

ABSTRACT
Macroautophagy/autophagy is a lysosomal degradation system which plays a protective role against kidney injury. RUBCN/Rubicon (RUN domain and cysteine-rich domain containing, Beclin 1-interacting protein) inhibits the fusion of autophagosomes and lysosomes. However, its physiological role in kidney proximal tubular epithelial cells (PTECs) remains uncertain. In the current study, we analyzed the phenotype of newly generated PTEC-specific rubcn-deficient (KO) mice. Additionally, we investigated the role of RUBCN in lipid metabolism using isolated rubcn-deficient PTECs. Although KO mice exhibited sustained high autophagic flux in PTECs, they were not protected from acute ischemic kidney injury. Unexpectedly, KO mice exhibited hallmark features of metabolic syndrome accompanied by expanded lysosomes containing multi-layered phospholipids in PTECs. RUBCN deficiency in cultured PTECs promoted the mobilization of phospholipids from cellular membranes to lysosomes via enhanced autophagy. Treatment of KO PTECs with oleic acid accelerated fatty acids transfer to mitochondria. Furthermore, KO PTECs promoted massive triglyceride accumulation in hepatocytes (BNL-CL2 cells) co-cultured in transwell, suggesting accelerated fatty acids efflux from the PTECs contributes to the metabolic syndrome in KO mice. This study shows that sustained high autophagic flux by RUBCN deficiency in PTECs leads to metabolic syndrome concomitantly with an accelerated mobilization of phospholipids from cellular membranes to lysosomes.
Abbreviations: ABC: ATP binding cassette; ACADM: acyl-CoA dehydrogenase medium chain; ACTB: actin, beta; ATG: autophagy related; AUC: area under the curve; Baf: bafilomycin A1; BAT: brown adipose tissue; BODIPY: boron-dipyrromethene; BSA: bovine serum albumin; BW: body weight; CAT: chloramphenicol acetyltransferase; CM: complete medium; CPT1A: carnitine palmitoyltransferase 1a, liver; CQ: chloroquine; CTRL: control; EGFP: enhanced green fluorescent protein; CTSD: cathepsin D; EAT: epididymal adipose tissue; EGFR: epidermal growth factor receptor; EIF4EBP1: eukaryotic translation initiation factor 4E binding protein 1; FA: fatty acid; FBS: fetal bovine serum; GTT: glucose tolerance test; HE: hematoxylin and eosin; HFD: high-fat diet; I/R: ischemia-reperfusion; ITT: insulin tolerance test; KAP: kidney androgen regulated protein; KO: knockout; LAMP1: lysosomal associated membrane protein 1; LD: lipid droplet; LRP2: low density lipoprotein receptor related protein 2; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; MAT: mesenteric adipose tissue; MS: mass spectrometry; MTOR: mechanistic target of rapamycin kinase; MTORC1: MTOR complex 1; NDRG1: N-myc downstream regulated 1; NDUFB5: NADH:ubiquinone oxidoreductase subunit B5; NEFA: non-esterified fatty acid; OA: oleic acid; OCT: optimal cutting temperature; ORO: Oil Red O; PAS: Periodic-acid Schiff; PFA: paraformaldehyde; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; PPARA: peroxisome proliferator activated receptor alpha; PPARGC1A: PPARG coactivator 1 alpha; PTEC: proximal tubular epithelial cell; RAB7A: RAB7A, member RAS oncogene family; RPS6: ribosomal protein S6; RPS6KB1: ribosomal protein S6 kinase B1; RT: reverse transcription; RUBCN: rubicon autophagy regulator; SAT: subcutaneous adipose tissue; SFC: supercritical fluid chromatography; SQSTM1: sequestosome 1; SREBF1: sterol regulatory element binding transcription factor 1; SV-40: simian virus-40; TFEB: transcription factor EB; TG: triglyceride; TS: tissue specific; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling; UN: urea nitrogen; UQCRB: ubiquinol-cytochrome c reductase binding protein; UVRAG: UV radiation resistance associated; VPS: vacuolar protein sorting; WAT: white adipose tissue.
KEYWORDS: Autophagic flux, autophagy, lipid efflux, lysosome, metabolic syndrome, RUBCN/Rubicon
Introduction
Macroautophagy/autophagy is one of the major degradation pathways in the cell along with the ubiquitin-proteasome system [1,2]. Cytoplasmic damaged organelles and aberrant macromolecules are engulfed in a double-membrane structure called an autophagosome. The autophagosome is transported to and fuses with the lysosome, followed by the degradation of sequestered materials. We have identified that autophagy in kidney proximal tubular epithelial cells (PTECs) protects them from acute kidney injury [3,4] and maintains homeostasis against chronic disorders such as aging, obesity-related nephropathy, and diabetic nephropathy [5–7]. These studies raise hope that enhancing autophagic activity in the PTECs could be a promising strategy to prevent the progression of kidney diseases.
BECN1/Beclin 1, a mammalian homolog of the yeast autophagy protein Vps30/Atg6 (vacuolar protein sorting 30), has been shown to not only serve as a tumor suppressor but also regulate autophagy [8]. BECN1 binds to PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), and resulting BECN1-PIK3C3 complex mediates multiple vesicle-trafficking processes including endocytosis and autophagy [9]. In 2009, 2 new BECN1-PIK3C3 complex-associated proteins, ATG14 (autophagy related 14) and RUBCN/Rubicon (RUN domain and cysteine-rich domain containing, Beclin 1-interacting protein), were identified [10,11]. ATG14 enhances PIK3C3 lipid kinase activity and upregulates autophagy. By contrast, RUBCN reduces the PIK3C3 activity and negatively regulates autophagic activity by inhibiting the fusion of autophagosomes and lysosomes. Although most autophagy-regulating proteins such as ATG family promote autophagy, RUBCN exceptionally restrains autophagy. Indeed, RUBCN deficiency results in the enhancement of autophagosome maturation in vitro [10,12]. Recently, several studies have shown that enhanced autophagic activity by genetic ablation of Rubcn alleviates the pathology of diseases; RUBCN deficiency prolongs survival and improves cardiac output in mice after lipopolysaccharide treatment [13]. Another paper reported that hepatocyte-specific rubcn-deficient mice exhibit high autophagic flux in the liver, which improves liver steatosis and injury upon high-fat diet (HFD) [14]. Thus, we hypothesized that genetic ablation of Rubcn in PTECs might alleviate kidney injury. To test this, we newly generated and analyzed PTEC-specific rubcn-deficient mice in this study.
Results
RUBCN deficiency increases autophagic flux in PTECs
To evaluate the effect of RUBCN deficiency in PTECs, we generated PTEC-specific rubcn-deficient mice. To this end, we used male transgenic mice that express Cre recombinase under the Kap (kidney androgen regulated protein) promotor. First, we assessed Cre recombinase expression in the Kap-Cre transgenic mice by crossing EGFP (enhanced green fluorescent protein)-CAT (chloramphenicol acetyltransferase) transgenic mice, in which EGFP can be detected only after Cre-mediated recombination [15]. The Kap-Cre+;EGFP-CAT male mice exhibited a PTEC-specific pattern of Cre recombinase activity as assessed by EGFP signals in cells with LRP2/MEGALIN (low density lipoprotein receptor related protein 2) staining, whereas EGFP-positive tubules were absent in Kap-Cre−;EGFP-CAT male mice (Figure S1). EGFP was not detected in liver, pancreas, adipose tissue, and muscle in Kap-Cre+;EGFP-CAT male mice (Figure S1), which agrees with previous reports [3,16].
We then crossed the Kap-Cre transgenic mice with mice bearing a Rubcnflox allele [14] and generated Rubcn-floxed Kap-Cre mice (rubcnF/F-TSKO [tissue-specific knockout]). To evaluate the autophagic flux precisely in vivo, we crossed RubcnF/F (RubcnF/F-CTRL [control]) or rubcnF/F-TSKO mice with GFP-MAP1LC3B (microtubule-associated protein 1 light chain 3 beta) transgenic mice, in which GFP-positive puncta reflect autophagosomes [17]. Chloroquine (CQ, 50 μg/g body weight [BW]), an inhibitor of intralysosomal acidification, was administered to the obtained RubcnF/F-CTRL;GFP-MAP1LC3B and rubcnF/F-TSKO;GFP-MAP1LC3B mice 6 h before euthanasia and the number of GFP-positive puncta on immunofluorescence microscopy was compared with and without CQ administration as we reported previously [5]. In RubcnF/F-CTRL;GFP-MAP1LC3B mice, the number of puncta in the LRP2-positive PTECs remained almost unchanged by CQ administration (Figure 1A,B). By contrast, CQ administration significantly increased the number of puncta in the PTECs of rubcnF/F-TSKO;GFP-MAP1LC3B mice (Figure 1A,B).
Figure 1.
