosomes, which is critical for efficient mitochondrial quality control and the resolution of inflammation mediated by Toll-like receptor 9 (TLR9). Blocking TLR9 activation prevents inflammation and myolysis in vitro and may suppress or attenuate rhabdomyolysis flares in patients.
Results
Lipin1 regulates the level of PI3P at the membrane of late endosomes in human myoblasts
To determine whether human lipin1 was implicated in autophagy, we knocked down its expression using a lentivirus expressing a short hairpin RNA (shRNA) targeting lipin1 (sh-LPIN1). In control myoblasts transduced with sh-LPIN1 and in patient myoblasts (see mutations in Table S1), lipin1 was barely detectable by confocal imaging (Figure 1A) or immunoblot (Figure 1B). PAP activity was also blunted, although not abolished, likely reflecting residual PAP activity of lipin2 and lipin3 (Figure 1C).
Figure 1.

Lipin1 localizes in proximity of late endosomes, autophagosomes, and mitochondria in human myoblasts
(A) The confocal images show a representative staining of endogenous lipin1 in primary myoblasts from 1 of 6 controls and 6 patients carrying LPIN1 mutations (Table S1) and in immortalized myoblasts from 1 healthy donor transduced with sh-LPIN1 or control scrambled (sh-Sc) shRNA.
(B) Immunoblot analysis of endogenous lipin1 expression as in (A) in 5 patients.
(C) Dot plots (mean of 3 technical replicates/dot) depict the means ± SDs of PAP activity quantified in myoblasts from 6 controls and 5 patients (Mann-Whitney U test).
(D) Lipin1 distribution within myoblast organelles of 5 controls by confocal microscopy. Dot plots (50–80 cells/dot) show the means ± SDs of the percentage of lipin1 in proximity with a given protein. The images are representative of 1 of at least 3 independent experiments.
Scale bars, 10 μm.
In 5 primary control myoblast lines, endogenous lipin1 was found in close proximity (50%–85%) of small punctate and tubular structures positive for Rab7 and the lysosomal-associated membrane protein 1 (LAMP1) and LAMP2 (Figure 1D) involved in the fusion of lysosomes with phagosomes and late endosomes.18 Lipin1 furthermore displayed proximity with the autophagosomal marker microtubule-associated protein 1A/1B-light chain 3 (LC3) (45%–50%). To a lesser extent, lipin1 overlapped with protein disulfide isomerase (PDI, 35%), a marker of the ER involved in autophagosome biogenesis19 and with the mitochondrial network as shown by 35% proximity with the translocase of the outer mitochondrial membrane (TOMM)20 and with the MitoSpy fluorescent membrane probe. By contrast, we found no relevant proximity with the early endosome antigen 1 (EEA1) marker (Figure 1D).
Next, we examined the consequence of a loss of lipin1 on the activity of Vps34. We used a green fluorescent protein (GFP) probe encoding the Fab1, YOTB, Vac1, and EEA1 (FYVE) domain, which specifically binds to membrane PI3P. Myoblasts transfected with a plasmid encoding GFP-2xFYVE displayed a 60% reduction in fluorescent dots in patient cells (Figure 2A) and sh-LPIN1-transduced control cells (Figure 2B), confirmed by probing with a PI3P-specific antibody (Figures S1A and S1B). Individual PI3P vesicles had a smaller volume in patient cells (Figure S1B). Consistent with the subcellular distribution of lipin1, this dearth of PI3P concerned vesicles staining for Rab7 and LC3 but not EEA1+ compartments (Figure 2C).
Figure 2.

Lipin1 deficiency results in a loss of PI3P close to Rab7 structures in myoblasts
(A) The confocal images show a representative 2xFYVE-GFP staining from 1 of 6 controls and patients. Dot plots (50 images/dot) show the means ± SDs number of 2xFYVE-GFP dots per patient versus control myoblast (Mann-Whitney U test).
(B) As in (A), but using immortalized myoblasts from 1 healthy donor treated with sh-Sc or sh-LPIN1. The plots (30–45 cells/condition) show the number of 2xFYVE-GFP dots (Mann-Whitney U test).
(C) Typical distribution of PI3P within the myoblast vesicles from 1 of 3 controls and patients. Box and whisker plots (50 images/condition) depict the percentage of proximity between PI3P and the given marker for 1 representative control and patient (Mann-Whitney U test).
(D) Myoblasts from 3 controls and 3 patients were stained for PI35P2 and late endosomal or lysosomal markers. Proximity was calculated as in (C) (unpaired t test). Results are from 1 representative of at least 3 independent experiments.
Scale bars, 10 μm.
See also Figure S1.
Examining the impact on other phosphoinositides (PIs) strongly expressed in late endosomal structures and produced by PI3P metabolism,20 we found a significant reduction in phosphatidylinositol 3,5-bisphosphate (PI35P2), which is produced by the phosphorylation of PI3P by the PI kinase PIKfyve, in the proximity of Rab7 vesicles (Figure 2D), but no effect on phosphatidylinositol (3,4)-bisphosphate (PI34P2) levels (Figure S1C). We concluded that lipin1 expression in human myoblasts regulates the levels of PI3P and its downstream product PI35P2 close to late endosomes and autophagosomes.
Lipin1 modulates Rab7 activity by promoting the recruitment of the Rab7 GAP Armus through PI3P production
To monitor the role of lipin1 in late endosome homeostasis, we examined the size and the distribution of Rab7+ endosomes. In control myoblasts, Rab7 localized to small vesicular structures at the cell periphery (Figures 2C and 3A), whereas in patient cells, slightly larger Rab7+ vesicles (Figure S2A) accumulated in the perinuclear region, presumably in close proximity to the microtubule-organizing center (MTOC) (Figure 3A), a feature reminiscent of Vps34-deficient mouse embryonic fibroblasts.21 Positioning of Rab7+ vesicles in the cell relies on the balance of Rab7 binding to 2 effectors: the FYVE coiled-coil domain-containing 1 (FYCO1) protein mediating kinesin-dependent anterograde transport and the Rab-interacting lysosomal protein (RILP) mediating retrograde transport via dynein motor proteins.22, 23, 24 FYCO1 depends on PI3P to promote the microtubule-dependent translocation to the periphery. We speculated that the reduction of PI3P levels compromised the recruitment of FYCO1 to Rab7+ vesicles and favored a perinuclear positioning. FYCO1 displayed a reduced ability to interact with Rab7 (Figure 3B) and showed less proximity with residual PI3P in patient myoblasts (Figure 2B).
Figure 3.

Lipin1-deficient myoblasts display alterations in Rab7 positioning and activation and impaired lysosomal function
(A) The distribution of endogenous Rab7 in 30 individual patient and control myoblasts was determined by calculating the intensity of Rab7 fluorescence along a longitudinal axis traversing the nucleus.
(B) The interaction between Rab7 and FYCO1 determined by fluorescence resonance energy transfer (FRET) after staining with Rab7 (green) only or for Rab7 (donor) and FYCO1 (acceptor) in red was depicted by line graphs (ns, nanoseconds).
(C) Myoblasts transfected with RFP-RILP were immunostained for Rab7. Box and whisker plots (30 images/condition) show the percentage of proximity of Rab7 with RFP-RILP (unpaired t test).
(D and E) Myoblasts transfected with a construct encoding RFP-Armus were stained for Rab7 or PI3P. Box and whisker plots (50 images/condition) show the percentage of proximity of Rab7 (D) or PI3P (E) with RFP-Armus (Mann-Whitney U test).
(F) Dot plot (mean of 2 technical replicates/dot) with lines depicts the relative level of residual EGFR as compared to β-actin upon incubation with EGF for indicated times.
(G) Myoblasts were loaded with DQ-OVA, and fluorescence resulting from the cleavage of DQ-OVA substrate was analyzed by flow cytometry. Dot plots (mean of 2 technical replicates/dot) show the means ± SDs of the value of the mean fluorescence intensity (MFI) of DQ-OVA 60 and 120 min after uptake (mean effect of interaction F(1,6) = 14.43, p = 0.009, of time F(1,6) = 53.46, p = 0.0003, of subjects F(1,6) = 11.23, p = 0.0154).
(H) Levels of immature and mature cathepsin D relative to β-actin expression in primary myoblasts were determined by immunoblot.
(I) Myoblasts cultured in GM or EBSS were loaded with the Lysotracker Red dye, then stained for endogenous Rab7, LAMP1, and LC3. Box and whisker plots (30 images/condition) show the percentage of proximity of Rab7 (mean effect of interaction F(1,116) = 22.01, p < 0,0001, of stimulus F(1,116) = 16.55, p < 0.0001, of subjects F(1,116) = 47.86, p < 0.0001), LAMP1 (mean effect of interaction F(1,116) = 0.2088, p = 0.6486, of stimulus F(1,116) = 4.252, p = 0.0414, of subjects F(1,116) = 0.7115, p = 0.4007), or LC3 (mean effect of interaction F(1,116) = 1.786, p = 0.1840, of stimulus F(1,116) = 7.002, p = 0.0093, of subjects F(1,116) = 11.76, p = 0.0008) with Lysotracker Red.
Scale bars, 10 μm (A–E and I). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001: adjusted p values as determined by within-subjects (G) or between-subjects (I) 2-way ANOVA and post hoc Sidak’s correction for multiple comparisons. Images and graphs show typical staining in myoblasts from 1 of 2 (B), 3 (C–F, H, and I), or 4 (A and G) patients and controls. Results are from 1 of at least 3 independent experiments.
See also Figure S2.
Considering that FYCO1 competes with RILP for binding to Rab7, we suspected enhanced RILP recruitment to patient late endosomes. Red fluorescent protein (RFP)-tagged RILP displayed enhanced proximity with endogenous Rab7 in patient cells (Figure 3C). Since only the guanosine triphosphate (GTP)-bound Rab7 can recruit RILP, this suggested that Rab7 was preferentially shifted toward its active GTP-bound form. Physiologic hydrolysis of Rab7-bound GTP to guanosine diphosphate (GDP) is mediated by the GTPase activating protein (GAP) Armus, which binds to late endosomal PI3P.21,25 An Armus-RFP fusion protein displayed extensive proximity with Rab7 and PI3P in control myoblasts, which was strongly reduced in patient cells (Figures 3D and 3E). Thus, lipin1 deficiency is associated with impaired recruitment of Armus, presumably because of reduced production of PI3P at late endosomes, suggesting that lipin1 regulates the balance between GTP- and GDP-bound Rab7.
Given the key role of Rab7 in regulating endosome homeostasis,26 we speculated that lipin1 deficiency may perturb a broad range of endosome functions. Confocal imaging revealed the presence of hybrid endosomes staining for EEA1 and Rab7 in patient myoblasts (Figure S2C), suggesting compromised endosome maturation. The enlarged vesicles positive for both Rab7 and LAMP1 in patient myoblasts (Figure S2D) suggest perturbation in the transition from late endosomes to endolysosomes. In contrast, we did not observe any increase in the proximity of Rab7 with the mannose 6 phosphate receptor (M6PR), indicating that M6PR trafficking between the trans-Golgi network (TGN) and late endosomes is not modified (Figure S2E). However, retrograde transport from late endosomes to the TGN was impaired as assessed by the compromised accumulation of internalized cholera toxin B-subunit in GM130+ (Golgi) structures after a 30-min chase (Figure S2F). Rab7 is required for the degradation of various cell surface receptors.27 Degradation of 2 model proteins, epidermal growth factor receptor (EGFR) (Figure 3F) and DQ-ovalbumin (DQ-OVA, a substrate becoming fluorescent upon degradation; Figure 3G), was delayed and/or reduced in patient cells. This correlated with a decreased proteolytic conversion of cathepsin D to the mature form (Figure 3H); yet, the quantity of acidic vesicles was similar between patient and control cells (Figure S2F). The proximity of Rab7+ with LC3+ vesicles, the structures affected by low PI3P levels (Figure S2C), but not LAMP1+ structures with LysoTracker Red, a dye labeling acidic structures, was also significantly increased in patient myoblasts (Figure 3I). Since acidification relies on the interaction of the V-ATPase subunit V1G1 with RILP,28 enhanced RILP recruitment to GTP-locked Rab7 likely explains this observation, as well as predominant perinuclear distribution of Rab7 vesicles in patient myoblasts.29 We concluded that compromised Rab7 inactivation in lipin1-deficient myoblasts leads to a perturbation in global endosome maturation and function, and ultimately to a decrease in cellular degradative capacities.
Lipin1 regulates the clearance of autophagosomes and of mitochondrial DNA (mtDNA) from damaged mitochondria via the GAP Armus
Paralleling Rab7 distribution, LC3 staining concentrated in the perinuclear region in patient myoblasts (Figure S3A). Considering the role of complexes formed by FYCO1, Rab7, and LC3 in the microtubule transport of autophagic vesicles,30 we reasoned that defective FYCO1 recruitment may disturb autophagosomes. The proximity between LC3 and FYCO1 was significantly reduced in patient myoblasts (Figure S3B), as was that between LC3 and Armus-RFP (Figure S3C).
