

ABSTRACT
SETX (senataxin) is an RNA/DNA helicase that has been implicated in transcriptional regulation and the DNA damage response through resolution of R-loop structures. Mutations in SETX result in either of two distinct neurodegenerative disorders. SETX dominant mutations result in a juvenile form of amyotrophic lateral sclerosis (ALS) called ALS4, whereas recessive mutations are responsible for ataxia called ataxia with oculomotor apraxia type 2 (AOA2). How mutations in the same protein can lead to different phenotypes is still unclear. To elucidate AOA2 disease mechanisms, we first examined gene expression changes following SETX depletion. We observed the effects on both transcription and RNA processing, but surprisingly observed decreased R-loop accumulation in SETX-depleted cells. Importantly, we discovered a strong connection between SETX and the macroautophagy/autophagy pathway, reflecting a direct effect on transcription of autophagy genes. We show that SETX depletion inhibits the progression of autophagy, leading to an accumulation of ubiquitinated proteins, decreased ability to clear protein aggregates, as well as mitochondrial defects. Analysis of AOA2 patient fibroblasts also revealed a perturbation of the autophagy pathway. Our work has thus identified a novel function for SETX in the regulation of autophagy, whose modulation may have a therapeutic impact for AOA2.
Abbreviations: 3ʹREADS: 3ʹ region extraction and deep sequencing; ACTB: actin beta; ALS4: amyotrophic lateral sclerosis type 4; AOA2: ataxia with oculomotor apraxia type 2; APA: alternative polyadenylation; AS: alternative splicing; ATG7: autophagy-related 7; ATP6V0D2: ATPase H+ transporting V0 subunit D2; BAF: bafilomycin A1; BECN1: beclin 1; ChIP: chromatin IP; Chloro: chloroquine; CPT: camptothecin; DDR: DNA damage response; DNMT1: DNA methyltransferase 1; DRIP: DNA/RNA IP; DSBs: double strand breaks; EBs: embryoid bodies; FTD: frontotemporal dementia; GABARAP: GABA type A receptor-associated protein; GO: gene ontology; HR: homologous recombination; HTT: huntingtin; IF: immunofluorescence; IP: immunoprecipitation; iPSCs: induced pluripotent stem cells; KD: knockdown; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MN: motor neuron; MTORC1: mechanistic target of rapamycin kinase complex 1; PASS: PolyA Site Supporting; PFA: paraformaldehyde; RNAPII: RNA polymerase II; SCA: spinocerebellar ataxia; SETX: senataxin; SMA: spinal muscular atrophy; SMN1: survival of motor neuron 1, telomeric; SQSTM1/p62: sequestosome 1; TFEB: transcription factor EB; TSS: transcription start site; TTS: transcription termination site; ULK1: unc-51 like autophagy activating kinase 1; WB: western blot; WIPI2: WD repeat domain, phosphoinositide interacting 2; XRN2: 5ʹ-3ʹ exoribonuclease 2.
KEYWORDS: AOA2, autophagy, DRIP, LC3, lysosomal degradation, R loop, R-loop, senataxin, SETX, transcription regulation
Introduction
SETX is the human homolog of the yeast superfamily I RNA/DNA helicase Sen1 [1]. Interest in SETX was heightened in 2004 when the SETX gene was discovered to be mutated in two distinct neurodegenerative diseases. One is a juvenile form of ataxia called ataxia with oculomotor apraxia type 2 (AOA2) and the other a juvenile form of amyotrophic lateral sclerosis named ALS4 [2,3]. This discovery has led to increased investigation of SETX function. SETX was found to interact with RNAPII (RNA polymerase II) and has been implicated in transcription termination [4–7]. More recent work revealed how SETX binding at promoter regions of antiviral genes prevents their transcription by promoting premature termination, providing a mechanism to inhibit their expression in the absence of viral infection [7]. Besides its role in premature termination, SETX also appears to function in “normal” termination on specific genes, by resolving R loops at G-rich pause sites located downstream of polyadenylation signals, thereby allowing degradation of the cleaved downstream RNA by the 5ʹ to 3ʹ exoribonuclease XRN2 [8].
R-loop resolution appears to be an evolutionarily conserved function of SETX [9]. R loops are hybrid structures formed during transcription between nascent RNA and the DNA template and are present at as much as 5% of mammalian genomes [10,11]. R loops have been found to form at the 5ʹ and 3ʹ ends of genes to influence epigenetic regulation [12]. While R loops are necessary for important biological functions such as immunoglobulin class switch and transcription termination [8,13,14], persistent R loops can create DNA breaks, mutations and chromosomal rearrangements leading to genome instability [10,15,16]. Not surprisingly, SETX has also been found to play a role in the DNA damage response (DDR) [4,17–19]. SETX colocalizes with TP53BP1 and γH2AX at transcription- and R loop-dependent replication stress foci during replication or in S/G2 phase [4]. Additionally, the tumor suppressor BRCA1 has been identified as a SETX-interacting protein that is necessary to prevent DNA damage caused by R loops at transcriptional pause sites of specific genes [20,21]. The SETX-BRCA1 complex has also been found to localize at sex chromosomes, maintaining the integrity of the genome during meiotic recombination by preventing R-loop accumulation as well as promoting effective meiotic sex chromosome inactivation in mice [18].
Recent data offer deeper insights into how SETX might link R loops to DNA double-strand breaks (DSBs). SETX KD has been shown to increase large DSB-induced deletions that are R loop-dependent near the site of the break [22]. ChIP-seq experiments showed that SETX localizes to sites of DSBs induced at transcriptionally active loci, where it resolves R loops and regulates γH2AX foci formation. Importantly, SETX appears to promote homologous recombination (HR) repair and prevent translocation, thus maintaining genome integrity [23]. While it is clear that SETX/Sen1 resolves R-loop structures formed upon collision of the transcription and replication machinery, in coordination with DNA repair factors [4,9,24], we and others extended these studies by showing that SETX interacts with EXOSC9/RRP45 (exosome component 9), a core component of the multisubunit exosome that functions in RNA quality control, processing and turnover. This is thought to facilitate recruitment of the exosome to collision-associated R loops to degrade the RNA moiety following SETX unwinding, which would otherwise be deleterious or create persistent R-loops [25–27]. While AOA2 or SETX-depleted cells have been associated with an increase of R loops and DSBs, ALS4 cells actually show a reduction in R loops compared to controls [28]. This reduction correlates with increased gene expression that seems to be the result of a decrease in promoter methylation. In fact, DNMT1 (DNA methyltransferase 1) binding to promoters is reduced by the presence of R loops, for which it has less affinity than double-strand DNA. These data show that SETX can regulate gene expression not only by resolving R loops but also by promoting R-loop formation at certain promoters, protecting them from extensive methylation.
Many neurodegenerative disorders have been linked to R-loop formation and resolution [29,30]. An interesting example is the autosomal recessive disorder spinal muscular atrophy (SMA) caused by loss of the SMN1 (survival of motor neuron 1, telomeric) gene, which encodes a protein that plays a crucial role in snRNP assembly and consequently splicing [31]. SMN KD cells also display an increase in R loops and DSBs [32]. Interestingly, SETX had been found to interact with SMN [5,31,33] and overexpression of SETX in SMA neurons that have reduced levels of SETX can rescue neurodegeneration [34]. It is thus very likely that SETX’s absence or dysfunction plays a role in several neurodegenerative diseases. ALS has also been characterized by an increase in R loops, particularly for patients carrying the G4C2 repeats in the first intron of the C9ORF72 gene. Cells expressing a high number of G4C2 RNA repeats display R loop-driven DSB accumulation that can be reduced by SETX overexpression [35]. Recent data showed that C9ORF72 protein is a component of the autophagy initiation complex, reflecting a direct interaction with ATG13 (autophagy-related 13), an essential subunit of the autophagy initiation complex [36]. In fact, C9ORF72 is one of a number of genes mutated in ALS that can be linked to autophagy [37].
Autophagy is an evolutionarily conserved mechanism by which cells eliminate unwanted cytosolic proteins, lipids or organelles, including mitochondria and the endoplasmic reticulum. Upon extra- or intracellular induction (for example serum starvation), a cup-shaped membrane called a phagophore elongates and encloses a portion of the cytoplasm containing the material to be degraded, resulting in formation of an autophagosome, which will fuse with lysosomes to form an autolysosome, leading to degradation of the enclosed materials together with the autophagosome [38,39]. Autophagy is initiated through changes in the phosphorylation status of components of the ULK1 (unc-51 like autophagy activating kinase 1) complex, which is regulated by mechanistic target of rapamycin kinase complex 1 (MTORC1). Cellular stress such as starvation (or rapamycin treatment) inhibits MTORC1 leading to activation of the ULK1 complex. Mutations of genes regulating autophagy and lysosomal degradation cause a variety of neurodegenerative and neuromuscular diseases. Neuronal autophagy is indeed essential for synaptic plasticity, anti-inflammatory function in glial cells, as well as the myelination process [40]. Currently, more than twenty different neurological disorders have been linked to autophagy defects, including Alzheimer and Parkinson diseases, ALS, spinal and bulbar muscular atrophy and spinocerebellar ataxia (SCA) type 3 [41]. Here, we present evidence that AOA2 is an additional neurological disorder triggered by a defect in autophagy, reflecting the previously unknown role of SETX in this process.
Results
SETX depletion impairs mRNA transcription and processing
Previous studies have implicated SETX in various functions ranging from gene expression to DNA repair and RNA processing. We therefore set out to examine the impact of SETX depletion on transcription and mRNA processing. To this end, we initially performed microarray analysis using an Affymetrix Exon array (Affymetrix GeneChip Exon 1.0 ST Arrays) to compare the transcriptome of U87 glioblastoma-astrocytoma cells treated with either a control siRNA or siSETX. SETX knockdown (KD) affected about 4,000 genes (t test, P < 0.05) and 70% of them (2,622) surprisingly displayed reduced expression (see gene list in Table S1). When considering a change of 2-fold or more and an adjusted p-value (t test, P < 0.01), we identified ~ 400 differentially regulated genes with a majority (62%) downregulated after SETX KD (Figure 1A and Table S2). Gene Ontology (GO) analysis identified various affected processes including cell cycle, DNA replication and cytoskeleton organization (Fig. S1A). We compared the dysregulated genes by SETX KD with the gene expression analysis performed by Fogel et al. in AOA2 patient fibroblasts [42] and found 102 genes in common (Table S3). Interestingly, we also found that SETX KD led to a global reduction in accumulation of transcripts encoding RNA binding proteins and 3ʹ end processing factors (Figure 1B and Table S1 pASet).
Figure 1.
