

SUMMARY
HIV-1-negative factor (Nef) protein antagonizes serine incorporator 5 (SERINC5) by redirecting this potent restriction factor to the endosomes and lysosomes for degradation. However, the precise mechanism remains unclear. Using affinity purification/mass spectrometry, we identify cyclin K (CycK) and cyclin-dependent kinase 13 (CDK13) as a Nef-associated kinase complex. CycK/CDK13 phosphorylates the serine at position 360 (S360) in SERINC5, which is required for Nef downregulation of SERINC5 from the cell surface and its counteractivity of the SERINC5 antiviral activity. To understand the role of S360 phosphorylation, we generate chimeric proteins between CD8 and SERINC5 to study their response to Nef. Nef not only downregulates but, importantly, also binds to this chimera in an S360-dependent manner. Thus, S360 phosphorylation increases interactions between Nef and SERINC5 and initiates the destruction of SERINC5 by the endocytic machinery.
Graphical abstract
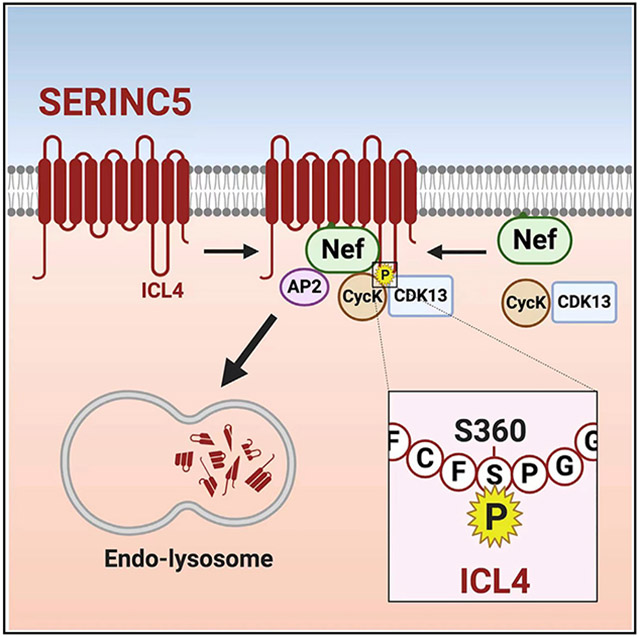
In brief
Chai et al. show that CycK/CDK13 is a Nef-associated serine kinase complex. They provide additional insights into how Nef antagonizes SERINC5 via the endocytic machinery for optimal HIV-1 infectivity by identifying S360 as the SERINC5 phosphorylation site, which is required for its binding to Nef and its subsequent downregulation.
INTRODUCTION
The accessory negative factor (Nef) protein is expressed by primate lentiviruses and functions to promote immune evasion, and subsequent viral fitness, by antagonizing a number of host cell-surface proteins, such as CD4 and major histocompatibility complex class I (MHC-I) (Kirchhoff, 2010). Recently, Nef was also found to antagonize three of the multi-pass transmembrane proteins representing the human serine incorporator (SERINC) family, whose five total members include SERINC1 through 5 (Inuzuka et al., 2005). SERINC5 and, to a lesser degree, SERINC3 are specifically targeted by Nef in order to increase HIV-1 infectivity (Rosa et al., 2015; Usami et al., 2015). SERINC4 also has a Nef-sensitive anti-HIV-1 activity, but it is rapidly turned over by the host cell proteasomes (Qiu et al., 2020).
In a manner that is conserved across different species, SERINC5 is structurally composed of 10 transmembrane domains (TMDs), in an arrangement that forms five extracellular loops (ECLs) and four intracellular loops (ICLs) (Pye et al., 2020). In the absence of Nef, SERINC5 is incorporated into budding HIV-1 virions and subsequently inhibits viral entry into host cells by attacking HIV-1 Env trimers, in their open confirmation, thereby blocking the fusion pore formation between virions and target cells (Beitari et al., 2017; Chen et al., 2020; Sood et al., 2017; Zhang et al., 2019). The potency of SERINC5 antiretroviral activity has been validated in SERINC5-null mice (Timilsina et al., 2020).
HIV-1 Nef antagonizes the antiretroviral effect of SERINC5 by downregulating it from the cell surface, thereby preventing its virion incorporation, in a manner that is analogous to Nef-mediated CD4 downregulation (Rosa et al., 2015; Usami et al., 2015). The process is dependent on binding between the C-terminal dileucine motif 160ExxxLL165 of Nef and adaptor protein 2 (AP-2) (Rosa et al., 2015) as well as an interaction of the Nef N-terminal 32–39 amino acids (aa’s) with SERINC5 (Ananth et al., 2019). These Nef N-terminal residues are located in a multifunctional hydrophobic pocket that is also required for binding to the cytoplasmic tails of CD4 and MHC-I (Kwon et al., 2020). The assembly of SERINC5 and CD4 with the Nef/AP-2 complex leads to their endocytosis and subsequent lysosome-mediated degradation (daSilva et al., 2009; Shi et al., 2018).
The importance of Nef-mediated antagonism of SERINC5 is illustrated by both its conservation across primate lentiviruses and the fact that the global spread of primate lentiviruses is directly correlated to the potency of Nef-mediated SERINC5 antagonism (Heigele et al., 2016). SERINC5 is also counteracted by murine leukemia virus (MLV) glycoGag and by the equine infectious anemia virus (EIAV) S2 protein (Ahi et al., 2016; Chande et al., 2016; Rosa et al., 2015). Although glycoGag and S2 do not share any homology with Nef, they also target SERINC5 to endosomes and lysosomes for degradation (Ahmad et al., 2019; Li et al., 2019). Finally, the clinical relevance of SERINC5 antagonism is underscored by the observation that Nef genes isolated from HIV-1 controllers antagonize SERINC5 poorly when compared to those from progressors (Jin et al., 2019). Thus, SERINC5 is an important restriction factor whose antiretroviral activity has been demonstrated in vitro and in vivo.
Cyclin-dependent kinases (CDKs) belong to a family of serine/threonine kinases whose activities are regulated by cyclin (Cyc) subunits (Lim and Kaldis, 2013). The human genome encodes 21 CDKs and 29 Cycs, the functions of which can be separated into two major categories. Whereas CDK1, CDK2, CDK4, and CDK6 regulate the cell cycle, CDK7, CDK8, CDK9, CDK11, CDK12, and CDK13 play an essential role in transcription (Fan et al., 2020). CDK12, in conjunction with the regulatory subunit CycK (Bartkowiak et al., 2010; Blazek et al., 2011), phosphorylates the C-terminal domain of RNA polymerase II (Bösken et al., 2014; Greifenberg et al., 2016) and consequently regulates genome stability and RNA processing. Although CDK13-mediated phosphorylation proceeds in a manner similar to CDK12 via CycK, the precise cellular function of CDK13 remains largely unknown (Greenleaf, 2019). In this study, we demonstrate that CycK/CDK13 phosphorylates SERINC5 and this phosphorylation is required for Nef antagonism of SERINC5.
RESULTS
Identification of CycK/CDK13 as a Nef-associated kinase complex via mass spectrometry
Initially, FLAG-tagged SERINC5 (Ser5) protein was expressed alone in HEK293T cells and purified by an anti-FLAG affinity column. When the eluted proteins were analyzed by SDS-PAGE, purified monomeric SERINC5 with a molecular mass of ~45 kDa was detected via Coomassie brilliant blue staining (Figure 1A). Next, SERINC5 was purified again after its co-expression with a wild-type (WT) or Nef-deficient (ΔNef) HIV-1 proviral vector. To avoid SERINC5 degradation by Nef, the amounts of proviral vectors were minimized. The monomeric SERINC5 was purified again but, notably, an ~170-kDa protein was co-purified with SERINC5 in the presence of Nef (Figure 1B, lane 3, labeled with an asterisk). To identify this large protein, purified proteins were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). CDK13 was one of six large proteins identified that ranged in size from 150 to 200 kDa (Figure 1C). Because CDK13 interacts with CycK and, importantly, CycK is a well-characterized Nef-binding protein (Khan and Mitra, 2011), CycK and CDK13 were selected for further study.
Figure 1. Identification of CycK/CDK13 as a Nef-associated kinase complex via mass spectrometry.

(A) FLAG-tagged SERINC5 protein was expressed in HEK293T cells and purified by anti-FLAG M2 affinity chromatography. After SDS-PAGE, proteins from total cell lysate and three eluted fractions were analyzed after being stained with Coomassie brilliant blue. The monomeric SERINC5 band is labeled.
(B) FLAG-tagged SERINC5 was expressed with a WT or ΔNef HIV-1 proviral vector in HEK293T cells and purified and analyzed similar to as in (A). The monomeric SERINC5 band is labeled and a protein band at ~170 kDa is indicated by an asterisk.
(C) purified proteins from (B) were analyzed by liquid chromatography-mass spectrometry (LC-MS). Control experiments were conducted using beads that were not conjugated with any antibodies. Six proteins with molecular masses of 150–200 kDa that are not found in the control experiments are listed.
(D) FLAG-tagged CycK was expressed with WT or ΔNef HIV-1 in HEK293T cells. Proteins were immunoprecipitated with an anti-FLAG antibody and analyzed by western blotting (WB). CycK was detected by an anti-FLAG antibody and Nef and GAPDH were detected by their specific antibodies. IP, immunoprecipitation; Input, cell lysate.
(E) FLAG-tagged CycK was expressed with HA-tagged CDK12 or CDK13 in HEK293T cells. Proteins were immunoprecipitated and analyzed as in (D). CDK12 and CDK13 were detected by an anti-HA antibody.