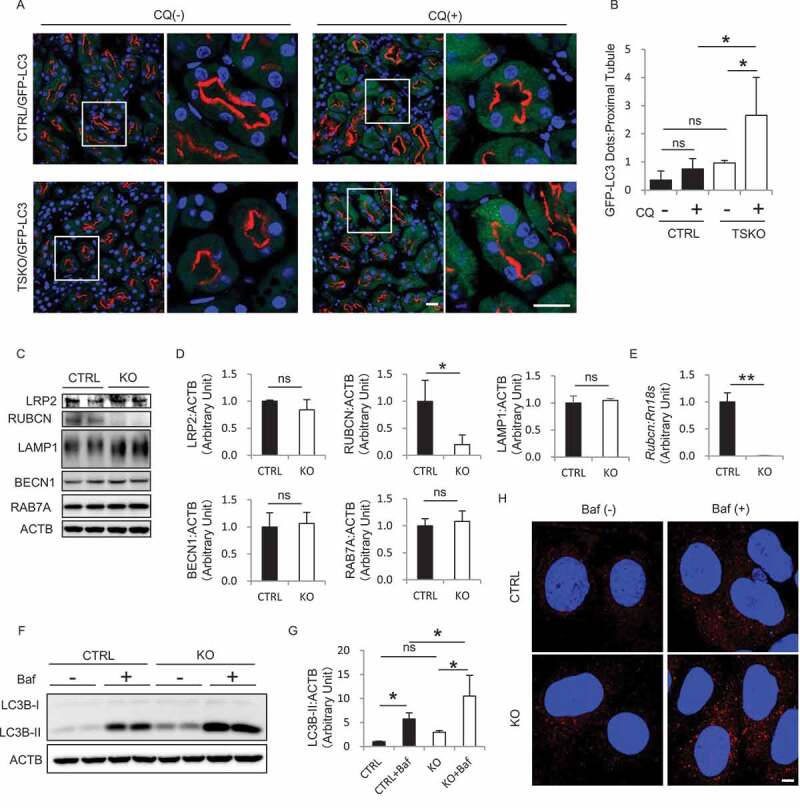
RUBCN deficiency increases autophagic flux in the PTECs. (A and B) In vivo evaluation of the autophagic flux in PTECs during the fed state. (A) Representative images of immunofluorescence analysis for LRP2 (red) and 4ʹ, 6-diamidino-2-phenylindole (DAPI; blue) as counterstaining on the kidney sections from 3-month-old RubcnF/F-CTRL;GFP-MAP1LC3B and rubcnF/F-TSKO;GFP-MAP1LC3B mice with or without CQ administration 6 h before euthanasia. The right panels show a magnification of the indicated areas (white squares) in the left panels. (B) Quantification of the number of GFP-positive puncta per proximal tubule. (C) Representative images of western blot analysis for LRP2, RUBCN, LAMP1, BECN1, and RAB7A in the cell lysate from cultured RUBCN CTRL and KO PTECs are shown. (D) Densitometric quantification of the protein levels in (C). (E) The mRNA expression of Rubcn in the cell lysate from cultured CTRL and KO PTECs was analyzed. (F-H) Evaluation of the autophagic flux in cultured PTECs. Representative images of western blot analysis (F) and immunofluorescence analysis for MAP1LC3B (red) and DAPI (blue) (H) of CTRL and KO PTECs with or without Baf. (C and F) ACTB (actin, beta) was used as a loading control. (G) Densitometric quantification of the MAP1LC3B-II protein levels in (F). n = 3 to 5 (B); 3 (D); 4 or 5 (E); 5 (G) in each group. Data are provided as the mean ± SD. ns, not significant. Statistically significant differences (*P < 0.05, **P < 0.01) are indicated. Bars: 20 µm (A) and 5 µm (H).
We then generated immortalized PTECs from 3-week-old RubcnF/F mice and established rubcn-deficient (KO) and -competent (CTRL) PTEC lines by transfecting pCAG (cytomegalovirus immediate-early enhancer and chicken β-actin promoter)-Cre and empty vectors, respectively. RUBCN deficiency in KO PTECs was confirmed by western blot and quantitative reverse transcription (RT)-PCR analysis (Figure 1C-E). RUBCN forms the complex with BECN1 and UVRAG (UV radiation resistance associated), and interacts with RAB7A (RAB7A, member RAS oncogene family), a Rab GTPase that localizes in the late endosome/lysosome [12]. The protein levels of LRP2, LAMP1 (lysosomal associated membrane protein 1), BECN1, and RAB7A in KO PTECs were comparable to those in CTRL PTECs (Figure 1C,D). During the process of autophagy, cytosolic MAP1LC3B/LC3B-I is converted to MAP1LC3B-II, a phagophore and autophagosome-associated phosphatidylethanolamine conjugate [18]. Treatment with bafilomycin A1 (Baf), a specific inhibitor of the vacuolar-type H+-translocating ATPase (V-ATPase), increased the level of MAP1LC3B-II in KO PTECs compared with CTRL PTECs, indicating high autophagic flux in KO PTECs (Figure 1F,G). Similarly, immunofluorescence analysis revealed that Baf increased the number of MAP1LC3B-positive puncta in KO PTECs (Figure 1H). Overall, these findings indicate that RUBCN deficiency in PTECs enhances high autophagic flux.
Rubcn-deficient PTECs exhibit phospholipids accumulation in lysosomes in autophagy-dependent manner
From the above observations, we expected that RUBCN deficiency in PTECs would protect them from ischemic injury via enhancing autophagic flux, since autophagy-deficiency predispose the PTECs to various stresses [3,6,7]. Then, we subjected 8-week-old RubcnF/F-CTRL and rubcnF/F-TSKO mice to ischemia-reperfusion (I/R) injury. However, no obvious alleviation was observed in rubcnF/F-TSKO mice compared with RubcnF/F-CTRL mice as assessed by histological injury score (Figure S2A and B) and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (Figure S2C and D).
Periodic-acid Schiff (PAS) staining demonstrated that 4-month-old rubcnF/F-TSKO mice showed no obvious change in the kidney under normal feeding conditions (Figure 2A). However, the PTECs from 12-month-old rubcnF/F-TSKO mice showed multiple cytosolic vacuolar formation, which was hardly observed in corresponding RubcnF/F-CTRL mice (Figure 2A). The vacuoles were negative for Oil Red O (ORO) staining (Figure 2B), indicating that they were not lipid droplets (LDs). Instead, the margins of the vacuoles were surrounded by the lysosomal transmembrane protein, LAMP1 (Figure 2C). The vacuoles were also positively stained with toluidine blue staining which can detect phospholipids (Figure 2D). Consistently, electron microscopy revealed that 12-month-old rubcnF/F-TSKO mice showed expanded lysosomes containing multi-layered, onion-like structures in PTECs (Figure 2E). To explore lysosomal function in KO PTECs, we assessed CTSD/cathepsin D activity in CTRL, KO, and Atg5-knocked down CTRL and KO PTECs by measuring the proportion of the levels of CTSD heavy chain (component of mature form) to pro-CTSD (Figure S3A). It was demonstrated that the lysosomal activity was higher in KO PTECs, which was attenuated by Atg5 knockdown. Moreover, Atg5 knockdown attenuated enlargement of lysosomes in KO PTECs (Figure S3B), suggesting that enlargement of lysosomes induced by RUBCN deficiency is the consequence of excessive autophagy. Morphology of mitochondria was well preserved and there was no accumulation of SQSTM1/p62 (sequestosome 1)-positive aggregates, a surrogate marker for autophagy stagnation, in PTECs of 12-month-old rubcnF/F-TSKO mice, suggesting that overall autophagic flux is maintained at least under basal condition (Figure S3C and D). There observed no significant differences between 12-month-old rubcnF/F-TSKO and RubcnF/F-CTRL mice in plasma urea nitrogen (UN) and urinary albumin to creatinine ratio; however, plasma creatinine level was higher in rubcnF/F-TSKO mice (Figure 2F).
Figure 2.
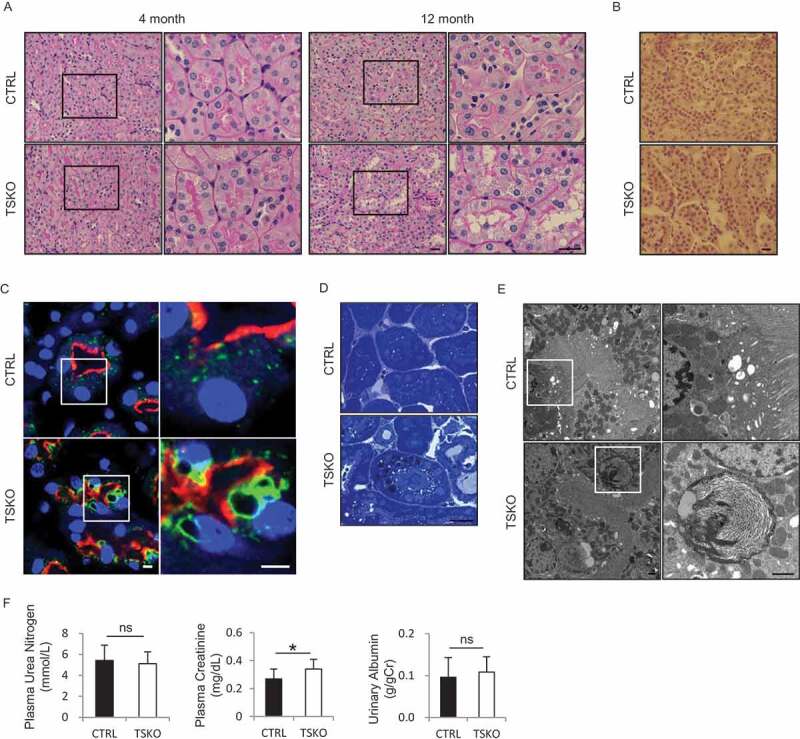
Phospholipids accumulation in PTEC lysosomes of rubcnF/F-TSKO mice. (A-E) Representative images of PAS (A) and ORO (B) staining, immunofluorescence analysis for LAMP1 (green), LRP2 (red), and DAPI (blue) as counterstaining (C), toluidine blue staining (D), and electron microscopic analysis (E) on the kidney sections from 4-month-old- (A) and 12-month-old- (A-E) RubcnF/F-CTRL and rubcnF/F-TSKO mice are shown. (F) The plasma UN, plasma creatinine, and the ratio of urinary albumin to creatinine were analyzed. (A, C, and E) The right panels show a magnification of the indicated areas (black or white squares) in the left panels. n = 12 or 13 in each group. Data are provided as the mean ± SD. ns, not significant. Bars: 20 µm (A, B, and D), 5 µm (C), and 1 µm (E).