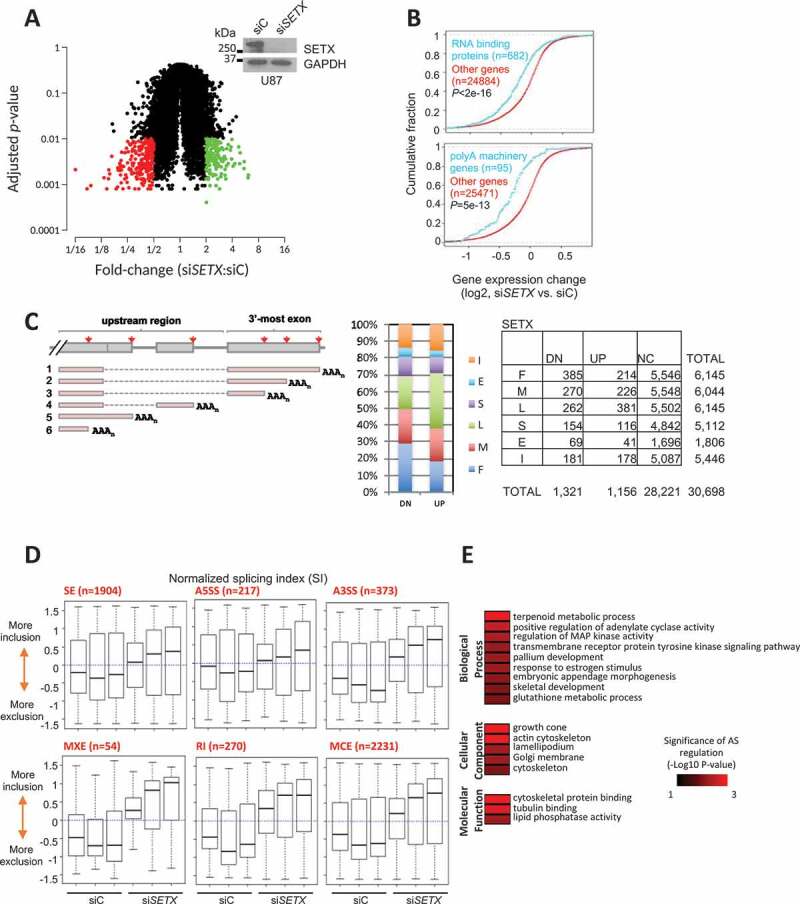
Gene expression and processing changes after SETX KD. (A) Fold-changes of gene expression after SETX KD (microarray). 250 genes were downregulated 2X or more (red dots) and 153 genes were upregulated at least 2X (green dots) (t-test, P < 0.01). The western blot (WB) shows SETX KD efficiency in U87 cells. GAPDH is used as a loading control. (B) Gene expression regulation (microarray) of genes encoding RNA binding proteins, polyA machinery genes. (C) APA analysis from 3ʹ READS analysis. PolyA type: 3ʹ-most exon: S, single; L, last (1); M, middle (2); F, first (3); Upstream region: I, intron (4,5); E, exon (6). DN, relatively downregulated; UP, relatively upregulated; NC, no change. (D) AS regulation in SETX KD (siSETX) cells vs. control (siC) organized by splicing type. Normalized SI is the mean-centered SI (SI – average SI of all samples). (SE, skipped exon; RI, retained intron; A5SS, alternative 5ʹ splice site; A3SS, alternative 3ʹ splice site; MXE, mutually exclusive exon; MCE, multiple cassette exons). (E) Gene ontology analysis of AS regulation after knockdown (microarray)
These data, together with prior evidence linking SETX to termination and 3ʹ end formation, led us to examine a possible function of SETX in 3ʹ end processing. Using microarray data, we first examined APA based on annotated pA sites (method illustrated in Fig. S2A). The result showed a global lengthening of mRNAs through APA (Fig. S2B), i.e., distal pA sites tend to be more used in KD cells compared to the control. To analyze 3ʹ end regulation more precisely, we performed 3ʹ region extraction and deep sequencing (3ʹREADS) [43] under the same conditions. We mapped 1,405,523 PolyA Site Supporting (PASS) reads in control siRNA-treated U87 cells and 1,568,989 in SETX KD cells. The data confirmed that the last pA site of a gene was generally upregulated as compared to the first polyA site in the 3ʹ-most exon, leading to 3ʹUTR lengthening (Figure 1C and Table S4).
SETX may also play a role in splicing. For example, SETX associates with several proteins implicated in splicing including SF3B1 (splicing factor 3b subunit 1), hnRNPs and SMN [5], and SETX colocalizes with SRSF2/SC35 (serine and arginine rich splicing factor 2) in SKNBE cells [44]. To determine whether SETX depletion affects splicing, we analyzed multiple types of alternative splicing (AS) (Fig. S3A) and calculated the splicing index using the microarray data (Fig. S3B). Splicing changes are detailed in Table S5 and Fig. S3C and S3D. We observed significant changes in AS events after SETX KD (Figure 1D and S3C). Notably, there were more changes involving inclusion of alternative exonic or intronic regions, such as alternative 3ʹ splice site usage, intron retention, multiple cassette exon, etc., as compared to those involving exclusion. Retained introns (RI), which often correlate with gene downregulation, also appeared as a hallmark of SETX depletion and are associated with a greater decrease in gene expression (Fig. S3D, P= 7e-6, Fisher’s Exact test). GO analysis revealed that AS of transcripts encoding cytoskeleton proteins was disrupted (Figure 1E). This data is in accordance with an earlier study that showed a defect in splicing in SETX-depleted cells [5]. Taken together, our analyses of mRNA processing in SETX-depleted cells indicate that SETX plays a role in modulation of both splicing and 3ʹ end processing activities.
R loops are reduced in abundance in SETX-depleted cells
We next investigated whether SETX depletion triggered changes in R-loop formation, as expected from its putative role in RNA:DNA hybrid resolution. To this end, we performed DNA/RNA IP coupled to sequencing (DRIP-seq) using the S9.6 antibody that specifically recognizes RNA:DNA hybrids [45,46]. Interestingly, SETX KD led to modest changes in the R-loop landscape, with a strong trend toward R-loop loss. Out of nearly 15,000 R-loop peaks genome-wide, about 1,500 loci showed significant loss of signal (> 2-fold, p-value < 0.05). The losses were observed over all types of genic regions including promoters, gene bodies and terminal regions (Figure 2A). Very few changes were observed over intergenic regions. Overall signal reduction upon SETX depletion could be easily validated in metagene plots (Figure 2B), indicating that these losses affected R-loop prone regions and led to consistent signal loss. R-loop gains, by contrast, were far fewer (150 loci; Table S6) and could not be readily visualized in metagene plots due to higher variability in position and signal amplitude (Fig. S4A).
Figure 2.
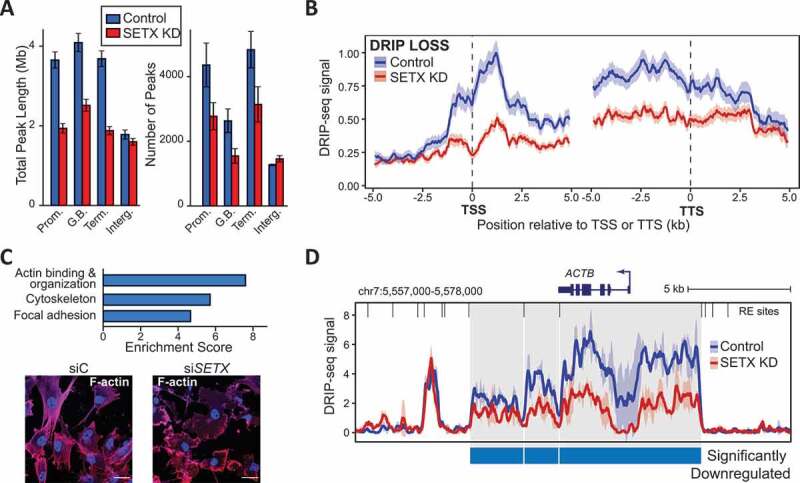
SETX depletion leads to loss of R loops at a subset of human genes. (A) Left; combined DRIP peak length over various gene features (Promoter [−2/+5 kb around TSS]/gene body [GB]/Terminal [−2/+5 kb around PAS]/Intergenic). Right; combined peak numbers over the same gene features. Data was normalized to represent the same number of mapped sequencing reads so these effects are not due to differential coverage. (B) Metagene plots of DRIP-seq signal over promoters and terminal regions that show significant R-loop loss in SETX-depleted cells. The lines represent the average signal surrounded by the standard error (shaded) for control and SETX-depleted cells, respectively. (C) Top; GO enrichment analyses for genes undergoing R-loop loss (PANTHER); Bottom; F-actin labeling with Phalloidin in U87 cells KD with siC or siSETX. Scale bar: 25 µm. (D) Screenshot of R-loop signal over the ACTB gene and surrounding regions. The signal is depicted as mean and standard error over two biological replicates for each condition. Regions with significant R-loop losses are highlighted and correspond to three restriction fragments (restriction enzyme sites used to fragment the genome before DRIP are indicated at the top)
GO analysis revealed that genes involved in the actin cytoskeleton and focal adhesion showed significant enrichment among genes showing lower R-loop signals after SETX depletion (Figure 2C top, Table S6; DAVID GO sheet). Since SETX depletion caused both downregulation, AS, and reduced R-loop levels of cytoskeleton protein-coding genes, we examined whether the actin cytoskeleton network was disturbed by SETX KD. To this end, we stained F-actin filaments in SETX KD and control U87 cells. Immunofluorescence (IF) analysis revealed a clear disorganization of the actin cytoskeleton after SETX KD in U87 cells (Figure 2C, bottom). Interestingly, ACTB (actin beta) was one of the genes showing the strongest R-loop loss genome-wide (Figure 2D).
Given that R loops form co-transcriptionally [10], it is possible that the overall trend toward R-loop loss observed upon SETX KD was due to gene expression downregulation. However, we observed only a 20% overlap between the ~ 400 downregulated genes (Table S2) and genes showing R-loop loss. Most of these overlapping genes corresponded to genes involved in cytoskeletal organization. This indicates that in the majority of cases, genes undergoing R-loop loss did not display similar reductions in steady-state mRNA pools. Vice versa, most genes showing lower gene expression at the mRNA level did not show lower R-loop levels (Fig. S4B), a trend that could be explained by the fact that the majority of these genes did not form R loops in control cells. Similarly, genes showing mRNA expression gains did not necessarily show R-loop gains (Fig. S4B), in agreement with the notion that R-loop formation not only requires transcription but also requires favorable DNA sequences and topological constraints [47]. Overall, our data confirm that SETX exerts a modest influence on the cellular R-loop landscape. Surprisingly, the dominant impact of SETX depletion corresponded to a decrease in R-loop levels, which is inconsistent with a role for SETX as a major R-loop resolving enzyme. The implications of these findings are discussed below.
Genes implicated in autophagy and lysosomal degradation are regulated by SETX
GO analysis revealed deregulation of various biological processes, and several indicated a significant impact on lysosome organization and biogenesis (Fig. S1). Because many diseases, and especially neurodegenerative disorders including ALS, have been linked to defects in the autophagy/lysosomal degradation pathway, we directed our efforts toward understanding the role of SETX in autophagy [41]. We first confirmed by RT-qPCR that several genes implicated in various stages of autophagy were up- or downregulated by SETX KD (Figure 3A). Among them, TFEB (transcription factor EB), upregulated ~ 4-fold after SETX KD, and ATP6V0D2 (ATPase H+ transporting V0 subunit D2), downregulated by ~ 90% after SETX KD, were especially noticeable. TFEB is a transcription factor that controls expression of crucial autophagy genes involved in autophagy initiation such as those encoding BECN1/Beclin1, ATG13 and in the degradation process such as V-ATPases including ATP6V0D2 [48–50]. V-ATPases are proton pumps that are critical components of the endosome/lysosome required for acidification, an essential mechanism for protein hydrolysis [51]. In fact, according to the microarray data, SETX KD led to decreased expression of nine genes encoding subunits of V-ATPases as well as several other genes that encode proteins having crucial roles in lysosomal biogenesis and function, including LAMP1 (lysosomal associated membrane protein 1) and many cathepsins (Fig. S1B and Table S1).
Figure 3.