(F) FLAG-tagged CDK13 was expressed with HA-tagged SERINC5 in the presence of WT or ΔNef HIV-1 in HEK293T cells. Proteins were immunoprecipitated and analyzed as in (D) and (E).
All experiments were repeated twice, and similar results were obtained.
First, we co-expressed FLAG-tagged CycK with a WT or ΔNef HIV-1 proviral vector. Proteins were pulled down by anti-FLAG antibodies and analyzed by western blotting (WB). CycK specifically pulled down Nef (Figure 1D, lane 1). The same CycK was also co-expressed with hemagglutinin (HA)-tagged CDK12 or CDK13. Three major CDK12 and CDK13 species were detected at 100–200 kDa. CycK pulled down all these CDK12 and CDK13 proteins, although proteins with the highest molecular mass were pulled down more effectively (Figure 1E, lanes 2 and 4). Thus, we confirmed the Nef and CycK, the CycK and CDK12, and the CycK and CDK13 interactions as reported previously.
Second, we expressed FLAG-tagged CDK13 with HA-tagged SERINC5 proteins in the presence of reduced amounts of a WT and ΔNef HIV-1 proviral vector. When proteins were pulled down again by anti-FLAG antibodies, CDK13 associated with SERINC5 only in the presence of Nef (Figure 1F, lane 1). Thus, Nef should serve as an adaptor that bridges SERINC5 with CycK/CDK13.
CycK/CDK13 is required for Nef downregulation of SERINC5
We examined how CycK/CDK13 affects Nef downregulation of SERINC5 by silencing the endogenous CycK gene (encoded by CCNK). Because CycK is essential for cell survival (Dai et al., 2012), we used CRISPR interference (CRISPRi) to knock down its expression. Catalytically dead Cas9 (dCas9) and CCNK-specific guide RNA (gRNA) were co-expressed in HEK293T cells, and their knockdown (KD) of CCNK was assessed by WB. Unlike the ectopic CycK protein that was detected at ~55 kDa (Figure 1D), endogenous CycK protein was detected at ~55 kDa and ~75 kDa (Figure S1A). Both of these CycK proteins were reduced by CCNK-CRISPRi in a dose-dependent manner (Figure S1A).
CCNK-KD was reported to stall the cell cycle in the G1 phase (Lei et al., 2018). Indeed, when dCas9/CCNK-gRNA were co-expressed in human CD4+ Jurkat T cells, the fraction of cells in G2 was decreased in a dose-dependent manner (Figure S1B). Importantly, under such CCNK-KD conditions, Nef was unable to decrease SERINC5 protein expression in HEK293T cells (Figure 2A, upper panel, lanes 1 and 3). We reported that Nef also decreases SERINC5 protein expression in Jurkat cells (Shi et al., 2018). When the endogenous CycK was similarly targeted by CCNK-CRISPRi in Jurkat cells, the Nef activity also diminished (Figure 2A, lower panel). Measurement of SERINC5 expression on the cell surface produced similar results. Nef effectively decreased both the number of SERINC5-positive cells (Figure 2B) and the mean fluorescence intensity (MFI) of these positive cell populations (Figure 2E). However, this Nef activity was disrupted by CCNK-KD. Nef also downregulates CD4 from the cell surface (Shi et al., 2018). However, CCNK-KD had no effect on Nef downregulation of CD4 (Figure S1C, lanes 1 and 3). Thus, the CycK effect is highly specific for Nef downregulation of SERINC5.
Figure 2. CycK/CDK13 is required for Nef downregulation of SERINC5.

(A) SERINC5 was expressed with Cas9 and CCNK-gRNA expression vectors in the presence of WT or ΔNef HIV-1 in HEK293T (upper gels) or SERINC3/5 knockout Jurkat-TAg cells (lower gels) and analyzed by WB. SERINC5 was detected by an anti-FLAG antibody.
(B) SERINC5 with an internal HA tag (Ser5-iHA) was expressed with a CCNK-shRNA expression vector in the presence of WT or ΔNef HIV-1 in HEK293T cells. SERINC5 expression on the cell surface was analyzed by flow cytometry using a fluorescent anti-HA antibody. Results are presented as histograms and levels of SERINC5-positive cells are indicated (%).
(C) SERINC5 was expressed with a CDK12- or CDK13-shRNA expression vector in the presence of WT or ΔNef HIV-1 in HEK293T cells and analyzed by WB. SERINC5 was detected by an anti-FLAG antibody.
(D) Ser5-iHA was expressed with a CDK12- or CDK13-shRNA expression vector as in (B) and analyzed similarly.
(E) Mean fluorescence intensity (MFI) values for the SERINC5-positive cell populations in (B) and (D) were statistically analyzed.
(F) SERINC5 was expressed with WT or ΔNef HIV-1 in the presence of an indicated shRNA or control (Ctrl) expression vector in HEK293T cells. After virions were collected and quantified by p24Gag ELISA, TZM-bI cells were infected with an equal amount of virions and viral infectivity was analyzed by measuring the intracellular firefly luciferase activity after 48 h of infection. Infectivity is presented as a relative value, with the WT HIV-1 infectivity in the absence of SERINC5 set to 100%.
(G) WT and ΔNef HIV-1 were produced from HEK293T cells in the presence of a CCNK-, CDK13-, or CDK12-shRNA expression vector and viral infectivity was analyzed and presented as in (F).
Error bars in (E)–(G) represent SEM from three independent experiments. Statistical analysis: *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant (p > 0.05).
Next, we examined the endogenous CDK13 activity using CDK12 as a control. Like CycK, both CDK12 and CDK13 are essential for cell survival, so we used previously reported short hairpin RNAs (shRNAs) to knock down their expression (Dai et al., 2012). CDK12-shRNA and CDK13-shRNA effectively reduced the expression of their target proteins (Figure S2). Notably, Nef could no longer decrease the SERINC5 expression in the presence of CDK13-KD (Figure 2C, lanes 1–4). In contrast, this Nef activity was not affected by CDK12-KD (Figure 2C, lanes 5–8). In addition, CDK13-KD disrupted Nef downregulation of SERINC5 from the cell surface, whereas CDK12-KD did not (Figures 2D and 2E).
Furthermore, we determined how CCNK, CDK12, and CDK13 KD affects the Nef counteractivity of the SERINC5 anti-HIV-1 activity using a viral infectivity assay. As expected, SERINC5 strongly reduced the ΔNef HIV-1 infectivity, which was counteracted by Nef (Figure 2F). This Nef counteractivity was disrupted by KD of CCNK and CDK13, but not CDK12 (Figure 2F). KD of CDK13 and CDK12 alone had very little effect on WT and ΔNef HIV-1 infectivity, and CCNK-KD slightly reduced the infectivity of both viruses (Figure 2G). Taken together, these KD experiments support a conclusion that CycK/CDK13 is required for Nef downregulation of SERINC5.
SERINC5 S360 is critical for Nef antagonism of SERINC5
CDKs preferentially phosphorylate serine or threonine residues in the motif [(S/T)Px(R/K)], in which only the (S/T)P residues are invariant (Lim and Kaldis, 2013). SERINC5 has two (S/T)P motifs at aa positions 249SP250 and 360SP361. Due to its presence in TMD6, S249 is unlikely to be accessible for phosphorylation by CycK/CDK13 (Figure 3A). In contrast, S360 is in ICL4, so it is a likely target for CycK/CDK13 (Figure 3A). In order to evaluate the importance of S249 and S360 for Nef downregulation of SERINC5, we independently substituted both residues to alanine and tested their respective response to Nef. When SERINC5, SERINC5S249A, and SERINC5S360A were co-expressed with WT or ΔNef HIV-1 proviral vectors in HEK293T cells, Nef selectively decreased the expression of SERINC5 and SERINC5S249A, but not SERINC5S360A, at steady-state levels (Figure 3B, lanes 2, 4, and 6).
Figure 3. SERINC5 S360 is critical for Nef antagonism of SERINC5.

(A) SERINC5 transmembrane topology and its ICL4 aa sequence are presented. Residues selected for mutagenesis are colored in red.
(B)SERINC5, SERINC5S249A, and SERINC5S360A were expressed with WT or ΔNef HIV-1 in HEK293T cells. Their expression was detected by WB with an anti-FLAG antibody.
(C) SERINC5, SERINC5S249A, and SERINC5S360A with an internal FLAG tag were expressed with WT or ΔNef HIV-1 in HeLa cells. Cells were stained with a fluorescent anti-FLAG antibody, and the antibody uptake was determined by confocal microscopy at 37°C (scale bars, 5 μm).
(D) SERINC5 and SERINC5S360A with an internal HA tag were expressed with WT or ΔNef HIV-1 in HEK293T cells and their surface expression was analyzed by flow cytometry.
(E) MFI values for the SERINC5-positive cell populations in (D) were statistically analyzed.
(F) The anti-HIV-1 activity of SERINC5 and SERINC5S360A was compared and presented similar to as previously.
Error bars in (E) and (F) represent SEM from three independent experiments. Statistical analysis: *p < 0.05, ****p < 0.0001; ns, p > 0.05.
Three more experiments were conducted to confirm the effect of the S360A substitution. First, SERINC5, SERINC5S249A, and SERINC5S360A were co-expressed with a WT or ΔNef HIV-1 proviral vector in HeLa cells, and SERINC5 endocytosis was determined by a confocal-microscopy-based antibody uptake assay (Shi et al., 2018). SERINC5 and SERINC5S249A were endocytosed in the presence of Nef, whereas SERINC5S360A was not (Figure 3C, top 12 panels versus bottom 6 panels). Second, Nef downregulation of SERINC5 and SERINC5S360A from the cell surface were compared by flow cytometry. Although SERINC5 was effectively downregulated in the presence of Nef, SERINC5S360A was not (Figures 3D and 3E). Third, the antiviral activity of SERINC5 was determined similar to as we did previously. Whereas SERINC5 strongly reduced ΔNef HIV-1 infectivity, SERINC5S360A strongly inhibited both WT and ΔNef HIV-1 infectivity (Figure 3F). These results demonstrate that the S360 residue is required for Nef downregulation and its counteractivity of SERINC5.
