

ABSTRACT
Burkholderia pseudomallei: which causes melioidosis with high mortality in humans, has become a global public health concern. Recently, infection-driven lipid droplet accumulation has been related to the progression of host-pathogen interactions, and its contribution to the pathogenesis of infectious disease has been investigated. Here, we demonstrated that B. pseudomallei infection actively induced a time-dependent increase in the number and size of lipid droplets in human lung epithelial cells and macrophages. We also found that lipid droplet accumulation following B. pseudomallei infection was associated with downregulation of PNPLA2/ATGL (patatin like phospholipase domain containing 2) and lipophagy inhibition. Functionally, lipid droplet accumulation, facilitated via PNPLA2 downregulation, inhibited macroautophagic/autophagic flux and, thus, hindered autophagy-dependent inhibition of B. pseudomallei infection in lung epithelial cells. Mechanistically, we further revealed that nuclear receptor NR1D2 might be involved in the suppression of PNPLA2 after cell exposure to B. pseudomallei. Taken together, our findings unraveled an evolutionary strategy, by which B. pseudomallei interferes with the host lipid metabolism, to block autophagy-dependent suppression of infection. This study proposes potential targets for clinical therapy of melioidosis.
Abbreviations: 3-MA: 3-methyladenine; ACTB: actin beta; ATG7: autophagy related 7; B. pseudomallei: Burkholderia pseudomallei; CFU: colony-forming unit; DG: diglyceride; FASN: fatty acid synthase; GFP: green fluorescent protein; LAMP1: lysosomal associated membrane protein 1; LC-MS/MS: liquid chromatography-tandem mass spectrometry; LD: lipid droplet; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MG: monoglyceride; MOI: multiplicity of infection; mRFP: monomeric red fluorescent protein; NR1D2: nuclear receptor subfamily 1 group D member 2; p.i., post-infection; PLIN2/ADRP: perilipin 2; PNPLA2/ATGL: patatin like phospholipase domain containing 2; Rapa: rapamycin; SQSTM1/p62: sequestosome 1; shRNA: short hairpin RNA; TEM: transmission electron microscopy; TG: triglyceride
KEYWORDS: Autophagy, Burkholderia pseudomallei, lipid metabolism, lipid droplet, lipophagy, NR1D2, PNPLA2/ATGL
Introduction
Lipid droplets are subcellular organelles that store neutral fat and are present in most cell types [1]. While the biological function of these lipid-rich organelles was initially circumscribed to lipid storage, numerous recent studies have indiated that they are involved in many other cellular functions. Interest in lipid droplet biogenesis and functioning has dramatically increased because of their association with metabolic and inflammatory diseases, including diabetes [2], cancer [3,4], neurodegenerative diseases [5], as well as numerous infectious diseases [6,7]. Recent studies have revealed increases in the number of lipid droplets during interactions of pathogenic bacteria with host cells, and the long-underestimated contribution of these organelles to the pathogenesis of infectious diseases has only begun to emerge [8,9]. The precise function of lipid droplets in host-pathogen interactions is not yet fully understood.
Lipid droplet formation during infection involves a highly specific, regulated cellular mechanism. Recently, significant advances have been achieved in the understanding of the molecular mechanisms underlying infection-driven lipid droplet formation. These new insights indicate that the accumulation of lipid droplets rapidly occurs in infections caused by a variety of pathogens and results from a balance between new lipid synthesis and regulated lipolysis, involving both transcription-dependent and -independent mechanisms [9]. Cytoplasmic lipid droplet-associated triglyceride (TG) hydrolysis mainly occurs via two pathways, namely, neutral lipolysis and acid lipolysis. It has been reported that PNPLA2/ATGL (patatin like phospholipase domain containing 2), LIPE/HSL (lipase E, hormone sensitive type), and MGLL (monoglyceride lipase) are involved in neutral lipolysis and hydrolyze TG, diglyceride (DG), and monoglyceride (MG), respectively [10,11]. Historically, the two distinct pathways were considered to be separate, whereas the 2009 discovery of lipophagy [12], a subtype of macroautophagy/autophagy, which is well known as one of the earliest defense responses encountered by intracellular pathogens [13,14], linked neutral and acid lipolysis [15]. Recently, crosstalk between lipid droplets and lysosomes through autophagy has been suggested [16]. Mycobacterium leprae-induced lipid droplets inhibit the maturation of M. leprae-containing phagosome to favor its intracellular survival in SW-10 cells [17]. Mycobacterium tuberculosis-induced lipid droplet formation inhibits activation of autophagic pathways [18]. And in response to Porphyromonas gingivalis infection, lipid droplets affect the formation of autolysosomes for its elimination in HepG2 cells [19]. The above evidence suggests that intracellular pathogens acquired the evolutionary ability to subvert autophagy by hijacking host lipid metabolism for intracellular survival.
Burkholderia pseudomallei is a gram-negative bacterium, which is commonly found in soil and surface groundwater in many tropical and subtropical regions [20]. B. pseudomallei is known as the causative agent of melioidosis and to have a broad host range, including humans, cattle, horses, dolphins, goats, and koalas. Infection occurs through inhalation of aerosols and exposure to contaminated water [21]. The typical clinical manifestations of melioidosis vary, mainly causing organ abscesses in the lung, liver, spleen, brain, with a mortality rate of up to 40% [22]. Furthermore, B. pseudomallei has been listed as a tier 1 select agent by the U.S. Centers for Disease Control and Prevention, owing to its potential as a biological weapon. To date, no effective vaccines have been developed against B. pseudomallei [23]. Many studies have shown that B. pseudomallei have evolved numerous strategies for intracellular survival [24]. In particular, it has been reported that B. pseudomallei can cause persistent infection by avoiding the host autophagy defense system [24,25]. However, it is unclear whether the underlying mechanism of B. pseudomallei persistent infection is related to host lipid metabolism.
In this study, we aimed to investigate the relationship between B. pseudomallei infection and lipid accumulation in human lung epithelial cells. We demonstrate that B. pseudomallei infection interferes with lipid metabolisms leading to lipid accumulation through PNPLA2 suppression and inhibition of lipophagy, which in turn inhibits autophagic flux for its own benefits. Furthermore, we revealed that nuclear receptor NR1D2 regulation mediates the PNPLA2 downregulation in response to B. pseudomallei exposure. These results shed light on a new facet of the intricate pathogen-host interaction network in which the host autophagic flux is diverted and manipulated by pathogens for survival.
Results
B. pseudomallei infection actively induces lipid droplets accumulation
To investigate the association between lipid metabolism and B. pseudomallei infection in host cells, we used transmission electron microscopy (TEM) to observe lipid droplets in B. pseudomallei-infected human lung epithelial A549 cells. As shown in Figure 1A, the typical structures of lipid droplets were observed, and their numbers gradually increased over time after B. pseudomallei infection at a multiplicity of infection (MOI) of 10:1, as compared with that in an uninfected control. The area of lipid droplets also gradually increased after infection, and both the number and area reached the maximum at 24 h post-infection (p.i.) (Figure 1B,C). Consistently, Nile red staining for neutral lipids showed that the number of lipid droplets gradually increased over time compared with that in uninfected A549 and BEAS-2B cells (Figure 1D,E, S1A, and S1B). Thus, it was apparent that B. pseudomallei infection increased the number and size of lipid droplets in infected cells. To investigate the content of the lipid droplets accumulated after B. pseudomallei infection, we detected the levels of total triglycerides in cells. Consistent with the above observations, total triglycerides were increased in response to B. pseudomallei infection (Fig. S1C and S1D). Furthermore, we determined the levels of TG, DG, and MG in A549 cells by LC-MS/MS, respectively. Interestingly, we found that the level of TG increased upon infection, while DG declined (Figure 1F), suggesting the underlying mechanisms involved in lipid accumulation after B. pseudomallei infection.
Figure 1.
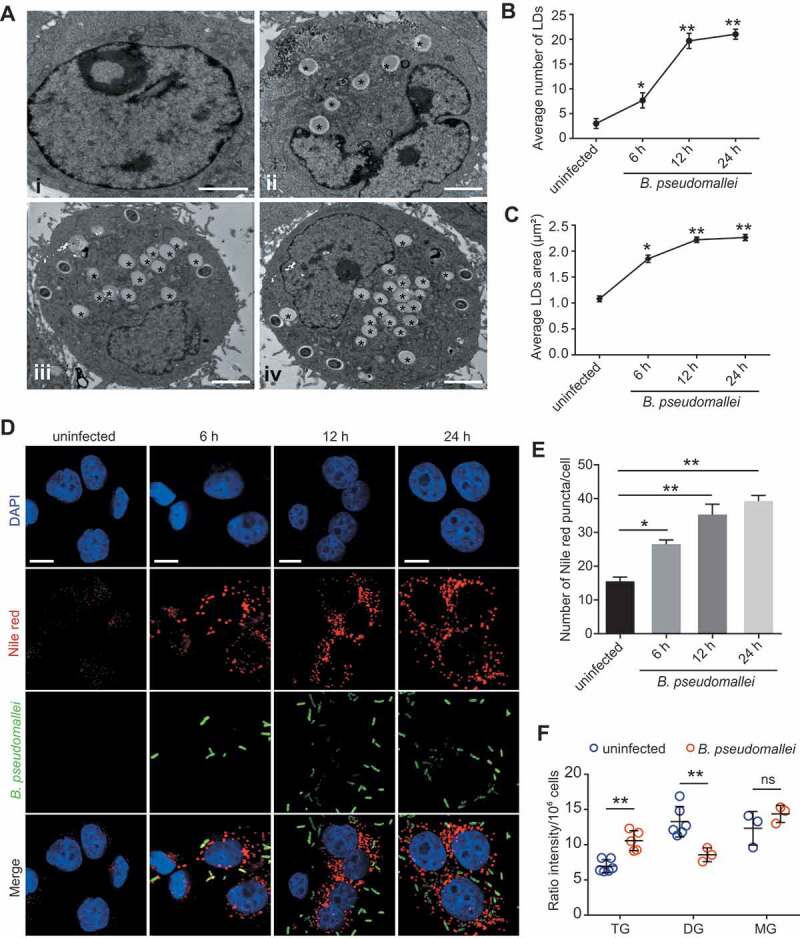
Lipid droplets accumulation in response to B. pseudomallei in A549 cells. (A) Representative TEM images of lipid droplets in A549 cells. A549 cells were uninfected (I) or treated with B. pseudomallei (MOI = 10) for 6 h (ii), 12 h (iii) and 24 h (iv). * indicates lipid droplets. Scale bars: 2 μm. (B and C) Quantification of the number and area of LDs per cell in TEM images. LDs, lipid droplets. (D and E) Lipid staining of B. pseudomallei infected A549 cells by Nile red as described above. Representative images are displayed. The number of lipid droplet puncta in each cell was counted. Scale bars: 10 μm. (F) Measurement of cellular lipid levels in control or B. pseudomallei-infected A549 cells by LC-MS/MS analysis. A549 cells were uninfected or B. pseudomallei-infected (MOI = 10) for 12 h. Data is shown as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. ns, not significant
To further characterize what triggers the increase of lipid abundance upon B. pseudomallei infection, we recapitulate the lipid droplet accumulation using THP-1 human pulmonary macrophages. We found that infection with live B. pseudomallei, but not heat-killed B. pseudomallei, induced the formation of lipid droplets in THP-1 cells (Fig. S2A and S2B). Consistent with the TEM observations, live B. pseudomallei induced the expression of adipose differentiation-related protein PLIN2/ADRP (perilipin 2), a marker of lipid droplet, whereas heat-killed B. pseudomallei did not affect the expression (Fig. S2D). Furthermore, heat-killed B. pseudomallei showed the same internalization rate as the live bacterium (Fig. S2C). Then, we further analyzed whether lipid accumulation was actively modulated by B. pseudomallei, and found that lipid droplet formation was significantly reduced upon chloramphenicol treatment in B. pseudomallei-infected THP-1 cells, which indicates that B. pseudomallei induced lipid droplet accumulation may be dependent on bacterial protein synthesis (Fig. S2E). Taken together, these results indicate lipid droplets accumulation in response to B. pseudomallei infection is an active process.