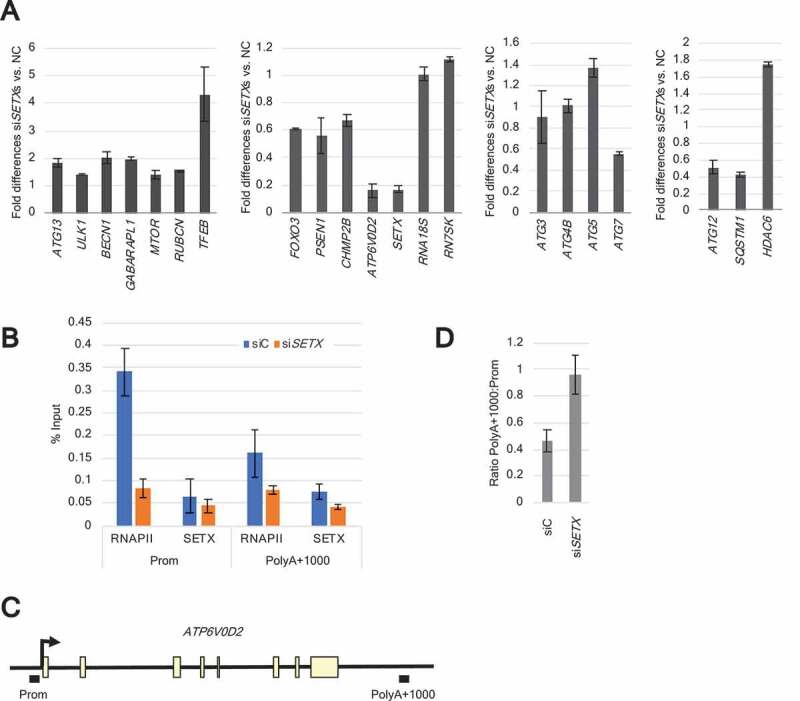
Autophagy/lysosomal degradation genes are regulated by SETX. (A) mRNA levels of autophagy/lysosomal degradation genes. n = 2, SE shown. RNA18S and RN7SK RNAs are used as controls. A pool of four siRNAs (2 nM each) was used for SETX KD (siSETXs). (B) RNAPII and SETX ChIP at the promoter (prom) and 3ʹ end regions (PolyA+1000) of ATP6V0D2. (C) Diagram of ATP6V0D2 and location of the probes used for q-PCR. (D) RNAPII occupancy at the 3ʹ end of ATP6V0D2 compared to the promoter region in siC and siSETX U87 cells
Increased levels of TFEB should lead to an increase in target gene expression, including those encoding V-ATPase subunits such as ATP6V0D2. Because ATP6V0D2 transcript levels did not increase after SETX KD but instead drastically decreased, we examined whether SETX has a direct impact on its transcription. To this end, we analyzed recruitment of RNAPII and SETX at ATP6V0D2 by ChIP assays. Strikingly, RNAPII recruitment to the promoter (Prom) and 3ʹ end regions (1 kb after the polyA site: PolyA+1000) was greatly reduced (Figure 3B,C). This indicates that the low level of ATP6V0D2 mRNA is, at least partially, a consequence of a transcriptional defect rather than enhanced RNA degradation. Interestingly, the ratio (signal polyA+1000):(signal Prom) of RNAPII occupancy revealed an accumulation of RNAPII at the ATP6V0D2 3ʹ end, which indicates a possible termination defect after SETX KD (Figure 3D). Despite the low efficiency of SETX antibodies in ChIP assays, we were able to detect SETX at the promoter and 3ʹ end regions of ATP6V0D2.
Altogether, our data indicate that SETX regulates transcription of genes involved in autophagy and lysosomal degradation, and that this regulation might, in some cases, occur through the direct recruitment of RNAPII to specific genes.
SETX KD affects autophagy flux
To investigate further a possible role for SETX in autophagy and lysosomal biogenesis, we first assayed the lipidation status of LC3, a marker of autophagy, in U87 cells transfected with siC or siSETX. LC3 is a ubiquitin-like protein that is important for elongation of the phagophore and formation of autophagosomes. Following translation, the unprocessed form of LC3 is proteolytically cleaved, resulting in LC3-I formation. Upon autophagy induction, cytosolic LC3-I is conjugated to the highly lipophilic phosphatidylethanolamine and forms LC3-II, which integrates into the outer and inner lipid membranes of phagophores and autophagosomes [52–54]. Detection of processed LC3-II by WB is a reliable indicator of autophagic flux. We detected less lipidation of LC3 (LC3-II) after SETX KD, both in normally growing cells (Ctrl; ~ 40% less LC3-II than siC) and in cells subjected to starvation for 24 h (0.1% FBS; ~ 55% less LC3-II than siC), indicating a defect in autophagy (Figure 4A). We confirmed this data by using each of the siRNAs from the siRNA pool separately (Fig. S5A) and by performing several similar experiments using medium without FBS for 6 h as starvation conditions (Fig. S5B). We also observed a similar defect in SETX KO cell lines generated using CRISPR-Cas9-based approaches (Fig. S5C; compare WT [A, B, C] to KO [X, Y, Z]; see Methods for details of construction of these cell lines).
Figure 4.
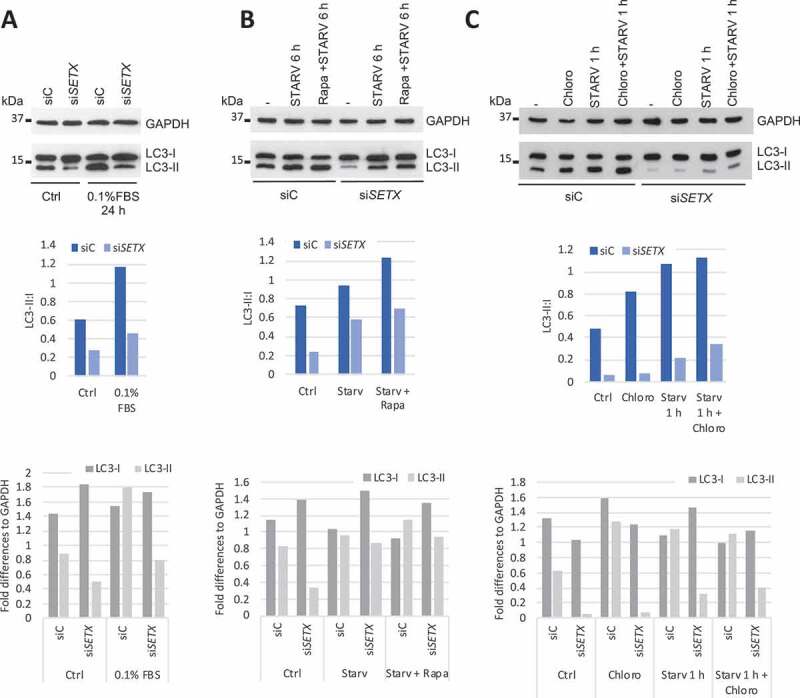
The autophagy flux is defective in SETX-depleted cells. (A) U87 cells were transfected with siC and siSETX for 3 d and grown in normal (Ctrl) or starvation condition for 24 h and 0.1% FBS. LC3 level (LC3-I and LC3-II) was analyzed by WB. GAPDH is used as loading control. (B) U87 cells were transfected as in A and grown under normal conditions (-), starvation in medium without FBS for 6 h (STARV) or in combination with rapamycin at 10 nM for 6 h (Rapa + STARV). (C) U87 cells were transfected as in A and grown under normal conditions (-), treated with chloroquine at 20 μM for 1 h (Chloro), starved in medium without FBS for 1 h or starved in combination with chloroquine treatment for 1 h (Chloro + STARV). Graphs show the quantifications of LC3-II:LC3-I and LC3-I and LC3-II levels normalized to GAPDH
The failure of LC3-II levels to increase indicates a possible defect prior to degradation at the autolysosome. According to the microarray data, expression of genes involved in the nucleation step of autophagosome formation was affected by SETX KD, and qRT-PCR analysis of RNAs from cells transfected with siC or siSETX confirmed that many of these genes were indeed deregulated (Figure 3A). In addition to starvation for 6 h (medium without FBS), we simultaneously or separately treated the cells with another activator of autophagy, the MTOR inhibitor rapamycin [55]. Using two different autophagy inducers, starvation and rapamycin treatment (alone or in combination with starvation) for 6 h, we observed the same defect in LC3-II accumulation (Figure 4B and S5D). Interestingly, while SETX KD led to a defect in the conversion of LC3-I to LC3-II, it also resulted in an increase in LC3-I (Figure 4A, bottom graph and Fig. S5B and S5D). This increase cannot be explained solely by blockage of the transition of LC3-I to LC3-II since the overall level of LC3 is often higher after SETX KD with or without autophagy stimulation (Fig. S5E). Indeed, SETX KD led to an increase of mRNAs that encode proteins that stimulate autophagy (Figure 3A).
The lack of LC3-II can result from a defect in autophagosome formation or from a more efficient and rapid degradation of autophagosomes. To investigate this, we used chloroquine (Chloro) and bafilomycin A1 (BAF), which impair lysosomal acidification and autophagosome/lysosome fusion, respectively, leading to lysosomal degradation inhibition [56]. Such treatments should lead to an accumulation of LC3-II that can no longer be degraded. If LC3-II degradation is extremely rapid after SETX KD, we should see an accumulation of LC3-II after Chloro or BAF treatments. However, accumulation of LC3-II after Chloro treatment (for 1 h (Figure 4C) or 6 h (Fig. S5B)) was still lower after SETX KD compared to control (siC) with or without autophagy induction through starvation (Figure 4C and S5B) and almost undetectable after BAF treatment for 6 h (Fig. S5B). We therefore conclude that the lack of LC3-II increase after SETX KD was the result of a defect in LC3-I conversion into LC3-II and most likely a defect in autophagosome formation. Consistent with this, transcripts encoding several proteins involved in the LC3-I to LC3-II transition were downregulated after SETX KD, including the ubiquitin-like activating enzyme ATG7 (autophagy-related 7) (Figure 3A).
We next wished to confirm that SETX depletion leads to a defect in autophagosome formation. To this end, we monitored autophagic flux by transfecting the mRFP-GFP-LC3 plasmid into U87 cells depleted of SETX (Figure 5A and S6A). Autophagosomes are visualized in yellow, a result of expression of both GFP and mRFP, while autolysosomes appear in red because of quenching of the GFP fluorescence by lysosome acidity [57]. While we detected a significant increase in autophagosomes after starvation in cells transfected with control siRNA (NC) compared to untreated cells (~ 2 times more), the number of autophagosomes after SETX KD in starved cells remained as low as in untreated cells (average of ~ 1 foci/cell in SETX KD cells compared to ~ 4 foci/cell in NC cells) (Figure 5B). Similarly, autophagy activation led to an increase in autolysosomes in control cells (NC), while these structures were barely detectable in SETX KD cells (Figure 5B), resulting from a lack of autophagosome accumulation. This data confirms a defect in the early stages of autophagy following SETX depletion. Additionally, starvation led to a ~ 50% increase in autophagosome size in NC cells while no difference was seen after SETX KD (Figure 5C). We obtained similar results after autophagy induction by rapamycin treatment for 6 h instead of starvation (Fig. S6A-C). To identify more precisely the defective step in autophagy activation after SETX KD, we performed IFs of endogenous WIPI2 in control (NC) and SETX KD U87 cells (Fig. S6D). Upon autophagy initiation WIPI2 is recruited to the phagophore assembly site to assist in formation of LC3-positive autophagosomes [58]. WIPI2 localizes at immature autophagosomes and dissociates before their maturation. Under SETX KD conditions, in untreated (NT) or starved cells, ~ 2-fold more WIPI2 cytoplasmic foci accumulate compared to control (NC) (Fig. S6E), confirming that SETX KD leads to autophagy initiation and activation. However, IF of both WIPI2 and LC3 showed that most WIPI2 puncta in SETX KD cells did not colocalize with LC3 (Figure 6A,B), indicating an accumulation of immature autophagosomes unable to form LC3-positive particles in control (NT) or autophagy-activating conditions (starvation or rapamycin treatment). Finally, we asked whether the autophagy defect triggered by SETX depletion could be linked to the accumulation of DNA damage observed after SETX KD [4,17]. In the absence of SETX, autophagy induction through starvation led to an accumulation of γH2AX, a marker of DSBs, comparable to that observed after treatment with the topoisomerase inhibitor camptothecin (CPT), as shown by WB (Fig. S5F). Additionally, WB of CYCS (cytochrome c) also showed an increase under starvation and SETX KD conditions, indicative of apoptosis (Fig. S5F).