RUBCN deficiency in PTECs leads to excessive weight gain with systemic fat accumulation
Unexpectedly, when fed standard mouse chow, rubcnF/F-TSKO mice began to exhibit a significant increase in weight compared with RubcnF/F-CTRL mice after 4 months of age (Figure 3A,B). At 12 months of age, a significant increase in wet weight of liver and adipose tissue (mesenteric, epididymal, and subcutaneous) was observed in rubcnF/F-TSKO mice (Figure 3C). LD formation (assessed by ORO staining) and triglyceride (TG) content were significantly increased in the liver of 12-month-old rubcnF/F-TSKO mice (Figure 3D,E). Histological analysis revealed that mesentery and subcutaneous adipocytes of 12-month-old rubcnF/F-TSKO mice had more abundant cytoplasm than those from RubcnF/F-CTRL mice (Figure 3F). These findings suggest that an increase in fat accumulation observed in rubcnF/F-TSKO mice was not limited to specific tissue, but rather a consequence of systemic metabolic alteration. Indeed, the mRNA expression of Srebf1 (sterol regulatory element binding transcription factor 1), a master lipogenic transcription factor, tends to increase in both liver and mesenteric adipose tissue of 12-month-old mice in rubcnF/F-TSKO mice compared with RubcnF/F-CTRL mice, suggesting systemic accelerated lipogenesis in rubcnF/F-TSKO mice (Figure 3G).
Figure 3.
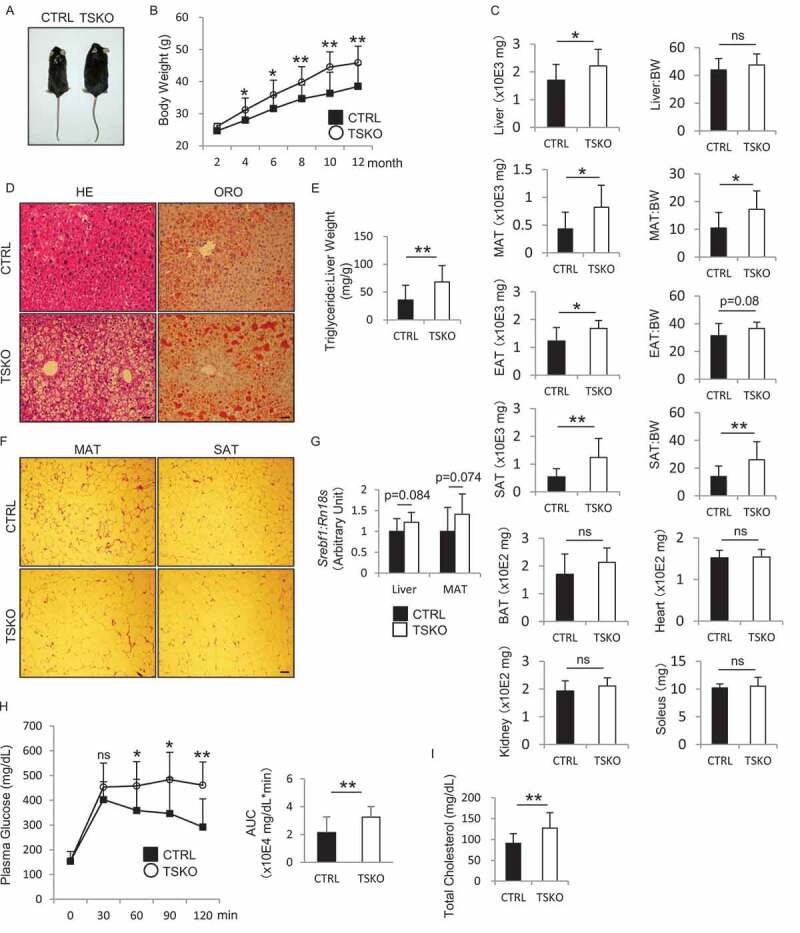
Fat accumulation and BW gain in rubcnF/F-TSKO mice. Metabolic effects of RUBCN deficiency in PTECs were analyzed. Appearance (A), BW curves (B), the weight and ratio relative to BW of liver, MAT, EAT, SAT, BAT, kidney, heart, and soleus (C), images of hematoxylin and eosin (HE) (left) and ORO (right) staining on the liver sections (D), the ratio of hepatic TG levels to weight (E), and HE staining on MAT (left) and SAT (right) sections (F), the mRNA expression of Srebf1 in the liver and MAT (G), blood glucose levels during GTT (H), and occasional plasma concentrations of total cholesterol (I) in RubcnF/F-CTRL and rubcnF/F-TSKO mice are shown. (A, D, and F) Representative images are presented. (H) AUCs were calculated by summation of trapezoids (right). (A, C-I) Data from 12-month-old mice (n = 10 to 13 in each group). Data are provided as the mean ± SD. ns, not significant. Statistically significant differences (*P < 0.05, **P < 0.01) are indicated. Bars: 20 µm (D) and 40 µm (F). MAT, mesenteric adipose tissue; EAT, epididymal adipose tissue; SAT, subcutaneous adipose tissue; BAT, brown adipose tissue; BW, body weight.
Next, we evaluated biochemical parameters related to metabolic syndrome in RubcnF/F-CTRL and rubcnF/F-TSKO mice. Insulin tolerance test (ITT) demonstrated reduced hypoglycemic response to insulin in 6-month-old rubcnF/F-TSKO mice compared with RubcnF/F-CTRL mice, although the difference did not reach significance (Figure S4). Glucose tolerance test (GTT) demonstrated that fasting blood glucose levels in 12-month-old rubcnF/F-TSKO mice were comparable to those in RubcnF/F-CTRL mice; however, rubcnF/F-TSKO mice maintained significant higher glucose levels for longer period than corresponding RubcnF/F-CTRL mice, indicating impaired glucose tolerance (Figure 3H). By contrast, circulating plasma insulin concentrations in 12-month-old rubcnF/F-TSKO mice were comparable to those in RubcnF/F-CTRL mice (Figure S5A). The plasma total cholesterol level was significantly increased in 12-month-old rubcnF/F-TSKO mice compared with RubcnF/F-CTRL mice, while plasma TG, phospholipids, and non-esterified fatty acids (FAs) (NEFAs) concentrations were indistinguishable between these mice (Figure 3I and S5B-D). We also measured fasting plasma concentration of NEFAs of 6-month-old mice, and found no significant difference (Figure S5E).
To clarify the cause of excessive weight gain observed in rubcnF/F-TSKO mice, we examined energy expenditure and daily food intake. Three-month-old rubcnF/F-TSKO mice showed similar oxygen consumption (VO2) and carbon dioxide release (VCO2) to corresponding RubcnF/F-CTRL mice (Figure S6A and B). This led to no significant difference in energy expenditure between these mice receiving standard mouse chow (Figure S6C). Moreover, there were no significant differences in food intake between these mice during the observational period (Figure S6D). These results suggest that the obesity of rubcnF/F-TSKO mice could neither be attributed to reduced energy expenditure nor to increased caloric intake.
To exclude the possibility of off-target expression of Cre recombinase in Kap-Cre mice, another strain of PTEC-specific tamoxifen-inducible mice (rubcn-floxed Ndrg1 [N-myc downstream regulated 1]-Cre+) was generated and analyzed. These mice similarly exhibit obesity after tamoxifen treatment, suggesting that metabolic effect observed in rubcnF/F-TSKO mice derives from RUBCN deficiency in PTECs (Figure S7).
RUBCN deficiency activates MTOR (mechanistic target of rapamycin) pathway in PTECs
We next evaluated the master growth regulator, MTOR complex 1 (MTORC1), which is activated at the lysosomal surface in response to nutrient inputs, such as free amino acids, glucose, and cholesterol [19], using kidney sections from 12-month-old RubcnF/F-CTRL and rubcnF/F-TSKO mice. MTORC1 directly phosphorylates the translational regulators eukaryotic translation initiation factor RPS6 (ribosomal protein S6), RPS6KB1/S6K1 (ribosomal protein S6 kinase B1), and EIF4EBP1/4E-BP1 (eukaryotic translation initiation factor 4E binding protein 1), all of which promote protein synthesis [20]. Immunohistochemical analysis for p-RPS6 revealed that MTORC1 signaling was hyperactivated in PTECs of rubcnF/F-TSKO mice compared with RubcnF/F-CTRL mice (Figure 4A).
Figure 4.
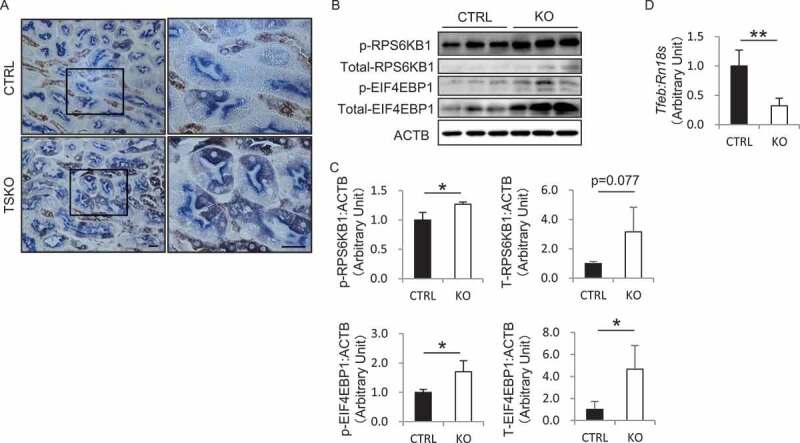
RUBCN deficiency activates MTOR pathway in PTECs. (A) Representative images of immunohistochemical analysis for p-RPS6 in the kidney sections from 12-month-old RubcnF/F-CTRL and rubcnF/F-TSKO mice are shown. Sections were co-immunostained for the proximal tubule marker, LRP2 in blue. The right panels show a magnification of the indicated areas (black squares) in the left panels. (B) The protein levels of p-RPS6KB1, p-EIF4EBP1, and total RPS6KB1 and EIF4EBP1 in the cell lysate from cultured CTRL and KO PTECs are shown. ACTB was used as a loading control. (C) Densitometric quantification of protein levels in (B). (D) The mRNA expression of Tfeb in the cell lysate from cultured CTRL and KO PTECs was analyzed. n = 3 (C); 4 or 5 (D) in each group. Data are provided as the mean ± SD. ns, not significant. Statistically significant differences (*P < 0.05, **P < 0.01) are indicated. Bars: 20 µm.