Inhibition of PNPLA2 results in lipid accumulation during B. pseudomallei infection
To explore the potential mechanisms involved in lipid accumulation, we analyzed lipid metabolism-related gene expression in B. pseudomallei-infected A549 cells (Figure 2A and S3A). As shown in Figure 2A, the PNPLA2 gene, which encodes a major TG hydrolase, was downregulated in B. pseudomallei-infected A549 cells. Therefore, we hypothesized that PNPLA2 deficiency might be a key factor contributing to lipid accumulation, in line with the observations of an increase in TG and a decrease in DG, after B. pseudomallei infection. To verify this hypothesis, we first determined the protein expression levels of PNPLA2 by western blot analysis and found that PNPLA2 expression largely decreased at 12 and 24 h.p.i. in B. pseudomallei-infected A549 cells (Figure 2B). Similar results were obtained when MOI-dependency was tested at 12 h.p.i. (Figure 2C). But we detected no significant change of FASN (fatty acid synthase) expression in time or dose-dependency tests (Figure 2B,C). Consistent results were obtained for other lung epithelial cells, including BEAS-2B, H460, and H358 cells, confirming that the effects were not cell type-specific (Figure 2D). Furthermore, to examine whether downregulation of PNPLA2 was specific to B. pseudomallei infection, we used Burkholderia thailandensis (an avirulent bacterium closely related to B. pseudomallei), Salmonella Typhimurium, and Escherichia coli as controls. As shown in Figure 2E, there were no significant changes in the expression levels of PNPLA2 after infection with B. thailandensis, S. Typhimurium, and E. coli. Besides, we measured the expression of PNPLA2 by using chloramphenicol and found that the protein levels of PNPLA2 were not downregulated in the presence of chloramphenicol, indicating that alterations of PNPLA2 expression levels were actively driven by B. pseudomallei infection (Fig. S3B). To further understand the functional role of PNPLA2 in lipid accumulation after B. pseudomallei infection, we constructed a PNPLA2-expressing plasmid (Figure 2F and S4A) and demonstrated that overexpression of PNPLA2 in A549 cells markedly reduced the number of lipid droplets by promoting TG turnover (Figure 2G,H). Meanwhile, quantification of bacteria by immunofluorescence staining implied that PNPLA2 might be involved in the regulation of B. pseudomallei infection (Figure 2I and S3D). Moreover, B. pseudomallei showed the same internalization rate in control or PNPLA2 overexpression conditions (Fig. S3C and S3D). Besides, as shown in Figure 2J, cellular lipid analysis showed B. pseudomallei infection-suppressed PNPLA2 could be fully rescued by overexpression of PNPLA2, and then lipid accumulation in A549 cells was completely prevented compared to the above data, which resulted in significant changes in the TG and DG measurements after B. pseudomallei infection. These results indicate that downregulation of PNPLA2 results in lipid droplet accumulation in response to B. pseudomallei infection.
Figure 2.
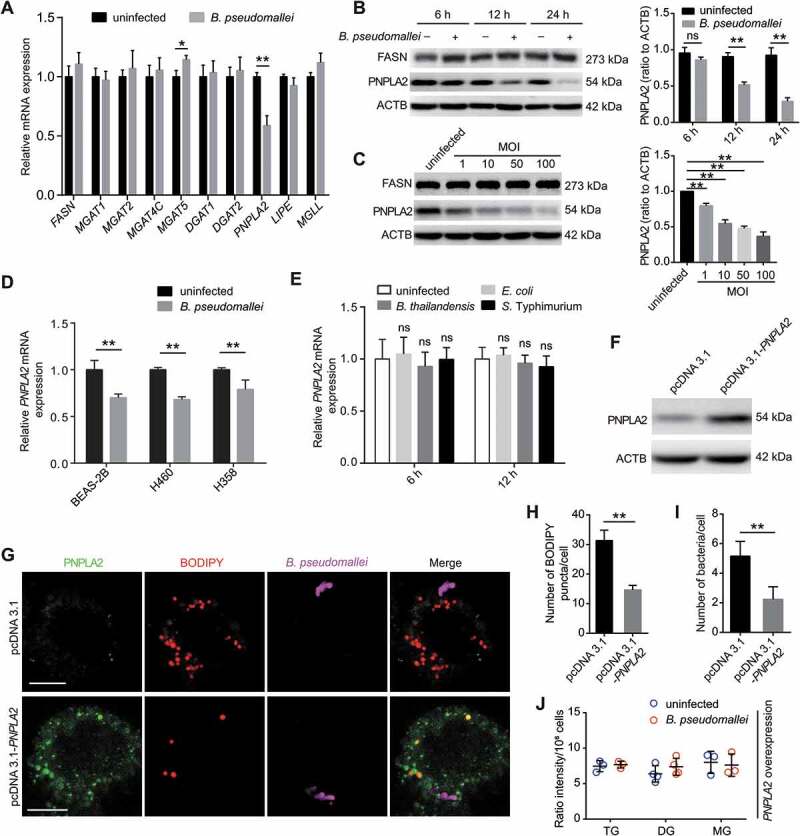
Inhibition of PNPLA2 contributes to lipid accumulation after B. pseudomallei infection. (A) Expression of lipid metabolism-related genes in A549 cells infected with B. pseudomallei (MOI = 10) for 12 h. The mRNA levels of FASN, MGAT1, MGAT2, MGAT4C, MGAT5, DGAT1, DGAT2, PNPLA2, LIPE and MGLL were measured by qRT-PCR. (B and C) Western blot analysis of PNPLA2 and FASN in A549 cells after B. pseudomallei infection (MOI = 10) for 6, 12 and 24 h or at indicated MOIs (1, 10, 50, 100) for 12 h. Densitometric analyses were performed to quantitate the fraction of PNPLA2 of at least three independent experiments. (D) qRT-PCR analysis of PNPLA2 expression in BEAS-2B, H460 and H358 cells after treated with B. pseudomallei (MOI = 10) for 12 h. (E) Measurement of PNPLA2 expression by qRT-PCR in A549 cells after infected with B. thailandensis, S. Typhimurium and E. coli (MOI = 10) for 12 h, respectively. (F) The protein level of PNPLA2 was determined by western blot. A549 cells were transfected with the pcDNA3.1 or pcDNA3.1-PNPLA2 for 24 h. (G) Lipid droplets were detected by confocal microscopy. A549 cells were transfected with pcDNA3.1 or pcDNA3.1-PNPLA2 for 24 h, and then infected with B. pseudomallei (MOI = 10) for 12 h. Scale bars: 10 μm. (H and I) The numbers of lipid droplet puncta and B. pseudomallei in each cell were counted. Lipid droplets were staining by BODIPY (red). (J) Cellular lipid levels were detected by LC-MS/MS analysis. A549 cells were transfected with pcDNA3.1-PNPLA2 plasmids for 24 h, then uninfected or B. pseudomallei-infected (MOI = 10) for 12 h. This experiment was repeated at least three times. *P < 0.05, **P < 0.01. ns, not significant
PNPLA2 works with lipophagy to regulate lipid accumulation during B. pseudomallei infection
Given that lipophagy contributes to lipid metabolism, we sought to dissect the link between PNPLA2-catalyzed lipolysis and lipophagy during B. pseudomallei infection. To investigate whether autophagy is involved in the regulation of lipid metabolism during B. pseudomallei infection, we examined the effects of pretreatment with the autophagy activator rapamycin (Rapa) or inhibitor 3-methyladenine (3-MA) on lipid droplet accumulation in A549 cells. As shown in Figure 3A-C, stimulation of autophagy by Rapa resulted in a significantly decreased amount of both lipid droplets and B. pseudomallei compared with the controls, while the numbers of lipid droplets and bacteria increased in 3-MA-treated A549 cells. Besides, stimulation of autophagy increased the lipid droplet turnover in Rapa-treated cells, but this effect was abrogated when autophagy was blocked via exposure to 3-MA or when ATG7 was knocked down (Figure 3D, S4B, and S4C). However, pharmacological alterations of autophagy by Rapa or 3-MA did not affect lipid droplet abundance in uninfected controls (Fig. S5A and S5B). These results indicate the involvement of lipophagy in the regulation of lipid metabolism upon B. pseudomallei infection.