A SERINC5 S360D phosphomimetic substitution reduces its expression
It was reported that two hydrophobic residues, L350 and I352, in SERINC5 ICL4 (Figure 3A) also determine SERINC5 sensitivity to Nef (Dai et al., 2018). We substituted both L350 and I352 residues for alanine to generate a double-mutant SERINC5L350A/I352A and tested its sensitivity to Nef, in comparison to SERINC5 and SERINC5S360A. When these SERINC5 proteins were expressed alone in Jurkat cells, they all had similar levels of cell-surface expression (Figures 4A and 4B). When they were expressed with a WT or ΔNef HIV-1 proviral vector, SERINC5 was sensitive, whereas SERINC5S360A and SERINC5L350A/I352A were both resistant to Nef-mediated downregulation from the cell surface (Figures 4C and 4D). These results were also confirmed in HEK293T cells (Figure S3), indicating that their resistance to Nef is not dependent on cell type.
Figure 4. A SERINC5 S360D phosphomimetic substitution reduces its expression.

(A) SERINC5, SERINC5S360A, SERINC5S360D, and SERINC5L350A/I352A With an internal HA tag were expressed in SERINC3/5 knockout Jurkat-TAg cells. SERINC5 expression on the cell surface was determined by flow cytometry.
(B) MFI values for the SERINC5-positive cell populations in (A) were statistically analyzed and presented.
(C) SERINC5, SERINC5S360A, SERINC5S360D, and SERINC5L350A/I352A with an internal HA tag were expressed with WT or ΔNef HIV-1 in SERINC3/5 knockout Jurkat-TAg cells. SERINC5 expression on the cell surface was determined by flow cytometry.
(D) MFI values for the SERINC5-positive cell populations in (C) were statistically analyzed and presented.
(E) The indicated SERINC5 proteins with an internal HA tag were expressed in HEK293T cells and their expression was determined by WB using an anti-HA antibody.
(F) The indicated SERINC5 proteins were expressed in HEK293T cells. Cells were treated with MG132, lactacystin (LA), bafilomycin A1 (Baf A1), or NH4Cl, and SERINC5 protein expression was determined by WB.
(G) The anti-HIV-1 activities of the indicated SERINC5 proteins were compared and presented similar to as previously.
Error bars in (B), (D), and (G) represent SEM from three independent experiments. Statistical analysis: *p < 0.05, **p < 0.01, ***p < 0.001; ns, p > 0.05.
Next, we replaced SERINC5 S360 with an aspartic acid (S360D) to create a phosphomimetic mutant, SERINC5S360D. When expressed in Jurkat cells, the SERINC5S360D level on the cell surface was very low even in the absence of Nef (Figures 4A-4D). When the expression level at steady state was compared in HEK293T cells by WB, SERINC5S360D also had a much lower expression than SERINC5, SERINC5S360A, and SERINC5L350A/I352A (Figure 4E). To determine whether SERINC5S360D was targeted for degradation, cells were treated with proteasomal inhibitors (MG132, lactacystin) and lysosomal inhibitors (bafilomycin A1, NH4Cl) during SERINC5 expression. The SERINC5S360D expression level was increased strongly by bafilomycin A1 and mildly by NH4Cl, whereas MG132 and lactacystin barely had any effect (Figure 4F, lanes 6–10). On the contrary, although the SERINC5 expression was also increased in the presence of all four inhibitors, the level of increase was much less than that of SERINC5S360D (Figure 4F, lanes 1–5). Thus, this SERINC5 phosphomimetic S360D mutant is selectively targeted to lysosomes for degradation.
It was reported that T366 in ICL4 of SERINC5 (Figure 3A) is phosphorylated by casein kinase II (Stoneham et al., 2020). We constructed a SERINC5T366A mutant to further complement our panel of SERINC5 mutants. When their anti-HIV-1 activity was compared, SERINC5, SERINC5S249A, or SERINC5T366A strongly reduced HIV-1 infectivity only in the absence of Nef, whereas SERINC5S360A and SERINC5L350A/I352A did so independent of Nef (Figure 4G). SERINC5S360D had a very marginal antiviral activity, which was due to its relatively low levels of expression. Collectively, these results confirm that residues S360, L350, and I352 determine the SERINC5 sensitivity to Nef and demonstrate that a negative charge at position 360 destabilizes SERINC5, due to its phosphomimetic property. Thus, S360 phosphorylation should play an important role in Nef antagonism of SERINC5.
CycK/CDK13 phosphorylates SERINC5 at S360
In the CDK13 kinase domain, D837 acts as a proton acceptor for phosphate transfer to the substrate, and D855 binds to two Mg2+ ions for coordinating the phosphate donor. Simultaneous disruption of these two residues by introducing D837A/D855N substitutions completely abrogates the CDK13 kinase activity (Greifenberg et al., 2016). To determine whether the S360 residue of SERINC5 is phosphorylated by CycK/CDK13, we generated CDK13D837A and CDK13D837A/D855N mutants and tested their activity on Nef downregulation of SERINC5. In addition, we also generated similar CDK12D858A and CDK12D858A/D876N mutants and tested them, too. CDK13D837A, CDK12D858A, and CDK12D858A/D876N had no effect on Nef-mediated downregulation of SERINC5 (Figure 5A, lanes 1, 2, and 5–8). However, CDK13D837A/D855N disrupted this Nef activity (Figure 5A, lanes 3 and 4). We then treated SERINC5 and HIV-1 expression cells with a CDK12/CDK13 covalent inhibitor, THZ531 (Zhang et al., 2016). We found that Nef could no longer decrease the SERINC5 expression in the presence of this inhibitor (Figure 5B, lane 3). Taken together, these results demonstrate that the CDK13 kinase activity is required for Nef downregulation of SERINC5.
Figure 5. CycK/CDK13 phosphorylates SERINC5 at S360.

(A) SERINC5 was expressed with the indicated CDK13 or CDK12 proteins in the presence of WT or ΔNef HIV-1 in HEK293T cells. SERINC5, CDK13, and CDK12 proteins were detected by WB using an anti-FLAG antibody.
(B) SERINC5 was expressed with WT or ΔNef HIV-1 in HEK293T cells and treated with THZ531 at 100 μM. SERINC5 expression was analyzed by WB using an anti-FLAG antibody.
(C) SERINC5 and SERINC5S360A were expressed with WT or ΔNef HIV-1 in HEK293T cells and purified similar to as previously. After SDS-PAGE, proteins were analyzed after being stained with Coomassie brilliant blue or Phos-tag.
(D) GST-ICL4 and GST-ICL4S360A recombinant proteins were expressed in E. coli and purified by glutathione resin. An in vitro kinase assay was conducted by incubating these proteins with affinity-purified CycK/CDK13. After SDS-PAGE, proteins were analyzed after being stained with Coomassie brilliant blue or Phos-tag.
(E) Peptide 357FCFSPGGEDTEEQQPGK373, its S360-phosphorylated version, and its T366K-substituted version were synthesized. They were analyzed by high-resolution MS via direct infusion or analyzed by CZE-MS after treatment with affinity-purified CycK/CDK13.
To determine whether SERINC5 is phosphorylated at S360 in cells, we purified SERINC5 and SERINC5S360A proteins from HEK293T cells in the presence of HIV-1 proviral vectors as described previously (Figure 5C, upper gel). When their phosphorylation status was analyzed by Phos-tag staining, SERINC5 and SERINC5S360A showed a similar level of Phos-tag staining in the absence of Nef (Figure 5C, lower gel, lanes 2 and 4). However, in the presence of Nef, SERINC5 had a much stronger Phos-tag staining than SERINC5S360A (Figure 5C, lower gel, lanes 1 and 3).
To confirm that S360 is phosphorylated by CycK/CDK13, we conducted an in vitro kinase assay as reported previously (Greifenberg et al., 2016). The ICL4 region of SERINC5 was fused to a glutathione S-transferase (GST) tag and an S360A mutant was also created in this fusion protein. Recombinant GST-ICL4 and GST-ICL4S360A proteins were expressed in E. coli and purified by glutathione-Sepharose beads (Figure 5D). In addition, the CycK/CDK13 complex was directly purified from HEK293T cells by anti-FLAG affinity chromatography. When these purified proteins were incubated with CycK/CDK13 and analyzed by Phos-tag staining, GST-ICL4 showed much stronger staining than GST-ICL4S360A (Figure 5D).
To further confirm this phosphorylation result, we conducted another in vitro kinase assay, using ICL4-derived 17-aa peptides as substrates, and measured the phosphorylation by high-resolution MS via direct infusion. Initially, we compared the unphosphorylated WT peptide (357FCFSPGGEDTEEQQPGK373) with the S360-phosphorylated version (Figure 5E, left panels). As expected, we found that the phosphorylated peptide had an 80-Da higher molecular mass than the WT peptide (1934.76 Da versus 1854.78 Da).