To exclude the possibility that the cellular environment in obese rubcnF/F-TSKO mice such as circulating abundant amino acids might activate the MTOR signaling in PTECs, we investigated the MTOR activity in vitro. Notably, western blot analysis revealed that the levels of phosphorylated RPS6KB1 and EIF4EBP1, and total EIF4EBP1 were significantly increased in cultured KO PTECs compared with CTRL PTECs under the complete medium (CM) with 5% fetal bovine serum (FBS) with the same amino acids, insulin, and glucose concentrations, suggesting that sustained high autophagic flux may transport excessive nutrients to the lysosomes (Figure 4B,C). TFEB (transcription factor EB), a master regulator of lysosomal biogenesis and autophagy, is phosphorylated and regulated in an MTORC1-dependent manner [21,22]. In concordance with these previous reports, the mRNA expression of Tfeb was significantly suppressed in cultured KO PTECs compared with CTRL PTECs (Figure 4D). Thus, RUBCN deficiency activates MTOR pathway in PTECs in a cell-intrinsic manner.
RUBCN deficiency promotes the mobilization of phospholipids from cellular membranes to lysosomes in cultured PTECs
Autophagy contributes to transfer phospholipids from plasma and organelle membranes to lysosomes. Additionally, FAs released from phospholipids by autophagy are stored in LDs [23]. To investigate how RUBCN deficiency modulates the distribution of FAs derived from phospholipids, we performed a FA pulse-chase labeling method using 2-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine (FL HPC), fluorescent-tagged phosphatidylcholine at its FA tail (Figure 5A). Cultured CTRL and KO PTECs were labeled for 16 h with trace amounts of FL HPC, which diffusely integrated into various cellular membranes, including the endoplasmic reticulum, mitochondria, and Golgi apparatus in accord with the previous report (Figure 5B, 0 h) [23]. FL HPC signal on cellular membranes was significantly decreased in KO PTECs chased for 6 h compared with CTRL PTECs (Figure 5B, 6 h). Instead, FL HPC redistribution into LysoTracker Red-positive lysosomes was significantly accelerated in KO PTECs (Figures 5B, 6 h, and C). In addition, the average size of LysoTracker Red-positive lysosomes colocalized with FL HPC was significantly increased in KO PTECs compared with CTRL PTECs chased for 24 h (Figure 5B, 24 h, and D). These results reflect that enhanced autophagy in KO PTECs promotes the mobilization of phospholipids from cellular membranes to lysosomes in PTECs, leading to lysosomal stress.
Figure 5.
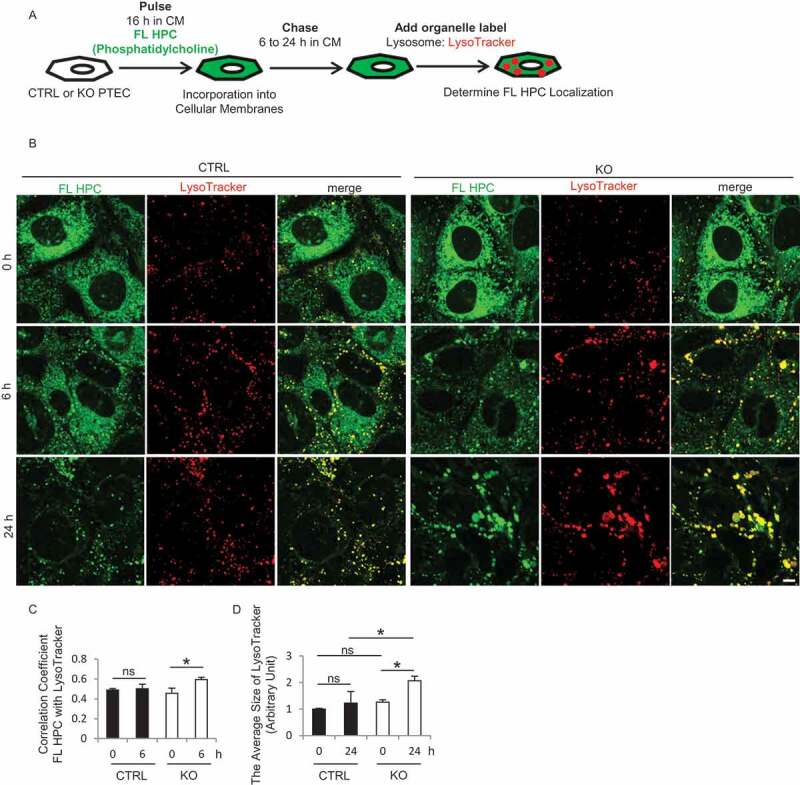
RUBCN deficiency promotes the mobilization of phospholipids from cellular membranes to lysosomes in cultured PTECs. (A) Schematic representation of the FA pulse-chase assay. PTECs were incubated with CM (DMEM with 5% FBS) containing 2 μM FL HPC for 16 h, and then chased for the indicated times. (B) Representative images of fluorescence analysis for LysoTracker Red (red) and DAPI (blue) as counterstaining in CTRL and KO PTECs. (C) Colocalization of FL HPC and LysoTracker Red in (B) was assessed by the Pearson correlation coefficient. (D) The average size of LysoTracker Red in (B) was quantified. n = 3 in each group. Data are provided as the mean ± SD. ns, not significant. Statistically significant differences (*P < 0.05) are indicated. Bar: 5 µm.
RUBCN deficiency promotes LD degradation in cultured PTECs
FAs are transferred into mitochondria and utilized for ATP synthesis through β-oxidation in PTECs [24,25]. To understand how RUBCN deficiency alters this FA trafficking process, we performed another pulse-chase assay using cultured CTRL and KO PTECs treated with oleic acid (OA)-bound albumin to mimic and magnify the LD-forming effect of FAs. We utilized 4,4-difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-dodecanoic acid (Red C12), a saturated FA analog with a tail composed of 12 carbons and a boron-dipyrromethene (BODIPY) 558/568 fluorophore covalently bound at the hydrophobic end (Figure 6A) [23]. Cultured PTECs were treated with trace amounts of Red C12 and 250 μM OA for 36 h, and then chased in OA-free CM in the absence of Red C12 for up to 48 h (0, 24, and 48 h), followed by labeling with 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene (BODIPY-LD) for LDs or MitoTracker Deep Red for mitochondria. At baseline (0 h), the dot-like Red C12 distribution was almost completely merged with BODIPY-LD in both CTRL and KO PTECs, suggesting that most of the Red C12 was incorporated into LDs (Figure 6B). In chased CTRL PTECs, nearly all Red C12 signal localized to LDs and little was detected in mitochondria at 24 h (Figure 6B,C). By contrast, in KO PTECs, a part of Red C12 signal was distributed reticularly in the cytosol with significant decrease in LD content at this time point, and significant colocalization of Red C12 with mitochondria was detected by co-staining with MitoTracker, which was confirmed by assessing with the Pearson correlation coefficient (Figure 6B,C). Consistently, both the measurement of TG content and ORO staining revealed significant rapid LD degradation in chased KO PTECs compared with CTRL PTECs (Figure 6D,E). We also performed ORO staining on the kidney sections of RubcnF/F-CTRL mice and rubcnF/F-TSKO mice after 18 h of starvation (Figure S8). It was demonstrated that positive area is decreased in rubcn-deficient kidney, but the difference did not reach significance. Acceleration of FA mobilization to mitochondria in KO PTECs was supported by the observation that mRNA levels of β-oxidation and oxidative phosphorylation-related molecules including Cpt1a (carnitine palmitoyltransferase 1a, liver), Acadm (acyl-CoA dehydrogenase medium chain), Ppargc1a (PPARG coactivator 1 alpha), Ndufb5 (NADH:ubiquinone oxidoreductase subunit B5), and Uqcrb (ubiquinol-cytochrome c reductase binding protein) were increased in KO PTECs in response to OA (Figure S9). Taken together, these data suggest the acceleration of FA mobilization from LDs to mitochondria in KO PTECs.
Figure 6.
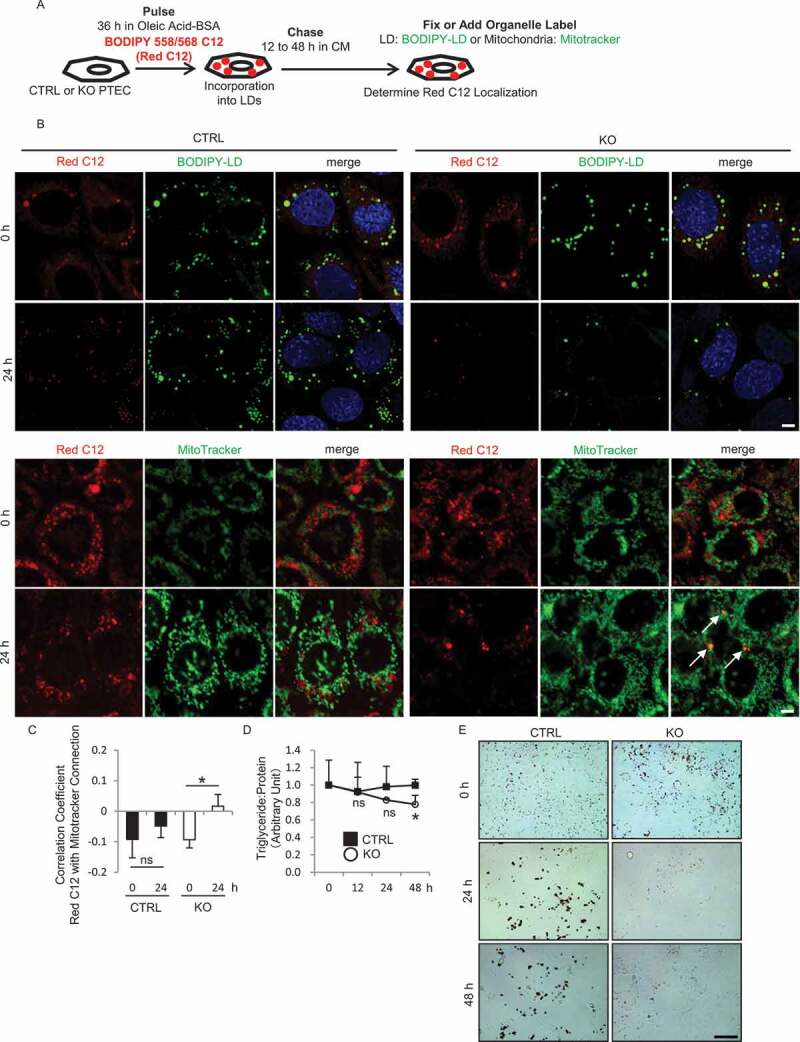
RUBCN deficiency promotes LD degradation in cultured PTECs. (A) Schematic representation of the FA pulse-chase assay. PTECs were incubated with CM containing 1 μM Red C12 and 250 μM OA for 36 h, and then chased for the indicated times. (B) Representative images of fluorescence analysis for BODIPY-LD (green) or MitoTracker Deep Red (green) and DAPI (blue) as counterstaining in chased CTRL and KO PTECs at 0 and 24 h. (C) Colocalization of Red C12 and MitoTracker Deep Red in (B) was assessed by the Pearson correlation coefficient. (D) The ratio of TG levels to protein in chased CTRL and KO PTECs was analyzed. (E) Representative images of ORO staining in chased CTRL and KO PTECs. n = 3 in each group. Data are provided as the mean ± SD. ns, not significant. Statistically significant differences (*P < 0.05) are indicated. Bars: 5 µm (B) and 200 µm (E).