Figure 3.
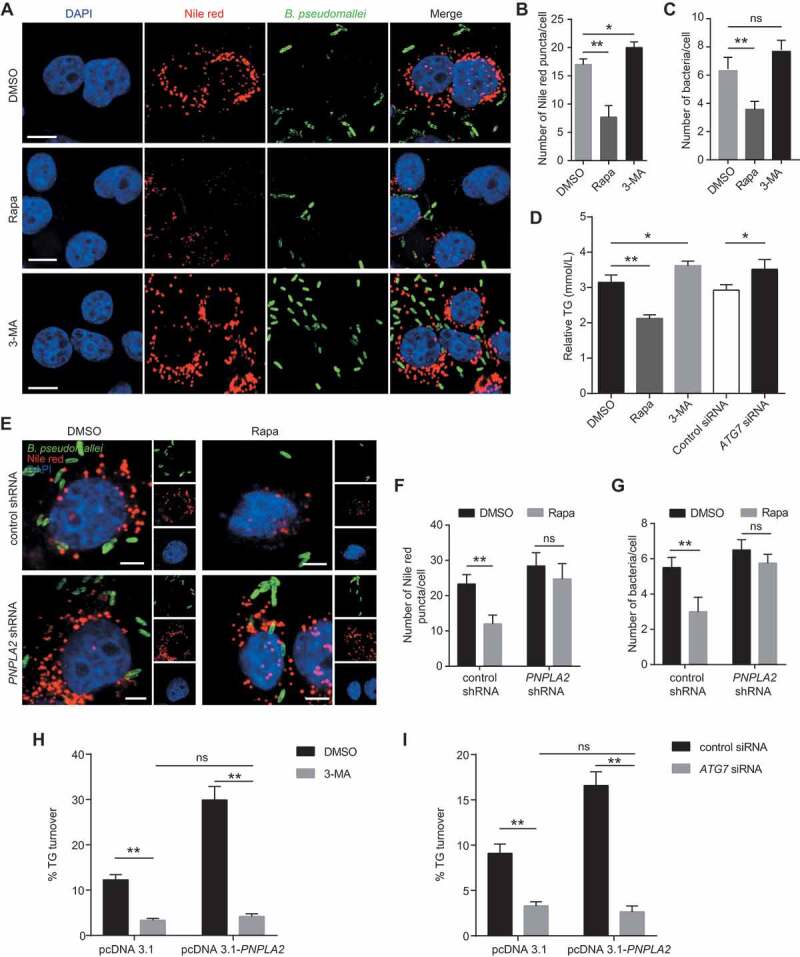
PNPLA2 and lipophagy regulate the lipid accumulation during B. pseudomallei infection. (A) Representative images show the number of lipid droplets in A549 cells infected with B. pseudomallei (MOI = 10) for 12 h in the presence of Rapa (200 nM) or 3-MA (10 mM). Scale bars: 10 μm. (B and C) ImageJ quantification of the numbers of lipid droplets and B. pseudomallei per cell in above confocal images. (D) Lipophagy is involved in the regulation of TG in A549 cells. After pretreatment by DMSO, Rapa and 3-MA, or transfected with ATG7 siRNA (100 nM), cells were infected with B. pseudomallei (MOI = 10) for 12 h. (E) Lipid droplet was visualized by immunostaining in A549 cells. After transfected with control or PNPLA2 shRNA (1 μg), cells were infected with B. pseudomallei (MOI = 10) for 12 h in the presence of DMSO or Rapa (200 nM). Scale bars: 5 μm. (F and G) Quantification of the numbers of Nile red puncta and B. pseudomalle in each cell. (H and I) Measurement of TG turnover in A549 cells. After transfected with the pcDNA3.1 or pcDNA3.1-PNPLA2, cells were treated with 3-MA or ATG7 siRNA and then infected with B. pseudomallei for 12 h. Experiments performed in triplicate showed consistent results. *P < 0.05, **P < 0.01. ns, not significant
To explore whether lipophagy involves PNPLA2 to mediate lipid metabolism upon B. pseudomallei infection, PNPLA2 expression was inhibited via short hairpin RNA (shRNA)-mediated knockdown (Fig. S4D). As shown in Figure 3E-G, Rapa treatment resulted in a significant decline in the lipid droplet and B. pseudomallei numbers, while PNPLA2 knockdown abrogated these alterations following stimulation of autophagy. The bacterial colony-forming unit (CFU) assay was also consistent with the quantification of B. pseudomallei numbers by immunofluorescence (Fig. S6), implying the importance of PNPLA2-mediated lipophagy in B. pseudomallei infection. To further verify whether lipophagy was responsible for mediating the effects of PNPLA2 on lipid accumulation, overexpression of PNPLA2 resulted in the TG turnover increase during B. pseudomallei infection (Figure 3H,I). However, this effect was significantly abrogated when 3-MA blocked autophagy or via ATG7 knockdown (Figure 3H,I, S4E, and S4F). Based on these results, we evaluated whether lipophagy was responsible for the reduced mRNA and protein expression of PNPLA2 during B. pseudomallei infection. Interestingly, the mRNA and protein expression of PNPLA2 was not significantly affected by the treatment with 3-MA or Rapa compared with that in the controls (Fig. S5C and S5D), which suggested the involvement of additional mechanisms upon B. pseudomallei infection. Taken together, these data show that both PNPLA2 and lipophagy are required for lipid metabolism during B. pseudomallei infection.
PNPLA2 overexpression restricts the B. pseudomallei infection by promoting autophagic flux
Next, we sought to explore the biological significance of lipid accumulation in B. pseudomallei infection. The data showed that overexpression of PNPLA2 increased the protein levels of LC3B-II and LAMP1 but decreased SQSTM1/p62, compared with their expression levels in the controls (Figure 4A). Additionally, overexpression of PNPLA2 increased colocalization of LysoTracker with LC3B during B. pseudomallei infection, implying that B. pseudomallei-mediated suppression of PNPLA2 impaired the autophagic flux (Figure 4B,C). To further measure the autophagic flux, a tandem monomeric red fluorescent protein (mRFP)-green fluorescent protein (GFP)-LC3B reporter was used. We observed that overexpression of PNPLA2 mimicked the Rapa treatment, resulting in increased mRFP+GFP− puncta, which indicated enhanced autophagic flux (Figure 4E,F). Meanwhile, quantification of immunofluorescent bacteria indicated that PNPLA2 overexpression decreased the bacterial load of B. pseudomallei by promoting autophagic flux (Figure 4D,G). Furthermore, to evaluate whether PNPLA2 overexpression, facilitating lipid droplet turnover, limits the B. pseudomallei infection, we transfected A549 cells with PNPLA2-expressing plasmids and counted the number of live internalized B. pseudomallei. As shown in Figure 4H-J, overexpression of PNPLA2 resulted in a decrease of lipid abundance and an enhanced ability of cells to control the infection, while no significant changes were observed in the lipid droplet and bacteria numbers when autophagy was blocked via ATG7 knockdown. Besides, the bacterial CFU assay also indicated a similar observation (Figure 4K,L). Taken together, our data show that PNPLA2 overexpression, facilitating lipid droplet turnover, promotes autophagic flux and limits the B. pseudomallei infection.
Figure 4.