We then used an ICL4-derived peptide and a control peptide containing a T366K substitution (357FCFSPGGEDKEEQQPGK373) to conduct the same experiment and analyzed peptide phosphorylation by capillary zone electrophoresis (CZE)-MS. In the WT peptide sample, a peptide with an ~81-Da higher mass, compared to the mock treatment control, was observed (Figure 5E, middle panels). This result was most likely due to the phosphorylation (+80 Da) and deamination (+1 Da) of the peptide. Moreover, the phosphorylated peptide migrated slower than the unphosphorylated control during CZE separation due to the addition of one negative charge on the peptide (28 versus 27 min), a result which is consistent with previous phosphoproteomic data of CZE-MS (Chen et al., 2019). In the T366K peptide sample, a phosphorylated version of the peptide was also detected (Figure 5E, right panels). Similarly, this peptide had an 80-Da higher mass than the unphosphorylated one (1961.74 Da versus 1881.77 Da) and migrated significantly slower than the unphosphorylated control (18 min versus 15.6 min). Due to the fact that the T366K substitution eliminates this residue as a potential phosphorylation target, this result demonstrates that the phosphorylation occurs at the S360 residue. Thus, we conclude that CycK/CDK13 phosphorylates the S360 residue of SERINC5.
Nef downregulates CD8-ICL4 fusion protein
CD8 is a type I membrane protein with a signal peptide (SP), an N-terminal extracellular domain (ECD), a TMD, and a short C-terminal cytoplasmic tail (CT) (Figure 6A). CD8 exists as an αα homodimer or an αβ heterodimer, and Nef selectively downregulates CD8β by interacting with its CT (Stove et al., 2005). Previously, we created a CD8α-Nef fusion protein to study Nef-mediated internalization of cell-surface molecules (Mandic et al., 2001). To further characterize the role of phosphorylation in Nef downregulation of SERINC5, we generated a WT CD8α-ICL4 fusion protein by both swapping the CD8α CT with SERINC5 ICL4 and inserting an HA tag immediately downstream of the SP (Figure 6A). Three CD8-ICL4 mutants containing the S360A, S360D, or L350A/I352A substitutions were also generated.
Figure 6. Nef downregulates CD8-ICL4 fusion protein.

(A) CD8-ICL4 fusion proteins with an internal HA tag were created by swapping the CD8α CT with SERINC5 ICL4 and introducing substitutions into ICL4. They were expressed in HEK293T cells and detected by WB using an anti-HA antibody. SP, signal peptide; ECD, extracellular domain; TM, transmembrane; CT, cytoplasmic tail.
(B) Expression of SERINC5 proteins with the indicated substitutions was compared by WB using an anti-FLAG antibody.
(C) The indicated CD8-ICL4 fusion proteins were expressed in HEK293T cells and their expression on the cell surface was detected by flow cytometry after staining with a fluorescent anti-HA antibody.
(D) MFI values for the CD8-positive cell populations in (C) were statistically analyzed and presented.
(E) The indicated CD8-ICL4 fusion proteins were expressed with WT or ΔNef HIV-1 in HEK293T cells and analyzed as in (C).
(F) MFI values for the CD8-positive cell populations in (E) were statistically analyzed and presented.
Error bars in (D) and (F) indicate SEM calculated from three independent experiments. Statistical analysis: *p < 0.05; ns, p > 0.05.
CD8-ICL4, CD8-ICL4S360A, CD8-ICL4S360D and CD8-ICL4L350A/I352A were expressed in HEK293T cells and their expression level at steady state was compared. CD8-ICL4S360A and CD8-ICL4L350A/I352A had a relatively higher level of expression than CD8-ICL4 when compared by WB (Figure 6A, lanes 1, 2, and 4) or flow cytometry (Figures 6C and 6D). Like SERINC5S360D (Figure 4E), the expression of CD8-ICL4S360D was poorly detectable by WB (Figure 6A, lane 3). Its low relative level of expression was also confirmed by flow cytometry (Figures 6C and 6D). These results suggest that S360 phosphorylation should also downregulate CD8-ICL4.
To further characterize the function of the L350/I352 residues, a triple L350A/I352A/S360D mutation was introduced into CD8-ICL4 and SERINC5. When CD8-ICL4L350A/I352A/S360D and SERINC5L350A/I352A/S360D were expressed in HEK293T cells, both of them were expressed as poorly as CD8-ICL4S360D and SERINC5S360D (Figures 6B-6D). Thus, the poor expression of the S360D mutants cannot be rescued by removing the L360/I352 residues.
We then tested the ability of Nef to downregulate each of these CD8-ICL4 fusion proteins. Nef effectively downregulated CD8-ICL4, but not CD8-ICL4S360A and CD8-ICL4L350A/I352A (Figures 6E and 6F). The relative expression levels of CD8-ICL4S360D and CD8-ICL4L350A/I352A/S360D on the cell surface were both low, regardless of Nef expression. Taken together, these results demonstrate that Nef downregulates CD8-ICL4 and SERINC5 via a similar mechanism.
Detection of Nef and CD8-ICL4 interaction by quantitative proteomics
To test whether Nef binds to SERINC5 via ICL4, CD8-ICL4, CD8-ICL4S360A, and CD8-ICL4L350A/I352A were expressed in HEK293T cells in the presence of a downregulation-deficient Nef, which contains an LL/AA substitution in its dileucine motif. Next, a co-immunoprecipitation (coIP) assay was carried out using an anti-Nef antibody. Approximately an equivalent level of each of the CD8-ICL4 fusion proteins, as well as the Nef protein, was detected in these cells by WB (Figure 7A). In addition, a similar level of Nef proteins was detected, as expected, whereas none of any CD8-ICL4 fusion proteins were detected, in these coIP fractions by WB (Figure 7A).
Figure 7. Detection of Nef and CD8-ICL4 interaction by quantitative proteomics.

(A) The indicated CD8-ICL4 fusion proteins were expressed with HIV-1 expressing Nef with a dileucine substitution (Nef LL/AA). Proteins were co-immunoprecipitated with a polyclonal anti-Nef antibody and detected by WB using the indicated antibodies.
(B) CoIP samples 1, 2, and 3 in (A) were analyzed by MS after trypsin digestion. Nef, CD8, and ICL4 peptides detected from these samples are underlined. Three ICL4 peptides are numbered (1, 2, 3).
(C) Levels of Nef, CD8, and ICL4 peptides detected by MS in (B) were quantified by label-free quantification (LFQ) and their intensity is presented as relative values, with the value of WT CD8-ICL4 set to 100%. Nef and CD8 were quantified by all detectable peptides and ICL4 was quantified by peptide-1 (SSSDALQGR). The error bars indicate SEM calculated from three independent experiments. Statistical analysis: *p < 0.05, **p < 0.01; ns, p > 0.05.
(D) A proposed model of how S360 phosphorylation promotes SERINC5 downregulation.
We then used MS to further analyze these coIP fractions. A total of 13 Nef, 3 CD8, and 3 ICL4 peptides were detected from these CD8-ICL4, CD8-ICL4S360A, or CD8-ICL4L350A/I352A samples (Figure 7B). Among the 3 ICL4 peptides, peptide-1 (336SSSDALQGR344) was detected in all three samples. However, peptide-2 with the L350A/I352A substitutions (345YAAPEAEAAR354) was only detectable from CD8-ICL4L350A/I352A, and peptide-3 with the S360A substitution (355CCFCFSPGGEDTEEQQPGKEGPR377) was only detectable from CD8-ICL4S360A (Figure S4). Thus, we could detect Nef-ICL4 binding via MS and validated the specificity of this binding by identifying each of the ICL4 substitutions in their appropriate coIP fraction.
Lastly, we compared the intensity of Nef binding to CD8-ICL4, CD8-ICL4S360A, and CD8-ICL4L350A/I352A. We calculated the label-free quantification (LFQ) intensity of the Nef protein, the three CD8 peptides, and ICL4 peptide-1 in each of these three IP fractions. ICL4 peptide-1 was selected because only this peptide could be detected from all three samples. Consistent with the WB result (Figure 7A), approximately an equivalent quantity of Nef proteins was detected from all three samples, excepting a slightly higher level in the L350A/I352A-derived sample (Figure 7C). Notably, both CD8 and ICL4 quantities from CD8-ICL4 were at least 2-fold higher than those from CD8-ICL4S360A and were also slightly higher than those from CD8-ICL4L350A/I352A (Figure 7C). These results demonstrate that S360, and to a lesser degree L350/I352, is required for Nef binding to SERINC5, which suggests that S360 phosphorylation increases SERINC5 binding to Nef.
DISCUSSION
In this study, we provide evidence that CycK/CDK13 is required for Nef antagonism of SERINC5. When the kinase activity of this complex was inhibited by KD of CCNK or CDK13, expression of the kinase inactive mutant CDK13D837A/D855N, or treatment of cells with the kinase inhibitor THZ531, Nef became unable to downregulate SERINC5 and/or counteract its antiviral activity. CycK/CDK13 phosphorylated SERINC5 at S360, and S360 phosphorylation was absolutely essential for these Nef activities. Thus, our findings not only reveal another role for CycK/CDK13 in eukaryotic biology but also extend the previous observation on the interaction between Nef and CycK (Khan and Mitra, 2011). Interestingly, although CycK partners with both CDK12 and CDK13, CDK12 had no effect on SERINC5. This finding could be due to different subcellular localizations of the two CDKs or to additional interactions between CycK/CDK13, Nef, and SERINC5. Our results are consistent with CDK12 and CDK13 regulating the expression of markedly different sets of genes (Greifenberg et al., 2016).