RUBCN deficiency increases FA expulsion from cultured PTECs
To investigate how RUBCN deficiency leads to metabolic syndrome observed in rubcnF/F-TSKO mice, PTECs were co-cultured with hepatocytes (BNL-CL2 cells) in transwell. Cultured CTRL and KO PTECs were initially treated with 500 μM OA for 36 h on cell culture inserts, and then co-cultured with indicator BNL-CL2 cells in OA-free CM for up to additional 36 h (Figure 7A). Notably, ORO staining and fluorescence analysis for BODIPY-LD showed that BNL-CL2 cells exhibited a significant increase in LD formation when co-cultured with KO PTECs compared with CTRL PTECs (Figure 7B-D). Previous work has shown that FAs are transferred into the lipid-depleted extracellular space from high-lipid AML12 cells, which are established from mouse hepatocytes [26]. To test this possibility of lipid expulsion, cultured PTECs were treated with trace amounts of Red C12 with OA for 36 h, and then co-cultured with BNL-CL2 cells for 36 h in Red C12- and OA-free CM (Figure 7E). Interestingly, fluorescent signal of Red C12 was more detectable in the LDs of BNL-CL2 cells co-cultured with KO PTECs (Figure 7F). To confirm an increased efflux of FAs from rubcn-deficient PTECs, FA levels in the cultured medium of CTRL and KO PTECs were determined by colorimetric assay and lipidomic analysis. Cultured CTRL and KO PTECs were initially treated with 500 μM OA for 36 h, and then cultured in OA-free CM for additional 12 h. Concentrations of total and most of respective FAs in KO PTECs were significantly increased compared with CTRL PTECs (Figure S10 and Table 1). Moreover, we performed lipidomic analysis using whole kidneys from RubcnF/F-CTRL and rubcnF/F-TSKO mice (Table 2). It was demonstrated that the amount of most FAs was significantly lower in the kidney of rubcnF/F-TSKO mice compared with RubcnF/F-CTRL mice, suggesting that FAs produced by an enhanced autophagy are rapidly expelled from the kidney, resulting in decreased FA content in kidney of rubcnF/F-TSKO mice. Finally, we demonstrated that protein expression of phospholipid and cholesterol transporters ABCG1 (ATP binding cassette [ABC] subfamily G member 1) and ABCA1 (ABC subfamily A member 1) increase in rubcn-deficient PTECs under OA overload (Figure S11). Collectively, these results suggest accelerated FA efflux from KO PTECs.
Figure 7.
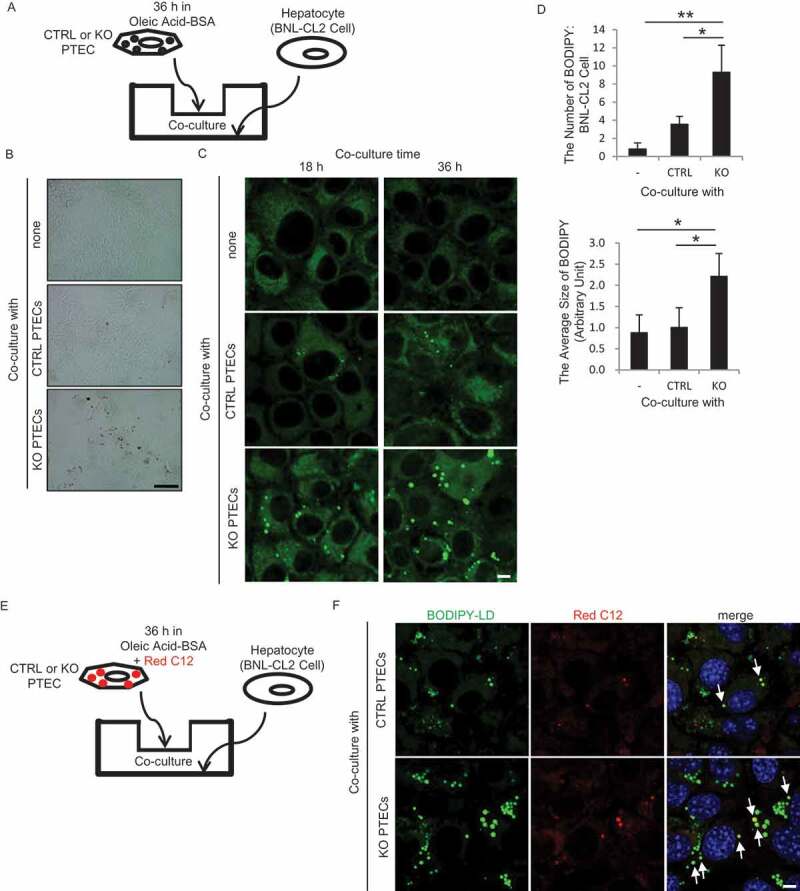
RUBCN deficiency promotes FA expulsion from the PTECs. (A) Schematic representation of the co-culture assay. PTECs treated with 500 µM OA for 36 h on cell culture inserts were co-cultured with BNL-CL2 cells in OA-free CM. (B and C) Representative images of ORO staining (B) and fluorescence analysis for BODIPY-LD (green) (C) of BNL-CL2 cells co-cultured for 18 h (C) or 36 h (B and C) with CTRL and KO PTECs. (D) The number and the average size of BODIPY-LD-positive puncta per BNL-CL2 cell co-cultured for 36 h in (C) were quantified. (E) Schematic representation of another co-culture assay. PTECs treated with 500 µM OA and 1 μM Red C12 for 36 h on cell culture inserts were co-cultured with BNL-CL2 cells in OA-free CM. (F) Representative images of fluorescence analysis for BODIPY-LD (green) in BNL-CL2 cells co-cultured for 36 h with Red C12-prelabeled CTRL and KO PTECs. n = 3 in each group. Data are provided as the mean ± SD. Statistically significant differences (*P < 0.05, **P < 0.01) are indicated. Bars: 200 µm (B) and 5 µm (C and F).
Table 1.
Increased efflux of FA from rubcn-deficient PTECs.
| CTRL |
KO |
|||||
|---|---|---|---|---|---|---|
| Lipid | Mean | SD | Mean | SD | KO:CTRL | P value |
| FA (12:0) | 275.6 | 310.1 | 252.6 | 293.2 | 0.9 | 0.90 |
| FA (13:0) | 1.0 | 2.5 | 146.3 | 170.7 | 145.7 | 0.09 |
| FA (14:0) | 0.0 | 0.0 | 0.0 | 0.0 | - | - |
| FA (14:1) | 118.8 | 27.3 | 96.4 | 24.0 | 0.8 | 0.16 |
| FA (16:0) | 177.1 | 433.8 | 156.1 | 382.3 | 0.9 | 0.93 |
| FA (16:1) | 48.6 | 118.9 | 285.1 | 87.5 | 5.9 | 0.00 |
| FA (17:0) | 0.0 | 0.0 | 0.0 | 0.0 | - | - |
| FA (18:0) | 0.0 | 0.0 | 0.0 | 0.0 | - | - |
| FA (18:1) | 2574.5 | 1044.3 | 5117.0 | 1476.3 | 2.0 | 0.01 |
| FA (18:2) | 54.4 | 133.2 | 74.9 | 119.3 | 1.4 | 0.78 |
| FA (20:1) | 116.6 | 98.3 | 152.0 | 42.2 | 1.3 | 0.44 |
| FA (20:2) | 122.9 | 75.8 | 155.7 | 42.6 | 1.3 | 0.38 |
| FA (20:3) | 95.0 | 54.5 | 246.9 | 58.5 | 2.6 | 0.00 |
| FA (20:4) | 46.9 | 78.4 | 79.6 | 29.5 | 1.7 | 0.36 |
| FA (20:5) | 119.5 | 67.1 | 107.1 | 28.4 | 0.9 | 0.69 |
Cultured CTRL and KO PTECs were treated with 500 μM OA for 36 h, and then cultured in OA-free CM for an additional 12 h. Difference in FA contents of the CM was analyzed by lipidomic analysis. Mean value and SD of each FA contents (pmol/mg protein), the ratio of the levels of KO to CTRL PTECs, and P value vs. CTRL PTECs are indicated. n = 6 in each group.
Table 2.