Armus depletion, mediating a shift to GTP-locked Rab7, significantly delays autophagic flux.31 To examine the fusion of autophagosomes with lysosomes, we transfected myoblasts with a vector encoding RFP-GFP-LC3, allowing the distinction of autophagosomes (RFP+GFP+) from autolysosomes (RFP+GFP−), due to the loss of GFP fluorescence in acidic compartments. Incubation in minimal medium lacking growth factors and amino acids (Earle’s balanced salt solution, EBSS) induced the formation of autophagosomes in both patient and control myoblasts. However, while supplementation with a nutrient-rich growth medium (GM) increased the percentage of autolysosomes strongly in control cells, it had no effect in patient cells, indicating defective autophagosome maturation in the absence of lipin1 (Figure 4A). This suggested that although late endosomes undergo enhanced acidification in patient myoblasts, acidification in autophagosome maturation is compromised.
Figure 4.

Autophagic clearance and mitochondrial quality and functions are impaired in human lipin1-deficient myoblasts
(A) Autolysosome formation (red puncta) in myoblasts transfected with the RFP-GFP-LC3 construct upon exposure to EBSS and refed or not with GM. Box and whisker plots (50 images/condition) represent the percentage of autolysosomes (mean effect of interaction F(1,196) = 44.4, p < 0.0001, of stimulus F(1,196) = 47.63, p < 0.0001, of subjects F(1,196) = 17.78, p < 0.0001).
(B) Myoblasts were cultured in GM or EBSS and refed or not with GM (starvation-induced mitophagy) or exposed to CCCP before immunostaining for TOMM20 and LC3. Box and whisker plots (50 images/condition) depict the percentage of proximity of TOMM20 with LC3 (mean effect of interaction F(3,392) = 11.09, p < 0.0001, of stimulus F(3,392) = 62.9, p < 0.0001, of subjects F(1,392) = 20.70, p < 0.0001).
(C) Box and whisker plots (30 images/condition) represent the number of mitochondrial nucleoids per myoblast after exposure to CCCP (mean effect of interaction F(3,116) = 9.554, p < 0.0001, of time F(3,116) = 19.99, p < 0.0001, of subjects F(1;116) = 26.22, p < 0.0001).
(D) Real-time analysis of oxygen consumption rate (OCR) in myoblasts under basal respiration and after addition of (1) oligomycin, (2) FCCP, or (3) antimycin A. Dot plot (mean of 4–6 technical replicates/dot) represents the means ± SDs of the OCR in myoblasts from 5 controls and 4 patients.
(E) Evaluation of the mitochondrial membrane potential by flow cytometry. Dot plot (mean of 2 technical replicates/dot) depicts the means ± SDs of the MFI of TMRM, expressed as a percentage of the MFI for GM condition (mean effect of interaction F(1,12) = 3.340, p = 0.0926, of stimulus F(1,12) = 7.313, p = 0.0192, of subjects F(1,12) = 0.2404, p = 0.6327).
(F) Distribution of oxidized DNA within the LC3 and LAMP1 structures of myoblasts cultured as in (B). Box and whisker plots (25 images/condition) show the percentage of proximity of 8OHDG with LC3 (mean effect of interaction F(2,144) = 1.504, p = 0.2258, of stimulus F(2,144) = 0.2997, p = 0.7415, of subjects F(1,144) = 100.8, p < 0.0001) or LAMP1 (mean effect interaction F(2,144) = 3.130, p = 0.0467, of stimulus F(2,144) = 18.41, p < 0.0001, of subjects F(1,144) = 96.93, p < 0.0001).
(G) Quantification of 12S mtDNA levels qPCR in cytosolic fractions from myoblasts of 4 patients and controls exposed to CCCP or EBSS (mean effect of interaction F(1,12) = 0.004867, p = 0.9455, of stimulus F(1,12) = 1.798, p = 0.2047, of subjects F(1,12) = 0.03755, p = 0.8496) and of the mtDNA motif DLOOP (mean effect of interaction F(1,12) = 0.2168, p = 0.6498, of stimulus F(1,12) = 0.3160, p = 0.5844, of subjects F(1,12) = 3.371, p = 0.0913). Dot plots (mean of 3 technical replicates/dot) depict the means ± SDs of the ratio of cycle threshold (CT) values of the given cytosolic fraction normalized to unfractioned cells.
Scale bars, 10 μm (A and B). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001: adjusted p values after between-subjects (A, B, and E–G) or within-subjects (C, as compared to H0 for each family) 2-way ANOVA and post hoc Sidak’s correction for multiple comparisons. Images and plots show typical staining and quantification for 1 of 3 patients and controls (A, B, C, and F). Results are representative from 1 of 2 (G), 3 (A–D and F), and 4 (E) independent experiments.
See also Figures S3 and S4.
Mitochondrial quality control depends on autophagy to remove damaged mitochondria.32 Deletion of the lipin ortholog in Drosophila results in abnormal mitochondrial and autophagosomal structures in fat body cells.33 Perturbed mitochondrial and autophagosomal structures and functions are also observed in muscle cells of lipin1-deficient mice.14 We hypothesized that damaged mitochondria may accumulate in patient myoblasts because of impaired mitophagy. The formation of mitophagosomes was not impaired (Figure 4B) in patient cells exposed to the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) or to EBSS. However, CCCP-induced mitophagosomes failed to be cleared at late time points (Figure 4C), as indicated by the increased number of nucleoids as compared to baseline (0 h) 16 h after exposure to CCCP. Considering the key role of mitophagy in mitochondrial homeostasis, we suspected that mitochondrial function may be perturbed in the absence of lipin1. Patient myoblasts displayed a significantly lower oxygen consumption rate (OCR) at baseline conditions. The lower OCR was more pronounced upon challenging with the uncoupler carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone (FCCP; Figures 4D, S3D, and S3E), while the maximal glycolytic capacity was significantly increased (Figure S3F). The total mass of mitochondrial structures remained unchanged between patient and control myoblasts (Figure S3G), suggesting that damaged or non-functional mitochondrial material accumulating at the steady state does not affect cell growth, as supported by a normal proliferation rate upon culture with different glucose concentrations (Figure S3H). Incubation with EBSS induced a substantial loss of mitochondrial transmembrane potential in patient myoblasts, as indicated by reduced tetramethylrhodamine methyl ester perchlorate (TMRM) sequestration (Figure 4E), resulting in an increased production of reactive oxygen species (ROS) (Figure S3I). These observations suggest that mitochondrial dysfunction is unmasked upon higher energetic demand and results from a higher proportion of damaged mitochondria in patient myoblasts.
Compromised mitophagy can alter the integrity of mtDNA and result in the accumulation of damaged mtDNA. Double-stranded mtDNA (dsDNA) and the transcriptional factor A mitochondrial (TFAM), an exclusively mitochondrial protein involved in the maintenance of intact mtDNA,34 showed normal proximity in patient myoblasts (Figure S4A). Consistently, 8-hydroxy-2′deoxyguanosine (8OHDG), a marker of oxidized mtDNA physiologically removed by mitophagy,35 showed limited proximity with TFAM and TOMM20 (Figures S4A and S4B). However, upon exposure to EBSS, the total cellular amount of oxidized mtDNA increased strongly in patient myoblasts, suggesting that oxidized mtDNA was properly released from mitochondria but not degraded upon metabolic distress (Figure S4C). Consistently, qPCR analysis demonstrated a significant increase in mtDNA (Figure S4D).
Oxidized mtDNA could accumulate within the endolysosomal pathway or in the cytosol. In patient myoblasts, 8OHDG localized mainly to LC3+ and LAMP1+ structures at the steady state and upon metabolic stress, which persisted after refeeding cells (Figure 4F). In contrast, the cytosolic fraction purified from stressed myoblasts did not contain increased amounts of 12S DNA and of D-loop, 2 mtDNA-related products34 (Figure 4G). We concluded that in the absence of lipin1, oxidized mtDNA accumulates in a non-degradative poorly acidified vesicular compartment, most likely mitophagosomes, and fails to be cleared as observed for autophagosomes (Figures 4A–4C). We propose that the loss of PI3P close to autophagosomes, similar to that of late endosomes, compromises the recruitment of Armus, which in turn inhibits the assembly of FYCO1/Rab7/LC3 complexes required for efficient autophagic/mitophagic clearance. This results in functional perturbation of mitochondria and accumulation of oxidized mtDNA in lipin1-deficient myoblasts.
Accumulation of mtDNA enhances inflammation in patient cells through a TLR9-dependent pathway
Fasting and metabolic stress precede flares of lipin1-related rhabdomyolysis. We reasoned that the accumulation of damaged mtDNA may be responsible for inflammation precipitating flares. mtDNA was readily detectable in the plasma of patients with chronic and residual elevated creatine kinase (CK) between flares, as were pro-inflammatory cytokines and chemokines (Figure 5A). We also identified a type I interferon (IFN) signature by quantifying 6 IFN-related genes (Figure 5B) in peripheral blood mononuclear cells (PBMCs) of patients. Since circulating inflammatory mediators are mainly produced by peripheral blood monocyte-derived cells activated by microbial products,36 we examined the production of inflammatory cytokines and IFN by dendritic cells (DCs) following stimulation by TLR ligands. DCs from patients displayed enhanced production of the pro-inflammatory cytokine IL-6 when stimulated by a synthetic TLR9 agonist, but not by ligands for other endosomal or plasma membrane TLRs (Figure 5C). In line with this, TLR9 stimulation induced an increased maturation of patient DC (Figure 5C) and a much stronger type I IFN response (Figure 5D). Patient and sh-LPIN1-transduced control myoblasts were also hypersensitive to TLR9 ligands (Figures 5E and 5F). Rescuing lipin1 expression (Figures S5A–S5D) abrogated hypersensitivity to TLR9 ligands of patient myoblasts (Figure 5G), confirming the causative role of lipin1 expression and/or mutation
Figure 5.

Accumulation of oxidized mitochondria enhances inflammation in lipin1-deficient cells through a TLR9 pathway
(A) Circulating mtDNA was quantified in plasma by qPCR and inflammatory molecules were measured in sera by flow cytometry, from 12 healthy donors (6 for mtDNA) and 6 patients (Mann-Whitney U test). Dot plots (mean of 3 technical replicates/dot) show the means ± SDs.
(B) IFN signature in peripheral blood mononuclear cells was identified by qPCR in 4 controls and 4 patients. Dot plots (mean of 3 technical replicates/dot) show the mean ± SD of the CT value of a given gene normalized to the CT value of BACT (Mann-Whitney U test).
(C) DC maturation reflected by CD83 expression (mean effect of interaction F(4,70) = 2.114, p = 0.0881, of stimulus F(4,70) = 36.27, p < 0.0001, of subjects F(1,70) = 14.21, p = 0.0003) and interleukin-6 (IL-6) production (mean effect of interaction F(3,64) = 2.847, p = 0.0444, of stimulus F(3,64) = 18.20, p < 0.0001, of subjects F(1,64) = 2.807, p = 0.0987) were evaluated by flow cytometry and ELISA, respectively, in 10 controls and 6 patients, after exposure to Poly:IC (PIC), lipopolysaccharide (LPS), imiquimod (Im), and CpG-A. Dot plots (mean of 2 [CD83] or 4 [IL-6] technical replicates/dot) show the means ± SDs.
(D) CpG-A-induced IFN responses in DCs from 3 controls and patients were evaluated by qPCR by quantifying SIGLEC1 (mean effect of interaction F(1,8) = 4.436, p = 0.0683, of stimulus F(1,8) = 9.255, p = 0.0160, of subjects F(1,8) = 20.78, p = 0.0019), ISG15 (mean effect of interaction F(1,8) = 0.3191, p = 0.5876, of stimulus F(1,8) = 21.75, p = 0.0016, of subjects F(1,8) = 41.33, p = 0.0002), and IFI27 (mean effect of interaction F(1,8) = 21.99, p = 0.0016, of stimulus F(1,8) = 25.45, p = 0.0010, of subjects F(1,8) = 91.20, p < 0.0001) expression. Dot plots (mean of 3 technical replicates/dot) show the means ± SDs of CT value calculated as in (B).
(E) Production of IL-6 (ELISA) by myoblasts from 8 controls and 6 patients (mean effect of interaction F(2,36) = 3.882, p = 0.0297, of stimulus F(2,36) = 45.49, p < 0.0001, of subjects F(1,36) = 15.28, p = 0.0004), IFNA (mean effect of interaction F(3,24) = 23.44, p < 0.0001, of stimulus F(3,24) = 74.13, p < 0.0001, of subjects F(1,24) = 42.33, p < 0.0001), and IFNB (mean effect of interaction F(3,24) = 2.207, p = 0.1133, of stimulus F(3,24) = 10.19, p = 0.0002, of subjects F(1,24) = 13.60, p = 0.0012).
(F) As in (E), but using sh-Sc or sh-LPIN1-transduced immortalized myoblasts from 1 healthy donor (1 different cell vial from the same single healthy donor/dot) and primary myoblasts from 4 patients. Dot plots (mean of 4 technical replicates/dot) show the means ± SDs of the IL-6 concentration (mean effect of interaction F(2,14) = 6.234, p = 0.0116, of stimulus F(1,14) = 0.0874, p = 0.3653, of subjects F(2,14) = 4.397, p = 0.0330).
(G) As in (F), but primary myoblasts from 4 controls and patients were transduced with a lentivirus expressing a plasmid encoding for a myc-DDK-tagged lipin1 protein (+LPIN1) or an empty vector (+vector) before being challenged (mean effect of interaction F(2,18) = 0.2196, p = 0.8050, of stimulus F(1,18) = 0.2906, p = 0.5965, of subjects F(2,18) = 14.80, p = 0.0002).