We have obtained identical results from SERINC5 and the CD8-ICL4 chimeric proteins in their response to Nef, which confirms that ICL4 is targeted by Nef (Dai et al., 2018). Among those 58 aa’s in ICL4, S360/L350/I352 have been identified to play a critical role in Nef downregulation of SERINC5. However, because the S360D phosphomimetic SERINC5 was rapidly internalized from the cell surface and degraded in lysosomes even in the absence of Nef, ICL4 contains an unknown phosphoserine-dependent endocytic motif. This situation resembles CD4 downregulation from the cell surface by phorbol esters that activate protein kinase C. Activated protein kinase C phosphorylates serine residues in a dileucine motif, 408SQIxxLLS415, in the CD4 cytoplasmic tail, which facilitates its binding to AP-2 (Pitcher et al., 1999). As recently reported, ICL4 binds to the μ subunit of AP-1 and AP-2 in a phosphorylation-dependent manner (Stoneham et al., 2020). We found that inactivating L350/I352 in conjunction with the S360D substitution did not rescue its expression, indicating that L350/I352 do not represent a bona fide dileucine internalization motif. Thus, in addition to S360/L350/I352, there are additional residues that play an important role in SERINC5 downregulation by interacting with adaptor proteins in a phosphorylation-dependent fashion.
We found that when L350/I352 were inactivated, the CD8-ICL4 interaction with Nef was reduced, indicating that these two hydrophobic residues are involved in SERINC5-Nef binding. Thus, although L350/I352 do not constitute a dileucine motif, they could still function as a Nef-binding site. L350/I352 should constitute a hydrophobic surface and bind to the multifunctional hydrophobic pocket of Nef, which also accommodates the CD4 dileucine motif (Kwon et al., 2020). However, because a single S360A substitution disrupts the Nef-binding ability, the Nef-SERINC5 binding is dependent on phosphoserine, which is different from the Nef-CD4 binding, which is independent of phosphoserine (Garcia and Miller, 1991).
We suggest that S360 phosphorylation is a dynamic process, which promotes SERINC5 downregulation via changing the ICL4 conformation (Figure 7D). Nef interacts with SERINC5 and CycK/CDK13, which induces a conformational change in ICL4 via phosphorylating S360. This conformational change makes L350/I352 more accessible to Nef and enhances the Nef-SERINC5 binding, resulting in SERINC5 endocytosis and degradation via the Nef dileucine motif. We also suggest that this conformational change is transient due to rapid dephosphorylation by cellular phosphatases and sustainable only in the presence of Nef and CycK/CD13 (Figure 7D). If this conformational change is maintained, for example, by introducing the phosphomimetic S360D substitution, the unknown endocytic motif in ICL4 is stably exposed, resulting in AP-2 recruitment and SERINC5 degradation in a Nef-independent manner.
In summary, our findings promise another direction in anti-HIV therapy, namely by interfering with CDK13 as a strategy to attenuate viral replication and spread. Besides our covalent chemical inhibitor, great efforts are being made to inhibit specific transcriptional CDKs and those that function independent of the cell cycle. Our study revealed one such attractive target for HIV-1, as CDK13 does not play an essential role in transcription or in the cell cycle.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Yong-Hui Zheng (zhengyo@msu.edu).
Materials availability
Further information and requests for resources and reagents should be directed to Yong-Hui Zheng (zhengyo@msu.edu).
Data and code availability
This paper does not report original code.
Original data for figures in paper available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
The human HEK293T cells were obtained from the American Type Culture Collection. TZM-bI cells were obtained from the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program. HEK293T and TZM-bI cells were maintained in Dulbecco modified Eagle medium (DMEM) with 10% bovine calf serum (BCS) (HyClone), at 37°C and 5% CO2. The SERINC3/5 knockout Jurkat-TAg (JTAg) cell line was provided by Heinrich Gottlinger and cultured in RPMI 1640 with 10% fetal bovine serum (FBS) (Sigma), at 37°C and 5% CO2.
Bacterial strains
Escherichia coli HB101 (Promega) was used as the recipient strain for preparation of HIV-1 proviral vectors, whereas all of the other plasmid vectors were prepared from Escherichia coli DH10B (NEB). GST-fusion proteins were prepared from Escherichia coli BL21 (NEB). All these bacteria were cultured in LB broth in a shaking incubator at 37°C.
METHOD DETAILS
Expression vectors
The HIV-1 proviral clones pH22 and pH22ΔN were reported previously (Zheng et al., 2003). pCMV6-Ser5-FLAG and pCMV6-CD4-FLAG were reported previously (Ahmad et al., 2019; Shi et al., 2018; Zhang et al., 2017). pBJ5-Ser5-HA and pBJ5-iHA-Ser5 were provided by Heinrich Gottlinger. pcDNA3.1-CDK12-FLAG, pcDNA3.1-CDK13-FLAG, prEF-FLAG-CCNK, pLKO.1-CCNKshRNA, pLKO.1-CDK12shRNA, pLKO.1-CDK13shRNA expression vectors were provided by Qintong Li. pCMV6-CCNK-FLAG was purchased from Origene. The Cas9 expression vector pMJ920 that has a GFP marker was obtained from Jennifer Doudna through Addgene. A CCNK gRNA (5′-AGGACTTGATCCAGCCACCGAGG-3′) targeting the 2nd exon was expressed from pGEM-T (Promega) as we did before (Zhou et al., 2014). pcDNA3.1-CDK12-HA and pcDNA3.1-CDK13-HA were created by cloning CDK12-HA and CDK13-HA into pEGPF-N1 via HindIII/KpnI digestion. To create pGEX-ICL4, ICL4 and vector fragments were amplified from SERINC5 cDNA or pGEX-6P-1 by PCR and ligated via homologous recombination using a CloneExpress II one-step cloning kit (Vazyme Biotech, China). The S249A, S360A, S360D, T366A, and L350A/I352A mutation in pCMV6-Ser5-FLAG or pBJ5-iHA-Ser5, the L350A/I352A/S360D mutation in pBJ5-iHA-Ser5, the S360A mutation in pGEX-ICL4, the D858A and D876N mutation in pcDNA3.1-CDK12-FLAG, and the D837A and D855N mutation in pcDNA3.1-CDK13-FLAG, were created by a site-directed mutagenesis kit (Agilent). CD8α-ICL4 and its mutants were synthesized from TWIST Bioscience and expressed from the pTwist-CMV-Puro vector. All mutations were confirmed by Sanger sequencing. Primers and cloning methods are available upon request.
Protein purification
To purify SERINC5 proteins, HEK293T cells were cultured in twenty 10-cm cell culture dishes at 2x106 cells in 10 mL medium per dish. Cells were transfected with 4.5 μg pCMV6-Ser5-FLAG and 4.5 μg HIV-1 proviral vector in 27 μg PEI and cultured for 48 h. After collecting cells and washing with PBS, cells were incubated with a lysis buffer [50 mM Tris, pH 7.5, 150 mM NaCl, 5% glycerol, 1% n-Dodecyl β-D-maltoside (DDM, Sigma), and EDTA-free protease inhibitor cocktail (Roche)] at 4°C for 2 h. Total cell lysate was spun at 11,000 rpm using a F13-14x50cy rotor in a SORVALL RC6+ centrifuge at 4°C for 30 min. Supernatant was collected and applied to a Poly-Prep chromatography column (BioRad) containing 0.5 mL anti-FLAG M2 affinity gel (Sigma). After incubating at 4°C for 2 h with gentle shaking, supernatant was drained away, SERINC5 proteins were eluted out by 3xFLAG peptide (Sigma) at 100 μg/ml in a purification buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 5% glycerol, 0.05% DDM, and protease inhibitor cocktail). SERINC5 proteins were analyzed by SDS-PAGE followed by mass spectrum and phosphorylation analyses. To purify CycK/CDK13 complex, HEK293T cells were transfected with an equal amount of pcDNA3.1-CDK13-FLAG and pCMV6-CCNK-FLAG. Proteins were purified similarly and used as enzymes for in vitro kinase assay.
To purify GST-ICL4 proteins, E. coli strain BL21 was transformed with pGEX-ICL4 vectors and cultured in 1 l of LB medium at 37°C with shaking to an OD600 of 0.5-1.0. GST-fusion protein expression was induced by adding IPTG to a final concentration of 0.1 mM for an additional 3 h at 37°C with shaking. After centrifugation at 3,500 g for 20 min at 4°C, bacterial pellets were subjected to three cycles of sonication. Supernatant was collected after centrifugation of the lysate at 12,000 g for 15 m at 4°C and incubated with glutathione-Sepharose beads (GE Healthcare). Fusion proteins were eluted out by 20 mM reduced glutathione in 50 mM Tris-Cl (pH 8.0) and used as substrates for in vitro kinase assay.
Immunoprecipitation (IP)
To detect protein interactions in Figures 1D, 1E, and 1F, FLAG-tagged proteins were expressed with their target proteins in HEK293T cells cultured in 10-cm dishes. Proteins were pulled down by an anti-FLAG M2 antibody (Sigma) using Pierce Classic IP Kit (Thermo Fisher Scientific) and analyzed by western blotting (WB). To detect CD8-ICL4 interactions with Nef in Figure 7A, these proteins were expressed in HEK293T cells cultured in 10-cm dishes. Cells were lysed in a lysis buffer [50 mM Tris, pH 7.5, 150 mM NaCl, 5% glycerol, 1% DDM, and EDTA-free protease inhibitor cocktail] at 4°C for 2 h. Cytosolic factions were collected and incubated with an anit-HIV-1 Nef polyclonal antibody (NIH AIDS Reagent Program, Cat. #2949) at 37°C for 2 h. Samples were then incubated with Protein G beads (ThermoFisher) at 4°C overnight and analyzed by WB.