Decreased FA content in kidney of rubcnF/F-TSKO mice.
| CTRL |
TSKO |
|||||
|---|---|---|---|---|---|---|
| Lipid | Mean | SD | Mean | SD | TSKO:CTRL | P value |
| FA (14:0) | 1.5 | 0.9 | 0.8 | 0.6 | 0.53 | 0.150 |
| FA (16:0) | 111.8 | 19.0 | 79.8 | 22.3 | 0.71 | 0.032 |
| FA (16:1) | 28.0 | 7.2 | 21.5 | 3.2 | 0.77 | 0.076 |
| FA (18:0) | 69.3 | 14.1 | 45.1 | 15.0 | 0.65 | 0.023 |
| FA (18:1) | 205.2 | 35.9 | 154.1 | 20.9 | 0.75 | 0.016 |
| FA (18:2) | 202.0 | 29.7 | 151.5 | 27.8 | 0.75 | 0.017 |
| FA (18:3) | 9.3 | 2.1 | 6.9 | 1.9 | 0.74 | 0.077 |
| FA (20:2) | 9.1 | 1.5 | 6.4 | 1.1 | 0.70 | 0.006 |
| FA (20:3) | 9.8 | 2.0 | 6.5 | 1.0 | 0.66 | 0.005 |
| FA (20:4) | 54.6 | 13.6 | 33.7 | 3.1 | 0.62 | 0.025 |
| FA (20:5) | 5.1 | 1.0 | 3.9 | 0.8 | 0.76 | 0.047 |
| FA (22:1) | 2.1 | 0.3 | 1.3 | 0.2 | 0.63 | 0.001 |
| FA (22:2) | 0.7 | 0.1 | 0.5 | 0.1 | 0.74 | 0.022 |
| FA (22:5) | 14.4 | 3.0 | 10.7 | 2.2 | 0.74 | 0.044 |
| FA (22:6) | 44.7 | 14.1 | 29.0 | 7.7 | 0.65 | 0.043 |
| FA total | 767.5 | 113.9 | 551.5 | 101.2 | 0.72 | 0.009 |
Difference in FA contents of the whole kidneys from 12-month-old RubcnF/F-CTRL and rubcnF/F-TSKO mice was analyzed by lipidomic analysis. Mean value and SD of each FA contents (pmol/mg kidney weight), the ratio of the levels of rubcnF/F-TSKO to RubcnF/F-CTRL mice, and P value vs. RubcnF/F-CTRL mice are indicated. n = 5 or 6 in each group.
Metabolic effects of RUBCN deficiency are dependent on autophagy but not on endocytosis
Since RUBCN negatively regulate both the endocytic and autophagic pathway [10,12], metabolic effects of RUBCN deficiency observed in rubcnF/F-TSKO mice or KO PTECs may come from activated endocytosis pathway. Indeed, EGFR (epidermal growth factor receptor) degradation assay demonstrated that the degradation of EGFR protein was promoted in KO PTECs compared with CTRL PTECs, suggesting that RUBCN deficiency activates endocytosis pathway in PTECs (Figure S12A and B). To determine the relative contribution of the endocytic and autophagic pathway to the metabolic effects of RUBCN deficiency, we generated PTEC-specific rubcn- and atg5-deficient mice (rubcnF/Fatg5F/F-TS double KO [TSDKO] mice). PAS staining and immunohistochemical analysis demonstrated that 6-month-old rubcnF/Fatg5F/F-TSDKO mice showed cellular hypertrophy with the accumulation of SQSTM1 in PTECs, hallmark features of autophagy deficiency (Figure S12C and D) [3]. On the other hand, expanded lysosomes observed in rubcnF/F-TSKO mice were not seen in rubcnF/Fatg5F/F-TSDKO mice (Figure S12C-E). Notably, rubcnF/Fatg5F/F-TSDKO mice did not exhibit excessive weight gain by 6 months of age (Figure S12F), suggesting that ablation of both Rubcn and Atg5 genes significantly reversed an obese phenotype. Together, metabolic effect of RUBCN deficiency is mainly dependent on an activation of autophagy rather than endocytosis.
Discussion
In this study, we found previously unknown relationship between RUBCN deficiency in PTECs and metabolic syndrome. Although RUBCN deficiency leads to sustained high autophagic flux, it does not protect PTECs from kidney injury. Instead, KO mice exhibit several hallmark features of metabolic syndrome concomitantly with expanded lysosomes containing phospholipids in PTECs. RUBCN deficiency-induced acceleration of autophagy in cultured PTECs promotes the mobilization of phospholipids from cellular membranes to lysosomes and FA transfer to mitochondria upon OA treatment. Furthermore, RUBCN deficiency accelerates FA efflux from cell to blood circulation. Although we cannot completely exclude the possibility of off-target expression of Cre recombinase in Kap-Cre mice and Ndrg1-Cre mice, FA efflux from PTECs may link RUBCN deficiency and metabolic syndrome.
From 2 recent studies demonstrating the effect of enhanced autophagy on disease progression using rubcn-deficient mice [13,14] and our previous reports that autophagy deficiency jeopardizes PTECs during acute and chronic kidney injuries, we speculated that RUBCN deficiency in PTECs might alleviate kidney injury. However, contrary to our hypothesis, RUBCN deficiency did not alleviate kidney injury including I/R injury and streptozotocin-induced diabetic nephropathy (Fig. S2 and data not shown), despite enhanced autophagic flux. Recently, several attempts have been made to explore autophagic pathways as a therapeutic strategy for a variety of pathological conditions, including neurodegeneration and kidney disease; however, our observation suggests that pharmacological manipulation that simply upregulates autophagic flux can be insufficient or harmful, since different steps in the autophagic process are altered in each disorder and that therapies that are tailored to autophagic status must be developed. This may be achieved by modulating the activity of TFEB, a master transcriptional modulator that influences both lysosomal biogenesis and autophagy, by making autophagy substrates more easily degraded, or by targeting more selective autophagy (e.g. mitophagy).
Our in vivo and in vitro experiments demonstrated that RUBCN deficiency in PTECs leads to expanded lysosomes containing multi-layered structures of phospholipids, suggesting that excessive mobilization of phospholipids from cellular membranes to lysosomes via enhanced autophagy overwhelmed the degradation capacity of lysosome. Although these findings may ultimately be reconciled by a better understanding of the diverse set of roles of RUBCN, RUBCN may serve as a safety for preventing excessive nutrient influx into lysosomes by modulating autophagic flux. A recent report that, under nutrient-rich condition, MTORC1 phosphorylates UVRAG, which enhances the UVRAG and RUBCN interaction, leading to an inhibition of autophagosome-lysosome fusion supports this notion [27]. Similar to the phenotypes of rubcnF/F-TSKO mice, we previously observed phospholipid accumulation in the lysosomes of PTECs of HFD-fed wild-type mice, which comes from stagnation of autophagy due to lysosomal dysfunction despite an increased need for autophagic degradation. Indeed, phospholipid accumulation in the lysosomes was synergistically exaggerated in PTECs in HFD-fed rubcnF/F-TSKO mice (data not shown), confirming that RUBCN alleviates lysosomal stress during nutritional excess.
Although plasma concentrations of FAs (Figure S5) did not show obvious difference between RubcnF/F-CTRL and rubcnF/F-TSKO mice, lipidomic analysis and transwell experiment (Figure 7) both suggest that acceleration of FA efflux may explain systemic fat accumulation and obesity in rubcnF/F-TSKO mice. We speculate that since FAs expelled by PTECs are rapidly absorbed by the liver and adipose tissue (and are converted into cholesterol) and plasma FAs, especially elevated during starvation, are mainly derived from lipolysis in the adipocyte, and therefore hardly reflect efflux from PTECs, we could not detect the difference between these mice. We cannot exclude the possibility that KO PTECs secrete cytokines or exosomes that may affect the metabolic statuses. FAs derived from phospholipid degradation are once stored as TG in the LDs, since excess free FAs are harmful [24]. Then, FAs are reproduced by lipolysis (via lypophagy and/or cytosolic lipase), which are utilized for β-oxidation in the mitochondria. It has been demonstrated that, in mouse embryonic fibroblasts, reduction in β-oxidation rates, induced by mitochondria fusion deficiency, increases rates of FA efflux out of cells [23]. Similarly, in rubcn-deficient PTECs, aberrant autophagy activation (and probably, increased lipolysis) accelerates FAs production in the lysosomes, some of which are delivered to mitochondria, but others are expelled out of the cells (Figure 8). Metabolic effects derived from RUBCN deficiency seem to be tissue-dependent, since in hepatocyte-specific rubcn-deficient mice, liver steatosis and injury upon HFD are attenuated without apparent BW gain [14]. Moreover, phospholipids accumulation in the lysosome is not observed in rubcn-deficient hepatocytes despite high autophagic flux. In another study, RUBCN knockdown in A549 cells does not lead to the upregulation MTOR pathway as assessed by phosphorylation of RPS6KB1 [10]. These discrepant effects of RUBCN deficiency in different organs may reflect multifaceted and highly heterogenous role of lysosomes; lysosomes in PTECs are highly vulnerable to quantitative and qualitative overload and feedback regulation by RUBCN is particularly important in PTECs. Indeed, HFD-fed wild-type mice, phospholipid accumulation in the lysosome was observed only in PTECs [6].
Figure 8.

Model of FA mobilization in RUBCN CTRL and KO PTECs. In KO PTECs, the mobilization of phospholipids to lysosomes is promoted by enhanced autophagy, followed by increased FA release (1). Excessive FAs distribute into mitochondria for β-oxidation, otherwise FAs are exported from PTECs (2).
The mechanism underlying FA efflux remains largely unknown [24], but ABC transporters may be involved in their transport. Among 48 ABC transporters, twenty have been implicated in the transport of lipids including phospholipid and cholesterol [28]. In macrophage, ultrasound-therapy triggers autophagy activation and promote FA efflux via increased expression of ABCA1 [29]. ABCA1 and ABCG1 are expressed in human kidney [30]. The protein levels of cholesterol transport proteins such as ABCA1, ABCG1 and SCARB1/SR-B1 are reduced in type 1 diabetic kidney [31,32] and acute kidney injury [33], which promote cholesterol accumulation and enhance kidney injury. Our observation that the expression of ABCA1 and ABCG1 are increased in rubcn-deficient PTECs (Figure S11) may support that excessive FA efflux via these transporters promotes metabolic syndrome.
In conclusion, sustained high autophagic flux by RUBCN deficiency leads to metabolic syndrome concomitantly with an accelerated mobilization of phospholipids from cellular membranes to lysosomes. Although it remains to be solved whether RUBCN modulates the lipid efflux from kidney, elucidating how RUBCN deficiency in the kidney can lead to metabolic syndrome will be a clue to understanding the pathogenesis of the metabolic syndrome, especially in relation to kidney diseases.