Figure 5.
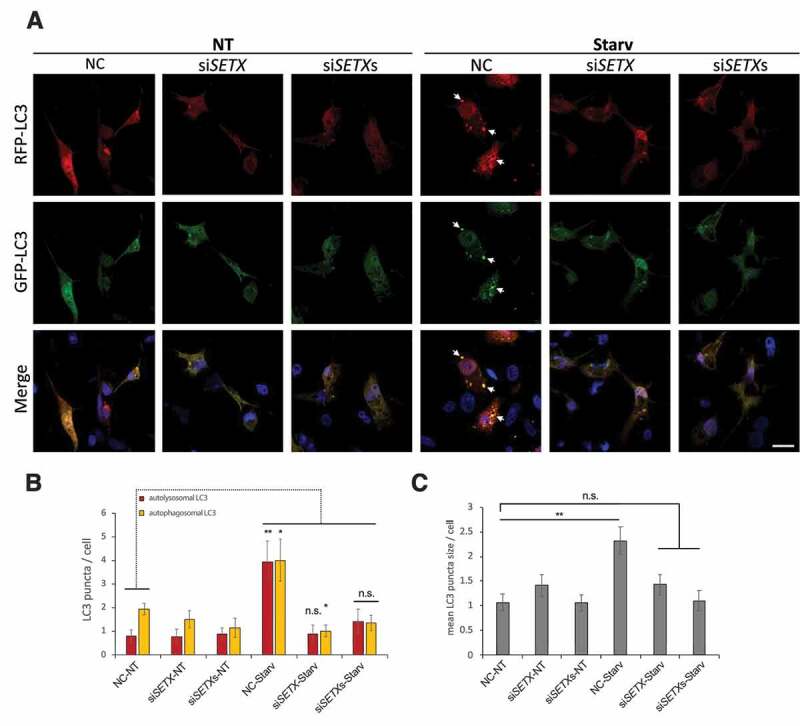
SETX KD compromises autophagosome formation. (A) mRFP-GFP-LC3 puncta formation assay in control (NT) and starved (Starv) (0% FBS for 6 h) U87 cells transfected with a siRNA control (NC), siSETX or a pool of siRNAs (siSETXs) for 48 h prior mRFP-GFP-LC3 transfection for 24 h. White arrows indicate the formation of autophagosomes (yellow signal). Scale bar: 15 µm. (B) Quantification of autophagosomes (yellow foci) and autolysosomes (red foci) per cell in control (NC) and SETX KD cells. Total cells analyzed: n = 88; Unpaired-test *p < 0.05, **p < 0.01, n.s.: no significance. (C) Quantification of mean autophagosome size per cell in NC and SETX KD cells. Total number of 223 LC3 puncta from 81 cells were analyzed. Data is displayed as mean +/- s.e.m. Unpaired t-test **p < 0.01, n.s.: no significance
Figure 6.
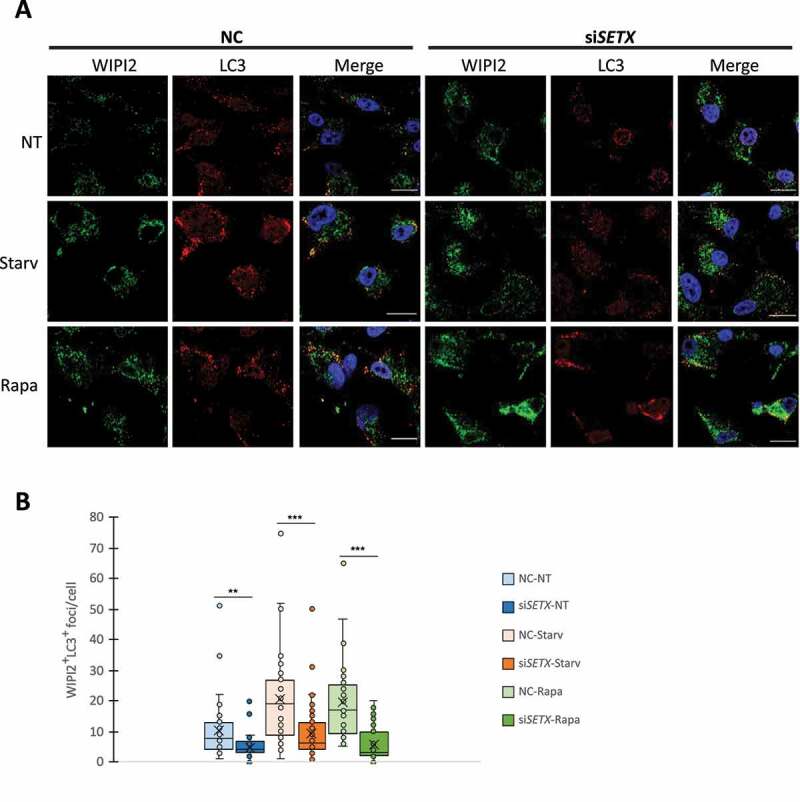
SETX KD leads to the accumulation of immature autophagosomes. (A) U87 cells were transfected with a siRNA control (NC) or siSETX for 72 h. Cells were untreated (NT), starved (Starv) or treated with rapamycin (Rapa) at 10 nM for 6 h and stained for endogenous WIPI2 and LC3. Scale bar: 15 µm. (B) Quantification of WIPI2 and LC3 colocalization-positive foci (yellow foci) per cell. Significance was analyzed by a student’s t-test, **p < 0.01, ***p < 0.001. At least 40 cells were analyzed in each condition
The above data together thus provides strong evidence that SETX plays an important regulatory role in initiation of autophagy and maturation of the phagophore, and that this occurs in parallel with the DSB accumulation associated with a lack of SETX.
SETX KD leads to accumulation of ubiquitinated proteins, protein aggregates and defective mitochondria
Since autophagy participates in the elimination of ubiquitinated proteins and aggregates, we next searched for a possible accumulation of ubiquitinated proteins after SETX KD. We transfected U87 cells with an HA-ubiquitin expression vector for 48 h and determined the level of ubiquitinated proteins by WB (Figure 7A). As expected, ubiquitinated protein levels after SETX KD (siSETX) were ~ 6 times higher than in control cells (siC), indicating a defect in degradation of ubiquitinated proteins.
Figure 7.
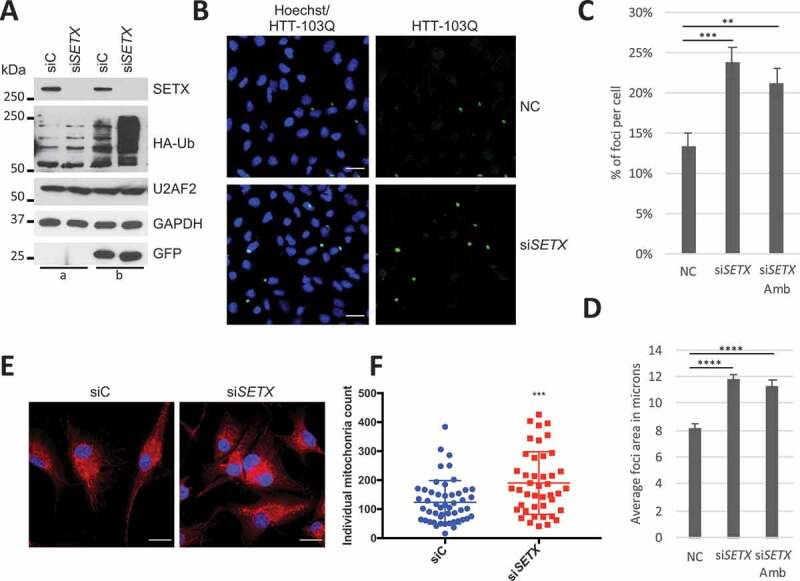
SETX depletion affects proteins and organelles clearance. (A) Ubiquitinated proteins accumulate after SETX KD. U87 cells were transfected with control (siC) and siSETX for 72 h without (a) or with (B) HA-Ub (4 μg) and a GFP (200 ng) plasmids (as transfection control) for 48 h. GAPDH and U2AF2 are used as loading controls. (B) IF of HTT-103Q-CFP in UC7 cells after 3 d of siRNA control (NC) and siSETX (KD). Scale bar: 25 µm. (C) Quantification of foci number in control cells (NC) and cells transfected with 2 different siRNA targeting SETX (siSETX and siSETX Amb). Cells count for NC = 1831, siSETX = 1843, siSETX Amb = 1803. SE shown. Significance was analyzed by a student’s t-test, **p < 0.01, ***p < 0.001. (D) Measurement of foci size in experiments as described in B. Area threshold: between 1 and 58 microns. Foci count for NC = 503, siSETX = 543, siSETX Amb = 522. n = 2, SE shown. Significance was analyzed by a student’s t-test, ****p < 0.0001. (E) U87 cells KD with siC and siSETX for 3 d and labeled with MitoTracker M7512. Scale bar: 25 µm. (F) Plot quantitating mitochondria using images as in (E). Each dot (blue or red) represents the number of mitochondria in that cell, with ~ 50 cells quantitated for each condition. Middle bar represents median with error bars representing standard deviation (SD). ***p ≤ 0.001. Significance was assessed via unpaired t-test
Because misfolded ubiquitinated protein aggregates are a hallmark of several neurodegenerative disorders [59], we also investigated whether SETX plays a role in aggregate clearance. To this end, we utilized a HeLa stable cell line conditionally expressing the first intron of HTT (huntingtin) fused with CFP (cyan fluorescent protein) and containing 103 glutamines (HTT-103Q-CFP), making the fusion protein prone to aggregation predominantly in the cytoplasm [60]. Huntington disease is a polyglutamine disorder caused by the expansion of CAG repeats leading to the accumulation and aggregation of the protein [61]. We depleted SETX in the HTT-103Q-CFP cells for 3 d and monitored the accumulation of CFP aggregates by IF (Figure 7B). Using two different siRNAs targeting SETX, we quantified the percentage of foci (aggregates) per cell (Figure 7C) as well as the average size of the foci (Figure 7D). In both cases, this analysis revealed a significant increase in cytoplasmic foci number (~ 1.7 times more) and size (~ 30% larger) after SETX KD. These data provide evidence that SETX plays a role in ubiquitinated protein aggregate clearance.
Defective mitochondria are also cleared by autophagy through a process called mitophagy [62]. Since mitochondrial damage is suspected to be one of the pathogenic mechanisms leading to several neurological disorders [63,64], we examined whether the mitochondrial network is disturbed by SETX depletion. We used a MitoTracker red probe that labels mitochondria in live cells and analyzed the mitochondrial network by IF (Figure 7E). Quantification of the mitochondrial network showed a 54% increase in mitochondria mass in SETX KD cells compared to controls (Figure 7F).