Viral antagonism of SERINC5
HEK293T cells were transfected with polyethylenimine (PEI) as we described previously (Zhang et al., 2017). Briefly, HEK293T cells were seeded in 6-well plates at 4x105 cells per well with 2 mL medium one day before. Each well was transfected with 10 μg PEI mixed with 2 μg HIV-1 proviral vector and 50 ng SERINC5 expression vector in the presence or absence of 1 μg silencing vectors (Cas9+sgRNA, or shRNA vectors) or expression vectors (CycK, CDK12, CDK13). Jurkat cells were electroporated by Amaxa Nucleofector II system on program 3 for Jurkat E6 cells. Briefly, 2x106 cells and 4 μg plasmid DNA were resuspended with 100 μL of Mirus electroporation buffer and electroporated. Cells were immediately transferred to 12-well plates containing 2 mL of RPMI 1640 with 10% FBS and cultured for 5 days. Relevant empty vectors were added to keep the same amounts of total DNAs for each transfection whenever necessary. Cells were collected for analyzing total protein expression by western blotting and flow cytometry. Virions in supernatants were quantified by p24Gag ELISA and viral infectivity was measured after infection of TZM-bI cells.
Western blotting (WB)
Cells were lysed with 1% NP-40 in PBS containing protease inhibitor cocktail (Sigma). After removal of nuclei by brief centrifugation, cytosolic fraction was collected and mixed with 4x Laemmli Sample Buffer (Bio-Rad). Proteins were separated in 10% Bis-Tris acrylamide gels, blotted on to nitrocellulose membrane (Bio-Rad), and detected by antibodies. Mouse monoclonal anti-Nef was described previously (Zheng et al., 2003). Horseradish peroxidase (HRP)-conjugated mouse monoclonal anti-FLAG, anti-HA antibodies, and anti-actin were purchased from Sigma; HRP-conjugated mouse monoclonal anti-GAPDH was purchased from Proteintech; rabbit polyclonal anti-CrkRS (CDK12) and anti-CDC2L5 (CDK13) were purchased from Novus; rabbit polyclonal anti-CycK was purchased from Abcam; HRP-conjugated anti-human, -rabbit, and -mouse immunoglobulin G secondary antibodies were purchased from Thermo Fisher. Immobilon Classico and Crescendo Western HRP Substrate was purchased from MilliporeSigma.
Viral infectivity
TZM-bI cells were seeded at 10,000 cells per well in 100 μL medium in 96-well black plates with half area transparent bottom (Greiner Bio-One). Each well was inoculated with 100 μL of viruses that were subjected to 10-fold serial dilution and infection was done in triplicate. After 48 h, viral infection was determined by a Firefly Luciferase Assay Kit (Biotium). Briefly, after removal of media, 20 μL of 1x Firefly Lysis Buffer were added to each well. After 5 m, 15 μL of Firefly Luciferase Assay Buffer 2.0 containing 0.3 μL of D-luciferin at 10 mg/ml were added to each well and luciferase activities were measured by Veritas Microplate luminometer (Turner Biosystem). Viral infectivity was calculated by normalizing luciferase activities with the amounts of p24Gag in viral inoculum.
Flow cytometry
After transfection of HEK293T and Jurkat cells with SERINC5 and other expression vectors, cells were treated with Fixation/Permeabilization Solution kit (BD Biosciences) for detecting intracellular proteins or remained untreated for detecting cell surface proteins. Cells were stained with Alexa Fluor 647- or pacific blue labeled anti-HA antibodies (BioLegend). After extensive washing, non-permeabilized cells were fixed with 4% paraformaldehyde (PFA), and all cells were analyzed by BD LSR II.
To analyze cell cycle, Jurkat cells were transfected with increasing amounts of CCNK silencing or expression vectors. After 24 h, cells were collected and held in 0.4 mL 50% FBS in PBS on ice. Cells were fixed by adding 1.2 mL 70% EtOH with gentle mixing over 20-30 s. After washing with PBS, 1 mL of Propidium Iodide Cell Cycle Reagent (Thermo Fisher) were added to stain cells for 15-30 min at 37°C and analyzed by BD LSR II. ModFit LT version 4.1.7 software was used to analyze the resulting DNA histograms and calculate the percentage of cells in each phase of the cell cycle, based on a total of 10,000 events per sample.
Endocytosis assay
An antibody uptake assay was used to measure Nef-mediated SERINC5 endocytosis as we did previously (Shi et al., 2018). Briefly, HeLa cells were transfected with SERINC5 and Nef expression vectors and cultured for 48 h. Cells were then stained with an anti-HA antibody to label SERINC5 proteins on the cell surface and incubated at either 37°C to allow endocytosis. One hour later, SERINC5 subcellular localization was determined by fluorescence microscopy.
Peptides
Three peptides FCFSPGGEDTEEQQPGK, FCF{pSer}PGGEDTEEQQPGK, and FCFSPGGEDKEEQQPGK derived from SERINC5 ICL4 were synthesized and purchased from GenScript.
In vitro kinase assay
Purified GST-ICL4 proteins or synthesized ICL4-derived peptides were pre-incubated with purified CycK/CDK13 for 10 min at room temperature in a kinase buffer [150 mM HEPES, pH 7.6, 34 mM KCl, 7 mM MgCl2, 2.5 mM dithiothreitol, 5 mM β-glycerol phosphate, 1xPhosSTOP (Roche)] as described (Greifenberg et al., 2016). Cold ATP was added to a final concentration of 2 mM, and the reaction mixture was incubated up to 60 min at 37°C. Reactions were stopped by adding EDTA to a final concentration of 50 mM. GST-ICL4 phosphorylation was determined by phos-tag staining buffer (see below) whereas peptide phosphorylation was analyzed by capillary zone electrophoresis (CZE)-MS.
Phos-tag gel assay
Phosphorylation of SERINC5 proteins purified from HEK293T cells in the presence of Nef, and GST-ICL4 proteins after in vitro kinase reaction were checked by a Phos-Tag Phosphoprotein Gel Stain kit (ABP Biosciences, Rockville, MD). Briefly, equal amounts of proteins were run on an SDS-polyacrylamide gel (PAGE) and fixed with 50% methanol and 10% acetic acid. After washing with water, the gel was incubated with staining reagent for 90 m on an orbital shaker. Background was removed by incubation with de-staining reagent and the gel was imaged using a 300-nm UV transilluminator.
LC (liquid chromatography)-MS/MS
Proteins associated with anti-FLAG M2 affinity gels or with control beads were washed three times with PBS, and treated with 20 μl of denaturing buffer (8 M urea, 100 mM NH4HCO3) at 37°C for 30 min. Proteins were then reduced with addition of 2 μl of 5 mM dithiothreitol (DTT) for 20 m at 37°C. After alkylation with addition of 5 μl of 5 mM iodoacetamide (IAA) for 10 min at room temperature in dark, the sample was diluted 4 times with addition of 60 μL of 100 mM NH4HCO3 to reduce the concentration of urea. After digestion with 0.2 μg of trypsin at 37°C for overnight, 1 μl of formic acid (FA) was added to quench the digestion. The sample tube was centrifuged at 1000 rpm for 30 s and supernatant was taken out for desalting using Zip-tip (Millipore, ZTC18S096). Protein digest was resuspended in 10 μl of buffer A [2% Acetonitrile (ACN), 0.1% FA] and 1 μl of sample was loaded for nanoRPLC-MS/MS analysis.
An EASY nanoLC-1200 system (Thermo Fisher) equipped with a C18 RPLC column (75 μm i.d. x 50 cm, C18, 2 μm, 100 Å, Thermo Fisher Scientific) was connected to a Q-Exactive HF mass spectrometer (Thermo Fisher) for nanoRPLC-MS/MS analysis. Buffer A containing 2% ACN and 0.1% FA, and buffer B containing 80% (v/v) ACN and 0.1% FA were used to generate gradient separation. The flow rate was 200 nl/min. The gradient for RPLC separation was as follows: from 8 to 30% (v/v) B in 50 min, from 30% to 50% (v/v) B in 30 min, from 50% to 80% (v/v) B in 10 min and maintain at 80% (v/v) B for 10 min. A Top 10 data dependent acquisition (DDA) method was used. The MS parameter was set as follows: the full MS resolution was 60,000 and the maximum injection time was 50 ms. The scan range was 300 – 1800 m/z. The AGC target was 3e6. The MS/MS resolution was 30,000 and the maximum injection time was 50 ms. The AGC target was set 1e5 for MS/MS. The isolation window was set 2.0 m/z. The intensity threshold for fragmentation was 5e4. The dynamic exclusion was set 30 s.
Database search for protein identification
Proteome Discoverer 2.2 (Thermo Fisher) with SEQUEST HT searching engine built in was used. The mass tolerance for precursor ion was set 20 ppm and for fragment ion was set 0.05 Da. UniProt human proteome database (UP000005640) was used for the database searching. Trypsin was set as the enzyme with two maximum missed cleavages. Oxidation on Methionine, acetylation on Protein N-terminal, phosphorylation on Serine, Tyrosine and Threonine and deamination on Asparagine or Glutamine were set as variable modifications. Carbamidomethylation on cysteine was set as the fixed modification. The peptides were filtered with confidence as high, corresponding to a 1% peptide-level false discovery rate. Protein grouping was enabled, and the strict parsimony principle was applied.
To determine the relative abundance of peptides and proteins across different samples using label-free quantification (LFQ), MaxQuant software (version 1.6.15.0) (Cox and Mann, 2008) was also employed for the identification and quantification of proteins and peptides using the UniProt human proteome database (UP000005640) and default settings. The false discovery rates of peptide and protein identifications were less than 1%. Specifically, the MaxLFQ function (Cox et al., 2014) integrated in the MaxQuant software was used for LFQ. The basic idea of LFQ is to compare peptide intensity obtained from MS measurement directly across different biological conditions to determine the relative abundance difference of the peptide and the corresponding protein in the samples. The major MaxQuant parameters related to the LFQ were turning on LFQ with the minimal ratio count as 2, peptides used for protein quantification as “razor,” match between runs as “true,” matching time window as 0.7 min, and alignment time window as 20 min.