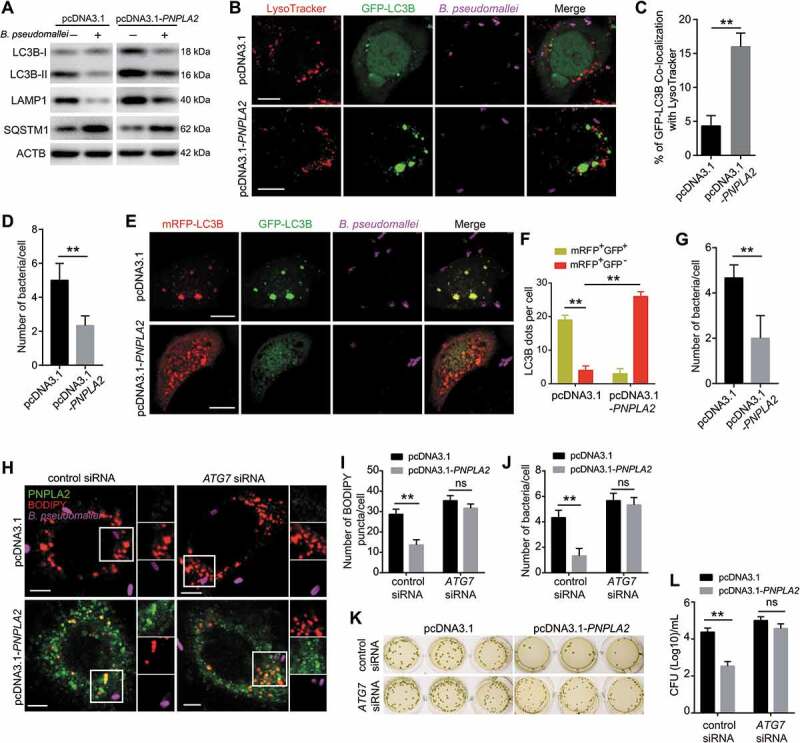
PNPLA2 overexpression promotes autophagic flux and restricts B. pseudomallei replication. (A) PNPLA2 overexpression increased the LC3B-II:LC3B-I ratio and LAMP1 protein levels and decreased SQSTM1 expression as analyzed via western blot. A549 cells were transiently transfected with the pcDNA3.1 or pcDNA3.1-PNPLA2 plasmids for 24 h and then infected with B. pseudomallei for 12 h (MOI = 10). (B) PNPLA2 overexpression increased lysosomal association with GFP-LC3B in A549 cells infected with B. pseudomallei (MOI = 10) for 12 h. Scale bars: 10 μm. (C and D) Quantitative analysis of GFP-LC3B associated with LysoTracker and B. pseudomallei numbers per cell in (B). (E) A549 cells transiently expressing mRFP-GFP-LC3B by adenoviruses transduction were transfected with control or pcDNA3.1-PNPLA2 plasmids, and then infected with B. pseudomallei (MOI = 10) for 12 h. Scale bars: 10 μm. (F and G) The graph shows the quantification of mRFP+GFP+ or mRFP+GFP− by taking the average number of puncta and B. pseudomallei numbers per cell in (E). (H) Representative images show lipid droplets by immunostaining. After transfected with the pcDNA3.1 or pcDNA3.1-PNPLA2, A549 cells were treated with ATG7 siRNA or control and then infected with B. pseudomallei (MOI = 10) for 12 h. Scale bars: 5 μm. (I and J) Quantification of the numbers of BODIPY puncta and B. pseudomallei in each cell in (H). (K and L) Intracellular growth of B. pseudomallei was detected by CFU assay. Cells were treated as described in (H). Data is shown as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. ns, not significant
Upregulation of NR1D2 expression during B. pseudomallei infection
To gain additional insight into the mechanism of PNPLA2 downregulation, we analyzed lipid-related gene expression differences in A549 cells after B. pseudomallei infection following our previously reported gene microarray analysis [26]. Consistent with our previous observations, the key components of the AMP-activated protein kinase (AMPK) pathway, which plays a vital role in the process of lipid metabolism, significantly changed (Fig. S7 and Table S2). Meanwhile, as shown in Figure 5A, NR1D2/ERV-REBβ (nuclear receptor subfamily 1 group D member 2), a member of the nuclear receptor subfamily, which also includes NR1D1/ERV-REBα, was upregulated in A549 cells after B. pseudomallei infection. It has recently been documented that NR1D2 functions as a repressor of genes involved in lipid metabolism [27,28]. Therefore, we presumed that upregulation of NR1D2 might be a key factor contributing to the lipid accumulation upon B. pseudomallei infection. To confirm the validity of the microarray data, the expression levels of NR1D2 were examined by qRT-PCR and western blot analysis. The dynamic expression of NR1D2 did not significantly change at 2 and 4 h.p.i. but increased at more than 6 h.p.i. (Figure 5B), and similar results were obtained when MOI dependency was tested (Figure 5C). The protein levels of NR1D2 also increased at 6 and 12 h.p.i. (Figure 5D). We further confirmed that the NR1D2 mRNA levels were also upregulated in B. pseudomallei-infected BEAS-2B, H460, and H358 cells (Figure 5E). Furthermore, to determine whether the upregulation of NR1D2 was specific to B. pseudomallei infection, we also used B. thailandensis, S. Typhimurium, and E. coli as controls. As shown in Figure 5F, there were no significant changes in the expression levels of NR1D2 after infection with B. thailandensis, S. Typhimurium, and E. coli. Besides, in the presence of chloramphenicol, B. pseudomallei infection did not upregulate NR1D2 expression, indicating that alterations of NR1D2 expression levels may be dependent on bacterial protein synthesis (Fig. S8A). Taken together, these results suggest that the expression level of NR1D2 increases in response to B. pseudomallei infection.
Figure 5.
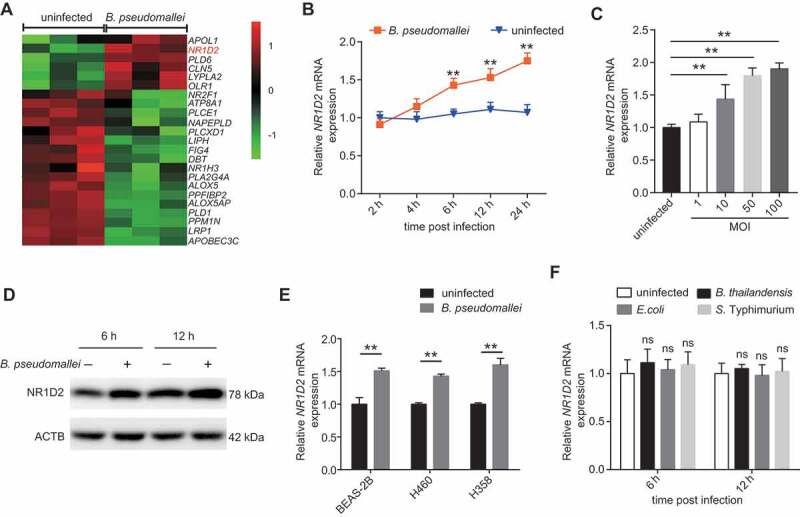
Upregulation of NR1D2 expression during B. pseudomallei infection. (A) Cluster analysis of mRNA expression profiles identified a group of altered lipid metabolism-related genes. (B and C) Confirmation of microarray results by qRT-PCR. qRT-PCR analysis of NR1D2 expression levels in A549 cells after infected with B. pseudomallei for the indicated time points (MOI = 10) or the MOIs (6 h p.i.). (D) Western blot analysis of NR1D2 protein levels in uninfected and B. pseudomallei-infected A549 cells for 6 and 12 h (MOI = 10). (E) The expression of NR1D2 in BEAS-2B, H460 and H358 cells after infected with B. pseudomallei (MOI = 10) for 6 h. (F) Expression of NR1D2 in A549 cells infected with B. thailandensis, E. coli and S. Typhimurium (MOI = 10) for 6 h. Data represent mean ± SD for at least 3 separate experiments. **P < 0.01. ns, not significant
NR1D2 promotes lipid accumulation through repression of the PNPLA2 expression in response to B. pseudomallei infection
Given that the expression levels of PNPLA2 and NR1D2 were inversely regulated in response to B. pseudomallei infection, we presumed that the upregulation of NR1D2 might be a key factor contributing to lipid accumulation by repressing PNPLA2 expression. To verify this hypothesis, we used an NR1D2-specific shRNA to evaluate the role of NR1D2 in the regulation of PNPLA2 expression and lipid accumulation after B. pseudomallei infection. We observed that the mRNA and protein levels of NR1D2 drastically decreased in A549 cells transfected with the shRNA (Figure 6A,B). Moreover, inhibition of NR1D2 significantly upregulated the mRNA and protein expression levels of PNPLA2 (Figure 6C,D), indicating that NR1D2 deficiency upregulates the expression of PNPLA2. To further confirm the regulation of PNPLA2 expression by NR1D2, we examined changes in the PNPLA2 mRNA expression in A549 cells following pretreatment with the REV-ERBs (including NR1D1 and NR1D2) agonist SR9009 and NR1D1 agonist GSK4112 (Fig. S8B and S8C). As shown in Figure 6E, compared with the controls, SR9009 dose-dependently suppressed the mRNA expression level of PNPLA2, while activation of NR1D1 by GSK4112 did not affect PNPLA2 expression. Moreover, we used a 2-kb PNPLA2 promoter-luciferase reporter vector and observed an approximately seven-fold lower luciferase activity than that in cells carrying the pGL3-Basic control vector upon B. pseudomallei infection (Figure 6F, S4A and S8D). In addition, co-transfection of a plasmid coding NR1D2 inhibited the PNPLA2 promoter-driven luciferase activity by ~50% (Figure 6G, S4A and S4G), suggesting that NR1D2 may directly regulate PNPLA2 transcription.
Figure 6.
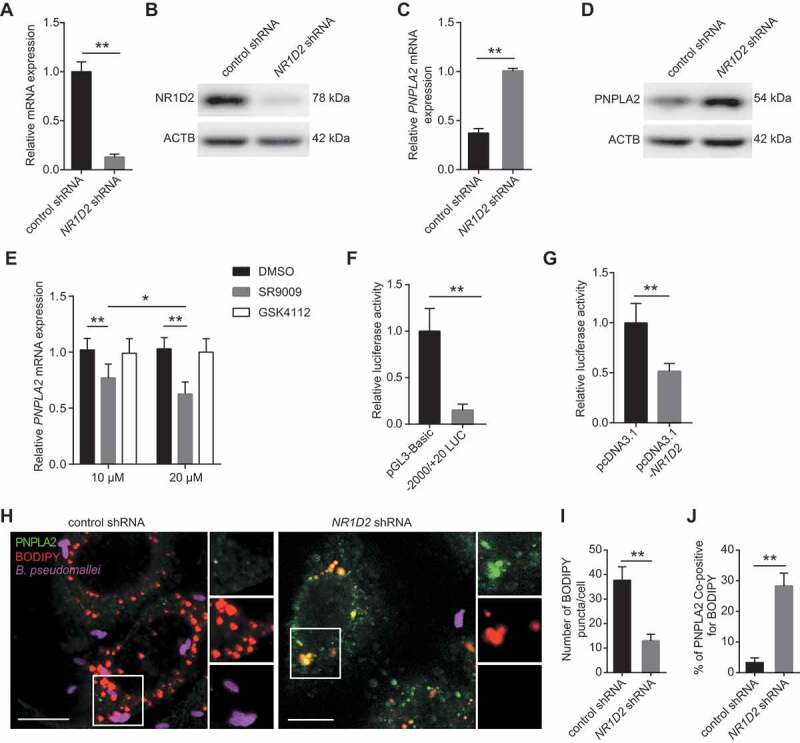
NR1D2 mediates lipid accumulation through inhibiting PNPLA2 expression following B. pseudomallei infection. (A and B) The mRNA and protein levels of NR1D2 were measured in A549 cells after transfection with control or NR1D2 shRNA (1 μg) for 24 h. (C and D) The mRNA and protein levels of PNPLA2 in control and NR1D2-silenced A549 cells after infected with B. pseudomallei (MOI = 10) for 12 h. (E) The expression of PNPLA2 was measured in A549 cells. After pretreatment with SR9009 or GSK4112 in different concentrations (10 μM and 20 μM) for 48 h, cells were infected with B. pseudomallei (MOI = 10) for 12 h. (F) A549 cells were transfected with either pGL3-Basic empty vector (200 ng) or −2000/+20-LUC PNPLA2 construct (200 ng) plus pRL-TK-Renilla (10 ng) and then infected with B. pseudomallei (MOI = 10) for 12 h. Luciferase activities were measured and normalized to Renilla internal control. (G) −2000/+20 LUC PNPLA2 (200 ng) plus pRL-TK-Renilla (10 ng) was transfected into A549 cells with control or pcDNA3.1-NR1D2 (200 ng) for 24 h. (H) Representative images of control and NR1D2-silenced A549 cells infected with B. pseudomallei for 12 h. Scale bar: 10 μm. (I and J) Quantitative analysis of the number of lipid droplets and the colocalization of PNPLA2 and BODIPY shown in (H). Data is shown as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01
To further examine whether NR1D2 is required for lipid accumulation, NR1D2 was knocked down, which significantly reduced the number of lipid droplets and increased the colocalization of PNPLA2 and lipid droplets in A549 cells compared with those in the control (Figure 6H-J). Besides, B. pseudomallei showed the same internalization rate in control or NR1D2 shRNA conditions (Fig. S8E and S8F). Altogether, these results indicate that NR1D2 may tightly control the lipid metabolism by repressing the PNPLA2 lipase upon B. pseudomallei infection.