Altogether, these experiments provide further evidence that the autophagy defect detected after SETX KD affects metabolic processes linked to autophagy that can contribute to neurological disorders.
Autophagy is perturbed in AOA2 patient cells
We next wanted to know whether the autophagy defect we identified after SETX KD in U87 cells also occurred in AOA2 patients’ cells. We obtained fibroblasts from a family of five, of which two members have been diagnosed with AOA2 (#032 and #083) and carry novel SETX mutations, specifically a missense mutation (6872 T > C, M2230 T) in exon 21 located in the helicase-encoding domain and an AT insertion between nucleotides 2930–2931 in exon 10 (Fig. S7A). mRNA and protein levels confirmed that the AOA2 patients expressed ~ 50% SETX mRNA and protein compared to their asymptomatic father (#034) and brother (#082), who carry the missense mutation only (Fig. S7B and S7C). The mother (#033), on the other hand, who has not been diagnosed with AOA2 but manifests some motor difficulties, carries the AT mutation and the expression of only one allele, similar to her two affected children. We monitored the autophagic flux in fibroblasts by assaying the lipidation status of LC3 and GABARAP after autophagy induction by rapamycin (Figure 8). While LC3 plays an important role in phagophore expansion, GABARAP appears to promote later stages such as fusion of autophagosomes with lysosomes [65,66]. Quantification of LC3-II and GABARAP-II levels by WB showed a significant decrease for AOA2 patient #083 but not for patient #032, compared to controls (#033, #034, #082). A similar result was observed for BECN1, an important factor in autophagy initiation [67] (Figure 8A).
Figure 8.
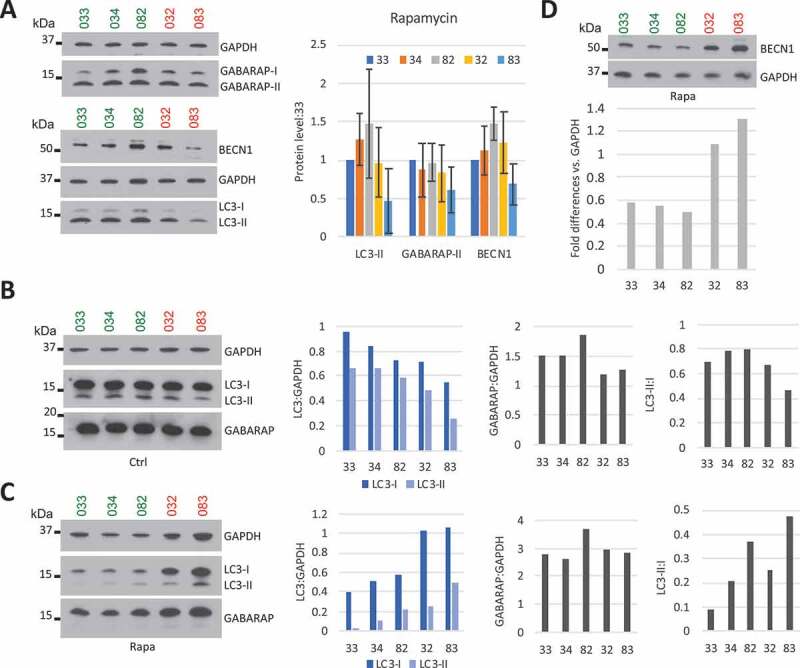
The autophagic flux in AOA2 cells is different from controls but also from SETX KD. (A) LC3, GABARAP and BECN1 visualization after rapamycin treatment (100 nM) of control (green) and AOA2 (red) fibroblasts for 6 h. GAPDH was used as loading control. Graph quantification appears on the right. n = 3, SD shown. (B) Autophagic flux in MNs derived from control (day 12 of differentiation) and AOA2 (day 13 of differentiation) fibroblasts by monitoring LC3 and GABARAP under normal conditions. GAPDH was used as loading control. Quantifications of proteins level appear on the right side of the corresponding WB. (C) Autophagic flux in MNs as in (B) by monitoring LC3 and GABARAP under rapamycin (100 nM, 6 h) condition. (D) BECN1 protein level in control (green) and AOA2 (red) MNs after rapamycin treatment at 100 nM for 6 h analyzed by WB. GAPDH is used as a loading control. The graph shows the protein quantifications of the WB
We also analyzed two immortalized AOA2 lymphoblastic cells that carry a homozygous nonsense mutation leading to a lack of SETX protein as determined by WB (Fig. S7D, bottom blot). Interestingly, the two different “KO” cell lines displayed a similar defect in LC3 as observed after SETX KD in U87 cells, with an accumulation of LC3-I and, consequently, a low LC3-II:I ratio (Fig. S7D, upper blot).
Because autophagy is a crucial process in post-mitotic cells, we monitored the autophagy flux in neurons derived from patient fibroblasts. To this end, we generated induced pluripotent stem cells (iPSCs) by reprogramming of fibroblasts, and the iPSCs were then differentiated into spinal motor neurons (MNs) (Fig. S7E-G). To determine if the MNs showed any pathological phenotype, we monitored their survival (Fig. S7H) and neurite outgrowth (data not shown) for 7 d in culture. We did not detect any significant differences between control and AOA2 MNs, indicating that the newly described AOA2 mutations did not affect their development. We then monitored LC3 and GABARAP levels in protein extracts prepared from the MNs under normal conditions or following rapamycin treatment. Under normal conditions, GABARAP and LC3-II levels were reduced in the MNs from AOA2 patients (#032 and #083) compared to controls (#033, #034, #082), a comparable phenotype to that observed in SETX KD cells (Figure 8B). However, after autophagy induction by rapamycin, LC3-I and LC3-II protein levels increased in AOA2 MNs while GABARAP levels were not significantly changed (Figure 8C). This result implies that the response to autophagy induction is different in AOA2 and control cells. But in contrast to SETX KD cells, the autophagy defect in AOA2 cells was not the result of an inhibition of the process but rather an activation compared to control cells. Hyper-activation of the autophagy pathway in the AOA2 patient-derived MNs is supported by higher levels of BECN1, which is involved in autophagy initiation, following rapamycin treatment (Figure 8D). The significance of these apparently discrepant results is discussed below.
Discussion
The experiments reported here provide new insights into SETX function, and how this might be relevant to disease. We first showed that SETX impacts transcription of specific genes, such as a number that encode proteins involved in autophagy and lysosomal degradation, which we showed translates into an autophagy flux defect. Importantly, we provided several lines of evidence indicating that SETX’s most crucial role in autophagy takes place at the early stage and most likely during autophagosome formation. We also found an increase in retained introns after SETX KD. Notably, retained introns are also a hallmark of SMN deficiency [32]. While we did not compare the retained intron data from our analysis with similar data from SMA mice or human cells depleted of SMN, such analysis might confirm the similarities and/or involvement of SETX and SMN in the same pathways, such as R-loop resolution at the 3ʹ ends of specific genes [33]. Indeed, retained introns increased in the SMA mouse model or in SMN KD human cells are R-loop hot spots that are associated with DSBs [32]. The lengthening trend of mRNAs after SETX KD we detected in both microarray and 3ʹREADS data can be the result of a direct role of SETX in transcription termination and/or R-loop regulation as well as possible indirect effects arising from SETX KD. These include altered expression of several RNA binding proteins and/or polyA machinery components such as FIP1L1 (factor interacting with PAPOLA and CPSF1) or NUDT21/CFIm25 (nudix hydrolase 21) (Table S1), which may affect APA [68]. We can also not exclude a role for SETX in influencing RNAPII elongation rate that could affect APA [69].
SETX depletion led to modest changes in R-loop distribution, with a general trend toward R-loop losses. This result appears to contradict studies that associated the loss of SETX with R-loop gains. It should be noted, however, that in HeLa cells depleted of SETX and in AOA2 patient cells, changes in R-loop distribution have not been ascertained genome-wide. Instead, a specific gain of R loops was reported at the 5ʹ pause region immediately downstream of ACTB [8,13,21]. Interestingly, other regions of the ACTB gene instead showed R-loop losses of similar magnitude to those reported here. Thus, our findings are in line with these observations. Unlike these previous studies, we did not observe any gain of R loops downstream of ACTB (Figure 2D), but it should be noted that DRIP-seq possesses lower spatial resolution and may have missed a hyper-localized gain. Analysis of murine germ cells knocked-out for SETX using S9.6 IF suggested that loss of SETX triggers broad R-loop gains [18]. However, significant concerns surround S9.6 IF given the affinity of S9.6 for dsRNA [70,71]. In contrast to AOA2/SETX KD cells, locus-specific decreases in R-loop signals were also reported in ALS4 patient cells [28]. In this case, it was proposed that SETX is required for R-loop formation at promoter regions and that such promoter R-loops protect genes from DNMT1-mediated DNA methylation and silencing. Our observation that a subset of promoters showed R-loop loss upon SETX depletion is consistent with these findings. Interestingly, most of the examples of R-loop loss did not correspond to genes displaying reduced transcript levels (except for cytoskeletal genes; see below). Given that R loops form co-transcriptionally, these observations could be explained if SETX depletion affects levels of nascent transcription without significantly impacting mature mRNA levels. Our finding that RNAPII recruitment at the ATP6V0D2 promoter was reduced upon SETX KD is compatible with the idea that SETX may fine-tune gene expression.
Overall, our data have established that the dominant impact of SETX depletion on R-loop accumulation is a loss of R loops at a subset of human genes. The fact that only few examples of R-loop gain could be observed further suggests that SETX does not function as a major R-loop resolving enzyme, as often stated. Instead, since the majority of genes showed no detectable changes in R-loop signals upon SETX depletion, it is likely that R-loop homeostasis is accomplished by a series of enzymes with redundant activities.
The changes in gene expression we observed after SETX KD affects various biological processes. Perhaps most notable was the autophagy/lysosomal degradation pathway, from its initiation to the degradation of autolysosomes. While several genes encoding proteins involved in autophagy and lysosomal biogenesis and function were downregulated, some playing an important role in autophagy activation, such as TFEB, BECN1 and ATG5 (autophagy-related 5), were upregulated. This is in accordance with the increase in LC3-I levels and WIPI2 foci observed after SETX KD. Notably, this autophagy/lysosomal degradation regulation may be evolutionarily conserved, as mutation of SEN1 has also been linked to mis-regulation of autophagy in yeast [72]. N terminal-truncated Sen1 leads to an accumulation of ubiquitinated proteins and lysosomes, an indication of a defect in the degradation process. The same Sen1 mutant also showed a loss of mitochondrial DNA and an increase in ROS. Several of those phenotypes are similar or comparable to what we observed in SETX KD and AOA2 cells. Interestingly, the R-loop landscape and AS at cytoskeleton/actin genes appear to be specifically affected by SETX KD. Cytoskeletal organization plays an important role in vesicle transport and autophagosome-lysosome fusion [73], and its deregulation might exacerbate the autophagy defect that occurs in SETX-depleted or mutant cells.