(H) Myoblasts from 6 controls and patients were exposed to a vehicle or ethidium bromide (Et. B.). Dot plots (mean of 3 technical replicates/dot) show the means ± SDs of the ratio of the CT value for 12S mitochondrial DNA normalized to the CT value for BACT (mean effect of interaction F(1,12) = 3.186, p = 0.0996, of stimulus F(1,12) = 101.6, p < 0.0001, of subjects F(1,12) = 5.268, p = 0.0405).
(I) Myoblasts from 6 controls and patients pre-treated with Et. B. for 5 days were exposed to EBSS and challenged or not with CpG-A for 16 h. Dot plots (mean of 4 technical replicates/dot) show the mean ± SD of IL-6 concentration in culture supernatants at the end of the experiment (mean effect of interaction F(3,24) = 4.989, p = 0.0079, of stimulus F(3,24) = 8.218, p = 0.0006, of subjects F(1,24) = 15.14, p = 0.0007).
(J) Myoblasts were transfected with a plasmid encoding HA-tagged TLR9 and exposed to GM or EBSS and with vehicle or CpG-A before refeeding (Refed) cells exposed to EBSS and CpG-A with GM. Cells were then stained for HA-TLR9 (anti-HA antibody, red) and 8OHDG (green). Box whisker plots (25 images/condition) show the percentage of proximity of TLR9 with 8OHDG (mean effect of interaction F(4,240) = 1.712, p = 0.1481, of stimulus F(4,240) = 25.07, p < 0.0001, of subjects F(1,240) = 68.55, p < 0.0001) in myoblasts from 1 of 3 controls and patients.
(K) As in (J) but myoblasts were immunostained for HA-TLR9 (green) and LC3 (red). Mean effect of interaction F(4,240) = 18.56, p < 0.0001, of stimulus F(4,240) = 125.8, p < 0.0001, of subjects F(1,240) = 61.39, p < 0.0001.
IL-6 concentration is presented after background (which corresponds to values obtained with a control vehicle) subtraction (C, E, F, H, and I). Scale bars, 10 μm (J and K). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001: adjusted p values after between-subjects 2-way ANOVA (B–K) and post hoc Sidak’s correction for multiple comparisons. Data are representative of 1 of at least 3 independent experiments.
See also Figure S5.
Febrile illness due to viral infection, another factor frequently preceding rhabdomyolysis flares, not only causes metabolic stress but also provides TLR ligands. We reasoned that the simultaneous presence of metabolic stress and microbial TLR9 ligands may enhance inflammation in a synergistic manner or through the cooperative binding of mtDNA and exogenous ligands to TLR9.37 We exposed myoblasts to EBSS alone or in combination with CpG-A to mimic an episode of febrile illness. Exposure to EBSS created proinflammatory conditions only in patient cells (Figure S5E), which was exacerbated by the addition of CpG-A. Depleting mitochondria (Figure 5H) significantly reduced the inflammatory response to a level close to control myoblasts (Figure 5I), indicating that mitochondria and probably mtDNA are mandatory for triggering inflammation upon starvation. mtDNA can also trigger inflammatory responses through the cytosolic stimulator of interferon genes (STING) pathway.38 We could not detect any significant difference in IFN expression upon exposure to EBSS combined to the STING ligand cyclic GMP-AMP (cGAMP; Figure S5F), consistent with the lack of an effect of EBSS and CCCP on the cytosolic concentration of mtDNA (Figure 4G).
These results suggested that mtDNA could reach endolysosomal compartments, where it could bind and activate TLR9. To verify this hypothesis, we studied the trafficking of TLR9 in myoblasts transfected with a plasmid encoding hemagglutinin (HA)-tagged TLR9. In patient myoblasts, a larger proportion of HA-TLR9 was observed in the proximity of 8OHDG at the steady state or upon exposure to CpG-A alone or in combination with EBSS and persisted after refeeding with GM (Figure 5J). Similarly, exposure to both CpG-A and EBSS but not CpG-A alone (Figure 5K) significantly increased the proximity of HA-TLR9 with LC3 exclusively in patient myoblasts. Our results suggest that mtDNA can accumulate in a LC3+ compartment together with TLR9 and trigger its activation in lipin1-deficient cells. Starvation and cell exogenous TLR9 ligands exacerbate this by favoring the transport or accumulation of mtDNA and/or the recruitment of TLR9 to endolysosomes. CpG-A may also increase TLR9 activation in patient myoblasts helped by the cooperative binding with 8OHDG.37
mtDNA accumulation triggers clinical inflammation and rhabdomyolysis in vivo
We next focused on identifying the pathogenic sequence leading to myoblast cell death. During rhabdomyolysis, skeletal muscle fibers release high concentrations of Ca2+ before activation of apoptosis and/or necrosis.39 Exposure of patient, but not control, myoblasts, to CpG-A was sufficient to trigger a Ca2+ flux (Figure 6A). This was TLR9 dependent since pre-incubation with ODN TTAGGG (A151), a synthetic TLR9 antagonist ligand, normalized Ca2+ profiles (Figure 6A). Cell death of patient myoblasts was triggered by exposure to EBSS alone and greatly enhanced by the addition of a TLR9 ligand (Figures 6B and 6C). Importantly, staurosporin triggered similarly cell death in primary patient and control myoblasts. Again, rescuing lipin1 expression (Figures 6B and 6C) in primary myoblasts from patients, knocking down lipin1 expression (Figure 6D), or depleting mitochondria (Figure 6E) was sufficient to prevent caspase-dependent cell death. Consistent with a role of inflammatory signaling, dexamethasone prevented cell death in vitro (Figure 6F), and the treatment of 2 patients with steroids during a flare up resulted in the rapid regression of muscle pain and normalization of plasma CK levels (Figure 6G). Pre-incubating patient myoblasts with a TLR9 antagonist or chloroquine (CLQ), a drug used to prevent acidification of endolysosomes and consequently TLR9 proteolytic activation and signaling, was sufficient to reverse inflammation (Figure 6H) and prevent cell death (Figure 6I). Thus, TLR9 signaling promotes mitochondrial caspase-dependent cell death in metabolically stressed patient myoblasts. Prompted by these promising results, we treated 6 patients, 5 of whom had previously undergone between 3 and 6 flares, with hydroxychloroquine sulfate (HS; daily dose 6.5 mg/kg) given orally for 12–36 months. Four patients were completely protected from flares while adhering to the treatment, whereas 2 other patients experienced a single episode (Table 1). In all 6 patients, HS was well tolerated, except for moderate abdominal pain in 3. Therefore, targeting inflammatory TLR9 signaling directly by inhibiting its activation, or indirectly using steroids, may be of significant clinical interest to prevent flare-ups and manage lipin1-related rhabdomyolysis.
Figure 6.

mtDNA accumulation is responsible for inflammation and rhabdomyolysis that can be reversed by steroids or chloroquine (CLQ) treatment in vivo
(A) Real-time calcium flux in myoblasts from 4 controls and patients challenged with CpG-A, after pre-incubation with ODN151, a synthetic TLR9 antagonist. Each dot plot with lines depicts the mean ± SD of the intensity of the fluo-4 calcium probe as normalized to basal intensity of 1 single individual (≥10 cells/condition).
(B) Death of myoblasts was evaluated by staining cells from 4 control individuals transduced with a lentivirus expressing a plasmid encoding for myc-DDK-tagged lipin1 protein (+LPIN1) and 4 patients transduced with a lentivirus expressing a plasmid encoding for myc-DDK-tagged lipin1 protein or an empty vector, for the apoptosis marker caspase3/7 green and propidium iodide or 7-actinomycin D (7-AAD), a dye that stains both apoptotic and necrotic cells. Staurosporin (ST): positive control. Fluorescence-activated cell sorting (FACS) plots show the codistribution of the fluorescence intensity of caspase3/7 and of 7-AAD in 1 of 4 controls and patients.
(C) Dot plots (≥1,000 single cells/dot) depict the mean ± SD of the proportion of dead cells (ie, cells positive for capsase3/7 and/or 7-AAD) from (B) (mean effect of interaction F(8,45) = 5.296, p = 0.0001, of stimulus F(4,45) = 70.03, p < 0.0001, of subjects F(2,45) = 6.607, p = 0.003).
(D) As in (C), but using immortalized myoblast cell line transduced with Sh-Sc or Sh-LPIN1 shRNA (mean effect of interaction F(4,20) = 6.154, p = 0.0021, of stimulus F(4,20) = 48.93, p < 0.0001, of subjects F(1,20) = 15.62, p = 0.0008).
(E and F) As in (C), but myoblasts were pre-treated with Et. B. or vehicle (mean effect of interaction F(3,24) = 21.28, p < 0.0001, of stimulus F(3,24) = 114.8, p < 0.0001, of subjects F(1,24) = 25.79, p < 0.0001) for 5 days (E) or dexamethasone (DXM) or vehicle (mean effect of interaction F(3,24) = 9.490, p = 0.0003, of stimulus F(3,24) = 33.95, p < 0.0001, of subjects F(1,24) = 11.56, p = 0.0024) for 8 h (F), before challenging myoblasts with CpG-A in the presence of GM or EBSS.
(G) CK levels (means ± SDs of 3 individual values obtained from independent blood samples drawn the same day) were measured in 2 patients during a flare before and after the administration of steroids.
(H) As in (C) and Figure 5, but myoblasts from 4 controls and patients were pre-treated with CLQ or ODN151, or vehicle for 8 h followed by challenging with CpG-A together with GM or EBSS for another 16 h (mean effect of interaction F(5,36) = 19.53, p < 0.0001, of stimulus F(5,36) = 38.32, p < 0.0001, of subjects F(1,36) = 67.60, p < 0.0001). Dot plots (mean of 4–6 technical replicates/dot) show the mean ± SD of IL-6 concentration at the end of the experiment.
(I) As in (H), but evaluating the proportion of cell death (mean effect of interaction F(5,36) = 17.51, p < 0.0001, of stimulus F(5,36) = 73.41, p < 0.0001, of subjects F(1,36) = 40.06, p < 0.0001). Dot plots (≥1,000 single cells/dot) show the means ± SDs. IL-6 concentration is presented after background (which corresponds to values obtained from culture supernatants of cells exposed to vehicle) subtraction.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001: adjusted p values as determined by a between-subjects 2-way ANOVA (C–F, H, and I) and post hoc Sidak’s correction for multiple comparisons. Results are from 1 representative of 2 (D) or at least 3 (A–C, E–I) independent experiments.
Table 1.
Follow-up of 6 patients treated by HS (6.5 mg/kg/day)
| Patient no. | Age at diagnosis, y | Age at the initiation of treatment, y | No. severe rhabdomyolysis (CK > 10,000 U/L) |
Mean (median) plasma concentration of HS (μg/L) | |
|---|---|---|---|---|---|
| Before treatment | During treatment (treatment duration, months) | ||||
| P1 | 5 | 7 | 7 | 0a (36) | 99 (101) |
| P2 | 10 | 13 | 3 | 0a (33) | 292 (318) |
| P3 | 7 | 17 | 1 | 0 (12) | 466 (597) |
| P4 | 12 | 12 | 1 | 0 (12) | 637 (593) |
| P5 | 3 | 9 | 3 | 1 triggered by gastroenteritis (12) | 1,060 (993) |
| P6 | 2 | 4 | 4 | 2 triggered by gastroenteritis (12) | 1,043 (922) |
CK, creatine kinase; HS, hydroxychloroquine sulfate.
Occurrence of 1 rhabdomyolysis flare 6 weeks after stopping HS (non-adherence to prescribed treatment), without any other identified trigger. Although the frequency of rhabdomyolysis events is known to decrease with patient age, this factor is unlikely to have contributed to the low number of flares in treated patients considering the limited duration of treatment.
Discussion
The results of this study suggest a key role of perturbed late endosome and autophagosome positioning and fusion in the pathogenesis of lipin1 disease. We find that lipin1 deficiency with a consequent lack of PI3P hampers recruitment of the Rab7 GAP Armus, thereby preventing the physiologic Rab7 cycling between the GTP/GDP-bound states and shifting the balance of Rab7 effector binding to RILP at the expense of FYCO1.31,40 It is well documented, although incompletely understood, that kinesin-mediated transport mediated by FYCO1 is required to complete the clearance of late endosome/autophagosome content.41 This phenomenon is in apparent contrast to the fact that peripheral late endosomes/lysosomes are known to be less degradative than those positioned at the MTOC.42 However, our observations suggest that lipin1-deficient late endosomes display poor proteolytic capacity despite their positioning close to the MTOC. In lipin1-deficient endosomes/lysosomes, efficient acidification contrasts with reduced maturation of cathepsin D, suggesting dissociation between vesicles containing hydrolases and others rich in V-ATPase. A plausible explanation for normal or even enhanced acidification is provided by recruitment of the V-ATPase subunit V1G1 by RILP.28 Conversely, immature cathepsins are stored in lysosomes and require activation by lysosome fusion with late endosomes, which relies on the kinase PIKfyve producing PI35P2,43 a compound with reduced abundance at lipin1-deficient late endosomes, likely because of the dearth of its precursor PI3P. Additional factors may contribute to deficient fusion between late endosomes and lysosomes, including an impaired activation of Wdfy3/Alfy,44,45 an enzyme expressed in skeletal muscle directly recruited through a PI3P-binding domain,44 and/or the enzyme EPG5, which requires activation by the PI3P phosphatase MTMR-3.46,47 We argue that lipin1-deficient myoblasts contain static late endosomes retained at the MTOC but are unable to fuse with hydrolase-containing lysosomes. Does lipin1 co-transcriptional activity play any role in the pathophysiology of lipin1-related rhabdomyolysis? Considering our previous report arguing against this hypothesis,10 as did findings in recent mouse models,8,9 there is strong support for the causative role of defective PAP-1 activity.