Capillary zone electrophoresis (CZE)-MS
The synthesized peptide samples from the CCNK/CDK13 incubation and mock treatment experiments were desalted using C18 ZipTips (MilliporeSigma), lyophilized, and dissolved in 50 mM ammonium bicarbonate. A commercialized electro-kinetically pumped sheath flow CE-MS interface (an EMASS-II CE-MS interface, CMP Scientific) was employed to couple CZE with MS (Sun et al., 2015). A 7100 CE System (Agilent) was used for the automated operation of CZE. The electrospray (ESI) emitters were pulled from borosilicate glass capillary (1.0 mm o.d., 0.75 mm i.d.) by a Sutter P-1000 flaming/brown micropipette puller. The opening of the emitter was 20-40 μm. Voltage for ESI was ~2.2 kV.
A 1-m-long fused silica capillary (50 μm i.d., 360 μm o.d.) was used for CZE separation. The inner wall of the capillary was coated with linear polyacrylamide (LPA) as reported (Chen et al., 2017). The background electrolyte (BGE) for CZE was 5% (v/v) acetic acid (pH 2.4) and the sheath buffer for electrospray was 10% (v/v) methanol and 0.2% (v/v) formic acid in water. For each run, about 500 nL sample was injected into the capillary. Then 30 kV was applied at the injection end and 100 mbar was applied at the mean time for CZE separation for 35 min.
A 6545XT AdvanceBio Q-TOF mass spectrometer (Agilent) was used for data acquisition. The gas temperature was 325°C. The drying gas was 1 l/min. The fragmentor and skimmer were set as 150 V and 65 V, respectively. The mass range was 800-1000 m/z. The acquisition rate was 0.4 spectra/s. The acquisition mode was Extended Dynamic Range (2 GHz) and the slicer mode was High sensitivity. The Agilent MassHunter Qualitative Analysis Navigator B.08.00 was used for data analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical tests were performed using GraphPad Prism 8. Variance was estimated by calculating the standard error of the mean (SEM) and represented by error bars. Significance of differences between samples was assessed using two-way ANOVA with Bonferroni post-test. All experiments were performed independently at least three times, with representative experiment being shown. *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant (p > 0.05). All of the statistical details of experiments can be found in the figure legends and figures.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FLAG M2 | Sigma Aldrich | Cat#F1804; RRID: AB_262044 |
| Anti-HIV Nef Polyclonal | NIH AIDS Reagent Program | Cat#2949; RRID: n/a. |
| Anti-HA-HRP | Sigma Aldrich | Cat#H6533-1VL; RRID: AB_439706 |
| Anti-FLAG-HRP | Sigma Aldrich | Cat#A8592-.2MG; RRID: AB_262044 |
| Anti-Actin-HRP | Sigma Aldrich | Cat#A3854-200UL; RRID: AB_262011 |
| Anti-GAPDH-HRP | Proteintech | Cat#HRP-60004; RRID: AB_2107436 |
| Rabbit polyclonal anti-CrkRS (CDK12) | Novus | Cat#NB100-87011; RRID: AB_1199396 |
| Rabbit polyclonal anti-CDC2L5 (CDK13) | Novus | Cat#NB100-68268; RRID: AB_1107830 |
| Rabbit polyclonal anti-Cyclin K | Abcam | Cat#Ab85854; RRID: AB_1860218 |
| Goat-anti-human-IgG-HRP | Thermo Fisher | Cat#31412; RRID: AB_228265 |
| Goat-anti-rabbit-IgG-HRP | Thermo Fisher | Cat#G21234; RRID: AB_2536530 |
| Goat-anti-mouse-IgG-HRP | Thermo Fisher | Cat#G21040; RRID: AB_2536527 |
| AF647 anti-HA | BioLegend | Cat#682404; RRID: AB_2566616 |
| Pacific Blue anti-HA | BioLegend | Cat#901525; RRID: AB_2734659 |
| Monoclonal anti-Nef | Zheng et al., 2003 | n/a |
| Bacterial and virus strains | ||
| HIV-1 Provirus pH22, pH22ΔN | Zheng et al., 2003 | n/a |
| BL21 E. coli | NEB | Cat#C2530H |
| DH10B E. coli | NEB | Cat#C3019H |
| HB101 E. coli | Promega | Cat#L2011 |
| Chemicals, peptides, and recombinant proteins | ||
| Propidum Iodide Cell Cycle Reagent | Thermo Fisher | Cat#BMS500PI |
| MG132 | APExBio | Cat#A2585 |
| Lactacystin | Enzo | Cat#BML-P1104-0200 |
| Bafilomycin A1 | APExBio | Cat#A8627 |
| NH4Cl | Sigma | Cat#A9434-500G |
| THZ531 | APExBio | Cat#A8736 |
| Protease Inhibitor Cocktail | Sigma | Cat#P8340 |
| FCFSPGGEDKEEQQPGK (T366K mutant) | GenScript | n/a |
| FCFSPGGEDTEEQQPGK | GenScript | n/a |
| FCF{pSer}PGGEDTEEQQPGK | GenScript | n/a |
| 3X FLAG peptide | Sigma | Cat#F4799-4MG |
| Critical commercial assays | ||
| CloneExpressII one-step cloning kit | Vazyme Biotech, China | Cat#C112 |
| Site-directed Mutagenesis Kit | Agilent | Cat#200521 |
| Firefly Luciferase Assay Kit | Biotium | Cat#30085-T |
| Phos-Tag Phosphoprotein Gel Stain Kit | ABP Biosciences | Cat#P005A |
| Pierce Classic IP Kit | Thermo Fisher | Cat#26146 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-3216 |
| TZM-bI | NIH AIDS Research and Reference Reagent Program | 8129 |
| SERINC3/5 knockout Jurkat-TAg | A gift from Heinrich Gottlinger, University of Massachusetts | n/a |
| Oligonucleotides | ||
| CCNK gRNA (5′-AGGACTTGATCCAGCCA CCGAGG-3′) (pGEM-T) | Promega | n/a |
| Recombinant DNA | ||
| pCMV6-Ser5-FLAG | Ahmad et al., 2019; Shi et al., 2018; Zhang et al., 2017 | n/a |
| pCMV6-CD4-FLAG | Ahmad et al., 2019; Shi et al., 2018; Zhang et al., 2017 | n/a |
| pBJ5-Ser5-HA | A gift from Heinrich Gottlinger, University of Massachusetts | n/a |
| pBJ5-iHA-Ser5 | A gift from Heinrich Gottlinger, University of Massachusetts | n/a |
| pcDNA3.1-CDK12-FLAG | A gift from Qintong Li, Sichuan University | n/a |
| pcDNA3.1-CDK13-FLAG | A gift from Qintong Li, Sichuan University | n/a |
| prEF-FLAG-CCNK | A gift from Qintong Li, Sichuan University | n/a |
| pLKO.1-CCNKshRNA | A gift from Qintong Li, Sichuan University | n/a |
| pLKO.1-CDK12shRNA | A gift from Qintong Li, Sichuan University | n/a |
| pLKO.1-CDK13shRNA | A gift from Qintong Li, Sichuan University | n/a |
| pCMV6-CCNK-FLAG | Origene | n/a |
| pMJ920 (GFP marker) | Jinek et al., 2013 | Jennifer Doudna, UC Berkeley, Addgene Cat#42234 |
| pcDNA3.1-CDK12-HA | This paper | n/a |
| pcDNA3.1-CDK13-HA | This paper | n/a |
| pGEX-ICL4 | This paper | n/a |
| pCMV6-Ser5-FLAG S249A, S360A, T366A, L350A/I352A | This paper | n/a |
| pBJ5-iHA-Ser5 S249A, S360A, T366A, L350A/I352A, L350A/I352A/S360D | This paper | n/a |
| pGEX-ICL4 S360A | This paper | n/a |
| pcDNA3.1-CDK12-FLAG D858A, D876N, D858A/D876N | This paper | n/a |
| pcDNA3.1-CDK13-FLAG D837A, D855N, D837A/D855N | This paper | n/a |
| pTWIST-CD8α-ICL4 and mutants | TWIST Bioscience | n/a |
| pNL4.3-LL-AA | A gift from Warner Greene, UCSF | n/a |
| Software and algorithms | ||
| ModFit LT version 4.1.7 | Verity | https://www.vsh.com/products/mflt/mfTrialVersions.asp |
| Proteome Discoverer 2.2 + SEQUEST HT Search Engine | Thermo Fisher OPTON-30945 | https://www.thermofisher.com/order/catalog/product/OPTON-30945#/OPTON-30945 |
| MassHunter Qualitative Analysis Navigator B.08.00 | Agilent | https://www.agilent.com/en/support/software-informatics/qual-b-0800-sp1 |
| FlowJo v10.7 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads |
| Other | ||
| Zip-tip | Millipore | Cat#ZTC18S096 |
| EASY nanoLC-1200 system | Thermo Fisher | Cat#LC140 |
| Protein G beads | Thermo Fisher | Cat#20398 |
| Anti-HA Affinity Matrix | Sigma | Cat#11815016001 |
| Anti-FLAG M2 Affinity Gel | Sigma | Cat#A2220 |
Highlights.
HIV-1 Nef interacts with SERINC5 and the CycK/CDK13 kinase complex
CycK/CDK13 phosphorylates S360 in intracellular loop 4 (ICL4) of SERINC5
S360 phosphorylation enhances SERINC5 binding to the Nef/AP-2 complex
SERINC5 is then targeted to endosomes and lysosomes for degradation
ACKNOWLEDGMENTS
We thank Heinrich Gottlinger, Qintong Li, Jennifer Doudna, and the NIH AIDS Research and Reference Reagent Program for providing reagents. The graphical abstract and the model in Figure 7D were created with BioRender. Y.-H.Z. is supported by a grant from the National Institutes of Health (AI145504).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109514.