An important question is the relevance to disease of the autophagy defects we observed. AOA2 is characterized by cerebellar atrophy, oculomotor apraxia, loss of movement coordination and peripheral neuropathy. Considerable evidence has pointed to the importance of autophagy regulation in Purkinje cells, the neurons located in the cerebellar cortex. For example, Atg5 and Atg7 deletion in mouse Purkinje cells leads to degeneration and cell death in the cerebellum [74,75]. Purkinje cells require a large supply of energy and thus need a large pool of healthy mitochondria [76]. Indeed, abnormal activation of autophagy leading to excess mitophagy has been observed in the Purkinje cells of Purkinje cell degeneration (pcd) mice [77]. Interestingly, pcd mice display very similar phenotypes to AOA2. These include gait ataxia, cerebellar degeneration and male sterility, but a normal lifespan [78,79]. Additionally, expression of a hyperactive form of MTOR in the mouse cerebellum also leads to cerebellar atrophy and shows increased mitochondrial biogenesis, confirming the importance of MTORC1 signaling in Purkinje cell survival [80]. Notably, these mice also exhibited ataxic gait. These data emphasize that maintenance of cerebellar homeostasis is crucial to prevent the neurodegeneration process that leads to ataxia and other related abnormalities. Too much autophagy can be as detrimental as too little.
Without fully functional autophagy and lysosomal degradation, neurons accumulate protein aggregates that are deleterious for the cells and the brain. Mutations of autophagy/lysosomal genes involved in neurodegenerative disorders can target any stage of the autophagy pathway, from phagophore formation to lysosomal degradation. For example, patients with ALS and/or frontotemporal dementia (FTD) carrying C9ORF72 expansions have abnormal neurons with SQSTM1/p62 (sequestosome 1)-positive inclusions [81,82]. The SQSTM1 gene has been found mutated in some ALS patients [83,84]. SQSTM1 interacts directly with LC3 to facilitate degradation of polyubiquitinated proteins and aggregates [85]. Pathologic inclusions containing ubiquitin and SQSTM1 are a common feature of ALS and ALS-FTD. Additionally, the C9ORF72 expansion can produce sense and antisense transcripts encoding dipeptide repeat proteins by repeat-associated non-ATG translation that are detected in SQSTM1-positive aggregates [86,87], although the relevance of these to disease remains unclear. Another finding suggesting that autophagy defects can play an important role in ALS is the fact that TARDBP/TDP-43 (TAR DNA-binding protein) turnover is enhanced by activation of autophagy, which leads to increased survival in neuronal ALS models [88]. Furthermore, TARDBP depletion has been shown to increase autophagosome formation in a TFEB-dependent manner but to inhibit autophagosome-lysosome fusion [89]. In contrast, we identified a defect in WIPI2- and LC3-positive foci and LC3-II formation after SETX KD, indicating impaired autophagosome formation. Overexpression of or mutations in FUS (FUS RNA binding protein), another RNA binding protein implicated in ALS and involved in several aspects of RNA biogenesis [90,91], has also been shown to inhibit autophagy, and this may also contribute to neurodegeneration [92]. In the present work, we did not investigate a potential autophagy defect in ALS4. We previously showed that AOA2- and ALS4-associated mutations located in the N-terminal domain of SETX do not affect the same function of the protein (e.g., interaction with the exosome) [25]. But since a number of genes that encode proteins that function in autophagy are mutated in ALS [37], it is very possible that ALS4-associated mutations lead to similar problems. While the autophagy defect linked to AOA2 is the result of SETX loss-of-function, it is conceivable that a gain-of-function in ALS4 affects the same pathway but most likely by a different mechanism.
AOA2 patient fibroblasts and MNs response to autophagy induction by rapamycin are different. It is not completely surprising that autophagy regulation differs depending on the cell and activation types [93]. While we derived iPSCs into MNs, it would be interesting to monitor the autophagic flux in iPSCs differentiated into cerebellar cells such as Purkinje cells. All the AOA2 patient cells tested, including lymphoblasts, do not share the exact same autophagic defect. It is important to note that despite the fact that they all have been diagnosed with AOA2, and present autophagic interference, AOA2 patients carry different mutations that will likely differentially affect SETX functions. AOA2 patients should probably be categorized in different clusters corresponding to specific pathways that lead to similar outcomes (such as a defect in autophagy). It is however difficult to establish such clusters with a limited number of patient samples.
Modulating autophagy is believed to be a promising therapeutic strategy for many neurodegenerative disorders [93]. Indeed, several drugs used to stimulate autophagy, such as rapamycin (selective inhibitor of TORC1) and lithium (activation of MTORC1-independent pathway), have been used with several neurodegenerative disorders. However, these drugs appear to have mixed effects on ALS models, sometimes beneficial and sometimes detrimental [94–97]. It is then possible, as suggested by our data in MNs and previous studies [89,94,98] that hyper-activation of the autophagy pathway can be in some cases and cell types as detrimental as its inhibition. It will be crucial to better understand the fine-tuning of the autophagy pathway in normal and disease cells to target the proper cells efficiently, precisely and safely.
Materials and methods
Gene expression analysis
Raw data from the Affymetrix GeneChip Human Exon 1.0 ST Array were normalized by the RMA method in the Affymetrix Power Tools (APT) program and probe sets with detection above background (DABG) P value < 0.05 in at least one sample group were used for further analysis [99]. To eliminate the potential effect of alternative polyadenylation (APA) in 3ʹ UTRs on expression analysis of a gene, we only used probe sets mapped to the coding sequence (CDS) to represent the expression level of a gene. We used t-test P value < 0.05 to select significantly regulated genes. Genes encoding RNA-binding proteins were defined by GO term GO:0003723. Genes encoding the core polyA machinery and associated factors, collectively called polyA machinery genes, were defined by Shi et al. [100]
Affymetrix microarray
SETX siRNA (20 nM) or control siRNA was transfected in U87 cells (ATCC, HTB-14) with RNAimax (Invitrogen, 13778150) for 72 h. RNA was purified using the RNeasy kit (Qiagen, 74104) followed by on-column DNAse treatment (Qiagen, 15200–50). The GeneChip WT Terminal Labeling and Controls Kit (Thermo Fisher Scientific, 901525), combined with the Ambion WT Expression Kit (Thermo Fisher Scientific, 4411973) were then used to generate biotin-labeled cDNA prepared with 1 μg of total RNA and hybridized to GeneChip Exon 1.0 ST Arrays (Affymetrix, 900650) according to standard protocols. Briefly, hybridization was performed using 5 μg of biotinylated target, which was incubated with the GeneChip Exon 1.0 ST Array at 45°C for 16 h. RNA quality was in the range of 8–10 RIN with Agilent Bioanalyzer. After hybridization, nonspecifically bound material was removed by washing and specifically bound target was detected using GeneChip Expression Wash, Stain and Scan kit (Thermo Fisher Scientific, 900720) and the GeneChIP Fluidics Station 450. Arrays were scanned using the GeneChip Scanner 3000 7 G (Affymetrix) and.CEL intensity files were produced using GeneChip Operating Software version 1.4 (Affymetrix). The data are available in the GEO database (accession #: GSE145677). Source code, detailed instructions, and intermediary data files related to the microarray analysis are accessible on GitHub (https://github.com/HKeyHKey/Richard_et_al_2020).
3ʹREADS
Total RNA was processed by the 3ʹ region extraction and deep sequencing (3ʹ READS) method as described in [43]. APA analysis of 3ʹ READS data was detailed in [101]. Briefly, we first removed 5ʹ adapter sequences from reads and reads with length < 15 nt after this step were discarded. We then mapped reads against the hg19 genome sequence using bowtie 2 (version 2.1.0) [102] with the following setting: “--local -5 4”. Reads with ≥ 2 unaligned Ts at the 5ʹ end are called poly(A) site supporting (PASS) reads, which were used to identify polyA sites. PolyA sites located within 24 nt from each other were clustered. The polyA site location with the highest read count in a cluster was used to represent the cluster. PolyA sites mapped to the genome were further assigned to genes, using gene models defined by RefSeq, Ensembl and UCSC Known Gene databases. The 3ʹ ends of the gene models were extended by 4 kb to include downstream polyA sites. To reduce false polyA sites, we further required (1) the number of PASS reads for a polyA site was ≥ 5% of all PASS reads for the gene; and (2) detected in at least two samples. PolyA sites were separated into different types based on the gene model. The microarray and 3ʹREADS data have been uploaded to the GEO database (accession #: GSE145677).
Gene ontology (GO) analysis
GO annotation of genes was obtained from the NCBI Gene database. The Fisher’s exact test was used to test whether the genes associated with a given GO term are more likely to be upregulated or downregulated as compared to genes associated with other GO terms.
DRIP-seq analysis
DRIP-seq was performed essentially as described [46] using two replicates for each sample (siC and siSETX) in U87 cells. DRIP was validated by qPCR over positive and negative test loci and sequencing libraries were built, verified and sequenced on an Illumina HiSeq 2500 (SE50). DRIP-seq data was mapped to the human genome using Bowtie2 using default parameters and normalized across samples by uniquely mapped reads. DRIP peaks were called using a Hidden Markov Model, as described [11]. Peaks were annotated to reflect their genomic feature (promoter/gene body/terminal/intergenic). When comparing DRIP signals across samples, peaks over any given locus were merged prior to calculating the average signal for each sample over the merged peak. Significantly higher/lower DRIP peaks in scramble vs SETX KD were identified using ANOVA one way; similar results were obtained when using DeSeq2. The RNA-seq data have been submitted to the GEO repository and are publicly available (accession #: GSE135349).
Microscopy
F-actin was labeled with CF dye phalloidin conjugate CF568 according to the manufacturer protocol (Biotium, 00044). MitoTracker Red CMXRos (Invitrogen, M7512) was added to live cells at 200 μM + Hoechst (Thermo Fisher Scientific, H3570) at 1 μg/ml for 1 h. Cells were fixed by adding paraformaldehyde (PFA) directly to the medium at 4% for 30 min. Images were acquired using Zeiss LSM 700 or 800 confocal microscope and 20x/1.4 and 40x/1.4 oil objectives were used. HTT-103Q-CFP foci signals have been analyzed after 3 d of siRNA transfection using ImageJ. For the mRFP-GFP-LC3 puncta formation assay, the ptfLC3 vector (Addgene, 21074; deposited by Tamotsu Yoshimori) was transfected into U87 cells (ATCC, HTB-14) at 0.5 μg/ml 48 h after siRNA transfection. WIPI2 and LC3 IFs were performed after fixation of the cells with 4% PFA and permeabilization with 0.1% Triton X-100 (Sigma-Aldrich, T8787) in PBS for 20 min. Primary antibodies (WIPI2 [Rabbit]: Proteintech, 15432-1-AP; LC3 [mouse]: Biolegend, A15143 K) were incubated at room temperature for 1 h at 1:500 followed by incubation with Alexa Fluor 488 goat anti-rabbit (to visualize WIPI2) and Alexa Fluor 568 anti-mouse (to visualize LC3) at 1:500 (Thermo Fisher Scientific, A-11031). Cells were mounted with DAPI (Abcam, ab104139) for imaging with Zeiss Zen LSM700 confocal microscope with a 63 X oil objective.