NR1D2 mediates the effects of PNPLA2 on autophagy-dependent inhibition of B. pseudomallei infection
Based on the above data, showing that PNPLA2 regulates autophagic flux and B. pseudomallei infection by inhibiting lipid droplet accumulation, we sought to investigate further whether upregulation of NR1D2 is also involved in these processes through targeting PNPLA2. We initially analyzed the effect of NR1D2 on lipophagy and found that NR1D2 knockdown in A549 cells increased the colocalization of GFP-LC3B and lipid droplet staining (Figure 7A,B). Additionally, NR1D2 knockdown induced an increase in LC3B-II and the lysosomal protein LAMP1 and a marked decrease in the SQSTM1 level, and Baf A1 challenge resulted in further accumulation of LC3B-II and SQSTM1 in A549 cells (Figure 7C). These results suggested that NR1D2 potently inhibited lipophagy. Furthermore, consistent with the above data, NR1D2 knockdown also enhanced the autophagic flux in A549 cells compared with that in the control, resulting in increased mRFP+GFP− puncta, but this effect was abrogated entirely when PNPLA2 was chemically inhibited by ATGListatin (Figure 7D,E). Similarly, the immunofluorescence staining indicated that NR1D2 knockdown resulted in a decrease in the bacterial load of B. pseudomallei, but this effect was also abolished when PNPLA2 was inhibited (Figure 7F,G). Meanwhile, bacterial CFU assay also showed a similar observation (Figure 7H,I). Overall, these data show that PNPLA2 acts downstream of NR1D2 to mediate autophagic flux and inhibit B. pseudomallei infection.
Figure 7.
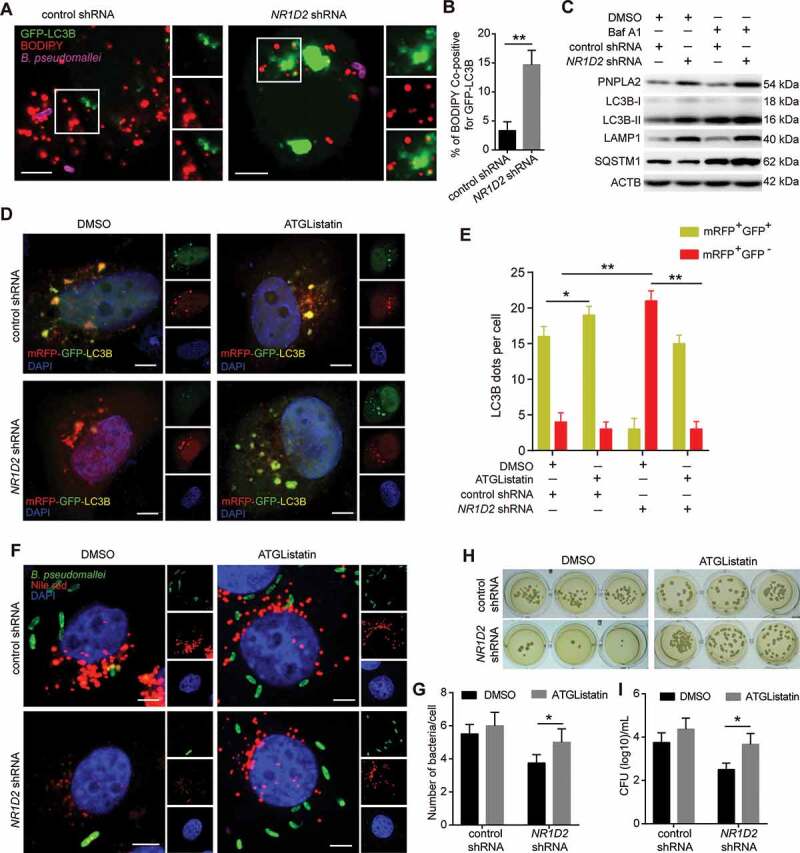
NR1D2 is required for PNPLA2-mediated effects on autophagic flux and regulation of B. pseudomallei infection. (A and B) Representative images of lipid droplet and GFP-LC3B puncta in A549 cells. After transfected with the control and NR1D2 shRNA, A549 cells were treated with GFP-LC3B and then infected with B. pseudomallei (MOI = 10) for 12 h. Quantitative analysis of the colocalization of BODIPY and GFP-LC3B. Scale bars: 5 μm. (C) The autophagic flux was detected by western blot. A549 cells were transiently transfected with the control and NR1D2 shRNA for 24 h, and then infected with B. pseudomallei (MOI = 10) for 12 h in the presence of DMSO or Baf A1 (10 nM). (D and E) A549 cells transiently expressing mRFP-GFP-LC3B were transfected with control or NR1D2 shRNA, and then treated with the DMSO or ATGListatin (30 μM) for 24 h and infected with B. pseudomallei (MOI = 10) for 12 h. The graph shows the quantification of mRFP+GFP+ or mRFP+GFP− by taking the average number of puncta per cell. Scale bars: 5 μm. (F and G) Representative images were visualized by confocal. After transfected with control or NR1D2 shRNA, A549 cells were treated with the DMSO or ATGListatin (30 μM) for 24 h and infected with B. pseudomallei (MOI = 10) for 12 h. Quantification shows the number of B. pseudomallei in each cell. Scale bar: 5 μm. (H and I) Intracellular growth of B. pseudomallei was detected by CFU assay. A549 cells were treated as described above. Data is shown as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01
Discussion
Accumulating evidence has revealed that lipid droplets participate in fundamental mechanisms of host-pathogen interactions. However, the exact mechanisms of their formation and function and how these relate to the pathophysiology of infectious diseases remain largely unknown. In the present study, we revealed that B. pseudomallei infection actively boosted lipid accumulation in human lung epithelial cells via PNPLA2 downregulation and lipophagy inhibition. In particular, we discovered that the underlying mechanism of lipid accumulation after B. pseudomallei exposure involved NR1D2-mediated suppression of the PNPLA2 expression. Consequently, the NR1D2- PNPLA2-mediated lipid droplet accumulation inhibited the autophagic flux, ultimately leading to the promotion of B. pseudomallei infection in lung epithelial cells. Additionally, B. pseudomallei-driven lipid accumulation may be dependent on the bacterial protein synthesis, which needs to be further investigated (Figure 8).
Figure 8.
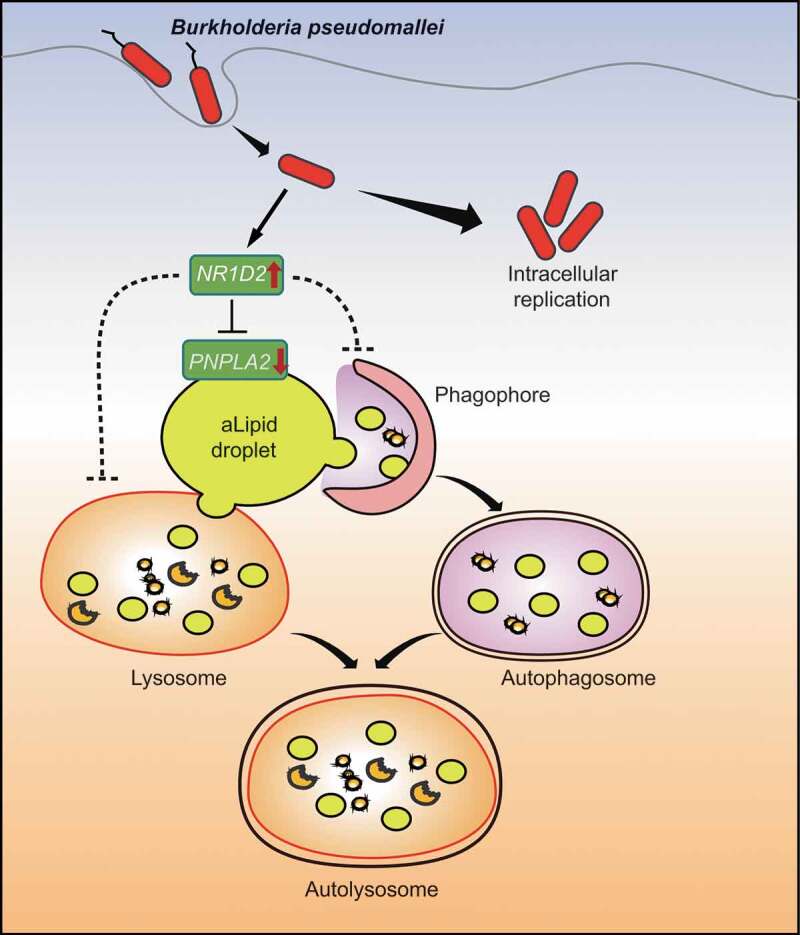
A Schematic of the inferred mechanism by which B. pseudomallei interferes with host lipid metabolism to block autophagy-dependent inhibition of infection in human lung epithelial cells. After entry into the host cells, B. pseudomallei infection upregulates the expression of NR1D2, which results in a decrease in PNPLA2 expression. Subsequently, PNPLA2-dependent reduction of TG turnover and inhibition of lipophagy leads to lipid droplets accumulation, which in turn inhibits autophagic flux and promotes B. pseudomallei infection
Recently, numerous studies have revealed that lipid droplet is a pivotal player in host-pathogen interactions. Moreover, the functional meaning of lipid droplet accumulation during infectious diseases, host defense, or pathogen survival strategy, has been inconclusive. Here, we revealed that the intracellular pathogen B. pseudomallei actively led to lipid accumulation in human lung epithelial cells and THP-1 macrophages. Interestingly, while the accumulation of lipid droplets was observed in the cytoplasm of cells with and without bacteria, the lipid droplet formation was highly significant in cells with internalized bacteria, suggesting the involvement of critical virulence factors of B. pseudomallei. Accumulating evidence indicates that the molecular mechanisms of infection-driven lipid droplet formation involve both pathogen and host factors. Expectedly, different pathogens have evolved different mechanisms to induce lipid accumulation. For instance, bacterial components, such as lipopolysaccharide (LPS) [29], and lipoarabinomannan (LAM) [30], can induce lipid droplet formation in macrophages. Additionally, virus capsid proteins, such as Dengue virus NS4A, exploit the acyltransferase activity of a lipid droplet membrane protein AUP1 to mediate lipophagy and virus replication [31]; M. tuberculosis-induced lipid droplet formation is correlated with virulence ESAT-6 protein of the infecting Mycobacterial strain [18]. Therefore, the precise mechanisms of B. pseudomallei infection-induced lipid droplet accumulation require further exploration.