Lipin1 deficiency can increase mitochondrial proteolysis48; however, this phenomenon is not likely sufficient to explain mitochondrial dysfunction, which may also relate to increased local PA levels. For example, the lack of dephosphorylation of mitochondrial phosphatidate by lipin1 may enhance mitochondrial fusion itself, inhibiting organelle elimination by autophagy. However, given the strong evidence for defective late endosome-lysosome fusion, we propose that mitochondrial dysfunction preferentially results from failed mitochondrial quality control. The low mitochondrial OCR at baseline may in part reflect dysfunctional free cytoplasmic mitochondria and/or mitochondria found in vesicles. A formal experimental demonstration would be difficult to achieve considering the tight connections between autophagic flux and mitochondrial homeostasis, but would likely include strategies to restore defective autophagic clearance.49 The finding that autophagosomes form normally in lipin1-deficient myoblasts indicates that the activation of Atg14-containing Vps34 complexes is independent of lipin1 and that LC3+ vesicles staining for lipin1 correspond to autophagosomes having fused with late endosomes (“autoendosomes”). Considering the strong overlap of 8OHDG with LC3 staining, it is likely that mitochondrial-derived vesicles (MDVs) delivering mtDNA directly to late endosomes account for a minor source of oxidized mtDNA in TLR9+ vesicles and that compromised resolution of mitophagy is its principal source.50 Compromised recruitment of FYCO1 and Armus to LC3+ structures suggests that the mechanisms responsible for blocking late endosome fusion with lysosomes also hinder fusion of autoendosomes with lysosomes and therefore degradation of mtDNA. This fusion event likely is also required for TLR9 inactivation. TLR9 associates with the chaperone Unc93b in the ER and travels to endosomes where it can be activated by proteolytic cleavage.51 Strong and prolonged proximity of TLR9 with 8OHDG in stressed myoblasts indicates that vesicles containing MDVs, which undergo fusion with TLR9-containing endosomes, are sufficiently degradative to activate TLR9, but not equipped to degrade either mtDNA or TLR9 to terminate signaling. We did not observe any difference in TLR9 expression by qRT-PCR between patient and control myoblasts or DCs (data not shown). The strong TLR9-dependent deleterious effect of mtDNA is not restricted to lipin1 deficiency; has been observed for renal tubular cells, cardiomyocytes, and neurons32,52,53; and acts both in a cell-autonomous manner and through release from cells upon cell death, formation of extracellular DNA traps, or exosome secretion.34 The presence of mtDNA in patient sera, together with this study, indicate that both mechanisms are at work in lipin1 patients.
The cell-autonomous effects of mtDNA include pro-inflammatory signaling through inflammasome or cGAMP synthase-STING pathway activation upon mtDNA release into the cytosol. Our results argue against a significant role of the latter pathways in lipin1 myoblast pathology. Damaged mitochondria and, more specifically, a reduction in mitochondrial membrane potential can directly engage caspase-dependent cell death. We show that both exogenous TLR9 ligands and starvation, conditions mimicking patient fasting and infections frequently triggering rhabdomyolysis episodes, induce inflammation and myoblast inflammation and death, with an additive effect. Exacerbation of stressed cell death by an exogenous TLR9 agonist has also been observed in cardiac muscle.34,54 The additive effect may involve the capacity of TLR9 to recognize multiple types of DNA through 2 specific binding sites that function in cooperation to boost TLR9 activation.37 Termination of TLR9 signaling requires the degradation of ligands and/or receptor in the lysosomes, which is impaired in lipin1-deficient muscle and/or immune cells. How precisely TLR9-containing late endosomes fuse with lysosomes containing specific hydrolase remains poorly investigated, but may depend on the regulation of the late endosomal/lysosomal Rab7b small GTPase55 and/or on the Vps33b subunit of the homotypic fusion and vacuole protein sorting (HOPS) complex,56 at least in human macrophages.
While our results highlight the pivotal role of mtDNA-driven inflammation in lipin1 pathology, mice lacking lipin1 display chronic myopathy and evidence suggesting a contribution of ER stress to pathology.8,57 Given that the dysregulation of autophagy can trigger ER stress and vice versa,58 it is conceivable that ER stress contributes to pathology. However, lipin1-deficient mice do not undergo acute episodes of massive and lethal rhabdomyolysis, limiting extrapolation to humans. Our observations documenting a direct role of mtDNA and TLR9 signaling in myoblast death led us to conclude that pro-inflammatory signaling by trapped mtDNA plays a key role in human lipin1 pathology, even though it may not be the sole pathogenic mechanism involved. Finally, human lipin1 deficiency may affect the crosstalk between TLR9 and Beclin-1, recently identified to be critical for muscle contraction during exercise.59 As a future direction, we propose investigating in vivo the capacity of natural TLR9 ligands to trigger hyperinflammation and acute rhabdomyolysis episodes in lipin1-deficient animal models.
The results of this study have provided therapeutic leads for lipin1 pathology. Attenuation of inflammation with steroids is strikingly efficient; however, they are not well suited as a preventive therapy because of the adverse effects on metabolism. Our in vitro and in vivo observations support the use of HS to block TLR9 activation and signaling as an alternative opportunity. Mauthe et al.60 reported that CLQ and hydroxychloroquine block the fusion of autophagosomes with degradative lysosomes in U2OS and HeLa cell lines. This effect may be different in muscle cells and depend on drug concentration. For example, Mauthe et al.60 observed that receptor-mediated versus fluid phase endocytosis were differentially affected. We speculate that, in lipin1 disease, HS may predominantly affect TLR9 activity while permitting residual autophagosome clearance.59,61 However, further experimentation is required to dissect the mechanism of defective clearance of TLR9-containing vesicles and to study the effect of HS on autophagic flux in myoblasts. In addition, HS can act on immune cells blocking TLR9 activation.62, 63, 64 Drugs restoring resolution of autophagy, shifting endosomal/autophagosome cargos toward alternative quality control mechanisms not affected by lipin1 deficiency or restoring efficient PI3P synthesis at late endosomes would seem sensible objectives of future research for the clinical management of lipin1 deficiency. As the pathophysiological sequence identified in this work may also be active in other inherited or sporadic forms of rhabdomyolysis, the therapeutic leads resulting from studying lipin1 disease may be of broader interest in the future.
Limitations of study
The main limitations are the limited number of patients treated with HS and the design of our small clinical trial, precluding formal demonstration of the efficacy of HS. Further directions should ideally include an international randomized control study to compare HS versus placebo, although this would be difficult to organize considering the very low incidence of the disease. Another limitation is the use of mono-DCs rather than plasmacytoid DCs to study TLR9 response in myeloid cells, motivated by the scarcity of plasmacytoid DCs in the limited blood sample volumes from pediatric patients. However, human mono-DCs express TLR9 and respond to TLR9 agonists.65 Finally, the number of myoblast cell lines was limited, as muscle biopsy is an invasive procedure no longer required for the diagnosis of lipin-1 disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-β-actin (clone C4), HRP-conjugated | Santa Cruz Biotechnology | Cat# sc-47778 HRP, RRID: AB_2714189 |
| Mouse monoclonal anti-ds DNA (clone HYB331-01) | Abcam | Cat# ab27156, RRID: AB_470907 |
| Rabbit polyclonal anti-cathepsin D | Santa Cruz Biotechnology | Cat# sc-6487, RRID: AB_2292414 |
| Mouse monoclonal anti-DDK (clone OTI4C5) | OriGene | Cat# TA50011-100, RRID:AB_2622345 |
| Goat polyclonal anti-EEA1 | Santa Cruz Biotechnology | Cat# sc-6415, RRID: AB_2096822 |
| Mouse monoclonal anti-EGF-R (clone 6F1) | MBL International | Cat# MI-12-1 |
| Rabbit polyclonal anti-FYCO1 | Sigma-Aldrich | Cat# HPA035526, RRID: AB_10672867 |
| Mouse Anti-GM130 Monoclonal Antibody (clone 35/GM130) | BD Biosciences | Cat# 610822, RRID: AB_10015242 |
| Mouse monoclonal anti-HA-Tag (clone HA.C5) | Applied Biological Materials | Cat# G036, RRID: AB_2616604 |
| Mouse monoclonal anti-LAMP-1 (clone 1D4B) | BD Biosciences | Cat# 553792, RRID: AB_2134499 |
| Rabbit monoclonal anti-HA-Tag (clone C29F4) | Cell signaling Technology | Cat# 3724S |
| Rabbit monoclonal anti-LAMP1 (clone D2D11) | Cell signaling Technology | Cat# 9091S |
| Mouse monoclonal anti-LAMP2 (clone H4B4) | Santa Cruz Biotechnology | Cat# sc-18822, RRID:AB_626858 |
| Rabbit monoclonal anti-TFAM (clone D5C8) | Cell signaling Technology | Cat# 8076S |
| Rabbit polyclonal anti-LC3 | MBL International | Cat# PM036, RRID: AB 2274121 |
| Mouse monoclonal anti-LC3 (clone 5F10) | Nanotools | Cat# 0231-100/LC3-5F10, RRID: AB_2722733 |
| Mouse monoclonal anti-Myc (clone 9E10) | OriGene | Cat# TA150121, RRID:AB_2622266 |
| Mouse monoclonal anti-PDI (clone C-2) | Santa Cruz Biotechnology | Cat# sc-74551, RRID: AB_2156462 |
| Rabbit polyclonal antibody anti-lipin1 | Cell Signaling Technology | Cat# 5195S, RRID: AB_10694491 |
| Mouse monoclonal 8OHDG (clone 15A3) | Santa Cruz Biotechnology | Cat# sc-66036, RRID: AB_832272 |
| Mouse monoclonal anti-PI3P | Echelon Biosciences | Cat# Z-P003, RRID: AB_427221 |
| Mouse monoclonal anti-PI34P2 | Echelon Biosciences | Cat# Z-P034B, RRID: AB_427223 |
| Mouse monoclonal anti-PI35P2 | Echelon Biosciences | Cat# Z-P035, RRID: AB_427224 |
| Mouse monoclonal anti- SQSTM1 (Clone 2C11) | Abnova Corporation | Cat# H00008878-M01, RRID: AB_437085 |
| Rabbit monoclonal anti-Rab7 (clone D95F2) | Cell Signaling | Cat# 9367S, RRID: AB_1904103) |
| Mouse monoclonal anti-Rab7 (clone Rab7-117) | Abcam | Cat# ab50533, RRID: AB_882241 |
| Rabbit polyclonal anti-STX6 | Proteintech Group | Cat# 10841-1-AP, RRID: AB_2196506 |
| Mouse monoclonal anti-TOMM20 (clone 29) | Santa Cruz Biotechnology | Cat# sc-136211, RRID: AB_2207538 |
| Mouse monoclonal anti-CD14 conjugated to microbeads | Miltenyi Biotec | Cat# 130-050-201, RRID: AB_2665482 |
| Mouse monoclonal anti-CD56 conjugated to microbeads (clone AF12-7H3) | Miltenyi Biotec | Cat# 130-050-401, RRID: AB_244333 |
| Mouse monoclonal anti-CD56 Allophycocyanin Conjugated (clone B159) | BD Biosciences | Cat# 555518, RRID: AB_398601 |
| Mouse monoclonal anti-CD83 Phycoerythrin Conjugated (clone HB15e) | BD Biosciences | Cat# 556855, RRID: AB_396526 |
| Goat Anti-Mouse polyclonal IgG, Human ads-HRP antibody | Southern Biotech | Cat# 1030-05, RRID: AB_2619742 |
| Goat Anti-Mouse IgG (H+L) secondary antibody Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A32723, RRID: AB_2633275 |
| Goat anti-Rabbit IgG (H+L) secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11008, RRID: AB_143165 |
| Donkey anti-Goat IgG (H+L) secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11055, RRID: AB_2534102 |
| Goat anti-Mouse IgG (H+L) secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11032, RRID: AB_2534091 |
| Goat anti-Rabbit IgG (H+L) secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11037, RRID: AB_2534095 |
| Goat anti-Mouse IgG (H+L secondary Antibody, Alexa Fluor Plus 647 | Thermo Fisher Scientific | Cat# A32728, RRID: AB_2633277 |
| Goat anti-Rabbit IgG (H+L) secondary Antibody, Alexa Fluor Plus 647 | Thermo Fisher Scientific | Cat# A32733, RRID: AB_2633282) |
| Bacterial and virus strains | ||
| LPIN1 - Human shRNA lentiviral particle | OriGene | Cat# TL303494V |
| Biological samples | ||