siRNAs transfection and western blots
U87 cells were transfected with the siRNA control NC or siC (NC: TTCTCCGAACGTGTCACGT, GenePharma, siC: UAGCGACUAAACACAUCAA, Dharmacon, D-001210-01), siSETX (AGCAAGAGAUGAAUUGCCA), siSETX1 (AUCUAACUCUGUACAACUUGC), siSETX2 (GAGUUCCUCUUCUUGAAAU), siSETX4 (GCCAGAUCGUAUACAAUUA), siSETXAmb (Ambion, 4427037) for 72 h at 20 nM with RNAiMAX (Invitrogen, 13778) prior to extraction. Cell culture medium was removed and 2X sample loading buffer (50 mM Tris HCl, pH 7, 4% SDS [Thermo Fisher Scientific, BP166-100], 20% glycerol [Pharmco, 349000000], 0.002% bromophenol blue, 5% beta-mercaptoethanol) (200 μl/6 wells plate) was used to scrape the cells. Cell extracts were boiled for 10 min and centrifuged for 5 min at 12,000 rpm. Soluble extracts were transferred to a fresh tube prior loading 5–20 μl onto SDS-PAGE gels (5% [for SETX] to 14% [for LC3]). Transfer was performed using nitrocellulose membrane (Bio-Rad, 1620115) except for LC3 western blot (WB), PVDF (Millipore, IPVH00010) was used. High molecular weight (HMW) transfer buffer (12.11 g Tris base, pH 8.6, 3 g glycine, 0.36 g SDS, 20% methanol) was used for HMW proteins (gel 5–6%) and Laemmli buffer was used for gels above 8%. Membranes were blocked in PBST (PBS [140 mM NaCl, 2.5 mM KCl, 1.6 mM KH2PO4, 15 mM Na2HPO4] + Tween-20 [Sigma-Aldrich, P7949] at 0.1%) + 5% milk (LabScientific, M-0841] for 15–20 min followed by overnight incubation with the primary antibody diluted in PBST + 5% milk (usually between 1:1000 to 1:5000). Secondary antibody was usually added at 1:20,000 for 30 min to 1 h in PBST + 5% milk. The following primary antibodies were used: GAPDH (Sigma, G9545), LC3 (BIO-RAD, AHP2167), GABARAP (GeneTex, GTX129710), BECN1 (Santa Cruz Biotechnology, E8: sc-48341), U2AF2/U2AF65 (Sigma, U4758), GFP (abm, G095), HA (abm, G166), SETX (Novus Biologicals, NBP1-94712), CSTF3/CSTF77 (Bethyl Laboratories, A301-096A). Each blot was incubated with the antibody(ies) of interest and GAPDH or U2AF2 used as loading controls. Protein bands were detected by chemiluminescence and exposed to X-ray films that were subsequently scanned. Signals were quantified using ImageJ. Multiple exposures were performed to ensure that blots were not over or underexposed. Each protein signal was normalized to GAPDH or U2AF2 from the same blot.
ChIP
ChIP assays were performed as described [103]. RNAPII and SETX were IPed overnight at 4°C with 2 μg (Santa Cruz Biotechnology, sc-899) or 4 μg (Bethyl Laboratories, A301-105A) of antibody, respectively. DNA was analyzed by qPCR using the following primers: ATP6V0D2 prom: F: AGCCACTGAAACCAGAGGAA, R: CCAAGGTAGGGTGAAACAGC; ATP6V0D2 PolyA+1000: F: AAAGCAGCCACTGACCAAGT, R: GCCTTAAACCCATCACCAGA.
RT-qPCR
Real-time PCR was performed in 96-well plates with power SYBR Green using StepOnePlus (Applied Biosystems, 4367659). RNA extraction was carried out with TRIZOL (Thermo Fisher Scientific, 15596026) followed by DNaseI treatment (Promega, M6101). cDNA was produced with Maxima reverse transcriptase (Thermo Fisher Scientific, EP0741) and a random hexamer following the manufacturer’s protocol and using 500–1000 ng of RNA. The following primers were used: ATG13 (F: GCGGAGTCTTAGGAGCAAAA, R: GGTTCCGGGCTCTCATTAC), ULK1 (F: ACACCATCAGGCTCTTCCTG, R: GATCTTGACGCGGATGCT), BECN1 (F: CTACCGGGAAGTCGCTGA, R: GTCACCCAAGTCCGGTCTAC), GABARAPL1 (F: CGGACGTTTCTGCAGCTATT, R: CTCAAAGGGATGGTCCTCCT), MTOR (F: AGGGCAAGATGCTTGGAAC, R: ATAGTGCTGGAGCTCCTTGG), RUBCN (F: ACGAGAACGACCAGAGCAGT, R: TACTGTCTATCCCCGAGCAG), TFEB (F: CCAGAAGCGAGAGCTCACAGAT, R: TGTGATTGTCTTTCTTCTGCCG), FOXO3 (F: CCTTGTACAGGGTGGTGAGC, R: AAACGTCCCATAAACCATCG), PSEN1 (F: TCACCTTTGGGCTTGTTTTC, R: TTTGTCCTCCCCAGATTTTG), CHMP2B (F: AGCCAAACAACTTGTGCATCT, R: ACATTGCTCCAGCCATCTTC), ATP6V0D2 (F: TTTTGGCTAATCACACAAATCC, R: AGGTGAGAAATGTGCTGAGG), SETX (F: CTTCATCCTCGGACATTTGAG, R: TTAATAATGGCACCACGCTTC), RNA18S (F: TACCTGGTTGATCCTGCCAGTAGC, R: AACTGATTTAATGAGCCATTCGC), RN7SK (F: ATCTGTCACCCCATTGATCG, R: AGACCGGTCCTCCTCTATCG), ATG3 (F: ACGGCAAGAGAGTGAGAAGG, R: GAAGGGAGACCTGAGGTGAG), ATG4B (F: ACTGCGTTTCCTGCAGATTC, R: ACGTAGGCCTCGTTGATGTC), ATG5 (F: GAGGGTGACTGGACTTGTGG, R: GTAGCAGGACTCCAGGAAGC), ATG7 (F: GGGGGATCCTGGACTCTCTA, R: GAGCTTCATCCAGCCGATAC), ATG12 (F: GGCGTTTTGGTTTCACATCT, R: TGTCCTATGTGCTTGCTCTCC), SQSTM1 (F: CCCGTCTACAGGTGAACTCC, R: ATGTGGGTACAAGGCAGCTT), HDAC6 (F: TTCTGAGGCCCACGATAG, R: TTGGGGAGTTGCAAAGGATA).
CRISPR cells generation
gRNA sequences were designed using the E-CRISP online tool (www.e-crisp.org/E-CRISP/) to excise SETX Exon 5 (rRNA targeting sequences are as followed: 5ʹ of Exon 5: GTCAGGTAAATATTGTTCCT and 3ʹ of Exon 5: TTCAGTATGTTGAATTGAAC) and cloned into the cas9 vector pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene, 62988; deposited by Feng Zhang) as detailed in [104]. Constructs were transfected in monoclonal U87 cells using lipofectamine 2000 (Thermo Fisher Scientific, 11668019). Clones were isolated after puromycin selection at 2 μg/ml.
Mitochondria quantification
Mitochondria quantitation was performed using a publicly available macro MiNA for ImageJ as described [105].
Fibroblast reprogramming into induced pluripotent stem cells (iPSCs)
Fibroblast were cultured in DMEM (Gibco, Thermo Fisher Scientific, 11885084) supplemented with 15% fetal bovine serum (Thermo Fisher Scientific, A3160402), 1% glutamine (Thermo Fisher Scientific, 25030081) 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific, 15070063). The reprogramming was performed using the CytoTune-iPS 2.0 Sendai Reprogramming Kit (Invitrogen, A16517). iPSCs were at first cultured on feeder and at passage two were plated on Cultrex (Trevigen, 3434–005-02).
Differentiation of iPSCs into motor neurons (MNs)
The iPSCs were differentiated into MNs using the method described by Nedelec et al. [106]. This protocol is a rapid method of differentiation through embryoid bodies (EBs). For all the differentiation process, neuronal medium N2B27 (DMEM/F12 medium [Gibco, Thermo Fisher Scientific, 11320033] + Neurobasal Medium [Gibco, Thermo Fisher Scientific, 21103049] + MEM Non-Essential Amino Acids [Gibco, Thermo Fisher Scientific, 11140035] + B-27 Supplement [Gibco, Thermo Fisher Scientific, 17504001] + N-2 Supplement [Gibco, Thermo Fisher Scientific, 17502001] + Penicillin-Streptomycin [Gibco, Thermo Fisher Scientific, 15070063] + 2-Mercaptoethanol [Gibco, Thermo Fisher Scientific, 31350–010]) was used. iPSCs were harvested at day 0 and plated at a concentration of 1.106 cells into low attachment plates (CORNING, 3471). The medium was changed on days 2, 4, 7, 9 and 11. Starting at day 2, the medium was supplemented with SB 431542 (Sigma-Aldrich, S4317), LDN193189 (STEMCELL Technologies, 72149) and CHIR99021 (Sigma-Aldrich, SML1046) until day 7. Starting at day 2 and day 4, respectively, Retinoic Acid (RA; Sigma-Aldrich, R3255) and Smoothened Agonist (SAG) (Calbiochem, 566660) were added to the medium. DAPT (Tocris Bioscience, 2634) was added at day 9. EBs were dissociated at day 12 and cells were resuspended in N2B27 supplemented with RA, SAG, BDNF (Peprotech, 450–02) and GDNF (Peprotech, 450–10) and plated on 24-well plates coated with polyornithine (Sigma-Aldrich, P3655) and laminin (Sigma-Aldrich, L2020). To perform survival and neurite outgrowth analysis, MNs were fixed with 4% paraformaldehyde (Thermo Fisher Scientific, AA433689 L) for 7 min on days 1, 3, 5 and 7 post-plating.
Survival and neurite outgrowth analysis
All five cell lines were stained with MNX1 (motor neuron and pancreas homeobox 1) (kindly provided by Dr. Wichterle, Columbia University) and SMI32 (1:100; Biolegend, 801701). The images were acquired with a Leica Confocal sp5. Images for MNs counting for the survival data were analyzed using Fiji software. For the neurite outgrowth analysis, the images were analyzed using Fiji software, NeuronJ plugin.
Statistical analysis
Data are expressed as the mean and standard deviation (SD) or standard error (SE) as indicated in the figure legends. Methods used to calculate significant differences between groups are indicated in the figure’s legend or in the main text. Asterisks indicate statistically significant differences. A p-value < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We would like to thank the National Ataxia Foundation (NAF) (NAF CU14-2541) for their support to PR and the NIH (R35 GM118136) and the Muscular Dystrophy Association (377780) for their support to PR and JLM. BT was supported by NIH R01 GM084089 and R01 GM129069. Research in the Chédin lab is supported by NIH R01 GM120607. Work in the Lotti lab is supported by NIH grant R21NS101575. Paola Rinchetti is supported by the European Union’s Horizon 2020 research and innovation program (MSCA-RISE-2017 778003) . PR would also like to thank Manley lab members, in particular Leroy Mensah for his assistance and hard work. PR also thanks Malgorzata Rak and Emanuel Rosonina for their helpful advice, discussion and support. We thank Ai Yamamoto for the UC7 cells, helpful discussion and input, and the Wichterle lab for the MNX1 antibody. We also thank Isabelle Le Ber and Christelle Dussert for providing the AOA2 lymphoblastic cells. Most importantly, we are exceptionally grateful to members of the family affected by AOA2 who offered their help and traveled across the country to provide samples for our research through skin biopsy procedures with Dr. Hirano.