The functions of newly formed lipid droplets in pathogen-infected host cells have begun to be deciphered. Several studies have shown that host cells accumulate lipid droplets in parallel with bacterial replication [17,32–34]. Mycobacterium leprae-induced lipid droplet formation favors M. leprae survival in SW-10 cells [17], and Chlamydia trachomatis has been found to acquire host-derived lipid droplets to transport to the lumen of the parasitophorous vacuole [33], indicating that the lipid droplet could offer physical shelter for bacteria benefits. Besides, pharmacological changes in lipid metabolism in host cells can affect the pathogens’ survival chances. Kumar et al. showed that triacsin C, which inhibits the activity of long-chain acyl-coenzyme A synthase, decreased the lipid biosynthesis and growth of Chlamydia trachomatis in HepG2 cells [35]. Similarly, pretreatment with another lipid metabolism inhibitor, C75, which inhibits FASN, inhibited not only Mycobacterium tuberculosis-induced lipid droplet formation but also the bacterial viability in Schwann cells [36]. Thus, lipid metabolism has been found to be crucial for the growth of intracellular pathogens. Nevertheless, its involvement in intracellular viability has not yet been demonstrated for B. pseudomallei. Together with the results of our previous study [26], here, we list various strategies used by B. pseudomallei to manipulate host autophagy for their own benefits. In particular, (1) Inhibition of autophagy: autophagy is inhibited by miRNA-mediated suppression of ATG10; (2) Modulation of autophagosomal maturation: autophagic flux is impaired by infection-driven lipid droplet accumulation. The findings of the present study deepened the current understanding and showed that lipid droplet turnover decreased through two mechanisms, namely, downregulation of PNPLA2 (neutral lipolysis) and inhibition of lipophagy (acid lipolysis). Recent findings have indicated the possibility of a functional link between autophagy and cytosolic lipolysis [15], whereby LC3B-II binds PNPLA2 on cytoplasmic lipid droplets and lipophagy acts downstream of PNPLA2 to promote the TG hydrolysis [37,38]. Here, we demonstrated that B. pseudomallei infection-induced decreases in PNPLA2 expression, which concomitantly disturbed the TG hydrolysis and promoted lipid accumulation. A previous study has demonstrated that the large size of cytoplasmic lipid droplets impedes their recruitment into lipoautophagosomes [12]. Here, we found that the B. pseudomallei infection-driven lipid droplet accumulation impaired the autophagic flux, which eventually facilitated the bacterial survival. We speculate that downregulation of PNPLA2 attenuates lipid droplet recruitment into lipoautophagosomes after B. pseudomallei infection, or the excessive size of lipid droplets may hinder autophagosome maturation by inhibiting fusion or acidification with lysosomes, which in turn quench the effect of lysosomes to impair the autophagic response and block lysosomal degradation for B. pseudomallei clearance. However, the potential mechanisms and recruitment factors involved in this process remain to be further elucidated.
Interestingly, the expression of PNPLA2 decreased at the transcriptional level in response to B. pseudomallei infection, and pharmacological activation or inhibition of autophagy was not required for the alteration of PNPLA2 expression. Therefore, there may be involved with the underlying mechanism to regulate PNPLA2 expression. Recent studies have indicated that many proteins directly or indirectly regulate PNPLA2 (neutral lipolysis). It has been reported that ABHD5 (abhydrolase domain containing 5), as a co-activator of PNPLA2, and SERPINF1/PEDF (serpin family F member 1) activate PNPLA2 [39–41], whereas G0S2 (G0/G1 switch 2) and CIDEC/FSP27 (cell death inducing DFFA like effector c) inhibit the enzyme [42,43]. In our study, we investigated the mRNA expression levels of the above four proteins (data not shown), but no significant changes were observed, indicating that they may not be responsible for the regulation of PNPLA2 upon B. pseudomallei infection. Furthermore, based on our previous gene microarray data of B. pseudomallei-infected A549 cells [26], we found that NR1D2 might be involved in the regulation of PNPLA2. Recent studies have documented that NR1D2 exerts redundant functions in regulating the circadian rhythm, inflammatory response, and lipid metabolism and is generally characterized as a repressor [27,28,44,45]. A previous study has demonstrated that ERV-REBs tightly control lipid metabolism by repressing several key enzymes in the liver and muscle tissues, including FASN, SCD1 (stearoyl-CoA desaturase 1), DGAT1 and DGAT2 (diacylglycerol O-acyltransferase 1 and 2) [27]. Here, we found that PNPLA2 was repressed by upregulated NR1D2, which resulted in decreased TG hydrolysis and lipid droplet accumulation in response to B. pseudomallei infection. However, the precise regulatory mechanism is currently unknown, which requires further exploration to determine whether NR1D2 interacts with the PNPLA2 promoter directly or indirectly combined with a transcription factor to inhibit its hydrolytic activity. Of note, the nuclear receptor NR1D2, one of the core clock genes, is reportedly involved in the control of glucose metabolism, as well as the regulation of insulin sensitivity and secretion [46]. It is universally acknowledged that approximately 80% of patients with melioidosis have known risk factors, mainly diabetes mellitus, which is the main comorbidity in more than 50% of melioidosis patients worldwide [47]. Collectively, the contribution of the present research suggests that NR1D2 may be a potential target for melioidosis therapy.
In conclusion, this study highlights that B. pseudomallei infection manipulates host lipid metabolism, triggering lipid droplet accumulation for their own benefits. These insights provide a more in-depth understanding of the host-pathogen relationship and suggest potential therapeutic strategies to treat melioidosis caused by B. pseudomallei.
Materials and methods
Antibodies and reagents
The GFP-LC3B plasmid was kindly provided by Dr. Tamotsu Yoshimori (Osaka University, Japan). The reagents 3-methyladenine (3-MA; M9281), rapamycin (R8781), dimethyl sulfoxide (DMSO; D2650), Nile red (N3013), SR9009 (55472), GSK4112 (G0673), ATGListatin (SML1075), 4ʹ6-diamidino-2-phenylindole dihydrochloride (DAPI; D9542), and chloramphenicol (R4408) were purchased from Sigma Aldrich. The primary antibodies used in this work are as follows: LC3B (L7543), ATG7 (A2856), NR1D2 (HPA054798) and ACTB (SAB4301137) were obtained from Sigma Aldrich. The antibodies against PNPLA2/ATGL (2138), FASN (3180) and SQSTM1 (5114) were obtained from Cell Signaling Technology. PLIN2/ADRP antibody (PA525042), BODIPY 558/568 (D3835) and LysoTracker Red (L12492) were obtained from Invitrogen. The antibody against LAMP1 (ab25245) was obtained from Abcam. The secondary antibody used for immunofluorescence studies conjugated with Alexa Fluor 405 (A31556, A31553), 488 (A11008) and 647 (A21244) were purchased from Invitrogen. The rabbit polyclonal anti-B. pseudomallei antibody was generated in our lab and used in our previous study [26].
Cell lines and bacterial strains
The cell lines A549 (TCHu150), H460 (TCHu205), H358 (TCHu151) and BEAS-2B (GNHu27) were purchased from the cell bank at the Chinese Academy of Sciences. A549, H460 and H358 cells were grown in DMEM medium (Gibco, 11965–092) containing 10% fetal bovine serum (Gibco, 10099–141) with 100 U/ml penicillin/streptomycin (Gibco, 15140–122). The normal lung bronchial epithelial BEAS-2B cells were cultured in RPMI 1640 medium (Gibco, 11875–093) supplemented with 10% FBS and penicillin/streptomycin. THP-1 cells (SCSP-567), which were also purchased from the cell bank at the Chinese Academy of Sciences, were grown in RPMI-1640 supplemented with 10% FBS, penicillin/streptomycin and 0.05 mM 2-mercaptoethanol (Gibco, 21985), and before infection, THP-1 cells were differentiated with 50 ng/mL phorbol-12-myristate 13-acetate (Sigma Aldrich, P8139) for 2 d. All the above cell lines were cultured at 37°C in 5% CO2. For all experiments, the B. pseudomallei strain used in all experiments is BPC006, a virulent clinical isolate from a melioidosis patient in China [48]. And E. coli K12 (29425), S. Typhimurium (14028) and B. thailandensis E264 (700388) were purchased from ATCC. Bacteria were grown in Luria-Bertani (LB) broth for 12 h at 37°C. After washing twice with phosphate-buffered saline (PBS; pH 7.4; Gibco, 10010023), the number of bacteria was estimated by measuring the absorbance of the bacterial suspension at 600 nm. In general, an absorbance of 0.33 to 0.35 was equivalent to approximately 108 CFU/ml of viable bacteria. The number of viable bacteria used in infection studies was determined by retrospective plating of serial 10-fold dilutions of the inoculum to LB agar.