| Human primary myoblasts from healthy individuals and patients | Department of biology and molecular genetics, Cochin Hospital, Paris, France | N/A |
| Human immortalized myoblasts from healthy donor | MYOBANK-AFM (Myology institute BB-0033), Paris, France | N/A |
| Blood samples from healthy individuals and patients | Department of Inherited Metabolic Diseases, Necker Hospital, Paris, France | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human IL-4 | Miltenyi Biotec | Cat# 130-095-915 |
| Recombinant human GM-CSF | Miltenyi Biotec | Cat# 130-093-372 |
| Animal-Free Recombinant Human EGF | Peprotec | Cat# AF-100-15 |
| Pam3CSK4 | Invivogen | Cat# tlr-pms |
| Poly(I:C) (HMW) | Invivogen | Cat# tlr-pic |
| LPS-B5 (LPS from E. coli 055:B5) | Invivogen | Cat# tlr-b5lps |
| FLA-BS, standard flagellin from B. subtilis | Invivogen | Cat# tlr-bsfla |
| Imiquimod (R837) | Invivogen | Cat# tlr-imqs |
| CpG-A (ODN2216) | Invivogen | Cat# tlr-2216 |
| TLR9 antagonist ODN TTAGGG (A151) | Invivogen | Cat# tlr-ttag151 |
| Calcium indicator Fluo-4 AM | Thermo Fisher Scientific | Cat# F14201 |
| DAPI (4’,6-diamidino-2-phenylindole) | Thermo Fisher Scientific | Cat# D1306 |
| DQ-Ovalbumin | Thermo Fisher Scientific | Cat# D12053 |
| Dulbecco’s Modified Eagle’s Medium - high glucose | Sigma-Aldrich | Cat# D6546 |
| Dulbecco’s Modified Eagle’s Medium - low glucose | Sigma-Aldrich | Cat# D5546 |
| Earle’s Balanced Salt Solution, EBSS, calcium, magnesium, phenol red | Thermo Fisher Scientific | Cat# 24010043 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 16000044 |
| Fluoromount-G | Thermo Fisher Scientific | Cat# 00-4958-02 |
| Ham’s F-10 Nutrient Mix | Thermo Fisher Scientific | Cat# 11550043 |
| Image-iT TMRM Reagent (mitochondrial membrane potential indicator) | Thermo Fisher Scientific | Cat# I34361 |
| LysoTracker Deep Red | Thermo Fisher Scientific | Cat# L12492 |
| MitoSpy Orange CMTMRos | Biolegend | Cat# 424804 |
| MitoTracker Green FM | Thermo Fisher Scientific | Cat# M22425 |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | Cat# 15140122 |
| Phosphate-Buffered Saline, PBS, pH 7.4 | Thermo Fisher Scientific | Cat# 10010023 |
| Puromycin | Thermo Fisher Scientific | Cat# A1113802 |
| Recombinant Cholera Toxin subunit B Alexa Fluor 594 | Thermo Fisher Scientific | Cat# C34777 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat# A2153; CAS Number: 9048-46-8 |
| Carbonyl cyanide 3-chlorophenylhydrazone, CCCP | Sigma-Aldrich | Cat# C2759; CAS Number: 555-60-2 |
| Dexamethasone | Sigma-Aldrich | Cat# D4902; CAS Number: 50-02-2 |
| Etidium bromide | Sigma-Aldrich | Cat# E7637; CAS Number: 1239-45-8 |
| Hexadimethrine bromide, Polybrene | Sigma-Aldrich | Cat# H9268; CAS Number: 28728-55-4 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148; CAS Number 30525-89-4 |
| RIPA Buffer | Sigma-Aldrich | Cat# R0278 |
| Saponin | Sigma-Aldrich | Cat# 47036; CAS Number: 8047-15-2 |
| Sodium pyruvate | Sigma-Aldrich | Cat# P5280; CAS Number: 113-24-6 |
| Staurosporin | Sigma-Aldrich | Cat# S4400; CAS Number: 62996-74-1 |
| Uridine | Sigma-Aldrich | Cat# U3003; CAS Number 58-96-8 |
| Propidium Iodide Solution | Biolegend | Cat# 421301 |
| 7-AAD Viability Staining Solution | Biolegend | Cat# 420404 |
| Critical commercial assays | ||
| Caspase 3/7 assay | Thermo Fisher Scientific | Cat# R37111 |
| Click-iT® EdU Alexa Fluor® 647 Flow Cytometry Assay Kit, 50 assays | Thermo Fisher Scientific | Cat# C10424 |
| Human Inflammatory Cytokine Kit | BD Bioscience | Cat# 551811 |
| ELISA MAX Deluxe Set Human IL-6 | Biolegend | Cat# 430504 |
| Cell ROS flow cytometry assay kit | Thermo Fisher Scientific | Cat# C100491 |
| Seahorse XFp cell mito stress test kit | Seahorse Bioscience | Cat#103010-10 |
| JetPrime Transfection assay | Polyplus transfection | Cat# 114-07 |
| Cell Fractionation Kit standard | Abcam | Cat# ab109719 |
| Oligonucleotides | ||
| 12S-forward 5′-TAGCCCTAAACCTCAACAGT-3′; reverse 5′-TGCGCTTACTTTGTAGCCTTCAT-3′ | Eurofins Genomics | N/A |
| BACTIN-forward 5′-CAGCGGAACCGCTCATTGCCAATGG-3′; reverse 5′-TCACCCACACTGTGCCCATCTACGA-3′ | Eurofins Genomics | N/A |
| DLOOP-forward 5′-ACCTGTGATCCATCGTGATGT-3′; reverse 5′-GCCCATAACACTTGGGGGTA-3′ | Eurofins Genomics | N/A |
| GAPDH-Hs03929097_g1 | Thermo Fisher Scientific | N/A |
| IFI27-Hs01086370_m1 | Thermo Fisher Scientific | N/A |
| IFI44-Hs00199115_ m1 | Thermo Fisher Scientific | N/A |
| IFIT1-Hs01675197_m1 | Thermo Fisher Scientific | N/A |
| IFNA-forward 5′-GAAGAATCTCTCCTTTCTCCTGCC-3′; reverse 5′-ATGGAGGACAGAGATGGCTTG-3′ | Eurofins Genomics | N/A |
| IFNB-forward 5′-CAGCAATTTTCAGTGTCAGAAGC-3′; reverse 5′-TCATCCTGTCCTTGAGGCAGT-3′ | Eurofins Genomics | N/A |
| ISG15- Hs01921425_s1 | Thermo Fisher Scientific | N/A |
| LPIN1-forward 5′-CCTTCTATGCTGCTTTTGGGAACC-3′; reverse 5′-CTGATCGACCACTTCGCAGAGC-3′ | Eurofins Genomics | N/A |
| RSAD2-Hs00369813_m1 | Thermo Fisher Scientific | N/A |
| SIGLEC1-Hs00988063_m1 | Thermo Fisher Scientific | N/A |
| Recombinant DNA | ||
| pGFP-2XFYVE | Ketel et al., 201666 | N/A |
| pRFP-GFP-LC3 | Kimura et al., 200767 | N/A |
| pRFP-RILP | Jongsma et al., 202068 | N/A |
| pRFP-ARMUS | Carroll et al., 201331 | N/A |
| pTLR9-HA | Invivogen | Cat# puno1ha-htlr9a |
| LPIN1 (Myc-DDK-tagged)-Human lipin1 (LPIN1) ORF clone (NM_145693) | OriGene | Cat# RC207138 |
| Software and algorithms | ||
| FIJI v2.00 | https://fiji.sc | RRID: SCR_002285 |
| FlowJo v9.3.0 (Mac) | https://www.flowjo.com/solutions/flowjo | RRID: SCR_008520 |
| Image Reader LAS-1000 v2.3 | http://www.imb.sinica.edu.tw/core/publicfacilities/pdf/LAS-1000plus_om_e_ver2.pdf | RRID: SCR_014585 |
| GraphPad Prism v6.0 | https://www.graphpad.com/ | RRID: SCR_002798 |
| Icy v1.9.5.1 | http://icy.bioimageanalysis.org/ | RRID: SCR_010587 |
| Imaris v8.4.2 | https://imaris.oxinst.com/packages | RRID: SCR_007370 |
| Soft Flow v3.00 | http://softflow.com/ | RRID: SCR_002599 |
| System Detector System (SDS) v2.3 | https://www.thermofisher.com/order/catalog/product/4444202 | RRID: SCR_015806 |
| Other | ||
| 3 Well Chamber, removable microscopy glass slide, sterilized | Ibidi | Cat# 80381 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yamina Hamel (yamina.hamel@inserm.fr).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Human subjects
Blood samples and skeletal muscle biopsies from six individuals carrying two recessively inherited mutations in LPIN1 and suffering from recurrent episodes of rhabdomyolysis, and from healthy control teenagers and children (30 for blood samples, 6 for skeletal muscle biopsies) were obtained at Necker Hospital, Paris, France. All patients had at least one flare of severe rhabdomyolysis and two causal mutations in the LPIN1 gene and were followed-up in the Department of inherited metabolic diseases of Necker hospital. Age range of subjects was 2-11 years, 4 patients were male and 2 female. Clinical and molecular characteristics of patients are shown in Table S1. Skeletal muscle biopsies were performed in the brachial region (deltoid muscle) for lipin1-deficient patients, or from the dorsal region (dorsal paravertebral muscle) for control individuals with no chronic muscle disease history.
Hydroxychloroquine sulfate treatment
The treatment of 6 patients with Hydroxychloroquine Sulfate treatment was approved by the ethical institutional review board “Ile-de-France II.” Patients received an oral daily dose of 6.5 mg/kg, based on the clinical guidelines for treated pediatric patients suffering from lupus or rheumatoid inflammatory disorders. Informed written consent was obtained from the legal representatives of the children and ophthalmological examination was performed to eliminate the presence of macular pathology, prior to the initiation of the treatment. Patients were followed at regular intervals: 4 weeks after the initiation of the treatment then every 3 months and in the presence of any clinical sign suggesting a flare. All patients underwent clinical examination, and blood samples were collected to measure Hydroxychloroquine Sulfate concentrations, creatine kinase (CK) levels, liver enzymes activity and renal function. Primary outcome was the number of severe rhabdomyolysis (defined by plasma CK levels > 10,000 U/L), upon treatment initiation during the follow up period.
Study approval
Written informed consent was obtained from all individuals in accordance with local regulations and with a human research protocol approved by the ethical institutional review board of the University Paris XI. Experiments using samples from human subjects were conducted in France in accordance with local regulations and with the approval of the ethical institutional review board of the University Paris XI. The biological collection (plasma, myoblasts) has been declared to the Ministère de la Santé.
Primary myoblasts from controls and patients
Primary myoblasts from patients and age- and gender-matched controls were obtained from the cell bank of Cochin Hospital, Paris, France. Primary myoblasts were isolated and grown as described.10 CD56+ myoblasts were isolated by magnetic cell sorting using CD56 MicroBeads (Miltenyi Biotec) and routinely maintained in F10 medium supplemented with 20% Fetal Bovine Serum (FBS) both from Thermo Fisher Scientific on gelatin (Sigma Aldrich) coated tissue culture flasks. After magnetic sorting, purity of cells was assessed by flow cytometry using anti-CD56 Allophycocyanin conjugated antibody (BD Biosciences). All cell lines tested negative for the presence of Mycoplasma at the time of the study.
Generation of monocyte-derived dendritic cells
Monocytes were purified using immunomagnetic CD14 MicroBeads (Miltenyi Biotec) from Peripheral Blood Mononuclear Cells (PBMC) and frozen at −80°C for further experiments. Monocytes were cultured for 7 days in RPMI 1640 medium containing 10% heat-inactivated FBS, 100 IU/mL Penicillin, and 100 μg/mL Streptomycin (all from Thermo Fisher Scientific), 50 ng/mL Granulocyte Macrophage Colony-Stimulating Factor (GM-CSF, Miltenyi Biotec) and 100 ng/mL interleukin (IL)-4 (Miltenyi Biotec) to generate immature DC as previously described.69
Generation of myoblasts deficient for lipin1
An immortalized human myoblast cell line from a healthy donor was obtained from the cell bank. Lentiviral cassettes containing 2 major functional elements, shRNA targeting LPIN1 (Sh-LPIN1) or a scramble sequence (Sh-Sc) and a puromycin selection marker (Origene), were transduced in immortalized myoblasts from 1 healthy control individual for 72 hr and selected with puromycin 5 μg/ml (Thermo Fisher Scientific). Efficacy of lipin1 knock-down was determined by real-time quantitative RT-PCR,10 by confocal imaging and by immunoblot. All cell lines were tested negative for the presence of Mycoplasma at the time of the study.
Generation of myoblasts overexpressing lipin1
A pCMV6 plasmid carrying a Myc-DDK-tagged-human lipin1 ORF clone that contains a puromycin resistance gene was purchased from Origene and used to produce VSV-G Pseudotyped Lentiviral Vector lentiviruses with an average titer of 9.46 × 109 TU/mL. Forty to fifty thousand primary myoblasts/well were harvested into 24-well flat bottom culture plate, exposed to lentivirus infection in the presence of polybrene at 8 μg/mL in a nutrient-rich growth medium at a multiplicity of infection of 100. Sixteen hr later, cells were washed once with culture medium and the day after washed twice and cultured in the presence of 10 μg/mL of puromycin. Two days later, medium was replaced by fresh medium supplemented with 5 μg/mL of puromycin for 2 to 3 days before using primary myoblasts for further experimentation.
Method details
Plasmids and transfection of myoblasts
pGFP-2XFYVE was a gift from Mario Pende (INSERM U1151, Paris, France) and described previously.66 pRFP-RILP was a gift from Jacques Neefjes (Netherlands Cancer Institute, Amsterdam, Netherlands). pRFP-Armus was a gift from Vania Braga (Imperial College London, London, UK) and described elsewhere.31 pRFP-GFP-LC3 was a gift from Patrice Codogno (INSERM U1151, Paris, France) and described previously.67 The plasmid encoding TLR9-HA was purchased from Invivogen.