Materials and Methods
Animals
Kap-Cre mice [3], GFP-MAP1LC3B transgenic mice [17], Ndrg1-Cre mice [5], Atg5-floxed mice [3], and EGFP-CAT mice [15,34] all with a C57BL/6N background have been described previously. To generate rubcnF/F-TSKO mice, Rubcn-floxed mice, in which exon 6 of the Rubcn gene is flanked by 2 lox P sequences [14], were crossed with the Kap-Cre transgenic mice. To generate PTEC-specific rubcn- and atg5-deficient mice, rubcnF/F-TSKO mice were crossed with Atg5-floxed mice. All mice were allowed free access to water and standard mouse chow (Oriental Yeast, OYC2103800). All animal experiments were approved by the institutional committee of the Animal Research Committee of Osaka University and were in accordance with the Japanese Animal Protection and Management Law (No. 25). Kidney I/R injury was induced as described previously [3]. The induction of Atg5 deletion in Atg5F/F;Ndrg1-Cre mice and in vivo autophagy flux assay using CQ (Sigma-Aldrich, C6628) were described previously [5].
Antibodies
We used the following antibodies: antibodies for LRP2 (a gift from T. Michigami, Department of Bone and Mineral Research, Osaka Medical Center and Research Institute for Maternal and Child Health, Japan), RUBCN (Cell Signaling Technology [CST], 8465), SQSTM1 (Medical and Biological Laboratory [MBL], PM045), BECN1 (MBL, PD017), RAB7A (CST, 9367), MAP1LC3B (CST, 2775 for western blot analysis; MBL, PM036 for immunofluorescence analysis), ACTB (Sigma-Aldrich, A5316), LAMP1 (BD Bioscience, 553792), p-RPS6 (Ser235/236; CST, 2211), p-RPS6KB1 (Thr389; CST, 9234), p-EIF4EBP1 (Thr37/46; CST, 2855), RPS6KB1 (CST, 2708), EIF4EBP1 (CST, 9452), Alexa Fluor 555-conjugated secondary antibody (Molecular Probes, A31572 [anti-rabbit IgG]), Alexa Fluor 488-conjugated secondary antibody (Molecular Probes, A21208 [anti-rat IgG]), biotinylated secondary antibody (Vector Laboratories, BA-1000 [anti-rabbit IgG]), horseradish peroxidase–conjugated secondary antibodies (Dako, P0447 [anti-mouse IgG], P0448 [anti-rabbit IgG], P0449 [anti-goat IgG], P0450 [anti-rat IgG]), CTSD (Santa Cruz Biotechnology, sc-6486), ABCG1 (Proteintech, 13578-1-AP), ABCA1 (Novus Biologicals, NB400-105), and EGFR (Santa Cruz Biotechnology, sc-03).
Quantitative RT-PCR analysis
Quantitative RT-PCR analysis was performed as previously described [34]. The sequences of primers used were as follows: Rubcn-F, 5ʹ-CTCATCCATGACCAGGTGTG-3ʹ; Rubcn-R, 5ʹ-GTCGCTCTCATGCAAACTGA-3ʹ; Srebf1-F, 5ʹ-GATCAAAGAGGAGCCAGTGC-3ʹ; Srebf1-R 5ʹ-TAGATGGTGGCTGCTGAGTG-3ʹ; Tfeb-F, 5ʹ-CCAGAAGCGAGAGCTCACAGAT-3ʹ; Tfeb-R 5ʹ-TGTGATTGTCTTTCTTCTGCCG-3ʹ; Ppara (peroxisome proliferator activated receptor alpha)-F,; 5ʹ-CGTCACGGAGCTCACAGAAT-3ʹ; Ppara-R; 5ʹ-ACTCGCGTGTGATAAAGCCA-3ʹ; Cpt1a-F, 5ʹ-ACCACTGGCCGAATGTCAAG-3ʹ; Cpt1a-R, 5ʹ-AGCGAGTAGCGCATGGTCAT-3ʹ; Acadm-F, 5ʹ-TAATCGGTGAAGGAGCAGGTTT-3ʹ; Acadm-R, 5ʹ-GGCATACTTCGTGGCTTCGT-3ʹ; Ppargc1a-F, 5ʹ-ATGTGTCGCCTTCTTGCTCT-3ʹ; Ppargc1a-R, 5ʹ-ATGTAGTGCCTGGGGACCTT-3ʹ; Ndufb5-F, 5ʹ-TCCTAGACTCGGAGTCGGAA-3ʹ; Ndufb5-R, 5ʹ-AACTTCCTGCTCCTTTAACC-3ʹ; Uqcrb-F, 5ʹ-ACTTACCCAGAAGGCAGCG-3ʹ; Uqcrb-R, 5ʹ-TGCCCACTCTTCTCTCTCCT-3ʹ.
Western blot analysis
Cultured PTECs were lysed in cell lysis buffer (CST, 9803) and a protease inhibitor cocktail, Complete Mini (Roche, 11836170001). The protein concentration was determined using Pierce BCA Protein Assay Reagent (Thermo Fisher Scientific, 23228). Each sample was subjected to immunoblot analysis as described previously [34].
Histological analysis
Histological analysis was performed as previously described with some modifications [34]. Mice were transcardially perfused using 4% paraformaldehyde (PFA) in phosphate-buffered saline (pH 7.4; composed of 137 mM NaCl [Wako, 191–01665], 2.7 mM KCl [Nacalai tesque, 28514–75], 8 mM Na2HPO4 [Wako, 196–02835], and 1.5 mM KH2PO4 [Wako, 169–04245])). Tissues were post-fixed and embedded in paraffin or frozen in optimal cutting temperature (OCT) compound (Sakura Finetek, 4583). Paraffin sections were stained with PAS (for kidney) or HE (for liver and adipose tissue) (both performed by Applied Medical Research Laboratory, Osaka, Japan). Paraffin-embedded or fixed-frozen sections were immunostained with the respective antibodies. Immunohistochemical double staining for LRP2 and p-RPS6 was performed as previously described [5]. Immunofluorescence images were obtained using an Olympus FV1000-D confocal laser scanning microscope (Olympus, Tokyo, Japan). To evaluate autophagic flux in vivo, the number of GFP-MAP1LC3B puncta per proximal tubule in at least 10 high-power fields (× 600) were counted. LDs were labeled with BODIPY-LD (Molecular Probes, D3922). The size and the number of BODIPY-LD-positive LDs were calculated using a digital image-analyzing software, ImageJ (available at http://rsbweb.nih.gov/ij/index.html; National Institutes of Health, Bethesda, MD). For each sample, at least 10 fields were analyzed. For electron microscopy, kidneys were fixed with 2% glutaraldehyde (Wako, 071–01931) and observed using a Hitachi H-7650 transmission electron microscope (Hitachi, Tokyo, Japan). Kidney injury after I/R was evaluated according to scoring system as previously described [3]. TUNEL assay was performed with in situ apoptosis detection kit (Takara, MK-500) according to the manufacturer’s instructions. Toluidine blue staining and ORO staining was performed as previously described [35].
Biochemical measurements
Plasma concentrations of total cholesterol, insulin, phospholipid, TG, NEFA, UN, and creatinine were measured using the Cholesterol E-Test (Wako, 439–17501), the Ultra Sensitive Mouse Insulin ELISA Kit (Morinaga Institute of Biological Science, M1104), the Phospholipid C-Test (Wako, 433–36201), the Triglyceride E-Test (Wako, 432–40201), the NEFA C-Test (Wako, 279–75401), the BUN-Test-Wako (Wako, 279–36201), and CRE-EN Kainos kit (Kainos, TKA7500), respectively. Total lipids in liver samples or cultured PTECs were extracted as described previously [36]. Prior to the extraction, the liver samples were weighed and homogenized by polytron (Kinematica, PT10-35). TGs were assayed using the Triglyceride E-test (as noted above), and then adjusted to the sample weight for the liver or to the protein level for cultured PTECs. The urinary albumin level was measured using the Microfluoral microalbumin test (Progen, PR2005), and then adjusted to the urinary creatinine level which was measured using the CRE-EN Kainos kit (as noted above). All kits were used in accordance with the manufacturer’s instruction.
ITT and GTT
ITT was performed by intraperitoneally injecting mice with 0.83 mU/g BW human insulin (Lilly, Humulin R, U100) as previously described [37]. As for GTT, after fasting for 5 h, mice received an intraperitoneal injection of D-glucose (2 g/kg BW, Nacalai Tescque, 16806–25). Blood glucose levels were measured using Glutest Every (Sanwa Kagaku Kenkyusho, Nagoya, Japan). Area under the curve (AUC) was calculated by summation of trapezoids.
Metabolism measurement
Whole body oxygen consumption was measured by using an O2/CO2 metabolism measuring system MK-5000RQ (Muromachi Kikai, Tokyo, Japan). Each mouse was placed in a sealed chamber (3220 ml volume) with an air flow of 0.5 l/min for 24 h at 24°C. Air was taken every 3 min, and the consumed oxygen concentration was converted to ml per min by multiplying it by the flow rate. Oxygen consumption was normalized by BW. Respiratory quotient was acquired by dividing carbon dioxide production by oxygen consumption.