Western blot analysis
Samples were collected and the cell pellet was lysed in RIPA lysis buffer, containing 50 mM Tris HCl, pH 7.4, 150 mM NaCl, 0.1% Nonidet P40 (Roche, 11754599001), 0.5% sodium deoxycholate (Sigma Aldrich, D6750), 1% SDS (Sigma Aldrich, L3771), 0.5% benzonase endonuclease (Merck Millipore, 71206) and protease and phosphatase inhibitor cocktails (Roche, 11697498001) for 10 min at RT. Protein concentration was determined by BCA Protein Assay according to the instructions of the supplier (Thermo Fisher Scientific, A53226). Equal amounts of protein in 1× Laemmli buffer were denatured at 95°C for 5 min and subjected to standard SDS-PAGE and were transferred to PVDF membranes (GE Healthcare, 10600023). The membranes were blocked with 5% nonfat dry milk (Sangon Biotech, A600669) in TBS (BOSTER, AR0031) containing 0.05% Tween 20 (Sigma Aldrich, P1379) and were incubated with incubated overnight with the respective primary antibodies at 4°C. The membranes were incubated at room temperature for 1 h HRP-conjugated secondary antibodies (Jackson Immuno Research Laboratories, 115–035-003, 111–035-003) according to the manufacturer’s instructions. A commercial protein marker was used for identification of protein size. Membranes were developed using ECL plus on ECL Hyper film (GE Healthcare, United States), scanned, and evaluated using ImageJ. ACTB was used as loading control.
Sample preparation and LC-MS/MS analysis
Lipid extraction was performed as described previously with some modifications [49–51]. Briefly, thawed samples were extracted by incubating with 2 volumes of chloroform:methanol (1:2, vol:vol). Internal standards selected for the experiment were spiked into the samples and vortexed over 10 min at room temperature. Carefully collecting the organic layer after further centrifugation. Another repeated extraction was processed as above and extracts were combined together. Then, samples were divided into two aliquots before drying under stable N2 stream, followed by adding 200 μL DCM to each extract. One batch of extracts were utilized for TG analyses, the others were derived by 4-dimethylaminopyridine (Sigma Aldrich, 107700) and 2, 4-difluorophenyl isocyanate (Sigma Aldrich, 250759) for DG and MG analyses. Extracted samples were separated by an AQUITY UPLC system (Waters). The Waters BEH-C18 (2.1 × 100 mm, 2.1 × 50 mm 1.7 μm) column and gradient program were optimized. A 10 μL aliquot of each sample was injected in the column. Mass spectrometry was performed on an AB SCIEX QTRAP-6500. Lipid extracts were detected in electrospray ion (ESI) mode. Samples were analyzed using full spectral scanning (Q1) and constant neutral loss scanning (NL). All samples were conducted in triplicate and kept at 4°C throughout the analyses. Final calculations of amounts of analyte per sample require isotope corrections and application of deisotope factors. Data acquisitions were performed using Analyst 1.6.2 software (Applied Biosystems). Rationing the peak area of targeted lipids from multiple reaction monitoring (MRM) to the internal standards was employed as relative quantitation method via Multiquant software (Applied Biosystems).
Biochemical analysis of triglyceride
The levels of TGs in A549 or BEAS-2B cells were determined by commercially available kits according to manufacturer’s instruction (Sigma Aldrich, MAK266). Briefly, for A549 cells, after being transfected with pcDNA 3.1-PNPLA2 and control plasmids or untreated, cells were infected with B. pseudomallei at an MOI of 10 for 12 h in the presence of Rapa (200 nM), 3-MA (10 mM) or ATG7 siRNA (100 nM); and for BEAS-2B cells, they were uninfected or infected with B. pseudomallei (MOI = 10) for 6, 12 and 24 h, then washed in PBS and scraped. Add 100 µL of lipid extraction buffer per sample, and incubate for 30 min at 100°C, then cool sample to room temperature and shake plate for 1 min to homogenize solution. Lysates were centrifuged at 12,000 x g for 10 min and bring samples to a final volume of 50 μL. Add 2 μL of lipase solution to each sample, and incubate for 10 min at room temperature. Set up the 50 μL master reaction mix according to manufacturer’s scheme. Then add 50 μL of the master reaction mix to each sample. Mix well using a horizontal shaker and incubate the plate for 30 min at room temperature. Samples were washed in PBS (Gibco, 10010023) and add 50 μL termination buffer. Protect the plate from light during the incubation. Finally, measure the absorbance at 570 nm (A570) when the reaction is stable.
Confocal microscopy
A549 cells were cotransfected the indicated shRNA, pcDNA 3.1-PNPLA2 or control with the GFP-LC3B plasmids for 24 h. Specially, for autophagic flux measurement, A549 cells were transduced with Ad-mRFP-GFP-LC3B adenoviruses (Hanbio, HB-AP2100001) for 36 h. Then cells were infected with B. pseudomallei (MOI = 10) for the indicated time. For lipid measurement, after infection, cells were washed with PBS, fixed for 10 min at 37°C in 4% paraformaldehyde, permeabilized with 0.1% (vol:vol) Triton X-100 (Thermo Fisher Scientific, 85112). Coverslips were incubated with anti-B. pseudomallei serum (1:200 dilution) for 1 h, or anti-PNPLA2 (1:1000 dilution) for 1 h, washed extensively with PBS buffer, and incubated with secondary antibodies for 1 h. Then coverslips were incubated with DAPI for 10 min, and incubated with Nile red for 5 min or BODIPY 558/568 (2 μM) for 20 min, then washed extensively with PBS buffer. Glass coverslips (NEST, 801011) were mounted on glass slides (Thermo Fisher Scientific, 3063–002) using Fluorescent mounting medium (Dako, S3023). All steps were carried out at room temperature. The cells were viewed using a laser-scanning confocal microscope (Zeiss, Germany).
Plasmid construction
The DNA oligonucleotides containing CDS of PNPLA2 and NR1D2 were synthesized with flanking Hind III and Xho I restriction enzyme digestion sites, respectively. The DNAs and pcDNA3.1 plasmids (Invitrogen, V79020) were used to build the overexpression vectors. The −2000 to +22 promoter region of PNPLA2 were synthesized with flanking KpnI and Hind III restriction enzyme digestion sites and then ligated to KpnI and Hind III sites in the pGL3-Basic vector (Promega, E1751) to obtain the reporter construct pGL3-2000/+ 20 LUC.
Dual-luciferase assay
A549 cells were transfected with expression vectors or luciferase reporter plasmids plus the internal control vector pRL-TK-Renilla for luciferase assays in 24-well plates. After overnight culture, cells were infected with B. pseudomallei for 12 h (MOI = 10). Then all of the cells were lysed via dual luciferase reporter assay system (Promega, E1910), and then the fluorescence activity was detected via GloMax 20/20 Luminometer (Promega, United States) and normalized to Renilla luciferase values.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using Trizol (Thermo Fisher Scientific, 15596026) according to the manufacturer’s instruction, and the concentration of which was measured with a spectrophotometer. qRT-PCR assays for the mRNA of lipid metabolism-related genes were performed by using the PrimeScript RT-PCR kit (Takara, RR037A) in a Bio-Rad IQ5 (Bio-Rad, United States). The reactions were performed using the following parameters: 95°C for 1 min followed by 40 cycles of 95°C for 5 s, 60°C for 5 s and 72°C for 5 s. The mRNA levels of ACTB were used as an internal control. The primers were shown in Supplementary information, Table S1.
A549 cell transfection
For siRNA or shRNA transfections, A549 cells were seeded at 1 × 106 per well in 6-well culture plates for RNA or protein extraction in antibiotic-free DMEM and were incubated overnight. Cells were transfected with ATG7 siRNA (sc-41447), NR1D2 shRNA (sc-61456-SH) and PNPLA2 shRNA (sc-60223-SH), which were purchased from Santa Cruz Biotechnology, using Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) for 24 h. The effects of ATG7 siRNA, NR1D2 shRNA and PNPLA2 shRNA were compared with those of a nontargeting control siRNA or control shRNA. For plasmid DNA transfections, A549 cells were seeded at 5 × 105 per well 1 d before the transfection according to the manufacturer’s protocol. Cells were transfected 16–20 h before further experiments. All experiments were performed in triplicate.
Transmission electron microscopy
A549 or THP-1 cells were collected and fixed in a solution containing 2.5% glutaraldehyde in 0.1 M sodium cacodylate for 2 h, post-fixed with 1% OsO4 for 1.5 h, and washed and stained in 3% aqueous uranyl acetate for 1 h. The samples were then washed again, dehydrated with a graded alcohol series, and embedded in Epon-Araldite resin (Canemco, 034). Ultrathin sections were cut using a Reichert ultramicrotome (Reichert, United States), counterstained with 0.3% lead citrate, and examined on a Philips EM420 electron microscope (Philips, United Kingdom).
Bacterial invasion and growth in A549 cells
Bacterial invasion of A549 cells was investigated by using the method described by Elsinghorst, except for the following modifications [52]. A549 cells were cotransfected the indicated pcDNA3.1-PNPLA2 or NR1D2 shRNA with the relative controls for 24 h, then treated with ATG7 siRNA, Rapa (200 nM) or ATGListatin (30 μM). Next, A549 cells were infected with B. pseudomallei at an MOI of 10. One hour after infection, cells were washed twice with PBS, and 2 ml of fresh culture medium containing 250 μg of kanamycin (TIANGEN, RT503) per ml was added, and the preparation was incubated to kill the extracellular bacteria. After infection with B. pseudomallei for 3 or 12 h, cells were washed three times with PBS and lysed with 1 ml of 0.1% Triton X-100 after infection. Serially diluted cell lysates were plated on Luria broth plates. Colonies were counted after 36 h. Experiments were performed at least three times in triplicates.