Vps34 activity and PI3P production assay
Vps34 activity was evaluated by visualization of PI3P production in myoblasts. Myoblasts were seeded on glass coverslips at 20x103 per coverslip and transfected with pGFP-2XFYVE using Jet prime reagent (Polyplus) according to the manufacturer’s instructions before being processed for confocal microscopy 16 hr later. GFP dots corresponding to PI3P were quantified in more than 80 cells per group.
Immunofluorescence and confocal microscopy
Cells plated on glass coverslips were fixed in 4% paraformaldehyde (Sigma Aldrich) in Phosphate Buffered-Saline (PBS) (Thermo Fisher Scientific) for 12 min at 37°C, washed three times with PBS and permeabilized with 0.2% saponin (Sigma Aldrich) in PBS for 10 min. After blocking in PBS 0.2% saponin 0.2% Bovine Serum Albumin 2% Donkey and 2% Goat serum (Sigma Aldrich) for 30 min, myoblasts were incubated with primary antibodies for 60 min at 37°C in 0.2% saponin, 0.2% Bovine Serum Albumin in PBS. After washing, cells further incubated with fluorophore-conjugated secondary antibodies in PBS with 0.2% saponin for 30 min at 37°C, further washed in PBS, stained for DAPI (1 μg/mL) and mounted using Fluoromount-G (Thermo Fisher Scientific). To detect acid compartments or mitochondria, myoblasts were incubated with a growth medium containing 100 mM LystoTracker™ Deep Red (Thermo Fisher Scientific) or 250 nM Mitospy™ Orange CMTMRos for 30 min, followed by incubation in dye-free medium for at least another 30 min and by further processing for the detection of endogenous proteins by confocal imaging. Images were recorded on a confocal Leica SP8 microscope, equipped with a 63X 1.3 numerical aperture (NA) oil immersion objective.
Image analysis
Images were processed using the Imaris software (Bitplane AG v8.4.2). A Gaussian Filter was applied to images using default value. Each cell was then segmented with the area selection tool and considered as a region of interest (ROI). We used the Imaris “Coloc” module to set the threshold for signal positivity based on signal intensity and calculate the overlapping percentage between the 2 signals of interest over the volume analyzed, reflecting a percentage of 3D proximity between the 2 proteins. Threshold calculation was based on manual pre-analysis of 10 cells, determined as compared to signal obtained with the use of appropriate isotype control (background) and applied to all the cells of a same set of experiments. The number of cells or images containing 1 cell or more used for calculation and the number of independent experiments is indicated in each legend. The number of fluorescent dots was quantified by analyzing cells using the Icy software (v1.9.5.1 BioImage Analysis unit, Institut Pasteur, France). After processing on Imaris, images were exported to TIFF files and opened with the FIJI software70 v2.00 (Wayne Rasband National Institutes of Health, USA) transformed into RGB color images. For each key illustration, a representative single z-level was selected, and images of single-color and merged channels were generated. TIFF illustrations for publication were generated by using the “Make montage” feature. The number of fluorescent dots was quantified by analyzing cells using the “Spot detector” module of the Icy software (v1.9.5.1 BioImage Analysis unit, Institut Pasteur, France). The number of fragments of PI3P, PI34P2 and PI35P2 among Rab7 structures was determined as follows. A mask for Rab7 and the given PI species was created using the Imaris Software and the “Add Surfaces” wizard module with the following parameters: a region of interest was segmented before processing entire Image finally; for each channel of fluorescence, thresholding was based on absolute intensity as compared to isotype control or cells stained with fluorochrome-conjugated secondary antibodies only and applied to all the images of actual experiment including patient and control individual cells. Then the number of PI3P, PI34P2 and PI35P2 binary objects within the Rab7 mask was determined using the Imaris “statistics” module.
Fluorescence resonance energy transfer (FRET) microscopy
FRET was measured by using the acceptor photobleaching technique71 where, upon irreversible photobleaching of the acceptor, we record if the donor fluorescence increases. Myoblasts grown for 2 days on Ibidi chambers were fixed and mounted in PBS/glycerol (1:1) for FRET measurements on an SP8 confocal microscope (Leica) with a 63X oil objective with a NA of 1.4. The donor is fused to CFP fluorophore (detected with 405-nm wavelength laser) and the acceptor is fused to YFP fluorophore (detected with 514-nm wavelength laser). For each analysis, a square of 3 × 2 mm was selectively photobleached.
Phosphatidic acid phosphatase (PAP) activity
PAP activity was measured using an enzymatic assay at 37°C for 20 min as described previously.57 Total phosphatidate phosphatase activity was measured in the presence of 1.5 mM MgCl2. Each sample was assayed in a total volume of 100 μL consisting of 100 mM Tris/HCl (pH 7.4) containing 0.6 mM DTT, 1.5 mM MgCl2, 2 mg/ml fat-free BSA, protease inhibitor cocktail, 30 nM microcystin-LR, 0.6 mM PA labeled with [3H]palmitate (approximately 6,000 dpm per assay), 1 mM EDTA/EGTA, 0.4 mM phosphatidylcholine, and 200 μM tetrahydrolipstatin to block lipase activity. Parallel measurements were performed in the absence of Mg2+ and in the presence of 8 mM N-ethylmaleimide to determine the contribution from lipid phosphate phosphatase (LPP) activity. PAP activity was calculated by subtracting the NEM-insensitive LPP activity from the total phosphatidate phosphatase activity. Each sample was assayed at 3 different protein concentrations to ensure a proportional response. One unit of PAP activity was defined as the amount of enzyme that catalyzed the formation of 1 nmol of product/min. Specific activity was defined as units/mg of protein.
EGFR degradation assays
Myoblasts were serum starved overnight then stimulated with 100 ng/ml EGF (Peprotech) in serum-free medium for the indicated time. Cells were rinsed twice with PBS and lysed in RIPA buffer plus 1% SDS, and EDTA (5 mM) all from Sigma Aldrich. The expression of EGFR in total cell lysates was determined by immunoblot using an anti-EGFR monoclonal antibody and normalized to the unstimulated condition. Fluorescence intensity was measured using Luminescent Image Reader LAS-1000 Pro software version 2.3 and an Intelligent Dark Box II (FUJIFILM). EGFR levels, proportional to fluorescence intensity, were then quantified using the FIJI software.
DQ-OVA assays
DQ Ovalbumin (DQ-OVA, Thermo Fisher Scientific) was added (200 μg/ml) to myoblasts in culture plates for 20 min at 4°C. Cells were washed 3 times with ice-cold PBS containing 2% FBS to remove unbound or nonspecifically associated DQ-OVA. Cells were incubated at 4°C for 10 min or for various time intervals at 37°C. Cells were then removed from the plates by trypsin, washed with ice-cold PBS, fixed with 2% paraformaldehyde. Flow cytometric measurements were performed on a BD LSR Fortessa cell analyzer from BD Biosciences. Data were analyzed with FlowJo software (version 9.8.3, Mac).
Multiplex cytokine assay
DC and myoblasts were harvested and transferred (respectively 50x103 and 10x103 cells/well) to 96-well plates and incubated with LipoPolySaccharide LPS (1 μg/mL), flagellin (10 ng/mL), zymosan (1 μg/mL), CpG-A (10 μg/mL), imiquimod (0.5 μg/mL), poly (I:C) (1 μg/mL), or Pam3CSK4 (100 ng/mL), all from Invivogen, for 16 hr. Cytokine concentrations were measured in culture supernatants and plasma using a ten-plex Cytometric Bead Assay (CBA) kit from BD Biosciences, according to the manufacturer’s instructions. In brief, 50 μL of plasma was mixed with 50 μL of mixed capture beads and a standard curve was generated. After 3 hr of incubation at room temperature, the samples were washed and suspended in PBS and then analyzed by flow cytometry (ARIA II; BD Biosciences) with CBA Analysis Software (FCAP Array version 3.0; Soft Flow, St Louis Park, Minnesota). The results are expressed in picograms per milliliter (pg/mL).
IL-6 ELISA assay
DC and myoblasts were harvested and transferred (respectively 50x103 and 10x103 cells/well) to 96-well plates and incubated with LipoPolySaccharide LPS (1 μg/mL), flagellin (10 ng/mL), zymosan (1 μg/mL), CpG-A (10 μg/mL), imiquimod (0.5 μg/mL), poly (I:C) (1 μg/mL), or Pam3CSK4 (100 ng/mL), all from Invivogen, for 16 hr. IL-6 concentration was measured in culture supernatants using a Human IL-6 ELISA MAX Deluxe, according to the manufacturer’s instructions and performing 3 to 4 technical replicates per condition. The results are expressed in picograms per milliliter (pg/mL). Unless otherwise specified in figures or legends, cytokine concentrations in vitro are presented after background subtraction, obtained with cells incubated with vehicle (DMSO or water). In some experiments the TLR9 antagonist ODN TTAGGG (A151) was added 1 hr before addition of stimuli at the concentration of 1 μmol/L.
Gene expression analysis by RT-qPCR
Total RNA was isolated from PBMC, DC and myoblasts using the RNeasy mini Kit and cDNA (QIAGEN) was prepared using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) after depleting genomic DNA. The expression of a set of 6 interferon-stimulated genes (ISGs) in PBMC, DC and myoblasts was assessed by RT-qPCR using Taqman Gene Expression Assays and normalized against GAPDH. Primer-Probes used were as follows IFI27 - Hs01086370_m1, IFI44LHs00199115_ m1, IFIT1 - Hs01675197_m1, RSAD2 - Hs00369813_m1, SIGLEC1- Hs00988063_m1, ISG15 – Hs01921425_s1, GAPDH- Hs03929097_g1 labeled with FAM dye and amplified in a universal reaction mix. Real Time quantitative PCR was performed in duplicate using the Light Cycler VIIA7 System (Roche). The RQ value is equal to 2ΔΔct where ΔΔct is calculated by (Ct target- Ct GAPDH) test sample - (Ct target- Ct GAPDH) calibrator sample. Each value is derived from 3 technical replicates. The expression of LPIN1, IFNA and IFNB genes in myoblasts was assessed by q-RT-PCR using Power SYBR® Green PCR Master Mix and normalized against β-actin (BACT). Reactions were performed in triplicate on an ABI Prism 7000 machine (PE Applied Biosystems) or on an ABI Prism 7900HT Fast Real-time PCR System, equipped with System Detector System (SDS) software v2.3. The RQ value is equal to 2ΔΔct where ΔΔct is calculated by (Ct target- Ct BACT) test sample - (Ct target- Ct β BACT) calibrator sample. Each value is derived from 3 technical replicates.
Mitochondrial DNA gene expression analysis by qPCR
Primary myoblasts were trypsinized and were re-suspended in 200 μL of PBS buffer containing Proteinase K (0.2mg/ml), SDS (0.2%), EDTA (5mM) for 3 hr at 56°C. After isopropanol precipitation, 50 ng of DNA was amplified by Q-PCR using specific primers for 12S, D-LOOP and BACT and Power SYBR® Green PCR Master Mix. Reactions were performed in triplicate and analyzed as described above with a normalization to BACT.
Cholera toxin uptake
Fifteen to twenty thousand primary myoblasts seeded on coverslips were deprived of FBS for 30 min and then loaded with 1 μg/ml cholera toxin subunit B (CTxB) CF568 (Thermo Fisher Scientific) for 45 min at 37°C, followed by a 30-min chase in nutrient-rich growth medium. Cells were then fixed and processed for confocal microscopy.
Autophagic flux analyses
Myoblasts seeded on glass coverslips were transfected as described above with pRFP-GFP-LC3,67 starved by replacing nutrient-rich growth medium with a starvation medium, EBSS medium (Thermo Fisher Scientific) for 3 hr to induce and evaluate autophagy initiation and maturation and refed for 6 hr with nutrient-rich growth medium to evaluate autophagic clearance. At the end of incubation with the different media, cells were washed in PBS, fixed and processed for confocal microscopy. Images were acquired with a Leica SP8 and with a 63x 1.4 NA oil objective. Puncta structures with GFP-RFP and/or RFP signals were quantified in more than 30 images per group, and the percentage of autolysosomes was expressed as the percent of puncta with red color.
Mitophagy flux analyses
To analyze mitophagy initiation, myoblasts were treated with 30 μM CCCP (Sigma-Aldrich) for 30 min and 2 hr. Mitophagosomes including mitochondrial fractions (TOMM20) were examined for recruited LC3 protein by confocal microscopy. To analyze mitophagy resolution, cells were treated with 30 μM CCCP for 8 and 16 hr, and then fixed and immunostained for dsDNA, TFAM. 8OHDG. Nucleoid number was quantified using Icy software, by segmenting and counting dsDNA dots.
Cell fractionation
Cytosolic, mitochondrial and nuclear fractions were prepared using Cell Fractionation Kit from Abcam according to the manufacturer’s instructions and analyzed by qPCR as described above.