Cell culture
Isolation and immortalization of PTECs was performed as previously described with some modifications [3]. In brief, PTECs were isolated from 3-week-old Rubcn-floxed mice and immortalized by transfecting pEF321-T (a gift from Sumio Sugano, University of Tokyo), a simian virus-40 (SV-40) large T antigen expression vector. Cells derived from a single colony were transfected with the pCAG-Cre vector (Addgene, 13775; a gift from Connie Cepko) to construct rubcn-deficient PTEC lines by electroporation method using Nucleofector I (Amaxa Biosystems, Colonge, Germany) and Basic Epithelial Cells Nucleofector Kit (Lonza, VPI-1005) in accordance with manufacturer’s instructions. We used Cre-negative Rubcn-floxed PTECs as a control. PTECs and BNL-CL2 cells, murine non-transformed hepatocyte cell line (a gift from S. Tanaka [Department of Gastroenterology and Hepatology, Osaka University Graduate School of Medicine], originally obtained from American Type Culture Collection [ATCC, TIB-73]), were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (1 g/l glucose; Nacalai tesque, 08456–36) with L-glutamine and sodium pyruvate (Nacalai Tesque, 08456–36) containing 5% FBS (Sigma-Aldrich, F7524) at 37°C under a humidified atmosphere of 5% CO2 and 95% air. To evaluate autophagic flux, PTECs were treated with 100 nM of Baf (Wako, 023–11641) in dimethyl sulfoxide (Sigma-Aldrich, D8418) 3 h before harvest and were subjected to western blot and immunofluorescence analysis for MAP1LC3B. We prepared lipid-containing media (OA-BSA) by conjugation of sodium OA (Sigma-Aldrich, O7501) with essentially FA-free, low endotoxin bovine serum albumin (BSA) (Sigma-Aldrich, A8806) at the final concentration of 250 or 500 µM as described previously [35]. Knockdown of Atg5 was performed by RNA interference using 3 unique 27 mer siRNA duplexes (OriGene Technologies, SR407903) with lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific, 13778100). Scrambled RNA (OriGene Technologies, SR30002) was used as a control. Kits were used in accordance with the manufacturer’s instruction.
Pulse-chase assay of phospholipid and FA
Fluorescent FA pulse-chase assay using FL HPC (Invitrogen, D3792) was performed as previously described [6]. PTECs were incubated with CM (DMEM with 5% FBS) containing 2 μM FL HPC for 16 h to allow to incorporate into cellular membranes, followed by washing 3 times with FL HPC-free CM, incubated for 1 h, and then chased for the indicated times. FL HPC-treated and chased PTECs were stained with 50 nM LysoTracker Red DND-99 (Invitrogen, L7528) for 30 min at 37°C immediately prior to imaging. As for fluorescent FA pulse-chase assay using Red C12 (Invitrogen, D3835), PTECs were incubated with CM containing 250 μM OA-BSA and 1 µM Red C12 for 36 h, followed by washing with CM, and chased for the indicated time periods under OA free condition. Chased PTECs were stained with 200 ng/ml BODIPY-LD or 25 nM of MitoTracker Deep Red FM (Invitrogen, M22426) for 15 min at 37°C, or fixed with 4% PFA for 15 min at room temperature for immunocytochemistry immediately prior to imaging.
Co-culture assay
Rubcn-deficient and -competent PTECs were seeded on 12-well cell culture inserts (0.4-µm pore size) (Corning, 353180) to form a monolayer. After treated with 500 µM OA for 36 h, PTECs were washed with CM and the inserts were moved onto 12-well plates in which BNL-CL2 cells were incubated. At each time point, BNL-CL2 cells were fixed with 4% PFA for 15 min at room temperature for ORO staining and fluorescence analysis for BODIPY-LD. Alternatively, PTECs were treated with 500 µM OA and 1 µM Red C12 for 36 h, were washed with CM and the inserts were moved onto 12-wells in which BNL-CL2 cells were incubated. At each time point, BNL-CL2 cells were fixed with 4% PFA for 15 min at room temperature for fluorescence analysis detecting Red C12.
EGFR assay
EGFR assay was performed as described previously [3]. In brief, PTECs were pre-cultured in DMEM without FBS for 5 h and incubated in DMEM containing 1% FBS with 40 μM BSA for 6 h. After adding 200 ng/ml EGF (Sigma-Aldrich, E4127), cells were cultured for the indicated times.
Lipidomic analysis
To assess an increased efflux of FAs from rubcn-deficient PTECs, we performed comparative lipidomic analysis of whole kidneys. Lipid extraction from mice kidney tissue and subsequent analysis were performed as described previously [35]. FA levels were quantified using supercritical fluid chromatography triple quadrupole mass spectrometry (SFC/MS/MS) in multiple reaction monitoring mode [38]. The SFC/MS/MS system using ACQUITY UPC2 Torus diethylamine column (100 × 3.0 mm inner diameter, particle size: 1.7 μm [Waters, Milford, MA, USA]) is composed of a SFC (Nexera UC [Shimadzu, Kyoto, Japan]) and a triple quadrupole mass spectrometer (LCMS-8060 [Shimadzu]). FA 16:0 (13C16) (0.22 μM) is used as internal standard. To assess the difference of the FA concentration in the cultured medium between CTRL and KO PTECs, cultured PTECs were initially treated with 500 μM OA for 36 h, and then incubated in OA-free CM for additional 12 h. FA levels were determined by colorimetric assay kit (Abcam, ab65341) and lipidomic analysis. The levels of FA in the medium were measured using SFC/MS/MS with ACQUITY UPC2 HSS C18 column (100 × 3.0 mm inner diameter, particle size: 1.8 μm [Waters]). FA 16:0 (13C16) (2.0 μM) was used as internal standard.
Statistics
All results are expressed as the mean ± standard deviation (SD). Statistical analyses were conducted using JMP software (SAS Institute, Cary, NC). Multiple-group comparisons were performed using analysis of variance with post-testing using the Tukey-Kramer test. Differences between 2 experimental values were assessed by the Student’s t-test. A P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank N. Mizushima (University of Tokyo) for the GFP-MAP1LC3B mice, T. Michigami (Osaka Medical Center and Research Institute) for LRP2 antibody, and N. Horimoto for technical assistance. This work was supported by MSD Life Science Foundation, Public Interest Incorporated Foundation (to A.T.), Ono Medical Research Foundation (to A.T.), and Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan (JP16K09614 [to A.T.], JP18K15975 [to R.F.], 15H06371 [to T.Y.], and JP18K08208 [to Y.T.]).
Funding Statement
This work was supported by the MSD Life Science Foundation, Public Interest Incorporated Foundation ;Ono Medical Research Foundation ;Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan [15H06371];Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan [JP18K15975];Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan [JP18K08208];Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan [JP16K09614].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Choi AMK, Ryter SW, Levine B.. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. [DOI] [PubMed] [Google Scholar]
- [3].Kimura T, Takabatake Y, Takahashi A, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22:902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Takahashi A, Kimura T, Takabatake Y, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012;180:517–525. [DOI] [PubMed] [Google Scholar]
- [5].Yamamoto T, Takabatake Y, Kimura T, et al. Time-dependent dysregulation of autophagy: implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy. 2016;12:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yamamoto T, Takabatake Y, Takahashi A, et al. High-fat diet-induced lysosomal dysfunction and impaired autophagic flux contribute to lipotoxicity in the kidney. J Am Soc Nephrol. 2017;28:1534–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Takahashi A, Takabatake Y, Kimura T, et al. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes. 2017;66:1359–1372. [DOI] [PubMed] [Google Scholar]
- [8].Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. [DOI] [PubMed] [Google Scholar]
- [9].Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, et al. DNA damage and autophagy. Mutat Res. 2011;711:158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. [DOI] [PubMed] [Google Scholar]
- [11].Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tabata K, Matsunaga K, Sakane A, et al. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol Biol Cell. 2010;21:4162–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zi Z, Song Z, Zhang S, et al. Rubicon deficiency enhances cardiac autophagy and protects mice from lipopolysaccharide-induced lethality and reduction in stroke volume. J Cardiovasc Pharmacol. 2015;65:252–261. [DOI] [PubMed] [Google Scholar]
- [14].Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64:1994–2014. [DOI] [PubMed] [Google Scholar]
- [15].Kawamoto S, Niwa H, Tashiro F, et al. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett. 2000;470:263–268. [DOI] [PubMed] [Google Scholar]
- [16].Ding Y, Davisson RL, Hardy DO, et al. The kidney androgen-regulated protein promoter confers renal proximal tubule cell-specific and highly androgen-responsive expression on the human angiotensinogen gene in transgenic mice. J Biol Chem. 1997;272:28142–28148. [DOI] [PubMed] [Google Scholar]
- [17].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Castellano BM, Thelen AM, Moldavski O, et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science. 2017;355:1306–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Laplante M, Sabatini DM.. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vega-Rubin-de-Celis S, Peña-Llopis S, Konda M, et al. Multistep regulation of TFEB by MTORC1. Autophagy. 2017;13:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32:678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens. 2010;19:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Uchida S, Endou H. Substrate specificity to maintain cellular ATP along the mouse nephron. Am J Physiol. 1988;255:F977–83. [DOI] [PubMed] [Google Scholar]
- [26].Herms A, Bosch M, Ariotti N, et al. Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Curr Biol. 2013;23:1489–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim Y-M, Jung CH, Seo M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015;57:207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Neumann J, Rose-Sperling D, Hellmich UA. Diverse relations between ABC transporters and lipids: an overview. Biochim Biophys Acta. 2017;1859:605–618. [DOI] [PubMed] [Google Scholar]
- [29].Li X, Zhang X, Zheng L, et al. Hypericin-mediated sonodynamic therapy induces autophagy and decreases lipids in THP-1 macrophage by promoting ROS-dependent nuclear translocation of TFEB. Cell Death Dis. 2016;7:e2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Klucken J, Büchler C, Orsó E, et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci U S A. 2000;97:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tang C, Kanter JE, Bornfeldt KE, et al. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J Lipid Res. 2010;51:1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Tsun JGS, Yung S, Chau MKM, et al. Cellular cholesterol transport proteins in diabetic nephropathy. PLoS One. 2014;9:e105787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zager RA, Johnson ACM, Hanson SY, et al. Acute tubular injury causes dysregulation of cellular cholesterol transport proteins. Am J Pathol. 2003;163:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Matsuda J, Namba T, Takabatake Y, et al. Antioxidant role of autophagy in maintaining the integrity of glomerular capillaries. Autophagy. 2018;14:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Minami S, Yamamoto T, Takabatake Y, et al. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy. 2017;13:1629–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- [37].Sakai S, Yamamoto T, Takabatake Y, et al. Proximal tubule autophagy differs in type 1 and 2 diabetes. J Am Soc Nephrol. 2019;30:929–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Takeda H, Izumi Y, Takahashi M, et al. Widely-targeted quantitative lipidomics method by supercritical fluid chromatography triple quadrupole mass spectrometry. J Lipid Res. 2018;59:1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.