Statistical analysis
All images were analyzed by ImageJ software (NIH, MD, USA). Images of the samples were acquired with blinding of the experimental conditions. Statistical analysis of images was based on ImageJ quantification of randomly selected fields of view (including infected cells, n > 50). The average numbers or diameters of the observed lipid droplets were calculated by averaging multiple number or diameter measurements with ImageJ software. The area of the lipid droplets was calculated by using the average diameter in the equation s = Πr2. The number of intracellular B. pseudomallei per cell was calculated by automated count with ImageJ software. The results are expressed as the mean ± SD of at least three separate experiments. The differences between the groups were determined with the SPSS 13.0 software. Student’s t-test was used to analyze the data. The differences were considered significant at P < 0.05. Statistically significant differences are indicated by asterisks (*P < 0.05, **P < 0.01).
Supplementary Material
Acknowledgments
We are grateful to Prof. Yongsheng Li (Xinqiao Hospital, Army Medical University, Chongqing, China) for technical assistance with LC-MS/MS analysis. We thank Dr. Tamotsu Yoshimori for providing the GFP-LC3B plasmid. We also thank Prof. Wenyue Xu (Department of Pathogenic Biology, Army Medical University, Chongqing, China) for providing the THP-1 cell line. This study was supported by grants from National Natural Science Foundation of China (No. 81971907 and No. 31970135) and Third Military Medical University Science Foundation of Outstanding Youth (2017YQRC-07).
Funding Statement
This study was supported by grants from National Natural Science Foundation of China [No. 81971907 and No. 31970135] and Third Military Medical University Science Foundation of Outstanding Youth [2017YQRC-07].
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bozza PT, Magalhaes KG, Weller PF.. Leukocyte lipid bodies - biogenesis and functions in inflammation. Biochim Biophys Acta. 2009;1791(6):540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Boren J, Taskinen M-R, Olofsson S-O, et al. Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med. 2013;274(1):25–40. [DOI] [PubMed] [Google Scholar]
- [3].Bozza PT, Viola JP. Lipid droplets in inflammation and cancer. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4–6):243–250. [DOI] [PubMed] [Google Scholar]
- [4].Xiang W, Shi R, Kang X, et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat Commun. 2018;9(1):2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liu L, Zhang K, Sandoval H, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160(1–2):177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].van der Meer-janssen YP, van Galen J, Batenburg JJ, et al. Lipids in host-pathogen interactions: pathogens exploit the complexity of the host cell lipidome. Prog Lipid Res. 2010;49(1):1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saka HA, Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol. 2012;28:411–437. [DOI] [PubMed] [Google Scholar]
- [8].Melo RC, Dvorak AM, Chitnis CE. Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog. 2012;8(7):e1002729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vallochi AL, Teixeira L, Oliveira KDS, et al. Lipid Droplet, a Key Player in Host-Parasite Interactions. Front Immunol. 2018;9:1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306(5700):1383–1386. [DOI] [PubMed] [Google Scholar]
- [11].Vaughan M, Berger JE, Steinberg D. Hormone-sensitive lipase and monoglyceride lipase activities in adipose tissue. J Biol Chem. 1964;239:401–409. [PubMed] [Google Scholar]
- [12].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kuo CJ, Hansen M, Troemel E. Autophagy and innate immunity: insights from invertebrate model organisms. Autophagy. 2018;14(2):233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. 2017;18(11):671–684. [DOI] [PubMed] [Google Scholar]
- [16].Cingolani F, Czaja MJ. Regulation and functions of autophagic lipolysis. Trends Endocrinol Metab. 2016;27(10):696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jin SH, An SK, Lee SB. The formation of lipid droplets favors intracellular Mycobacterium leprae survival in SW-10, non-myelinating Schwann cells. PLoS Negl Trop Dis. 2017;11(6):e0005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Singh V, Jamwal S, Jain R, et al. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. 2012;12(5):669–681. [DOI] [PubMed] [Google Scholar]
- [19].Zaitsu Y, Iwatake M, Sato K, et al. Lipid droplets affect elimination of Porphyromonas gingivalis in HepG2 cells by altering the autophagy-lysosome system. Microbes Infect. 2016;18(9):565–571. . [DOI] [PubMed] [Google Scholar]
- [20].Kaestli M, Schmid M, Mayo M, et al. Out of the ground: aerial and exotic habitats of the melioidosis bacterium Burkholderia pseudomallei in grasses in Australia. Environ Microbiol. 2012;14(8):2058–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wiersinga WJ, Currie BJ, Peacock SJ. Melioidosis. N Engl J Med. 2012;367(11):1035–1044. [DOI] [PubMed] [Google Scholar]
- [22].Galyov EE, Brett PJ, DeShazer D. Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu Rev Microbiol. 2010;64:495–517. [DOI] [PubMed] [Google Scholar]
- [23].Wiersinga WJ, van der Poll T, White NJ, et al. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol. 2006;4(4):272–282. [DOI] [PubMed] [Google Scholar]
- [24].Allwood EM, Devenish RJ, Prescott M, et al. Strategies for intracellular survival of Burkholderia pseudomallei. Front Microbiol. 2011;2:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cullinane M, Gong L, Li X, et al. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy. 2008;4(6):744–753. [DOI] [PubMed] [Google Scholar]
- [26].Li Q, Fang Y, Zhu P, et al. Burkholderia pseudomallei survival in lung epithelial cells benefits from miRNA-mediated suppression of ATG10. Autophagy. 2015;11(8):1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Solt LA, Wang Y, Banerjee S, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature. 2012;485(7396):62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sulli G, Rommel A, Wang X, et al. Pharmacological activation of REV-ERBs is lethal in cancer and oncogene-induced senescence. Nature. 2018;553(7688):351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pacheco P, Bozza FA, Gomes RN, et al. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular Loci involved in eicosanoid metabolism. J Immunol. 2002;169(11):6498–6506. [DOI] [PubMed] [Google Scholar]
- [30].D’Avila H, Melo RCN, Parreira GG, et al. Mycobacterium bovis bacillus calmette-guérin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol. 2006;176(5):3087–3097. [DOI] [PubMed] [Google Scholar]
- [31].Zhang J, Lan Y, Li MY, et al. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe. 2018;23(6):819–831. e5. [DOI] [PubMed] [Google Scholar]
- [32].Rank RG, Whittimore J, Bowlin AK, et al. In vivo ultrastructural analysis of the intimate relationship between polymorphonuclear leukocytes and the chlamydial developmental cycle. Infect Immun. 2011;79(8):3291–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cocchiaro JL, Kumar Y, Fischer ER, et al. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci U S A. 2008;105(27):9379–9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Daniel J, Maamar H, Deb C, et al. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7(6):e1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16(16):1646–1651. [DOI] [PubMed] [Google Scholar]
- [36].Mattos KA, Oliveira VGC, D’Avila H, et al. TLR6-driven lipid droplets in mycobacterium leprae- infected schwann cells: immunoinflammatory platforms associated with bacterial persistence. J Immunol. 2011;187(5):2548–2558. [DOI] [PubMed] [Google Scholar]
- [37].Martinez-Lopez N, Garcia-Macia M, Sahu S, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23(1):113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep. 2017;19(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lass A, Zimmermann R, Haemmerle G, et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in chanarin-dorfman syndrome. Cell Metab. 2006;3(5):309–319. [DOI] [PubMed] [Google Scholar]
- [40].Chung C, Doll JA, Gattu AK, et al. Anti-angiogenic pigment epithelium-derived factor regulates hepatocyte triglyceride content through adipose triglyceride lipase (ATGL). J Hepatol. 2008;48(3):471–478. [DOI] [PubMed] [Google Scholar]
- [41].Borg ML, Andrews ZB, Duh EJ, et al. Pigment epithelium-derived factor regulates lipid metabolism via adipose triglyceride lipase. Diabetes. 2011;60(5):1458–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang X, Lu X, Lombès M, et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11(3):194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Singh M, Kaur R, Lee M-J, et al. Fat-specific protein 27 inhibits lipolysis by facilitating the inhibitory effect of transcription factor Egr1 on transcription of adipose triglyceride lipase. J Biol Chem. 2014;289(21):14481–14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lam MT, Cho H, Lesch HP, et al. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498(7455):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ramakrishnan SN, Lau P, Burke LJ, et al. Rev-erbbeta regulates the expression of genes involved in lipid absorption in skeletal muscle cells: evidence for cross-talk between orphan nuclear receptors and myokines. J Biol Chem. 2005;280(10):8651–8659. [DOI] [PubMed] [Google Scholar]
- [46].Stenvers DJ, Scheer FAJL, Schrauwen P, et al. Circadian clocks and insulin resistance. Nat Rev Endocrinol. 2019;15(2):75–89. [DOI] [PubMed] [Google Scholar]
- [47].Wiersinga WJ, Virk HS, Torres AG, et al. Melioidosis. Nat Rev Dis Primers. 2018;4:17107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fang Y, Huang Y, Li Q, et al. First genome sequence of a Burkholderia pseudomallei Isolate in China, strain BPC006, obtained from a melioidosis patient in Hainan. J Bacteriol. 2012;194(23):6604–6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. [DOI] [PubMed] [Google Scholar]
- [50].Leiker TJ, Barkley RM, Murphy RC. Analysis of diacylglycerol molecular species in cellular lipid extracts by normal-phase LC-electrospray mass spectrometry. Int J Mass Spectrom. 2011;305(2–3):103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Murphy RC, Leiker TJ, Barkley RM. Glycerolipid and cholesterol ester analyses in biological samples by mass spectrometry. Biochim Biophys Acta. 2011;1811(11):776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Elsinghorst EA. Measurement of invasion by gentamicin resistance. Methods Enzymol. 1994;236:405–420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.