Measurements of mitochondrial mass, potential and ROS
Mitochondrial mass and membrane potential (Ψm) were measured by flow cytometry performed on 15,000-20,000 primary myoblasts labeled with the mitochondrion selective dyes MitoTracker Green FM and TMRM (Thermo Fisher Scientific). ROS production in live cells was assessed using the CellROS assay kit (Thermo Fisher Scientific) following the manufacturer’s instructions. After labeling with dyes, myoblasts were detached with trypsin digestion, washed using culture medium and analyzed by flow cytometry 5 min after the addition of 7-AAD. and analyzed by flow cytometry.
Muscle oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurements
Primary myoblasts were plated in cell culture XF96 microplates (Agilent technologies) at 2x104 cells/well and the cellular OCR or ECAR was determined by a Seahorse Bioscience XF96e extracellular flux analyzer (Agilent technologies) according to the manufacturer’s instructions. Cells were balanced for 1 h in unbuffered XF assay media (Agilent Technologies) supplemented for OCR analysis with either 2 mM Glutamine, 10 mM Glucose and 1 mM Sodium Pyruvate or just 2 mM Glutamine for ECAR measurement. For OCR measurements, compounds were injected during the assay at the following final concentrations: Oligomycin (ATP synthase inhibitor, 1 μM), FCCP (uncoupling agent measuring the maximal respiration capacity; 1 μM), Rotenone and Antimycin A (ETC inhibitors; 1 μM). For ECAR measurements, Glucose (10 mM), Oligomycin (1 μM), and 2-Deoxyglucose (2-DG, glycolytic inhibitor; 500 mM) were injected. For each cell line 4 to 6 technical replicates were evaluated. All OCR and ECAR measurements were normalized to the protein concentration dosed at the end of every experiment.
MtDNA depletion
MtDNA depletion was achieved by incubating myoblasts in complete medium with ethidium bromide (100 ng/ml), uridine (50 μg/ml) and sodium pyruvate (110 μg/ml) all from Sigma Aldrich for 5 days to reduce mtDNA levels. Depletion was evaluated by performing qPCR, as described above.
Ca2+-mobilization
Myoblasts seeded on Ibidi flow chambers (Ibidi) were stained with 5 μM Fluo-4 AM (Thermo Fisher Scientific) for 30 min at 37°C in 5% CO2 incubator. After washing cells were cultured in Krebs medium and the flow chambers were then transferred to a temperature-controlled stage (Harvard Apparatus, Holliston) on a confocal Leica SP8 SMD inverted microscope. Cells were visualized using a 20x and 0.7 NA dry objective. In some experiments a TLR9 antagonist ODN TTAGGG (A151) was added 1 hr before addition of stimuli at the concentration of 1 μmol/L.
Caspase 3/7-cell death assay
Myoblasts were cultured with various conditions with a potential for cell death induction at 37°C, in a 5% CO2 incubator, including: EBSS and/or TLR ligands, or staurosporin (Sigma-Aldrich), which was used as a positive control. In some experiments the TLR9 antagonist ODN TTAGGG (A151) was added 1 hr before addition of stimuli at the concentration of 1 μmol/L. Cells were then detached by trypsin, transferred into 5 mL FACS tubes and stained in culture medium at room temperature for 1 hr with 5 μM of the Caspase3/7 Green Detection Reagent (Thermo Fisher Scientific). Five min before fluorescence acquisition, Propidium Iodide (Biolegend) or 7-AAD (Biolegend) according to the manufacturer’s instructions, to discriminate between necrotic and apoptotic cells. The fluorescence was recorded on a BD LSR Fortessa flow cytometer and analyzed using the FlowJo software.
Myoblasts were gated on based on FSC and SSC characteristics and single cells were further identified based on successive gating on homogeneous FSC-A and FSC-H characteristics as well as SSC-A and SSC-H characteristics. A dot plot showing the fluorescence intensity for 7-AAD or Propidium Iodide versus caspase3/7 activity was then constructed and a quadrant gate to discriminate live (7-AAD-/caspase3/7-) from apoptotic (7-AAD-/caspase3/7+) and necrotic (7-AAD+/caspase3/7+/−) cells was drawn. Dead cells were identified as cells contained within apoptotic and/or necrotic cell gates.
Cell proliferation assay
Primary myoblasts were cultured in Dulbecco’s Modified Eagle’s Medium – high (4,500 mg/mL) or low glucose (1,000 mg/mL), all from Sigma-Aldrich, supplemented with 20% FBS, 100 IU/mL Penicillin and 100 μg/mL Streptomycin for 5 days. The proliferation of myoblasts was then evaluated using the Click-iT Plus EdU Flow Cytometry Assay Kits according to the manufacturer’s instructions. Myoblasts were exposed to 5 μM EdU for 1 hr. The fluorescence was recorded on a BD LSR Fortessa flow cytometer and analyzed using the FlowJo software.
Quantification and statistical analysis
Data are presented as mean ± SD (n ≤ 10 values per group; dot plots) or median, 10th percentile and 90th percentile (n > 10 values per group; box and whisker plots) for continuous variables. Statistical analysis was performed with GraphPad Prism v6.0 (GraphPad Software). The normality of the data distribution was tested in series with n ≥ 20 values by using the Shapiro-Wilk test. To determine statistical significance for comparison of 1 continuous variable between 2 groups, a non-parametric unpaired two-tailed Mann-Whitney test was applied to series with n ≤ 20 values per group or to larger series with a non-normal distribution and a parametric unpaired two-tailed t test was applied to series with n > 20 values and with a normal distribution of the data. When possible exact p values were indicated. The alpha value for threshold of significance was pre-fixed to 0.05. Two-tailed two-way analysis of variance (ANOVA) test with post hoc Sidak’s correction for multiple comparison was performed to determine statistical significance for comparison of 2 or more factors between 2 or 3 series, by indicating for the mean effect of interaction, the mean effect of factors the F-statistics, degrees of freedom and exact p value. In addition, paired two-way ANOVA test was used when within-subjects factor was under evaluation (time). In all experiments, post hoc comparison was performed to test statistical significance, with adjusted p values denoted directly on the figures. Family-wise significance and confidence level were pre-fixed to 0.05 and 95% respectively. All relevant details about statistical tests and analysis can be found in figure legends. For the small clinical trial, no sample size was pre-determined, as dealing with a very rare disorder and all patients suffering from lipin1-related rhabdomyolysis and followed up in the Department of Inherited Metabolic Disorders, Necker hospital, Paris, France were proposed for enrolment. No subject or data was excluded from analysis of the data of the clinical trial. FACS data with less than 500 events in the final gates analyzed were excluded from analysis. Immunofluorescence data with less than 100 cells visible on the slides were excluded from analysis. For in vitro experiments, randomization was not applicable in comparing patient versus control individuals. Evaluation of the results of the small open clinical trial in which all the patients enrolled received hydroxychloroquine sulfate was based on before/after observations, so that randomization procedure was not applicable. Flow cytometry and immunofluorescence experiments data were gathered blinded to the status of the cells (i.e., control or patient cells).
Additional resources
The clinical trial has been registered on clinicaltrials.gov under the number NCT04007562 (https://clinicaltrials.gov/ct2/show/NCT04007562).
Acknowledgments
This work was supported by grants to P.d.L. from Agence Nationale de la Recherche (ANR-13-BSV1-0020-01), the Fondation Bettencourt (2012), the Fondation Lejeune (grant 2014), and patient associations (Nos Anges, AMMI, Hyperinsulinisme, OPPH, Noa Luû); grants to P.d.L. and Y.H. from the Association Française contre les Myopathies (grant nos. 13988 and 15947); grant 14-CE11-0014 from the ANR to P.v.E.; and grant AO2017-5 from the patient association Vaincre les Maladies Lysosomales, to F.-X.M. F.-X.M. was supported by a Poste d’Accueil Inserm.We thank Arnaud Hubas from the Department of Biology and Molecular Genetics, Cochin Hospital, for assistance in isolating human myoblasts; Sylvie Fabrega (Viral Vector for Gene Transfer core facility, INSERM US24/CNRS UMS3633) for production of the VSV-G pseudotyped lentiviral vector particles; Meriem Garfa-Traoré from the cell imaging platform, for assistance in confocal imaging; Gillian Rice and Yanick Crow for their help in qPCR experiments and analysis of type I interferon-related genes; Patrice Codogno, Nicolas Dupont, Vania Braga, and Jacques Neefjes for providing plasmids; the MYOBANK-AFM (myology institute BB-0033); Laure Cacavelli of the Department of Biotherapy, Necker Hospital, for technical assistance; and Fatima Djouadi and Jean Bastin for providing myoblasts. The authors are grateful to Mario Pende, Timothy Wai, Erika Brunet, Patrice Codogno, and Nicolas Dupont for helpful discussions. The authors thank Salvatore Cisternino, Philippe Henri Secretan, and Joel Schlatter from the Department of Pharmacy, Necker Hospital, for the preparation of hydroxychloroquine sulfate; Antoine Legendre from the Cardiology Department, Necker Hospital, for the monitoring of patients; and Benoit Blanchet from the Department of Pharmacology, Cochin Hospital, and Noël Zahr from the Department of Pharmacology, Pitié-Salpétrière Hospital, for dosing of HS.
Author contributions
Conceptualization, Y.H., F.-X.M., P.d.L., and P.v.E. Methodology, Y.H., F.-X.M., C.L., X.T., D.N.B., P.v.E., and P.d.L. Investigation, Y.H., F.-X.M., M.M., P.R., C.L., N.G., I.N., M.P.R., X.T., O.P., and C.T.-D. Resources, I.N., M.P.R., X.T., D.N.B., and P.N. Writing – original draft, Y.H., F.-X.M., and P.v.E. Writing – review & editing, Y.H., F.-X.M., P.v.E. and P.d.L. Supervision, Y.H., F.-X.M., P.v.E., and P.d.L. Funding acquisition, Y.H., F.-X.M., P.v.E., and P.d.L.
Declaration of interests
Y.H., F.-X.M., C.L., P.v.E., and P.d.L. have filed PCT (WO/2017/085115; EP3377095; PCT/ EP2016/077843) and US patent applications (US20180325890). Y.H., F.-X.M., M.M., P.v.E., and P.d.L. have filed PCT (WO/2019/020732; PCT/EP2018/070256) patent applications. The remaining authors declare no competing interests.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this article self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: August 17, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100370.
Supplemental information
Data and code availability
This study did not generate/analyze datasets/code.