Funding Statement
This work was supported by the Muscular Dystrophy Association [377780]; National Ataxia Foundation [NAF CU14-2541]; National Institutes of Health [R01 GM120607]; National Institutes of Health [R01 GM084089]; National Institutes of Health [R21NS101575]; National Institutes of Health [R35 GM118136]; ; National Institutes of Health [R01 GM129069]; European Union’s Horizon 2020 research and innovation program[778003].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kim HD, Choe J, Seo YS.. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 1999;38:14697–14710. [DOI] [PubMed] [Google Scholar]
- [2].Moreira MC, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–227. [DOI] [PubMed] [Google Scholar]
- [3].Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yuce O, West SC. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol. 2013;33:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Suraweera A, Lim Y, Woods R, et al. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet. 2009;18:3384–3396. [DOI] [PubMed] [Google Scholar]
- [6].Wagschal A, Rousset E, Basavarajaiah P, et al. Microprocessor, SETX, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell. 2012;150:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Miller MS, Rialdi A, Ho JS, et al. Senataxin suppresses the antiviral transcriptional response and controls viral biogenesis. Nat Immunol. 2015;16:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell. 2011;42:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mischo HE, Gomez-Gonzalez B, Grzechnik P, et al. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell. 2011;41:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–124. [DOI] [PubMed] [Google Scholar]
- [11].Sanz LA, Hartono SR, Lim YW, et al. Prevalent, dynamic, and conserved R-Loop structures associate with specific epigenomic signatures in mammals. Mol Cell. 2016;63:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ginno PA, Lim YW, Lott PL, et al. GC skew at the 5ʹ and 3ʹ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013;23:1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Skourti-Stathaki K, Kamieniarz-Gdula K, Proudfoot NJ. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 2014;516:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu K, Chedin F, Hsieh CL, et al. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442–451. [DOI] [PubMed] [Google Scholar]
- [15].Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365–378. [DOI] [PubMed] [Google Scholar]
- [16].Wahba L, Amon JD, Koshland D, et al. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell. 2011;44:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Suraweera A, Becherel OJ, Chen P, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol. 2007;177:969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Becherel OJ, Yeo AJ, Stellati A, et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet. 2013;9:e1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sollier J, Stork CT, Garcia-Rubio ML, et al. Transcription-coupled nucleotide excision repair factors promote R-Loop-induced genome instability. Mol Cell. 2014;56:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hill SJ, Rolland T, Adelmant G, et al. Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 2014;28:1957–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hatchi E, Skourti-Stathaki K, Ventz S, et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell. 2015;57:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Brustel J, Kozik Z, Gromak N, et al. Large XPF-dependent deletions following misrepair of a DNA double strand break are prevented by the RNA:DNA helicase Senataxin. Sci Rep. 2018;8:3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Cohen S, Puget N, Lin YL, et al. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat Commun. 2018;9:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Alzu A, Bermejo R, Begnis M, et al. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell. 2012;151:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Richard P, Feng S, Manley JL. A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev. 2013;27:2227–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bennett CL, Chen Y, Vignali M, et al. Protein interaction analysis of senataxin and the ALS4 L389S mutant yields insights into senataxin post-translational modification and uncovers mutant-specific binding with a brain cytoplasmic RNA-encoded peptide. PLoS One. 2013;8:e78837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Richard P, Manley JL. SETX sumoylation: A link between DNA damage and RNA surveillance disrupted in AOA2. Rare Dis. 2014;2:e27744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Grunseich C, Wang IX, Watts JA, et al. Senataxin mutation reveals how R-Loops promote transcription by blocking DNA methylation at gene promoters. Mol Cell. 2018;69:426–437 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Richard P, Manley JL. R Loops and links to human disease. J Mol Biol. 2017;429:3168–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Groh M, Gromak N. Out of balance: R-loops in human disease. PLoS Genet. 2014;10:e1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp Cell Res. 2004;296:51–56. [DOI] [PubMed] [Google Scholar]
- [32].Jangi M, Fleet C, Cullen P, et al. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc Natl Acad Sci USA. 2017;114:E2347–E2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhao DY, Gish G, Braunschweig U, et al. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature. 2016;529:48–53. [DOI] [PubMed] [Google Scholar]
- [34].Kannan A, Bhatia K, Branzei D, et al. Combined deficiency of senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018;46:8326–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Walker C, Herranz-Martin S, Karyka E, et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci. 2017;20:1225–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ho WY, Tai YK, Chang JC, et al. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy. 2019;15:827–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nguyen DKH, Thombre R, Wang J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci Lett. 2019;697:34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. [DOI] [PubMed] [Google Scholar]
- [39].Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109–122. [DOI] [PubMed] [Google Scholar]
- [40].Kesidou E, Lagoudaki R, Touloumi O, et al. Autophagy and neurodegenerative disorders. Neural Regen Res. 2013;8:2275–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. [DOI] [PubMed] [Google Scholar]
- [42].Fogel BL, Cho E, Wahnich A, et al. Mutation of senataxin alters disease-specific transcriptional networks in patients with ataxia with oculomotor apraxia type 2. Hum Mol Genet. 2014;23:4758–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hoque M, Ji Z, Zheng D, et al. Analysis of alternative cleavage and polyadenylation by 3ʹ region extraction and deep sequencing. Nat Methods. 2013;10:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vantaggiato C, Bondioni S, Airoldi G, et al. Senataxin modulates neurite growth through fibroblast growth factor 8 signalling. Brain. 2011;134:1808–1828. [DOI] [PubMed] [Google Scholar]
- [45].Boguslawski SJ, Smith DE, Michalak MA, et al. Characterization of monoclonal antibody to DNA.RNA and its application to immunodetection of hybrids. J Immunol Methods. 1986;89:123–130. [DOI] [PubMed] [Google Scholar]
- [46].Ginno PA, Lott PL, Christensen HC, et al. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell. 2012;45:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stolz R, Sulthana S, Hartono SR, et al. Interplay between DNA sequence and negative superhelicity drives R-loop structures. Proc Natl Acad Sci USA. 2019;116:6260–6269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by mTORC1. Embo J. 2011;30:3242–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. [DOI] [PubMed] [Google Scholar]
- [51].Marshansky V, Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pyo JO, Nah J, Jung YK. Molecules and their functions in autophagy. Exp Mol Med. 2012;44:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. [DOI] [PubMed] [Google Scholar]
- [56].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. [DOI] [PubMed] [Google Scholar]
- [58].Polson HE, de Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. [DOI] [PubMed] [Google Scholar]
- [59].Aguzzi A, O’Connor T. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 2010;9:237–248. [DOI] [PubMed] [Google Scholar]
- [60].Yamamoto A, Cremona ML, Rothman JE. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J Cell Biol. 2006;172:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. [DOI] [PubMed] [Google Scholar]
- [62].Tolkovsky AM. Mitophagy. Biochim Biophys Acta. 2009;1793:1508–1515. [DOI] [PubMed] [Google Scholar]
- [63].Cozzolino M, Ferri A, Valle C, et al. Mitochondria and ALS: implications from novel genes and pathways. Mol Cell Neurosci. 2013;55:44–49. [DOI] [PubMed] [Google Scholar]
- [64].Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. [DOI] [PubMed] [Google Scholar]
- [65].Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016;215:857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang H, Sun HQ, Zhu X, et al. GABARAPs regulate PI4P-dependent autophagosome: lysosomefusion. Proc Natl Acad Sci USA. 2015;112:7015–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Sahni S, Merlot AM, Krishan S, et al. Gene of the month: BECN1. J Clin Pathol. 2014;67:656–660. [DOI] [PubMed] [Google Scholar]
- [68].Li W, You B, Hoque M, et al. Systematic profiling of poly(A)+ transcripts modulated by core 3ʹ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015;11:e1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu X, Freitas J, Zheng D, et al. Transcription elongation rate has a tissue-specific impact on alternative cleavage and polyadenylation in Drosophila melanogaster. RNA. 2017;23:1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vanoosthuyse V. Strengths and weaknesses of the current strategies to map and characterize R-Loops. Noncoding RNA. 2018;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hartono SR, Malapert A, Legros P, et al. The affinity of the S9.6 antibody for double-stranded RNAs impacts the accurate mapping of R-loops in fission yeast. J Mol Biol. 2018;430:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Sariki SK, Sahu PK, Golla U, et al. Sen1, the homolog of human Senataxin, is critical for cell survival through regulation of redox homeostasis, mitochondrial function, and the TOR pathway in Saccharomyces cerevisiae. Febs J. 2016;283:4056–4083. [DOI] [PubMed] [Google Scholar]
- [73].Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nishiyama J, Miura E, Mizushima N, et al. Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy. 2007;3:591–596. [DOI] [PubMed] [Google Scholar]
- [75].Komatsu M, Wang QJ, Holstein GR, et al. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA. 2007;104:14489–14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. [DOI] [PubMed] [Google Scholar]
- [77].Chakrabarti L, Eng J, Ivanov N, et al. Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain. 2009;2:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci USA. 1976;73:208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang T, Morgan JI. The Purkinje cell degeneration (pcd) mouse: an unexpected molecular link between neuronal degeneration and regeneration. Brain Res. 2007;1140:26–40. [DOI] [PubMed] [Google Scholar]
- [80].Sakai Y, Kassai H, Nakayama H, et al. Hyperactivation of mTORC1 disrupts cellular homeostasis in cerebellar Purkinje cells. Sci Rep. 2019;9:2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Murray ME, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122:673–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Al-Sarraj S, King A, Troakes C, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. [DOI] [PubMed] [Google Scholar]
- [83].Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446. [DOI] [PubMed] [Google Scholar]
- [84].Rubino E, Rainero I, Chio A, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. [DOI] [PubMed] [Google Scholar]
- [86].Gendron TF, Bieniek KF, Zhang YJ, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126:829–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Zu T, Liu Y, Banez-Coronel M, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110:E4968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Barmada SJ, Serio A, Arjun A, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol. 2014;10:677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Xia Q, Wang H, Hao Z, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. Embo J. 2016;35:121–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Kwiatkowski TJ Jr., Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- [92].Ling SC, Dastidar SG, Tokunaga S, et al. Overriding FUS autoregulation in mice triggers gain-of-toxic dysfunctions in RNA metabolism and autophagy-lysosome axis. eLife. 2019;8. DOI: 10.7554/eLife.40811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Pierzynowska K, Gaffke L, Cyske Z, et al. Autophagy stimulation as a promising approach in treatment of neurodegenerative diseases. Metab Brain Dis. 2018;33:989–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zhang X, Li L, Chen S, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:412–425. [DOI] [PubMed] [Google Scholar]
- [95].Staats KA, Hernandez S, Schonefeldt S, et al. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol Neurodegener. 2013;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Fornai F, Longone P, Cafaro L, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2008;105:2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Pizzasegola C, Caron I, Daleno C, et al. Treatment with lithium carbonate does not improve disease progression in two different strains of SOD1 mutant mice. Amyotroph Lateral Scler. 2009;10:221–228. [DOI] [PubMed] [Google Scholar]
- [98].Ching JK, Elizabeth SV, Ju JS, et al. mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum Mol Genet. 2013;22:1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. [DOI] [PubMed] [Google Scholar]
- [100].Shi Y, Di Giammartino DC, Taylor D, et al. Molecular architecture of the human pre-mRNA 3ʹ processing complex. Mol Cell. 2009;33:365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Di Giammartino DC, Li W, Ogami K, et al. RBBP6 isoforms regulate the human polyadenylation machinery and modulate expression of mRNAs with AU-rich 3ʹ UTRs. Genes Dev. 2014;28:2248–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Richard P, Ogami K, Chen Y, et al. NRDE-2, the human homolog of fission yeast Nrl1, prevents DNA damage accumlation in human cells. RNA Biol. 2018;15:868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Valente AJ, Maddalena LA, Robb EL, et al. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017;119:315–326. [DOI] [PubMed] [Google Scholar]
- [106].Maury Y, Come J, Piskorowski RA, et al. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol. 2015;33:89–96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.