REFERENCES
- Ahi YS, Zhang S, Thappeta Y, Denman A, Feizpour A, Gummuluru S, Reinhard B, Muriaux D, Fivash MJ, and Rein A (2016). Functional interplay between murine leukemia virus glycogag, Serinc5, and surface glycoprotein governs virus entry, with opposite effects on gammaretroviral and Ebolavirus glycoproteins. mBio 7, e01985–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad I, Li S, Li R, Chai Q, Zhang L, Wang B, Yu C, and Zheng YH (2019). The retroviral accessory proteins S2, Nef, and glycoMA use similar mechanisms for antagonizing the host restriction factor SERINC5. J. Biol. Chem 294, 7013–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananth S, Morath K, Trautz B, Tibroni N, Shytaj IL, Obermaier B, Stolp B, Lusic M, and Fackler OT (2019). Multifunctional roles of the N-terminal region of HIV-1SF2Nef are mediated by three independent protein interaction sites. J. Virol 94, e01398–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, and Greenleaf AL (2010). CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 24, 2303–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beitari S, Ding S, Pan Q, Finzi A, and Liang C (2017). Effect of HIV-1 Env on SERINC5 antagonism. J. Virol 91, e02214–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, and Peterlin BM (2011). The cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 25, 2158–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bösken CA, Farnung L, Hintermair C, Merzel Schachter M, Vogel-Bachmayr K, Blazek D, Anand K, Fisher RP, Eick D, and Geyer M (2014). The structure and substrate specificity of human Cdk12/cyclin K. Nat. Commun 5, 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chande A, Cuccurullo EC, Rosa A, Ziglio S, Carpenter S, and Pizzato M (2016). S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 113, 13197–13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Shen X, and Sun L (2017). Capillary zone electrophoresis-mass spectrometry with microliter-scale loading capacity, 140 min separation window and high peak capacity for bottom-up proteomics. Analyst (Lond.) 142, 2118–2127. [DOI] [PubMed] [Google Scholar]
- Chen D, Ludwig KR, Krokhin OV, Spicer V, Yang Z, Shen X, Hummon AB, and Sun L (2019). Capillary zone electrophoresis-tandem mass spectrometry for large-scale phosphoproteomics with the production of over 11,000 phosphopeptides from the colon carcinoma HCT116 cell line. Anal. Chem 91, 2201–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Sood C, Marin M, Aaron J, Gratton E, Salaita K, and Meikyan GB (2020). Super-resolution fluorescence imaging reveals that serine incorporator protein 5 inhibits human immunodeficiency virus fusion by disrupting envelope glycoprotein clusters. ACS Nano 14, 10929–10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, and Mann M (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q, Lei T, Zhao C, Zhong J, Tang YZ, Chen B, Yang J, Li C, Wang S, Song X, et al. (2012). Cyclin K-containing kinase complexes maintain self-renewal in murine embryonic stem cells. J. Biol. Chem 287, 25344–25352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Usami Y, Wu Y, and Göttlinger H (2018). A long cytoplasmic loop governs the sensitivity of the anti-viral host protein SERINC5 to HIV-1 Nef. Cell Rep. 22, 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- daSilva LL, Sougrat R, Burgos PV, Janvier K, Mattera R, and Bonifacino JS (2009). Human immunodeficiency virus type 1 Nef protein targets CD4 to the multivesicular body pathway. J. Virol 83, 6578–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Devlin JR, Hogg SJ, Doyle MA, Harrison PF, Todorovski I, Cluse LA, Knight DA, Sandow JJ, Gregory G, et al. (2020). CDK13 co-operates with CDK12 to control global RNA polymerase II processivity. Sci. Adv 6, eaaz5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JV, and Miller AD (1991). Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350, 508–511. [DOI] [PubMed] [Google Scholar]
- Greenleaf AL (2019). Human CDK12 and CDK13, multi-tasking CTD kinases for the new millenium. Transcription 10, 91–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greifenberg AK, Hönig D, Pilarova K, Düster R, Bartholomeeusen K, Bösken CA, Anand K, Blazek D, and Geyer M (2016). Structural and functional analysis of the Cdk13/cyclin K complex. Cell Rep. 14, 320–331. [DOI] [PubMed] [Google Scholar]
- Heigele A, Kmiec D, Regensburger K, Langer S, Peiffer L, Stürzel CM, Sauter D, Peeters M, Pizzato M, Learn GH, et al. (2016). The potency of Nef-mediated SERINC5 antagonism correlates with the prevalence of primate lentiviruses in the wild. Cell Host Microbe 20, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka M, Hayakawa M, and Ingi T (2005). Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J. Biol. Chem 280, 35776–35783. [DOI] [PubMed] [Google Scholar]
- Jin SW, Alsahafi N, Kuang XT, Swann SA, Toyoda M, Göttlinger H, Walker BD, Ueno T, Finzi A, Brumme ZL, and Brockman MA (2019). Natural HIV-1 Nef polymorphisms impair SERINC5 downregulation activity. Cell Rep. 29, 1449–1457.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, and Doudna J (2013). RNA-programmed genome editing in human cells. eLife 2, e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SZ, and Mitra D (2011). Cyclin K inhibits HIV-1 gene expression and replication by interfering with cyclin-dependent kinase 9 (CDK9)-cyclin T1 interaction in Nef-dependent manner. J. Biol. Chem 286, 22943–22954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff F (2010). Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8, 55–67. [DOI] [PubMed] [Google Scholar]
- Kwon Y, Kaake RM, Echeverria I, Suarez M, Karimian Shamsabadi M, Stoneham C, Ramirez PW, Kress J, Singh R, Sali A, et al. (2020). Structural basis of CD4 downregulation by HIV-1 Nef. Nat. Struct. Mol. Biol 27, 822–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei T, Zhang P, Zhang X, Xiao X, Zhang J, Qiu T, Dai Q, Zhang Y, Min L, Li Q, et al. (2018). Cyclin K regulates prereplicative complex assembly to promote mammalian cell proliferation. Nat. Commun 9, 1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Ahmad I, Shi J, Wang B, Yu C, Zhang L, and Zheng YH (2019). Murine leukemia virus glycosylated Gag reduces murine SERINC5 protein expression at steady-state levels via the endosome/lysosome pathway to counteract SERINC5 antiretroviral activity. J. Virol 93, e01651–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, and Kaldis P (2013). Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093. [DOI] [PubMed] [Google Scholar]
- Mandic R, Fackler OT, Geyer M, Linnemann T, Zheng YH, and Peterlin BM (2001). Negative factor from SIV binds to the catalytic subunit of the V-ATPase to internalize CD4 and to increase viral infectivity. Mol. Biol. Cell 12, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher C, Höning S, Fingerhut A, Bowers K, and Marsh M (1999). Cluster of differentiation antigen 4 (CD4) endocytosis and adaptor complex binding require activation of the CD4 endocytosis signal by serine phosphorylation. Mol. Biol. Cell 10, 677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye VE, Rosa A, Bertelli C, Struwe WB, Maslen SL, Corey R, Liko I, Hassall M, Mattiuzzo G, Ballandras-Colas A, et al. (2020). A bipartite structural organization defines the SERINC family of HIV-1 restriction factors. Nat. Struct. Mol. Biol 27, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Eke IE, Johnson SF, Ding C, and Zheng YH (2020). Proteasomal degradation of human SERINC4: a potent host anti-HIV-1 factor that is antagonized by nef. Curr. Res. Virol. Sci 1, 100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, et al. (2015). HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Xiong R, Zhou T, Su P, Zhang X, Qiu X, Li H, Li S, Yu C, Wang B, et al. (2018). HIV-1 Nef antagonizes SERINC5 restriction by downregulation of SERINC5 via the endosome/lysosome system. J. Virol 92, e00196–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood C, Marin M, Chande A, Pizzato M, and Melikyan GB (2017). SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J. Biol. Chem 292, 6014–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoneham CA, Ramirez PW, Singh R, Suarez M, Debray A, Lim C, Jia X, Xiong Y, and Guatelli J (2020). A conserved acidic-cluster motif in SERINC5 confers partial resistance to antagonism by HIV-1 Nef. J. Virol 94, e01554–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stove V, Van de Walle I, Naessens E, Coene E, Stove C, Plum J, and Verhasselt B (2005). Human immunodeficiency virus Nef induces rapid internalization of the T-cell coreceptor CD8alphabeta. J. Virol 79, 11422–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zhu G, Zhang Z, Mou S, and Dovichi NJ (2015). Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. J. Proteome Res 14, 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timilsina U, Umthong S, Lynch B, Stablewski A, and Stavrou S (2020). SERINC5 potently restricts retrovirus infection in vivo. mBio 11, e00588–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usami Y, Wu Y, and Göttlinger HG (2015). SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, et al. (2016). Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol 12, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhou T, Yang J, Lin Y, Shi J, Zhang X, Frabutt DA, Zeng X, Li S, Venta PJ, and Zheng YH (2017). Identification of SERINC5-001 as the predominant spliced isoform for HIV-1 restriction. J. Virol 91, e00137–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi J, Qiu X, Chai Q, Frabutt DA, Schwartz RC, and Zheng YH (2019). CD4 expression and Env conformation are critical for HIV-1 restriction by SERINC5. J. Virol 93, e00544–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Plemenitas A, Fielding CJ, and Peterlin BM (2003). Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc. Natl. Acad. Sci. USA 100, 8460–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Dang Y, and Zheng YH (2014). The mitochondrial translocator protein, TSPO, inhibits HIV-1 envelope glycoprotein biosynthesis via the endoplasmic reticulum-associated protein degradation pathway. J. Virol 88, 3474–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original code.
Original data for figures in paper available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.