References
- 1.Cervellin G., Comelli I., Lippi G. Rhabdomyolysis: historical background, clinical, diagnostic and therapeutic features. Clin. Chem. Lab. Med. 2010;48:749–756. doi: 10.1515/CCLM.2010.151. [DOI] [PubMed] [Google Scholar]
- 2.Luck R.P., Verbin S. Rhabdomyolysis: a review of clinical presentation, etiology, diagnosis, and management. Pediatr. Emerg. Care. 2008;24:262–268. doi: 10.1097/PEC.0b013e31816bc7b7. [DOI] [PubMed] [Google Scholar]
- 3.Michot C., Hubert L., Brivet M., De Meirleir L., Valayannopoulos V., Müller-Felber W., Venkateswaran R., Ogier H., Desguerre I., Altuzarra C. LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum. Mutat. 2010;31:E1564–E1573. doi: 10.1002/humu.21282. [DOI] [PubMed] [Google Scholar]
- 4.Baba T., Kashiwagi Y., Arimitsu N., Kogure T., Edo A., Maruyama T., Nakao K., Nakanishi H., Kinoshita M., Frohman M.A. Phosphatidic acid (PA)-preferring phospholipase A1 regulates mitochondrial dynamics. J. Biol. Chem. 2014;289:11497–11511. doi: 10.1074/jbc.M113.531921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donkor J., Sariahmetoglu M., Dewald J., Brindley D.N., Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J. Biol. Chem. 2007;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- 6.Pascual F., Carman G.M. Phosphatidate phosphatase, a key regulator of lipid homeostasis. Biochim. Biophys. Acta. 2013;1831:514–522. doi: 10.1016/j.bbalip.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasser T., Qiu Q.S., Karunakaran S., Padolina M., Reyes A., Flood B., Smith S., Gonzales C., Fratti R.A. Yeast lipin 1 orthologue pah1p regulates vacuole homeostasis and membrane fusion. J. Biol. Chem. 2012;287:2221–2236. doi: 10.1074/jbc.M111.317420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rashid T., Nemazanyy I., Paolini C., Tatsuta T., Crespin P., de Villeneuve D., Brodesser S., Benit P., Rustin P., Baraibar M.A. Lipin1 deficiency causes sarcoplasmic reticulum stress and chaperone-responsive myopathy. EMBO J. 2019;38:e99576. doi: 10.15252/embj.201899576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schweitzer G.G., Collier S.L., Chen Z., McCommis K.S., Pittman S.K., Yoshino J., Matkovich S.J., Hsu F.F., Chrast R., Eaton J.M. Loss of lipin 1-mediated phosphatidic acid phosphohydrolase activity in muscle leads to skeletal myopathy in mice. FASEB J. 2019;33:652–667. doi: 10.1096/fj.201800361R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michot C., Mamoune A., Vamecq J., Viou M.T., Hsieh L.S., Testet E., Lainé J., Hubert L., Dessein A.F., Fontaine M. Combination of lipid metabolism alterations and their sensitivity to inflammatory cytokines in human lipin-1-deficient myoblasts. Biochim. Biophys. Acta. 2013;1832:2103–2114. doi: 10.1016/j.bbadis.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelosi M., Testet E., Le Lay S., Dugail I., Tang X., Mabilleau G., Hamel Y., Madrange M., Blanc T., Odent T. Normal human adipose tissue functions and differentiation in patients with biallelic LPIN1 inactivating mutations. J. Lipid Res. 2017;58:2348–2364. doi: 10.1194/jlr.P075440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finck B.N., Gropler M.C., Chen Z., Leone T.C., Croce M.A., Harris T.E., Lawrence J.C., Jr., Kelly D.P. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Peterson T.R., Sengupta S.S., Harris T.E., Carmack A.E., Kang S.A., Balderas E., Guertin D.A., Madden K.L., Carpenter A.E., Finck B.N., Sabatini D.M. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang P., Verity M.A., Reue K. Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab. 2014;20:267–279. doi: 10.1016/j.cmet.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meana C., Peña L., Lordén G., Esquinas E., Guijas C., Valdearcos M., Balsinde J., Balboa M.A. Lipin-1 integrates lipid synthesis with proinflammatory responses during TLR activation in macrophages. J. Immunol. 2014;193:4614–4622. doi: 10.4049/jimmunol.1400238. [DOI] [PubMed] [Google Scholar]
- 16.Bergounioux J., Brassier A., Rambaud C., Bustarret O., Michot C., Hubert L., Arnoux J.B., Laquerriere A., Bekri S., Galene-Gromez S. Fatal rhabdomyolysis in 2 children with LPIN1 mutations. J. Pediatr. 2012;160:1052–1054. doi: 10.1016/j.jpeds.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 17.Kim H.B., Kumar A., Wang L., Liu G.H., Keller S.R., Lawrence J.C., Jr., Finck B.N., Harris T.E. Lipin 1 represses NFATc4 transcriptional activity in adipocytes to inhibit secretion of inflammatory factors. Mol. Cell. Biol. 2010;30:3126–3139. doi: 10.1128/MCB.01671-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyttinen J.M., Niittykoski M., Salminen A., Kaarniranta K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim. Biophys. Acta. 2013;1833:503–510. doi: 10.1016/j.bbamcr.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 19.Bissa B., Deretic V. Autophagosome Formation: Cutting the Gordian Knot at the ER. Curr. Biol. 2018;28:R347–R349. doi: 10.1016/j.cub.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 20.Schink K.O., Tan K.W., Stenmark H. Phosphoinositides in Control of Membrane Dynamics. Annu. Rev. Cell Dev. Biol. 2016;32:143–171. doi: 10.1146/annurev-cellbio-111315-125349. [DOI] [PubMed] [Google Scholar]
- 21.Jaber N., Mohd-Naim N., Wang Z., DeLeon J.L., Kim S., Zhong H., Sheshadri N., Dou Z., Edinger A.L., Du G. Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase-activating protein Armus. J. Cell Sci. 2016;129:4424–4435. doi: 10.1242/jcs.192260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong Z., Pedersen N.M., Wang L., Torgersen M.L., Stenmark H., Raiborg C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J. Cell Biol. 2017;216:4217–4233. doi: 10.1083/jcb.201611073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krauß M., Haucke V. A grab to move on: ER-endosome contacts in membrane protrusion formation and neurite outgrowth. EMBO J. 2015;34:1442–1444. doi: 10.15252/embj.201591553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pankiv S., Alemu E.A., Brech A., Bruun J.A., Lamark T., Overvatn A., Bjørkøy G., Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemmon M.A. Phosphoinositide recognition domains. Traffic. 2003;4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 26.Kümmel D., Ungermann C. Principles of membrane tethering and fusion in endosome and lysosome biogenesis. Curr. Opin. Cell Biol. 2014;29:61–66. doi: 10.1016/j.ceb.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 27.Guerra F., Bucci C. Multiple Roles of the Small GTPase Rab7. Cells. 2016;5:E34. doi: 10.3390/cells5030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Luca M., Bucci C. A new V-ATPase regulatory mechanism mediated by the Rab interacting lysosomal protein (RILP) Commun. Integr. Biol. 2014;7:e971572. doi: 10.4161/cib.29616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cabukusta B., Neefjes J. Mechanisms of lysosomal positioning and movement. Traffic. 2018;19:761–769. doi: 10.1111/tra.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pankiv S., Johansen T. FYCO1: linking autophagosomes to microtubule plus end-directing molecular motors. Autophagy. 2010;6:550–552. doi: 10.4161/auto.6.4.11670. [DOI] [PubMed] [Google Scholar]
- 31.Carroll B., Mohd-Naim N., Maximiano F., Frasa M.A., McCormack J., Finelli M., Thoresen S.B., Perdios L., Daigaku R., Francis R.E. The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev. Cell. 2013;25:15–28. doi: 10.1016/j.devcel.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K., Klionsky D.J. Mitochondria removal by autophagy. Autophagy. 2011;7:297–300. doi: 10.4161/auto.7.3.14502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ugrankar R., Liu Y., Provaznik J., Schmitt S., Lehmann M. Lipin is a central regulator of adipose tissue development and function in Drosophila melanogaster. Mol. Cell. Biol. 2011;31:1646–1656. doi: 10.1128/MCB.01335-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West A.P., Shadel G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017;17:363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caielli S., Athale S., Domic B., Murat E., Chandra M., Banchereau R., Baisch J., Phelps K., Clayton S., Gong M. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016;213:697–713. doi: 10.1084/jem.20151876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roers A., Hiller B., Hornung V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity. 2016;44:739–754. doi: 10.1016/j.immuni.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Ohto U., Ishida H., Shibata T., Sato R., Miyake K., Shimizu T. Toll-like Receptor 9 Contains Two DNA Binding Sites that Function Cooperatively to Promote Receptor Dimerization and Activation. Immunity. 2018;48:649–658.e4. doi: 10.1016/j.immuni.2018.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Fang C., Wei X., Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. 2016;7:11–16. doi: 10.1007/s13238-015-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torres P.A., Helmstetter J.A., Kaye A.M., Kaye A.D. Rhabdomyolysis: pathogenesis, diagnosis, and treatment. Ochsner J. 2015;15:58–69. [PMC free article] [PubMed] [Google Scholar]
- 40.Jordens I., Fernandez-Borja M., Marsman M., Dusseljee S., Janssen L., Calafat J., Janssen H., Wubbolts R., Neefjes J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001;11:1680–1685. doi: 10.1016/s0960-9822(01)00531-0. [DOI] [PubMed] [Google Scholar]
- 41.Saridaki T., Nippold M., Dinter E., Roos A., Diederichs L., Fensky L., Schulz J.B., Falkenburger B.H. FYCO1 mediates clearance of α-synuclein aggregates through a Rab7-dependent mechanism. J. Neurochem. 2018;146:474–492. doi: 10.1111/jnc.14461. [DOI] [PubMed] [Google Scholar]
- 42.Johnson D.E., Ostrowski P., Jaumouillé V., Grinstein S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016;212:677–692. doi: 10.1083/jcb.201507112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakamura S., Yoshimori T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017;130:1209–1216. doi: 10.1242/jcs.196352. [DOI] [PubMed] [Google Scholar]
- 44.Filimonenko M., Isakson P., Finley K.D., Anderson M., Jeong H., Melia T.J., Bartlett B.J., Myers K.M., Birkeland H.C., Lamark T. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell. 2010;38:265–279. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Napoli E., Song G., Panoutsopoulos A., Riyadh M.A., Kaushik G., Halmai J., Levenson R., Zarbalis K.S., Giulivi C. Beyond autophagy: a novel role for autism-linked Wdfy3 in brain mitophagy. Sci. Rep. 2018;8:11348. doi: 10.1038/s41598-018-29421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cullup T., Kho A.L., Dionisi-Vici C., Brandmeier B., Smith F., Urry Z., Simpson M.A., Yau S., Bertini E., McClelland V. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 2013;45:83–87. doi: 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hori I., Otomo T., Nakashima M., Miya F., Negishi Y., Shiraishi H., Nonoda Y., Magara S., Tohyama J., Okamoto N. Defects in autophagosome-lysosome fusion underlie Vici syndrome, a neurodevelopmental disorder with multisystem involvement. Sci. Rep. 2017;7:3552. doi: 10.1038/s41598-017-02840-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacVicar T., Ohba Y., Nolte H., Mayer F.C., Tatsuta T., Sprenger H.G., Lindner B., Zhao Y., Li J., Bruns C. Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature. 2019;575:361–365. doi: 10.1038/s41586-019-1738-6. [DOI] [PubMed] [Google Scholar]
- 49.Kameoka S., Adachi Y., Okamoto K., Iijima M., Sesaki H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2018;28:67–76. doi: 10.1016/j.tcb.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugiura A., McLelland G.L., Fon E.A., McBride H.M. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142–2156. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park B., Brinkmann M.M., Spooner E., Lee C.C., Kim Y.M., Ploegh H.L. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol. 2008;9:1407–1414. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oka T., Hikoso S., Yamaguchi O., Taneike M., Takeda T., Tamai T., Oyabu J., Murakawa T., Nakayama H., Nishida K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shintani Y., Kapoor A., Kaneko M., Smolenski R.T., D’Acquisto F., Coppen S.R., Harada-Shoji N., Lee H.J., Thiemermann C., Takashima S. TLR9 mediates cellular protection by modulating energy metabolism in cardiomyocytes and neurons. Proc. Natl. Acad. Sci. USA. 2013;110:5109–5114. doi: 10.1073/pnas.1219243110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao M., Liu X., Li D., Chen T., Cai Z., Cao X. Late endosome/lysosome-localized Rab7b suppresses TLR9-initiated proinflammatory cytokine and type I IFN production in macrophages. J. Immunol. 2009;183:1751–1758. doi: 10.4049/jimmunol.0900249. [DOI] [PubMed] [Google Scholar]
- 56.Akbar M.A., Mandraju R., Tracy C., Hu W., Pasare C., Krämer H. ARC Syndrome-Linked Vps33B Protein Is Required for Inflammatory Endosomal Maturation and Signal Termination. Immunity. 2016;45:267–279. doi: 10.1016/j.immuni.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kok B.P., Kienesberger P.C., Dyck J.R., Brindley D.N. Relationship of glucose and oleate metabolism to cardiac function in lipin-1 deficient (fld) mice. J. Lipid Res. 2012;53:105–118. doi: 10.1194/jlr.M019430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Senft D., Ronai Z.A. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem. Sci. 2015;40:141–148. doi: 10.1016/j.tibs.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y., Nguyen P.T., Wang X., Zhao Y., Meacham C.E., Zou Z., Bordieanu B., Johanns M., Vertommen D., Wijshake T. TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature. 2020;578:605–609. doi: 10.1038/s41586-020-1992-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K.J., Coppes R.P., Engedal N., Mari M., Reggiori F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaanine A.H., Gordon R.E., Nonnenmacher M., Kohlbrenner E., Benard L., Hajjar R.J. High-dose chloroquine is metabolically cardiotoxic by inducing lysosomes and mitochondria dysfunction in a rat model of pressure overload hypertrophy. Physiol. Rep. 2015;3:e12413. doi: 10.14814/phy2.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuznik A., Bencina M., Svajger U., Jeras M., Rozman B., Jerala R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 2011;186:4794–4804. doi: 10.4049/jimmunol.1000702. [DOI] [PubMed] [Google Scholar]
- 63.Sacre K., Criswell L.A., McCune J.M. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther. 2012;14:R155. doi: 10.1186/ar3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Torigoe M., Sakata K., Ishii A., Iwata S., Nakayamada S., Tanaka Y. Hydroxychloroquine efficiently suppresses inflammatory responses of human class-switched memory B cells via Toll-like receptor 9 inhibition. Clin. Immunol. 2018;195:1–7. doi: 10.1016/j.clim.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Hoene V., Peiser M., Wanner R. Human monocyte-derived dendritic cells express TLR9 and react directly to the CpG-A oligonucleotide D19. J. Leukoc. Biol. 2006;80:1328–1336. doi: 10.1189/jlb.0106011. [DOI] [PubMed] [Google Scholar]
- 66.Ketel K., Krauss M., Nicot A.S., Puchkov D., Wieffer M., Müller R., Subramanian D., Schultz C., Laporte J., Haucke V. A phosphoinositide conversion mechanism for exit from endosomes. Nature. 2016;529:408–412. doi: 10.1038/nature16516. [DOI] [PubMed] [Google Scholar]
- 67.Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 68.Jongsma M.L., Bakker J., Cabukusta B., Liv N., van Elsland D., Fermie J. SKIP-HOPS recruits TBC1D15 for a Rab7-to-Arl8b identity switch to control late endosome transport. EMBO J. 2020;39 doi: 10.15252/embj.2019102301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamel Y., Blake N., Gabrielsson S., Haigh T., Jooss K., Martinache C., Caillat-Zucman S., Rickinson A.B., Hacein-Bey S., Fischer A., Cavazzana-Calvo M. Adenovirally transduced dendritic cells induce bispecific cytotoxic T lymphocyte responses against adenovirus and cytomegalovirus pp65 or against adenovirus and Epstein-Barr virus EBNA3C protein: a novel approach for immunotherapy. Hum. Gene Ther. 2002;13:855–866. doi: 10.1089/10430340252899028. [DOI] [PubMed] [Google Scholar]
- 70.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kenworthy A.K., Edidin M. Imaging fluorescence resonance energy transfer as probe of membrane organization and molecular associations of GPI-anchored proteins. Methods Mol. Biol. 1999;116:37–49. doi: 10.1385/1-59259-264-3:37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.